
UNITED NATIONS ENVIRONMENT PROGRAMME
INTERNATIONAL LABOUR ORGANISATION
WORLD HEALTH ORGANIZATION
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
Environmental Health Criteria 216
DISINFECTANTS AND DISINFECTANT BY-PRODUCTS
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation or the World Health Organization.
First draft prepared by G. Amy, University of Colorado, Boulder,
Colorado, USA; R. Bull, Battelle Pacific Northwest Laboratory,
Richland, Washington, USA; G.F. Craun, Gunther F. Craun and
Associates, Staunton, Virginia, USA; R.A. Pegram, US Environmental
Protection Agency, Research Triangle Park, North Carolina, USA; and M.
Siddiqui, University of Colorado, Boulder, Colorado, USA
Published under the joint sponsorship of the United Nations
Environment Programme, the International Labour Organisation and the
World Health Organization, and produced within the framework of the
Inter-Organization Programme for the Sound Management of Chemicals.
World Health Organization
Geneva, 2000
The International Programme on Chemical Safety (IPCS),
established in 1980, is a joint venture of the United Nations
Environment Programme (UNEP), the International Labour Organisation
(ILO) and the World Health Organization (WHO). The overall objectives
of the IPCS are to establish the scientific basis for assessment of
the risk to human health and the environment from exposure to
chemicals, through international peer review processes, as a
prerequisite for the promotion of chemical safety, and to provide
technical assistance in strengthening national capacities for the
sound management of chemicals.
The Inter-Organization Programme for the Sound Management of
Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and
Agriculture Organization of the United Nations, WHO, the United
Nations Industrial Development Organization and the Organisation for
Economic Co-operation and Development (Participating Organizations),
following recommendations made by the 1992 UN Conference on
Environment and Development to strengthen cooperation and increase
coordination in the field of chemical safety. The purpose of the IOMC
is to promote coordination of the policies and activities pursued by
the Participating Organizations, jointly or separately, to achieve the
sound management of chemicals in relation to human health and the
environment.
WHO Library Cataloguing-in-Publication Data
Disinfectants and disinfectant by-products.
(Environmental health criteria ; 216)
1.Disinfectants - chemistry 2.Disinfectants - toxicity
3.Drinking water 4.Risk assessment
5.Epidemiologic studies I.Series
ISBN 92 4 157216 7 (NLM Classification: QV 220)
ISSN 0250-863X
The World Health Organization welcomes requests for permission to
reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made to
the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 2000
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material in
this publication do not imply the expression of any opinion whatsoever
on the part of the Secretariat of the World Health Organization
concerning the legal status of any country, territory, city or area or
of its authorities, or concerning the delimitation of its frontiers or
boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar nature
that are not mentioned. Errors and omissions excepted, the names of
proprietary products are distinguished by initial capital letters.
CONTENTS
ENVIRONMENTAL HEALTH CRITERIA FOR DISINFECTANTS AND DISINFECTANT
BY-PRODUCTS
PREAMBLE
ACRONYMS AND ABBREVIATIONS
1. SUMMARY AND EVALUATION
1.1. Chemistry of disinfectants and disinfectant by-products
1.2. Kinetics and metabolism in laboratory animals and humans
1.2.1. Disinfectants
1.2.2. Trihalomethanes
1.2.3. Haloacetic acids
1.2.4. Haloaldehydes and haloketones
1.2.5. Haloacetonitriles
1.2.6. Halogenated hydroxyfuranone derivatives
1.2.7. Chlorite
1.2.8. Chlorate
1.2.9. Bromate
1.3. Toxicology of disinfectants and disinfectant by-products
1.3.1. Disinfectants
1.3.2. Trihalomethanes
1.3.3. Haloacetic acids
1.3.4. Haloaldehydes and haloketones
1.3.5. Haloacetonitriles
1.3.6. Halogenated hydroxyfuranone derivatives
1.3.7. Chlorite
1.3.8. Chlorate
1.3.9. Bromate
1.4. Epidemiological studies
1.4.1. Cardiovascular disease
1.4.2. Cancer
1.4.3. Adverse pregnancy outcomes
1.5. Risk characterization
1.5.1. Characterization of hazard and dose-response
1.5.1.1 Toxicological studies
1.5.1.2 Epidemiological studies
1.5.2. Characterization of exposure
1.5.2.1 Occurrence of disinfectants and disinfectant
by-products
1.5.2.2 Uncertainties of water quality data
1.5.2.3 Uncertainties of epidemiological data
2. CHEMISTRY OF DISINFECTANTS AND DISINFECTANT BY-PRODUCTS
2.1. Background
2.2. Physical and chemical properties of common disinfectants and
inorganic disinfectant by-products
2.2.1. Chlorine
2.2.2. Chlorine dioxide
2.2.3. Ozone
2.2.4. Chloramines
2.3. Analytical methods for disinfectant by-products and
disinfectants
2.3.1. Trihalomethanes, haloacetonitriles, chloral hydrate,
chloropicrin and haloacetic acids
2.3.2. Inorganic disinfectant by-products
2.3.3. Total organic carbon and UV absorbance at 254 nm
2.3.4. Chloramines
2.4. Mechanisms involved in the formation of disinfectant
by-products
2.4.1. Chlorine reactions
2.4.2. Chlorine dioxide reactions
2.4.3. Chloramine reactions
2.4.4. Ozone reactions
2.5. Formation of organohalogen disinfectant by-products
2.5.1. Chlorine organohalogen by-products
2.5.2. Chloramine organohalogen by-products
2.5.3. Chlorine dioxide organohalogen by-products
2.5.4. Ozone organohalogen by-products
2.6. Formation of inorganic disinfectant by-products
2.6.1. Chlorine inorganic by-products
2.6.2. Chloramine inorganic by-products
2.6.3. Chlorine dioxide inorganic by-products
2.6.4. Ozone inorganic by-products
2.7. Formation of non-halogenated organic disinfectant
by-products
2.7.1. Chlorine organic by-products
2.7.2. Chloramine organic by-products
2.7.3. Chlorine dioxide organic by-products
2.7.4. Ozone organic by-products
2.8. Influence of source water characteristics on the amount and
type of by-products produced
2.8.1. Effect of natural organic matter and UV absorbance
at 254 nm
2.8.2. Effect of pH
2.8.3. Effect of bromide
2.8.4. Effect of reaction rates
2.8.5. Effect of temperature
2.8.6. Effect of alkalinity
2.9. Influence of water treatment variables on the amount and
type of by-products produced
2.9.1. Effect of ammonia
2.9.2. Effect of disinfectant dose
2.9.3. Effect of advanced oxidation processes
2.9.4. Effect of chemical coagulation
2.9.5. Effect of pre-ozonation
2.9.6. Effect of biofiltration
2.10. Comparative assessment of disinfectants
2.11. Alternative strategies for disinfectant by-product control
2.11.1. Source control
2.11.2. Organohalogen by-products
2.11.3. Inorganic by-products
2.11.4. Organic by-products
2.12. Models for predicting disinfectant by-product formation
2.12.1. Factors affecting disinfectant by-product formation
and variables of interest in disinfectant by-product
modelling
2.12.2. Empirical models for disinfectant by-product
formation
2.12.3. Models for predicting disinfectant by-product
precursor removal
2.13. Summary
3. TOXICOLOGY OF DISINFECTANTS
3.1. Chlorine and hypochlorite
3.1.1. General toxicological properties and information on
dose-response in animals
3.1.2. Reproductive and developmental toxicity
3.1.3. Toxicity in humans
3.1.4. Carcinogenicity and mutagenicity
3.1.5. Comparative pharmacokinetics and metabolism
3.1.6. Mode of action
3.2. Chloramine
3.2.1. General toxicological properties and information on
dose-response in animals
3.2.2. Reproductive and developmental toxicity
3.2.3. Toxicity in humans
3.2.4. Carcinogenicity and mutagenicity
3.2.5. Comparative pharmacokinetics and metabolism
3.3. Chlorine dioxide
3.3.1. General toxicological properties and information on
dose-response in animals
3.3.2. Reproductive and developmental toxicity
3.3.3. Toxicity in humans
3.3.4. Carcinogenicity and mutagenicity
3.3.5. Comparative pharmacokinetics and metabolism
4. TOXICOLOGY OF DISINFECTANT BY-PRODUCTS
4.1. Trihalomethanes
4.1.1. Chloroform
4.1.1.1 General toxicological properties and
information on dose-response in animals
4.1.1.2 Toxicity in humans
4.1.1.3 Carcinogenicity and mutagenicity
4.1.1.4 Comparative pharmacokinetics and metabolism
4.1.1.5 Mode of action
4.1.2. Bromodichloromethane
4.1.2.1 General toxicological properties and
information on dose-response in animals
4.1.2.2 Reproductive and developmental toxicity
4.1.2.3 Neurotoxicity
4.1.2.4 Toxicity in humans
4.1.2.5 Carcinogenicity and mutagenicity
4.1.2.6 Comparative phamacokinetics and metabolism
4.1.2.7 Mode of action
4.1.3. Dibromochloromethane
4.1.3.1 General toxicological properties and
information on dose-response in animals
4.1.3.2 Reproductive and developmental toxicity
4.1.3.3 Neurotoxicity
4.1.3.4 Toxicity in humans
4.1.3.5 Carcinogenicity and mutagenicity
4.1.3.6 Comparative pharmacokinetics and metabolism
4.1.3.7 Mode of action
4.1.4. Bromoform
4.1.4.1 General toxicological properties and
information on dose-response in animals
4.1.4.2 Reproductive and developmental toxicity
4.1.4.3 Neurotoxicity
4.1.4.4 Toxicity in humans
4.1.4.5 Carcinogenicity and mutagenicity
4.1.4.6 Comparative pharmacokinetics and metabolism
4.1.4.7 Mode of action
4.2. Haloacids
4.2.1. Dichloroacetic acid (dichloroacetate)
4.2.1.1 General toxicological properties and
information on dose-response in animals
4.2.1.2 Reproductive effects
4.2.1.3 Developmental effects
4.2.1.4 Neurotoxicity
4.2.1.5 Toxicity in humans
4.2.1.6 Carcinogenicity and mutagenicity
4.2.1.7 Comparative pharmacokinetics and metabolism
4.2.1.8 Mode of action
4.2.2. Trichloroacetic acid (trichloroacetate)
4.2.2.1 General toxicological properties and
information on dose-response in animals
4.2.2.2 Reproductive effects
4.2.2.3 Developmental effects
4.2.2.4 Neurotoxicity
4.2.2.5 Toxicity in humans
4.2.2.6 Carcinogenicity and mutagenicity
4.2.2.7 Comparative pharmacokinetics and metabolism
4.2.2.8 Mode of action
4.2.3. Brominated haloacetic acids
4.2.3.1 General toxicological properties and
information on dose-response in animals
4.2.3.2 Reproductive effects
4.2.3.3 Neurotoxicity
4.2.3.4 Toxicity in humans
4.2.3.5 Carcinogenicity and mutagenicity
4.2.3.6 Comparative pharmacokinetics and metabolism
4.2.3.7 Mode of action
4.2.4. Higher molecular weight halogenated acids
4.3. Haloaldehydes and haloketones
4.3.1. Chloral hydrate (trichloroacetaldehyde, chloral)
4.3.1.1 General toxicological properties and
information on dose-response in animals
4.3.1.2 Toxicity in humans
4.3.1.3 Carcinogenicity and mutagenicity
4.3.1.4 Comparative metabolism and pharmacokinetics
4.3.1.5 Mode of action
4.3.2. Halogenated aldehydes and ketones other than chloral
hydrate
4.3.2.1 General toxicological properties and
information on dose-response in animals
4.3.2.2 Toxicity in humans
4.3.2.3 Carcinogenicity and mutagenicity
4.3.2.4 Comparative pharmacokinetics and metabolism
4.3.2.5 Mode of action
4.4. Haloacetonitriles
4.4.1. General toxicological properties and information on
dose-response in animals and humans
4.4.2. Reproductive and developmental toxicity
4.4.3. Carcinogenicity and mutagenicity
4.4.4. Comparative pharmacokinetics and metabolism
4.4.5. Mode of action
4.5. Halogenated hydroxyfuranone derivatives
4.5.1. General toxicological properties and information on
dose-response in animals
4.5.2. Toxicity in humans
4.5.3. Carcinogenicity and mutagenicity
4.5.3.1 Studies in bacteria and mammalian cells
in vitro
4.5.3.2 Studies in experimental animals
4.5.4. Comparative pharmacokinetics and metabolism
4.6. Chlorite
4.6.1. General toxicological properties and information on
dose-response in animals
4.6.2. Reproductive and developmental toxicity
4.6.3. Toxicity in humans
4.6.4. Carcinogenicity and mutagenicity
4.6.5. Comparative pharmacokinetics and metabolism
4.6.6. Mode of action
4.7. Chlorate
4.7.1. General toxicological properties and information on
dose-response in animals
4.7.2. Reproductive and developmental toxicity
4.7.3. Toxicity in humans
4.7.4. Carcinogenicity and mutagenicity
4.7.5. Mode of action
4.8. Bromate
4.8.1. General toxicological properties and information on
dose-response in animals
4.8.2. Toxicity in humans
4.8.3. Carcinogenicity and mutagenicity
4.8.4. Comparative pharmacokinetics and metabolism
4.8.5. Mode of action
4.9. Other disinfectant by-products
5. EPIDEMIOLOGICAL STUDIES
5.1. Epidemiological study designs and causality of
epidemiological associations
5.1.1. Experimental studies
5.1.2. Observational studies
5.1.3. Random and systematic error
5.1.4. Causality of an epidemiological association
5.2. Epidemiological associations between disinfectant
use and adverse health outcomes
5.2.1. Epidemiological studies of cancer and disinfected
drinking-water
5.2.1.1 Cancer associations in ecological studies
5.2.1.2 Cancer associations in analytical studies
5.2.1.3 Meta-analysis of cancer studies
5.2.1.4 Summary of results of cancer studies
5.2.2. Epidemiological studies of cardiovascular disease and
disinfected drinking-water
5.2.2.1 Summary of results of cardiovascular studies
5.2.3. Epidemiological studies of adverse
reproductive/developmental outcomes and disinfected
drinking-water
5.2.3.1 Summary of results of
reproductive/developmental studies
5.3. Epidemiological associations between disinfectant
by-products and adverse health outcomes
5.3.1. Epidemiological studies of cancer and disinfectant
by-products
5.3.1.1 Cancer associations in ecological studies
5.3.1.2 Cancer associations in analytical studies
5.3.1.3 Summary of results of cancer studies
5.3.2. Epidemiological studies of cardiovascular disease and
disinfectant by-products
5.3.2.1 Summary of results of cardiovascular studies
5.3.3. Epidemiological studies of adverse
reproductive/developmental outcomes and disinfectant
by-products
5.3.3.1 Summary of results of
reproductive/developmental studies
5.4. Summary
6. RISK CHARACTERIZATION
6.1. Characterization of hazard and dose-response
6.1.1. Toxicological studies
6.1.1.1 Chlorine
6.1.1.2 Monochloramine
6.1.1.3 Chlorine dioxide
6.1.1.4 Trihalomethanes
6.1.1.5 Haloacetic acids
6.1.1.6 Chlorate hydrate
6.1.1.7 Haloacetonitriles
6.1.1.8 MX
6.1.1.9 Chlorite
6.1.1.10 Chlorate
6.1.1.11 Bromate
6.1.2. Epidemiological studies
6.2. Characterization of exposure
6.2.1. Occurrence of disinfectants and disinfectant
by-products
6.2.2. Uncertainties of water quality data
6.2.3. Uncertainties of epidemiological data
7. RISK CONCLUSIONS AND COMPARISONS
7.1. Epidemiological studies
7.2. Toxicological studies
7.2.1. Diversity of by-products
7.2.2. Diversity of modes of action
7.2.3. Reproductive, developmental and neurotoxic effects
7.3. Risks associated with mixtures of disinfectant by-products
8. CONCLUSIONS AND RECOMMENDATIONS
8.1. Chemistry
8.2. Toxicology
8.3. Epidemiology
9. RESEARCH NEEDS
9.1. Chemistry of disinfectants and disinfectant by-products
9.2. Toxicology
9.3. Epidemiology
PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
REFERENCES
RESUME ET EVALUATION
RESUMEN Y EVALUACION
NOTE TO READERS OF THE CRITERIA MONOGRAPHS
Every effort has been made to present information in the criteria
monographs as accurately as possible without unduly delaying their
publication. In the interest of all users of the Environmental Health
Criteria monographs, readers are requested to communicate any errors
that may have occurred to the Director of the International Programme
on Chemical Safety, World Health Organization, Geneva, Switzerland, in
order that they may be included in corrigenda.
* * *
A detailed data profile and a legal file can be obtained from the
International Register of Potentially Toxic Chemicals, Case postale
356, 1219 Châtelaine, Geneva, Switzerland (telephone no. + 41 22 -
9799111, fax no. + 41 22 - 7973460, E-mail irptc@unep.ch).
Environmental Health Criteria
PREAMBLE
Objectives
In 1973, the WHO Environmental Health Criteria Programme was
initiated with the following objectives:
(i) to assess information on the relationship between exposure to
environmental pollutants and human health, and to provide
guidelines for setting exposure limits;
(ii) to identify new or potential pollutants;
(iii) to identify gaps in knowledge concerning the health effects of
pollutants;
(iv) to promote the harmonization of toxicological and
epidemiological methods in order to have internationally
comparable results.
The first Environmental Health Criteria (EHC) monograph, on
mercury, was published in 1976, and since that time an ever-increasing
number of assessments of chemicals and of physical effects have been
produced. In addition, many EHC monographs have been devoted to
evaluating toxicological methodology, e.g., for genetic, neurotoxic,
teratogenic and nephrotoxic effects. Other publications have been
concerned with epidemiological guidelines, evaluation of short-term
tests for carcinogens, biomarkers, effects on the elderly and so
forth.
Since its inauguration, the EHC Programme has widened its scope,
and the importance of environmental effects, in addition to health
effects, has been increasingly emphasized in the total evaluation of
chemicals.
The original impetus for the Programme came from World Health
Assembly resolutions and the recommendations of the 1972 UN Conference
on the Human Environment. Subsequently, the work became an integral
part of the International Programme on Chemical Safety (IPCS), a
cooperative programme of UNEP, ILO and WHO. In this manner, with the
strong support of the new partners, the importance of occupational
health and environmental effects was fully recognized. The EHC
monographs have become widely established, used and recognized
throughout the world.
The recommendations of the 1992 UN Conference on Environment and
Development and the subsequent establishment of the Intergovernmental
Forum on Chemical Safety with the priorities for action in the six
programme areas of Chapter 19, Agenda 21, all lend further weight to
the need for EHC assessments of the risks of chemicals.
Scope
The criteria monographs are intended to provide critical reviews
on the effects on human health and the environment of chemicals and of
combinations of chemicals and physical and biological agents. As such,
they include and review studies that are of direct relevance for the
evaluation. However, they do not describe every study carried out.
Worldwide data are used and are quoted from original studies, not from
abstracts or reviews. Both published and unpublished reports are
considered, and it is incumbent on the authors to assess all the
articles cited in the references. Preference is always given to
published data. Unpublished data are used only when relevant published
data are absent or when they are pivotal to the risk assessment. A
detailed policy statement is available that describes the procedures
used for unpublished proprietary data so that this information can be
used in the evaluation without compromising its confidential nature
(WHO (1990) Revised Guidelines for the Preparation of Environmental
Health Criteria Monographs. PCS/90.69, Geneva, World Health
Organization).
In the evaluation of human health risks, sound human data,
whenever available, are preferred to animal data. Animal and in
vitro studies provide support and are used mainly to supply evidence
missing from human studies. It is mandatory that research on human
subjects is conducted in full accord with ethical principles,
including the provisions of the Helsinki Declaration.
The EHC monographs are intended to assist national and
international authorities in making risk assessments and subsequent
risk management decisions. They represent a thorough evaluation of
risks and are not, in any sense, recommendations for regulation or
standard setting. These latter are the exclusive purview of national
and regional governments.
Content
The layout of EHC monographs for chemicals is outlined below.
* Summary -- a review of the salient facts and the risk evaluation
of the chemical
* Identity -- physical and chemical properties, analytical methods
* Sources of exposure
* Environmental transport, distribution and transformation
* Environmental levels and human exposure
* Kinetics and metabolism in laboratory animals and humans
* Effects on laboratory mammals and in vitro test systems
* Effects on humans
* Effects on other organisms in the laboratory and field
* Evaluation of human health risks and effects on the environment
* Conclusions and recommendations for protection of human health
and the environment
* Further research
* Previous evaluations by international bodies, e.g., IARC, JECFA,
JMPR
Selection of chemicals
Since the inception of the EHC Programme, the IPCS has organized
meetings of scientists to establish lists of priority chemicals for
subsequent evaluation. Such meetings have been held in: Ispra, Italy,
1980; Oxford, United Kingdom, 1984; Berlin, Germany, 1987; and North
Carolina, USA, 1995. The selection of chemicals has been based on the
following criteria: the existence of scientific evidence that the
substance presents a hazard to human health and/or the environment;
the possible use, persistence, accumulation or degradation of the
substance shows that there may be significant human or environmental
exposure; the size and nature of populations at risk (both human and
other species) and risks for the environment; international concern,
i.e., the substance is of major interest to several countries;
adequate data on the hazards are available.
If an EHC monograph is proposed for a chemical not on the
priority list, the IPCS Secretariat consults with the cooperating
organizations and all the Participating Institutions before embarking
on the preparation of the monograph.
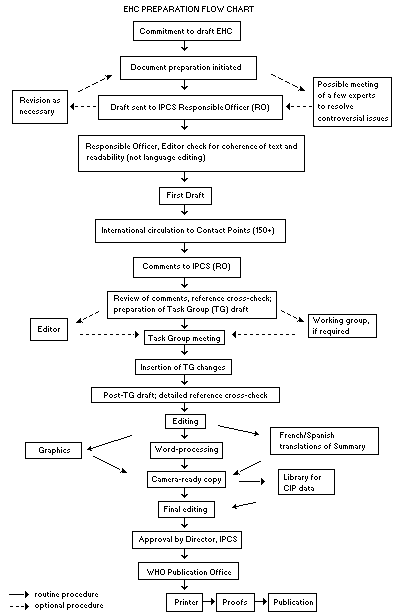
Procedures
The order of procedures that result in the publication of an EHC
monograph is shown in the flow chart. A designated staff member of
IPCS, responsible for the scientific quality of the document, serves
as Responsible Officer (RO). The IPCS Editor is responsible for layout
and language. The first draft, prepared by consultants or, more
usually, staff from an IPCS Participating Institution, is based
initially on data provided from the International Register of
Potentially Toxic Chemicals and from reference databases such as
Medline and Toxline.
The draft document, when received by the RO, may require an
initial review by a small panel of experts to determine its scientific
quality and objectivity. Once the RO finds the document acceptable as
a first draft, it is distributed, in its unedited form, to well over
150 EHC contact points throughout the world who are asked to comment
on its completeness and accuracy and, where necessary, provide
additional material. The contact points, usually designated by
governments, may be Participating Institutions, IPCS Focal Points or
individual scientists known for their particular expertise. Generally,
some four months are allowed before the comments are considered by the
RO and author(s). A second draft incorporating comments received and
approved by the Director, IPCS, is then distributed to Task Group
members, who carry out the peer review, at least six weeks before
their meeting.
The Task Group members serve as individual scientists, not as
representatives of any organization, government or industry. Their
function is to evaluate the accuracy, significance and relevance of
the information in the document and to assess the health and
environmental risks from exposure to the chemical. A summary and
recommendations for further research and improved safety aspects are
also required. The composition of the Task Group is dictated by the
range of expertise required for the subject of the meeting and by the
need for a balanced geographical distribution.
The three cooperating organizations of the IPCS recognize the
important role played by nongovernmental organizations.
Representatives from relevant national and international associations
may be invited to join the Task Group as observers. While observers
may provide a valuable contribution to the process, they can speak
only at the invitation of the Chairperson. Observers do not
participate in the final evaluation of the chemical; this is the sole
responsibility of the Task Group members. When the Task Group
considers it to be appropriate, it may meet in camera.
All individuals who as authors, consultants or advisers
participate in the preparation of the EHC monograph must, in addition
to serving in their personal capacity as scientists, inform the RO if
at any time a conflict of interest, whether actual or potential, could
be perceived in their work. They are required to sign a conflict of
interest statement. Such a procedure ensures the transparency and
probity of the process.
When the Task Group has completed its review and the RO is
satisfied as to the scientific correctness and completeness of the
document, the document then goes for language editing, reference
checking and preparation of camera-ready copy. After approval by the
Director, IPCS, the monograph is submitted to the WHO Office of
Publications for printing. At this time, a copy of the final draft is
sent to the Chairperson and Rapporteur of the Task Group to check for
any errors.
It is accepted that the following criteria should initiate the
updating of an EHC monograph: new data are available that would
substantially change the evaluation; there is public concern for
health or environmental effects of the agent because of greater
exposure; an appreciable time period has elapsed since the last
evaluation.
All Participating Institutions are informed, through the EHC
progress report, of the authors and institutions proposed for the
drafting of the documents. A comprehensive file of all comments
received on drafts of each EHC monograph is maintained and is
available on request. The Chairpersons of Task Groups are briefed
before each meeting on their role and responsibility in ensuring that
these rules are followed.
 WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR DISINFECTANTS AND
DISINFECTANT BY-PRODUCTS
Members
Dr G. Amy, Department of Civil, Environmental, and Architectural
Engineering, University of Colorado, Boulder, Colorado, USA
Mr J. Fawell, Water Research Centre, Marlow, Buckinghamshire, United
Kingdom (Co-Rapporteur)
Dr B. Havlik, Ministry of Health, National Institute of Public Health,
Prague, Czech Republic
Dr C. Nokes, Water Group, Institute of Environmental Science and
Research, Christchurch, New Zealand (Co-Rapporteur)
Dr E. Ohanian, Office of Water/Office of Science and Technology,
United States Environmental Protection Agency, Washington, DC,
USA (Chairman)
Dr E. Soderlund, Department of Environmental Medicine, National
Institute of Public Health, Torshov, Oslo
Secretariat
Dr J. Bartram, Water, Sanitation and Health Unit, Division of
Operational Support in Environment Health, World Health
Organization, Geneva, Switzerland
Dr R. Bull, Battelle Pacific Northwest Laboratory, Richland,
Washington, USA
Mr G.F. Craun, Gunther F. Craun and Associates, Staunton, Virginia,
USA
Dr H. Galal-Gorchev, Chevy Chase, Maryland, USA (Secretary)
Mr N. Nakashima, Assessment of Risk and Methodologies,
International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland
Dr R.A. Pegram, United States Environmental Protection Agency,
Research Triangle Park, North Carolina, USA
Mr S.T. Yamamura, Water, Sanitation and Health Unit, Division of
Operational Support in Environment Health, World Health
Organization, Geneva, Switzerland
Representatives/Observers
Dr N. Drouot, Dept Toxicologie Industrielle, Paris, France
(representing European Centre for Ecotoxicology and Toxicology
of Chemicals)
Mr O. Hydes, Drinking Water Inspectorate, London, United Kingdom
Dr B.B. Sandel, Olin Corporation, Norwalk, Connecticut, USA
(representing American Industrial Health Council/International
Life Sciences Institute)
IPCS TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR DISINFECTANTS AND
DISINFECTANT BY-PRODUCTS
A WHO Task Group on Environmental Health Criteria for
Disinfectants and Disinfectant By-products met in Geneva from 17 to 21
August 1998. Dr Peter Toft, Associate Director, IPCS, welcomed the
participants on behalf of the three IPCS cooperating organizations
(UNEP/ILO/WHO). The Task Group reviewed and revised the draft document
and made an evaluation of risks for human health from exposure to
certain disinfectants and disinfectant by-products.
The first draft of the chemistry section was prepared by G. Amy
and M. Siddiqui, University of Colorado, Boulder, Colorado, USA; the
toxicology section was prepared by R. Bull, Battelle Pacific Northwest
Laboratory, Richland, Washington, USA, and R.A. Pegram, US
Environmental Protection Agency, Research Triangle Park, North
Carolina, USA; and the epidemiology section was prepared by G.F.
Craun, Gunther F. Craun and Associates, Staunton, Virginia, USA.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
* * *
The preparation of the first draft of this Environmental Health
Criteria monograph was made possible by the financial support afforded
to IPCS by the International Life Sciences Institute.
A financial contribution from the United States Environmental
Protection Agency for the convening of the Task Group is gratefully
acknowledged.
ACRONYMS AND ABBREVIATIONS
ALAT alanine aminotransferase
AP alkaline phosphatase
ARB atypical residual bodies
ASAT aspartate aminotransferase
AWWA American Water Works Association
BAN bromoacetonitrile
BCA bromochloroacetic acid/bromochloroacetate
BCAN bromochloroacetonitrile
BDCA bromodichloroacetic acid/bromodichloroacetate
BDCM bromodichloromethane
BUN blood urea nitrogen
bw body weight
CAN chloroacetonitrile
CHO Chinese hamster ovary
CI confidence interval
CoA coenzyme A
Cmax maximum concentration
CMCF 3-chloro-4-(chloromethyl)-5-hydroxy-2(5H)-furanone
2-CP 2-chloropropionate
CPN chloropropanone
CT computerized tomography
CYP cytochrome P450
DBA dibromoacetic acid/dibromoacetate
DBAC dibromoacetone
DBAN dibromoacetonitrile
DBCM dibromochloromethane
DBP disinfectant by-product
DCA dichloroacetic acid/dichloroacetate
DCAN dichloroacetonitrile
DCPN dichloropropanone
DHAN dihaloacetonitrile
DOC dissolved organic carbon
ECD electron capture detector
ECG electrocardiogram
EEG electroencephalogram
EHEN N-ethyl- N-hydroxyethylnitrosamine
EPA Environmental Protection Agency (USA)
ESR electron spin resonance
FAO Food and Agriculture Organization of the United Nations
GAC granular activated carbon
GC gas chromatography
GGT gamma-glutamyl transpeptidase
GOT glutamate-oxalate transaminase
GPT glutamate-pyruvate transaminase
GSH glutathione-SH
GST glutathione- S-transferase
HAA haloacetic acid
HAN haloacetonitrile
HDL high-density lipoprotein
HPLC high-performance liquid chromatography
hprt hypoxanthine phosphoribosyl transferase
IARC International Agency for Research on Cancer
IC ion chromatography
i.p. intraperitoneal
IPCS International Programme on Chemical Safety
JECFA Joint FAO/WHO Expert Committee on Food Additives
LD50 median lethal dose
LDH lactate dehydrogenase
LDL low-density lipoprotein
LOAEL lowest-observed-adverse-effect level
MA 3,4-(dichloro)-5-hydroxy-2(5H)-furanone
MBA monobromoacetic acid/monobromoacetate
MCA monochloroacetic acid/monochloroacetate
MNU methylnitrosourea
MOR mortality odds ratio
MRI magnetic resonance imaging
MTBE methyl tert-butyl ether
MX 3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone
NADP nicotinamide adenine dinucleotide phosphate
NOAEL no-observed-adverse-effect level
NOEL no-observed-effect level
NOM natural organic matter
NTP National Toxicology Program (USA)
8-OH-dG 8-hydroxy-2-deoxyguanosine
OR odds ratio
PAS periodic acid/Schiff's reagent
PBPK physiologically based pharmacokinetic model
PFBHA O-(2,3,4,5,6-pentafluorobenzyl)-hydroxylamine
p Ka log acid dissociation constant
PPAR peroxisome proliferator activated receptor
PPRE peroxisome proliferator responsive element
RR relative risk
SCE sister chromatid exchange
SD standard deviation
SDH sorbitol dehydrogenase
SE standard error
SGOT serum glutamate-oxaloacetate transaminase
SGPT serum glutamate-pyruvate transaminase
SMR standardized mortality ratio
SSB single strand breaks
TBA tribromoacetic acid/tribromoacetate
TBARS thiobarbituric acid reactive substances
TCA trichloroacetic acid/trichloroacetate
TCAN trichloroacetonitrile
TCPN trichloropropanone
TDI tolerable daily intake
TGF transforming growth factor
THM trihalomethane
TOC total organic carbon
TOX total organic halogen
TPA 12- O-tetradecanoylphorbol-13-acetate
UDS unscheduled DNA synthesis
UV ultraviolet
UVA254 UV absorbance at 254 nm
Vmax maximum rate of metabolism
WHO World Health Organization
1. SUMMARY AND EVALUATION
Chlorine (Cl2) has been widely used throughout the world as a
chemical disinfectant, serving as the principal barrier to microbial
contaminants in drinking-water. The noteworthy biocidal attributes of
chlorine have been somewhat offset by the formation of disinfectant
by-products (DBPs) of public health concern during the chlorination
process. As a consequence, alternative chemical disinfectants, such as
ozone (O3), chlorine dioxide (ClO2) and chloramines (NH2Cl,
monochloramine), are increasingly being used; however, each has been
shown to form its own set of DBPs. Although the microbiological
quality of drinking-water cannot be compromised, there is a need to
better understand the chemistry, toxicology and epidemiology of
chemical disinfectants and their associated DBPs in order to develop a
better understanding of the health risks (microbial and chemical)
associated with drinking-water and to seek a balance between microbial
and chemical risks. It is possible to decrease the chemical risk due
to DBPs without compromising microbiological quality.
1.1 Chemistry of disinfectants and disinfectant by-products
The most widely used chemical disinfectants are chlorine, ozone,
chlorine dioxide and chloramine. The physical and chemical properties
of disinfectants and DBPs can affect their behaviour in
drinking-water, as well as their toxicology and epidemiology. The
chemical disinfectants discussed here are all water-soluble oxidants,
which are produced either on-site (e.g., ozone) or off-site (e.g.,
chlorine). They are administered as a gas (e.g., ozone) or liquid
(e.g., hypochlorite) at typical doses of several milligrams per litre,
either alone or in combination. The DBPs discussed here are measurable
by gas or liquid chromatography and can be classified as organic or
inorganic, halogenated (chlorinated or brominated) or non-halogenated,
and volatile or non-volatile. Upon their formation, DBPs can be stable
or unstable (e.g., decomposition by hydrolysis).
DBPs are formed upon the reaction of chemical disinfectants with
DBP precursors. Natural organic matter (NOM), commonly measured by
total organic carbon (TOC), serves as the organic precursor, whereas
bromide ion (Br-) serves as the inorganic precursor. DBP formation is
influenced by water quality (e.g., TOC, bromide, pH, temperature,
ammonia, carbonate alkalinity) and treatment conditions (e.g.,
disinfectant dose, contact time, removal of NOM before the point of
disinfectant application, prior addition of disinfectant).
Chlorine in the form of hypochlorous acid/hypochlorite ion
(HOCl/OCl-) reacts with bromide ion, oxidizing it to hypobromous
acid/hypobromite ion (HOBr/OBr-). Hypochlorous acid (a more powerful
oxidant) and hypobromous acid (a more effective halogenating agent)
react collectively with NOM to form chlorine DBPs, including
trihalomethanes (THMs), haloacetic acids (HAAs), haloacetonitriles
(HANs), haloketones, chloral hydrate and chloropicrin. The dominance
of chlorine DBP groups generally decreases in the order of THMs, HAAs
and HANs. The relative amounts of TOC, bromide and chlorine will
affect the species distribution of THMs (four species: chloroform,
bromoform, bromodichloromethane [BDCM] and dibromochloromethane
[DBCM]), HAAs (up to nine chlorinated/brominated species) and HANs
(several chlorinated/brominated species). Generally, chlorinated THM,
HAA and HAN species dominate over brominated species, although the
opposite may be true in high-bromide waters. Although many specific
chlorine DBPs have been identified, a significant percentage of the
total organic halogens still remain unaccounted for. Another reaction
that occurs with chlorine is the formation of chlorate (ClO3-) in
concentrated hypochlorite solutions.
Ozone can directly or indirectly react with bromide to form
brominated ozone DBPs, including bromate ion (BrO3-). In the
presence of NOM, non-halogenated organic DBPs, such as aldehydes,
ketoacids and carboxylic acids, are formed during ozonation, with
aldehydes (e.g., formaldehyde) being dominant. If both NOM and bromide
are present, ozonation forms hypobromous acid, which, in turn, leads
to the formation of brominated organohalogen compounds (e.g.,
bromoform).
The major chlorine dioxide DBPs include chlorite (ClO2-) and
chlorate ions, with no direct formation of organohalogen DBPs. Unlike
the other disinfectants, the major chlorine dioxide DBPs are derived
from decomposition of the disinfectant as opposed to reaction with
precursors.
Use of chloramine as a secondary disinfectant generally leads to
the formation of cyanogen chloride (CNCl), a nitrogenous compound, and
significantly reduced levels of chlorine DBPs. A related issue is the
presence of nitrite (NO2-) in chloraminated distribution systems.
From the present knowledge of occurrence and health effects, the
DBPs of most interest are THMs, HAAs, bromate and chlorite.
The predominant chlorine DBP group has been shown to be THMs,
with chloroform and BDCM as the first and second most dominant THM
species. HAAs are the second predominant group, with dichloroacetic
acid (DCA) and trichloroacetic acid (TCA) being the first and second
most dominant species.
Conversion of bromide to bromate upon ozonation is affected by
NOM, pH and temperature, among other factors. Levels may range from
below detection (2 µg/litre) to several tens of micrograms per litre.
Chlorite levels are generally very predictable, ranging from about 50%
to 70% of the chlorine dioxide dose administered.
DBPs occur in complex mixtures that are a function of the
chemical disinfectant used, water quality conditions and treatment
conditions; other factors include the combination/sequential use of
multiple disinfectants/oxidants. Moreover, the composition of these
mixtures may change seasonally. Clearly, potential chemically related
health effects will be a function of exposure to DBP mixtures.
Other than chlorine DBPs (in particular THMs), there are very few
data on the occurrence of DBPs in finished water and distribution
systems. Based on laboratory databases, empirical models have been
developed to predict concentrations of THMs (total THMs and THM
species), HAAs (total HAAs and HAA species) and bromate. These models
can be used in performance assessment to predict the impact of
treatment changes and in exposure assessment to simulate missing or
past data (e.g., to predict concentrations of HAAs from THM data).
DBPs can be controlled through DBP precursor control and removal
or modified disinfection practice. Coagulation, granular activated
carbon, membrane filtration and ozone biofiltration can remove NOM.
Other than through the use of membranes, there is little opportunity
to effectively remove bromide. Source water protection and control
represent non-treatment alternatives to precursor control. Removal of
DBPs after formation is not viable for organic DBPs, whereas bromate
and chlorite can be removed by activated carbon or reducing agents. It
is expected that the optimized use of combinations of disinfectants,
functioning as primary and secondary disinfectants, can further
control DBPs. There is a trend towards combination/sequential use of
disinfectants; ozone is used exclusively as a primary disinfectant,
chloramines exclusively as a secondary disinfectant, and both chlorine
and chlorine dioxide in either role.
1.2 Kinetics and metabolism in laboratory animals and humans
1.2.1 Disinfectants
Residual disinfectants are reactive chemicals that will react
with organic compounds found in saliva and stomach content, resulting
in the formation of by-products. There are significant differences in
the pharmacokinetics of 36Cl depending on whether it is obtained from
chlorine, chloramine or chlorine dioxide.
1.2.2 Trihalomethanes
The THMs are absorbed, metabolized and eliminated rapidly by
mammals after oral or inhalation exposure. Following absorption, the
highest tissue concentrations are attained in the fat, liver and
kidneys. Half-lives generally range from 0.5 to 3 h, and the primary
route of elimination is via metabolism to carbon dioxide. Metabolic
activation to reactive intermediates is required for THM toxicity, and
the three brominated species are all metabolized more rapidly and to a
greater extent than chloroform. The predominant route of metabolism
for all the THMs is oxidation via cytochrome P450 (CYP) 2E1, leading
to the formation of dihalocarbonyls (i.e., phosgene and brominated
congeners), which can be hydrolysed to carbon dioxide or bind to
tissue macromolecules. Secondary metabolic pathways are reductive
dehalogenation via CYP2B1/2/2E1 (leading to free radical generation)
and glutathione (GSH) conjugation via glutathione- S-transferase
(GST) T1-1, which generates mutagenic intermediates. The brominated
THMs are much more likely than chloroform to proceed through the
secondary pathways, and GST-mediated conjugation of chloroform to GSH
can occur only at extremely high chloroform concentrations or doses.
1.2.3 Haloacetic acids
The kinetics and metabolism of the dihaloacetic and trihaloacetic
acids differ significantly. To the extent they are metabolized, the
principal reactions of the trihaloacetic acids occur in the microsomal
fraction, whereas more than 90% of the dihaloacetic acid metabolism,
principally by glutathione transferases, is observed in the cytosol.
TCA has a biological half-life in humans of 50 h. The half-lives of
the other trihaloacetic acids decrease significantly with bromine
substitution, and measurable amounts of the dihaloacetic acids can be
detected as products with brominated trihaloacetic acids. The
half-lives of the dihaloacetic acids are very short at low doses but
can be drastically increased as dose rates are increased.
1.2.4 Haloaldehydes and haloketones
Limited kinetic data are available for chloral hydrate. The two
major metabolites of chloral hydrate are trichloroethanol and TCA.
Trichloroethanol undergoes rapid glucuronidation, enterohepatic
circulation, hydrolysis and oxidation to TCA. Dechlorination of
trichloroethanol or chloral hydrate would lead to the formation of
DCA. DCA may then be further transformed to monochloroacetate (MCA),
glyoxalate, glycolate and oxalate, probably through a reactive
intermediate. No information was found on the other haloaldehydes and
haloketones.
1.2.5 Haloacetonitriles
The metabolism and kinetics of HANs have not been studied.
Qualitative data indicate that the products of metabolism include
cyanide, formaldehyde, formyl cyanide and formyl halides.
1.2.6 Halogenated hydroxyfuranone derivatives
3-Chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX) is the
member of the hydroxyfuranone class that has been most extensively
studied. From animal studies, it appears that the 14C label of MX is
rapidly absorbed from the gastrointestinal tract and reaches systemic
circulation. MX itself has not been measured in blood. The MX label is
largely excreted in urine and faeces, urine being the major route of
excretion. Very little of the initial radiolabel is retained in the
body after 5 days.
1.2.7 Chlorite
The 36Cl from chlorite is rapidly absorbed. Less than half the
dose is found in the urine as chloride, and a small proportion as
chlorite. A significant proportion probably enters the chloride pool
of the body, but a lack of analytical methods to characterize chlorite
in biological samples means that no detailed information is available.
1.2.8 Chlorate
Chlorate behaves similarly to chlorite. The same analytical
constraints apply.
1.2.9 Bromate
Bromate is rapidly absorbed and excreted, primarily in urine, as
bromide. Bromate is detected in urine at doses of 5 mg/kg of body
weight and above. Bromate concentrations in urine peak at about 1 h,
and bromate is not detectable in plasma after 2 h.
1.3 Toxicology of disinfectants and disinfectant by-products
1.3.1 Disinfectants
Chlorine gas, chloramine and chlorine dioxide are strong
respiratory irritants. Sodium hypochlorite (NaOCl) is also used as
bleach and is frequently involved in human poisoning. These exposures,
however, are not relevant to exposures in drinking-water. There have
been relatively few evaluations of the toxic effects of these
disinfectants in drinking-water in experimental animals or humans.
Evidence from these animal and human studies suggests that chlorine,
hypochlorite solutions, chloramine and chlorine dioxide themselves
probably do not contribute to the development of cancer or any toxic
effects. Attention has focused on the wide variety of by-products that
result from reactions of chlorine and other disinfectants with NOM,
which is found in virtually all water sources.
1.3.2 Trihalomethanes
THMs induce cytotoxicity in the liver and kidneys of rodents
exposed to doses of about 0.5 mmol/kg of body weight. The vehicle of
administration significantly affects the toxicity of the THMs. The
THMs have little reproductive and developmental toxicity, but BDCM has
been shown to reduce sperm motility in rats consuming 39 mg/kg of body
weight per day in drinking-water. Like chloroform, BDCM, when
administered in corn oil, induces cancer in the liver and kidneys
after lifetime exposures to high doses. Unlike chloroform and DBCM,
BDCM and bromoform induce tumours of the large intestine in rats
exposed by corn oil gavage. BDCM induces tumours at all three target
sites and at lower doses than the other THMs. Since the publication of
the 1994 WHO Environmental Health Criteria monograph on chloroform,
additional studies have added to the weight of evidence indicating
that chloroform is not a direct DNA-reactive mutagenic carcinogen. In
contrast, the brominated THMs appear to be weak mutagens, probably as
a result of GSH conjugation.
1.3.3 Haloacetic acids
The HAAs have diverse toxicological effects in laboratory
animals. Those HAAs of most concern have carcinogenic, reproductive
and developmental effects. Neurotoxic effects are significant at the
high doses of DCA that are used therapeutically. Carcinogenic effects
appear to be limited to the liver and to high doses. The bulk of the
evidence indicates that the tumorigenic effects of DCA and TCA depend
on modifying processes of cell division and cell death rather than
their very weak mutagenic activities. Oxidative stress is also a
feature of the toxicity of the brominated analogues within this class.
Both DCA and TCA cause cardiac malformations in rats at high doses.
1.3.4 Haloaldehydes and haloketones
Chloral hydrate induces hepatic necrosis in rats at doses equal
to or greater than 120 mg/kg of body weight per day. Its depressant
effect on the central nervous system in humans is probably related to
its metabolite trichloroethanol. Limited toxicity data are available
for the other halogenated aldehydes and ketones. Chloroacetaldehyde
exposure causes haematological effects in rats. Exposure of mice to
1,1-dichloropropanone (1,1-DCPN), but not 1,3-dichloropropanone
(1,3-DCPN), results in liver toxicity.
Chloral hydrate was negative in most but not all bacterial tests
for point mutations and in in vivo studies on chromosomal damage.
However, it has been shown that chloral hydrate may induce structural
chromosomal aberrations in vitro and in vivo. Chloral hydrate has
been reported to cause hepatic tumours in mice. It is not clear if it
is the parent compound or its metabolites that are involved in the
carcinogenic effect. The two chloral hydrate metabolites, TCA and DCA,
have induced hepatic tumours in mice.
Some halogenated aldehydes and ketones are potent inducers of
mutations in bacteria. Clastogenic effects have been reported for
chlorinated propanones. Liver tumours were noted in a lifetime
drinking-water study with chloroacetaldehyde. Other halogenated
aldehydes, e.g., 2-chloropropenal, have been identified as tumour
initiators in the skin of mice. The haloketones have not been tested
for carcinogenicity in drinking-water. However, 1,3-DCPN acted as a
tumour initiator in a skin carcinogenicity study in mice.
1.3.5 Haloacetonitriles
Testing of these compounds for toxicological effects has been
limited to date. Some of the groups are mutagenic, but these effects
do not relate well to the activity of the chemicals as tumour
initiators in the skin. There are only very limited studies on the
carcinogenicity of this class of substances. Early indications of
developmental toxicity of members of this class appear to be largely
attributable to the vehicle used in treatment.
1.3.6 Halogenated hydroxyfuranone derivatives
Based on experimental studies, the critical effects of MX appear
to be its mutagenicity and carcinogenicity. Several in vitro studies
have revealed that MX is mutagenic in bacterial and mammalian test
systems. MX caused chromosomal aberrations and induced DNA damage in
isolated liver and testicular cells and sister chromatid exchanges in
peripheral lymphocytes from rats exposed in vivo. An overall
evaluation of the mutagenicity data shows that MX is mutagenic
in vitro and in vivo. A carcinogenicity study in rats showed
increased tumour frequencies in several organs.
1.3.7 Chlorite
The toxic action of chlorite is primarily in the form of
oxidative damage to red blood cells at doses as low as 10 mg/kg of
body weight. There are indications of mild neurobehavioural effects in
rat pups at 5.6 mg/kg of body weight per day. There are conflicting
data on the genotoxicity of chlorite. Chlorite does not increase
tumours in laboratory animals in chronic exposure studies.
1.3.8 Chlorate
The toxicity of chlorate is similar to that of chlorite, but
chlorate is less effective at inducing oxidative damage. It does not
appear to be teratogenic or genotoxic in vivo. There are no data
from long-term carcinogenicity studies.
1.3.9 Bromate
Bromate causes renal tubular damage in rats at high doses. It
induces tumours of the kidney, peritoneum and thyroid in rats at doses
of 6 mg/kg of body weight and above in chronic studies. Hamsters are
less sensitive, and mice are considerably less sensitive. Bromate is
also genotoxic in vivo in rats at high doses. Carcinogenicity
appears to be secondary to oxidative stress in the cell.
1.4 Epidemiological studies
1.4.1 Cardiovascular disease
Epidemiological studies have not identified an increased risk of
cardiovascular disease associated with chlorinated or chloraminated
drinking-water. Studies of other disinfectants have not been
conducted.
1.4.2 Cancer
The epidemiological evidence is insufficient to support a causal
relationship between bladder cancer and long-term exposure to
chlorinated drinking-water, THMs, chloroform or other THM species. The
epidemiological evidence is inconclusive and equivocal for an
association between colon cancer and long-term exposure to chlorinated
drinking-water, THMs, chloroform or other THM species. The information
is insufficient to allow an evaluation of the observed risks for
rectal cancer and risks for other cancers observed in single
analytical studies.
Various types of epidemiological studies have attempted to assess
the cancer risks that may be associated with exposure to chlorinated
drinking-water. Chloraminated drinking-water was considered in two
studies. Several studies have attempted to estimate exposures to total
THMs or chloroform and the other THM species, but the studies did not
consider exposures to other DBPs or other water contaminants, which
may differ for surface water and groundwater sources. One study
considered the mutagenicity of drinking-water as measured by the
Salmonella typhimurium assay. Assessments of possible cancer risks
that may be associated with drinking-water disinfected with ozone or
chlorine dioxide have not been performed.
Ecological and death certificate-based case-control studies have
provided hypotheses for further evaluation by analytical studies that
consider an individual's exposure to drinking-water and possible
confounding factors.
Analytical studies have reported weak to moderate increased
relative risks of bladder, colon, rectal, pancreatic, breast, brain or
lung cancer associated with long-term exposure to chlorinated
drinking-water. Single studies reported associations for pancreatic,
breast or brain cancer; however, the evaluation of a possible causal
relationship for epidemiological associations requires evidence from
more than a single study. In one study, a small increased relative
risk of lung cancer was associated with the use of surface water
sources, but the magnitude of risk was too small to rule out residual
confounding.
A case-control study reported a moderately large association
between rectal cancer and long-term exposure to chlorinated
drinking-water or cumulative THM exposure, but cohort studies have
found either no increased risk or a risk too weak to rule out residual
confounding.
Decreased bladder cancer risk was associated with increased
duration of exposure to chloraminated drinking-water, but there is no
biological basis for assuming a protective effect of chloraminated
water.
Although several studies found increased risks of bladder cancer
associated with long-term exposure to chlorinated drinking-water and
cumulative exposure to THMs, inconsistent results were reported among
the studies for bladder cancer risks between smokers and non-smokers
and between men and women. Estimated exposure to THMs was considered
in three of these studies. In one study, no association was found
between estimated cumulative exposure to THMs. In another study, a
moderately strong increased relative risk was associated with
increased cumulative exposure to THMs in men but not in women. The
third study reported a weak increased relative risk associated with an
estimated cumulative exposure of 1957-6425 µg of THMs per litre-year;
weak to moderate associations were also reported for exposure to THM
concentrations greater than 24, greater than 49 and greater than 74
µg/litre. No increased relative risk of bladder cancer was associated
with exposure to chlorinated municipal surface water supplies,
chloroform or other THM species in a cohort of women, but the
follow-up period of 8 years was very short, resulting in few cases for
study.
Because inadequate attention has been paid to assessing exposure
to water contaminants in epidemiological studies, it is not possible
to properly evaluate the increased relative risks that were reported.
Specific risks may be due to other DBPs, mixtures of by-products or
other water contaminants, or they may be due to other factors for
which chlorinated drinking-water or THMs may serve as a surrogate.
1.4.3 Adverse pregnancy outcomes
Studies have considered exposures to chlorinated drinking-water,
THMs or THM species and various adverse outcomes of pregnancy. A
scientific panel recently convened by the US Environmental Protection
Agency reviewed the epidemiological studies and concluded that the
results of currently published studies do not provide convincing
evidence that chlorinated water or THMs cause adverse pregnancy
outcomes.
Results of early studies are difficult to interpret because of
methodological limitations or suspected bias.
A recently completed but not yet published case-control study has
reported moderate increased relative risks for neural tube defects in
children whose mothers' residence in early pregnancy was in an area
where THM levels were greater than 40 µg/litre. Replication of the
results in another area is required before this association can be
properly evaluated. A previously conducted study in the same
geographic area reported a similar association, but the study suffered
from methodological limitations.
A recently reported cohort study found an increased risk of early
miscarriage associated with heavy consumption of water (five or more
glasses of cold tapwater per day) containing high levels (>75
µg/litre) of THMs. When specific THMs were considered, only heavy
consumption of water containing BDCM (>18 µg/litre) was associated
with a risk of miscarriage. As this is the first study to suggest an
adverse reproductive effect associated with a brominated by-product, a
scientific panel recommended that another study be conducted in a
different geographic area to attempt to replicate these results and
that additional efforts be made to evaluate exposures of the cohort to
other water contaminants.
1.5 Risk characterization
It should be noted that the use of chemical disinfectants in
water treatment usually results in the formation of chemical
by-products, some of which are potentially hazardous. However, the
risks to health from these by-products at the levels at which they
occur in drinking-water are extremely small in comparison with the
risks associated with inadequate disinfection. Thus, it is important
that disinfection not be compromised in attempting to control such
by-products.
1.5.1 Characterization of hazard and dose-response
1.5.1.1 Toxicological studies
1) Chlorine
A WHO Working Group for the 1993 Guidelines for drinking-water
quality considered chlorine. This Working Group determined a
tolerable daily intake (TDI) of 150 µg/kg of body weight for free
chlorine based on a no-observed-adverse-effect level (NOAEL) of
approximately 15 mg/kg of body weight per day in 2-year studies in
rats and mice and incorporating an uncertainty factor of 100 (10 each
for intra- and interspecies variation). There are no new data that
indicate that this TDI should be changed.
2) Monochloramine
A WHO Working Group for the 1993 Guidelines for drinking-water
quality considered monochloramine. This Working Group determined a
TDI of 94 µg/kg of body weight based on a NOAEL of approximately 9.4
mg/kg of body weight per day, the highest dose tested, in a 2-year
bioassay in rats and incorporating an uncertainty factor of 100 (10
each for intra- and interspecies variation). There are no new data
that indicate that this TDI should be changed.
3) Chlorine dioxide
The chemistry of chlorine dioxide in drinking-water is complex,
but the major breakdown product is chlorite. In establishing a
specific TDI for chlorine dioxide, data on both chlorine dioxide and
chlorite can be considered, given the rapid hydrolysis to chlorite.
Therefore, an oral TDI for chlorine dioxide is 30 µg/kg of body
weight, based on the NOAEL of 2.9 mg/kg of body weight per day for
neurodevelopmental effects of chlorite in rats.
4) Trihalomethanes
Cancer following chronic exposure is the primary hazard of
concern for this class of DBPs. Because of the weight of evidence
indicating that chloroform can induce cancer in animals only after
chronic exposure to cytotoxic doses, it is clear that exposures to low
concentrations of chloroform in drinking-water do not pose
carcinogenic risks. The NOAEL for cytolethality and regenerative
hyperplasia in mice was 10 mg/kg of body weight per day after
administration of chloroform in corn oil for 3 weeks. Based on the
mode of action evidence for chloroform carcinogenicity, a TDI of 10
µg/kg of body weight was derived using the NOAEL for cytotoxicity in
mice and applying an uncertainty factor of 1000 (10 each for inter-
and intraspecies variation and 10 for the short duration of the
study). This approach is supported by a number of additional studies.
This TDI is similar to the TDI derived in the 1998 WHO Guidelines
for drinking-water quality, which was based on a 1979 study in which
dogs were exposed for 7.5 years.
Among the brominated THMs, BDCM is of particular interest because
it produces tumours in rats and mice and at several sites (liver,
kidneys, large intestine) after corn oil gavage. The induction of
colon tumours in rats by BDCM (and by bromoform) is also interesting
because of the epidemiological associations with colo-rectal cancer.
BDCM and the other brominated THMs are also weak mutagens. It is
generally assumed that mutagenic carcinogens will produce linear
dose-response relationships at low doses, as mutagenesis is generally
considered to be an irreversible and cumulative effect.
In a 2-year bioassay, BDCM given by corn oil gavage induced
tumours (in conjunction with cytotoxicity and increased proliferation)
in the kidneys of mice and rats at doses of 50 and 100 mg/kg of body
weight per day, respectively. The tumours in the large intestine of
the rat occurred after exposure to both 50 and 100 mg/kg of body
weight per day. Using the incidence of kidney tumours in male mice
from this study, quantitative risk estimates have been calculated,
yielding a slope factor of 4.8 × 10-3 [mg/kg of body weight per
day]-1 and a calculated dose of 2.1 µg/kg of body weight per day for
a risk level of 10-5. A slope factor of 4.2 × 10-3 [mg/kg of body
weight per day]-1 (2.4 µg/kg of body weight per day for a 10-5 risk)
was derived based on the incidence of large intestine carcinomas in
the male rat. The International Agency for Research on Cancer (IARC)
has classified BDCM in Group 2B (possibly carcinogenic to humans).
DBCM and bromoform were studied in long-term bioassays. In a
2-year corn oil gavage study, DBCM induced hepatic tumours in female
mice, but not in rats, at a dose of 100 mg/kg of body weight per day.
In previous evaluations, it had been suggested that the corn oil
vehicle may play a role in the induction of tumours in female mice. A
small increase in tumours of the large intestine in rats was observed
in the bromoform study at a dose of 200 mg/kg of body weight per day.
The slope factors based on these tumours are 6.5 × 10-3 [mg/kg of
body weight per day]-1 for DBCM, or 1.5 µg/kg of body weight per day
for a 10-5 risk, and 1.3 × 10-3 [mg/kg of body weight per day]-1 or
7.7 µg/kg of body weight per day for a 10-5 risk for bromoform.
These two brominated THMs are weakly mutagenic in a number of
assays, and they were by far the most mutagenic DBPs of the class in
the GST-mediated assay system. Because they are the most lipophilic
THMs, additional concerns about whether corn oil may have affected
their bioavailability in the long-term studies should be considered. A
NOAEL for DBCM of 30 mg/kg of body weight per day has been established
based on the absence of histopathological effects in the liver of rats
after 13 weeks of exposure by corn oil gavage. IARC has classified
DBCM in Group 3 (not classifiable as to its carcinogenicity to
humans). A TDI for DBCM of 30 µg/kg of body weight was derived based
on the NOAEL for liver toxicity of 30 mg/kg of body weight per day and
an uncertainty factor of 1000 (10 each for inter- and intraspecies
variation and 10 for the short duration of the study and possible
carcinogenicity).
Similarly, a NOAEL for bromoform of 25 mg/kg of body weight per
day can be derived on the basis of the absence of liver lesions in
rats after 13 weeks of dosing by corn oil gavage. A TDI for bromoform
of 25 µg/kg of body weight was derived based on this NOAEL for liver
toxicity and an uncertainty factor of 1000 (10 each for inter- and
intraspecies variation and 10 for the short duration of the study and
possible carcinogenicity). IARC has classified bromoform in Group 3
(not classifiable as to its carcinogenicity to humans).
5) Haloacetic acids
The induction of mutations by DCA is very improbable at the low
doses that would be encountered in chlorinated drinking-water. The
available data indicate that DCA differentially affects the
replication rates of normal hepatocytes and hepatocytes that have been
initiated. The dose-response relationships are complex, with DCA
initially stimulating division of normal hepatocytes. However, at the
lower chronic doses used in animal studies (but still very high
relative to those that would be derived from drinking-water), the
replication rate of normal hepatocytes is eventually sharply
inhibited. This indicates that normal hepatocytes eventually
down-regulate those pathways that are sensitive to stimulation by DCA.
However, the effects in altered cells, particularly those that express
high amounts of a protein that is immunoreactive to a c-Jun antibody,
do not seem to be able to down-regulate this response. Thus, the rates
of replication in the pre-neoplastic lesions with this phenotype are
very high at the doses that cause DCA tumours to develop with a very
low latency. Preliminary data would suggest that this continued
alteration in cell birth and death rates is also necessary for the
tumours to progress to malignancy. This interpretation is supported by
studies that employ initiation/promotion designs as well.
On the basis of the above considerations, it is suggested that
the currently available cancer risk estimates for DCA be modified by
incorporation of newly developing information on its comparative
metabolism and modes of action to formulate a biologically based
dose-response model. These data are not available at this time, but
they should become available within the next 2-3 years.
The effects of DCA appear to be closely associated with doses
that induce hepatomegaly and glycogen accumulation in mice. The
lowest-observed-adverse-effect level (LOAEL) for these effects in
an 8-week study in mice was 0.5 g/litre, corresponding to
approximately 100 mg/kg of body weight per day, and the NOAEL was
0.2 g/litre, or approximately 40 mg/kg of body weight per day. A TDI
of 40 µg/kg of body weight has been calculated by applying an
uncertainty factor of 1000 to this NOAEL (10 each for inter- and
intraspecies variation and 10 for the short duration of the study and
possible carcinogenicity). IARC has classified DCA in Group 3 (not
classifiable as to its carcinogenicity to humans).
TCA is one of the weakest activators of the peroxisome
proliferator activated receptor (PPAR) known. It appears to be only
marginally active as a peroxisome proliferator, even in rats.
Furthermore, treatment of rats with high levels of TCA in
drinking-water does not induce liver tumours. These data strongly
suggest that TCA presents little carcinogenic hazard to humans at the
low concentrations found in drinking-water.
From a broader toxicological perspective, the developmental
effects of TCA are the end-point of concern. Animals appear to
tolerate concentrations of TCA in drinking-water of 0.5 g/litre
(approximately 50 mg/kg of body weight per day) with little or no
signs of adverse effect. At 2 g/litre, the only sign of adverse effect
appears to be hepatomegaly. Hepatomegaly is not observed in mice at
doses of 0.35 g of TCA per litre in drinking-water, estimated to be
equivalent to 40 mg/kg of body weight per day.
In another study, soft tissue anomalies were observed at
approximately 3 times the control rate at the lowest dose
administered, 330 mg/kg of body weight per day. At this dose, the
anomalies were mild and would clearly be in the range where
hepatomegaly (and carcinogenic effects) would occur. Considering the
fact that the PPAR interacts with cell signalling mechanisms that can
affect normal developmental processes, a common mechanism underlying
hepatomegaly and the carcinogenic effects and developmental effects of
this compound should be considered.
The TDI for TCA is based on a NOAEL estimated to be 40 mg/kg of
body weight per day for hepatic toxicity in a long-term study in mice.
Application of an uncertainty factor of 1000 (10 each for inter- and
intraspecies variation and 10 for possible carcinogenicity) to the
estimated NOAEL gives a TDI of 40 µg/kg of body weight. IARC has
classified TCA in Group 3 (not classifiable as to its carcinogenicity
to humans).
Data on the carcinogenicity of brominated acetic acids are too
preliminary to be useful in risk characterization. Data available in
abstract form suggest, however, that the doses required to induce
hepatocarcinogenic responses in mice are not dissimilar to those of
the chlorinated acetic acids. In addition to the mechanisms involved
in the induction of cancer by DCA and TCA, it is possible that
increased oxidative stress secondary to their metabolism might
contribute to their effects.
There are a significant number of data on the effects of
dibromoacetic acid (DBA) on male reproduction. No effects were
observed in rats at doses of 2 mg/kg of body weight per day for
79 days, whereas an increased retention of step 19 spermatids was
observed at 10 mg/kg of body weight per day. Higher doses led to
progressively more severe effects, including marked atrophy of the
seminiferous tubules with 250 mg/kg of body weight per day, which was
not reversed 6 months after treatment was suspended. A TDI of 20 µg/kg
of body weight was determined by allocating an uncertainty factor of
100 (10 each for inter- and intraspecies variation) to the NOAEL of
2 mg/kg of body weight per day.
6) Chloral hydrate
Chloral hydrate at 1 g/litre of drinking-water (166 mg/kg of body
weight per day) induced liver tumours in mice exposed for 104 weeks.
Lower doses have not been evaluated. Chloral hydrate has been shown to
induce chromosomal anomalies in several in vitro tests but has been
largely negative when evaluated in vivo. It is probable that the
liver tumours induced by chloral hydrate involve its metabolism to TCA
and/or DCA. As discussed above, these compounds are considered to act
as tumour promoters. IARC has classified chloral hydrate in Group 3
(not classifiable as to its carcinogenicity to humans).
Chloral hydrate administered to rats for 90 days in
drinking-water induced hepatocellular necrosis at concentrations of
1200 mg/litre and above, with no effect being observed at 600 mg/litre
(approximately 60 mg/kg of body weight per day). Hepatomegaly was
observed in mice at doses of 144 mg/kg of body weight per day
administered by gavage for 14 days. No effect was observed at 14.4
mg/kg of body weight per day in the 14-day study, but mild
hepatomegaly was observed when chloral hydrate was administered in
drinking-water at 70 mg/litre (16 mg/kg of body weight per day) in a
90-day follow-up study. The application of an uncertainty factor of
1000 (10 each for inter- and intraspecies variation and 10 for the use
of a LOAEL instead of a NOAEL) to this value gives a TDI of 16 µg/kg
of body weight.
7) Haloacetonitriles
Without appropriate human data or an animal study that involves a
substantial portion of an experimental animal's lifetime, there is no
generally accepted basis for estimating carcinogenic risk from the
HANs.
Data developed in subchronic studies provide some indication of
NOAELs for the general toxicity of dichloroacetonitrile (DCAN) and
dibromoacetonitrile (DBAN). NOAELs of 8 and 23 mg/kg of body weight
per day were identified in 90-day studies in rats for DCAN and DBAN,
respectively, based on decreased body weights at the next higher doses
of 33 and 45 mg/kg of body weight per day, respectively.
A WHO Working Group for the 1993 Guidelines for drinking-water
quality considered DCAN and DBAN. This Working Group determined a
TDI of 15 µg/kg of body weight for DCAN based on a NOAEL of 15 mg/kg
of body weight per day in a reproductive toxicity study in rats and
incorporating an uncertainty factor of 1000 (10 each for intra- and
interspecies variation and 10 for the severity of effects).
Reproductive and developmental effects were observed with DBAN only at
doses that exceeded those established for general toxicity (about 45
mg/kg of body weight per day). A TDI of 23 µg/kg of body weight was
calculated for DBAN based on the NOAEL of 23 mg/kg of body weight per
day in the 90-day study in rats and incorporating an uncertainty
factor of 1000 (10 each for intra- and interspecies variation and 10
for the short duration of the study). There are no new data indicating
that these TDIs should be changed.
LOAELs for trichloroacetonitrile (TCAN) of 7.5 mg/kg of body
weight per day for embryotoxicity and 15 mg/kg of body weight per day
for developmental effects were identified. However, later studies
suggest that these responses were dependent upon the vehicle used. No
TDI can be established for TCAN.
There are no data useful for risk characterization purposes for
other members of the HANs.
8) MX
The mutagen MX has recently been studied in a long-term study in
rats in which some carcinogenic responses were observed. These data
indicate that MX induces thyroid and bile duct tumours. An increased
incidence of thyroid tumours was seen at the lowest dose of MX
administered (0.4 mg/kg of body weight per day). The induction of
thyroid tumours with high-dose chemicals has long been associated with
halogenated compounds. The induction of thyroid follicular tumours
could involve modifications in thyroid function or a mutagenic mode of
action. A dose-related increase in the incidence of cholangiomas and
cholangiocarcinomas was also observed, beginning at the low dose in
female rats, with a more modest response in male rats. The increase in
cholangiomas and cholangiocarcinomas in female rats was utilized to
derive a slope factor for cancer. The 95% upper confidence limit for a
10-5 lifetime risk based on the linearized multistage model was
calculated to be 0.06 µg/kg of body weight per day.
9) Chlorite
The primary and most consistent finding arising from exposure to
chlorite is oxidative stress resulting in changes in the red blood
cells. This end-point is seen in laboratory animals and, by analogy
with chlorate, in humans exposed to high doses in poisoning incidents.
There are sufficient data available with which to estimate a TDI for
humans exposed to chlorite, including chronic toxicity studies and a
two-generation reproductive toxicity study. Studies in human
volunteers for up to 12 weeks did not identify any effect on blood
parameters at the highest dose tested, 36 µg/kg of body weight per
day. Because these studies do not identify an effect level, they are
not informative for establishing a margin of safety.
In a two-generation study in rats, a NOAEL of 2.9 mg/kg of body
weight per day was identified based on lower auditory startle
amplitude, decreased absolute brain weight in the F1 and F2
generations, and altered liver weights in two generations. Application
of an uncertainty factor of 100 (10 each for inter- and intraspecies
variation) to this NOAEL gives a TDI of 30 µg/kg of body weight. This
TDI is supported by the human volunteer studies.
10) Chlorate
Like chlorite, the primary concern with chlorate is oxidative
damage to red blood cells. Also like chlorite, 0.036 mg of chlorate
per kg of body weight per day for 12 weeks did not result in any
adverse effect in human volunteers. Although the database for chlorate
is less extensive than that for chlorite, a recent well conducted
90-day study in rats identified a NOAEL of 30 mg/kg of body weight per
day based on thyroid gland colloid depletion at the next higher dose
of 100 mg/kg of body weight per day. A TDI is not derived because a
long-term study is in progress, which should provide more information
on chronic exposure to chlorate.
11) Bromate
Bromate is an active oxidant in biological systems and has been
shown to cause an increase in renal tumours, peritoneal mesotheliomas
and thyroid follicular cell tumours in rats and, to a lesser extent,
hamsters, and only a small increase in kidney tumours in mice. The
lowest dose at which an increased incidence of renal tumours was
observed in rats was 6 mg/kg of body weight per day.
Bromate has also been shown to give positive results for
chromosomal aberrations in mammalian cells in vitro and in vivo
but not in bacterial assays for point mutation. An increasing body of
evidence, supported by the genotoxicity data, suggests that bromate
acts by generating oxygen radicals in the cell.
In the 1993 WHO Guidelines for drinking-water quality, the
linearized multistage model was applied to the incidence of renal
tumours in a 2-year carcinogenicity study in rats, although it was
noted that if the mechanism of tumour induction is oxidative damage in
the kidney, application of the low-dose cancer model may not be
appropriate. The calculated upper 95% confidence interval for a 10-5
risk was 0.1 µg/kg of body weight per day.
The no-effect level for the formation of renal cell tumours in
rats is 1.3 mg/kg of body weight per day. If this is used as a point
of departure from linearity and if an uncertainty factor of 1000 (10
each for inter- and intraspecies variation and 10 for possible
carcinogenicity) is applied, a TDI of 1 µg/kg of body weight can be
calculated. This compares with the value of 0.1 µg/kg of body weight
per day associated with an excess lifetime cancer risk of 10-5.
At present, there are insufficient data to permit a decision on
whether bromate-induced tumours are a result of cytotoxicity and
reparative hyperplasia or a genotoxic effect.
IARC has assigned potassium bromate to Group 2B (possibly
carcinogenic to humans).
1.5.1.2 Epidemiological studies
Epidemiological studies must be carefully evaluated to ensure
that observed associations are not due to bias and that the design is
appropriate for an assessment of a possible causal relationship.
Causality can be evaluated when there is sufficient evidence from
several well designed and well conducted studies in different
geographic areas. Supporting toxicological and pharmacological data
are also important. It is especially difficult to interpret
epidemiological data from ecological studies of disinfected
drinking-water, and these results are used primarily to help develop
hypotheses for further study.
Results of analytical epidemiological studies are insufficient to
support a causal relationship for any of the observed associations. It
is especially difficult to interpret the results of currently
published analytical studies because of incomplete information about
exposures to specific water contaminants that might confound or modify
the risk. Because inadequate attention has been paid to assessing
exposures to water contaminants in epidemiological studies, it is not
possible to properly evaluate the increased relative risks that were
reported. Risks may be due to other water contaminants or to other
factors for which chlorinated drinking-water or THMs may serve as a
surrogate.
1.5.2 Characterization of exposure
1.5.2.1 Occurrence of disinfectants and disinfectant by-products
Disinfectant doses of several milligrams per litre are typically
employed, corresponding to doses necessary to inactivate
microorganisms (primary disinfection) or doses necessary to maintain a
residual in the distribution system (secondary disinfection).
A necessary ingredient for an exposure assessment is DBP
occurrence data. Unfortunately, there are few published international
studies that go beyond case-study or regional data.
Occurrence data suggest, on average, exposure to about 35-50 µg
of total THMs per litre in chlorinated drinking-water, with chloroform
and BDCM being the first and second most dominant species. Exposure to
total HAAs can be approximated by a total HAA concentration (sum of
five species) corresponding to about one-half of the total THM
concentration (although this ratio can vary significantly); DCA and
TCA are the first and second most dominant species. In waters with a
high bromide to TOC ratio or a high bromide to chlorine ratio, greater
formation of brominated THMs and HAAs can be expected. When a
hypochlorite solution (versus chlorine gas) is used, chlorate may also
occur during chlorination.
DBP exposure in chloraminated water is a function of the mode of
chloramination, with the sequence of chlorine followed by ammonia
leading to the formation of (lower levels of) chlorine DBPs (i.e.,
THMs and HAAs) during the free-chlorine period; however, the
suppression of chloroform and TCA formation is not paralleled by a
proportional reduction in DCA formation.
All factors being equal, bromide concentration and ozone dose are
the best predictors of bromate formation during ozonation, with about
a 50% conversion of bromide to bromate. A study of different European
water utilities showed bromate levels in water leaving operating water
treatment plants ranging from less than the detection limit (2
µg/litre) up to 16 µg/L. The brominated organic DBPs formed upon
ozonation generally occur at low levels. The formation of chlorite can
be estimated by a simple percentage (50-70%) of the applied chlorine
dioxide dose.
1.5.2.2 Uncertainties of water quality data
A toxicological study attempts to extrapolate a laboratory
(controlled) animal response to a potential human response; one
possible outcome is the estimation of cancer risk factors. An
epidemiological study attempts to link human health effects (e.g.,
cancer) to a causative agent or agents (e.g., a DBP) and requires an
exposure assessment.
The chemical risks associated with disinfected drinking-water are
potentially based on several routes of exposure: (i) ingestion of DBPs
in drinking-water; (ii) ingestion of chemical disinfectants in
drinking-water and the concomitant formation of DBPs in the stomach;
and (iii) inhalation of volatile DBPs during showering. Although the
in vivo formation of DBPs and the inhalation of volatile DBPs may be
of potential health concern, the following discussion is based on the
premise that the ingestion of DBPs present in drinking-water is the
most significant route of exposure.
Human exposure is a function of both DBP concentration and
exposure time. More specifically, human health effects are a function
of exposure to complex mixtures of DBPs (e.g., THMs versus HAAs,
chlorinated versus brominated species) that can change
seasonally/temporally (e.g., as a function of temperature, nature and
concentration of NOM) and spatially (i.e., throughout a distribution
system). Each individual chemical disinfectant can form a mixture of
DBPs; combinations of chemical disinfectants can form even more
complex mixtures. Upon their formation, most DBPs are stable, but some
may undergo transformation by, for example, hydrolysis. In the absence
of DBP data, surrogates such as chlorine dose (or chlorine demand),
TOC (or ultraviolet absorbance at 254 nm [UVA254]) or bromide can be
used to indirectly estimate exposure. While TOC serves as a good
surrogate for organic DBP precursors, UVA254 provides additional
insight into NOM characteristics, which can vary geographically. Two
key water quality variables, pH and bromide, have been identified as
significantly affecting the type and concentrations of DBPs that are
produced.
An exposure assessment should first attempt to define the
individual types of DBPs and resultant mixtures likely to form, as
well as their time-dependent concentrations, as affected by their
stability and transport through a distribution system. For
epidemiological studies, some historical databases exist for
disinfectant (e.g., chlorine) doses, possibly DBP precursor (e.g.,
TOC) concentrations and possibly total THM (and, in some cases, THM
species) concentrations. In contrast to THMs, which have been
monitored over longer time frames because of regulatory scrutiny,
monitoring data for HAAs (and HAA species), bromate and chlorite are
much more recent and hence sparse. However, DBP models can be used to
simulate missing or past data. Another important consideration is
documentation of past changes in water treatment practice.
1.5.2.3 Uncertainties of epidemiological data
Even in well designed and well conducted analytical studies,
relatively poor exposure assessments were conducted. In most studies,
duration of exposure to disinfected drinking-water and the water
source were considered. These exposures were estimated from
residential histories and water utility or government records. In only
a few studies was an attempt made to estimate a study participant's
water consumption and exposure to either total THMs or individual
species of THMs. In only one study was an attempt made to estimate
exposures to other DBPs. In evaluating some potential risks, i.e.,
adverse outcomes of pregnancy, that may be associated with relatively
short term exposures to volatile by-products, it may be important to
consider the inhalation as well as the ingestion route of exposure
from drinking-water. In some studies, an effort was made to estimate
both by-product levels in drinking-water for etiologically relevant
time periods and cumulative exposures. Appropriate models and
sensitivity analysis such as Monte Carlo simulation can be used to
help estimate these exposures for relevant periods.
A major uncertainty surrounds the interpretation of the observed
associations, as exposures to a relatively few water contaminants have
been considered. With the current data, it is difficult to evaluate
how unmeasured DBPs or other water contaminants may have affected the
observed relative risk estimates.
More studies have considered bladder cancer than any other
cancer. The authors of the most recently reported results for bladder
cancer risks caution against a simple interpretation of the observed
associations. The epidemiological evidence for an increased relative
risk of bladder cancer is not consistent -- different risks are
reported for smokers and non-smokers, for men and women, and for high
and low water consumption. Risks may differ among various geographic
areas because the DBP mix may be different or because other water
contaminants are also present. More comprehensive water quality data
must be collected or simulated to improve exposure assessments for
epidemiological studies.
2. CHEMISTRY OF DISINFECTANTS AND DISINFECTANT BY-PRODUCTS
2.1 Background
The use of chlorine (Cl2) as a water disinfectant has come under
scrutiny because of its potential to react with natural organic matter
(NOM) and form chlorinated disinfectant by-products (DBPs). Within
this context, NOM serves as the organic DBP precursor, whereas bromide
ion (Br-) serves as the inorganic precursor. Treatment strategies
generally available to water systems exceeding drinking-water
standards include removing DBP precursors and using alternative
disinfectants for primary and/or secondary (distribution system)
disinfection. Alternative disinfectant options that show promise are
chloramines (NH2Cl, monochloramine), chlorine dioxide (ClO2) and
ozone (O3). While ozone can serve as a primary disinfectant only and
chloramines as a secondary disinfectant only, both chlorine and
chlorine dioxide can serve as either primary or secondary
disinfectants.
Chloramine presents the significant advantage of virtually
eliminating the formation of chlorination by-products and, unlike
chlorine, does not react with phenols to create taste- and
odour-causing compounds. However, the required contact time for
inactivation of viruses and Giardia cysts is rarely obtainable by
chloramine post-disinfection at existing water treatment facilities
(monochloramine is significantly less biocidal than free chlorine).
More recently, the presence of nitrifying bacteria and nitrite
(NO2-) and nitrate (NO3-) production in chloraminated distribution
systems as well as the formation of organic chloramines have raised
concern.
The use of chlorine dioxide, like chloramine, can reduce the
formation of chlorinated by-products during primary disinfection.
However, production of chlorine dioxide, its decomposition and
reaction with NOM lead to the formation of by-products such as
chlorite (ClO2-), a compound that is of health concern.
If used as a primary disinfectant followed by a chloramine
residual in the distribution system, ozone can eliminate the need for
contact between DBP precursors and chlorine. Ozone is known to react
both with NOM to produce organic DBPs such as aldehydes and increase
levels of assimilable organic carbon and with bromide ion to form
bromate.
A thorough understanding of the mechanisms of DBP formation
allows microbial inactivation goals and DBP control goals to be
successfully balanced. This chapter examines a range of issues
affecting DBP formation and control to provide guidance to utilities
considering the use of various disinfecting chemicals to achieve
microbial inactivation with DBP control.
2.2 Physical and chemical properties of common disinfectants and
inorganic disinfectant by-products
The important physical and chemical properties of commonly used
disinfectants and inorganic DBPs are summarized in Table 1.
2.2.1 Chlorine
Chlorine, a gas under normal pressure and temperature, can be
compressed to a liquid and stored in cylindrical containers. Because
chlorine gas is poisonous, it is dissolved in water under vacuum, and
this concentrated solution is applied to the water being treated. For
small plants, cylinders of about 70 kg are used; for medium to large
plants, tonne containers are common; and for very large plants,
chlorine is delivered by railway tank cars or road (truck) tankers.
Chlorine is also available in granular or powdered form as calcium
hypochlorite (Ca(OCl)2) or in liquid form as sodium hypochlorite
(NaOCl; bleach).
Chlorine is used in the form of gaseous chlorine or hypochlorite
(OCl-). In either form, it acts as a potent oxidizing agent and often
dissipates in side reactions so rapidly that little disinfection is
accomplished until amounts in excess of the chlorine demand have been
added. As an oxidizing agent, chlorine reacts with a wide variety of
compounds, in particular those that are considered reducing agents
(hydrogen sulfide [H2S], manganese(II), iron(II), sulfite [SO32-],
Br-, iodide [I-], nitrite). From the point of view of DBP formation
and disinfection, these reactions may be important because they may be
fast and result in the consumption of chlorine.
Chlorine gas hydrolyses in water almost completely to form
hypochlorous acid (HOCl):
Cl2 + H2O -> HOCl + H+ + Cl-
The hypochlorous acid dissociates into hydrogen ions (H+) and
hypochlorite ions in the reversible reaction:
HOCl <-> H+ + OCl-
Hypochlorous acid is a weak acid with a p Ka of approximately
7.5 at 25°C. Hypochlorous acid, the prime disinfecting agent, is
therefore dominant at a pH below 7.5 and is a more effective
disinfectant than hypochlorite ion, which dominates above pH 7.5.
The rates of the decomposition reactions of chlorine increase as
the solution becomes more alkaline, and these reactions can
theoretically produce chlorite and chlorate (ClO3-); they occur
during the electrolysis of chloride (Cl-) solutions when the anodic
and cathodic compartments are not separated, in which case the
chlorine formed at the anode can react with the alkali formed at the
cathode. On the other hand, hypochlorous acid/hypochlorite (or
hypobromous acid/hypobromite, HOBr/OBr-) can be formed by the action
of chlorine (or bromine) in neutral or alkaline solutions. The
Table 1. Physical and chemical properties of commonly used disinfectants and inorganic disinfectant by-products
Chemicala Eo (V)b Oxidation number lambamax (nm)c e (mol-1 litre-1 cm-1)d p epsilono e pKa f
of Cl or Br
HOCl/Cl- +1.49 +1 254 60 +25.2 7.5
292 (OCl-) 419
ClO2/ClO2- +0.95 +4 359 1250 +16.1 -
NH2Cl - +1 245 416 - -
O3/O2 +2.07 - 254 3200 +35.0
HOBr/Br- +1.33 +1 330 50 +22.5 8.7
ClO2-/Cl- +0.76 +3 262 - +12.8 1.96
ClO3-/Cl- +0.62 +5 360 - +10.5 1.45
BrO3-/Br- +0.61 +5 195 - 0.72
a Half-cell reactants/products.
b Eo = standard electrode potential (redox potential) in water at 25 °C. The oxidation-reduction state of an aqueous
environment at equilibrium can be stated in terms of its redox potential. In the chemistry literature, this is generally
expressed in volts, E, or as the negative logarithm of the electron activity, p epsilon. When p epsilon is large, the
electron activity is low and the system tends to be an oxidizing one: i.e., half-reactions tend to be driven to the left.
When p epsilon is small, the system is reducing, and reactions tend to be driven to the right.
c lambdamax = maximum absorbance wavelength of that particular solution in nm.
d e = molar absorptivity (molar extinction coefficient), in mol-1 litre-1 cm-1. This can be used for quantitative
determination of the various species of chemicals and is the only direct physical measurement. There is often some
background absorbance that may interfere with the measurement in natural waters that should be considered.
e p epsilono = - log {e-} where {e-} = electron activity.
f pKa = negative logarithm of the acid ionization constant (e.g., at pH 7.5, the molar concentration of HOCl is same as that
of OCl-). As this parameter is dependent upon temperature, the values listed were determined at 25 °C.
decomposition of hypohalites (XO-) is favoured in alkaline solutions
(2XO- -> X- + XO2-) and is such that there is no longer any
domain of thermodynamic stability for the hypohalite ions. These
oxyhalites are further converted to stable oxyhalates as follows:
XO- + XO2- -> X- + XO3-
Another reaction that occurs in waters containing bromide ion and
hypochlorite is the production of hypobromous acid:
HOCl + Br- -> HOBr + Cl-
This reaction is irreversible, and the product hypobromous acid is a
better halogenating agent than hypochlorous acid and interferes with
common analytical procedures for free chlorine. The presence of
bromide in hypochlorite solutions can ultimately lead to the formation
of bromate (BrO3-).
Hypobromous acid is a weak acid (p Ka = 8.7); like
hypochlorite, hypobromite is metastable. In alkaline solution, it
decomposes to give bromate and bromide:
3OBr- -> BrO3- + 2Br-
Bromic acid (HBrO3) is a strong acid (p Ka = 0.7). Bromic acid
and bromate can be obtained by the electrolytic oxidation of bromide
solutions or bromine water using chlorine. Bromic acid and bromate are
powerful oxidizing agents, but the speed of their oxidation reactions
is generally slow (Mel et al., 1953).
2.2.2 Chlorine dioxide
Chlorine dioxide is one of the few compounds that exists almost
entirely as monomeric free radicals. Concentrated chlorine dioxide
vapour is potentially explosive, and attempts to compress and store
this gas, either alone or in combination with other gases, have been
commercially unsuccessful. Because of this, chlorine dioxide, like
ozone, must be manufactured at the point of use. Chlorine dioxide in
water does not hydrolyse to any appreciable extent. Neutral or acidic
dilute aqueous solutions are quite stable if kept cool, well sealed
and protected from sunlight.
Chlorine dioxide represents an oxidation state (+4) intermediate
between those of chlorite (+3) and chlorate (+5). No acid or ion of
the same degree of oxidation is known. Chlorine dioxide is a powerful
oxidizing agent that can decompose to chlorite; in the absence of
oxidizable substances and in the presence of alkali, it dissolves in
water, decomposing with the slow formation of chlorite and chlorate:
2ClO2 + H2O -> ClO2- + ClO3- + 2H+
Chlorine dioxide has an absorption spectrum with a maximum at 359
nm, with a molar absorptivity of 1250 mol-1 litre-1 cm-1. This
extinction coefficient is independent of temperature, pH, chloride and
ionic strength. Chlorine dioxide is readily soluble in water, forming
a greenish-yellow solution. It can be involved in a variety of redox
reactions, such as oxidation of iodide ion, sulfide ion, iron(II) and
manganese(II). When chlorine dioxide reacts with aqueous contaminants,
it is usually reduced to chlorite ion. The corresponding electron
transfer reactions are comparable to those occurring when singlet
oxygen acts as an oxidant (Tratnyek & Hoigne, 1994).
Bromide (in the absence of sunlight) is not oxidized by chlorine
dioxide. Therefore, water treatment with chlorine dioxide will not
transform bromide ion into hypobromite and will not give rise to the
formation of bromoform (CHBr3) or bromate. This is an important
difference between the use of chlorine dioxide as an oxidant and the
use of chlorine or ozone as an oxidant.
2.2.3 Ozone
Ozone is a strong oxidizing agent ( Eo = 2.07 V). Oxidation
reactions initiated by ozone in water are generally rather complex; in
water, only part of the ozone reacts directly with dissolved solutes.
Another part may decompose before reaction. Such decomposition is
catalysed by hydroxide ions (OH-) and other solutes. Highly reactive
secondary oxidants, such as hydroxyl radicals (OH.), are thereby
formed. These radicals and their reaction products can additionally
accelerate the decomposition of ozone. Consequently, radical-type
chain reactions may occur, which consume ozone concurrently with the
direct reaction of ozone with dissolved organic material.
Many oxidative applications of ozone have been developed,
including disinfection, control of algae, removal of tastes and
odours, removal of colour, removal of iron and manganese,
microflocculation, removal of turbidity by oxidative flocculation,
removal of organics by oxidation of phenols, detergents and some
pesticides, partial oxidation of dissolved organics and control of
halogenated organic compounds. For disinfection and for oxidation of
many organic and inorganic contaminants in drinking-water, the
kinetics of ozone reactions are favourable; on the other hand, for
many difficult-to-oxidize organic compounds, such as chloroform
(CHCl3), the kinetics of ozone oxidation are very slow (Hoigne et
al., 1985).
2.2.4 Chloramines
Monochloramine has much higher CT values1 than free chlorine
and is therefore a poor primary disinfectant. Additionally, it is a
poor oxidant and is not effective for taste and odour control or for
oxidation of iron and manganese. However, because of its persistence,
1 The CT value is the product of the disinfectant concentration C
in mg/litre and the contact time T in minutes required to inactivate
a specified percentage (e.g., 99%) of microorganisms.
it is an attractive secondary disinfectant for the maintenance of a
stable distribution system residual. The use of disinfectants such as
ozone or chlorine dioxide combined with chloramines as a secondary
disinfectant appears to be attractive for minimizing DBP formation
(Singer, 1994b).
Monochloramine is the only useful ammonia-chloramine
disinfectant. Dichloramine (NHCl2) and nitrogen trichloride (NCl3)
are too unstable to be useful and highly malodorous. Conditions
practically employed for chloramination are designed to produce only
monochloramine.
2.3 Analytical methods for disinfectant by-products and disinfectants
Analytical methods for various DBPs and their detection limits
are summarized in Table 2. Methods for disinfectants are summarized in
APHA (1995).
2.3.1 Trihalomethanes, haloacetonitriles, chloral hydrate,
chloropicrin and haloacetic acids
Gas chromatographic (GC) techniques are generally employed
for organic DBPs. Detection and quantification of haloacetonitriles
(HANs) and chloral hydrate in chlorinated natural waters are
complicated by (i) hydrolysis of dihaloacetonitriles and chloral
hydrate to dihaloacetic acids and chloroform, respectively; (ii)
degradation of HANs by dechlorinating agents such as sodium sulfite
and sodium thiosulfate; (iii) low purge efficiency for the HANs and
chloral hydrate in the purge-and-trap technique; and (iv) low
extraction efficiency for chloral hydrate with pentane in the
liquid-liquid extraction normally used. Although chloral hydrate is
not efficiently extracted from water with pentane, it can be extracted
with an efficiency of approximately 36% when the ratio by volume of
methyl tert-butyl ether (MTBE) to water is 1 : 5 (Amy et al., 1998).
MTBE quantitatively extracts HANs, trihalomethanes (THMs), chloral
hydrate and chloropicrin, permitting simultaneous analysis for all of
these DBPs. Chloral hydrate decomposes on packed columns to
trichloroacetaldehyde, resulting in considerable band broadening,
although this does not appear to be a significant problem with DB-1
and DB-5 columns.
The extraction of THMs can be accomplished using MTBE (EPA Method
551) or pentane. Method 551 also permits simultaneous extraction and
measurement of chloral hydrate, HANs, THMs, chloropicrin and
haloketones. The pentane method can be used to extract THMs, HANs,
haloketones and chloropicrin but not chloral hydrate in the same run
(APHA, 1995).
The haloacetic acid (HAA) analytical method involves using an
acidic salted ether or acidic methanol liquid-liquid extraction,
requiring esterification with diazomethane prior to analysis on a gas
chromatograph equipped with an electron capture detector (ECD). THMs
and HANs can be analysed by extraction with pentane prior to analysis
on a capillary-column GC equipped with an ECD. The analysis of
Table 2. Summary of analytical methods for various DBPs and their minimum detection limits
DBPs Analytical method APHAa Minimum detection Major References
method limit (µg/litre) interferences
THMs MTBE extraction - 0.4 None AWWARF (1991)
Pentane extraction 6232B 0.1
HAAs Salted MTBE extraction and 6233B 0.5-1.0 None AWWARF (1991)
derivatization with diazomethane
HANs Pentane extraction 6232B 0.05 None Koch et al. (1988)
Cyanogen chloride MTBE extraction 6233A 0.5 None AWWARF (1991)
Chloramine Derivatization with - - None Lukasewycz et al. (1989)
2-mercaptobenzothiazole
Haloketonesb Pentane extraction 6232B 0.2 None Krasner et al. (1995)
AWWARF (1991)
Chloral hydrate MTBE extraction - 0.5 None AWWARF (1991)
Aldehydes Extraction with hexane and - 1.0 PFBHA sulfate Sclimenti et al. (1990)
derivatization with PFBHA
Bromate Ion chromatography (H3BO3/NaOH) 4500 2.0c Cl- Siddiqui et al. (1996a)
Krasner et al. (1993)
0.2 Weinberg et al. (1998)
Chlorate Ion chromatography (H3BO3/NaOH) 4500 5 Cl-, acetate Siddiqui (1996)
Chlorite Ion chromatography (NaHCO3/Na2CO3) 4500 10 Cl-, acetate AWWARF (1991)
TOC UV/persulfate or combustion 5310 200 Metals APHA (1995)
a American Public Health Association.
b Sum of 1,1-DCPN and 1,1,1-TCPN.
c 1.0 µg/litre with high-capacity column.
cyanogen compounds involves extraction with MTBE prior to injection
into GC-ECD. Aldehydes require derivatization with
O-(2,3,4,5,6-pentafluorobenzyl)-hydroxylamine (PFBHA) (to form an
oxime), extraction with hexane and GC-ECD analysis [(C6F5)-CH2ONH2
+ RCHO -> (C6F5)-CH2ON=CHR + H2O]. It should be noted that PFBHA
peaks are very large relative to other peaks in the chromatogram from
a purge-and-trap system, whereas the peaks are comparable to other
peaks in a GC-ECD chromatogram (Trehy et al., 1986).
2.3.2 Inorganic disinfectant by-products
An ion chromatography (IC) method (EPA Method 300) has been
developed to determine inorganic by-products. The elution order is
fluoride, chlorite, bromate, chloride, nitrite, bromide, chlorate,
nitrate and sulfate ion. The eluent is a carbonate buffer.
Ethylenediamine is used to preserve chlorite samples and to minimize
the potential for chlorite ion reaction on the IC separating column.
EPA Method 300 involves measurement by an IC system using a separating
column (e.g., Ion Pac AS9-SC) fitted with an anion micromembrane
suppressor column. An eluent containing 2.0 mmol of sodium carbonate
(Na2CO3) per litre / 0.75 mmol of sodium bicarbonate (NaHCO3) per
litre is used for bromide determination, and an eluent containing 40
mmol of boric acid (H3BO3) per litre / 20 mmol of sodium hydroxide
(NaOH) per litre is used for bromate and chlorate determination. The
analytical minimum detection limits for bromate and chlorate using a
borate eluent have been reported as 2 µg/litre and 5 µg/litre,
respectively (Siddiqui, 1996; Siddiqui et al., 1996a). For samples
with high chloride ion content, a silver cartridge can be used to
remove chloride prior to IC analysis to minimize its interference with
bromate measurement. It should be noted that for natural sources and
waters with high total organic carbon (TOC) levels, detection limits
will be slightly different because of the masking effect of NOM and
high concentrations of carbonate/bicarbonate ions that may interfere
with bromate/chlorate measurement.
2.3.3 Total organic carbon and UV absorbance at 254 nm
TOC is the primary surrogate parameter for the measurement of NOM
in water supplies. Several investigators have reported that the
ultraviolet (UV)/persulfate oxidation method underestimates the TOC
concentration in natural waters as compared with the combustion method
because of the inability of the persulfate method to oxidize highly
polymerized organic matter. It is generally assumed that the
calibration of a TOC analyser with a potassium hydrogen phthalate
standard is sufficient for the measurement of TOC in natural waters,
but potassium hydrogen phthalate has a simple molecular structure and
is easy to oxidize. Dissolved organic carbon (DOC) is operationally
defined by a (0.45-µm) filtration step. UV absorbance at 254 nm
(UVA254) is used to describe the type and character of NOM, whereas
TOC describes just the amount of NOM.
2.3.4 Chloramines
Knowledge of the amine content of the water during water
treatment processes involving chloramination is important to define
more adequately the content of a matrix described only as a combined
chlorine residual. The presence of organic nitrogen and the
instability of many organic chloramines continue to challenge the
analyst. Lukasewycz et al. (1989) developed a technique for the
analysis of chloramines and organic chloramines present in water using
2-mercaptobenzothiazole as a derivatizing agent. The resulting
sulfanilamides are stable and can be conveniently analysed by
high-performance liquid chromatography (HPLC) using UV or
electrochemical detection. This method appears to be superior to the
use of diazotization or phenylarsine oxide as a method of detection.
Organic chloramines are much weaker disinfectants than inorganic
monochloramine but are indistinguishable by the common analytical
methods.
2.4 Mechanisms involved in the formation of disinfectant by-products
2.4.1 Chlorine reactions
Chlorine reacts with humic substances (dissolved organic matter)
present in most water supplies, forming a variety of halogenated DBPs,
such as THMs, HAAs, HANs, chloral hydrate and chloropicrin, as
follows:
HOCl + DOC -> DBPs
It is generally accepted that the reaction between chlorine and
humic substances, a major component of NOM, is responsible for the
production of organochlorine compounds during drinking-water
treatment. Humic and fulvic acids show a high reactivity towards
chlorine and constitute 50-90% of the total DOC in river and lake
waters (Thurman, 1985). Other fractions of the DOC comprise the
hydrophilic acids (up to 30%), carbohydrates (10%), simple carboxylic
acids (5%) and proteins/amino acids (5%). The reactivity of
carbohydrates and carboxylic acids towards chlorine is low, and they
are not expected to contribute to the production of organochlorine
compounds. However, hydrophilic acids such as citric acid and amino
acids will react with chlorine to produce chloroform and other
products and may contribute towards total organochlorine production
(Larson & Rockwell, 1979).
Free chlorine reacts with water constituents by three general
pathways: oxidation, addition and substitution (Johnson & Jensen,
1986). Chlorine can undergo an addition reaction if the organic
compound has a double bond. For many compounds with double bonds, this
reaction is too slow to be of importance in water treatment. The
oxidation reactions with water constituents such as carbohydrates or
fatty acids (e.g., oleic acid) are generally slow.
Most chlorine DBPs are formed through oxidation and substitution
reactions. THMs have the general formula CHX3, where X can be Cl or
Br. Chloroform may be produced through a series of reactions with
functional groups of humic substances. The major functional groups of
humic substances include acetyl, carboxyl, phenol, alcohol, carbonyl
and methoxyl. The reactions proceed much more rapidly at high pH than
at low pH.
Rook (1977) proposed resorcinol structures to be the major
precursor structure in humic material for chloroform formation. In
accordance with this hypothesis in the chlorination of terrestrial and
aquatic humic substances, a series of intermediates were detected that
contained a trichloromethyl group and that could be converted to
chloroform by further oxidation or substitution reactions (Stevens et
al., 1976).
However, the production of chlorinated compounds such as
dichloropropanedoic acid, 2,2-dichlorobutanedoic acid, cyanogen
chloride (CNCl), HANs or the cyano-substituted acids cannot be
explained on the basis of resorcinol structures, and possible
production pathways require protein-type precursors (De Leer et al.,
1986). The reaction pathway for amino acids involves initial rapid
formation of the monochloramine and dichloramine, which can react
further to form aldehyde or HANs, respectively. Trehy et al. (1986)
demonstrated the formation of chloral hydrate along with HANs after
chlorination of amino acids by substitution reactions, and aldehydes
were shown to be the oxidation products. Luknitskii (1975) provided a
detailed chemistry of chloral hydrate formation.
Christman et al. (1983) also identified chloroform, chloral
hydrate, dichloroacetic acid (DCA), trichloroacetic acid (TCA) and
2,2-dichlorobutanedoic acid as the major products, accounting for 53%
of the total organic halogen (TOX). A number of other minor products
have been detected, including several chlorinated alkanoic acids and
non-chlorinated benzene carboxylic acids. De Leer et al. (1985)
extended these studies to incorporate chloroform intermediates,
chlorinated aromatic acids and cyano-compounds as potential products
in drinking-water. The presence of unhalogenated aldehydes and HANs in
chlorinated natural waters can be attributed in part to the presence
of amino acids or peptides in natural waters. Humic acids may also
contribute to the presence of amino acids in natural waters, as they
have amino acids associated with them either in a free or in a
combined form. Several studies regarding the chlorination of amino
acids have shown that the primary amino group on the amino acids can
be converted to either an aldehyde or a nitrile group (Morris et al.,
1980; Isaac & Morris, 1983). These studies indicate that with an
equimolar amount of halogenating agent, the major product is an
aldehyde. However, if an excess of halogenating agent is added, then
the corresponding nitrile can also be formed, with the ratio of the
aldehyde to nitrile formed increasing with pH.
Many treated waters contain not only chlorinated but also
brominated compounds, such as bromoform. These compounds form because
aqueous chlorine converts bromide in the water to hypobromous acid.
The bromine can then react with the organic matter in the same way as
hypochlorous acid to form various bromochlorinated DBPs. However,
compared with hypochlorous acid, hypobromous acid is a weaker oxidant
and stronger halogenating agent.
Chlorate, an inorganic by-product of chlorine, is formed in
concentrated hypochlorite solutions during their production and
storage through the following reactions (Gordon et al., 1997):
OCl- + OCl- -> ClO2- + Cl-
OCl- + ClO2- -> ClO3- + Cl-
The first reaction proceeds at a much slower rate and is rate
limiting, hence the generally observed second-order kinetics. Sodium
hypochlorite is stored at pH greater than 12 to prevent rapid
decomposition, and most of the sodium hypochlorite is present as
hypochlorite ion. The average rate constant for the formation of
chlorate is 85 × 10-5 mol-1 litre-1 d-1 (Gordon et al., 1995).
2.4.2 Chlorine dioxide reactions
The major chlorine dioxide by-products of concern are chlorite
and chlorate. Chlorine dioxide reacts generally as an electron
acceptor, and hydrogen atoms present in activated organic C-H or N-H
structures are thereby not substituted by chlorine (Hoigne & Bader,
1994). Moreover, in contrast to chlorine, chlorine dioxide's
efficiency for disinfection does not vary with pH or in the presence
of ammonia, and it does not oxidize bromide. As opposed to chlorine,
which reacts via oxidation and electrophilic substitution, chlorine
dioxide reacts only by oxidation; this explains why it does not
produce organochlorine compounds. In addition to this, chlorine
dioxide is more selective in typical water treatment applications, as
evidenced by its somewhat lower disinfectant demand as compared with
chlorine.
Chlorine dioxide is generally produced by reacting aqueous
(sodium) chlorite with chlorine (Gordon & Rosenblatt, 1996):
2ClO2- + HOCl + H+ -> 2ClO2(aq) + Cl- + H2O
However, under conditions of low initial reactant concentrations
or in the presence of excess chlorine, the reactant produces chlorate
ion:
ClO2- + HOCl -> ClO3- + Cl- + H+
This reaction scenario is common in generators that
overchlorinate to achieve high reaction yields based on chlorite ion
consumption.
An alternative approach to chlorine dioxide generation is with
hydrochloric acid (HCl), a process that results in less chlorate
during production:
5NaClO2 + 4HCl -> 4ClO2 + 5NaCl + 2H2O
Chlorite ion is also produced when chlorine dioxide reacts with
organics (Gordon & Rosenblatt, 1996):
ClO2 + NOM -> Products + ClO2-
Chlorine dioxide can also undergo a series of photochemically
initiated reactions resulting in the formation of chlorate ion (Gordon
et al., 1995).
While bromide is not generally oxidized by chlorine dioxide,
bromate can be formed in the presence of sunlight over a wide range of
pH values (Gordon & Emmert, 1996). Utilities need to be concerned with
bromate ion in the chlorine dioxide treatment of drinking-water if the
water contains bromide and is exposed to sunlight. Practically, this
means minimizing exposure to sunlight when chlorine dioxide is applied
in the presence of bromide ion. There appears to be a problem with
chlorine dioxide producing odour-causing compounds at the tap. This
has been linked to chlorine dioxide reacting with volatile organic
compounds derived from new carpets and office products (Hoehn et al.,
1990).
Hoigne & Bader (1994) described the kinetics of reaction between
chlorine dioxide and a wide range of organic and inorganic compounds
that are of concern in water treatment. Measured rate constants were
high for nitrite, hydrogen peroxide, ozone, iodide, iron(II), phenolic
compounds, tertiary amines and thiols. Bromide, ammonia, structures
containing olefinic double bonds, aromatic hydrocarbons, primary and
secondary amines, aldehydes, ketones and carbohydrates are unreactive
under the conditions of water treatment. Chlorine dioxide rapidly
oxidizes substituted phenoxide anions and many phenols, and
second-order rate constants have been measured (Rav-Acha & Choshen,
1987).
2.4.3 Chloramine reactions
Chloramination of drinking-water produces THMs (if chloramine is
formed by chlorination followed by ammonia addition), HAAs, chloral
hydrate, hydrazine, cyanogen compounds, nitrate, nitrite, organic
chloramines and 1,1-dichloropropanone (1,1-DCPN) (Dlyamandoglu &
Selleck, 1992; Kirmeyer et al., 1993, 1995).
In the presence of even small quantities of organic nitrogen, it
is possible for chloramination to produce organic chloramines. Several
researchers have shown that monochloramine readily transfers its
chlorine at a comparatively rapid rate to organic amines to form
organohalogen amines (Isaac & Morris, 1983; Bercz & Bawa, 1986).
Monochloramine was shown to cause binding of radiolabelled halogen to
nutrients such as tyrosine and folic acid; the amount of binding
varied with pH but was generally less at neutral pH than at higher pH
(Bercz & Bawa, 1986). Organic chloramines are much weaker
disinfectants than inorganic monochloramine but are indistinguishable
by common analytical methods. Organic chloramine formation may
necessitate changing chloramination conditions (e.g., ammonia and
chlorine addition order, chlorine-to-ammonia ratios and contact time).
HANs and non-halogenated acetonitriles are produced when
chloramines are reacted with humic materials and amino acids (Trehy et
al., 1986). The reaction pathway for these products is quite
complicated and very similar to that for chlorine, with many
intermediates and by-products formed. In the case of aspartic acid, De
Leer et al. (1986) demonstrated the presence of at least 11 other
significant products.
2.4.4 Ozone reactions
Ozone has been shown to oxidize bromide to hypobromite and
bromate, and hypochlorite to chlorate (Glaze et al., 1993; Siddiqui et
al., 1995; Siddiqui, 1996).
Bromate generally forms through a combination of molecular ozone
attack and reaction of bromide with free radical species. The
molecular ozone mechanism does not account for hydroxyl radicals
always formed as secondary oxidants from decomposed ozone during water
treatment. Siddiqui et al. (1995) indicated that there is a radical
pathway that is influenced by both pH and alkalinity. The hydroxyl
radical and, to a lesser degree, the carbonate radical (CO3Ê) pathway
may be more important than the molecular ozone pathway. Oxidants such
as hydroxyl and carbonate radicals may interact with intermediate
bromine species, leading to the formation of hypobromite radicals
(BrOÊ), which eventually undergo disproportionation to form
hypobromite and bromite (BrO2-). Bromate is then formed through
oxidation of bromite by ozone. The radical mechanism for the formation
of bromate includes two decisive reaction steps still involving
molecular ozone: the formation of hypobromite and oxidation of
bromite.
Bromate ion formed through reactions with molecular ozone
contributes in the range of 30-80% to the overall bromate ion
formation in NOM-containing waters (von Gunten and Hoigne, 1994).
Siddiqui et al. (1995) reported up to 65% and 100% bromate ion
formation through the radical pathway in NOM-free and NOM-containing
waters, respectively. Differences in NOM-containing waters can be
attributed to differences in the characteristics of the NOM present. A
change in mechanism as a function of pH and the competitive roles of
the free radical (one electron transfer) mechanism above pH 7 versus
oxygen atom (two electron transfer) mechanism help explain both the
large variations in bromate ion yield and the sensitivity to reactor
design, concentration of organic precursors and ozone/bromide ion
concentrations (Gordon, 1993).
The presence of bromide ion in a source water further complicates
the reaction of ozone and leads to the formation of additional DBPs,
such as bromoform, dibromoacetonitrile (DBAN) and dibromoacetone
(DBAC) (Siddiqui, 1992; Amy et al., 1993, 1994).
2.5 Formation of organohalogen disinfectant by-products
Table 3 summarizes the DBPs identified as being formed from the
use of chlorine, chlorine dioxide, chloramine and ozone.
The formation of organochlorine and organobromine compounds
during drinking-water treatment is a cause of health concern in many
countries. These compounds include THMs, HAAs, HANs, chloral hydrate,
chloropicrin, acetohalides, halogenated furanones and other compounds.
2.5.1 Chlorine organohalogen by-products
Table 4 summarizes the range of concentrations of chlorinated
DBPs formed from the reaction of chlorine with NOM, from various
sources.
The major chlorination DBPs identified are THMs, HAAs, HANs,
haloketones, chloropicrin and chloral hydrate. HAAs represent a major
portion of the non-THM halogenated organic compounds (Miller & Uden,
1983; Reckhow & Singer, 1985). Many researchers have identified HANs
and haloketones as other important DBPs (Trehy & Bieber, 1981; Miller
& Uden, 1983; Oliver, 1983; Reckhow & Singer, 1985). According to an
AWWARF (1991) study, for all eight utilities tested,
1,1,1-trichloropropanone (1,1,1-TCPN) was the more prevalent of the
two measured haloketone compounds. In addition, Kronberg et al. (1988)
identified the extremely mutagenic compound, MX.
Despite the fact that HAA formation and THM formation have very
different pH dependencies, HAA formation correlates strongly with THM
formation when treatment conditions are relatively uniform and when
the water has a low bromide concentration (Singer, 1993). DBP
formation and requisite chlorine dosage for disinfection strongly
correspond to the concentration of TOC at the point of chlorine
addition, suggesting that optimized or enhanced removal of organic
carbon prior to chlorination will decrease the formation of DBPs.
HAA formation can be appreciable when drinking-water is
chlorinated under conditions of slightly acidic pH and low bromide
concentrations. The concentrations of DCA and TCA are similar to the
concentrations of chloroform, and the total HAA concentration can be
as much as 50% greater than the THM concentration in the finished
water on a weight basis.
McGuire & Meadow (1988) reported that the national average THM
concentration in the USA was 42 µg/litre for drinking-water utilities
serving more than 100 000 persons, and only 3% of systems were above
the US maximum contaminant level of 100 µg/litre. Amy et al. (1993)
estimated that the national average THM concentration in the USA was
40 µg/litre, with an average TOC concentration of 3.0 mg/litre. The
median annual average THM concentration found for utilities among the
American Water Works Association's (AWWA) Water Industry Database was
35 µg/litre, as compared with 50 µg/litre for the non-database
utilities (Montgomery Watson, Inc., 1993).
Table 3. Disinfectant by-products present in disinfected waters
Disinfectant Significant Significant Significant
organo-halogen inorganic non-halogenated
products products products
Chlorine/ THMs, HAAs, HANs, Chlorate (mostly Aldehydes, cyanoalkanoic
hypochlorous acid chloral hydrate, from hypochlorite acids, alkanoic acids,
chloropicrin, use) benzene, carboxylic acids
chlorophenols,
N-chloramines,
halofuranones,
bromohydrins
Chlorine dioxide chlorite, chlorate unknown
Chloramine HANs, cyanogen chloride, nitrate, nitrite, aldehydes, ketones
organic chloramines, chlorate, hydrazine
chloramino acids,
chloral hydrate,
haloketones
Ozone bromoform, MBA, DBA, chlorate, iodate, aldehydes, ketoacids,
DBAC, cyanogen bromide bromate, hydrogen ketones, carboxylic
peroxide, hypobromous acids
acid, epoxides,
ozonates
Table 4. Concentration range of chlorinated disinfectant by-products in drinking-watera
DBPs Peters et al. (1990); Krasner et al. Nieminski et al. Koch et al. Reckhow et al.
Peters (1991) (1989) (1993) (1991) (1990)
THMs 3.1-49.5 30.0-44.0 17.0-51.0 49.0-81.0 201-1280
HAAs <0.5-14.7 13.0-21.0 5.0-25.0 22.0-32.0 118-1230
HANs 0.04-1.05 2.5-4.0 0.5-5.0 2.0-2.6 3.0-12.0
Haloketones - 0.9-1.8 0.2-1.6 1.0-2.0 4.8-25.3
Chlorophenols - - 0.5-1.0 - -
Chloral hydrate - 1.7-3.0 - - -
Chloropicrin - 0.1-0.16 <0.1-0.6 - -
TOC 1.7-5.6 2.9-3.2 1.5-6.0 2.5-3.0 4.8-26.6
Bromide 100-500 70-100 - 170-420 -
a All values shown in µg/litre, except TOC (mg/litre).
In Germany, 10% of the utilities produced disinfected
drinking-water with a THM concentration above 10 µg/litre; the median
annual average concentration was between 1 and 4 µg/litre, depending
on raw water quality and size of facility (Haberer, 1994).
Total THM levels in treated drinking-water were reported in one
survey in the United Kingdom (Water Research Centre, 1980):
chlorinated water derived from a lowland river contained a mean level
of 89.2 µg/litre, and that from an upland reservoir, 18.7 µg/litre.
The study also showed that chlorinated groundwater was contaminated by
THMs to a significantly lesser extent than chlorinated surface waters.
In a national survey of the water supplies of 70 communities
serving about 38% of the population in Canada, conducted in the winter
of 1976-1977, chloroform concentrations in treated water of the
distribution system 0.8 km from the treatment plant, determined by the
gas sparge technique, averaged 22.7 µg/litre. Levels of the other THMs
were considerably lower, averaging 2.9 µg/litre for
bromodichloromethane (BDCM), 0.4 µg/litre for dibromochloromethane
(DBCM) and 0.1 µg/litre for bromoform. Using direct aqueous injection
techniques, average concentrations of most of the THMs were higher
(Health Canada, 1993).
Samples collected from the distribution systems of eight major
cities in Saudi Arabia showed that THMs occurred in all the water
supplies, at concentrations ranging between 0.03 and 41.7 µg/litre.
Median total THM concentrations in several cities were higher during
the summer than during the winter. In addition, THM concentrations
were low in cities that did not mix groundwater and desalinated water.
Brominated THMs dominated (with bromoform the most abundant) and
existed at the highest concentration levels, whereas chloroform was
the least prevalent compound. This is the opposite of the occurrence
pattern found in almost all water distribution systems worldwide
(Fayad, 1993).
The concentrations of chloral hydrate in drinking-water in the
USA were summarized by IARC (1995) and varied from 0.01 to
28 µg/litre. The highest values were found in drinking-water prepared
from surface water.
Chlorination of water as well as the combination of ozonation and
chlorination can lead to the formation of chloropicrin (Merlet et al.,
1985). In a study conducted for over 25 utilities, very low levels of
chloropicrin were observed, and chlorination produced maximum
concentrations of less than 2 µg/litre (AWWARF, 1991). The
chloropicrin appeared to form slowly during the incubation period,
with concentrations tending to level off at approximately 40 h.
Dichloroacetonitrile (DCAN) is by far the most predominant HAN
species detected in water sources with bromide levels of 20 µg/litre
or less. For sources with higher bromide levels (50-80 µg/litre),
bromochloroacetonitrile (BCAN) was the second most prevalent compound.
However, none of these sources had a DBAN concentration exceeding 0.5
µg/litre, including one source water that had a much higher bromide
level, 170 µg/litre. Thus, it appears that ambient bromide
concentration is not the only factor influencing the speciation of HAN
compounds.
Chlorine can react with phenols to produce mono-, di- or
trichlorophenols, which can impart tastes and odours to waters. The
control of chlorophenolic tastes and odours produced when phenol-laden
water is treated with chlorine is essential. The sources of phenolic
compounds in water supplies are reported to be industrial wastes.
In natural waters, one of the most important sources of organic
nitrogen is proteins and their hydrolysis products. The reaction of
aqueous chlorine or monochloramine with organic nitrogen may form
complex organic chloramines (Feng, 1966; Morris et al., 1980; Snyder &
Margerum, 1982). The formation of N-chloramines resulting from the
reaction of amines and chlorine has been reported (Weil & Morris,
1949; Gray et al., 1979; Morris et al., 1980). Likewise, the
chlorination of amides has been reported (Morris et al., 1980).
Nieminski et al. (1993) reported the occurrence of DBPs for Utah
(USA) water treatment plants. All plants used chlorine for primary and
secondary disinfection purposes. Overall, THMs and HAAs represented
75% of the total specific DBPs analysed for the survey; however, total
DBPs represented only 25-50% of the TOX concentration. THMs
constituted 64% of the total DBPs by weight; HAAs were 30% of the
total DBPs by weight and approximately one-half of the total THM
concentrations. (However, in some waters, HAA concentrations may
approach or possibly exceed THM concentrations.) HANs, haloketones,
chlorophenols and chloropicrin represented 3%, 1.5%, 1.0% and 0.5%,
respectively, of the total surveyed DBPs.
The occurrence of DBPs in drinking-waters in the USA was
evaluated at 35 water treatment facilities that had a broad range of
source water qualities and treatment processes (Krasner et al., 1989).
THMs were the largest class of DBPs, and HAAs were the next most
significant class. Aldehydes, by-products of ozonation, were also
produced by chlorination. Over four quarterly sampling periods, median
total THM concentrations ranged from 30 to 44 µg/litre, with
chloroform, BDCM, DBCM and bromoform ranges of 9.6-15, 4.1-10, 2.6-4.5
and 0.33-0.88 µg/litre, respectively. Median total HAA concentrations
ranged from 13 to 21 µg/litre, with TCA, DCA, monochloroacetic acid
(MCA), dibromoacetic acid (DBA) and monobromoacetic acid (MBA) ranges
of 4.0-6.0, 5.0-7.3, <1-1.2, 0.9-1.5 and <0.5 µg/litre,
respectively.
Concentrations of DCA and TCA measured in various water sources
have been summarized by IARC (1995): in Japan, chlorinated
drinking-water contained 4.5 and 7.5 µg of DCA and TCA per litre,
respectively; rainwater in Germany contained 1.35 µg of DCA per litre
and 0.1-20 µg of TCA per litre, whereas groundwater contained 0.05 µg
of TCA per litre; in Australia, a maximum concentration of 200
µg/litre was found for DCA and TCA in chlorinated treated water; and
chlorinated water in Switzerland contained 3.0 µg of TCA per litre.
In a survey of 20 drinking-waters prepared from different source
waters in the Netherlands, HAAs were found in all drinking-waters
prepared from surface water, whereas they could not be detected in
drinking-waters prepared from groundwaters. Brominated acetic acids
accounted for 65% of the total acid concentration (Peters et al.,
1991). In another survey of Dutch drinking waters, the average
concentration of dihaloacetonitriles was about 5% of the average THM
concentration (Peters, 1990).
2.5.2 Chloramine organohalogen by-products
Chloramine treatment practice involves three potential
approaches: free chlorine followed by ammonia addition, ammonia
addition followed by chlorine addition (in situ production) and
pre-formed (off-line formation) chloramines. Generally, the objective
is monochloramine formation. Chlorine followed by ammonia is a common
approach, and, during the free-chlorine period, DBP formation may
mimic that of chlorine. Chloramination results in the production of
THMs (predominantly formed by chlorination followed by ammonia
addition), HAAs, chloral hydrate, hydrazine, cyanogen compounds,
organic chloramines and 1,1-DCPN (Dlyamandoglu & Selleck, 1992;
Singer, 1993; Kirmeyer et al., 1993, 1995). Chloramination
significantly reduces but does not eliminate THM formation; cyanogen
chloride and TOX represent the major DBP issues with respect to
chloramines.
Scully et al. (1990) identified chloramino acids such as
N-chloroglycine, N-chloroleucine and N-chlorophenylalanine as
by-products after chlorination of water containing nitrogen or after
chloramination.
2.5.3 Chlorine dioxide organohalogen by-products
Chlorine-free chlorine dioxide does not form THMs (Noack & Doerr,
1978; Symons et al., 1981). Several studies show that the TOX formed
with chlorine dioxide is 1-15% of the TOX formed with chlorine under
the same reaction conditions (Chow & Roberts, 1981; Symons et al.,
1981; Fleischacker & Randtke, 1983).
Treatment of phenol-laden source waters with chlorine dioxide
does not produce the typical chlorophenolic taste and odour compounds
that are produced when the water is treated using chlorine and is
effective in removing existing tastes and odours of this type.
2.5.4 Ozone organohalogen by-products
Ozonation of drinking-water containing bromide ion has been shown
to produce hypobromous acid/hypobromite, with hypobromite ion serving
as an intermediate to bromate formation. In the presence of NOM,
hypobromous acid produces a host of brominated organic compounds, such
as bromoform, MBA, DBA, DBAN, cyanogen bromide and DBAC (Glaze et al.,
1993; Siddiqui & Amy, 1993). Cavanagh et al. (1992) and Glaze et al.
(1993) reported the identification of bromohydrins, a new group of
labile brominated organic compounds from the ozonation of a natural
water in the presence of enhanced levels of bromide. However, results
by Kristiansen et al. (1994) strongly suggest that the bromohydrins,
such as 3-bromo-2-methyl-2-butanol, in extracts of unquenched
disinfected water are artefacts formed from the reaction of excessive
hypobromous acid with traces of olefins in the extraction solvents and
not novel DBPs.
Table 5 compares the median concentrations of various DBPs after
ozonation and chlorination.
2.6 Formation of inorganic disinfectant by-products
Although organic DBPs have been the subject of study over a
longer time frame, the formation of many inorganic by-products is
coming under increasing scrutiny.
2.6.1 Chlorine inorganic by-products
Chlorite and chlorate are inorganic by-products formed in some
chlorine solutions. This is of interest because many small
drinking-water utilities use hypochlorite solutions as a source of
free chlorine for disinfection. Bolyard & Fair (1992) examined the
occurrence of chlorate in samples of untreated source water,
drinking-water and hypochlorite solutions from 14 sites that use
hypochlorite solutions. The hypochlorite solutions used were found to
contain significant levels of chlorate. Chlorite and bromate were also
found in hypochlorite solutions from these same water utilities.
Chlorate was present in drinking-water, either as a manufacturing
by-product or from decomposition reactions occurring during storage.
Approximately 0.2 mg of chlorate per litre was observed in water
following the addition of chlorine as sodium hypochlorite at a dose
sufficient to maintain a residual of 0.45 mg/litre (Andrews &
Ferguson, 1995). The concentration of chlorite in commercial bleach
solutions typically ranges from 0.002 to 0.0046 mol/litre; similarly,
the chlorate concentration ranges from about 0.02 to 0.08 mol/litre
(Gordon et al., 1995).
A detailed study by Bolyard & Fair (1992) demonstrated that
hypochlorite solutions used to disinfect drinking-water contain
significant levels of chlorite and chlorate. The concentration of
chlorite ranged from <2 to 130 mg/litre for free available chlorine
concentrations ranging from 3 to 110 g/litre. The concentration of
chlorate varied over the range 0.19-50 g/litre, with a median of 12
g/litre. These solutions also contained bromate levels ranging from
<2 to 51 mg/litre. The concentrations of chlorate in treated source
waters ranged from 11 to 660 µg/litre. In another study involving 25
samples from plants using gaseous chlorine, no chlorate was detected,
indicating that the use of gaseous chlorine does not produce chlorate
(Bolyard & Fair, 1992). Nieminski et al. (1993) measured chlorate and
chlorite for six water treatment plants that use liquid chlorine
(i.e., hypochlorite) and found chlorate concentrations ranging from 40
to 700 µg/litre, with no chlorite or bromate detected in finished
waters. These chlorate concentrations may be attributed to high
Table 5. Median concentrations of organic disinfectant by-products
in drinking-water
DBPs Median concentration Median concentration
(µg/litre): chlorinationa (µg/litre): ozonationb
THMs 40 <1.0
Chloroform 15 -
BDCM 10 -
DBCM 4.5 -
Bromoform 0.57 <1.0
HANs 2.5 <1.0
TCAN <0.012 -
DCAN 1.1 -
BCAN 0.58 -
DBAN 0.48 <1.0
Haloketones 0.94 -
DCPN 0.46 -
TCPN 0.35 -
HAAs 20 <5.0
MCA 1.2 -
DCA 6.8 -
TCA 5.8 -
MBA <0.5 <1.0
DBA 1.5 <5.0
Aldehydes 7.8 45
Formaldehyde 5.1 20
Acetaldehyde 2.7 11
Glyoxal - 9
Methylglyoxal - 5
Chloral hydrate 3.0 -
Ketoacids - 75
Trichlorophenol <0.4 -
a Krasner et al. (1989).
b Siddiqui et al. (1993).
concentrations of chlorate, ranging from 1000 to 8000 mg/litre,
detected in a bleach used for disinfection and resulting from the
decomposition of hypochlorite stock solution. However, no chlorite or
chlorate was detected in any of the samples of finished water of the
treatment plants that apply gaseous chlorine. Chlorate formation is
expected to be minimal in low-strength hypochlorite solutions freshly
prepared from calcium hypochlorite, because of the low hypochlorite
concentration and only mildly alkaline pH.
2.6.2 Chloramine inorganic by-products
Inorganic by-products of chloramination include nitrate, nitrite,
hydrazine and, to some extent, chlorate (Dlyamandoglu & Selleck, 1992;
Kirmeyer et al., 1995).
2.6.3 Chlorine dioxide inorganic by-products
The major inorganic by-products of chlorine dioxide disinfection
have been identified as chlorite and chlorate. Andrews & Ferguson
(1995) measured a chlorate concentration of 0.38 mg/litre when a
chlorine dioxide residual of 0.33 mg/litre was maintained. The
application of chlorine dioxide produces about 0.5-0.7 mg of chlorite
and 0.3 mg of chlorate per mg of chlorine dioxide consumed or applied
(Andrews & Ferguson, 1995).
2.6.4 Ozone inorganic by-products
When bromide or iodide ions are present in waters, some of the
halogen-containing oxidants that can be produced during ozonation
include free bromine, hypobromous acid, hypobromite ion, bromate ion,
free iodine, hypoiodous acid and iodate ion.
During the oxidation or chemical disinfection of natural waters
containing bromide ion with ozone, bromate ion can be formed at
concentrations ranging from 0 to 150 µg/litre under normal water
treatment conditions (Siddiqui, 1992). Chlorate formation with an
initial total chlorine concentration of 0.6 mg/litre was evaluated at
pH levels of 8.0, 7.0 and 6.0, and chlorate concentrations ranging
from 10 to 106 µg/litre were formed after ozonation (Siddiqui et al.,
1996a).
It has been reported that ozone reacts with many metal ions and
with cyanide ion (Hoigne et al., 1985; Yang & Neely, 1986). Bailey
(1978) discussed the formation of ozonates, compounds of metal cations
having the general formula M+O3-. Hydrogen peroxide has been
identified as a by-product of ozonation of organic unsaturated
compounds (Bailey, 1978).
Table 6 provides the range of bromate concentrations normally
encountered in drinking-waters with a variety of source water
characteristics after ozonation.
Table 6. Summary of bromate ion formation potentials in different source waters under different conditions following
ozonation
Na Bromide Ozone pH Alkalinity DOC Bromate Reference
(µg/litre) (mg/litre) (mg/litre) (mg/litre) (µg/litre)
18 10-800 1-9.3 5.6-9.4 20-132 2.2-8.2 <5-60 Krasner et al. (1992)
4 60-340 3-12 6.5-8.5 90-230 3-7 <5-40 Siddiqui & Amy (1993)
28 10-100 2-4 6.8-8.8 20-120 0.3-11 <5-100 Amy et al. (1993, 1994)
4 12-37 0-3.97 7.8 N/A N/A <7-35 Hautman & Bolyard (1993)
1 500 2.3-9.5 7.2-8.3 N/A N/A 13-293 Yamada (1993)
23 12-207 0.3-4.3 5.7-8.2 14-246 0.5-6.8 <2-16 Legube et al. (1993)
8 107-237 1-5 6.8-8.0 N/A 2-5 <5-50 Kruithof & Meijers (1993)
a N = number of sources studied.
2.7 Formation of non-halogenated organic disinfectant by-products
2.7.1 Chlorine organic by-products
Lykins & Clark (1988) conducted a 1-year pilot plant study of the
effects of ozone and chlorine and determined that the concentration of
aldehydes increased by 144% upon ozonation. In the chlorinated stream,
the concentration of these aldehydes increased by 56%. This study
indicates that aldehyde formation, although greater with ozone, is not
unique to ozonation, but is associated with chlorination and other
oxidants as well.
2.7.2 Chloramine organic by-products
When Suwannee River (USA) fulvic acid was reacted with aqueous
solutions of 15N-labelled chloramine and 15N-labelled ammonia,
lyophilized products exhibiting nuclear magnetic resonances between 90
and 120 ppm were observed, denoting the formation of amides,
enaminones and aminoquinones (Ginwalla & Mikita, 1992). This
represents evidence for the formation of nitrogen-containing compounds
from the chloramination of NOM in natural waters.
Amino acids, peptides and amino sugars were chlorinated under
various chlorine/nitrogen ratios (Bruchet et al., 1992). Six natural
amino acids (alanine, methionine, valine, phenylalanine, leucine and
isoleucine) were shown to induce tastes and odours at concentrations
in the range of 10-20 µg/litre. Detectable odours were consistently
induced in a multicomponent mixture containing each of these amino
acids after a 2-h contact time with chlorine. Investigation of the
by-products indicated that the odours generated were systematically
linked to the aliphatic aldehydes formed. The peptides investigated
had varying degrees of odour formation potential, while the amino
sugars did not impart any odour. Chlorinous odours occasionally
detected during these experiments were found to be due to organic
chloramines and other oxidation by-products.
2.7.3 Chlorine dioxide organic by-products
Gilli (1990) showed the formation of carbonyl compounds
(34 µg/litre) such as n-valeraldehyde (7-15 µg/litre), formaldehyde
(3.4-9 µg/litre), acetaldehyde (4.5 µg/litre) and acetone (3.2
µg/litre) after using chlorine dioxide.
2.7.4 Ozone organic by-products
Ozone aliphatic oxidation products from organic impurities in
water are usually acids, ketones, aldehydes and alcohols. So-called
ultimate oxidation products of organic materials are carbon dioxide,
water, oxalic acid and acetic acid. However, ozonation conditions
generally employed in treating drinking-water are rarely sufficient to
form high concentrations of these ultimate products.
When source waters containing NOM and unsaturated organic
compounds are ozonated, ozonides, peroxides, diperoxides, triperoxides
and peroxy acids, for example, can be produced. The limited research
that has been conducted in aqueous solutions indicates that these
intermediates decompose readily in water to form products such as
aldehydes, ketones, carboxylic acids and ketoacids.
Coleman et al. (1992) identified numerous compounds in addition
to the following in ozonated humic samples: monocarboxylic acids up to
C-24, dicarboxylic acids up to C-10, ketoacids, furan carboxylic
acids, and benzene mono-, di- and tricarboxylic acids. Among the
various aldehydes, Paode et al. (1997) found four (formaldehyde,
acetaldehyde, glyoxal and methylglyoxal) to be dominant. Table 7
provides a range of concentrations for aldehydes from the ozonation of
a variety of source waters.
2.8 Influence of source water characteristics on the amount and type
of by-products produced
The extensive literature pertaining to DBP levels in disinfected
source waters and control of DBPs by various treatment processes
attests to the wide variety of factors influencing DBP formation and
the complex interrelationships between these factors. Variables
including the concentration and characteristics of precursor material,
pH, chlorine concentration, bromide level, presence of
chlorine-demanding substances such as ammonia, temperature and contact
time all play a role in DBP formation reactions.
2.8.1 Effect of natural organic matter and UV absorbance at 254 nm
NOM consists of a mixture of humic substances (humic and fulvic
acids) and non-humic (hydrophilic) material. Both the amount (as
indicated by TOC or UVA254) and the character (as described by UVA254)
of NOM can affect DBP formation. NOM provides the precursor material
from which organic DBPs are formed; consequently, increasing
concentrations of NOM lead to increasing concentrations of
by-products. This relationship has led to the use of TOC and UVA254
measurements as surrogate parameters for estimating the extent of DBP
formation.
The removal of NOM is strongly influenced by those properties
embodying the size, structure and functionality of this heterogeneous
mixture. The humic acids are more reactive than fulvic acids with
chlorine (Reckhow et al., 1990) and ozone, in terms of both
oxidant/disinfectant demand and DBP formation. Processes such as
coagulation, adsorption and membrane filtration are separation
processes that remove NOM intact, while ozonation transforms part of
the NOM into biodegradable organic matter, potentially removable by
biofiltration. Coagulation preferentially removes humic/higher
molecular weight NOM; the selectivity of membranes for NOM removal is
largely dictated by the molecular weight cutoff of the membrane; the
use of granular activated carbon (GAC) requires a significant empty
bed contact time; biofiltration can remove only the rapidly
biodegradable NOM fraction.
Table 7. Effect of ozone dose and TOC on non-halogenated organic by-products
Ozone dose TOC Formal Acetal Glyoxal Methyl-glyoxal Reference
(mg/litre) (mg/litre) (µg/litre) (µg/litre) (µg/litre) (µg/litre)
1.2-4.4 2.66 8-24 2-4 4-11 4-15 Miltner et al. (1992)
1.0-9.2 1.0-25.9 3-30 7-65 3-15 3-35 Weinberg et al. (1993)
5.5-28.5 5.4-17.4 58-567 6-28 15-166 17-54 Schechter & Singer (1995)
In an investigation of the nature of humic and fulvic acids
isolated from a variety of natural waters, Reckhow et al. (1990) found
that the fulvic fractions had a lower aromatic content and smaller
molecular size than the humic fractions. UV absorbance was
correspondingly higher for the humic fractions, owing to the higher
aromatic content and larger size. These researchers also found that
for all of the organic material investigated, the production of
chloroform, TCA, DCA and DCAN was higher upon chlorination of the
humic fractions than upon chlorination of the corresponding fulvic
fractions. These findings support the findings of other researchers
and show that the UV absorbance measurement is an indicator of the
nature of the precursor material present in a sample. This
measurement, in conjunction with the TOC (or DOC) measurement, can be
employed in the evaluation of data to provide an indication of the
reactivity of NOM towards forming DBPs.
The reaction of ozone with NOM can occur directly or by radical
processes. The disappearance of disinfecting chemical is influenced by
the type and concentration of NOM present in natural waters. Direct
consumption of these chemicals is greater when the UV absorbance (due
to electrophilic and nucleophilic sites of NOM) of the source water is
significant, resulting in decreased DBP formation potential.
It appears that the nature of the organic material in a source
water may have some impact on the relative concentrations of THMs and
HAAs formed upon chlorination. Treatment techniques that lower the
levels of DOC without affecting bromide levels have been implicated in
a shift from chlorinated to brominated THM compounds. This is of
concern because the theoretical risk to humans varies for the
individual THMs, with the brominated species generally being of more
concern (Bull & Kopfler, 1991).
2.8.2 Effect of pH
The impact of pH on THM concentrations has been reported by a
number of researchers since THMs in drinking-water first came to the
attention of the water industry (Stevens et al., 1976; Lange &
Kawczynski, 1978; Trussell & Umphres, 1978). More recently, the impact
of pH on a number of other chlorination by-products has been reported
(Miller & Uden, 1983; Reckhow & Singer, 1985). The rate of THM
formation increases with the pH (Stevens et al., 1976). Kavanaugh et
al. (1980) reported a 3-fold increase in the reaction rate per unit
pH.
In general, increasing pH has been associated with increasing
concentrations of THMs and decreasing concentrations of HAAs (pH
primarily impacting TCA), HANs and haloketones. The concentrations of
TCA tend to be higher in waters with pH levels less than 8.0 than in
waters with pH levels greater than 8.0; a less marked trend is
observed for DCA. Other researchers have reported similar findings
with respect to the pH dependency of HAA concentrations. For example,
Stevens et al. (1976) found that TCA concentrations were significantly
lower at a pH of 9.4 than at pH levels of approximately 5 and 7. TCA
was by far the most predominant of the measured HAA species at six of
the eight utilities surveyed. Carlson & Hardy (1998) reported that at
pH levels greater than 9.0, THM formation decreased with increasing
pH. It is possible that the shift in chlorine species from
hypochlorous acid to hypochlorite affects THM formation during short
reaction times.
AWWARF (1991) observed no relationship between pH and the
concentrations of THMs at eight utilities over time, suggesting that
although THM concentrations for a particular water are known to be pH
dependent, factors other than pH influence THM concentrations over a
variety of source waters. Nieminski et al. (1993) reported that
treatment plants with a pH of about 5.5 in finished water produced
equal amounts of THMs and HAAs, whereas plants with pHs greater than
7.0 in finished water produced higher amounts of THMs as compared with
HAAs.
No strong relationship has been observed between HAN
concentration and pH over time. Within the approximate pH range 7-8.5,
HAN concentrations increased slightly over time. In general, a trend
of decreasing HAN concentrations with increasing pH would be expected,
since these compounds are known to undergo base-catalysed hydrolysis
and have been identified as intermediates in the formation of
chloroform (Reckhow & Singer, 1985). Therefore, these compounds may be
unstable in the presence of free chlorine or under basic conditions.
In general, after an initial formation period, HAN and haloketone
concentrations level off or begin to decline over the remainder of the
reaction period. This indicates that base-catalysed hydrolysis may not
be a significant mechanism of reaction for the relatively low pH
sources.
Stevens et al. (1989) evaluated the effects of pH and reaction
time (4, 48 and 144 h) on the formation of chloral hydrate. Chloral
hydrate formation increased over time at pH 5 and 7, whereas chloral
hydrate that had formed within 4 h at pH 9.4 decayed over time at the
elevated pH.
The pH of the source water can also affect the formation of
by-products after chloramine addition. The disproportionation of
monochloramine, which is an important reaction leading to an oxidant
loss, has been shown by several researchers to be catalysed by
hydrogen ion, phosphate, carbonate and silicate (Valentine & Solomon,
1987).
Humic acids have shown reaction rates with chlorine dioxide that
increased by a factor of 3 per pH unit (pH 4-8) (Hoigne & Bader,
1994).
In addition to the impact of pH on THM and HAA formation noted
above, overall TOX formation decreases with increasing pH. Many of the
halogenated DBPs tend to hydrolyse at alkaline pH levels (>8.0)
(Singer, 1994a). This has significant implications, for example, for
precipitative softening facilities.
pH has a strong effect on aldehyde formation (Schechter & Singer,
1995). Higher ozonation pH values produced lower amounts of aldehydes,
supporting the theory that these DBPs are formed primarily through the
direct molecular ozone reaction pathway, as opposed to the radical
pathway. These results may also reflect greater destruction of
aldehydes by hydroxyl radicals at elevated pH levels.
2.8.3 Effect of bromide
The presence of bromide ion during water treatment disinfection
can lead to the formation of DBPs such as brominated organics and
bromate ion. Low but significant levels of bromide, the ultimate
precursor to bromate and other brominated compounds, may occur in
drinking-water sources as a result of pollution and saltwater
intrusion in addition to bromide from natural sources. An
understanding of the sources and levels of bromide ion in different
source waters is crucial for an understanding of the bromate ion
formation potential in drinking-waters. There are no known treatment
techniques available for economically removing bromide ion present in
source waters during drinking-water treatment.
The impact of bromide on the speciation of DBPs within a class of
compounds such as THMs or HAAs has been discussed by Cooper et al.
(1983, 1985) and Amy et al. (1998). Rook et al. (1978) reported that
bromine is more effective than chlorine in participating in
substitution reactions with organic molecules; furthermore, precursor
materials may differ in their susceptibility to bromination versus
chlorination reactions. Hypobromous acid formed from bromide may also
react with ammonia to form bromamines (Galal-Gorchev & Morris, 1965).
2.8.4 Effect of reaction rates
After chlorine addition, there is a period of rapid THM formation
for the initial few hours (e.g., 4 h), followed by a decline in the
rate of THM formation, suggesting fast and slow NOM reactive sites.
Many authors have indicated that the concentration of chloroform
appears to increase slowly even after 96 h, suggesting that as long as
low concentrations of free chlorine are present, chloroform continues
to form. Bromochlorinated THM species have been found to form more
rapidly than chloroform. Further data from many sources indicate that
bromoform formation slows at approximately 7-8 h and levels off almost
completely after 20 h (AWWARF, 1991; Koch et al., 1991). The same
general kinetic trend observed for THMs also appears to apply to HAAs.
A period of rapid formation occurs during the first 4-8 h, followed by
a reduction in the formation rate. In general, for most sources,
concentrations of chlorinated HAAs appear to slowly increase even
after 96 h, while the formation of DBA levels off after about 18-20 h.
Miller & Uden (1983) observed that nearly 90% of the final
concentrations of THMs, TCA and DCA form within the first 24 h of
chlorine addition to waters containing NOM. Reckhow et al. (1990)
found that although waters containing precursor materials isolated
from six different water sources differed in their yields of
chlorinated organic by-products, the formation curves for chloroform,
TCA and DCA had the same general shapes for all six precursor
materials. Some researchers have suggested that DCA may be an
intermediate in TCA formation; however, for all eight source waters
studied, both DCA and TCA concentrations increased or remained stable
throughout the 96-h reaction period, suggesting that DCA was an
end-product (AWWARF, 1991). Carlson & Hardy (1998) indicated that HAA
formation followed a pattern similar to that of THM formation. As with
the THMs, HAA formation rate appeared to be rapid for the first 30
min; after 30 min, HAAs formed at nearly a constant rate in four of
the source waters studied.
Different trends were observed in the HAN concentrations of
different source waters. For two source waters, HAN levels formed
rapidly for the first 8 h and continued to increase slowly or levelled
off after 96 h (AWWARF, 1991). DBAN levels remained relatively stable
over the 96 h, as did BCAN and DCAN levels. For other sources, levels
of HANs consisting mostly of DCAN increased rapidly up to 4-8 h and
began to decline by the end of the 96-h period. For these sources,
BCAN appeared to be slightly more stable than DCAN.
Very low levels of chloropicrin formation have been observed by
many researchers (AWWARF, 1991). The highest concentration observed
was 4.0 µg/litre. Chloropicrin appears to form slowly during the
incubation period, with concentrations tending to level off at
approximately 40 h.
2.8.5 Effect of temperature
The formation rates of THMs, HAAs, bromate ion and HANs have been
shown to increase with temperature (AWWARF, 1991; Siddiqui & Amy,
1993). Both haloketone and chloropicrin levels were found to be higher
at a lower temperature, while the concentrations of other DBP species
were similar or not significantly different. These results suggest
that a higher temperature allows for more rapid progression of the
transformation of haloketones to other by-products. In studies on the
effect of temperature on THMs, Peters et al. (1980) found an Arrhenius
dependency between the rate constant and temperature with an
activation energy of 10-20 kJ/mol.
The impact of temperature on THMs was strongest at longer contact
times (Carlson & Hardy, 1998). On a conceptual basis, it may be that
rapidly forming compounds are more reactive and form DBPs regardless
of temperature. On the other hand, slowly forming compounds require
higher activation energy, and an increase in the temperature supplies
the energy. In addition to reaction kinetics, the temperature of a
source water can also affect disinfection efficiency. The biocidal
effectiveness of monochloramine is significantly less than that of
free chlorine and is dependent on temperature, pH and residual
concentration.
2.8.6 Effect of alkalinity
Although pH is a very influential variable and alkalinity affects
pH, alkalinity itself does not appear to directly affect the formation
of THMs and HAAs (by chlorination) and has only a slight effect on
aldehydes and other organic by-products following ozonation (Andrews
et al., 1996). However, the majority of studies on the effect of
alkalinity on the formation of bromate during ozonation indicate that
increased alkalinity increases bromate formation (Siddiqui et al.,
1995). The quantity of aldehydes produced remains approximately
constant for similar changes in alkalinity and pH; however, deviation
from equivalent changes in pH and alkalinity results in increased
aldehyde concentrations. Therefore, conditions of high alkalinity and
low pH or low alkalinity and high pH produce greater quantities of
aldehydes than do intermediate values of these parameters (Andrews et
al., 1996).
2.9 Influence of water treatment variables on the amount and type of
by-products produced
Since DBPs are formed by all of the above chemical disinfectants,
the adoption of alternative disinfectants for DBP control often means
only a trade-off between one group of DBPs and another. The most
effective DBP control strategy is organic precursor (NOM) removal
through enhanced coagulation, biofiltration, GAC or membrane
filtration. There has been little success with bromide removal. Other
DBP control options include water quality modifications -- for
example, acid or ammonia addition for bromate minimization.
2.9.1 Effect of ammonia
The presence of ammonia in source waters during disinfection can
cause chlorine and ozone demand and participation in the formation of
by-products such as nitrate, cyanogen chloride and other nitrogenous
compounds.
Ammonia also does not consume chlorine dioxide. In contrast to
chlorine, chlorine dioxide can therefore be considered as a virucide
when ammonia is present. This might be one of the historical reasons
why chlorine dioxide has been adopted as a disinfectant by some
treatment plants using well oxidized waters but containing changing
ammonia concentrations. The addition of ammonia has been shown to
reduce the formation of bromate after ozonation (Siddiqui et al.,
1995), and the ammonia has been shown to participate in the formation
of HANs and cyanogen bromide (CNBr) (Siddiqui & Amy, 1993).
The growth of nitrifying bacteria is a potential problem in
chloraminated water supplies or chlorination of sources containing
nitrogen. In a study conducted by Cunliffe (1991), nitrifying bacteria
were detected in 64% of samples collected from five chloraminated
water supplies in South Australia and in 21% of samples that contained
more than 5 mg of monochloramine per litre. Increased numbers of the
bacteria were associated with monochloramine decay within the
distribution systems.
2.9.2 Effect of disinfectant dose
Chlorine dose is a factor affecting the type and concentration of
DBPs formed. The THM level rises with increasing chlorine dose
(Kavanaugh et al., 1980). However, there is some disagreement
regarding the quantitative relations between chlorine concentration
and THM levels (or the rate of THM production). Most investigators
found a linear relationship between chlorine consumption and THM
production, with an order of reaction greater than or equal to unity
(Trussell & Umphres, 1978; Kavanaugh et al., 1980). However, it is
also possible that the order of reaction changes during the course of
the reaction.
Reckhow & Singer (1985) found that the concentration of DBP
intermediates such as DCAN and 1,1,1-TCPN formed after 72 h of
reaction time was dependent on chlorine dose. DCAN, which was measured
at a concentration of approximately 5 µg/litre at a chlorine dose of
10 mg/litre, was not detected in samples dosed with 50 mg of chlorine
per litre. The concentration of chloroform was about 150 µg/litre in a
sample dosed with 10 mg of chlorine per litre but was approximately
200 µg/litre in a sample dosed with 20 mg of chlorine per litre. Thus,
it is imperative to have uniform chlorine doses for performing DBP
formation kinetic measurements.
Since chloramine residuals are longer-lasting than free chlorine
residuals, the doses for each set of chlorinated and chloraminated
samples will be different in order to achieve the prescribed target
residual. The disappearance of chloramines can be explained
approximately by a second-order reaction. However, as the chlorine
dose increased, the observed rate constant was found to decrease, then
increased after reaching a minimum value (Dlyamandoglu & Selleck,
1992). Below the chlorine dose at the minimum value of the observed
rate constant, the rate constant was proportional to the 1.4 power of
the chlorine dose, regardless of the ammonia concentration (Yamamoto
et al., 1988).
2.9.3 Effect of advanced oxidation processes
Water utilities can add treatment processes that remove DBP
precursors or DBPs. Many utilities will be using both approaches. The
hydrogen peroxide/UV process, an advanced oxidation process, offers
small water utilities a treatment process with the potential to
provide primary disinfection and a method of DBP control (Symons &
Worley, 1995). This process has been shown to oxidize dissolved
organic halogens and decrease TOC. TOC removal as a function of UV
dose has also been demonstrated by Worley (1994), with TOC removals of
between 0% and 80%. Andrews et al. (1996) evaluated the effect of the
hydrogen peroxide/UV process on THM formation and concluded that this
process is only slightly effective in reducing the formation of DBPs.
However, using hydrogen peroxide at 1 mg/litre in combination with UV
effectively reduced or prevented the formation of aldehydes. Other
advanced oxidation processes (e.g., hydrogen peroxide/ozone, ozone/UV)
involving hydroxyl (and hydroperoxyl) radical formation may provide
similar opportunities.
2.9.4 Effect of chemical coagulation
Enhanced coagulation and softening will remove TOC. Enhanced
coagulation is characterized by coagulant doses greater than those
required for optimum turbidity removal; as an alternative to higher
doses, a combination of acid (pH depression) and coagulant addition
can be practised.
All organic DBPs were reduced by the addition of commonly used
coagulants. Iron-based coagulants, such as ferric chloride, were
consistently more effective than alum in removing NOM (Crozes et al.,
1995). Alum coagulation removed all DBP precursors to a significant
extent. The percentage removals showed the same trends as, but were
not identical to, the percentage removals of TOC and UV absorbance. UV
absorbance was removed to a somewhat greater extent than TOC. Hence,
TOC and UV absorbance can serve as surrogate parameters for DBP
formation potential. A fairly good correlation was observed between
the ratio of HAAs to THMs and the ratio of UV absorbance to TOC,
indicating that the relative concentrations of HAAs and THMs do to
some extent depend on the nature of the precursor material. However,
more data from waters of different qualities would be required to
evaluate the validity of this relationship.
The effectiveness of coagulants in removing DBP precursors is
dependent upon the molecular size of the dissolved organic matter.
Normally, higher molecular weight fractions are effectively removed
through coagulation. In a study conducted by Teng & Veenstra (1995),
water containing dissolved organic matter with molecular weights in
the range 1000-10 000 daltons generally produced the largest amounts
of THMs and HAAs under conditions of free chlorination. Coagulation
and ozonation shift a proportionately greater amount of the THM and
HAA formation potential to the smallest molecular weight range (<1000
daltons).
Coagulation and filtration remove NOM but not bromide, hence
increasing the ratio of bromide to TOC. As a result, the subsequent
use of chlorine generally favours the formation of brominated organic
DBPs.
2.9.5 Effect of pre-ozonation
Several studies of ozone oxidation followed by chlorination
showed increased, rather than decreased, levels of THMs (Trussell &
Umphres, 1978). This is attributed, at least partially, to the
formation of aldehydes by ozonation. Another possibility is
hydroxylation of aromatic compounds to produce m-dihydroxy aromatic
derivatives, which are known THM precursors (Lykins & Clark, 1988).
Although the aldehydes produced contain polar groupings, they are
nevertheless not easily removed during the flocculation step by
complexation with aluminium or iron salts. A convenient and more
appropriate method for the removal of the aldehydes formed during
ozonation is the incorporation of a biological treatment step
(biofiltration) in the water treatment process following ozone
oxidation.
Pre-ozonation can have both positive and negative effects on DBP
formation. Pre-ozonation with typical water treatment dosages and
bicarbonate levels has been shown to remove TCA and DCAN precursors.
However, such treatment can result in no net change in the DCA
precursors and may lead to an increase in 1,1,1-TCPN precursors
(Reckhow & Singer, 1985). According to Teng & Veenstra (1995),
pre-ozonation resulted in enhanced formation of DCA during
chlorination and chloramination in the presence of precursors in the
<1000 dalton molecular weight range. They also indicated that
pre-ozonation plus chloramination controlled the overall production of
THMs and HAAs. However, the use of pre-ozonation coupled with free
chlorination increased the yield of DCA for both the hydrophilic and
hydrophobic fractions of NOM as compared with free chlorination alone.
With ozone-chlorine treatment, chloral hydrate formation can be
enhanced. This behaviour, which has also been observed for DCA,
suggests that the reaction that produces chloral hydrate is
accelerated under the conditions of ozonation in combination with
prechlorination and warm water temperatures (LeBel et al., 1995).
Ozonation in the presence of traces of hypochlorite ion can form
inorganic by-products such as chlorate. Siddiqui et al. (1996a) showed
that if there is any residual chlorine present, ozone can potentially
oxidize hypochlorite ion to chlorate.
Coleman et al. (1992) suggested that brominated MX analogues and
other mixed bromochlorinated by-products formed after ozonation and
chlorination can possibly increase mutagenic activity.
2.9.6 Effect of biofiltration
Biofiltration (ozone-sand filtration or ozone-GAC) can
potentially reduce TOC, organic by-products and the formation of
halogenated DBPs.
Passage of ozonated water samples through a rapid sand filter
reduced the concentration of aldehydes by 62% (Lykins & Clark, 1988).
Chlorinated samples experienced a 26% reduction in aldehyde
concentrations under the same conditions. These reductions in aldehyde
levels are attributable to biological activity in the sand filters. If
GAC filtration follows sand filtration, ozone oxidation can be
expected to promote more bioactivity in the GAC filter, because a
better colonization environment is provided for microorganisms on GAC
particles than on sand. Thus, the biological conversion of oxidized
water impurities to carbon dioxide and water will be greater during
passage through GAC media. Similar aldehyde removals have been
observed by several researchers (Van Hoof et al., 1985; Sketchell et
al., 1995).
Drinking-water treatment techniques that remove organic
contaminants without affecting bromide concentrations cause a shift in
the formation of DBPs towards brominated DBPs. Sketchell et al. (1995)
studied three sources containing three different DOC levels and
ambient bromides, which were filtered through biologically active GAC
filters. Analysis of treated waters showed no removal of bromide ion
and a shift towards more brominated organo-DBPs. THM levels after
treatment with GAC with no added ozone decreased from 900-1700 to
100-700 µg/litre. These water sources contained DOC levels ranging
from 10 to 25 mg/litre and high concentrations of biodegradable DOC
(DOC removals ranged from 60% to 80% after GAC treatment).
Table 8 summarizes the effects of ozonation and biofiltration on
the formation of DBPs from various sources.
2.10 Comparative assessment of disinfectants
A comparative assessment (Table 9) of various disinfecting
chemicals for pre-disinfection (or oxidation) and post-disinfection
and maintaining a residual for 5 days to simulate concentrations in
the distribution system showed that the use of free chlorine produces
the largest concentration of halogenated DBPs (Clark et al., 1994).
The concentration of DBPs may be reduced by adding ozone or chlorine
dioxide as a preoxidant, although enhanced formation has been
observed.
Table 10 summarizes the effects of water quality and treatment
variables on the formation of DBPs.
2.11 Alternative strategies for disinfectant by-product control
The concern about chlorite, bromate, chlorate and other DBPs in
drinking-water following treatment with disinfectants has stimulated
research into ways to eliminate the production or enhance the removal
of DBPs. Strategies for DBP control include source control, precursor
removal, use of alternative disinfectants and removal of DBPs by
technologies such as air stripping, activated carbon, UV light and
advanced oxidation technologies. For DBPs that can arise in
hypochlorite solutions (e.g., chlorate), the purity and storage
conditions of the solutions are important concerns.
2.11.1 Source control
Source control options involve controlling nutrient inputs to
waters (e.g., algae growth control) (Hoehn et al., 1990) that are used
as drinking-water sources, watershed management (e.g., constructing
stormwater detention basins), saltwater intrusion control (e.g.,
development of structural or hydrodynamic barriers to control TOC and
bromide), and using the concept known as aquifer storage and recovery
(e.g., drawing water during seasons when the quality of the water is
best) (Singer, 1994a).
2.11.2 Organohalogen by-products
Strategies for control of organohalogen by-products include
removal of DBPs that are formed using technologies such as oxidation,
aeration and carbon adsorption (Clark et al., 1994); and removal of
precursors using treatment techniques such as conventional treatment,
Table 8. Effects of ozonation and biofiltration on chlorine organic by-products
DBPs Ozonation Biofiltration Ozonation + biofiltration Reference
(% change) (% change) (% change)
THMs -20 -20 -40 (chlorine) Speitel et al. (1993)
HAAs -10 -13 -25 (chlorine) Speitel et al. (1993)
Chloropicrin +50 to +250 -50 to -100 (chlorine) Miltner et al. (1992)
Aldehydes +425 to +1300 -40 to -50 -92 to -98 Miltner et al. (1992)
TOX -30 -51 (chlorine) Miltner et al. (1992)
TOX -10 -38 -47 (chlorine) Shukairy & Summers (1992)
TOX +32 -69 -60 (monochloramine) Shukairy & Summers (1992)
Table 9. Comparative assessment of organic disinfectant by-products (µg/litre) in distribution systemsa,b
DBPs Sand-Cl2 Cl2-Sand-Cl2 O3-Sand-Cl2 NH2Cl-Sand-NH2Cl O3-Sand-NH2Cl ClO2-Sand-Cl2
THMs 236.0 225.0 154.0 9.0 3.2 138.0
HAAs 60.0 146.0 82.0 14.0 9.0 44.0
HANs 3.1 2.9 2.7 <0.1 <0.1 <0.1
Haloketones 2.1 2.6 2.6 <0.1 <0.1 4.2
Chloropicrin 1.3 1.3 7.7 <0.1 <0.1 1.4
Chloral hydrate 79.0 75.0 55.0 <0.1 <0.1 45.0
TOX 557.0 540.0 339.0 59.0 27.0 379.0
a Clark et al. (1994).
b TOC = 3.0 mg/litre; pH = 7.6.
Table 10. Summary of impact of water quality and treatment variables on disinfectant by-product formation
Variable Impact on THMs Impact on HAAs Impact on aldehydes Impact on Impact on bromate
chlorate/chlorite
Contact time Curvilinear increase Curvilinear increase Linear increase as long Linear increase in Curvilinear increase
with increasing with increasing as residual chemical bleach solutions with most bromate
contact time contact time present No discernible effects forming in <5 min
Rapid formation <5 h Rapid formation <5 h Secondary reactions in dilute solutions Formation is a function
90% formation in 24 h 90% formation in 24 h between disinfectants If oxidation of of ozone residual and
Levels off at 96 h Levels off at 150 h and aldehydes possible hypochlorite, contact bromide
time has a positive
effect
Disinfectant Rapid and curvilinear Curvilinear increase Curvilinear with Concentrations related Linear increase after
dose increase after TOC after TOC demand with increasing ozone dose to hypochlorite doses TOC demand and then
demand with dose, increasing dose, or chlorine dose applied levelling off after
levelling off at 2.0 levelling off at 2.0 No appreciable effect Ozone oxidation of ozone residual
mg/litre for TOC of mg/litre after ozone/DOC = 2 : 1 hypochlorite increases disappearance
2.0 mg/litre with dose
pH Curvilinear increase Mixed, possible pH Negative effect (forms Positive effect Strong linear positive
with increasing pH to maximum for DCAA mostly through molecular Decomposition of effect
pH 7.0 and possible and DBAA ozone) hypochlorite increases Hydroxyl radical
pH maximum TCAA decreases up 25% decrease for pH with pH generation efficiency
No positive effect at to pH > 9 7-8.5 Oxidation of increases
pH > 9.5 DCAA maximum at pH hypochlorite by ozone
7-7.5 increases
Temperature Linear increase with Linear increase with Terminal products such Positive effect Curvilinear increase
increasing temperature increasing temperature as carbon dioxide Decomposition of 20-30% increase for
(10-30 °C; 15-25% (10-30 °C; 20-30% increase and total hypochlorite increases 15-25 °C
increase) increase) aldehydes slightly
decrease
Table 10. (continued)
Variable Impact on THMs Impact on HAAs Impact on aldehydes Impact on Impact on bromate
chlorate/chlorite
TOC Increase with Increase with Positive effect Negative effect if Decreases with
increasing TOC; increasing TOC; (hydrophobic fraction ozone is used for increasing TOC;
precursor content precursor content mostly responsible) hypochlorite oxidation precursor content
important important Doubles for every Most likely no effect important
Humic acids more Humic acids more 2 mg/litre with hypochlorite Non-humic acid being
reactive than fulvic reactive than fulvic less reactive
acids acids with ozone
UVA254 Increase with Increase with Positive effect Negative effect if Decreases with
increasing UV increasing UV Ozone demand ozone is used for increasing UV
absorbance; precursor absorbance; increases with UV hypochlorite oxidation absorbance; precursor
content important precursor content (UV absorbance is Probable negative content important
Aromaticity of TOC important mostly due to effect with Humic acid being
being more important Aromaticity of TOC aromaticity and hypochlorous acid more reactive with
being more important hydrophobic fraction) ozone
Bromide Shift towards Shift towards Independent of bromide Shift towards more Bromide threshold
brominated species brominated species at <0.25 mg/litre toxic bromate in Curvilinear increase
At >0.25 mg/litre, hypochlorite solutions and dependent upon
aldehydes can decrease ozone residual
due to ozone-bromide
oxidation
Alkalinity No discernible effect No discernible effect Slight positive effect Unknown Positive effect
Minimization TOC removal, TOC removal, pH control, TOC Avoid hypochlorite pH depression,
strategies minimizing chlorine minimizing chlorine removal by coagulation, dosing solution ammonia addition,
residual, alternative residual, alternative GAC, optimizing doses, Minimize storage radical scavengers,
disinfectants, pH disinfectants, pH contact time Properly tune minimizing and
control, minimizing control, minimizing generators optimizing ozone
contact time contact time Use freshly made residual
solutions
Table 10. (continued)
Variable Impact on THMs Impact on HAAs Impact on aldehydes Impact on Impact on bromate
chlorate/chlorite
Removal GAC, electron beam, GAC, electron beam Biofiltration, advanced Ferrous sulfate, GAC, Ferrous sulfate, UV
strategies air stripping oxidation, GAC, electron beam, UV irradiation,
nanofilters irradiation, high-energy electron
nanofilters beam, GAC
oxidation, membrane processes, carbon adsorption and biological
degradation. For many organic compounds that are difficult to oxidize,
such as chloroform, the kinetics of ozone oxidation are generally very
slow but are faster if used in combination with UV irradiation. GAC
adsorption and membrane filtration are relatively expensive processes;
moreover, NOM removal by GAC cannot be accomplished to any significant
degree in a filter/adsorber (i.e., GAC filter cap) mode but requires a
separate post-filtration adsorber bed. The use of membranes requires
pretreatment to prevent fouling, as well as processing of waste brine.
The use of ozone in combination with biologically active GAC filters
is a promising alternative to reduce DBP precursors.
2.11.3 Inorganic by-products
Properly designed and operated chlorine and chlorine dioxide
generator systems can minimize some of the production of chlorate ion.
Removal of chlorite and chlorate has been reported using reduction by
Fe2+ or sulfite or by GAC (Voudrias et al., 1983; Lykins & Clark,
1988). GAC is seen as problematic because of chlorate production and a
short bed life. A chemical process using an appropriate agent such as
reduced iron (e.g., ferrous sulfate) appears to be a more promising
approach (Kraft & van Eldick, 1989; Gordon et al., 1990).
If bromate is present in treated water entering the coagulation
process (i.e., formed during pre-ozonation), several options exist for
its removal. An aqueous-phase reducing agent (e.g., Fe2+) can be
added at the rapid mix step. Powdered activated carbon can likewise be
added as a solid-phase reductant to remove bromate and DBP precursors.
Not all utilities contemplating ozone application intend to employ
pre-ozonation. Rather, they may use intermediate ozonation prior to
the filtration process; in this situation, removal of bromate by
activated carbon is possible. This approach has potential relevance to
integration of GAC columns into a process train or, more
realistically, to retrofitting of rapid sand filters with GAC filters.
For groundwaters that require no coagulation, bromate can be removed
after ozonation using a GAC filter, UV irradiation or high-energy
electron beam irradiation (Siddiqui et al., 1994, 1996a,b,c).
Brominated or bromochlorinated amines formed during the oxidation
step of the process train using chlorine can potentially be removed
using a suitable activated carbon before terminal chlorination.
However, carbon that has an accumulation of surface oxides, which
develop through reaction of amines, will have a diminished capacity to
reduce halogenated amines to nitrogen. Organic amines can potentially
be removed by activated carbon adsorption.
2.11.4 Organic by-products
There are some technologies for removing organic contaminants
formed after chlorination and chloramination, a less viable option
than minimizing their formation through DBP precursor removal or use
of alternative disinfectants. Studies of ozone oxidation have shown
that aromatic compounds, alkenes and certain pesticides (some of which
have structural similarities to certain organic DBPs) are removed well
by ozone treatment, but that alkanes are poorly removed. Also, removal
efficiency improves for the alkenes and aromatic compounds with
increasing ozone dosage and for some alkanes with increasing pH. For
most compounds, the efficacy of ozone is not affected by the
background water matrix if the ozone is used after coagulation.
Andrews et al. (1996) showed that using hydrogen peroxide at 1.0
mg/litre in combination with UV effectively reduced or prevented the
formation of aldehydes.
2.12 Models for predicting disinfectant by-product formation
The regulation of THMs and other halogenated DBPs has been
complicated by findings that alternative disinfectants to free
chlorine may also form by-products that are of potential health
concern. Additional complicating factors impacting the regulation of
DBPs have been the emergence of Giardia and Cryptosporidium as
major waterborne pathogens.
In view of the finding that water chlorination produces DBPs,
some of which are carcinogenic, mutagenic or possibly teratogenic,
several countries have recently laid down standards for various DBP
levels. This stimulated the search for mathematical models to describe
or predict DBP formation in disinfected water and to evaluate the
effectiveness of water treatment technologies designed to reduce DBP
levels so as to comply with the standards. Most of these models are
based on fitting mathematical equations to various empirical
observations, rather than mechanistic and kinetic considerations. This
is mainly due to the complexity of the reactions between organic
precursors and disinfecting chemicals, which usually involve several
parallel pathways leading to a great variety of products. The
complexity of the DBP formation reactions also makes it difficult to
develop universally applicable models for simulating DBP formation
potential associated with disinfection of a diverse array of natural
source waters. However, the analysis presented by many models suggests
that many waters exhibit comparable general responses to changes in a
given parameter (i.e., responses lending themselves to simulation by a
particular mathematical functionality), although specific responses
associated with individual waters may vary. The multiple regression
models developed by many researchers represent a rational framework
for modelling DBP formation in many sources. Another potential
application is the modelling of DBP mixtures, e.g., predicting HAA
levels from THM and water quality data.
2.12.1 Factors affecting disinfectant by-product formation and
variables of interest in disinfectant by-product modelling
The information on the factors controlling DBP formation, which
is available in the literature, is briefly summarized below.
The extent of formation of DBPs is dependent on several water
quality parameters, such as TOC concentration, UVA254, bromide
concentration and temperature. It is also dependent on chlorination
conditions, such as chlorine dose, pH, ammonia concentration and
contact time. After the various statistically significant factors were
identified, mathematical equations were developed to describe the
formation of various DBPs. A least squares method was used to
determine the optimum equation coefficients that best describe the
experimental data. The optimum coefficients have been defined as those
that produce a minimum residual error between the mathematical
predictions and the experimental data.
2.12.2 Empirical models for disinfectant by-product formation
Numerous models for predicting THM formation through chlorination
have been reported (Moore et al., 1979a; Kavanaugh et al., 1980;
Engerholm & Amy, 1983; Urano et al., 1983; Amy et al., 1987, 1998;
Morrow & Minear, 1987; AWWARF, 1991; Hutton & Chung, 1992). Of these,
models reported by AWWARF (1991) and Amy et al. (1998) are more recent
and were derived from a variety of natural source waters and more
realistic treatment conditions. Not much information has been reported
on the formation of other chlorination DBPs. Only Amy et al. (1998)
summarized empirical models for THMs, HAAs and chloral hydrate. These
chlorination by-product models can be used to assess both in-plant and
distribution system formation of THMs, HAAs and chloral hydrate. Water
quality conditions such as DOC, pH, temperature and bromide are needed
as inputs to the models; such data then allow assessment of
chlorination DBP formation as a function of reaction time:
DBP concentration (total THMs or THM species, total HAAs or HAA
species, or chloral hydrate) =
f(TOC, bromide, chlorine, pH, temperature, time)
Relatively little is known about the kinetics of the formation of
bromate and other DBPs during ozonation and the quantitative effects
of water quality factors (temperature, pH, etc.); such an
understanding is crucial for evaluating various bromate control
strategies. Siddiqui & Amy (1993) and Amy et al. (1998) developed
statistical relations to predict the concentrations of various ozone
DBPs, including bromate, as a function of water treatment variables.
Correlation matrix analysis has shown that ozone dose, dissolved ozone
concentration, bromide concentration, pH and reaction time all have a
positive influence on bromate formation. Von Gunten & Hoigne (1994)
developed kinetic models for bromate formation.
Ozone, as a result of its strong oxidizing power, produces a
variety of organic by-products, such as aldehydes and ketoacids, when
used to treat natural source waters. These by-products -- especially
aldehydes -- are highly biodegradable, and there is concern for
regrowth of microorganisms following ozone treatment. They are also
potentially hazardous and may produce increased amounts of chlorinated
by-products upon chlorination. Siddiqui et al. (1997) developed a
model to estimate the potential for total aldehyde formation in source
waters upon ozonation.
2.12.3 Models for predicting disinfectant by-product precursor
removal
It is recognized that chlorination will continue to be the most
common disinfection process; hence, enhanced removal of DBP precursors
present in raw sources represents a valuable option for reducing the
potential for by-product formation. The removal of NOM can be achieved
either by providing additional processes, such as GAC and
nanofiltration, or by enhancing the existing coagulation, flocculation
and sedimentation processes. Predictive models have been developed for
assessing coagulation efficiency in removing NOM and reducing DBP
precursor levels (AWWARF, 1991; Amy et al., 1998).
Coagulation can reduce DOC and DBP precursors but not bromide
levels; hence, a greater proportion of brominated DBP species can
potentially be produced in the finished water.
The effects of precursor removal by chemical coagulation can be
assessed through the use of treated water models. One can either
predict DBPs formed under a given degree of precursor removal or
define the degree of precursor removal required to meet DBP
regulations. The impact of bromide ion on meeting regulations can also
be assessed. If one makes the assumption that precursor reactivity
(i.e., DBP/DOC) does not change, one can also assess other precursor
removal processes, such as GAC or membrane processes, through use of
the raw/untreated water models. Care should be exercised when using
models to approximate post-chlorination DBPs following an ozonation
step.
2.13 Summary
* The primary and most important role of drinking-water treatment is
to remove or inactivate harmful microorganisms. Another role is to
minimize the concentrations of disinfectants and DBPs without
compromising in any way the removal or inactivation of pathogens.
* Drinking-water utility managers must be more knowledgeable about
options to meet regulations. It is often more practical to use
treatment methods that control the concentration of several
contaminants than to modify treatment practices for each new
standard that is promulgated.
* A thorough understanding of DBP formation would help the successful
balancing of appropriate microbial inactivation with the
minimization of DBPs. Water quality variables affect DBP formation
and must be considered when developing a strategy to control DBPs
with various disinfectants.
* The chemistry of chlorine and its by-products has been well
studied, and ozone and its by-products have recently received much
attention. Studies of chlorine dioxide and chloramines and their
by-products are relatively few, although more work in these areas
is now being undertaken.
* One of the simplest processes to minimize halogenated DBP formation
is limiting the free chlorine contact time by using monochloramine
to maintain a distribution system residual following primary
disinfection by chlorine or ozone. Chloramines are an effective
means of controlling DBPs. However, the growth of nitrifying
bacteria (and related production of nitrite) is a potential problem
in chloraminated water supplies.
* Various nitrogen-containing organic compounds may be present in
source waters after chlorination and chloramination. Because of
analytical complexities, very few detailed studies have been
undertaken to determine the individual compounds present and their
concentrations.
* Many factors between the source and the tap can influence the DBPs
to which consumers are exposed. Although THMs and HAAs continue to
form with increasing contact time, some other halogenated DBPs,
such as HANs and haloketones, form rapidly but then decay in the
distribution system as a result of hydrolysis. This has major
implications regarding exposure to these DBPs, depending upon their
proximity to the treatment plant. For treated source waters, median
levels of HAAs are often approximately one-half of the median THM
levels.
* For low-bromide source waters, chloroform is normally the dominant
THM species; DCA and TCA are the most prevalent HAA species; DCAN
is the most prevalent HAN species; and 1,1,1-TCPN is the most
prevalent of the two measured haloketones. Very low levels of
chloropicrin have been observed by various researchers; this
compound appears to form slowly during the incubation period, with
concentrations tending to level off at 40 h. For high-bromide
waters, increased levels of brominated DBPs are observed.
* Chlorine dioxide is a strong oxidant that under certain conditions
surpasses chlorine in its ability to destroy pathogenic organisms.
When chlorine dioxide is prepared and administered without excess
free chlorine, THMs and other chlorinated by-products are not
produced, but inorganic by-products are formed.
* TOC levels have been found to be correlated with halogenated DBP
formation. The nature of this relationship varies with the source.
TOC removal can be used as a surrogate for the reduction of DBP
formation.
* Although the presence of chloral hydrate and HANs in chlorinated
samples may be attributed to precursors other than amino acids, the
potential for amino acids to be present in natural sources is well
documented. Surface waters, but not groundwaters, tend to contain
amino acids. However, the removal of these precursors during
conventional water treatment is not well understood.
* The amount of chlorate that is present in delivered hypochlorite
solutions depends on many factors. Freshly made hypochlorite
solutions will contain less chlorate than hypochlorite that is
stored without concern for temperature and pH. If a utility is
using a single tank to store hypochlorite, it is likely that the
level of chlorate is increasing in the tank. Thus, storage tanks
should be periodically flushed and cleaned, and, if possible, the
storage time should be reduced.
* Models have been developed that can be used to simulate the fate
and movement of DBP precursors in distribution systems. The models
can be designed as a planning tool for evaluating the impacts of
source water management strategies and estimating DBP exposures.
Some limitations of existing models include calibration with a
limited database, application to only a specific water source or
group of related sources, lack of terms to simulate important
parameters, such as reaction time, and inadequate validation.
3. TOXICOLOGY OF DISINFECTANTS
In assessing the hazards associated with drinking-water
disinfection, it is important not to neglect the disinfectants
themselves. Adding disinfectant in excess of the demand has several
practical benefits. First, it ensures that reaction of the
disinfectant with DBP precursors (largely organic material and
ammonia) does not shorten contact time to the point of ineffective
disinfection. Second, residual disinfectant helps to prevent regrowth
of organisms in the remaining portions of the treatment and
distribution systems.
The result of this practice, however, is that one of the
chemicals that is present in the finished water at the highest
concentration is the disinfectant. In the present regulatory climate
in many countries, chemicals that are introduced as direct additives
to food would be subjected to a significant amount of toxicological
screening before they could be used. Since the major disinfectants
were introduced almost 100 years ago, they were subjected to much less
thorough toxicological evaluations than would be required today.
However, many of these data gaps have been addressed in the past
decade.
3.1 Chlorine and hypochlorite
3.1.1 General toxicological properties and information on
dose-response in animals
Chlorine gas has long been recognized as a lung irritant. This
topic will not be reviewed in the present document, as it appears to
be largely irrelevant to the small amounts of chlorine that are
volatilized from chlorinated water in showers or other points of use
in the household. In water treatment plants, however, there is a
possibility of occupational exposures that could have severe sequelae.
For information on these higher-level exposures, the interested reader
is referred to a recent review by Das & Blanc (1993). The effects of
chlorine gas that have been observed in humans will be discussed in
section 3.1.3.
Sodium hypochlorite (NaOCl) or calcium hypochlorite (Ca(OCl)2)
solutions have also been utilized extensively in the disinfection of
drinking-water. The stock solutions used for this purpose are highly
caustic and are a clear concern for occupational exposures. The
concentration required to produce irritation and decreased basal cell
viability in the skin of guinea-pigs after an application period of
2 weeks was 0.5% sodium hypochlorite (Cotter et al., 1985). Reducing
the concentration to 0.1% resulted in no effect on basal cell
viability relative to control animals. Yarington (1970) demonstrated
that instillation of bleach into the oesophagus of dogs produced
irritation. The minimal exposure that produced oesophageal burns was
10 ml of commercial bleach with a 5-min exposure. It should be noted
that the highly alkaline pH (about pH 11) of sodium hypochlorite is
not likely to be encountered in drinking-water.
There have been relatively few evaluations of the effects of
chlorine or hypochlorite in drinking-water. The present review will
focus on studies with treatment periods longer than 4 weeks where
drinking-water was the primary route of exposure. Reference to earlier
studies of shorter duration and less general applicability to a safety
evaluation can be found in previous reviews (Bull, 1980, 1982a,b,
1992; Bull & Kopfler, 1991).
Daniel et al. (1990a) evaluated the toxicity of solutions of
chlorine prepared by bubbling chlorine gas into distilled water and
adjusting the pH to 9.4. The nominal concentrations of chlorine used
were 0, 25, 100, 175 or 250 mg/litre in distilled water (approximately
0, 3, 10, 16 or 21 mg/kg of body weight per day). These solutions were
provided as drinking-water to both male and female Sprague-Dawley rats
(10 per sex per dose) for 90 days. No deaths occurred in any treatment
group. However, there were statistically significant decreases in
drinking-water consumption in females treated with 100 mg/litre and
higher, probably due to decreased palatability. There were no
consistent effects of chlorine treatment on organ to body weight
ratios or clinical chemistry parameters. A no-observed-effect level
(NOEL) of 10 mg/kg of body weight per day was identified by the
authors based on reduced body weight gain. However, since this was
associated with reduced palatability of the drinking-water, it is not
considered to be a true toxicological end-point.
The study in rats was followed up with another study in B6C3F1
mice (Daniel et al., 1991a). Male and female B6C3F1 mice (10 per sex
per group) were administered 12.5, 25, 50, 100 or 200 mg of chlorine
per litre of drinking-water for 90 days (calculated mean daily doses
were 2.7, 5.1, 10.3, 19.8 or 34.4 mg/kg of body weight in males and
2.8, 5.8, 11.7, 21.2 or 39.2 mg/kg of body weight in females). Spleen
and liver weights were depressed in males, but not in females, at the
highest dose rates (100 and 200 mg/litre). There were no other
consistent indications of target organ effects based on serum enzyme
concentrations. No gross or microscopic lesions could be related to
treatment with chlorine.
Several of the following studies utilized solutions of sodium
hypochlorite as the treatment chemical. It is now known that such
solutions can contain very high concentrations of chlorate within a
short time of their preparation (Bolyard et al., 1993). The extent of
this contamination has not been reported.
Hasegawa et al. (1986) examined the effects of much higher
concentrations (0.025, 0.05, 0.1, 0.2 or 0.4%) of sodium hypochlorite
(equivalent to 7, 14, 28, 55 and 111 mg/kg of body weight per day)
administered in drinking-water to male and female F344 rats for
13 weeks. Twenty rats of each sex were assigned to each experimental
group. Significant suppression of body weight (as a result of
decreased consumption of water and food) occurred at 0.2% and above.
The authors noted slight damage to the liver as indicated by increased
levels of serum enzymes (not specified) at 0.2% and 0.4% sodium
hypochlorite in both sexes. No evidence of treatment-related pathology
was observed in this study or in a 2-year study in which males were
subjected to 0.05% or 0.1% (13.5 or 27.7 mg/kg of body weight per day)
and females to 0.1% or 0.2% (34 or 63 mg/kg of body weight per day)
sodium hypochlorite. The extended exposures were conducted with
50 animals of each sex per treatment group. Analysis of dosing
solutions was not reported.
In a 2-year bioassay, the National Toxicology Program (NTP)
examined chlorine at 0, 70, 140 or 275 mg/litre (expressed as atomic
chlorine, Cl) in drinking-water of F344 rats and B6C3F1 mice (70 per
sex per group) (NTP, 1992). These solutions were prepared from gaseous
chlorine and neutralized to pH 9 by the addition of sodium hydroxide.
Stability studies indicated that 85% of the initial target
concentration remained after 3 days of preparation. Stock solutions
(concentrations not specified) were prepared once weekly, and
solutions for drinking were prepared 4 times weekly. Based on body
weight and water consumption, doses in these studies were
approximately 0, 4, 7 or 14 mg/kg of body weight per day for male
rats; 0, 4, 8 or 14 mg/kg of body weight per day for female rats; 0,
7, 14 or 24 mg/kg of body weight per day for male mice; and 0, 8, 14
or 24 mg/kg of body weight per day for female mice. The only
treatment-related non-tumour pathology was found to be a dilatation of
renal tubules in male mice receiving 275 mg/litre for more than 66
weeks. No non-neoplastic lesions were observed in either male or
female rats.
A number of immunological changes have been associated with the
treatment of rodents with sodium hypochlorite in drinking-water. Water
containing 25-30 mg of sodium hypochlorite per litre was found to
reduce the mean number of peritoneal exudate cells recovered from
female C57BL/6N mice after 2 weeks of treatment. This was, in turn,
associated with a significant decrease in macrophage-mediated
cytotoxicity to melanoma and fibrosarcoma cell lines (Fidler, 1977).
The treatment period was increased to 4 weeks in a subsequent study,
which demonstrated that 25 mg of sodium hypochlorite per litre
decreased the ability of peritoneal macrophages to phagocytose
51Cr-labelled sheep red blood cells. Macrophages obtained from the
mice treated with hypochlorite were found to be less effective in
destroying B16-BL6 melanoma cells in vitro. Mice so treated were
also found to have increased pulmonary metastasis of B16-BL6 cells
when they were introduced by subcutaneous injection (Fidler et al.,
1982).
Exon et al. (1987) examined the immunotoxicological effects of
sodium hypochlorite at 5, 15 or 30 mg/litre (0.7, 2.1 or 4.2 mg/kg of
body weight per day) in the drinking-water of male Sprague-Dawley rats
(12 per dose) for 9 weeks. Delayed hypersensitivity reaction to bovine
serum albumin was observed at the highest dose administered. Oxidative
metabolism by adherent resident peritoneal cells was decreased at 15
and 30 mg/litre, and the prostaglandin E2 levels of these cells were
found to be significantly elevated. No effects on natural killer cell
cytotoxicity, antibody responses, interleukin 2 production or
phagocytic activity were observed. The effects on macrophage activity
suggest that some impairment does occur at relatively low levels of
sodium hypochlorite. As pointed out by the authors, these were
relatively mild effects, the significance of which was unknown. It is
not clear that these effects would be translated into a significant
impairment of the immune response to a particular infectious agent.
However, modification of macrophage function appears to be one of the
most sensitive responses identified in studies of chlorine or
hypochlorite in experimental animals. A study in which female C57BL/6
mice were administered hypochlorite in their drinking-water (7.5, 15
or 30 mg of hypochlorite per litre) for 2 weeks showed no effects on
the immune system as measured by spleen and thymus weight,
plaque-forming cell response, haemagglutination titre and lymphocyte
proliferation (French et al., 1998).
Altered liver lipid composition has been observed as a result of
acute intragastric administration of sodium hypochlorite (5 ml of a 1%
solution) to rats (Chang et al., 1981). These data do not provide a
clear indication of whether these effects might give rise to
pathology. The concentrations of hypochlorite utilized were much
greater than those that would be encountered in drinking-water.
The effects of hypochlorous acid and hypochlorite on the skin
have received relatively little attention despite the current interest
in bathing as a significant source of chemical exposure from
drinking-water. Robinson et al. (1986) examined the effects of both
hypochlorite and hypochlorous acid solutions applied to the skin of
the entire body of female Sencar mice except for the head. Exposures
were to 1, 100, 300 or 1000 mg/litre as hypochlorous acid at pH 6.5
for 10 min on 4 consecutive days. Hypochlorite (formed by raising the
pH to 8.5) was studied only at 1000 mg/litre. Significant increases in
epidermal thickness and cell counts within the epidermal layer were
observed at concentrations of hypochlorous acid (pH 6.5) of 300
mg/litre and above, but the thickness of the skin was not
significantly different from that in animals at 100 mg/litre. The
increases in skin thickness were associated with an epidermis whose
thickness was increased to 4-6 cells as compared with the normal 1-2
cells seen in mice. The effects of hypochlorite were much less marked.
Following a single application, the increased thickness of the skin
observed in mice exposed to hypochlorous acid (i.e., pH 6.5) did not
appear until 4 days after the treatment. This differed from
hypochlorite, other disinfectants and the positive control,
12- O-tetradecanoylphorbol-13-acetate (TPA). In the latter cases, the
maximal response was observed within 24-48 h after treatment. The
hyperplastic response to hypochlorous acid required 12 days to return
to normal. This study suggests a considerable margin of safety between
the concentrations of chlorine required to produce hyperplasia and
those that are found in drinking-water.
The reactive nature of chlorine always raises questions of
whether it is chlorine or a by-product that is responsible for any
effect. Several studies have examined the formation of by-products in
the gastrointestinal tract following the administration of chlorine.
Invariably, these studies have involved the administration of chlorine
or hypochlorite by gavage at very high concentrations relative to the
amounts that would be encountered in chlorinated drinking-water. As a
consequence, the by-products formed following gavage dosing of high
concentrations may not be representative of the by-products that would
be seen following the consumption of modest to moderate levels of
chlorine in larger volumes of water. A particular issue is that the
high organic carbon concentration relative to chlorine that would be
encountered in the gastrointestinal tract when water is consumed at
low concentrations should dissipate disinfectant before sufficient
oxidative power would be present to break down substrates to small
molecules. Despite these design flaws, the data do indicate that
by-products are formed. The bulk of them remain as higher molecular
weight products, which may have little toxicological importance.
Vogt et al. (1979) reported that chloroform could be measured in
the blood, brain, liver, kidneys and fat of rats to which sodium
hypochlorite was administered by gavage at doses of 20, 50 or 80 mg in
5 ml of water. Thus, the by-product chloroform can be formed by the
reaction of chlorine with stomach contents.
Mink and co-workers (1983) pursued this observation and found
that other by-products could be detected in the stomach contents and
plasma of rats that had been administered sodium hypochlorite
solutions neutralized to pH 7.9. In addition to chloroform, DCAN, DCA
and TCA were detected in the stomach contents analysis. DCA and TCA
were also detected in blood plasma.
The third group of compounds identified as by-products of
chlorination in stomach contents of the rat are the organic
N-chloramines (Scully et al., 1990). N-Chloroglycine,
N-chloroleucine or N-chloroisoleucine and N-chlorophenylalanine
were confirmed products of reactions with normal amino acids that
would ordinarily be found in the gastrointestinal tract.
N-Chlorovaline and N-chloroserine were also tentatively
identified. Organic chloramines are reactive and could be responsible
for toxic effects that may be attributed to chlorine in toxicological
studies. The chlorine demand of free amino acids in stomach contents
was found to be only about 4% of the total. Consequently, this process
may be substrate-limited at concentrations of chlorine found as
residuals in drinking-water. However, use of higher concentrations of
chlorine would also lead to breakdown of proteins present in the
stomach fluid. Thus, as concentrations are increased to levels that
would be used in animal studies, these products would be formed at a
much higher concentration, similar to the phenomena noted with THM and
HAA by-products.
3.1.2 Reproductive and developmental toxicity
In general, animal studies have demonstrated no reproductive or
teratogenic effects of chlorine. Druckrey (1968) examined the effects
of water chlorinated to a level of 100 mg/litre (approximately 10
mg/kg of body weight per day) in BDII rats for seven generations. No
effects were observed on fertility, growth or survival.
A number of subsequent studies have studied the effects of
chlorine or hypochlorite on more specific aspects of reproduction or
development. Meier et al. (1985b) reported that oral administration of
sodium hypochlorite (pH 8.5) prepared from chlorine gas and
administered at 4 or 8 mg/kg of body weight per day for 5 weeks
increased the incidence of sperm head abnormalities in B6C3F1 mice
(10 animals per group). The effect was not observed when the solutions
were administered at pHs at which hypochlorous acid was the
predominant species (pH 6.5). However, other studies have not been
able to associate adverse reproductive outcomes with the
administration of chlorine or sodium hypochlorite.
Carlton et al. (1986) found no evidence of sperm head
abnormalities or adverse reproductive outcomes in Long-Evans rats.
Male rats were treated for 56 days prior to mating and female rats
from 14 days prior to mating through gestation. Each experimental
group consisted of 11-12 males and 23-24 females. Solutions of
chlorine were prepared at pH 8.5, so the study evaluated hypochlorite
as the dominant form in the drinking-water. Doses were as high as
5 mg/kg of body weight per day.
3.1.3 Toxicity in humans
There have been significant human exposures to chlorine and
hypochlorite solutions. Much of that experience is with inhalation of
chlorine gas, which is known to be a strong respiratory irritant.
Chlorine gas is also the largest single component involved in toxic
release incidents. A third major source of exposure is solutions of
sodium hypochlorite, usually marketed as bleach. Bleach is frequently
involved in human poisonings. These exposures are not particularly
relevant to exposures to chlorine or hypochlorite in drinking-water.
Therefore, only a few case reports are identified that illustrate the
types of problems that have been encountered. There was no attempt to
make this review comprehensive.
The irritating effects of chlorine gas have been well documented
because of its use as a chemical warfare agent during World War I (Das
& Blanc, 1993). In a follow-up of survivors of gassing, it was
concluded that there was no evidence of permanent lung damage;
however, these studies clearly indicated that survivors had breath
sounds that suggested bronchitis and limited chest and diaphragmatic
movement, even emphysema. Most studies suggested that there were high
incidences of acute respiratory disease and a lesser prevalence of
chronic sequelae. Similar sequelae have been identified following
exposure of humans to accidental releases of chlorine gas. In these
more modern characterizations, the acute signs and symptoms included a
high incidence of pulmonary oedema and severe bronchitis. These signs
and symptoms are of generally short duration and resolve themselves
over the course of about 1-4 weeks. However, chronic sequelae are
observed in some individuals, depending in part upon the severity of
the exposure. In such cases, a decrease in the forced expiratory
volume is the most consistently reported clinical sign.
Two recent reports suggest that chronic sequelae to acute
exposures to chlorine gas may be more prevalent than previously
appreciated. Moore & Sherman (1991) reported on an individual who was
previously asymptomatic and who developed chronic, recurrent asthma
after exposure to chlorine gas in an enclosed place. Schwartz et al.
(1990) followed 20 individuals who had been exposed to chlorine gas in
a 1975 incident. The prevalence of low residual lung volume was
increased during the follow-up period. Sixty-seven per cent of those
tested were found to have residual volumes below 80% of their
predicted values. Five of 13 subjects tested for airway reactivity to
methacholine were found to have a greater than 15% decline in forced
expiratory volume.
Controlled studies have been conducted in healthy, non-smoking
men exposed to chlorine gas at 1.5 and 2.9 mg/m3 (0.5 and 1.0 ppm)
for 4 or 8 h (reviewed in Das & Blanc, 1993). Four hours of exposure
to 2.9 mg/m3 (1.0 ppm) produced significant decreases in the forced
expiratory volume. One individual who was found to be experiencing
more difficulty than other subjects at this dose and who was later
exposed to 1.5 mg/m3 (0.5 ppm) experienced a significant decrease in
forced expiratory volume. While 1.5 mg/m3 (0.5 ppm) appears
protective for most people, some more sensitive individuals may in
fact have more significant responses to chlorine gas.
The effects of chronic exposure to chlorine gas have received
only limited study. In one study of paper mill workers, a more rapid
age-related decrease in lung volumes of workers exposed to chlorine
relative to those exposed to sulfur dioxide was noted, but the trend
was not statistically significant (Das & Blanc, 1993). Other studies
failed to identify chronic sequelae.
There are frequent reports of human poisonings from bleach. Most
often these exposures result from the mixing of bleach with acidic
products or ammonia. Acidification converts hypochlorite to
hypochlorous acid, which dissociates to chlorine gas, offgasses very
rapidly from the solutions and presents an inhalation exposure (MMWR,
1991). Mixing bleach with ammonia results in the formation of
monochloramine and dichloramine, both of which are effective
respiratory irritants (MMWR, 1991).
Any potential effects of chlorine or hypochlorite in
drinking-water are obscured by the fact that by-products inevitably
coexist with the residual chlorine. One series of studies in which
by-products formed were minimized by dissolving chlorine in distilled
water attempted to identify effects of chlorine in drinking-water on
humans. Chlorine in drinking-water was administered in a rising-dose
tolerance study beginning with 0.1 mg/litre in two 500-ml portions and
rising to a concentration of 24 mg/litre, equivalent to 0.34 mg/kg of
body weight per day (Lubbers & Bianchine, 1984). No clinically
important changes were observed. No findings of clinical importance
were identified in a follow-up treatment with repeated dosing with
500-ml portions of a solution containing 5 mg of chlorine per litre
for a 12-week period (Lubbers et al., 1984a).
Another study attempted to determine whether consumption of
chlorinated drinking-water affected blood cholesterol levels (Wones et
al., 1993a). The impetus for this study was a toxicological study in
pigeons that suggested that chlorine raised blood cholesterol levels
and modified serum thyroid levels (Revis et al., 1986a,b) and an
epidemiological study that associated small increases in cholesterol
of women with residence in communities having chlorinated water
(Zeighami et al., 1990a,b; described in detail in section 5.2.2). A
prior study (quoted in Wones et al., 1993a) was conducted that
examined men who consumed water containing 2, 5 or 10 mg of chlorine
per litre and found a small increase in serum cholesterol levels at
the highest dose group. However, no control group was studied, so the
changes could have been attributed to the change in diet imposed as
part of the study protocol (Wones & Glueck, 1986). The longer-term
study was composed of 30 men and 30 women who received a controlled
diet for the duration of the study. The first 4 weeks represented an
acclimatization period during which all subjects received distilled
water. Half the subjects were assigned to a group that consumed 1.5
litres of water containing 20 mg of chlorine per litre for the
following 4 weeks. At the end of each 4-week period, blood was
analysed for cholesterol, triglycerides, high-density lipoprotein
(HDL) cholesterol, low-density lipoprotein (LDL) cholesterol or
apolipoproteins A1, A2 and B. There were no significant effects. There
was a slight trend towards lower thyroid hormone levels in men
consuming chlorine, but this was not clinically significant (Wones et
al., 1993a). These data suggest that observations obtained previously
in pigeons could not be repeated under comparable conditions of
chlorine consumption. It is notable that the animals utilized in the
original pigeon study had consumed a modified diet (Revis et al.,
1986a) that was deficient in calcium and other trace metals. A
subsequent study failed to replicate the previous results in pigeons
(Penn et al., 1990).
3.1.4 Carcinogenicity and mutagenicity
The International Agency for Research on Cancer (IARC) has
evaluated the carcinogenicity of hypochlorite salts and concluded that
there were no data available from studies in humans on their
carcinogenicity and inadequate evidence for their carcinogenicity in
experimental animals. Hypochlorite salts were assigned to Group 3: the
compounds are not classifiable as to their carcinogenicity to humans
(IARC, 1991).
Several studies have shown that sodium hypochlorite produces
mutagenic responses in bacterial systems and mammalian cells
in vitro. However, there is no evidence of activity in mammalian
test systems in vivo. It is not clear to what extent this is
influenced by the formation of mutagenic by-products as a result of
reactions with components of the incubation media. Wlodkowski &
Rosenkranz (1975) used short-term exposures of Salmonella
typhimurium strain TA1530 followed by ascorbic acid-induced
decomposition to reduce the cytotoxic effects of hypochlorite. The
investigators applied 0.14 µmol per tube and added ascorbic acid after
intervals of 5, 10 and 15 min. At 5 min, a clear positive response was
observed with minimal cytotoxicity. Significant responses were also
observed in strain TA1535, but not in strain TA1538.
Rosenkranz (1973) and Rosenkranz et al. (1976) also demonstrated
a positive mutagenic response in DNA polymerase A deficient
Escherichia coli to 0.006 µmol of sodium hypochlorite. This response
was unaffected by the addition of catalase, suggesting that the
response was not related to the generation of hydrogen peroxide.
Matsuoka et al. (1979) reported that sodium hypochlorite at a
concentration of 6.7 mmol/litre (0.5 mg/ml) produced chromosomal
aberrations in Chinese hamster ovary (CHO) cells in the presence of S9
mix. This concentration was cytotoxic in the absence of S9. Some
concern must be expressed about whether responses observed with such
high and clearly cytotoxic concentrations in an in vitro system
represent specific clastogenic effects. The authors report only one
concentration tested with and without S9. It is probable that the
positive response in the presence of S9, if it is a specific response,
was a result of detoxifying hypochlorite. This protection could be
non-specific as well, in that it may not have depended upon any
catalytic activities present in the S9 fraction (i.e., the added
protein may have acted as a reactive sink to dissipate excess
hypochlorite). Consequently, it is difficult to use these data in
interpreting the effects of chlorine or hypochlorite in vivo.
Ishidate (1987) studied the induction of chromosomal aberrations
in cultures of Chinese hamster CHL cells at sodium hypochlorite
concentrations ranging from 125 to 500 µg/ml without exogenous
metabolic activation and from 31 to 125 µg/ml with and without rat
liver S9 mix. A clear increase in the number of cells with structural
chromosomal aberrations was observed at 500 µg/ml without S9 mix,
while the results obtained in the other series, showing weakly
positive responses, were considered inconclusive.
Meier et al. (1985b) evaluated the ability of hypochlorite and
hypochlorous acid to induce chromosomal damage or micronuclei in the
bone marrow of CD-1 mice. The samples to be tested were generated by
bubbling chlorine gas into water and then adjusting the pH to 6.5
(predominantly hypochlorous acid) or 8.5 (predominantly hypochlorite).
The doses administered were 1.6, 4 or 8 mg/kg of body weight for 5
consecutive days. There was no evidence of increased micronuclei or
chromosomal abnormalities in bone marrow cells. Significant positive
responses were observed with positive control chemicals in both
assays. As reported in section 3.1.2, these authors detected a
positive response in the sperm head abnormality assay in mice treated
at these same doses of hypochlorite in two separate experiments. This
assay is used primarily as a mutagenicity assay rather than as an
assay for reproductive toxicities. Hypochlorous acid had no effect in
the sperm head abnormality assay.
Tests of the ability of hypochlorite to induce cancer in rodents
were conducted in F344 rats by Hasegawa et al. (1986). Sodium
hypochlorite concentrations of 0, 500 or 1000 mg/litre (males) and 0,
1000 or 2000 mg/litre (females) were administered in the
drinking-water for 104 weeks (equivalent to 13.5 and 27.7 mg/kg of
body weight per day for males and 34.3 and 63.2 mg/kg of body weight
per day for females). There were 50 male and 50 female rats assigned
to each experimental group. No tumours could be attributed to sodium
hypochlorite administration.
NTP (1992) conducted a 2-year bioassay of chlorine in F344 rats
and B6C3F1 mice. The concentrations administered in drinking-water
were 0, 70, 140 or 275 mg/litre, and there were 70 animals of each sex
assigned to each group (approximately 0, 4, 8 or 14 mg/kg of body
weight per day for rats and 0, 7, 14 or 24 mg/kg of body weight per
day for mice). There was an apparent positive trend in the induction
of stromal polyps of the uterus of female mice treated with chlorine,
but this was considered unlikely to be treatment-related because the
incidence was below those observed in historical controls. In female
rats, there was an increase in mononuclear cell leukaemia at both 140
and 275 mg/litre (8 and 14 mg/kg of body weight per day). However, the
response was not considered treatment-related because it fell within
the range of historical controls, there was no apparent dose-response,
and there was no evidence for such an increase in male F344 rats.
A single study suggested that sodium hypochlorite could act as a
promoter of skin tumours following initiation with
4-nitroquinoline-1-oxide in female ddN mice (Hayatsu et al., 1971). A
solution of sodium hypochlorite that contained 10% effective
concentrations of chlorine was utilized. Skin tumours were produced in
9 of 32 mice given 45 applications of sodium hypochlorite following
initiation. Sodium hydroxide solutions were utilized as a control for
the alkaline pH of sodium hypochlorite and produced no tumours. No
tumours were observed with 60 applications of sodium hypochlorite
solution in non-initiated mice. Pfeiffer (1978) conducted a much
larger experiment that utilized 100 mice per group. This author found
that a 1% sodium hypochlorite solution applied alternately with
benzo [a]pyrene for 128 weeks was ineffective in producing skin
tumours in female NMRI mice above those that had been initiated with
benzo [a]pyrene alone at doses of 750 or 1500 µg. Pretreatment with
the sodium hypochlorite solution before application of the
benzo [a]pyrene actually reduced tumour yields at 128 weeks with
doses of either 750 or 1500 µg of benzo [a]pyrene. Sodium
hypochlorite used in a more traditional initiation/promotion study
(i.e., sodium hypochlorite treatment following initiation with
benzo [a]pyrene) produced a decrease in the tumour yield with the 750
µg dose of benzo [a]pyrene, but had no effect following 1500 µg.
Thus, the ability of sodium hypochlorite to act as a tumour promoter
may depend upon the initiator used, or the smaller experiment of
Hayatsu et al. (1971) may simply be a false result.
As pointed out in section 3.1.1, application of solutions of
hypochlorous acid to the skin of Sencar mice results in the
development of hyperplasia. The concentrations required are
considerably lower (300 mg/litre) (Robinson et al., 1986) than those
used in the studies of either Hayatsu et al. (1971) or Pfeiffer
(1978). Sodium hypochlorite was also effective at lower doses, but
less so than equivalent concentrations of hypochlorous acid. These
results suggest that these prior evaluations may have been conducted
at too high a dose. There appear to be no reports on the effectiveness
of hypochlorous acid as a tumour promoter, but the lack of activity at
doses of less than 300 mg/litre would suggest that this is of no
concern.
3.1.5 Comparative pharmacokinetics and metabolism
A series of pharmacokinetic studies using 36Cl-labelled
hypochlorous acid were conducted by Abdel-Rahman and co-workers
(1983). These studies are of limited value because the form of 36Cl
could not be determined in various body compartments.
3.1.6 Mode of action
There are no specific toxicities of chlorine for which a
mechanism needs to be proposed. It is a strong oxidizing agent, and it
must be presumed that damage induced at high doses by either gaseous
chlorine or solutions of hypochlorite is at least partially related to
this property. In studies in which sodium hypochlorite is used without
neutralization, a strong alkaline pH can also contribute to its
effects. There is always the possibility that chlorine is inducing
subtle effects by virtue of its reaction with organic compounds that
are found in the stomach. Such reactions have been demonstrated, but
there is no convincing evidence to date that any specific toxicity can
be attributed to these by-products.
3.2 Chloramine
3.2.1 General toxicological properties and information on
dose-response in animals
There have been relatively few evaluations of the toxic
properties of chloramine in experimental animals. In large part this
is because it is not marketed as a product but is created for
disinfection purposes on-site and in situ. Chloramine is primarily
used as a residual disinfectant in the distribution system. The final
solution consists of mostly monochloramine, with traces of other
chloramines, such as dichloramine. Chloramines, as a group, are
generally recognized as potent respiratory irritants, because the
formation of these compounds when household bleach and ammonia are
mixed results in a number of poisoning cases each year (MMWR, 1991).
In spite of this, there has been no attempt to quantify dose-response
relationships in animals.
Eaton et al. (1973) investigated concerns about
chloramine-induced methaemoglobin formation in kidney patients
dialysed with chloramine-containing water. This was done by examining
the ability of relatively large volumes of tapwater to oxidize
haemoglobin in dilute suspensions of red blood cells. This
circumstance is reflective of dialysis, but not of the concentrated
suspension of these cells in vivo. Nevertheless, the authors were
able to show that methaemoglobin formation occurred in a dose-related
manner when 1 volume of red blood cells (human) was suspended in 100
volumes of tapwater containing 1 mg of chloramine per litre or above.
This effect was not produced by comparable concentrations of sodium
hypochlorite. The ability to induce methaemoglobin formation was
eliminated by treating the water by reverse osmosis followed by carbon
filtration. Clearly, chloramine is capable of inducing
methaemoglobinaemia at low concentrations when there is a large
reservoir of chloramine. This is a decidedly different exposure
pattern from that of normal humans and other mammals, as they consume
small volumes of water relative to the volume of red blood cells that
are exposed.
Moore et al. (1980a) studied alterations of blood parameters in
male A/J mice treated with 0, 2.5, 25, 50, 100 or 200 mg of
monochloramine per litre in carbonate/bicarbonate-buffered (pH 8.9)
drinking-water. Twelve animals were assigned to each group, and
treatments were maintained over a 30-day period. Consistent with the
interpretation provided above, there were no treatment-related effects
on osmotic fragility, methaemoglobin levels, haemoglobin
concentrations, reticulocyte counts or a number of other derived
parameters. Haematocrits of mice treated with 50, 100 or 200 mg/litre
were actually higher than those observed in control mice. White blood
cell counts were not altered in these animals.
Daniel et al. (1990a) conducted a more traditional 90-day study
of monochloramine in Sprague-Dawley rats (10 animals per sex per
dose). Treatment concentrations were 0, 25, 50, 100 or 200 mg/litre,
corresponding to doses of 0, 1.8, 3.4, 5.8 or 9.0 mg/kg of body weight
per day in males and 0, 2.6, 4.3, 7.7 or 12.1 mg/kg of body weight per
day in females. Controls received carbonated, pH-adjusted
drinking-water. A large number of haematological and clinical
chemistry measures were included in the evaluation. Body weights were
significantly depressed in both sexes at treatment concentrations in
the 50-200 mg/litre range, but this appeared to be related to
depressed water and food consumption. There were minor changes in
organ to body weight ratios at the highest dose, but no evidence of
treatment-related pathology was observed. Male rats were found to have
decreased haematocrits at 100 mg/litre, and red blood cell counts were
slightly depressed at 100 and 200 mg/litre. The authors concluded that
monochloramine was more toxic than chlorine or chlorine dioxide.
However, it must be noted that the changes in blood parameters were
small and in themselves of no clinical significance. Other measures
were not related to specific toxic reactions. Based on the decrease in
organ and body weights observed in both sexes, the authors concluded
that the no-observed-adverse-effect level (NOAEL) was 100 mg/litre,
equivalent to 5.8 mg/kg of body weight per day.
The work in rats was followed up with a second study in B6C3F1
mice (Daniel et al., 1991a). Male and female B6C3F1 mice (10 per sex
per group) were administered 0, 12.5, 25, 50, 100 or 200 mg of
chloramine per litre of drinking-water for 90 days (calculated mean
daily dose was 0, 2.5, 5.0, 8.6, 11.1 or 15.6 mg/kg of body weight for
males and 0, 2.8, 5.3, 9.2, 12.9 or 15.8 mg/kg of body weight for
females). Water consumption significantly decreased at 100 and 200
mg/litre in males and at 25-200 mg/litre in females. Weight gain was
significantly depressed in both sexes at 100 and 200 mg/litre.
Neutrophil concentrations in blood were significantly depressed in
both male and female mice at the two highest doses, but other white
blood cell counts were unaltered. Absolute and relative spleen and
liver weights were depressed at both 100 and 200 mg/litre. No gross or
microscopic evidence of target organ toxicity was observed that could
be related to treatment. Based on decreased organ weights, weight
gain, and food and water consumption, the authors concluded that the
NOAEL was 50 mg/litre, equivalent to 8.6 mg/kg of body weight per day.
A 13-week study in which groups of 10 Sprague-Dawley rats were
given drinking-water containing 200 mg of monochloramine per litre or
buffered water as a control, ad libitum or restricted to the same
consumption as the monochloramine group, was designed to resolve some
of the outstanding toxicological questions. The results of this study
indicated that the reduced body weight gain and the minor biochemical,
haematological, immunological and histopathological changes associated
with exposure to 200 mg of monochloramine per litre (equivalent to
21.6 mg/kg of body weight per day) in drinking-water were largely
related to reduced water and food consumption (Poon et al., 1997).
In a 9-week study, Exon et al. (1987) examined the ability of
monochloramine to modify immunological parameters in male
Sprague-Dawley rats (12 per dose) exposed to concentrations of 0, 9,
19 or 38 mg of monochloramine per litre, equivalent to 0, 1.3, 2.6 or
5.3 mg/kg of body weight per day. At the middle and highest dose,
chloramine treatment was observed to increase prostaglandin E2
synthesis by adherent resident peritoneal cells (which include
macrophages) in response to lipopolysaccharide stimulation. No attempt
was made to relate this finding to other indices of modified
macrophage function. A small depression in spleen weights was observed
at the highest dose. The implications of these data for immune
function are not clear. Other measures of immune function did not
reveal statistically significant changes with treatment. These
included a decrease in antibody formation at the lowest and middle
doses in response to keyhole limpet haemocyanin injection or
delayed-type hypersensitivity reactions to bovine serum albumin
injected into the footpad.
The effect of monochloramine on skin irritation was tested by
immersing Sencar mice into water containing chloramine at
concentrations ranging from 1 to 1000 mg/litre for 10 min a day
(Robinson et al., 1986). Unlike hypochlorous acid (pH 6.5) or
hypochlorite (pH 8.5), chloramine did not produce hyperplasia of the
skin.
3.2.2 Reproductive and developmental toxicity
Studies in laboratory animals have indicated no reproductive or
developmental effects associated with chloramine. Abdel-Rahman et al.
(1982a) administered monochloramine to female Sprague-Dawley rats at
concentrations of 0, 1, 10 or 100 mg/litre (0, 0.15, 1.5 or 15 mg/kg
of body weight per day) for 2.5 months prior to breeding and through
gestation. Only six animals were assigned to each treatment group.
Reproductive performance was comparable between groups, and fetal
weights were not adversely affected by treatment. Between 50 and 60
fetuses were available (male and female combined) for evaluation.
There was no evidence of treatment-related skeletal or soft tissue
anomalies.
Carlton et al. (1986) examined the effects of monochloramine
administered by gavage at doses of 0, 2.5, 5 or 10 mg/kg of body
weight per day on the reproductive performance of Long-Evans rats.
Males (12 per group) were treated from 56 days prior to and through
mating, and females (24 per group) from 14 days prior to mating and
throughout the mating period. No statistically significant effects on
sperm morphology, concentration or motility were observed, nor were
there any effects on fertility, viability, litter size, pup weights,
day of eye opening or day of vaginal patency.
3.2.3 Toxicity in humans
The primary harmful effects of chloramine have been documented in
humans poisoned by chloramine formed when household bleach was mixed
with ammonia for use as a cleaning solution. Chloramine is a strong
respiratory irritant. These effects were discussed in section 3.1.3.
Forty-eight men completed an 8-week protocol during which diet
and other factors known to affect lipid metabolism were controlled.
During the first 4 weeks of the protocol, all subjects consumed
distilled water. During the second 4 weeks, one-third of the subjects
were assigned randomly to drink 1.5 litres of water containing 0, 2 or
15 mg of monochloramine per litre each day. At 2 mg/litre, no
significant changes were observed in total, HDL or LDL cholesterol,
triglycerides or apolipoproteins A1, A2 or B. Parameters of thyroid
function were unchanged. However, an increase in the level of
apolipoprotein B was observed at 15 mg/litre (Wones et al., 1993b).
3.2.4 Carcinogenicity and mutagenicity
Shih & Lederberg (1976) first demonstrated that monochloramine
induced mutation in a Bacillus subtilis reversion assay. The
concentration range studied extended from 18 to 74 µmol/litre. A
positive dose-response was observed through 56 µmol/litre, but 74
µmol/litre was cytotoxic. Repair-deficient mutants of B. subtilis,
rec3, recA and polyA5, were consistently more sensitive to the
cytotoxic effects of chloramine, while the uvr and recB mutants were
not. The sensitivity of the polyA5 mutants parallels the observations
of Rosenkranz (1973) with sodium hypochlorite and suggests that DNA
polymerase A is involved in the repair of DNA lesions produced by both
chemicals. Thus, it is possible that a common intermediate or
mechanism is involved in the mutagenic effects of hypochlorite and
chloramine.
A broader list of chloramines was tested by Thomas et al. (1987)
in Salmonella typhimurium tester strains TA97a, TA100 and TA102. The
chloramines tested included those that could be formed at low levels
from natural substrates in drinking-water or in the stomach. TA100 was
found to consistently be the most sensitive strain. The most potent
mutagens were the lipophilic dichloramines formed with histamine,
ethanolamine and putrescine. The corresponding monochloramines were
less potent. The more hydrophilic chloramines, such as
taurine-chloramine, had little activity. Monochloramine was active in
the 50 µmol/litre range, remarkably consistent with the data of Shih &
Lederberg (1976). Hypochlorous acid was inactive at all concentrations
that were tested, up to and including concentrations that induced
cytotoxicity.
Ashby and co-workers (1987) were unable to induce clastogenic
effects in the mouse bone marrow micronucleus assay when chloramine
was administered orally. They suggested that the in vitro
clastogenic effects were probably attributable to non-specific
cytotoxic effects that are secondary to the release of hypochlorite to
the media.
Meier et al. (1985b) found that intraperitoneal administration of
monochloramine to CD-1 mice at doses of up to 8 mg/kg of body weight
was without significant effect on either micronuclei or chromosomal
aberrations in the bone marrow. These data would appear to be
consistent with the findings of Ashby et al. (1987).
Studies on the carcinogenicity of chloramine are limited to a
single set of 2-year experiments conducted by the NTP (1992).
Drinking-water containing 0, 50, 100 or 200 mg of chloramine per litre
was provided to F344 rats and B6C3F1 mice. Seventy animals of each
species and of each sex within a species were assigned to each
experimental and control group. Doses in rats were 0, 2.1, 4.8 or
8.7 mg/kg of body weight per day in males and 0, 2.8, 5.3 or 9.5 mg/kg
of body weight per day in females; doses in mice were 0, 5.0, 8.9 or
15.9 mg/kg of body weight per day in males and 0, 4.9, 9.0 or
17.2 mg/kg of body weight per day in females. Of some interest was the
finding that two renal cell adenomas were found in male B6C3F1 mice
treated with the high dose of chloramine. In addition, one renal
adenoma was found in one male mouse treated with 100 mg/litre and in
one female mouse treated with 200 mg/litre. While this tumour site is
rare in both species, there was no real dose-response trend, nor were
the differences between the control and treatment groups statistically
significant. A second finding of some concern was an increase in the
incidence of mononuclear cell leukaemia in F344 rats. This pathology
was increased in rats treated with chloramine or hypochlorite,
although the effects were not clearly dose-dependent. The incidence of
mononuclear cell leukaemia was significantly greater than in
concurrent controls and was elevated above the historical incidence as
well. Nevertheless, these increases were not considered to be
treatment-related. In part, this conclusion arose from the lack of a
clear dose-response. It was also based on the fact that there was no
comparable trend in male rats.
3.2.5 Comparative pharmacokinetics and metabolism
The pharmacokinetics of 36Cl derived from monochloramine have
been examined in male Sprague-Dawley rats (Abdel-Rahman et al., 1983).
These data are difficult to interpret because the specific form of the
label is not known.
3.3 Chlorine dioxide
3.3.1 General toxicological properties and information on
dose-response in animals
Despite its use as a disinfectant, there have been very few
general toxicological evaluations of chlorine dioxide, because most
studies have focused on its major by-product, chlorite, which is
considered in section 4.6. The present review will first focus on the
limited characterization of chlorine dioxide's general toxicology,
then follow up with a discussion of its haematological and thyroid
effects.
Some very cursory investigations of chlorine dioxide's effects as
a respiratory irritant were published by Haller & Northgraves (1955)
in an article dealing with the general chemical properties of the
compound. In essence, these data suggested that exposure to chlorine
dioxide in air at a concentration of more than 420 mg/m3 (150 ppm)
for longer than 15 min was fatal to guinea-pigs. The total study
involved six guinea-pigs.
Rats (3-5 per group) were exposed to chlorine dioxide in various
concentrations (0.28-9520 mg/m3 [0.1-3400 ppm]) and for various
periods (3 min-10 weeks). All the rats exposed daily to chlorine
dioxide at 28 mg/m3 (10 ppm) died in less than 14 days. Purulent
bronchitis and disseminated bronchopneumonia were found at necroscopy.
No such changes were demonstrable in rats exposed to approximately
0.28 mg/m3 (0.1 ppm) for about 10 weeks (Dalhamn, 1957).
The LC50 of chlorine dioxide in rats (5 per sex per group)
exposed by inhalation for 4 h was 90 mg/m3 (32 ppm) (Ineris, 1996).
Robinson et al. (1986) studied the ability of chlorine and
alternative disinfectants to induce epidermal hyperplasia in the skin
of Sencar mice. The thickness of the interfollicular epidermis was
significantly increased by 10-min daily exposures to water containing
up to 1000 mg of chlorine dioxide per litre for 4 days. The thickness
of the epidermis was similar to that induced by an equivalent dose of
hypochlorous acid. Unlike hypochlorous acid, however, there was no
significant increase in skin thickness at concentrations of 300
mg/litre or less.
The study of Daniel et al. (1990a) was the first subchronic study
that adhered to modern expectations of toxicological studies. These
authors provided male and female Sprague-Dawley rats (10 per sex per
treatment group) with 0, 25, 50, 100 or 200 mg of chlorine dioxide per
litre of drinking-water for 90 days, equivalent to 0, 2, 4, 6 or 12
and 0, 2, 5, 8 or 15 mg/kg of body weight per day for males and
females, respectively. Conventional measures of body weight, organ
weights, a broad battery of clinical chemistry parameters and
histopathological examinations were all included in the study design.
Body and organ weights were significantly depressed at 200 mg/litre in
both sexes. This appeared to be secondary to depressed water
consumption, which is known to be tightly coupled to food consumption
in rats. The only significant histopathological damage found was
goblet cell hyperplasia and inflammation. This was observed at all
doses of chlorine dioxide in both male and female rats. Presumably
this inflammation occurs as a result of volatilization of chlorine
dioxide from the water bottle. The amount of chlorine dioxide actually
inhaled as a result of volatilization from the drinking-water
containing the lowest dose of chlorine dioxide (25 mg/litre) must be
extremely low. This suggests that there might be some concern for
sensitive individuals showering with water containing chlorine
dioxide.
The ability of chlorine dioxide to induce methaemoglobinaemia and
haemolytic anaemia has received extensive study. Abdel-Rahman et al.
(1980) found decreased red blood cell glutathione (GSH) concentrations
and decreased osmotic fragility in Sprague-Dawley rats and white
leghorn chickens given drinking-water containing chlorine dioxide
concentrations of 1, 10, 100 and 1000 mg/litre for up to 4 months, but
the changes were not consistently dose-related. However, the authors
found that the morphology of red blood cells was modified (codocytes
and echinocytes) in all dose groups, the severity increasing with
increased treatment concentration. Methaemoglobin was not detected
throughout these studies. However, there was no formal statistical
evaluation of these results, and only four rats were assigned to each
experimental group. Administration of acute doses of as little as 1
mg/kg of body weight by gavage decreased red blood cell GSH
concentrations. This response was not increased as dose was increased
to 4 mg/kg of body weight.
Abdel-Rahman and co-workers extended these observations to longer
treatment periods in a subsequent publication (Abdel-Rahman et al.,
1984a). Again, only four animals were assigned to each treatment
group. At 7 and 9 months of treatment, red blood cells appeared to
become resistant to osmotic shock at all treatment concentrations
(1-1000 mg/litre). These data did not display a clear dose-response
despite the large variation in the dose administered.
The study of Abdel-Rahman et al. (1984a) also reported changes in
the incorporation of 3H-thymidine into the DNA of various organs.
Incorporation was significantly inhibited in testes and apparently
increased in the intestinal mucosa. The effect on apparent DNA
synthesis was particularly marked in the testes at 100 mg/litre,
amounting to about 60% inhibition. These data are difficult to
interpret for several reasons. First, rats were sacrificed 8 h after
being injected with 3H-thymidine. Ordinarily, sacrifices are made
30-60 min after injection because the blood is essentially depleted of
3H-thymidine in an hour. Thus, it is not possible to determine if the
lowered amount of label is related to decreased synthesis or to
increased turnover of DNA. Second, the result was based on total
counts in DNA, which makes it impossible to determine what cell type
is affected or whether the change was associated with replicative or
repair synthesis. Third, only four animals were used per experimental
group.
In a rising-dose protocol study, Bercz et al. (1982) evaluated
the effects of chlorine dioxide on African green monkeys. These
animals were provided chlorine dioxide in drinking-water at
concentrations of 0, 30, 100 or 200 mg/litre, corresponding to doses
of 0, 3.5, 9.5 or 11 mg/kg of body weight per day. Each dose was
maintained for 30-60 days. Animals showed signs of dehydration at the
highest dose (11 mg/kg of body weight per day), so exposure at that
dose was discontinued. No effect was observed on any haematological
parameter, including methaemoglobinaemia. However, statistically
significant depressions in serum thyroxine levels were observed when
animals were dosed with chlorine dioxide at a concentration of
100 mg/litre. No effect had been observed in a prior exposure of the
same animals to 30 mg/litre for 30 days. The NOAEL in this study was
3.5 mg/kg of body weight per day.
The effects of chlorine dioxide on thyroid function were followed
up by Harrington et al. (1986). Thyroxine levels in African green
monkeys administered drinking-water containing 100 mg of chlorine
dioxide per litre (4.6 mg/kg of body weight per day) were again found
to be depressed at 4 weeks of treatment, but rebounded to above-normal
levels after 8 weeks of treatment. These investigators also found
significantly depressed thyroxine levels in rats treated with 100 or
200 mg of chlorine dioxide per litre in drinking-water (equivalent to
14 and 28 mg/kg of body weight per day) for 8 weeks. This change was
dose-related. Lower doses were not examined in the rat study. The
authors indicated that the results were based on 12 determinations; it
was not clear if these measurements were made on individual animals.
A set of in vivo and in vitro experiments was conducted in an
attempt to explain the effects of chlorine dioxide on serum thyroid
hormone concentrations. The authors demonstrated that chlorine dioxide
oxidizes iodide to reactive iodine species that would bind to the
stomach and oesophageal epithelium. Rat chow that was treated with
chlorine dioxide at approximately 80 mg/litre was found to increase
the binding of iodine to chow constituents. This activation of iodine
resulted in retention of labelled iodine in the ileum and colon and
reduced uptake by the thyroid gland. Previous work demonstrated that
chlorine dioxide was more effective than chlorine in activating iodide
to a form that would covalently bind with a variety of natural
foodstuffs (Bercz et al., 1986). Based on these observations, the
authors concluded that the effects of chlorine dioxide were probably
due to altered gastrointestinal absorption of iodide and reduced
uptake into the thyroid gland.
3.3.2 Reproductive and developmental toxicity
A number of reproductive effects have been reported in studies
with laboratory animals, but the relevance for humans of these
findings remains uncertain. The reproductive effects of chlorine
dioxide in Long-Evans rats were studied by Carlton et al. (1991).
Chlorine dioxide was administered by gavage at doses of 2.5, 5 or 10
mg/kg of body weight per day to male rats (12 per group) for 56 days
prior to and through mating and to female rats (24 per group) from 14
days prior to mating and through pregnancy. Fertility measures were
not significantly different among the dose groups. There were no
dose-related changes in sperm parameters (i.e., concentration,
motility, progressive movement or morphology). Thyroid hormone levels
were altered significantly, but not in a consistent pattern. The only
significant difference was significantly depressed vaginal weights in
female pups whose dams had been treated with 10 mg/kg of body weight
per day.
An evaluation of the effects of chlorine dioxide on the fetal
development of Sprague-Dawley rats was conducted by Suh et al. (1983).
Chlorine dioxide was administered at 0, 1, 10 or 100 mg/litre (0, 0.1,
1 or 10 mg/kg of body weight per day) for 2.5 months prior to mating
and throughout gestation. The total number of implants per dam was
significantly reduced at the highest concentration of chlorine
dioxide. The percentage of anomalous fetuses was increased in a
dose-related manner, but the response was not statistically
significant. These anomalies arose primarily as the percentage of
abnormal or incomplete sternebrae in treated rats relative to
controls. The lack of statistical significance was undoubtedly related
to the relatively few female rats that were included in the study (6-8
females per treatment group). As a consequence, the results of this
study must be considered inconclusive.
Orme et al. (1985) found that chlorine dioxide administered in
the drinking-water of female Sprague-Dawley rats (13-16 per dose) at
concentrations of 0, 2, 20 or 100 mg/litre (0, 1, 3 or 14 mg/kg of
body weight per day) throughout pregnancy and through weaning
decreased thyroxine levels in the serum of the pups at 100 mg/litre.
This was associated with delayed development of exploratory behaviour
in the pups away from their dams, and this, in turn, was probably due
to an indirect effect on iodine uptake. In a second experiment, pups
given 14 mg of chlorine dioxide per kg of body weight per day directly
by gavage on postnatal days 5-20 showed significantly depressed
activity and a decrease in serum thyroxine levels. Studies by the same
group (Taylor & Pfohl, 1985) indicated that cerebellar and forebrain
cell counts (based on DNA measurements) were depressed in 11-day-old
pups that had been treated with chlorine dioxide at 14 mg/kg of body
weight per day by gavage from 5 days of age. Cerebellar cell counts
remained depressed in rats at 21 days, but forebrain counts were
essentially the same as in controls. At 50-60 days of age, the
locomotor activity (measured by wheel-running) of these animals was
depressed relative to control animals.
The effects of chlorine dioxide on brain development were
examined further by Toth et al. (1990). These authors administered
chlorine dioxide by gavage at 14 mg/kg of body weight per day from
postnatal day 1 to 20. Body weight was reduced, but cerebellar weight
was unaltered at any age. Forebrain weight and protein content were
reduced on postnatal days 21 and 35. DNA content was depressed on
postnatal day 35, and the number of dendritic spines on cerebral
cortical pyramidal cells was significantly reduced. No
histopathological changes in the forebrain, cerebellum or brain stem
were observed. There were no consistent changes in serum thyroxine or
triiodothyronine levels in treated animals.
Collectively, these data suggest some effects of chlorine dioxide
on brain development. In most studies, there are suggestions of
modified thyroid function associated with these effects. It must be
pointed out that the changes in thyroid hormone levels are modest,
much less than are produced with classical antithyroid drugs such as
propylthiouracil (Toth et al., 1990).
3.3.3 Toxicity in humans
The effects of chlorine dioxide were assessed in a two-phase
study in 10 healthy male volunteers. The first study was a rising-dose
tolerance study (Lubbers & Bianchine, 1984) in which doses of chlorine
dioxide were increased from 0.1 to 24 mg/litre, administered in two
500-ml portions. The maximum dose for a 70-kg person was 0.34 mg/kg of
body weight. The details of this study were described in section
3.1.3. Some small changes in a variety of clinical chemistry
parameters were observed, but none was found to be outside the
accepted range of normal. The second phase of the experiment involved
the daily administration of a 500-ml portion of a solution containing
5 mg/litre to 10 healthy volunteers for a period of 12 weeks (Lubbers
et al., 1984a). Again, measurement of a large battery of clinical
chemistry parameters and routine physical examination failed to
identify any effects of chlorine dioxide that fell outside of the
normal range. Parameters yielding significant differences appeared to
be primarily a result of parallel drift of values with the control
group.
As with other disinfectants, it is important to recognize that
chlorine dioxide is a potent respiratory irritant. No quantitative
data can be used to construct a dose-response relationship for this
effect.
3.3.4 Carcinogenicity and mutagenicity
The mutagenic or clastogenic effects of chlorine dioxide have
received little attention. Ishidate et al. (1984) found chlorine
dioxide to be positive in Salmonella typhimurium tester strain
TA100. A linear dose-response was observed at concentrations between 2
and 20 µg per plate. Chlorine dioxide was ineffective as a clastogenic
agent in a CHO system.
Meier et al. (1985b) evaluated the ability of chlorine dioxide to
induce chromosomal aberrations and micronuclei in bone marrow of CD-1
mice or sperm head anomalies. Chlorine dioxide failed to produce such
damage following gavage doses of up to 16 mg/kg of body weight for 5
days.
With the exception of a 1949 study by Haag (cited in TERA, 1998),
which has serious limitations, no tests of the carcinogenic activity
of chlorine dioxide in experimental animals were identified in the
scientific literature.
3.3.5 Comparative pharmacokinetics and metabolism
There are significant differences in the pharmacokinetics of
36Cl obtained from different disinfectants. The absorption rate for
36Cl-labelled chlorine dioxide was at least 10 times that observed
with chlorine, chloramine or chloride. The relative amount of 36Cl
that is eliminated in the urine and faeces at 24 h has a distinct
pattern from that observed with other disinfectants and sodium
chloride. However, the terminal half-life of the 36Cl appears similar
for all disinfectants. These data suggest that the form of 36Cl that
is being absorbed differs chemically with the different disinfectants.
In the case of chlorine dioxide, this is supported by the observation
that measurable amounts of chlorite (about 3% of the original dose)
are eliminated in the urine during the first 24 h, and chlorite
comprises about 20% of the label present in blood 72 h after
administration of the test dose (Abdel-Rahman et al., 1980). However,
this higher absorption rate is not explained by the absorption rates
of chlorite and chlorate, which are about one-tenth as rapid
(Abdel-Rahman et al., 1982b). This suggests that some of the
absorption could be as chlorine dioxide itself. On the surface, this
hypothesis would seem to be incompatible with the high reactivity of
this disinfectant. As with other disinfectants, the terminal
elimination phases observed for 36Cl from chlorine dioxide seem
compatible with the hypothesis that the bulk of the elimination is as
chloride ion.
4. TOXICOLOGY OF DISINFECTANT BY-PRODUCTS
4.1 Trihalomethanes
As a class, the THMs are generally the most prevalent by-products
of drinking-water disinfection by chlorine. A variety of
non-neoplastic toxic effects have been associated with short-term and
long-term exposure of experimental animals to high doses of THMs, and
each of the four most common THMs -- chloroform, BDCM, DBCM and
bromoform -- has been shown to be carcinogenic to rodents in high-dose
chronic studies. Chloroform is generally the predominant THM in
chlorinated water and is also the most extensively studied chemical of
this class. Because the World Health Organization (WHO) recently
published an Environmental Health Criteria monograph on chloroform
(IPCS, 1994), this section will only update the information contained
in that publication. A thorough review of findings relevant to the
toxicology of the brominated THMs is included.
As with the other DBPs, the chemical and physical properties of
the THMs influence their potential routes of human exposure, their
pharmacokinetic behaviour, their toxicity and methods for conducting
toxicological studies with these compounds. The THMs are volatile
liquids at room temperature; therefore, as these chemicals vaporize
during water usage (e.g., showering), inhalation becomes an important
exposure route in addition to ingestion. Volatility decreases somewhat
with bromine substitution, but each of the brominated THMs evaporates
from drinking-water. Like that of other alkanes, the water solubility
of THMs is poor, although adequate to permit dissolution of the low
levels generated via water disinfection. When administered at higher
levels to animals in toxicity experiments, THMs are often either
emulsified in aqueous solutions or dissolved in oils. The use of oils
as vehicles of administration can significantly alter the
pharmacokinetics and toxicity of the THMs. Bromine substitution
enhances the lipid solubility of the halomethanes (and, consequently,
uptake into tissues) and generally increases their chemical reactivity
and the likelihood of biotransformation to a reactive intermediate.
Because toxicity is dependent upon a reactive compound actually
reaching a sensitive target site, greater bromine substitution may not
necessarily translate into greater in vivo toxicity (i.e., innocuous
reactions may occur, preventing arrival at target sites). Perhaps to a
greater extent than with other chemicals in this class, BDCM appears
to reach a variety of target tissues where it can be readily
metabolized to several intermediates, leading to adverse effects in
experimental animals.
4.1.1 Chloroform
In 1994, the WHO published an Environmental Health Criteria
monograph on chloroform (IPCS, 1994). The following sections will
update that document with the most recent findings from health-related
chloroform research. Another evaluation of chloroform was included in
the 1998 Addendum to the WHO Guidelines for drinking-water quality
(WHO, 1998).
4.1.1.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
Keegan et al. (1998) determined the
lowest-observed-adverse-effect level (LOAEL) and NOAEL for the
induction of acute hepatotoxicity following oral administration of
chloroform in an aqueous vehicle to male F344 rats. Based on
elevations of serum clinical chemistry indicators of liver damage, a
LOAEL of 0.5 mmol/kg of body weight (60 mg/kg of body weight) and a
NOAEL of 0.25 mmol/kg of body weight (30 mg/kg of body weight) were
established. In a corn oil gavage study of single-dose chloroform
effects, an increase in renal cell proliferation was observed at doses
as low as 10 mg/kg of body weight in male Osborne-Mendel rats and
90 mg/kg of body weight in male F344 rats (Templin et al., 1996a). The
only increase in the hepatic labelling index was in F344 rats given
477 mg/kg of body weight. Effects in the nasal passages of both rat
strains at 90 mg/kg of body weight and above included oedema and
periosteal hypercellularity. Gemma et al. (1996) dosed male B6C3F1
mice with chloroform (150 mg/kg of body weight) by gavage and observed
increases in cell proliferation in both the liver and kidneys. The
effect was more dramatic in the kidneys, where severe necrosis was
also noted.
Nephrotoxicity of chloroform was evaluated in male Sprague-Dawley
rats treated orally with single doses of chloroform using corn oil or
an aqueous preparation (5%) of Emulphor or Tween 85 as vehicle (10
ml/kg of body weight). Comparison between gavage vehicles indicated
clear trends for enhanced potency and severity of nephrotoxic effects
with corn oil administration of chloroform (Raymond & Plaa, 1997).
2) Short-term toxicity
Chloroform was administered by corn oil gavage to male B6C3F1
mice at doses of 0, 34, 90, 138 or 277 mg/kg of body weight for 4 days
or 3 weeks (5 days per week) (Larson et al., 1994a). Mild degenerative
changes in centrilobular hepatocytes were noted in mice given 34 and
90 mg/kg of body weight per day after 4 days of treatment, but these
effects were absent at 3 weeks. At 138 and 277 mg/kg of body weight
per day, centrilobular necrosis was observed at 4 days and with
increased severity at 3 weeks. Hepatic cell proliferation was
increased in a dose-dependent manner at all chloroform doses after 4
days, but only in the 277 mg/kg of body weight dose group at 3 weeks.
Renal tubular necrosis was observed in all dose groups after 4 days,
while 3 weeks of exposure produced severe nephropathy at the highest
dose and regenerating tubules at the lower doses. The nuclear
labelling index was increased in the proximal tubules at all doses
after 4 days of treatment, but was elevated only in the two highest
dose groups after 3 weeks.
In a similar study (Larson et al., 1994b), female B6C3F1 mice
were administered chloroform dissolved in corn oil by gavage at doses
of 0, 3, 10, 34, 238 or 477 mg/kg of body weight per day for 4 days or
3 weeks (5 days per week). Dose-dependent changes included
centrilobular necrosis and markedly elevated labelling index in mice
given 238 and 477 mg/kg of body weight per day. The NOAEL for
histopathological changes was 10 mg/kg of body weight per day, and for
induced cell proliferation, 34 mg/kg of body weight per day.
In an inhalation study, Templin et al. (1996b) exposed BDF1 mice
to chloroform vapour at concentrations of 0, 149 or 446 mg/m3 (0, 30
or 90 ppm) 6 h per day for 4 days or 2 weeks (5 days per week). In the
kidneys of male mice exposed to 149 and 446 mg/m3 (30 and 90 ppm),
degenerative lesions and 7- to 10-fold increases in cell proliferation
were observed. Liver damage and an increased hepatic labelling index
were noted in male mice exposed to 149 and 446 mg/m3 (30 and 90 ppm)
and in female mice exposed to 446 mg/m3 (90 ppm). Both doses were
lethal in groups exposed for 2 weeks (40% and 80% mortality at 149 and
446 mg/m3 [30 and 90 ppm], respectively).
Female F344 rats were given chloroform by corn oil gavage for 4
consecutive days or 5 days per week for 3 weeks (Larson et al.,
1995b). In the liver, mild degenerative centrilobular changes and
dose-dependent increases in hepatocyte proliferation were noted at
doses of 100, 200 and 400 mg/kg of body weight per day. At 200 and
400 mg/kg of body weight per day, degeneration and necrosis of the
renal cortical proximal tubules were observed. Increased regenerative
proliferation of epithelial cells lining proximal tubules was seen at
doses of 100 mg/kg of body weight per day or more. Lesions of the
olfactory mucosa lining the ethmoid region of the nose (new bone
formation, periosteal hypercellularity and increased cell replication)
were seen at all doses, including the lowest dose of 34 mg/kg of body
weight per day. Larson et al. (1995a) also administered chloroform to
male F344 rats by corn oil gavage (0, 10, 34, 90 or 180 mg/kg of body
weight per day) or in the drinking-water (0, 60, 200, 400, 900 or
1800 mg/litre) for 4 days or 3 weeks. Gavage of 90 or 180 mg/kg of
body weight per day for 4 days induced mild to moderate degeneration
of renal proximal tubules and centrilobular hepatocytes -- changes
that were no longer present after 3 weeks. Increased cell
proliferation in the kidney was noted only at the highest gavage dose
after 4 days. The labelling index was elevated in the livers of the
high-dose group at both time points. With drinking-water
administration, rats consuming the water containing 1800 mg/litre were
dosed at a rate of 106 mg/kg of body weight per day, but no increase
in renal or hepatic cell proliferation was observed at this or any
lower dose.
In a study carried out to evaluate whether exposure to chloroform
in drinking-water would interact with the activity of chloroform when
administered by gavage in corn oil, female B6C3F1 mice were exposed
to chloroform in drinking-water for 33 days at 0, 300 or 1800 mg/litre
or for 31 days at 0, 120, 240 or 480 mg/litre. Three days prior to
termination, mice also received a daily dose of 263 mg of chloroform
per kg of body weight per day, administered by gavage in corn oil.
Exposure to chloroform in drinking-water reduced both the
hepatotoxicity and the enhanced cell proliferation elicited in
response to chloroform administered by gavage in corn oil (Pereira &
Grothaus, 1997).
The cardiotoxicity of chloroform was examined in male Wistar rats
given daily doses of 37 mg/kg of body weight (0.31 mmol/kg) by gavage
in olive oil for 4 weeks (Muller et al., 1997). Chloroform caused
arrhythmogenic and negative chronotropic and dromotropic effects as
well as extension of the atrioventricular conduction time and
depressed myocardial contractility.
A 90-day chloroform inhalation study was conducted using male and
female B6C3F1 mice and exposure concentrations of 0, 1.5, 10, 50, 149
and 446 mg/m3 (0, 0.3, 2, 10, 30 and 90 ppm) for 6 h per day, 7 days
per week (Larson et al., 1996). Large, sustained increases in
hepatocyte proliferation were seen in the 446 mg/m3 (90 ppm) groups
at all time points (4 days and 3, 6 and 13 weeks). In the more
sensitive female mice, a NOAEL of 50 mg/m3 (10 ppm) for this effect
was established. Renal histopathology and regenerative hyperplasia
were noted in male mice at 50, 149 and 446 mg/m3 (10, 30 and 90 ppm).
In another 90-day inhalation study, F344 rats were exposed to
chloroform at concentrations of 0, 10, 50, 149, 446 or 1490 mg/m3 (0,
2, 10, 30, 90 or 300 ppm) for 6 h per day, 7 days per week. The 1490
mg/m3 (300 ppm) level was extremely toxic and deemed by the authors
to be inappropriate for chronic studies. Increases in renal epithelial
cell proliferation in cortical proximal tubules were observed at
concentrations of 149 mg/m3 (30 ppm) and above. Hepatic lesions and
increased proliferation were noted only at the highest exposure level.
In the ethmoid turbinates of the nose, enhanced bone growth and
hypercellularity in the lamina propria were observed at concentrations
of 50 mg/m3 (10 ppm) and above, and a generalized atrophy of the
turbinates was seen at all exposure levels after 90 days (Templin et
al., 1996c).
3) Reproductive and developmental toxicity
Rat embryo culture was used to assess the developmental effects
of chloroform (Brown-Woodman et al., 1998). The effect and
no-effect culture medium concentrations of chloroform were 2.06 and
1.05 µmol/ml. The authors estimated that fatal or near-fatal blood
levels would be required in the mother for the embryotoxic level to be
reached.
4.1.1.2 Toxicity in humans
Fatal acute chloroform intoxication via inhalation was reported
to cause cardiomyocyte fragmentation and waviness indicative of acute
heart failure possibly caused by arrhythmias or cardiac depression
(Harada et al., 1997). These observations are consistent with the
results of the short-term rat study (Templin et al., 1996b) described
above in section 4.1.1.1.
4.1.1.3 Carcinogenicity and mutagenicity
Jamison et al. (1996) reported that F344 rats exposed to a high
concentration of chloroform vapour (1490 mg/m3 [300 ppm]) for 90 days
developed atypical glandular structures lined by intestinal-like
epithelium and surrounded by dense connective tissue in their livers.
These lesions appeared to arise from a population of cells remote from
the bile ducts. The authors also observed a treatment-related increase
in transforming growth factor-alpha (TGF-alpha) immunoreactivity in
hepatocytes, bile duct epithelium, bile canaliculi and oval cells and
an increase in transforming growth factor-beta (TGF-ß)
immunoreactivity in hepatocytes, bile duct epithelium and intestinal
crypt-like ducts. The lesions occurred only in conjunction with
significant hepatocyte necrosis, regenerative cell proliferation and
increased growth factor expression or uptake.
Chloroform was tested for mutagenicity and clastogenicity by Le
Curieux et al. (1995) and was negative in the SOS chromotest, the Ames
fluctuation test and the newt micronucleus test. It appeared to these
authors that the presence of bromine substituents was needed for
genotoxic activity in the THM class. Pegram et al. (1997) examined
chloroform mutagenicity in a strain of Salmonella typhimurium TA1535
transfected with rat glutathione- S-transferase (GST) T1-1.
Chloroform was negative in this assay over a range of concentrations
(992-23 800 mg/m3 [200-4800 ppm]) that produced large dose-dependent
increases in revertants with BDCM. A doubling of revertants was
induced by chloroform in the GST-transfected strain only at the two
highest concentrations tested (95 200 and 127 000 mg/m3 [19 200 and
25 600 ppm]). Brennan & Schiestl (1998) found that chloroform induced
intrachromosomal recombination in the yeast strain Saccharomyces
cerevisiae at culture medium concentrations of 3-5.6 mg/ml.
In an in vivo study, Potter et al. (1996) found that chloroform
did not induce DNA strand breaks in the kidneys of male F344 rats
following seven daily doses of 1.5 mmol/kg of body weight. In
long-term mutagenicity studies with chloroform in female lacI
transgenic B6C3F1 mice conducted by Butterworth et al. (1998), the
mice were exposed daily by inhalation to chloroform concentrations of
0, 50, 149 or 446 mg/m3 (0, 10, 30 or 90 ppm) for 6 h per day, 7 days
per week, and lacI mutant frequency was determined after 10, 30, 90
and 180 days of exposure. No increase in lacI mutant frequency was
observed in the liver at any dose or time point with chloroform.
4.1.1.4 Comparative pharmacokinetics and metabolism
The percutaneous absorption of 14C-chloroform was examined
in vivo using human volunteers and in vitro using fresh, excised
human skin in a flow-through diffusion cell system (Dick et al.,
1995). Aqueous and ethanol solutions of chloroform were applied to the
forearm (16 and 81 g/cm) of volunteers, and absorption was determined
to be 7.8% from the water vehicle and 1.6% from ethanol. More than 95%
of the absorbed dose was excreted via the lungs (88% as carbon
dioxide), and maximum pulmonary excretion occurred between 15 min and
2 h after dosing. In vitro, 5.6% of a low dose and 7.1% of a high
dose were absorbed (skin plus perfusate).
The systemic uptake of chloroform during dermal exposure was also
studied in hairless rats (Islam et al., 1996). Animals were immersed
in water containing chloroform for 30 min, and the compound was
detected in blood as early as 4 min following exposure. About 10 mg of
chloroform were systemically absorbed after dermal exposure of a rat
to an aqueous solution of 0.44 mg/ml.
Absorption and tissue dosimetry of chloroform were evaluated
after gavage administration in various vehicles to male Fischer 344
rats and female B6C3F1 mice (Dix et al., 1997). Animals received a
single dose of chloroform (15-180 and 70-477 mg/kg of body weight for
rats and mice, respectively) in corn oil, water or aqueous 2% Emulphor
(dose volumes of 2 and 10 ml/kg of body weight for rats and mice,
respectively). Blood, liver and kidney chloroform concentration-time
courses were determined. Gavage vehicle had minimal effects on
chloroform dosimetry in rats. In mice, however, tissue chloroform
concentrations were consistently greater for aqueous versus corn oil
vehicle. At the low vehicle volume used for rats (2 ml/kg of body
weight), gavage vehicle may not play a significant role in chloroform
absorption and tissue dosimetry; at the higher vehicle volume used for
mice (10 ml/kg of body weight), however, vehicle may be a critical
factor.
Because chloroform metabolism was reviewed in detail in the
recent Environmental Health Criteria monograph (IPCS, 1994), the
primary discussion of the comparative metabolism of the THMs as a
class can be found in section 4.1.2.6 of the present report.
The contributions of cytochromes P450 (CYP) 2E1 and 2B1/2 to
chloroform hepatotoxicity were investigated in male Wistar rats
(Nakajima et al., 1995). The severity of toxicity observed in
differentially induced rats suggests that CYP2E1 is a low
Michaelis-Menten constant ( Km) isoform and CYP2B1/2 is a high
Km isoform for chloroform activation. A high dose of chloroform
(0.5 ml/kg of body weight) induced CYP2E1 but decreased CYP2B1/2.
Testai et al. (1996) generated similar results in a study examining
the involvement of these isozymes in in vitro chloroform metabolism.
At a low substrate concentration (0.1 mmol/litre), oxidative
metabolism by liver microsomes was dependent primarily on CYP2E1; at 5
mmol/litre, on the other hand, CYP2B1/2 was the major participant
responsible for chloroform activation, although CYP2E1 and CYP2C11
were also significantly involved. The reductive pathway was expressed
only at 5 mmol/litre and was not significantly increased by any CYP
inducer tested.
The reductive metabolism of chloroform by rat liver microsomes
was examined by Testai et al. (1995). In hypoxic (1% oxygen partial
pressure) and anoxic (0% oxygen partial pressure) incubations using
microsomes from phenobarbital-induced animals, no evidence of
formation of monochloromethyl carbene was found. Dichloromethane was
detected as a metabolite of chloroform under variable oxygenation
conditions using microsomes from phenobarbital-induced animals. With
uninduced microsomes, significant levels of dichloromethane were
formed only in hypoxic or anoxic incubations. In an in vivo study of
chloroform reductive metabolism, Knecht & Mason (1991) detected no
free radical adducts in the bile of rats treated with chloroform,
while radicals were detected from bromoform. Lipid adducts resulting
from the reductive metabolism of chloroform by hepatocytes appeared to
be generated by the unspecific attack of the radical on the
phospholipid fatty acyl chains (Guastadisegni et al., 1996). The
primary lipid adduct has now been identified as a modified
phosphatidylethanolamine, with the phosgene-derived carbonyl bound to
the amine of the head group (Guastadisegni et al., 1998). Waller &
McKinney (1993) found that chloroform had a lower theoretical
potential to undergo reductive metabolism than the brominated THMs.
Ade et al. (1994) reported that microsomes from the renal cortex of
DBA/2J mice can metabolize chloroform through the reductive and
oxidative pathways, as had been previously described using hepatic
microsomes. However, cytolethality of chloroform to freshly isolated
rodent hepatocytes was not increased under reduced oxygen tension,
indicating that reductive metabolism does not contribute to
chloroform-induced toxicity (Ammann et al., 1998).
The potential of chloroform to participate in the recently
discovered GSH conjugation pathway for brominated THMs has been
investigated (Pegram et al., 1997). The GST examined in this study has
a very low affinity for chloroform compared with the brominated THMs.
Chloroform conjugation with GSH occurred only at extremely high
substrate concentrations.
4.1.1.5 Mode of action
Direct DNA reactivity and mutagenicity cannot be considered to be
key factors in chloroform-induced carcinogenesis in experimental
animals. A substantial body of data demonstrates a lack of direct
in vivo or in vitro genotoxicity of chloroform. If THMs produce
their genotoxic effects primarily via the GSH conjugation mechanism,
the results of Pegram et al. (1997) indicate that chloroform would be
mutagenic in mammals only at lethal doses.
There is, however, compelling evidence to support a mode of
action for tumour induction based on metabolism of chloroform by the
target cell population, followed by cytotoxicity of oxidative
metabolites and regenerative cell proliferation. The evidence for the
link with cytotoxicity is strongest for hepatic and renal tumours in
the mouse and more limited for renal tumours in the rat (ILSI, 1997).
A number of recent studies support the hypothesis that chloroform acts
to produce cancer in rodents through a non-genotoxic/cytotoxic mode of
action, with carcinogenesis resulting from events secondary to
chloroform-induced cytolethality and regenerative cell proliferation
(Larson et al., 1994a,b, 1996; Pereira, 1994; Templin et al.,
1996a,b,c, 1998). These studies have shown that organ toxicity and
regenerative hyperplasia are associated with the tumorigenicity of
chloroform and are apparently the key steps in its carcinogenic mode
of action. Thus, sustained toxicity would result in tumour
development. Chloroform induces liver and kidney tumours in long-term
rodent cancer bioassays only at doses that induce frank cytotoxicity
in these target organs. Furthermore, there are no instances of
chloroform-induced tumours that are not preceded by this pattern of
dose-dependent toxic responses (Golden et al., 1997).
The organ toxicity and carcinogenicity of chloroform are
dependent on oxidative metabolism and levels of CYP2E1. Numerous
studies have also shown that oxidative metabolism by CYP2E1 generates
highly reactive metabolites (phosgene and hydrogen chloride), which
would lead to cytotoxicity and regenerative hyperplasia.
4.1.2 Bromodichloromethane
4.1.2.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
The acute oral lethality of the brominated THMs has been
determined in ICR Swiss mice (Bowman et al., 1978) and Sprague-Dawley
rats (Chu et al., 1980). The resulting LD50s for BDCM were 450 and
900 mg/kg of body weight for male and female mice, respectively, and
916 and 969 mg/kg of body weight for male and female rats,
respectively. Clinical observations of animals dosed with high levels
of BDCM in these studies and others (NTP, 1987) included ataxia,
sedation, laboured breathing and anaesthesia (500 mg/kg of body
weight), as well as gross evidence of liver and kidney damage. Hewitt
et al. (1983) gave single doses of BDCM to male Sprague-Dawley rats by
corn oil gavage and found doses of 1980 mg/kg of body weight and above
to be lethal. Little clinical evidence of hepatic or renal toxicity
was observed at doses below 1980 mg/kg of body weight.
Acute hepatotoxic and nephrotoxic responses to orally dosed BDCM
and various factors affecting these toxicities (e.g., gavage vehicle
and glutathione status) have been studied in male F344 rats. Lilly et
al. (1994, 1997a) examined the time course of toxicity and
dose-response relationships following oral administration of aqueous
solutions of BDCM. BDCM-induced liver toxicity was maximal at 24 h
after dosing with 1-3 mmol/kg of body weight (164-492 mg/kg of body
weight), as indicated by elevations in serum levels of aspartate
aminotransferase (ASAT), alanine aminotransferase (ALAT), sorbitol
dehydrogenase (SDH) and lactate dehydrogenase (LDH) and
histopathological observations of centrilobular vacuolar degeneration
and hepatocellular necrosis. Significant abatement of hepatic toxicity
was noted by 48 h post-dosing. The acute oral NOAEL and LOAEL for
liver toxicity following aqueous delivery of BDCM, based on elevations
in serum enzymes, were determined to be 0.25 and 0.5 mmol of BDCM per
kg of body weight (41 and 82 mg/kg of body weight), respectively
(Keegan et al., 1998). BDCM and chloroform appear to be equipotent
hepatotoxicants in rats at 24 h after exposure, but BDCM causes more
persistent damage to the liver, based on observations at 48 h
post-dosing (Lilly et al., 1997a; Keegan et al., 1998).
Kidney toxicity after corn oil or aqueous dosing of BDCM
(1.5-3 mol/kg of body weight) peaked between 24 and 48 h, as indicated
by elevations in kidney weight, urinary N-acetyl-ß-glucosaminidase,
ASAT, ALAT, LDH and protein, serum urea and creatinine, and
histopathological findings of renal tubule degeneration and necrosis
(Lilly et al., 1994, 1997a). The actual time of peak renal effects was
dose-dependent; in contrast to findings in the liver, toxicity was
increasingly prolonged in the kidney with increasing dose.
Nephrotoxicity has been noted in rats given single BDCM doses as low
as 200 mg or 1.2 mmol/kg of body weight (Lilly et al., 1994, 1997a),
and BDCM is a slightly more potent acute oral renal toxicant than
chloroform (based on the magnitude of the responses), especially at
lower doses (Lilly et al., 1997a). Kroll et al. (1994a,b) found that
among the THMs that occur in drinking-water, BDCM was the most potent
inducer of renal dysfunction in rats following intraperitoneal
injection of single 3 mmol/kg of body weight doses. Glomerular
filtration, renal concentrating ability, and proximal tubular
secretion and reabsorption were all more severely affected by BDCM
than by chloroform.
Several factors have been found to influence dose-response
relationships for BDCM toxicity. Acute hepatotoxicity and
nephrotoxicity were more severe after administration of 400 mg of BDCM
per kg of body weight in corn oil than when the same dose was given in
an aqueous vehicle (Lilly et al., 1994). However, vehicle differences
at a lower dose (200 mg/kg of body weight), although less pronounced,
were reversed: greater renal toxicity at this dose was associated with
the aqueous vehicle. The adverse renal and hepatic effects of BDCM
were also exacerbated in GSH-depleted rats (Gao et al., 1996) and in
rats that were dosed during the active period of their diurnal cycle
(Pegram et al., 1993). Induction of the cytochrome P-450 isozymes
CYP2E1 and CYP2B1/2 also potentiated acute liver toxicity, but not
renal toxicity, following dosing with BDCM (Thornton-Mannning et al.,
1994).
2) Short-term toxicity
Studies employing repeated daily BDCM dosing regimens have also
yielded results demonstrating liver and kidney toxicity.
Thornton-Manning et al. (1994) administered five consecutive daily
BDCM doses to female F344 rats and female C57BL/6J mice by aqueous
gavage and found that BDCM is both hepatotoxic and nephrotoxic to
female rats (150-300 mg/kg of body weight per day), but only
hepatotoxic to female mice (75-150 mg/kg of body weight per day).
Hepatic cytochrome P450 activities were decreased in rats, but not in
mice, in this study. Munson et al. (1982) administered BDCM (50, 125
or 250 mg/kg of body weight per day) to male and female CD-1 mice by
aqueous gavage for 14 days and reported evidence for hepatic and renal
toxicity as well as effects on the humoral immune system.
Nephrotoxicity, as reflected by significant elevations of blood urea
nitrogen (BUN), occurred only at the highest dose in both males and
females. Male mice appeared more sensitive than females to
BDCM-induced hepatotoxicity; 2- to 3-fold elevations in ASAT and ALAT
(although not significant, according to the authors' statistical
analysis) occurred at the lowest dose only in males. Based on the
degree of these elevations, BDCM was the most potent hepatotoxicant
compared with chloroform, DBCM and bromoform, which were also tested
in this study. Immunotoxic effects described in the study included
decreases in both antibody-forming cells and haemagglutination titres
at the 125 and 250 mg/kg of body weight per day doses, although a
recent investigation found no effects of BDCM on immune function
(French et al., 1999). Condie et al. (1983) conducted a similar 14-day
comparative dosing study with THMs and male CD-1 mice, but used corn
oil as the vehicle of administration for doses of BDCM of 37, 74 and
147 mg/kg of body weight per day. Evidence of renal damage was
observed at the mid and high doses, whereas liver toxicity occurred
only at the high dose. A 14-day corn oil gavage study by NTP (1987)
demonstrated the greater sensitivity of the B6C3F1 mouse to BDCM: all
male mice that received 150 or 300 mg/kg of body weight per day died
before the end of the study. Aida et al. (1992a) incorporated
microencapsulated BDCM into the diet of Wistar rats for 1 month, and a
LOAEL of 66 mg/kg of body weight per day and a NOAEL of 21 mg/kg of
body weight per day were determined based on histopathological
findings of hepatocellular vacuolization.
In a 13-week corn oil gavage study, NTP (1987) administered BDCM
doses of 0, 19, 38, 75, 150 or 300 mg/kg of body weight per day, 5
days per week, to F344/N rats (10 per sex per dose). The highest dose
was lethal to 50% of males and 20% of females, and body weight
depression was observed at the two highest doses. BDCM-induced lesions
were found only at 300 mg/kg of body weight per day; these included
hepatic centrilobular degeneration in both sexes and renal tubular
degeneration and necrosis in males. Additional findings included mild
bile duct hyperplasia and atrophy of the thymus, spleen and lymph
nodes in both sexes. B6C3F1 mice were also dosed with BDCM in this
study, and doses of 50 mg/kg of body weight per day and below produced
no compound-related effects. Degeneration and necrosis of the kidney
were observed in male mice at 100 mg/kg of body weight per day,
whereas centrilobular degeneration of the liver was noted in females
at 200 and 400 mg/kg of body weight per day.
3) Chronic toxicity
Moore et al. (1994) administered BDCM in drinking-water
(containing 0.25% Emulphor) to male F344 rats and B6C3F1 mice for
1 year and evaluated clinical indicators of kidney toxicity. Water
containing BDCM concentrations of 0.08, 0.4 and 0.8 g/litre for rats
and 0.06, 0.3 and 0.6 g/litre for mice resulted in average daily doses
of 4.4, 21 and 39 mg/kg of body weight for rats and 5.6, 24 and 49
mg/kg of body weight for mice. A urinary marker for renal proximal
tubule damage, N-acetyl-ß-glucosaminidase, was elevated above
controls in each dose group in rats and at the highest treatment level
in mice. Significant increases in urinary protein, indicative of
glomerular damage, were also noted in low- and mid-dose rats as well
as high-dose mice.
In an NTP (1987) study, BDCM was administered by corn oil gavage
for 102 weeks, 5 days per week, to F344/N rats (50 per sex per dose)
at doses of 0, 50 or 100 mg/kg of body weight per day and to B6C3F1
mice at doses of 0, 25 or 50 mg/kg of body weight per day (50 males
per group) and 0, 75 or 150 mg/kg of body weight per day (50 females
per group). In male rats, compound-related non-neoplastic lesions
included renal cytomegaly and tubular cell hyperplasia and hepatic
necrosis and fatty metamorphosis. Kidney tubule cell hyperplasia was
also observed in female rats, as well as eosinophilic cytoplasmic
change, clear cell change, focal cellular change and fatty
metamorphosis of the liver. Histopathological changes were noted at
both doses in rats. BDCM-induced non-neoplastic lesions in male mice
included hepatic fatty metamorphosis, renal cytomegaly and follicular
cell hyperplasia of the thyroid gland, all observed in both dose
groups. In female mice, hyperplasia of the thyroid gland was observed
at both doses.
Microencapsulated BDCM was fed in the diet to Wistar rats for 24
months, resulting in average daily doses of 6, 26 or 138 mg/kg of body
weight for males and 8, 32 or 168 mg/kg of body weight for females
(Aida et al., 1992b). Relative liver weight was increased in both
sexes of all dose groups, as was relative kidney weight in the
high-dose group. BDCM induced hepatic fatty degeneration and granuloma
in all dose groups and cholangiofibrosis in the high-dose groups.
Therefore, this study identified a LOAEL for chronic liver toxicity of
6 mg/kg of body weight per day.
4.1.2.2 Reproductive and developmental toxicity
Klinefelter et al. (1995) studied the potential of BDCM to alter
male reproductive function in F344 rats. BDCM was consumed in the
drinking-water for 52 weeks, resulting in average dose rates of 22 and
39 mg/kg of body weight per day. No gross lesions in the reproductive
organs were revealed by histological examination, but exposure to the
high BDCM dose significantly decreased the mean straight-line, average
path and curvilinear velocities of sperm recovered from the cauda
epididymis. These effects of BDCM on sperm motility occurred at a
lower exposure level than was observed for other DBPs that compromised
sperm motility.
A teratological assessment of BDCM was conducted in
Sprague-Dawley rats by administering the compound by gavage from day 6
to day 15 of gestation (Ruddick et al., 1983). Doses of 50, 100 and
200 mg of BDCM per kg of body weight per day did not produce any
teratogenic effects or dose-related histopathological changes in
either the dams or fetuses, but sternebra aberrations were observed
with a dose-dependent incidence in all dose groups. The increased
incidence of these variations appeared to be significant, but no
statistical analysis of the data was performed. Maternal weight gain
was depressed in the high-dose group, and maternal liver and kidney
weights were increased. Narotsky et al. (1997) employed a similar
experimental model to test BDCM in F344 rats using doses of 0, 25, 50
or 75 mg/kg of body weight per day in aqueous or oil gavage vehicles.
BDCM induced full-litter resorptions in the 50 and 75 mg/kg of body
weight per day dose groups with either vehicle of administration. For
dams receiving corn oil, full-litter resorptions were noted in 8% and
83% of the litters at 50 and 75 mg/kg of body weight per day,
respectively. With the aqueous vehicle, 17% and 21% of the litters
were fully resorbed at 50 and 75 mg/kg of body weight per day,
respectively. All vehicle control litters and litters from the group
given 25 mg/kg of body weight per day survived the experimental
period. BDCM had been shown to cause maternal toxicity at these doses
in a previous study (Narotsky et al., 1992).
4.1.2.3 Neurotoxicity
Neurotoxicological findings for the brominated THMs are limited
to various observations of anaesthesia associated with acute high-dose
exposures and results from a behavioural study conducted by Balster &
Borzelleca (1982). Adult male ICR mice were dosed by aqueous gavage
for up to 90 days. Treatments of 1.2 or 11.6 mg/kg of body weight per
day were without effect in various behavioural tests, and dosing for
30 days with 100 mg/kg of body weight per day did not affect passive
avoidance learning. Animals dosed with either 100 or 400 mg/kg of body
weight per day for 60 days exhibited decreased response rates in an
operant behaviour test; these effects were greatest early in the
regimen, with no evidence of progressive deterioration.
4.1.2.4 Toxicity in humans
Clinical case findings resulting from human exposure to BDCM have
not been reported.
4.1.2.5 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of BDCM and concluded that
there is sufficient evidence for its carcinogenicity in experimental
animals and inadequate evidence for its carcinogenicity in humans. On
this basis, BDCM was assigned to Group 2B: the agent is possibly
carcinogenic to humans (IARC, 1991, 1999).
Among the four THMs commonly found in drinking-water, BDCM
appears to be the most potent rodent carcinogen. BDCM caused cancer at
lower doses and at more target sites than for any of the other THMs.
In the NTP (1987) 2-year bioassay, a corn oil gavage study (50 animals
per sex per group, dosed 5 days per week), compound-related tumours
were found in the liver, kidneys and large intestine. Daily doses were
0, 50 or 100 mg/kg of body weight (male and female rats), 0, 25 or 50
mg/kg of body weight (male mice) and 0, 75 or 150 mg/kg of body weight
(female mice). NTP (1987) concluded that there was clear evidence of
carcinogenic activity for both sexes of F344 rats and B6C3F1 mice, as
shown by increased incidences of tubular cell adenomas and
adenocarcinomas in the kidney and adenocarcinomas and adenomatous
polyps in the large intestine of male and female rats, increased
incidences of tubular cell adenomas and adenocarcinomas in the kidney
of male mice, and increased incidences of hepatocellular adenomas and
carcinomas in female mice (Table 11).
Table 11. Tumour frequencies in rats and mice exposed to bromodichloromethane
in corn oil for 2 yearsa
Animal/tissue/tumour Tumour frequency at control, low and high
doses (mg/kg of body weight per day)
Male rat 0 50 100
Large intestineb
Adenomatous polyp 0/50 3/49 33/50
Adenocarcinoma 0/50 11/49 38/50
Combined 0/50 13/49 45/50
Kidneyb
Tubular cell adenoma 0/50 1/49 3/50
Tubular cell adenocarcinoma 0/50 0/49 10/50
Combined 0/50 1/49 13/50
Large intestine and/or kidney 0/50 13/49 46/50
combinedb
Female rat 0 50 100
Large intestinec
Adenomatous polyp 0/46 0/50 7/47
Adenocarcinoma 0/46 0/50 6/47
Combined 0/46 0/50 12/47
Kidney
Tubular cell adenoma 0/50 1/50 6/50
Tubular cell adenocarcinoma 0/50 0/50 9/50
Combined 0/50 1/50 15/50
Large intestine and/or kidney 0/46 1/50 24/48
combinedd
Male mouse 0 25 100
Kidneye
Tubular cell adenoma 1/46 2/49 6/50
Tubular cell adenocarcinoma 0/46 0/49 4/50
Combined 1/46 2/49 9/50
Female mouse 0 75 150
Liver
Hepatocellular adenoma 1/50 13/48 23/50
Hepatocellular carcinoma 2/50 5/48 10/50
Combined 3/50 18/48 29/50
Table 11. (continued)
a Adapted from NTP (1987).
b One rat died at week 33 in the low-dose group and was eliminated from
the cancer risk calculation.
c Intestine not examined in four rats from the control group and three
rats from the high-dose group.
d One rat in the high-dose group was not examined for intestinal tumours
and kidney tumours.
e In the control group, two mice died during the first week, one mouse
died during week 9 and one escaped in week 79. One mouse in the
low-dose group died in the first week. All of these mice were
eliminated from the cancer risk calculations.
Aida et al. (1992b) maintained Slc:Wistar rats on diets
containing microencapsulated BDCM for 24 months and examined the
animals for neoplastic lesions. The only significant finding was a
slight increase in the incidence of liver tumours in females receiving
the high dose (168 mg/kg of body weight per day). These included
cholangiocarcinomas and hepatocellular adenomas.
To date, effects following chronic BDCM administration via
drinking-water have not been described in the literature. However, two
separate drinking-water studies are currently being conducted by the
US Environmental Protection Agency (EPA) and the NTP.
Although BDCM has given mixed results in bacterial assays for
genotoxicity, the results have tended to be positive in tests
employing closed systems to overcome the problem of the compound's
volatility (IARC, 1991, 1999; Pegram et al., 1997). Pegram et al.
(1997) tested the THMs using a TA1535 strain transfected with rat GST
T1-1 and found that the mutagenicity of the brominated THMs, but not
chloroform, was greatly enhanced by the expression of the transferase.
BDCM was mutagenic in this assay at medium concentrations below 0.1
mmol/litre. Mutation spectra of the brominated THMs at the hisG46
allele were characterized by DeMarini et al. (1997) using revertants
induced in the GST-transfected strain. The overwhelming majority
(96-100%) of the mutations induced by the brominated THMs were GC to
AT transitions, and 87-100% of these were at the second position of
the CCC/GGG target. BDCM produced primary DNA damage in the SOS
chromotest (Escherichia coli PQ37), but was negative in the Ames
fluctuation test with Salmonella typhimurium TA100 (Le Curieux et
al., 1995). A mixture of BDCM and benzo [a]pyrene was tested in an
Ames mutagenicity test with S. typhimurium strains TA98 and TA100
plus S9 (Kevekordes et al., 1998). BDCM in combination with
benzo [a]pyrene caused a 25% increase in revertants in both strains
compared with benzo [a]pyrene alone.
BDCM was also positive in the majority of in vitro genotoxicity
tests employing eukaryotic systems, but the responses with and without
an exogenous metabolizing system are less consistent. This may be due
to the fact that the reactive intermediates suspected to be involved
in THM mutagenicity must be generated within the target cells (Thier
et al., 1993; Pegram et al., 1997). Moreover, extensive metabolism of
the THMs by the supplemental S9 outside of the cells would greatly
diminish the intracellular dose. Many of the positive studies are for
the induction of sister chromatid exchange (SCE) (IARC, 1991, 1999).
Morimoto & Koizumi (1983) found that BDCM induced SCEs in human
lymphocytes in vitro in the absence of S9 activation at
concentrations greater than or equal to 0.4 mmol/litre, and Fujie et
al. (1993) reported increased SCEs in rat erythroblastic leukaemia
cells under similar conditions. Metabolically activated BDCM also
increased SCEs in vitro in human lymphocytes (at 1 mmol/litre) and
in rat hepatocytes (at 100 mmol/litre) (Sobti, 1984).
In vivo, doses of 50 mg of BDCM per kg of body weight and above
produced SCEs in male CR/SJ mice (Morimoto & Koizumi, 1983). BDCM was
negative in in vivo clastogenicity tests (micronucleus formation) in
mice and rats (Ishidate et al., 1982; Hayashi et al., 1988). Fujie et
al. (1990) reported that BDCM induced bone marrow chromosomal
aberrations (primarily chromatid and chromosome breaks) in Long-Evans
rats following oral or intraperitoneal dosing at doses as low as 16.4
mg/kg of body weight. Potter et al. (1996) found that BDCM did not
induce DNA strand breaks in the kidney of male F344 rats following
seven daily doses of 1.5 mmol/kg of body weight (246 mg/kg of body
weight). Stocker et al. (1997) studied the effect of gastric
intubation of aqueous solutions of BDCM on unscheduled DNA synthesis
(UDS) in the liver of male rats. BDCM did not cause UDS in hepatocytes
isolated after administration of single doses of 135 or 450 mg/kg of
body weight. The in vivo mutagenicity studies of BDCM and the other
brominated THMs are summarized in Table 12.
In comparison with other chemicals known to produce mutations via
direct DNA reactivity such as aflatoxin B1 and ethylene dibromide,
BDCM is a relatively weak mutagen.
4.1.2.6 Comparative pharmacokinetics and metabolism
Bromine substitution would be expected to confer greater
lipophilicity on the brominated THMs compared with chloroform, which
would affect tissue solubility and other factors that can influence
pharmacokinetics. Because metabolism of each THM is qualitatively
similar (with one known exception), this section addresses key
features of the metabolism of all four THMs.
The absorption, distribution and elimination of BDCM have been
studied in rats and mice, and more recent work has led to the
development of a physiologically based pharmacokinetic (PBPK) model
for BDCM in rats. Mink et al. (1986) compared the pharmacokinetics of
orally administered 14C-BDCM in male B6C3F1 mice and Sprague-Dawley
rats. The animals received single doses of 100 mg/kg of body weight
(rats) or 150 mg/kg of body weight (mice) in corn oil by gavage, and
tissue levels of radioactivity were determined after 8 h. Absorption
of BDCM appeared to be rapid and fairly complete, as would be expected
for small halocarbons. This was especially true in the mouse, where
93% of the dose was recovered within 8 h as carbon dioxide (81%), as
Table 12. Dose information for selected in vivo mutagenicity studies of brominated trihalomethanes
End-point Assay system Dosea Result Reference
Sister chromatid exchange Male CR/SJ mice, gavage, 4 days 50 mg BDCM/kg bw per day positive Morimoto & Koizumi (1983)
Sister chromatid exchange Male CR/SJ mice, gavage, 4 days 25 mg DBCM/kg bw per day positive Morimoto & Koizumi (1983)
Sister chromatid exchange Male CR/SJ mice, gavage, 4 days 25 mg bromoform/kg bw positive Morimoto & Koizumi (1983)
per day
Sister chromatid exchange B6C3F1 mice, i.p.b 200 mg bromoform/kg bw positive NTP (1989a)
Micronucleus formation ddY mice, MS mice, Wistar rats, 500 mg BDCM/kg bw per day negative Ishidate et al. (1982)
i.p. in olive oil
Micronucleus formation ddY mice, MS mice, Wistar rats, 500 mg DBCM/kg bw per day negative Ishidate et al. (1982)
i.p. in olive oil
Micronucleus formation ddY mice, MS mice, Wistar rats, 500 mg bromoform/kg bw negative Ishidate et al. (1982)
i.p. in olive oil per day
Micronucleus formation ddY mice, i.p., single dose in 500 mg BDCM/kg bw negative Hayashi et al. (1988)
corn oil
Micronucleus formation ddY mice, i.p., single dose in 1000 mg DBCM/kg bw negative Hayashi et al. (1988)
corn oil
Micronucleus formation ddY mice, i.p., single dose in 1400 mg bromoform/kg bw negative Hayashi et al. (1988)
corn oil
Chromosomal aberrations Long-Evans rats, bone marrow, 16.4 mg BDCM/kg bw positive Fujie et al. (1990)
i.p., single dose
Chromosomal aberrations Long-Evans rats, bone marrow, 20.8 mg DBCM/kg bw positive Fujie et al. (1990)
i.p., single dose
Table 12. (continued)
End-point Assay system Dosea Result Reference
Chromosomal aberrations Long-Evans rats, bone marrow, 25.3 mg bromoform/kg bw positive Fujie et al. (1990)
i.p., single dose
Chromosomal aberrations Long-Evans rats, bone marrow 253 mg bromoform/kg bw positive Fujie et al. (1990)
per day
Unscheduled DNA synthesis Rat liver, gavage 450 mg BDCM/kg bw per negative Stocker et al. (1997)
day
Unscheduled DNA synthesis Rat liver, gavage 2000 mg DBCM/kg bw per negative Stocker et al. (1997)
day
Unscheduled DNA synthesis Rat liver, gavage 1080 mg bromoform/kg bw negative Stocker et al. (1997)
per day
Micronucleus formation Mouse, bone marrow, gavage, 1000 mg bromoform/kg bw negative Stocker et al. (1997)
single dose
DNA strand break Male F344 rats, kidney, gavage, 1.5 mmol BDCM/kg bw per negative Potter et al. (1996)
7 days day
DNA strand break Male F344 rats, kidney, gavage, 1.5 mmol DBCM/kg bw per negative Potter et al. (1996)
7 days day
DNA strand break Male F344 rats, kidney, gavage, 1.5 mmol bromoform/kg bw negative Potter et al. (1996)
7 days per day
Sex-linked recessive Drosophila 1000 ppm solution positive NTP (1989a)
mutation
a Doses listed are the lowest at which an effect was observed or, in the case of negative results, the highest dose tested.
bw = body weight.
b Intraperitoneal.
expired volatile organics assumed to be unmetabolized parent compound
(7.2%), in urine (2.2%) or in organs (3.2%). Much more of the 14C
dose was expired as the assumed parent compound by the rat (42%) than
by the mouse, but total recovery after 8 h was less (63%) because of
lower conversion to carbon dioxide (14%). The liver, stomach and
kidneys were the organs with the highest residual radioactivity
levels. The authors estimated BDCM half-lives of 1.5 and 2.5 h in the
rat and mouse, respectively.
Mathews et al. (1990) studied the disposition of 14C-BDCM in
male F344 rats after single oral (corn oil gavage) doses of 1, 10, 32
or 100 mg/kg of body weight and 10-day repeat oral dosing of 10 or
100 mg/kg of body weight per day. The doses of BDCM were well absorbed
from the gastrointestinal tract, as demonstrated by 24-h recoveries
exceeding 90% in non-faecal excreta samples and tissues. Persistence
of radiolabelled residues in tissues after 24 h was low (3-4% of
dose), with the most marked accumulation in the liver (1-3% of dose).
The kidneys, particularly cortical regions, also contained significant
concentrations of radiolabelled residues. Approximately 3-6% of the
dose was eliminated as volatile organics in the breath (primarily the
parent compound, presumably), much less than in Sprague-Dawley rats
(Mink et al., 1986). Urinary and faecal elimination were low at all
dose levels, accounting for 4% and 1-3% of the administered doses,
respectively. Repeated doses had no effect on the tissue distribution
of BDCM, and significant bioaccumulation was not observed (0.9-1.1%
total retention of the label).
Lilly et al. (1998) examined absorption and tissue dosimetry of
BDCM in male F344 rats after doses of 50 or 100 mg/kg of body weight
were given orally using either an aqueous emulsion or corn oil as the
vehicle of administration. After delivery in the aqueous vehicle,
concentrations of BDCM in venous blood peaked at about 6 min, with
maximum concentration ( Cmax) values of 16 and 26 mg/litre for the
low and high doses. With corn oil dosing, Cmax occurred at 15-30
min, with lower peak blood levels attained (5 and 9 mg/litre). The
time required for blood concentrations to decline to half- Cmax was
about 1 h with the 50 mg/kg of body weight dose and 1.5 h with the 100
mg/kg of body weight dose. Tissue partition coefficient determinations
confirmed the anticipated effect of bromine substitution on THM tissue
solubility. BDCM partition coefficients for fat and liver were 526 and
30.6 (Lilly et al., 1997b), compared with 203 and 21.1 for chloroform
(Corley et al., 1990). Lilly et al. (1998) found slightly higher
maximum concentrations of BDCM in the liver and kidneys after aqueous
administration compared with corn oil delivery. With the 100 mg/kg of
body weight aqueous dose, hepatic and renal levels peaked at about 15
mg/litre at 5 min after dosing in the liver and at 5-30 min in the
kidneys. At 6 h after dosing, concentrations of BDCM in the liver and
kidneys were less than 1 mg/litre. More of the parent compound was
eliminated unmetabolized via exhaled breath after aqueous dosing
(8.9%, low dose; 13.2%, high dose) than after corn oil gavage (5.3%,
low dose; 5.8%, high dose).
The elimination kinetics of BDCM have been studied in humans who
had swum in chlorinated pools (Lindstrom et al., 1997; Pleil &
Lindstrom, 1997). BDCM half-lives of 0.45-0.63 min for blood were
estimated using breath elimination data.
The deleterious effects of the THMs result from reactive
metabolites generated by biotransformation. Two isoenzyme groups of
cytochrome P450, CYP2E1 and CYP2B1/2, as well as a theta-class GST,
have been implicated in the metabolism of BDCM to toxic species in
rats (Thornton-Manning et al., 1993; Pegram et al., 1997). No specific
information is available regarding human metabolism of brominated
THMs. In rats, P450-mediated metabolism of BDCM (Figure 1) is believed
to proceed by the same two pathways established for chloroform:
oxidation with phosgene the proposed active metabolite, and reduction
with the dichloromethyl free radical proposed as the reactive product
(Tomasi et al., 1985; IPCS, 1994; Gao et al., 1996). Most
investigations of THM metabolism and reaction mechanisms have focused
on chloroform, and a detailed review of chloroform biotransformation
has been published recently (IPCS, 1994). Although the qualitative
aspects of the cytochrome P450-mediated metabolism of brominated THMs
are similar to those for chloroform, numerous studies have
demonstrated that brominated THMs are metabolized to a greater extent
and at faster rates than chloroform. There is evidence to suggest that
both CYP2E1 and CYP2B1/2 can catalyse the oxidative pathway and that
CYP2B1/2 catalyses the reductive metabolism of haloforms, but it has
also been postulated that either isoform can catalyse both routes
(Tomasi et al., 1985; Testai et al., 1996). CYP2E1 is clearly involved
in the hepatotoxicity induced by BDCM in rats, but its role in the
nephrotoxic response is less certain (Thornton-Manning et al., 1993).
Because CYP2E1 is highly conserved across mammalian species, it seems
likely that this isoform metabolizes BDCM in humans, although this has
not yet been demonstrated. CYP2B1/2 are not expressed in humans, and
there is no direct analogue for their catalytic activity; however,
based on substrate similarities with CYP2B1/2, human forms CYP2A6,
CYP2D6 and CYP3A4 appear to be possibilities.
Gao et al. (1996) demonstrated that GSH affords protection
against the toxicity and macromolecular binding of BDCM, indicating
that oxidative metabolism of BDCM, like that of chloroform, generates
phosgene, which can then react with GSH (Stevens & Anders, 1981).
In vitro binding of a BDCM-derived intermediate to microsomal
protein under aerobic conditions and the prevention of this binding by
GSH supplementation provide further evidence for the production of
phosgene from BDCM (Gao et al., 1996). The reaction of GSH with
phosgene forms S-(chlorocarbonyl)-GSH, which may react with a second
GSH molecule to produce diglutathionyl dithiocarbonate (Pohl et al.,
1981) or to give glutathione disulfide and carbon monoxide as minor
products (Stevens & Anders, 1981). Anders et al. (1978) and Mathews et
al. (1990) reported that carbon monoxide is a product of BDCM
metabolism. The most likely outcome for phosgene is hydrolysis to
carbon dioxide and hydrogen chloride (Brown et al., 1974), and, in
fact, 70-80% of 14C-BDCM doses administered to F344 rats or B6C3F1
mice appeared as expired 14C-labelled carbon dioxide (Mink et al.,
1986; Mathews et al., 1990), which also shows the predominance of
WHO TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR DISINFECTANTS AND
DISINFECTANT BY-PRODUCTS
Members
Dr G. Amy, Department of Civil, Environmental, and Architectural
Engineering, University of Colorado, Boulder, Colorado, USA
Mr J. Fawell, Water Research Centre, Marlow, Buckinghamshire, United
Kingdom (Co-Rapporteur)
Dr B. Havlik, Ministry of Health, National Institute of Public Health,
Prague, Czech Republic
Dr C. Nokes, Water Group, Institute of Environmental Science and
Research, Christchurch, New Zealand (Co-Rapporteur)
Dr E. Ohanian, Office of Water/Office of Science and Technology,
United States Environmental Protection Agency, Washington, DC,
USA (Chairman)
Dr E. Soderlund, Department of Environmental Medicine, National
Institute of Public Health, Torshov, Oslo
Secretariat
Dr J. Bartram, Water, Sanitation and Health Unit, Division of
Operational Support in Environment Health, World Health
Organization, Geneva, Switzerland
Dr R. Bull, Battelle Pacific Northwest Laboratory, Richland,
Washington, USA
Mr G.F. Craun, Gunther F. Craun and Associates, Staunton, Virginia,
USA
Dr H. Galal-Gorchev, Chevy Chase, Maryland, USA (Secretary)
Mr N. Nakashima, Assessment of Risk and Methodologies,
International Programme on Chemical Safety, World Health
Organization, Geneva, Switzerland
Dr R.A. Pegram, United States Environmental Protection Agency,
Research Triangle Park, North Carolina, USA
Mr S.T. Yamamura, Water, Sanitation and Health Unit, Division of
Operational Support in Environment Health, World Health
Organization, Geneva, Switzerland
Representatives/Observers
Dr N. Drouot, Dept Toxicologie Industrielle, Paris, France
(representing European Centre for Ecotoxicology and Toxicology
of Chemicals)
Mr O. Hydes, Drinking Water Inspectorate, London, United Kingdom
Dr B.B. Sandel, Olin Corporation, Norwalk, Connecticut, USA
(representing American Industrial Health Council/International
Life Sciences Institute)
IPCS TASK GROUP ON ENVIRONMENTAL HEALTH CRITERIA FOR DISINFECTANTS AND
DISINFECTANT BY-PRODUCTS
A WHO Task Group on Environmental Health Criteria for
Disinfectants and Disinfectant By-products met in Geneva from 17 to 21
August 1998. Dr Peter Toft, Associate Director, IPCS, welcomed the
participants on behalf of the three IPCS cooperating organizations
(UNEP/ILO/WHO). The Task Group reviewed and revised the draft document
and made an evaluation of risks for human health from exposure to
certain disinfectants and disinfectant by-products.
The first draft of the chemistry section was prepared by G. Amy
and M. Siddiqui, University of Colorado, Boulder, Colorado, USA; the
toxicology section was prepared by R. Bull, Battelle Pacific Northwest
Laboratory, Richland, Washington, USA, and R.A. Pegram, US
Environmental Protection Agency, Research Triangle Park, North
Carolina, USA; and the epidemiology section was prepared by G.F.
Craun, Gunther F. Craun and Associates, Staunton, Virginia, USA.
The efforts of all who helped in the preparation and finalization
of the monograph are gratefully acknowledged.
* * *
The preparation of the first draft of this Environmental Health
Criteria monograph was made possible by the financial support afforded
to IPCS by the International Life Sciences Institute.
A financial contribution from the United States Environmental
Protection Agency for the convening of the Task Group is gratefully
acknowledged.
ACRONYMS AND ABBREVIATIONS
ALAT alanine aminotransferase
AP alkaline phosphatase
ARB atypical residual bodies
ASAT aspartate aminotransferase
AWWA American Water Works Association
BAN bromoacetonitrile
BCA bromochloroacetic acid/bromochloroacetate
BCAN bromochloroacetonitrile
BDCA bromodichloroacetic acid/bromodichloroacetate
BDCM bromodichloromethane
BUN blood urea nitrogen
bw body weight
CAN chloroacetonitrile
CHO Chinese hamster ovary
CI confidence interval
CoA coenzyme A
Cmax maximum concentration
CMCF 3-chloro-4-(chloromethyl)-5-hydroxy-2(5H)-furanone
2-CP 2-chloropropionate
CPN chloropropanone
CT computerized tomography
CYP cytochrome P450
DBA dibromoacetic acid/dibromoacetate
DBAC dibromoacetone
DBAN dibromoacetonitrile
DBCM dibromochloromethane
DBP disinfectant by-product
DCA dichloroacetic acid/dichloroacetate
DCAN dichloroacetonitrile
DCPN dichloropropanone
DHAN dihaloacetonitrile
DOC dissolved organic carbon
ECD electron capture detector
ECG electrocardiogram
EEG electroencephalogram
EHEN N-ethyl- N-hydroxyethylnitrosamine
EPA Environmental Protection Agency (USA)
ESR electron spin resonance
FAO Food and Agriculture Organization of the United Nations
GAC granular activated carbon
GC gas chromatography
GGT gamma-glutamyl transpeptidase
GOT glutamate-oxalate transaminase
GPT glutamate-pyruvate transaminase
GSH glutathione-SH
GST glutathione- S-transferase
HAA haloacetic acid
HAN haloacetonitrile
HDL high-density lipoprotein
HPLC high-performance liquid chromatography
hprt hypoxanthine phosphoribosyl transferase
IARC International Agency for Research on Cancer
IC ion chromatography
i.p. intraperitoneal
IPCS International Programme on Chemical Safety
JECFA Joint FAO/WHO Expert Committee on Food Additives
LD50 median lethal dose
LDH lactate dehydrogenase
LDL low-density lipoprotein
LOAEL lowest-observed-adverse-effect level
MA 3,4-(dichloro)-5-hydroxy-2(5H)-furanone
MBA monobromoacetic acid/monobromoacetate
MCA monochloroacetic acid/monochloroacetate
MNU methylnitrosourea
MOR mortality odds ratio
MRI magnetic resonance imaging
MTBE methyl tert-butyl ether
MX 3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone
NADP nicotinamide adenine dinucleotide phosphate
NOAEL no-observed-adverse-effect level
NOEL no-observed-effect level
NOM natural organic matter
NTP National Toxicology Program (USA)
8-OH-dG 8-hydroxy-2-deoxyguanosine
OR odds ratio
PAS periodic acid/Schiff's reagent
PBPK physiologically based pharmacokinetic model
PFBHA O-(2,3,4,5,6-pentafluorobenzyl)-hydroxylamine
p Ka log acid dissociation constant
PPAR peroxisome proliferator activated receptor
PPRE peroxisome proliferator responsive element
RR relative risk
SCE sister chromatid exchange
SD standard deviation
SDH sorbitol dehydrogenase
SE standard error
SGOT serum glutamate-oxaloacetate transaminase
SGPT serum glutamate-pyruvate transaminase
SMR standardized mortality ratio
SSB single strand breaks
TBA tribromoacetic acid/tribromoacetate
TBARS thiobarbituric acid reactive substances
TCA trichloroacetic acid/trichloroacetate
TCAN trichloroacetonitrile
TCPN trichloropropanone
TDI tolerable daily intake
TGF transforming growth factor
THM trihalomethane
TOC total organic carbon
TOX total organic halogen
TPA 12- O-tetradecanoylphorbol-13-acetate
UDS unscheduled DNA synthesis
UV ultraviolet
UVA254 UV absorbance at 254 nm
Vmax maximum rate of metabolism
WHO World Health Organization
1. SUMMARY AND EVALUATION
Chlorine (Cl2) has been widely used throughout the world as a
chemical disinfectant, serving as the principal barrier to microbial
contaminants in drinking-water. The noteworthy biocidal attributes of
chlorine have been somewhat offset by the formation of disinfectant
by-products (DBPs) of public health concern during the chlorination
process. As a consequence, alternative chemical disinfectants, such as
ozone (O3), chlorine dioxide (ClO2) and chloramines (NH2Cl,
monochloramine), are increasingly being used; however, each has been
shown to form its own set of DBPs. Although the microbiological
quality of drinking-water cannot be compromised, there is a need to
better understand the chemistry, toxicology and epidemiology of
chemical disinfectants and their associated DBPs in order to develop a
better understanding of the health risks (microbial and chemical)
associated with drinking-water and to seek a balance between microbial
and chemical risks. It is possible to decrease the chemical risk due
to DBPs without compromising microbiological quality.
1.1 Chemistry of disinfectants and disinfectant by-products
The most widely used chemical disinfectants are chlorine, ozone,
chlorine dioxide and chloramine. The physical and chemical properties
of disinfectants and DBPs can affect their behaviour in
drinking-water, as well as their toxicology and epidemiology. The
chemical disinfectants discussed here are all water-soluble oxidants,
which are produced either on-site (e.g., ozone) or off-site (e.g.,
chlorine). They are administered as a gas (e.g., ozone) or liquid
(e.g., hypochlorite) at typical doses of several milligrams per litre,
either alone or in combination. The DBPs discussed here are measurable
by gas or liquid chromatography and can be classified as organic or
inorganic, halogenated (chlorinated or brominated) or non-halogenated,
and volatile or non-volatile. Upon their formation, DBPs can be stable
or unstable (e.g., decomposition by hydrolysis).
DBPs are formed upon the reaction of chemical disinfectants with
DBP precursors. Natural organic matter (NOM), commonly measured by
total organic carbon (TOC), serves as the organic precursor, whereas
bromide ion (Br-) serves as the inorganic precursor. DBP formation is
influenced by water quality (e.g., TOC, bromide, pH, temperature,
ammonia, carbonate alkalinity) and treatment conditions (e.g.,
disinfectant dose, contact time, removal of NOM before the point of
disinfectant application, prior addition of disinfectant).
Chlorine in the form of hypochlorous acid/hypochlorite ion
(HOCl/OCl-) reacts with bromide ion, oxidizing it to hypobromous
acid/hypobromite ion (HOBr/OBr-). Hypochlorous acid (a more powerful
oxidant) and hypobromous acid (a more effective halogenating agent)
react collectively with NOM to form chlorine DBPs, including
trihalomethanes (THMs), haloacetic acids (HAAs), haloacetonitriles
(HANs), haloketones, chloral hydrate and chloropicrin. The dominance
of chlorine DBP groups generally decreases in the order of THMs, HAAs
and HANs. The relative amounts of TOC, bromide and chlorine will
affect the species distribution of THMs (four species: chloroform,
bromoform, bromodichloromethane [BDCM] and dibromochloromethane
[DBCM]), HAAs (up to nine chlorinated/brominated species) and HANs
(several chlorinated/brominated species). Generally, chlorinated THM,
HAA and HAN species dominate over brominated species, although the
opposite may be true in high-bromide waters. Although many specific
chlorine DBPs have been identified, a significant percentage of the
total organic halogens still remain unaccounted for. Another reaction
that occurs with chlorine is the formation of chlorate (ClO3-) in
concentrated hypochlorite solutions.
Ozone can directly or indirectly react with bromide to form
brominated ozone DBPs, including bromate ion (BrO3-). In the
presence of NOM, non-halogenated organic DBPs, such as aldehydes,
ketoacids and carboxylic acids, are formed during ozonation, with
aldehydes (e.g., formaldehyde) being dominant. If both NOM and bromide
are present, ozonation forms hypobromous acid, which, in turn, leads
to the formation of brominated organohalogen compounds (e.g.,
bromoform).
The major chlorine dioxide DBPs include chlorite (ClO2-) and
chlorate ions, with no direct formation of organohalogen DBPs. Unlike
the other disinfectants, the major chlorine dioxide DBPs are derived
from decomposition of the disinfectant as opposed to reaction with
precursors.
Use of chloramine as a secondary disinfectant generally leads to
the formation of cyanogen chloride (CNCl), a nitrogenous compound, and
significantly reduced levels of chlorine DBPs. A related issue is the
presence of nitrite (NO2-) in chloraminated distribution systems.
From the present knowledge of occurrence and health effects, the
DBPs of most interest are THMs, HAAs, bromate and chlorite.
The predominant chlorine DBP group has been shown to be THMs,
with chloroform and BDCM as the first and second most dominant THM
species. HAAs are the second predominant group, with dichloroacetic
acid (DCA) and trichloroacetic acid (TCA) being the first and second
most dominant species.
Conversion of bromide to bromate upon ozonation is affected by
NOM, pH and temperature, among other factors. Levels may range from
below detection (2 µg/litre) to several tens of micrograms per litre.
Chlorite levels are generally very predictable, ranging from about 50%
to 70% of the chlorine dioxide dose administered.
DBPs occur in complex mixtures that are a function of the
chemical disinfectant used, water quality conditions and treatment
conditions; other factors include the combination/sequential use of
multiple disinfectants/oxidants. Moreover, the composition of these
mixtures may change seasonally. Clearly, potential chemically related
health effects will be a function of exposure to DBP mixtures.
Other than chlorine DBPs (in particular THMs), there are very few
data on the occurrence of DBPs in finished water and distribution
systems. Based on laboratory databases, empirical models have been
developed to predict concentrations of THMs (total THMs and THM
species), HAAs (total HAAs and HAA species) and bromate. These models
can be used in performance assessment to predict the impact of
treatment changes and in exposure assessment to simulate missing or
past data (e.g., to predict concentrations of HAAs from THM data).
DBPs can be controlled through DBP precursor control and removal
or modified disinfection practice. Coagulation, granular activated
carbon, membrane filtration and ozone biofiltration can remove NOM.
Other than through the use of membranes, there is little opportunity
to effectively remove bromide. Source water protection and control
represent non-treatment alternatives to precursor control. Removal of
DBPs after formation is not viable for organic DBPs, whereas bromate
and chlorite can be removed by activated carbon or reducing agents. It
is expected that the optimized use of combinations of disinfectants,
functioning as primary and secondary disinfectants, can further
control DBPs. There is a trend towards combination/sequential use of
disinfectants; ozone is used exclusively as a primary disinfectant,
chloramines exclusively as a secondary disinfectant, and both chlorine
and chlorine dioxide in either role.
1.2 Kinetics and metabolism in laboratory animals and humans
1.2.1 Disinfectants
Residual disinfectants are reactive chemicals that will react
with organic compounds found in saliva and stomach content, resulting
in the formation of by-products. There are significant differences in
the pharmacokinetics of 36Cl depending on whether it is obtained from
chlorine, chloramine or chlorine dioxide.
1.2.2 Trihalomethanes
The THMs are absorbed, metabolized and eliminated rapidly by
mammals after oral or inhalation exposure. Following absorption, the
highest tissue concentrations are attained in the fat, liver and
kidneys. Half-lives generally range from 0.5 to 3 h, and the primary
route of elimination is via metabolism to carbon dioxide. Metabolic
activation to reactive intermediates is required for THM toxicity, and
the three brominated species are all metabolized more rapidly and to a
greater extent than chloroform. The predominant route of metabolism
for all the THMs is oxidation via cytochrome P450 (CYP) 2E1, leading
to the formation of dihalocarbonyls (i.e., phosgene and brominated
congeners), which can be hydrolysed to carbon dioxide or bind to
tissue macromolecules. Secondary metabolic pathways are reductive
dehalogenation via CYP2B1/2/2E1 (leading to free radical generation)
and glutathione (GSH) conjugation via glutathione- S-transferase
(GST) T1-1, which generates mutagenic intermediates. The brominated
THMs are much more likely than chloroform to proceed through the
secondary pathways, and GST-mediated conjugation of chloroform to GSH
can occur only at extremely high chloroform concentrations or doses.
1.2.3 Haloacetic acids
The kinetics and metabolism of the dihaloacetic and trihaloacetic
acids differ significantly. To the extent they are metabolized, the
principal reactions of the trihaloacetic acids occur in the microsomal
fraction, whereas more than 90% of the dihaloacetic acid metabolism,
principally by glutathione transferases, is observed in the cytosol.
TCA has a biological half-life in humans of 50 h. The half-lives of
the other trihaloacetic acids decrease significantly with bromine
substitution, and measurable amounts of the dihaloacetic acids can be
detected as products with brominated trihaloacetic acids. The
half-lives of the dihaloacetic acids are very short at low doses but
can be drastically increased as dose rates are increased.
1.2.4 Haloaldehydes and haloketones
Limited kinetic data are available for chloral hydrate. The two
major metabolites of chloral hydrate are trichloroethanol and TCA.
Trichloroethanol undergoes rapid glucuronidation, enterohepatic
circulation, hydrolysis and oxidation to TCA. Dechlorination of
trichloroethanol or chloral hydrate would lead to the formation of
DCA. DCA may then be further transformed to monochloroacetate (MCA),
glyoxalate, glycolate and oxalate, probably through a reactive
intermediate. No information was found on the other haloaldehydes and
haloketones.
1.2.5 Haloacetonitriles
The metabolism and kinetics of HANs have not been studied.
Qualitative data indicate that the products of metabolism include
cyanide, formaldehyde, formyl cyanide and formyl halides.
1.2.6 Halogenated hydroxyfuranone derivatives
3-Chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX) is the
member of the hydroxyfuranone class that has been most extensively
studied. From animal studies, it appears that the 14C label of MX is
rapidly absorbed from the gastrointestinal tract and reaches systemic
circulation. MX itself has not been measured in blood. The MX label is
largely excreted in urine and faeces, urine being the major route of
excretion. Very little of the initial radiolabel is retained in the
body after 5 days.
1.2.7 Chlorite
The 36Cl from chlorite is rapidly absorbed. Less than half the
dose is found in the urine as chloride, and a small proportion as
chlorite. A significant proportion probably enters the chloride pool
of the body, but a lack of analytical methods to characterize chlorite
in biological samples means that no detailed information is available.
1.2.8 Chlorate
Chlorate behaves similarly to chlorite. The same analytical
constraints apply.
1.2.9 Bromate
Bromate is rapidly absorbed and excreted, primarily in urine, as
bromide. Bromate is detected in urine at doses of 5 mg/kg of body
weight and above. Bromate concentrations in urine peak at about 1 h,
and bromate is not detectable in plasma after 2 h.
1.3 Toxicology of disinfectants and disinfectant by-products
1.3.1 Disinfectants
Chlorine gas, chloramine and chlorine dioxide are strong
respiratory irritants. Sodium hypochlorite (NaOCl) is also used as
bleach and is frequently involved in human poisoning. These exposures,
however, are not relevant to exposures in drinking-water. There have
been relatively few evaluations of the toxic effects of these
disinfectants in drinking-water in experimental animals or humans.
Evidence from these animal and human studies suggests that chlorine,
hypochlorite solutions, chloramine and chlorine dioxide themselves
probably do not contribute to the development of cancer or any toxic
effects. Attention has focused on the wide variety of by-products that
result from reactions of chlorine and other disinfectants with NOM,
which is found in virtually all water sources.
1.3.2 Trihalomethanes
THMs induce cytotoxicity in the liver and kidneys of rodents
exposed to doses of about 0.5 mmol/kg of body weight. The vehicle of
administration significantly affects the toxicity of the THMs. The
THMs have little reproductive and developmental toxicity, but BDCM has
been shown to reduce sperm motility in rats consuming 39 mg/kg of body
weight per day in drinking-water. Like chloroform, BDCM, when
administered in corn oil, induces cancer in the liver and kidneys
after lifetime exposures to high doses. Unlike chloroform and DBCM,
BDCM and bromoform induce tumours of the large intestine in rats
exposed by corn oil gavage. BDCM induces tumours at all three target
sites and at lower doses than the other THMs. Since the publication of
the 1994 WHO Environmental Health Criteria monograph on chloroform,
additional studies have added to the weight of evidence indicating
that chloroform is not a direct DNA-reactive mutagenic carcinogen. In
contrast, the brominated THMs appear to be weak mutagens, probably as
a result of GSH conjugation.
1.3.3 Haloacetic acids
The HAAs have diverse toxicological effects in laboratory
animals. Those HAAs of most concern have carcinogenic, reproductive
and developmental effects. Neurotoxic effects are significant at the
high doses of DCA that are used therapeutically. Carcinogenic effects
appear to be limited to the liver and to high doses. The bulk of the
evidence indicates that the tumorigenic effects of DCA and TCA depend
on modifying processes of cell division and cell death rather than
their very weak mutagenic activities. Oxidative stress is also a
feature of the toxicity of the brominated analogues within this class.
Both DCA and TCA cause cardiac malformations in rats at high doses.
1.3.4 Haloaldehydes and haloketones
Chloral hydrate induces hepatic necrosis in rats at doses equal
to or greater than 120 mg/kg of body weight per day. Its depressant
effect on the central nervous system in humans is probably related to
its metabolite trichloroethanol. Limited toxicity data are available
for the other halogenated aldehydes and ketones. Chloroacetaldehyde
exposure causes haematological effects in rats. Exposure of mice to
1,1-dichloropropanone (1,1-DCPN), but not 1,3-dichloropropanone
(1,3-DCPN), results in liver toxicity.
Chloral hydrate was negative in most but not all bacterial tests
for point mutations and in in vivo studies on chromosomal damage.
However, it has been shown that chloral hydrate may induce structural
chromosomal aberrations in vitro and in vivo. Chloral hydrate has
been reported to cause hepatic tumours in mice. It is not clear if it
is the parent compound or its metabolites that are involved in the
carcinogenic effect. The two chloral hydrate metabolites, TCA and DCA,
have induced hepatic tumours in mice.
Some halogenated aldehydes and ketones are potent inducers of
mutations in bacteria. Clastogenic effects have been reported for
chlorinated propanones. Liver tumours were noted in a lifetime
drinking-water study with chloroacetaldehyde. Other halogenated
aldehydes, e.g., 2-chloropropenal, have been identified as tumour
initiators in the skin of mice. The haloketones have not been tested
for carcinogenicity in drinking-water. However, 1,3-DCPN acted as a
tumour initiator in a skin carcinogenicity study in mice.
1.3.5 Haloacetonitriles
Testing of these compounds for toxicological effects has been
limited to date. Some of the groups are mutagenic, but these effects
do not relate well to the activity of the chemicals as tumour
initiators in the skin. There are only very limited studies on the
carcinogenicity of this class of substances. Early indications of
developmental toxicity of members of this class appear to be largely
attributable to the vehicle used in treatment.
1.3.6 Halogenated hydroxyfuranone derivatives
Based on experimental studies, the critical effects of MX appear
to be its mutagenicity and carcinogenicity. Several in vitro studies
have revealed that MX is mutagenic in bacterial and mammalian test
systems. MX caused chromosomal aberrations and induced DNA damage in
isolated liver and testicular cells and sister chromatid exchanges in
peripheral lymphocytes from rats exposed in vivo. An overall
evaluation of the mutagenicity data shows that MX is mutagenic
in vitro and in vivo. A carcinogenicity study in rats showed
increased tumour frequencies in several organs.
1.3.7 Chlorite
The toxic action of chlorite is primarily in the form of
oxidative damage to red blood cells at doses as low as 10 mg/kg of
body weight. There are indications of mild neurobehavioural effects in
rat pups at 5.6 mg/kg of body weight per day. There are conflicting
data on the genotoxicity of chlorite. Chlorite does not increase
tumours in laboratory animals in chronic exposure studies.
1.3.8 Chlorate
The toxicity of chlorate is similar to that of chlorite, but
chlorate is less effective at inducing oxidative damage. It does not
appear to be teratogenic or genotoxic in vivo. There are no data
from long-term carcinogenicity studies.
1.3.9 Bromate
Bromate causes renal tubular damage in rats at high doses. It
induces tumours of the kidney, peritoneum and thyroid in rats at doses
of 6 mg/kg of body weight and above in chronic studies. Hamsters are
less sensitive, and mice are considerably less sensitive. Bromate is
also genotoxic in vivo in rats at high doses. Carcinogenicity
appears to be secondary to oxidative stress in the cell.
1.4 Epidemiological studies
1.4.1 Cardiovascular disease
Epidemiological studies have not identified an increased risk of
cardiovascular disease associated with chlorinated or chloraminated
drinking-water. Studies of other disinfectants have not been
conducted.
1.4.2 Cancer
The epidemiological evidence is insufficient to support a causal
relationship between bladder cancer and long-term exposure to
chlorinated drinking-water, THMs, chloroform or other THM species. The
epidemiological evidence is inconclusive and equivocal for an
association between colon cancer and long-term exposure to chlorinated
drinking-water, THMs, chloroform or other THM species. The information
is insufficient to allow an evaluation of the observed risks for
rectal cancer and risks for other cancers observed in single
analytical studies.
Various types of epidemiological studies have attempted to assess
the cancer risks that may be associated with exposure to chlorinated
drinking-water. Chloraminated drinking-water was considered in two
studies. Several studies have attempted to estimate exposures to total
THMs or chloroform and the other THM species, but the studies did not
consider exposures to other DBPs or other water contaminants, which
may differ for surface water and groundwater sources. One study
considered the mutagenicity of drinking-water as measured by the
Salmonella typhimurium assay. Assessments of possible cancer risks
that may be associated with drinking-water disinfected with ozone or
chlorine dioxide have not been performed.
Ecological and death certificate-based case-control studies have
provided hypotheses for further evaluation by analytical studies that
consider an individual's exposure to drinking-water and possible
confounding factors.
Analytical studies have reported weak to moderate increased
relative risks of bladder, colon, rectal, pancreatic, breast, brain or
lung cancer associated with long-term exposure to chlorinated
drinking-water. Single studies reported associations for pancreatic,
breast or brain cancer; however, the evaluation of a possible causal
relationship for epidemiological associations requires evidence from
more than a single study. In one study, a small increased relative
risk of lung cancer was associated with the use of surface water
sources, but the magnitude of risk was too small to rule out residual
confounding.
A case-control study reported a moderately large association
between rectal cancer and long-term exposure to chlorinated
drinking-water or cumulative THM exposure, but cohort studies have
found either no increased risk or a risk too weak to rule out residual
confounding.
Decreased bladder cancer risk was associated with increased
duration of exposure to chloraminated drinking-water, but there is no
biological basis for assuming a protective effect of chloraminated
water.
Although several studies found increased risks of bladder cancer
associated with long-term exposure to chlorinated drinking-water and
cumulative exposure to THMs, inconsistent results were reported among
the studies for bladder cancer risks between smokers and non-smokers
and between men and women. Estimated exposure to THMs was considered
in three of these studies. In one study, no association was found
between estimated cumulative exposure to THMs. In another study, a
moderately strong increased relative risk was associated with
increased cumulative exposure to THMs in men but not in women. The
third study reported a weak increased relative risk associated with an
estimated cumulative exposure of 1957-6425 µg of THMs per litre-year;
weak to moderate associations were also reported for exposure to THM
concentrations greater than 24, greater than 49 and greater than 74
µg/litre. No increased relative risk of bladder cancer was associated
with exposure to chlorinated municipal surface water supplies,
chloroform or other THM species in a cohort of women, but the
follow-up period of 8 years was very short, resulting in few cases for
study.
Because inadequate attention has been paid to assessing exposure
to water contaminants in epidemiological studies, it is not possible
to properly evaluate the increased relative risks that were reported.
Specific risks may be due to other DBPs, mixtures of by-products or
other water contaminants, or they may be due to other factors for
which chlorinated drinking-water or THMs may serve as a surrogate.
1.4.3 Adverse pregnancy outcomes
Studies have considered exposures to chlorinated drinking-water,
THMs or THM species and various adverse outcomes of pregnancy. A
scientific panel recently convened by the US Environmental Protection
Agency reviewed the epidemiological studies and concluded that the
results of currently published studies do not provide convincing
evidence that chlorinated water or THMs cause adverse pregnancy
outcomes.
Results of early studies are difficult to interpret because of
methodological limitations or suspected bias.
A recently completed but not yet published case-control study has
reported moderate increased relative risks for neural tube defects in
children whose mothers' residence in early pregnancy was in an area
where THM levels were greater than 40 µg/litre. Replication of the
results in another area is required before this association can be
properly evaluated. A previously conducted study in the same
geographic area reported a similar association, but the study suffered
from methodological limitations.
A recently reported cohort study found an increased risk of early
miscarriage associated with heavy consumption of water (five or more
glasses of cold tapwater per day) containing high levels (>75
µg/litre) of THMs. When specific THMs were considered, only heavy
consumption of water containing BDCM (>18 µg/litre) was associated
with a risk of miscarriage. As this is the first study to suggest an
adverse reproductive effect associated with a brominated by-product, a
scientific panel recommended that another study be conducted in a
different geographic area to attempt to replicate these results and
that additional efforts be made to evaluate exposures of the cohort to
other water contaminants.
1.5 Risk characterization
It should be noted that the use of chemical disinfectants in
water treatment usually results in the formation of chemical
by-products, some of which are potentially hazardous. However, the
risks to health from these by-products at the levels at which they
occur in drinking-water are extremely small in comparison with the
risks associated with inadequate disinfection. Thus, it is important
that disinfection not be compromised in attempting to control such
by-products.
1.5.1 Characterization of hazard and dose-response
1.5.1.1 Toxicological studies
1) Chlorine
A WHO Working Group for the 1993 Guidelines for drinking-water
quality considered chlorine. This Working Group determined a
tolerable daily intake (TDI) of 150 µg/kg of body weight for free
chlorine based on a no-observed-adverse-effect level (NOAEL) of
approximately 15 mg/kg of body weight per day in 2-year studies in
rats and mice and incorporating an uncertainty factor of 100 (10 each
for intra- and interspecies variation). There are no new data that
indicate that this TDI should be changed.
2) Monochloramine
A WHO Working Group for the 1993 Guidelines for drinking-water
quality considered monochloramine. This Working Group determined a
TDI of 94 µg/kg of body weight based on a NOAEL of approximately 9.4
mg/kg of body weight per day, the highest dose tested, in a 2-year
bioassay in rats and incorporating an uncertainty factor of 100 (10
each for intra- and interspecies variation). There are no new data
that indicate that this TDI should be changed.
3) Chlorine dioxide
The chemistry of chlorine dioxide in drinking-water is complex,
but the major breakdown product is chlorite. In establishing a
specific TDI for chlorine dioxide, data on both chlorine dioxide and
chlorite can be considered, given the rapid hydrolysis to chlorite.
Therefore, an oral TDI for chlorine dioxide is 30 µg/kg of body
weight, based on the NOAEL of 2.9 mg/kg of body weight per day for
neurodevelopmental effects of chlorite in rats.
4) Trihalomethanes
Cancer following chronic exposure is the primary hazard of
concern for this class of DBPs. Because of the weight of evidence
indicating that chloroform can induce cancer in animals only after
chronic exposure to cytotoxic doses, it is clear that exposures to low
concentrations of chloroform in drinking-water do not pose
carcinogenic risks. The NOAEL for cytolethality and regenerative
hyperplasia in mice was 10 mg/kg of body weight per day after
administration of chloroform in corn oil for 3 weeks. Based on the
mode of action evidence for chloroform carcinogenicity, a TDI of 10
µg/kg of body weight was derived using the NOAEL for cytotoxicity in
mice and applying an uncertainty factor of 1000 (10 each for inter-
and intraspecies variation and 10 for the short duration of the
study). This approach is supported by a number of additional studies.
This TDI is similar to the TDI derived in the 1998 WHO Guidelines
for drinking-water quality, which was based on a 1979 study in which
dogs were exposed for 7.5 years.
Among the brominated THMs, BDCM is of particular interest because
it produces tumours in rats and mice and at several sites (liver,
kidneys, large intestine) after corn oil gavage. The induction of
colon tumours in rats by BDCM (and by bromoform) is also interesting
because of the epidemiological associations with colo-rectal cancer.
BDCM and the other brominated THMs are also weak mutagens. It is
generally assumed that mutagenic carcinogens will produce linear
dose-response relationships at low doses, as mutagenesis is generally
considered to be an irreversible and cumulative effect.
In a 2-year bioassay, BDCM given by corn oil gavage induced
tumours (in conjunction with cytotoxicity and increased proliferation)
in the kidneys of mice and rats at doses of 50 and 100 mg/kg of body
weight per day, respectively. The tumours in the large intestine of
the rat occurred after exposure to both 50 and 100 mg/kg of body
weight per day. Using the incidence of kidney tumours in male mice
from this study, quantitative risk estimates have been calculated,
yielding a slope factor of 4.8 × 10-3 [mg/kg of body weight per
day]-1 and a calculated dose of 2.1 µg/kg of body weight per day for
a risk level of 10-5. A slope factor of 4.2 × 10-3 [mg/kg of body
weight per day]-1 (2.4 µg/kg of body weight per day for a 10-5 risk)
was derived based on the incidence of large intestine carcinomas in
the male rat. The International Agency for Research on Cancer (IARC)
has classified BDCM in Group 2B (possibly carcinogenic to humans).
DBCM and bromoform were studied in long-term bioassays. In a
2-year corn oil gavage study, DBCM induced hepatic tumours in female
mice, but not in rats, at a dose of 100 mg/kg of body weight per day.
In previous evaluations, it had been suggested that the corn oil
vehicle may play a role in the induction of tumours in female mice. A
small increase in tumours of the large intestine in rats was observed
in the bromoform study at a dose of 200 mg/kg of body weight per day.
The slope factors based on these tumours are 6.5 × 10-3 [mg/kg of
body weight per day]-1 for DBCM, or 1.5 µg/kg of body weight per day
for a 10-5 risk, and 1.3 × 10-3 [mg/kg of body weight per day]-1 or
7.7 µg/kg of body weight per day for a 10-5 risk for bromoform.
These two brominated THMs are weakly mutagenic in a number of
assays, and they were by far the most mutagenic DBPs of the class in
the GST-mediated assay system. Because they are the most lipophilic
THMs, additional concerns about whether corn oil may have affected
their bioavailability in the long-term studies should be considered. A
NOAEL for DBCM of 30 mg/kg of body weight per day has been established
based on the absence of histopathological effects in the liver of rats
after 13 weeks of exposure by corn oil gavage. IARC has classified
DBCM in Group 3 (not classifiable as to its carcinogenicity to
humans). A TDI for DBCM of 30 µg/kg of body weight was derived based
on the NOAEL for liver toxicity of 30 mg/kg of body weight per day and
an uncertainty factor of 1000 (10 each for inter- and intraspecies
variation and 10 for the short duration of the study and possible
carcinogenicity).
Similarly, a NOAEL for bromoform of 25 mg/kg of body weight per
day can be derived on the basis of the absence of liver lesions in
rats after 13 weeks of dosing by corn oil gavage. A TDI for bromoform
of 25 µg/kg of body weight was derived based on this NOAEL for liver
toxicity and an uncertainty factor of 1000 (10 each for inter- and
intraspecies variation and 10 for the short duration of the study and
possible carcinogenicity). IARC has classified bromoform in Group 3
(not classifiable as to its carcinogenicity to humans).
5) Haloacetic acids
The induction of mutations by DCA is very improbable at the low
doses that would be encountered in chlorinated drinking-water. The
available data indicate that DCA differentially affects the
replication rates of normal hepatocytes and hepatocytes that have been
initiated. The dose-response relationships are complex, with DCA
initially stimulating division of normal hepatocytes. However, at the
lower chronic doses used in animal studies (but still very high
relative to those that would be derived from drinking-water), the
replication rate of normal hepatocytes is eventually sharply
inhibited. This indicates that normal hepatocytes eventually
down-regulate those pathways that are sensitive to stimulation by DCA.
However, the effects in altered cells, particularly those that express
high amounts of a protein that is immunoreactive to a c-Jun antibody,
do not seem to be able to down-regulate this response. Thus, the rates
of replication in the pre-neoplastic lesions with this phenotype are
very high at the doses that cause DCA tumours to develop with a very
low latency. Preliminary data would suggest that this continued
alteration in cell birth and death rates is also necessary for the
tumours to progress to malignancy. This interpretation is supported by
studies that employ initiation/promotion designs as well.
On the basis of the above considerations, it is suggested that
the currently available cancer risk estimates for DCA be modified by
incorporation of newly developing information on its comparative
metabolism and modes of action to formulate a biologically based
dose-response model. These data are not available at this time, but
they should become available within the next 2-3 years.
The effects of DCA appear to be closely associated with doses
that induce hepatomegaly and glycogen accumulation in mice. The
lowest-observed-adverse-effect level (LOAEL) for these effects in
an 8-week study in mice was 0.5 g/litre, corresponding to
approximately 100 mg/kg of body weight per day, and the NOAEL was
0.2 g/litre, or approximately 40 mg/kg of body weight per day. A TDI
of 40 µg/kg of body weight has been calculated by applying an
uncertainty factor of 1000 to this NOAEL (10 each for inter- and
intraspecies variation and 10 for the short duration of the study and
possible carcinogenicity). IARC has classified DCA in Group 3 (not
classifiable as to its carcinogenicity to humans).
TCA is one of the weakest activators of the peroxisome
proliferator activated receptor (PPAR) known. It appears to be only
marginally active as a peroxisome proliferator, even in rats.
Furthermore, treatment of rats with high levels of TCA in
drinking-water does not induce liver tumours. These data strongly
suggest that TCA presents little carcinogenic hazard to humans at the
low concentrations found in drinking-water.
From a broader toxicological perspective, the developmental
effects of TCA are the end-point of concern. Animals appear to
tolerate concentrations of TCA in drinking-water of 0.5 g/litre
(approximately 50 mg/kg of body weight per day) with little or no
signs of adverse effect. At 2 g/litre, the only sign of adverse effect
appears to be hepatomegaly. Hepatomegaly is not observed in mice at
doses of 0.35 g of TCA per litre in drinking-water, estimated to be
equivalent to 40 mg/kg of body weight per day.
In another study, soft tissue anomalies were observed at
approximately 3 times the control rate at the lowest dose
administered, 330 mg/kg of body weight per day. At this dose, the
anomalies were mild and would clearly be in the range where
hepatomegaly (and carcinogenic effects) would occur. Considering the
fact that the PPAR interacts with cell signalling mechanisms that can
affect normal developmental processes, a common mechanism underlying
hepatomegaly and the carcinogenic effects and developmental effects of
this compound should be considered.
The TDI for TCA is based on a NOAEL estimated to be 40 mg/kg of
body weight per day for hepatic toxicity in a long-term study in mice.
Application of an uncertainty factor of 1000 (10 each for inter- and
intraspecies variation and 10 for possible carcinogenicity) to the
estimated NOAEL gives a TDI of 40 µg/kg of body weight. IARC has
classified TCA in Group 3 (not classifiable as to its carcinogenicity
to humans).
Data on the carcinogenicity of brominated acetic acids are too
preliminary to be useful in risk characterization. Data available in
abstract form suggest, however, that the doses required to induce
hepatocarcinogenic responses in mice are not dissimilar to those of
the chlorinated acetic acids. In addition to the mechanisms involved
in the induction of cancer by DCA and TCA, it is possible that
increased oxidative stress secondary to their metabolism might
contribute to their effects.
There are a significant number of data on the effects of
dibromoacetic acid (DBA) on male reproduction. No effects were
observed in rats at doses of 2 mg/kg of body weight per day for
79 days, whereas an increased retention of step 19 spermatids was
observed at 10 mg/kg of body weight per day. Higher doses led to
progressively more severe effects, including marked atrophy of the
seminiferous tubules with 250 mg/kg of body weight per day, which was
not reversed 6 months after treatment was suspended. A TDI of 20 µg/kg
of body weight was determined by allocating an uncertainty factor of
100 (10 each for inter- and intraspecies variation) to the NOAEL of
2 mg/kg of body weight per day.
6) Chloral hydrate
Chloral hydrate at 1 g/litre of drinking-water (166 mg/kg of body
weight per day) induced liver tumours in mice exposed for 104 weeks.
Lower doses have not been evaluated. Chloral hydrate has been shown to
induce chromosomal anomalies in several in vitro tests but has been
largely negative when evaluated in vivo. It is probable that the
liver tumours induced by chloral hydrate involve its metabolism to TCA
and/or DCA. As discussed above, these compounds are considered to act
as tumour promoters. IARC has classified chloral hydrate in Group 3
(not classifiable as to its carcinogenicity to humans).
Chloral hydrate administered to rats for 90 days in
drinking-water induced hepatocellular necrosis at concentrations of
1200 mg/litre and above, with no effect being observed at 600 mg/litre
(approximately 60 mg/kg of body weight per day). Hepatomegaly was
observed in mice at doses of 144 mg/kg of body weight per day
administered by gavage for 14 days. No effect was observed at 14.4
mg/kg of body weight per day in the 14-day study, but mild
hepatomegaly was observed when chloral hydrate was administered in
drinking-water at 70 mg/litre (16 mg/kg of body weight per day) in a
90-day follow-up study. The application of an uncertainty factor of
1000 (10 each for inter- and intraspecies variation and 10 for the use
of a LOAEL instead of a NOAEL) to this value gives a TDI of 16 µg/kg
of body weight.
7) Haloacetonitriles
Without appropriate human data or an animal study that involves a
substantial portion of an experimental animal's lifetime, there is no
generally accepted basis for estimating carcinogenic risk from the
HANs.
Data developed in subchronic studies provide some indication of
NOAELs for the general toxicity of dichloroacetonitrile (DCAN) and
dibromoacetonitrile (DBAN). NOAELs of 8 and 23 mg/kg of body weight
per day were identified in 90-day studies in rats for DCAN and DBAN,
respectively, based on decreased body weights at the next higher doses
of 33 and 45 mg/kg of body weight per day, respectively.
A WHO Working Group for the 1993 Guidelines for drinking-water
quality considered DCAN and DBAN. This Working Group determined a
TDI of 15 µg/kg of body weight for DCAN based on a NOAEL of 15 mg/kg
of body weight per day in a reproductive toxicity study in rats and
incorporating an uncertainty factor of 1000 (10 each for intra- and
interspecies variation and 10 for the severity of effects).
Reproductive and developmental effects were observed with DBAN only at
doses that exceeded those established for general toxicity (about 45
mg/kg of body weight per day). A TDI of 23 µg/kg of body weight was
calculated for DBAN based on the NOAEL of 23 mg/kg of body weight per
day in the 90-day study in rats and incorporating an uncertainty
factor of 1000 (10 each for intra- and interspecies variation and 10
for the short duration of the study). There are no new data indicating
that these TDIs should be changed.
LOAELs for trichloroacetonitrile (TCAN) of 7.5 mg/kg of body
weight per day for embryotoxicity and 15 mg/kg of body weight per day
for developmental effects were identified. However, later studies
suggest that these responses were dependent upon the vehicle used. No
TDI can be established for TCAN.
There are no data useful for risk characterization purposes for
other members of the HANs.
8) MX
The mutagen MX has recently been studied in a long-term study in
rats in which some carcinogenic responses were observed. These data
indicate that MX induces thyroid and bile duct tumours. An increased
incidence of thyroid tumours was seen at the lowest dose of MX
administered (0.4 mg/kg of body weight per day). The induction of
thyroid tumours with high-dose chemicals has long been associated with
halogenated compounds. The induction of thyroid follicular tumours
could involve modifications in thyroid function or a mutagenic mode of
action. A dose-related increase in the incidence of cholangiomas and
cholangiocarcinomas was also observed, beginning at the low dose in
female rats, with a more modest response in male rats. The increase in
cholangiomas and cholangiocarcinomas in female rats was utilized to
derive a slope factor for cancer. The 95% upper confidence limit for a
10-5 lifetime risk based on the linearized multistage model was
calculated to be 0.06 µg/kg of body weight per day.
9) Chlorite
The primary and most consistent finding arising from exposure to
chlorite is oxidative stress resulting in changes in the red blood
cells. This end-point is seen in laboratory animals and, by analogy
with chlorate, in humans exposed to high doses in poisoning incidents.
There are sufficient data available with which to estimate a TDI for
humans exposed to chlorite, including chronic toxicity studies and a
two-generation reproductive toxicity study. Studies in human
volunteers for up to 12 weeks did not identify any effect on blood
parameters at the highest dose tested, 36 µg/kg of body weight per
day. Because these studies do not identify an effect level, they are
not informative for establishing a margin of safety.
In a two-generation study in rats, a NOAEL of 2.9 mg/kg of body
weight per day was identified based on lower auditory startle
amplitude, decreased absolute brain weight in the F1 and F2
generations, and altered liver weights in two generations. Application
of an uncertainty factor of 100 (10 each for inter- and intraspecies
variation) to this NOAEL gives a TDI of 30 µg/kg of body weight. This
TDI is supported by the human volunteer studies.
10) Chlorate
Like chlorite, the primary concern with chlorate is oxidative
damage to red blood cells. Also like chlorite, 0.036 mg of chlorate
per kg of body weight per day for 12 weeks did not result in any
adverse effect in human volunteers. Although the database for chlorate
is less extensive than that for chlorite, a recent well conducted
90-day study in rats identified a NOAEL of 30 mg/kg of body weight per
day based on thyroid gland colloid depletion at the next higher dose
of 100 mg/kg of body weight per day. A TDI is not derived because a
long-term study is in progress, which should provide more information
on chronic exposure to chlorate.
11) Bromate
Bromate is an active oxidant in biological systems and has been
shown to cause an increase in renal tumours, peritoneal mesotheliomas
and thyroid follicular cell tumours in rats and, to a lesser extent,
hamsters, and only a small increase in kidney tumours in mice. The
lowest dose at which an increased incidence of renal tumours was
observed in rats was 6 mg/kg of body weight per day.
Bromate has also been shown to give positive results for
chromosomal aberrations in mammalian cells in vitro and in vivo
but not in bacterial assays for point mutation. An increasing body of
evidence, supported by the genotoxicity data, suggests that bromate
acts by generating oxygen radicals in the cell.
In the 1993 WHO Guidelines for drinking-water quality, the
linearized multistage model was applied to the incidence of renal
tumours in a 2-year carcinogenicity study in rats, although it was
noted that if the mechanism of tumour induction is oxidative damage in
the kidney, application of the low-dose cancer model may not be
appropriate. The calculated upper 95% confidence interval for a 10-5
risk was 0.1 µg/kg of body weight per day.
The no-effect level for the formation of renal cell tumours in
rats is 1.3 mg/kg of body weight per day. If this is used as a point
of departure from linearity and if an uncertainty factor of 1000 (10
each for inter- and intraspecies variation and 10 for possible
carcinogenicity) is applied, a TDI of 1 µg/kg of body weight can be
calculated. This compares with the value of 0.1 µg/kg of body weight
per day associated with an excess lifetime cancer risk of 10-5.
At present, there are insufficient data to permit a decision on
whether bromate-induced tumours are a result of cytotoxicity and
reparative hyperplasia or a genotoxic effect.
IARC has assigned potassium bromate to Group 2B (possibly
carcinogenic to humans).
1.5.1.2 Epidemiological studies
Epidemiological studies must be carefully evaluated to ensure
that observed associations are not due to bias and that the design is
appropriate for an assessment of a possible causal relationship.
Causality can be evaluated when there is sufficient evidence from
several well designed and well conducted studies in different
geographic areas. Supporting toxicological and pharmacological data
are also important. It is especially difficult to interpret
epidemiological data from ecological studies of disinfected
drinking-water, and these results are used primarily to help develop
hypotheses for further study.
Results of analytical epidemiological studies are insufficient to
support a causal relationship for any of the observed associations. It
is especially difficult to interpret the results of currently
published analytical studies because of incomplete information about
exposures to specific water contaminants that might confound or modify
the risk. Because inadequate attention has been paid to assessing
exposures to water contaminants in epidemiological studies, it is not
possible to properly evaluate the increased relative risks that were
reported. Risks may be due to other water contaminants or to other
factors for which chlorinated drinking-water or THMs may serve as a
surrogate.
1.5.2 Characterization of exposure
1.5.2.1 Occurrence of disinfectants and disinfectant by-products
Disinfectant doses of several milligrams per litre are typically
employed, corresponding to doses necessary to inactivate
microorganisms (primary disinfection) or doses necessary to maintain a
residual in the distribution system (secondary disinfection).
A necessary ingredient for an exposure assessment is DBP
occurrence data. Unfortunately, there are few published international
studies that go beyond case-study or regional data.
Occurrence data suggest, on average, exposure to about 35-50 µg
of total THMs per litre in chlorinated drinking-water, with chloroform
and BDCM being the first and second most dominant species. Exposure to
total HAAs can be approximated by a total HAA concentration (sum of
five species) corresponding to about one-half of the total THM
concentration (although this ratio can vary significantly); DCA and
TCA are the first and second most dominant species. In waters with a
high bromide to TOC ratio or a high bromide to chlorine ratio, greater
formation of brominated THMs and HAAs can be expected. When a
hypochlorite solution (versus chlorine gas) is used, chlorate may also
occur during chlorination.
DBP exposure in chloraminated water is a function of the mode of
chloramination, with the sequence of chlorine followed by ammonia
leading to the formation of (lower levels of) chlorine DBPs (i.e.,
THMs and HAAs) during the free-chlorine period; however, the
suppression of chloroform and TCA formation is not paralleled by a
proportional reduction in DCA formation.
All factors being equal, bromide concentration and ozone dose are
the best predictors of bromate formation during ozonation, with about
a 50% conversion of bromide to bromate. A study of different European
water utilities showed bromate levels in water leaving operating water
treatment plants ranging from less than the detection limit (2
µg/litre) up to 16 µg/L. The brominated organic DBPs formed upon
ozonation generally occur at low levels. The formation of chlorite can
be estimated by a simple percentage (50-70%) of the applied chlorine
dioxide dose.
1.5.2.2 Uncertainties of water quality data
A toxicological study attempts to extrapolate a laboratory
(controlled) animal response to a potential human response; one
possible outcome is the estimation of cancer risk factors. An
epidemiological study attempts to link human health effects (e.g.,
cancer) to a causative agent or agents (e.g., a DBP) and requires an
exposure assessment.
The chemical risks associated with disinfected drinking-water are
potentially based on several routes of exposure: (i) ingestion of DBPs
in drinking-water; (ii) ingestion of chemical disinfectants in
drinking-water and the concomitant formation of DBPs in the stomach;
and (iii) inhalation of volatile DBPs during showering. Although the
in vivo formation of DBPs and the inhalation of volatile DBPs may be
of potential health concern, the following discussion is based on the
premise that the ingestion of DBPs present in drinking-water is the
most significant route of exposure.
Human exposure is a function of both DBP concentration and
exposure time. More specifically, human health effects are a function
of exposure to complex mixtures of DBPs (e.g., THMs versus HAAs,
chlorinated versus brominated species) that can change
seasonally/temporally (e.g., as a function of temperature, nature and
concentration of NOM) and spatially (i.e., throughout a distribution
system). Each individual chemical disinfectant can form a mixture of
DBPs; combinations of chemical disinfectants can form even more
complex mixtures. Upon their formation, most DBPs are stable, but some
may undergo transformation by, for example, hydrolysis. In the absence
of DBP data, surrogates such as chlorine dose (or chlorine demand),
TOC (or ultraviolet absorbance at 254 nm [UVA254]) or bromide can be
used to indirectly estimate exposure. While TOC serves as a good
surrogate for organic DBP precursors, UVA254 provides additional
insight into NOM characteristics, which can vary geographically. Two
key water quality variables, pH and bromide, have been identified as
significantly affecting the type and concentrations of DBPs that are
produced.
An exposure assessment should first attempt to define the
individual types of DBPs and resultant mixtures likely to form, as
well as their time-dependent concentrations, as affected by their
stability and transport through a distribution system. For
epidemiological studies, some historical databases exist for
disinfectant (e.g., chlorine) doses, possibly DBP precursor (e.g.,
TOC) concentrations and possibly total THM (and, in some cases, THM
species) concentrations. In contrast to THMs, which have been
monitored over longer time frames because of regulatory scrutiny,
monitoring data for HAAs (and HAA species), bromate and chlorite are
much more recent and hence sparse. However, DBP models can be used to
simulate missing or past data. Another important consideration is
documentation of past changes in water treatment practice.
1.5.2.3 Uncertainties of epidemiological data
Even in well designed and well conducted analytical studies,
relatively poor exposure assessments were conducted. In most studies,
duration of exposure to disinfected drinking-water and the water
source were considered. These exposures were estimated from
residential histories and water utility or government records. In only
a few studies was an attempt made to estimate a study participant's
water consumption and exposure to either total THMs or individual
species of THMs. In only one study was an attempt made to estimate
exposures to other DBPs. In evaluating some potential risks, i.e.,
adverse outcomes of pregnancy, that may be associated with relatively
short term exposures to volatile by-products, it may be important to
consider the inhalation as well as the ingestion route of exposure
from drinking-water. In some studies, an effort was made to estimate
both by-product levels in drinking-water for etiologically relevant
time periods and cumulative exposures. Appropriate models and
sensitivity analysis such as Monte Carlo simulation can be used to
help estimate these exposures for relevant periods.
A major uncertainty surrounds the interpretation of the observed
associations, as exposures to a relatively few water contaminants have
been considered. With the current data, it is difficult to evaluate
how unmeasured DBPs or other water contaminants may have affected the
observed relative risk estimates.
More studies have considered bladder cancer than any other
cancer. The authors of the most recently reported results for bladder
cancer risks caution against a simple interpretation of the observed
associations. The epidemiological evidence for an increased relative
risk of bladder cancer is not consistent -- different risks are
reported for smokers and non-smokers, for men and women, and for high
and low water consumption. Risks may differ among various geographic
areas because the DBP mix may be different or because other water
contaminants are also present. More comprehensive water quality data
must be collected or simulated to improve exposure assessments for
epidemiological studies.
2. CHEMISTRY OF DISINFECTANTS AND DISINFECTANT BY-PRODUCTS
2.1 Background
The use of chlorine (Cl2) as a water disinfectant has come under
scrutiny because of its potential to react with natural organic matter
(NOM) and form chlorinated disinfectant by-products (DBPs). Within
this context, NOM serves as the organic DBP precursor, whereas bromide
ion (Br-) serves as the inorganic precursor. Treatment strategies
generally available to water systems exceeding drinking-water
standards include removing DBP precursors and using alternative
disinfectants for primary and/or secondary (distribution system)
disinfection. Alternative disinfectant options that show promise are
chloramines (NH2Cl, monochloramine), chlorine dioxide (ClO2) and
ozone (O3). While ozone can serve as a primary disinfectant only and
chloramines as a secondary disinfectant only, both chlorine and
chlorine dioxide can serve as either primary or secondary
disinfectants.
Chloramine presents the significant advantage of virtually
eliminating the formation of chlorination by-products and, unlike
chlorine, does not react with phenols to create taste- and
odour-causing compounds. However, the required contact time for
inactivation of viruses and Giardia cysts is rarely obtainable by
chloramine post-disinfection at existing water treatment facilities
(monochloramine is significantly less biocidal than free chlorine).
More recently, the presence of nitrifying bacteria and nitrite
(NO2-) and nitrate (NO3-) production in chloraminated distribution
systems as well as the formation of organic chloramines have raised
concern.
The use of chlorine dioxide, like chloramine, can reduce the
formation of chlorinated by-products during primary disinfection.
However, production of chlorine dioxide, its decomposition and
reaction with NOM lead to the formation of by-products such as
chlorite (ClO2-), a compound that is of health concern.
If used as a primary disinfectant followed by a chloramine
residual in the distribution system, ozone can eliminate the need for
contact between DBP precursors and chlorine. Ozone is known to react
both with NOM to produce organic DBPs such as aldehydes and increase
levels of assimilable organic carbon and with bromide ion to form
bromate.
A thorough understanding of the mechanisms of DBP formation
allows microbial inactivation goals and DBP control goals to be
successfully balanced. This chapter examines a range of issues
affecting DBP formation and control to provide guidance to utilities
considering the use of various disinfecting chemicals to achieve
microbial inactivation with DBP control.
2.2 Physical and chemical properties of common disinfectants and
inorganic disinfectant by-products
The important physical and chemical properties of commonly used
disinfectants and inorganic DBPs are summarized in Table 1.
2.2.1 Chlorine
Chlorine, a gas under normal pressure and temperature, can be
compressed to a liquid and stored in cylindrical containers. Because
chlorine gas is poisonous, it is dissolved in water under vacuum, and
this concentrated solution is applied to the water being treated. For
small plants, cylinders of about 70 kg are used; for medium to large
plants, tonne containers are common; and for very large plants,
chlorine is delivered by railway tank cars or road (truck) tankers.
Chlorine is also available in granular or powdered form as calcium
hypochlorite (Ca(OCl)2) or in liquid form as sodium hypochlorite
(NaOCl; bleach).
Chlorine is used in the form of gaseous chlorine or hypochlorite
(OCl-). In either form, it acts as a potent oxidizing agent and often
dissipates in side reactions so rapidly that little disinfection is
accomplished until amounts in excess of the chlorine demand have been
added. As an oxidizing agent, chlorine reacts with a wide variety of
compounds, in particular those that are considered reducing agents
(hydrogen sulfide [H2S], manganese(II), iron(II), sulfite [SO32-],
Br-, iodide [I-], nitrite). From the point of view of DBP formation
and disinfection, these reactions may be important because they may be
fast and result in the consumption of chlorine.
Chlorine gas hydrolyses in water almost completely to form
hypochlorous acid (HOCl):
Cl2 + H2O -> HOCl + H+ + Cl-
The hypochlorous acid dissociates into hydrogen ions (H+) and
hypochlorite ions in the reversible reaction:
HOCl <-> H+ + OCl-
Hypochlorous acid is a weak acid with a p Ka of approximately
7.5 at 25°C. Hypochlorous acid, the prime disinfecting agent, is
therefore dominant at a pH below 7.5 and is a more effective
disinfectant than hypochlorite ion, which dominates above pH 7.5.
The rates of the decomposition reactions of chlorine increase as
the solution becomes more alkaline, and these reactions can
theoretically produce chlorite and chlorate (ClO3-); they occur
during the electrolysis of chloride (Cl-) solutions when the anodic
and cathodic compartments are not separated, in which case the
chlorine formed at the anode can react with the alkali formed at the
cathode. On the other hand, hypochlorous acid/hypochlorite (or
hypobromous acid/hypobromite, HOBr/OBr-) can be formed by the action
of chlorine (or bromine) in neutral or alkaline solutions. The
Table 1. Physical and chemical properties of commonly used disinfectants and inorganic disinfectant by-products
Chemicala Eo (V)b Oxidation number lambamax (nm)c e (mol-1 litre-1 cm-1)d p epsilono e pKa f
of Cl or Br
HOCl/Cl- +1.49 +1 254 60 +25.2 7.5
292 (OCl-) 419
ClO2/ClO2- +0.95 +4 359 1250 +16.1 -
NH2Cl - +1 245 416 - -
O3/O2 +2.07 - 254 3200 +35.0
HOBr/Br- +1.33 +1 330 50 +22.5 8.7
ClO2-/Cl- +0.76 +3 262 - +12.8 1.96
ClO3-/Cl- +0.62 +5 360 - +10.5 1.45
BrO3-/Br- +0.61 +5 195 - 0.72
a Half-cell reactants/products.
b Eo = standard electrode potential (redox potential) in water at 25 °C. The oxidation-reduction state of an aqueous
environment at equilibrium can be stated in terms of its redox potential. In the chemistry literature, this is generally
expressed in volts, E, or as the negative logarithm of the electron activity, p epsilon. When p epsilon is large, the
electron activity is low and the system tends to be an oxidizing one: i.e., half-reactions tend to be driven to the left.
When p epsilon is small, the system is reducing, and reactions tend to be driven to the right.
c lambdamax = maximum absorbance wavelength of that particular solution in nm.
d e = molar absorptivity (molar extinction coefficient), in mol-1 litre-1 cm-1. This can be used for quantitative
determination of the various species of chemicals and is the only direct physical measurement. There is often some
background absorbance that may interfere with the measurement in natural waters that should be considered.
e p epsilono = - log {e-} where {e-} = electron activity.
f pKa = negative logarithm of the acid ionization constant (e.g., at pH 7.5, the molar concentration of HOCl is same as that
of OCl-). As this parameter is dependent upon temperature, the values listed were determined at 25 °C.
decomposition of hypohalites (XO-) is favoured in alkaline solutions
(2XO- -> X- + XO2-) and is such that there is no longer any
domain of thermodynamic stability for the hypohalite ions. These
oxyhalites are further converted to stable oxyhalates as follows:
XO- + XO2- -> X- + XO3-
Another reaction that occurs in waters containing bromide ion and
hypochlorite is the production of hypobromous acid:
HOCl + Br- -> HOBr + Cl-
This reaction is irreversible, and the product hypobromous acid is a
better halogenating agent than hypochlorous acid and interferes with
common analytical procedures for free chlorine. The presence of
bromide in hypochlorite solutions can ultimately lead to the formation
of bromate (BrO3-).
Hypobromous acid is a weak acid (p Ka = 8.7); like
hypochlorite, hypobromite is metastable. In alkaline solution, it
decomposes to give bromate and bromide:
3OBr- -> BrO3- + 2Br-
Bromic acid (HBrO3) is a strong acid (p Ka = 0.7). Bromic acid
and bromate can be obtained by the electrolytic oxidation of bromide
solutions or bromine water using chlorine. Bromic acid and bromate are
powerful oxidizing agents, but the speed of their oxidation reactions
is generally slow (Mel et al., 1953).
2.2.2 Chlorine dioxide
Chlorine dioxide is one of the few compounds that exists almost
entirely as monomeric free radicals. Concentrated chlorine dioxide
vapour is potentially explosive, and attempts to compress and store
this gas, either alone or in combination with other gases, have been
commercially unsuccessful. Because of this, chlorine dioxide, like
ozone, must be manufactured at the point of use. Chlorine dioxide in
water does not hydrolyse to any appreciable extent. Neutral or acidic
dilute aqueous solutions are quite stable if kept cool, well sealed
and protected from sunlight.
Chlorine dioxide represents an oxidation state (+4) intermediate
between those of chlorite (+3) and chlorate (+5). No acid or ion of
the same degree of oxidation is known. Chlorine dioxide is a powerful
oxidizing agent that can decompose to chlorite; in the absence of
oxidizable substances and in the presence of alkali, it dissolves in
water, decomposing with the slow formation of chlorite and chlorate:
2ClO2 + H2O -> ClO2- + ClO3- + 2H+
Chlorine dioxide has an absorption spectrum with a maximum at 359
nm, with a molar absorptivity of 1250 mol-1 litre-1 cm-1. This
extinction coefficient is independent of temperature, pH, chloride and
ionic strength. Chlorine dioxide is readily soluble in water, forming
a greenish-yellow solution. It can be involved in a variety of redox
reactions, such as oxidation of iodide ion, sulfide ion, iron(II) and
manganese(II). When chlorine dioxide reacts with aqueous contaminants,
it is usually reduced to chlorite ion. The corresponding electron
transfer reactions are comparable to those occurring when singlet
oxygen acts as an oxidant (Tratnyek & Hoigne, 1994).
Bromide (in the absence of sunlight) is not oxidized by chlorine
dioxide. Therefore, water treatment with chlorine dioxide will not
transform bromide ion into hypobromite and will not give rise to the
formation of bromoform (CHBr3) or bromate. This is an important
difference between the use of chlorine dioxide as an oxidant and the
use of chlorine or ozone as an oxidant.
2.2.3 Ozone
Ozone is a strong oxidizing agent ( Eo = 2.07 V). Oxidation
reactions initiated by ozone in water are generally rather complex; in
water, only part of the ozone reacts directly with dissolved solutes.
Another part may decompose before reaction. Such decomposition is
catalysed by hydroxide ions (OH-) and other solutes. Highly reactive
secondary oxidants, such as hydroxyl radicals (OH.), are thereby
formed. These radicals and their reaction products can additionally
accelerate the decomposition of ozone. Consequently, radical-type
chain reactions may occur, which consume ozone concurrently with the
direct reaction of ozone with dissolved organic material.
Many oxidative applications of ozone have been developed,
including disinfection, control of algae, removal of tastes and
odours, removal of colour, removal of iron and manganese,
microflocculation, removal of turbidity by oxidative flocculation,
removal of organics by oxidation of phenols, detergents and some
pesticides, partial oxidation of dissolved organics and control of
halogenated organic compounds. For disinfection and for oxidation of
many organic and inorganic contaminants in drinking-water, the
kinetics of ozone reactions are favourable; on the other hand, for
many difficult-to-oxidize organic compounds, such as chloroform
(CHCl3), the kinetics of ozone oxidation are very slow (Hoigne et
al., 1985).
2.2.4 Chloramines
Monochloramine has much higher CT values1 than free chlorine
and is therefore a poor primary disinfectant. Additionally, it is a
poor oxidant and is not effective for taste and odour control or for
oxidation of iron and manganese. However, because of its persistence,
1 The CT value is the product of the disinfectant concentration C
in mg/litre and the contact time T in minutes required to inactivate
a specified percentage (e.g., 99%) of microorganisms.
it is an attractive secondary disinfectant for the maintenance of a
stable distribution system residual. The use of disinfectants such as
ozone or chlorine dioxide combined with chloramines as a secondary
disinfectant appears to be attractive for minimizing DBP formation
(Singer, 1994b).
Monochloramine is the only useful ammonia-chloramine
disinfectant. Dichloramine (NHCl2) and nitrogen trichloride (NCl3)
are too unstable to be useful and highly malodorous. Conditions
practically employed for chloramination are designed to produce only
monochloramine.
2.3 Analytical methods for disinfectant by-products and disinfectants
Analytical methods for various DBPs and their detection limits
are summarized in Table 2. Methods for disinfectants are summarized in
APHA (1995).
2.3.1 Trihalomethanes, haloacetonitriles, chloral hydrate,
chloropicrin and haloacetic acids
Gas chromatographic (GC) techniques are generally employed
for organic DBPs. Detection and quantification of haloacetonitriles
(HANs) and chloral hydrate in chlorinated natural waters are
complicated by (i) hydrolysis of dihaloacetonitriles and chloral
hydrate to dihaloacetic acids and chloroform, respectively; (ii)
degradation of HANs by dechlorinating agents such as sodium sulfite
and sodium thiosulfate; (iii) low purge efficiency for the HANs and
chloral hydrate in the purge-and-trap technique; and (iv) low
extraction efficiency for chloral hydrate with pentane in the
liquid-liquid extraction normally used. Although chloral hydrate is
not efficiently extracted from water with pentane, it can be extracted
with an efficiency of approximately 36% when the ratio by volume of
methyl tert-butyl ether (MTBE) to water is 1 : 5 (Amy et al., 1998).
MTBE quantitatively extracts HANs, trihalomethanes (THMs), chloral
hydrate and chloropicrin, permitting simultaneous analysis for all of
these DBPs. Chloral hydrate decomposes on packed columns to
trichloroacetaldehyde, resulting in considerable band broadening,
although this does not appear to be a significant problem with DB-1
and DB-5 columns.
The extraction of THMs can be accomplished using MTBE (EPA Method
551) or pentane. Method 551 also permits simultaneous extraction and
measurement of chloral hydrate, HANs, THMs, chloropicrin and
haloketones. The pentane method can be used to extract THMs, HANs,
haloketones and chloropicrin but not chloral hydrate in the same run
(APHA, 1995).
The haloacetic acid (HAA) analytical method involves using an
acidic salted ether or acidic methanol liquid-liquid extraction,
requiring esterification with diazomethane prior to analysis on a gas
chromatograph equipped with an electron capture detector (ECD). THMs
and HANs can be analysed by extraction with pentane prior to analysis
on a capillary-column GC equipped with an ECD. The analysis of
Table 2. Summary of analytical methods for various DBPs and their minimum detection limits
DBPs Analytical method APHAa Minimum detection Major References
method limit (µg/litre) interferences
THMs MTBE extraction - 0.4 None AWWARF (1991)
Pentane extraction 6232B 0.1
HAAs Salted MTBE extraction and 6233B 0.5-1.0 None AWWARF (1991)
derivatization with diazomethane
HANs Pentane extraction 6232B 0.05 None Koch et al. (1988)
Cyanogen chloride MTBE extraction 6233A 0.5 None AWWARF (1991)
Chloramine Derivatization with - - None Lukasewycz et al. (1989)
2-mercaptobenzothiazole
Haloketonesb Pentane extraction 6232B 0.2 None Krasner et al. (1995)
AWWARF (1991)
Chloral hydrate MTBE extraction - 0.5 None AWWARF (1991)
Aldehydes Extraction with hexane and - 1.0 PFBHA sulfate Sclimenti et al. (1990)
derivatization with PFBHA
Bromate Ion chromatography (H3BO3/NaOH) 4500 2.0c Cl- Siddiqui et al. (1996a)
Krasner et al. (1993)
0.2 Weinberg et al. (1998)
Chlorate Ion chromatography (H3BO3/NaOH) 4500 5 Cl-, acetate Siddiqui (1996)
Chlorite Ion chromatography (NaHCO3/Na2CO3) 4500 10 Cl-, acetate AWWARF (1991)
TOC UV/persulfate or combustion 5310 200 Metals APHA (1995)
a American Public Health Association.
b Sum of 1,1-DCPN and 1,1,1-TCPN.
c 1.0 µg/litre with high-capacity column.
cyanogen compounds involves extraction with MTBE prior to injection
into GC-ECD. Aldehydes require derivatization with
O-(2,3,4,5,6-pentafluorobenzyl)-hydroxylamine (PFBHA) (to form an
oxime), extraction with hexane and GC-ECD analysis [(C6F5)-CH2ONH2
+ RCHO -> (C6F5)-CH2ON=CHR + H2O]. It should be noted that PFBHA
peaks are very large relative to other peaks in the chromatogram from
a purge-and-trap system, whereas the peaks are comparable to other
peaks in a GC-ECD chromatogram (Trehy et al., 1986).
2.3.2 Inorganic disinfectant by-products
An ion chromatography (IC) method (EPA Method 300) has been
developed to determine inorganic by-products. The elution order is
fluoride, chlorite, bromate, chloride, nitrite, bromide, chlorate,
nitrate and sulfate ion. The eluent is a carbonate buffer.
Ethylenediamine is used to preserve chlorite samples and to minimize
the potential for chlorite ion reaction on the IC separating column.
EPA Method 300 involves measurement by an IC system using a separating
column (e.g., Ion Pac AS9-SC) fitted with an anion micromembrane
suppressor column. An eluent containing 2.0 mmol of sodium carbonate
(Na2CO3) per litre / 0.75 mmol of sodium bicarbonate (NaHCO3) per
litre is used for bromide determination, and an eluent containing 40
mmol of boric acid (H3BO3) per litre / 20 mmol of sodium hydroxide
(NaOH) per litre is used for bromate and chlorate determination. The
analytical minimum detection limits for bromate and chlorate using a
borate eluent have been reported as 2 µg/litre and 5 µg/litre,
respectively (Siddiqui, 1996; Siddiqui et al., 1996a). For samples
with high chloride ion content, a silver cartridge can be used to
remove chloride prior to IC analysis to minimize its interference with
bromate measurement. It should be noted that for natural sources and
waters with high total organic carbon (TOC) levels, detection limits
will be slightly different because of the masking effect of NOM and
high concentrations of carbonate/bicarbonate ions that may interfere
with bromate/chlorate measurement.
2.3.3 Total organic carbon and UV absorbance at 254 nm
TOC is the primary surrogate parameter for the measurement of NOM
in water supplies. Several investigators have reported that the
ultraviolet (UV)/persulfate oxidation method underestimates the TOC
concentration in natural waters as compared with the combustion method
because of the inability of the persulfate method to oxidize highly
polymerized organic matter. It is generally assumed that the
calibration of a TOC analyser with a potassium hydrogen phthalate
standard is sufficient for the measurement of TOC in natural waters,
but potassium hydrogen phthalate has a simple molecular structure and
is easy to oxidize. Dissolved organic carbon (DOC) is operationally
defined by a (0.45-µm) filtration step. UV absorbance at 254 nm
(UVA254) is used to describe the type and character of NOM, whereas
TOC describes just the amount of NOM.
2.3.4 Chloramines
Knowledge of the amine content of the water during water
treatment processes involving chloramination is important to define
more adequately the content of a matrix described only as a combined
chlorine residual. The presence of organic nitrogen and the
instability of many organic chloramines continue to challenge the
analyst. Lukasewycz et al. (1989) developed a technique for the
analysis of chloramines and organic chloramines present in water using
2-mercaptobenzothiazole as a derivatizing agent. The resulting
sulfanilamides are stable and can be conveniently analysed by
high-performance liquid chromatography (HPLC) using UV or
electrochemical detection. This method appears to be superior to the
use of diazotization or phenylarsine oxide as a method of detection.
Organic chloramines are much weaker disinfectants than inorganic
monochloramine but are indistinguishable by the common analytical
methods.
2.4 Mechanisms involved in the formation of disinfectant by-products
2.4.1 Chlorine reactions
Chlorine reacts with humic substances (dissolved organic matter)
present in most water supplies, forming a variety of halogenated DBPs,
such as THMs, HAAs, HANs, chloral hydrate and chloropicrin, as
follows:
HOCl + DOC -> DBPs
It is generally accepted that the reaction between chlorine and
humic substances, a major component of NOM, is responsible for the
production of organochlorine compounds during drinking-water
treatment. Humic and fulvic acids show a high reactivity towards
chlorine and constitute 50-90% of the total DOC in river and lake
waters (Thurman, 1985). Other fractions of the DOC comprise the
hydrophilic acids (up to 30%), carbohydrates (10%), simple carboxylic
acids (5%) and proteins/amino acids (5%). The reactivity of
carbohydrates and carboxylic acids towards chlorine is low, and they
are not expected to contribute to the production of organochlorine
compounds. However, hydrophilic acids such as citric acid and amino
acids will react with chlorine to produce chloroform and other
products and may contribute towards total organochlorine production
(Larson & Rockwell, 1979).
Free chlorine reacts with water constituents by three general
pathways: oxidation, addition and substitution (Johnson & Jensen,
1986). Chlorine can undergo an addition reaction if the organic
compound has a double bond. For many compounds with double bonds, this
reaction is too slow to be of importance in water treatment. The
oxidation reactions with water constituents such as carbohydrates or
fatty acids (e.g., oleic acid) are generally slow.
Most chlorine DBPs are formed through oxidation and substitution
reactions. THMs have the general formula CHX3, where X can be Cl or
Br. Chloroform may be produced through a series of reactions with
functional groups of humic substances. The major functional groups of
humic substances include acetyl, carboxyl, phenol, alcohol, carbonyl
and methoxyl. The reactions proceed much more rapidly at high pH than
at low pH.
Rook (1977) proposed resorcinol structures to be the major
precursor structure in humic material for chloroform formation. In
accordance with this hypothesis in the chlorination of terrestrial and
aquatic humic substances, a series of intermediates were detected that
contained a trichloromethyl group and that could be converted to
chloroform by further oxidation or substitution reactions (Stevens et
al., 1976).
However, the production of chlorinated compounds such as
dichloropropanedoic acid, 2,2-dichlorobutanedoic acid, cyanogen
chloride (CNCl), HANs or the cyano-substituted acids cannot be
explained on the basis of resorcinol structures, and possible
production pathways require protein-type precursors (De Leer et al.,
1986). The reaction pathway for amino acids involves initial rapid
formation of the monochloramine and dichloramine, which can react
further to form aldehyde or HANs, respectively. Trehy et al. (1986)
demonstrated the formation of chloral hydrate along with HANs after
chlorination of amino acids by substitution reactions, and aldehydes
were shown to be the oxidation products. Luknitskii (1975) provided a
detailed chemistry of chloral hydrate formation.
Christman et al. (1983) also identified chloroform, chloral
hydrate, dichloroacetic acid (DCA), trichloroacetic acid (TCA) and
2,2-dichlorobutanedoic acid as the major products, accounting for 53%
of the total organic halogen (TOX). A number of other minor products
have been detected, including several chlorinated alkanoic acids and
non-chlorinated benzene carboxylic acids. De Leer et al. (1985)
extended these studies to incorporate chloroform intermediates,
chlorinated aromatic acids and cyano-compounds as potential products
in drinking-water. The presence of unhalogenated aldehydes and HANs in
chlorinated natural waters can be attributed in part to the presence
of amino acids or peptides in natural waters. Humic acids may also
contribute to the presence of amino acids in natural waters, as they
have amino acids associated with them either in a free or in a
combined form. Several studies regarding the chlorination of amino
acids have shown that the primary amino group on the amino acids can
be converted to either an aldehyde or a nitrile group (Morris et al.,
1980; Isaac & Morris, 1983). These studies indicate that with an
equimolar amount of halogenating agent, the major product is an
aldehyde. However, if an excess of halogenating agent is added, then
the corresponding nitrile can also be formed, with the ratio of the
aldehyde to nitrile formed increasing with pH.
Many treated waters contain not only chlorinated but also
brominated compounds, such as bromoform. These compounds form because
aqueous chlorine converts bromide in the water to hypobromous acid.
The bromine can then react with the organic matter in the same way as
hypochlorous acid to form various bromochlorinated DBPs. However,
compared with hypochlorous acid, hypobromous acid is a weaker oxidant
and stronger halogenating agent.
Chlorate, an inorganic by-product of chlorine, is formed in
concentrated hypochlorite solutions during their production and
storage through the following reactions (Gordon et al., 1997):
OCl- + OCl- -> ClO2- + Cl-
OCl- + ClO2- -> ClO3- + Cl-
The first reaction proceeds at a much slower rate and is rate
limiting, hence the generally observed second-order kinetics. Sodium
hypochlorite is stored at pH greater than 12 to prevent rapid
decomposition, and most of the sodium hypochlorite is present as
hypochlorite ion. The average rate constant for the formation of
chlorate is 85 × 10-5 mol-1 litre-1 d-1 (Gordon et al., 1995).
2.4.2 Chlorine dioxide reactions
The major chlorine dioxide by-products of concern are chlorite
and chlorate. Chlorine dioxide reacts generally as an electron
acceptor, and hydrogen atoms present in activated organic C-H or N-H
structures are thereby not substituted by chlorine (Hoigne & Bader,
1994). Moreover, in contrast to chlorine, chlorine dioxide's
efficiency for disinfection does not vary with pH or in the presence
of ammonia, and it does not oxidize bromide. As opposed to chlorine,
which reacts via oxidation and electrophilic substitution, chlorine
dioxide reacts only by oxidation; this explains why it does not
produce organochlorine compounds. In addition to this, chlorine
dioxide is more selective in typical water treatment applications, as
evidenced by its somewhat lower disinfectant demand as compared with
chlorine.
Chlorine dioxide is generally produced by reacting aqueous
(sodium) chlorite with chlorine (Gordon & Rosenblatt, 1996):
2ClO2- + HOCl + H+ -> 2ClO2(aq) + Cl- + H2O
However, under conditions of low initial reactant concentrations
or in the presence of excess chlorine, the reactant produces chlorate
ion:
ClO2- + HOCl -> ClO3- + Cl- + H+
This reaction scenario is common in generators that
overchlorinate to achieve high reaction yields based on chlorite ion
consumption.
An alternative approach to chlorine dioxide generation is with
hydrochloric acid (HCl), a process that results in less chlorate
during production:
5NaClO2 + 4HCl -> 4ClO2 + 5NaCl + 2H2O
Chlorite ion is also produced when chlorine dioxide reacts with
organics (Gordon & Rosenblatt, 1996):
ClO2 + NOM -> Products + ClO2-
Chlorine dioxide can also undergo a series of photochemically
initiated reactions resulting in the formation of chlorate ion (Gordon
et al., 1995).
While bromide is not generally oxidized by chlorine dioxide,
bromate can be formed in the presence of sunlight over a wide range of
pH values (Gordon & Emmert, 1996). Utilities need to be concerned with
bromate ion in the chlorine dioxide treatment of drinking-water if the
water contains bromide and is exposed to sunlight. Practically, this
means minimizing exposure to sunlight when chlorine dioxide is applied
in the presence of bromide ion. There appears to be a problem with
chlorine dioxide producing odour-causing compounds at the tap. This
has been linked to chlorine dioxide reacting with volatile organic
compounds derived from new carpets and office products (Hoehn et al.,
1990).
Hoigne & Bader (1994) described the kinetics of reaction between
chlorine dioxide and a wide range of organic and inorganic compounds
that are of concern in water treatment. Measured rate constants were
high for nitrite, hydrogen peroxide, ozone, iodide, iron(II), phenolic
compounds, tertiary amines and thiols. Bromide, ammonia, structures
containing olefinic double bonds, aromatic hydrocarbons, primary and
secondary amines, aldehydes, ketones and carbohydrates are unreactive
under the conditions of water treatment. Chlorine dioxide rapidly
oxidizes substituted phenoxide anions and many phenols, and
second-order rate constants have been measured (Rav-Acha & Choshen,
1987).
2.4.3 Chloramine reactions
Chloramination of drinking-water produces THMs (if chloramine is
formed by chlorination followed by ammonia addition), HAAs, chloral
hydrate, hydrazine, cyanogen compounds, nitrate, nitrite, organic
chloramines and 1,1-dichloropropanone (1,1-DCPN) (Dlyamandoglu &
Selleck, 1992; Kirmeyer et al., 1993, 1995).
In the presence of even small quantities of organic nitrogen, it
is possible for chloramination to produce organic chloramines. Several
researchers have shown that monochloramine readily transfers its
chlorine at a comparatively rapid rate to organic amines to form
organohalogen amines (Isaac & Morris, 1983; Bercz & Bawa, 1986).
Monochloramine was shown to cause binding of radiolabelled halogen to
nutrients such as tyrosine and folic acid; the amount of binding
varied with pH but was generally less at neutral pH than at higher pH
(Bercz & Bawa, 1986). Organic chloramines are much weaker
disinfectants than inorganic monochloramine but are indistinguishable
by common analytical methods. Organic chloramine formation may
necessitate changing chloramination conditions (e.g., ammonia and
chlorine addition order, chlorine-to-ammonia ratios and contact time).
HANs and non-halogenated acetonitriles are produced when
chloramines are reacted with humic materials and amino acids (Trehy et
al., 1986). The reaction pathway for these products is quite
complicated and very similar to that for chlorine, with many
intermediates and by-products formed. In the case of aspartic acid, De
Leer et al. (1986) demonstrated the presence of at least 11 other
significant products.
2.4.4 Ozone reactions
Ozone has been shown to oxidize bromide to hypobromite and
bromate, and hypochlorite to chlorate (Glaze et al., 1993; Siddiqui et
al., 1995; Siddiqui, 1996).
Bromate generally forms through a combination of molecular ozone
attack and reaction of bromide with free radical species. The
molecular ozone mechanism does not account for hydroxyl radicals
always formed as secondary oxidants from decomposed ozone during water
treatment. Siddiqui et al. (1995) indicated that there is a radical
pathway that is influenced by both pH and alkalinity. The hydroxyl
radical and, to a lesser degree, the carbonate radical (CO3Ê) pathway
may be more important than the molecular ozone pathway. Oxidants such
as hydroxyl and carbonate radicals may interact with intermediate
bromine species, leading to the formation of hypobromite radicals
(BrOÊ), which eventually undergo disproportionation to form
hypobromite and bromite (BrO2-). Bromate is then formed through
oxidation of bromite by ozone. The radical mechanism for the formation
of bromate includes two decisive reaction steps still involving
molecular ozone: the formation of hypobromite and oxidation of
bromite.
Bromate ion formed through reactions with molecular ozone
contributes in the range of 30-80% to the overall bromate ion
formation in NOM-containing waters (von Gunten and Hoigne, 1994).
Siddiqui et al. (1995) reported up to 65% and 100% bromate ion
formation through the radical pathway in NOM-free and NOM-containing
waters, respectively. Differences in NOM-containing waters can be
attributed to differences in the characteristics of the NOM present. A
change in mechanism as a function of pH and the competitive roles of
the free radical (one electron transfer) mechanism above pH 7 versus
oxygen atom (two electron transfer) mechanism help explain both the
large variations in bromate ion yield and the sensitivity to reactor
design, concentration of organic precursors and ozone/bromide ion
concentrations (Gordon, 1993).
The presence of bromide ion in a source water further complicates
the reaction of ozone and leads to the formation of additional DBPs,
such as bromoform, dibromoacetonitrile (DBAN) and dibromoacetone
(DBAC) (Siddiqui, 1992; Amy et al., 1993, 1994).
2.5 Formation of organohalogen disinfectant by-products
Table 3 summarizes the DBPs identified as being formed from the
use of chlorine, chlorine dioxide, chloramine and ozone.
The formation of organochlorine and organobromine compounds
during drinking-water treatment is a cause of health concern in many
countries. These compounds include THMs, HAAs, HANs, chloral hydrate,
chloropicrin, acetohalides, halogenated furanones and other compounds.
2.5.1 Chlorine organohalogen by-products
Table 4 summarizes the range of concentrations of chlorinated
DBPs formed from the reaction of chlorine with NOM, from various
sources.
The major chlorination DBPs identified are THMs, HAAs, HANs,
haloketones, chloropicrin and chloral hydrate. HAAs represent a major
portion of the non-THM halogenated organic compounds (Miller & Uden,
1983; Reckhow & Singer, 1985). Many researchers have identified HANs
and haloketones as other important DBPs (Trehy & Bieber, 1981; Miller
& Uden, 1983; Oliver, 1983; Reckhow & Singer, 1985). According to an
AWWARF (1991) study, for all eight utilities tested,
1,1,1-trichloropropanone (1,1,1-TCPN) was the more prevalent of the
two measured haloketone compounds. In addition, Kronberg et al. (1988)
identified the extremely mutagenic compound, MX.
Despite the fact that HAA formation and THM formation have very
different pH dependencies, HAA formation correlates strongly with THM
formation when treatment conditions are relatively uniform and when
the water has a low bromide concentration (Singer, 1993). DBP
formation and requisite chlorine dosage for disinfection strongly
correspond to the concentration of TOC at the point of chlorine
addition, suggesting that optimized or enhanced removal of organic
carbon prior to chlorination will decrease the formation of DBPs.
HAA formation can be appreciable when drinking-water is
chlorinated under conditions of slightly acidic pH and low bromide
concentrations. The concentrations of DCA and TCA are similar to the
concentrations of chloroform, and the total HAA concentration can be
as much as 50% greater than the THM concentration in the finished
water on a weight basis.
McGuire & Meadow (1988) reported that the national average THM
concentration in the USA was 42 µg/litre for drinking-water utilities
serving more than 100 000 persons, and only 3% of systems were above
the US maximum contaminant level of 100 µg/litre. Amy et al. (1993)
estimated that the national average THM concentration in the USA was
40 µg/litre, with an average TOC concentration of 3.0 mg/litre. The
median annual average THM concentration found for utilities among the
American Water Works Association's (AWWA) Water Industry Database was
35 µg/litre, as compared with 50 µg/litre for the non-database
utilities (Montgomery Watson, Inc., 1993).
Table 3. Disinfectant by-products present in disinfected waters
Disinfectant Significant Significant Significant
organo-halogen inorganic non-halogenated
products products products
Chlorine/ THMs, HAAs, HANs, Chlorate (mostly Aldehydes, cyanoalkanoic
hypochlorous acid chloral hydrate, from hypochlorite acids, alkanoic acids,
chloropicrin, use) benzene, carboxylic acids
chlorophenols,
N-chloramines,
halofuranones,
bromohydrins
Chlorine dioxide chlorite, chlorate unknown
Chloramine HANs, cyanogen chloride, nitrate, nitrite, aldehydes, ketones
organic chloramines, chlorate, hydrazine
chloramino acids,
chloral hydrate,
haloketones
Ozone bromoform, MBA, DBA, chlorate, iodate, aldehydes, ketoacids,
DBAC, cyanogen bromide bromate, hydrogen ketones, carboxylic
peroxide, hypobromous acids
acid, epoxides,
ozonates
Table 4. Concentration range of chlorinated disinfectant by-products in drinking-watera
DBPs Peters et al. (1990); Krasner et al. Nieminski et al. Koch et al. Reckhow et al.
Peters (1991) (1989) (1993) (1991) (1990)
THMs 3.1-49.5 30.0-44.0 17.0-51.0 49.0-81.0 201-1280
HAAs <0.5-14.7 13.0-21.0 5.0-25.0 22.0-32.0 118-1230
HANs 0.04-1.05 2.5-4.0 0.5-5.0 2.0-2.6 3.0-12.0
Haloketones - 0.9-1.8 0.2-1.6 1.0-2.0 4.8-25.3
Chlorophenols - - 0.5-1.0 - -
Chloral hydrate - 1.7-3.0 - - -
Chloropicrin - 0.1-0.16 <0.1-0.6 - -
TOC 1.7-5.6 2.9-3.2 1.5-6.0 2.5-3.0 4.8-26.6
Bromide 100-500 70-100 - 170-420 -
a All values shown in µg/litre, except TOC (mg/litre).
In Germany, 10% of the utilities produced disinfected
drinking-water with a THM concentration above 10 µg/litre; the median
annual average concentration was between 1 and 4 µg/litre, depending
on raw water quality and size of facility (Haberer, 1994).
Total THM levels in treated drinking-water were reported in one
survey in the United Kingdom (Water Research Centre, 1980):
chlorinated water derived from a lowland river contained a mean level
of 89.2 µg/litre, and that from an upland reservoir, 18.7 µg/litre.
The study also showed that chlorinated groundwater was contaminated by
THMs to a significantly lesser extent than chlorinated surface waters.
In a national survey of the water supplies of 70 communities
serving about 38% of the population in Canada, conducted in the winter
of 1976-1977, chloroform concentrations in treated water of the
distribution system 0.8 km from the treatment plant, determined by the
gas sparge technique, averaged 22.7 µg/litre. Levels of the other THMs
were considerably lower, averaging 2.9 µg/litre for
bromodichloromethane (BDCM), 0.4 µg/litre for dibromochloromethane
(DBCM) and 0.1 µg/litre for bromoform. Using direct aqueous injection
techniques, average concentrations of most of the THMs were higher
(Health Canada, 1993).
Samples collected from the distribution systems of eight major
cities in Saudi Arabia showed that THMs occurred in all the water
supplies, at concentrations ranging between 0.03 and 41.7 µg/litre.
Median total THM concentrations in several cities were higher during
the summer than during the winter. In addition, THM concentrations
were low in cities that did not mix groundwater and desalinated water.
Brominated THMs dominated (with bromoform the most abundant) and
existed at the highest concentration levels, whereas chloroform was
the least prevalent compound. This is the opposite of the occurrence
pattern found in almost all water distribution systems worldwide
(Fayad, 1993).
The concentrations of chloral hydrate in drinking-water in the
USA were summarized by IARC (1995) and varied from 0.01 to
28 µg/litre. The highest values were found in drinking-water prepared
from surface water.
Chlorination of water as well as the combination of ozonation and
chlorination can lead to the formation of chloropicrin (Merlet et al.,
1985). In a study conducted for over 25 utilities, very low levels of
chloropicrin were observed, and chlorination produced maximum
concentrations of less than 2 µg/litre (AWWARF, 1991). The
chloropicrin appeared to form slowly during the incubation period,
with concentrations tending to level off at approximately 40 h.
Dichloroacetonitrile (DCAN) is by far the most predominant HAN
species detected in water sources with bromide levels of 20 µg/litre
or less. For sources with higher bromide levels (50-80 µg/litre),
bromochloroacetonitrile (BCAN) was the second most prevalent compound.
However, none of these sources had a DBAN concentration exceeding 0.5
µg/litre, including one source water that had a much higher bromide
level, 170 µg/litre. Thus, it appears that ambient bromide
concentration is not the only factor influencing the speciation of HAN
compounds.
Chlorine can react with phenols to produce mono-, di- or
trichlorophenols, which can impart tastes and odours to waters. The
control of chlorophenolic tastes and odours produced when phenol-laden
water is treated with chlorine is essential. The sources of phenolic
compounds in water supplies are reported to be industrial wastes.
In natural waters, one of the most important sources of organic
nitrogen is proteins and their hydrolysis products. The reaction of
aqueous chlorine or monochloramine with organic nitrogen may form
complex organic chloramines (Feng, 1966; Morris et al., 1980; Snyder &
Margerum, 1982). The formation of N-chloramines resulting from the
reaction of amines and chlorine has been reported (Weil & Morris,
1949; Gray et al., 1979; Morris et al., 1980). Likewise, the
chlorination of amides has been reported (Morris et al., 1980).
Nieminski et al. (1993) reported the occurrence of DBPs for Utah
(USA) water treatment plants. All plants used chlorine for primary and
secondary disinfection purposes. Overall, THMs and HAAs represented
75% of the total specific DBPs analysed for the survey; however, total
DBPs represented only 25-50% of the TOX concentration. THMs
constituted 64% of the total DBPs by weight; HAAs were 30% of the
total DBPs by weight and approximately one-half of the total THM
concentrations. (However, in some waters, HAA concentrations may
approach or possibly exceed THM concentrations.) HANs, haloketones,
chlorophenols and chloropicrin represented 3%, 1.5%, 1.0% and 0.5%,
respectively, of the total surveyed DBPs.
The occurrence of DBPs in drinking-waters in the USA was
evaluated at 35 water treatment facilities that had a broad range of
source water qualities and treatment processes (Krasner et al., 1989).
THMs were the largest class of DBPs, and HAAs were the next most
significant class. Aldehydes, by-products of ozonation, were also
produced by chlorination. Over four quarterly sampling periods, median
total THM concentrations ranged from 30 to 44 µg/litre, with
chloroform, BDCM, DBCM and bromoform ranges of 9.6-15, 4.1-10, 2.6-4.5
and 0.33-0.88 µg/litre, respectively. Median total HAA concentrations
ranged from 13 to 21 µg/litre, with TCA, DCA, monochloroacetic acid
(MCA), dibromoacetic acid (DBA) and monobromoacetic acid (MBA) ranges
of 4.0-6.0, 5.0-7.3, <1-1.2, 0.9-1.5 and <0.5 µg/litre,
respectively.
Concentrations of DCA and TCA measured in various water sources
have been summarized by IARC (1995): in Japan, chlorinated
drinking-water contained 4.5 and 7.5 µg of DCA and TCA per litre,
respectively; rainwater in Germany contained 1.35 µg of DCA per litre
and 0.1-20 µg of TCA per litre, whereas groundwater contained 0.05 µg
of TCA per litre; in Australia, a maximum concentration of 200
µg/litre was found for DCA and TCA in chlorinated treated water; and
chlorinated water in Switzerland contained 3.0 µg of TCA per litre.
In a survey of 20 drinking-waters prepared from different source
waters in the Netherlands, HAAs were found in all drinking-waters
prepared from surface water, whereas they could not be detected in
drinking-waters prepared from groundwaters. Brominated acetic acids
accounted for 65% of the total acid concentration (Peters et al.,
1991). In another survey of Dutch drinking waters, the average
concentration of dihaloacetonitriles was about 5% of the average THM
concentration (Peters, 1990).
2.5.2 Chloramine organohalogen by-products
Chloramine treatment practice involves three potential
approaches: free chlorine followed by ammonia addition, ammonia
addition followed by chlorine addition (in situ production) and
pre-formed (off-line formation) chloramines. Generally, the objective
is monochloramine formation. Chlorine followed by ammonia is a common
approach, and, during the free-chlorine period, DBP formation may
mimic that of chlorine. Chloramination results in the production of
THMs (predominantly formed by chlorination followed by ammonia
addition), HAAs, chloral hydrate, hydrazine, cyanogen compounds,
organic chloramines and 1,1-DCPN (Dlyamandoglu & Selleck, 1992;
Singer, 1993; Kirmeyer et al., 1993, 1995). Chloramination
significantly reduces but does not eliminate THM formation; cyanogen
chloride and TOX represent the major DBP issues with respect to
chloramines.
Scully et al. (1990) identified chloramino acids such as
N-chloroglycine, N-chloroleucine and N-chlorophenylalanine as
by-products after chlorination of water containing nitrogen or after
chloramination.
2.5.3 Chlorine dioxide organohalogen by-products
Chlorine-free chlorine dioxide does not form THMs (Noack & Doerr,
1978; Symons et al., 1981). Several studies show that the TOX formed
with chlorine dioxide is 1-15% of the TOX formed with chlorine under
the same reaction conditions (Chow & Roberts, 1981; Symons et al.,
1981; Fleischacker & Randtke, 1983).
Treatment of phenol-laden source waters with chlorine dioxide
does not produce the typical chlorophenolic taste and odour compounds
that are produced when the water is treated using chlorine and is
effective in removing existing tastes and odours of this type.
2.5.4 Ozone organohalogen by-products
Ozonation of drinking-water containing bromide ion has been shown
to produce hypobromous acid/hypobromite, with hypobromite ion serving
as an intermediate to bromate formation. In the presence of NOM,
hypobromous acid produces a host of brominated organic compounds, such
as bromoform, MBA, DBA, DBAN, cyanogen bromide and DBAC (Glaze et al.,
1993; Siddiqui & Amy, 1993). Cavanagh et al. (1992) and Glaze et al.
(1993) reported the identification of bromohydrins, a new group of
labile brominated organic compounds from the ozonation of a natural
water in the presence of enhanced levels of bromide. However, results
by Kristiansen et al. (1994) strongly suggest that the bromohydrins,
such as 3-bromo-2-methyl-2-butanol, in extracts of unquenched
disinfected water are artefacts formed from the reaction of excessive
hypobromous acid with traces of olefins in the extraction solvents and
not novel DBPs.
Table 5 compares the median concentrations of various DBPs after
ozonation and chlorination.
2.6 Formation of inorganic disinfectant by-products
Although organic DBPs have been the subject of study over a
longer time frame, the formation of many inorganic by-products is
coming under increasing scrutiny.
2.6.1 Chlorine inorganic by-products
Chlorite and chlorate are inorganic by-products formed in some
chlorine solutions. This is of interest because many small
drinking-water utilities use hypochlorite solutions as a source of
free chlorine for disinfection. Bolyard & Fair (1992) examined the
occurrence of chlorate in samples of untreated source water,
drinking-water and hypochlorite solutions from 14 sites that use
hypochlorite solutions. The hypochlorite solutions used were found to
contain significant levels of chlorate. Chlorite and bromate were also
found in hypochlorite solutions from these same water utilities.
Chlorate was present in drinking-water, either as a manufacturing
by-product or from decomposition reactions occurring during storage.
Approximately 0.2 mg of chlorate per litre was observed in water
following the addition of chlorine as sodium hypochlorite at a dose
sufficient to maintain a residual of 0.45 mg/litre (Andrews &
Ferguson, 1995). The concentration of chlorite in commercial bleach
solutions typically ranges from 0.002 to 0.0046 mol/litre; similarly,
the chlorate concentration ranges from about 0.02 to 0.08 mol/litre
(Gordon et al., 1995).
A detailed study by Bolyard & Fair (1992) demonstrated that
hypochlorite solutions used to disinfect drinking-water contain
significant levels of chlorite and chlorate. The concentration of
chlorite ranged from <2 to 130 mg/litre for free available chlorine
concentrations ranging from 3 to 110 g/litre. The concentration of
chlorate varied over the range 0.19-50 g/litre, with a median of 12
g/litre. These solutions also contained bromate levels ranging from
<2 to 51 mg/litre. The concentrations of chlorate in treated source
waters ranged from 11 to 660 µg/litre. In another study involving 25
samples from plants using gaseous chlorine, no chlorate was detected,
indicating that the use of gaseous chlorine does not produce chlorate
(Bolyard & Fair, 1992). Nieminski et al. (1993) measured chlorate and
chlorite for six water treatment plants that use liquid chlorine
(i.e., hypochlorite) and found chlorate concentrations ranging from 40
to 700 µg/litre, with no chlorite or bromate detected in finished
waters. These chlorate concentrations may be attributed to high
Table 5. Median concentrations of organic disinfectant by-products
in drinking-water
DBPs Median concentration Median concentration
(µg/litre): chlorinationa (µg/litre): ozonationb
THMs 40 <1.0
Chloroform 15 -
BDCM 10 -
DBCM 4.5 -
Bromoform 0.57 <1.0
HANs 2.5 <1.0
TCAN <0.012 -
DCAN 1.1 -
BCAN 0.58 -
DBAN 0.48 <1.0
Haloketones 0.94 -
DCPN 0.46 -
TCPN 0.35 -
HAAs 20 <5.0
MCA 1.2 -
DCA 6.8 -
TCA 5.8 -
MBA <0.5 <1.0
DBA 1.5 <5.0
Aldehydes 7.8 45
Formaldehyde 5.1 20
Acetaldehyde 2.7 11
Glyoxal - 9
Methylglyoxal - 5
Chloral hydrate 3.0 -
Ketoacids - 75
Trichlorophenol <0.4 -
a Krasner et al. (1989).
b Siddiqui et al. (1993).
concentrations of chlorate, ranging from 1000 to 8000 mg/litre,
detected in a bleach used for disinfection and resulting from the
decomposition of hypochlorite stock solution. However, no chlorite or
chlorate was detected in any of the samples of finished water of the
treatment plants that apply gaseous chlorine. Chlorate formation is
expected to be minimal in low-strength hypochlorite solutions freshly
prepared from calcium hypochlorite, because of the low hypochlorite
concentration and only mildly alkaline pH.
2.6.2 Chloramine inorganic by-products
Inorganic by-products of chloramination include nitrate, nitrite,
hydrazine and, to some extent, chlorate (Dlyamandoglu & Selleck, 1992;
Kirmeyer et al., 1995).
2.6.3 Chlorine dioxide inorganic by-products
The major inorganic by-products of chlorine dioxide disinfection
have been identified as chlorite and chlorate. Andrews & Ferguson
(1995) measured a chlorate concentration of 0.38 mg/litre when a
chlorine dioxide residual of 0.33 mg/litre was maintained. The
application of chlorine dioxide produces about 0.5-0.7 mg of chlorite
and 0.3 mg of chlorate per mg of chlorine dioxide consumed or applied
(Andrews & Ferguson, 1995).
2.6.4 Ozone inorganic by-products
When bromide or iodide ions are present in waters, some of the
halogen-containing oxidants that can be produced during ozonation
include free bromine, hypobromous acid, hypobromite ion, bromate ion,
free iodine, hypoiodous acid and iodate ion.
During the oxidation or chemical disinfection of natural waters
containing bromide ion with ozone, bromate ion can be formed at
concentrations ranging from 0 to 150 µg/litre under normal water
treatment conditions (Siddiqui, 1992). Chlorate formation with an
initial total chlorine concentration of 0.6 mg/litre was evaluated at
pH levels of 8.0, 7.0 and 6.0, and chlorate concentrations ranging
from 10 to 106 µg/litre were formed after ozonation (Siddiqui et al.,
1996a).
It has been reported that ozone reacts with many metal ions and
with cyanide ion (Hoigne et al., 1985; Yang & Neely, 1986). Bailey
(1978) discussed the formation of ozonates, compounds of metal cations
having the general formula M+O3-. Hydrogen peroxide has been
identified as a by-product of ozonation of organic unsaturated
compounds (Bailey, 1978).
Table 6 provides the range of bromate concentrations normally
encountered in drinking-waters with a variety of source water
characteristics after ozonation.
Table 6. Summary of bromate ion formation potentials in different source waters under different conditions following
ozonation
Na Bromide Ozone pH Alkalinity DOC Bromate Reference
(µg/litre) (mg/litre) (mg/litre) (mg/litre) (µg/litre)
18 10-800 1-9.3 5.6-9.4 20-132 2.2-8.2 <5-60 Krasner et al. (1992)
4 60-340 3-12 6.5-8.5 90-230 3-7 <5-40 Siddiqui & Amy (1993)
28 10-100 2-4 6.8-8.8 20-120 0.3-11 <5-100 Amy et al. (1993, 1994)
4 12-37 0-3.97 7.8 N/A N/A <7-35 Hautman & Bolyard (1993)
1 500 2.3-9.5 7.2-8.3 N/A N/A 13-293 Yamada (1993)
23 12-207 0.3-4.3 5.7-8.2 14-246 0.5-6.8 <2-16 Legube et al. (1993)
8 107-237 1-5 6.8-8.0 N/A 2-5 <5-50 Kruithof & Meijers (1993)
a N = number of sources studied.
2.7 Formation of non-halogenated organic disinfectant by-products
2.7.1 Chlorine organic by-products
Lykins & Clark (1988) conducted a 1-year pilot plant study of the
effects of ozone and chlorine and determined that the concentration of
aldehydes increased by 144% upon ozonation. In the chlorinated stream,
the concentration of these aldehydes increased by 56%. This study
indicates that aldehyde formation, although greater with ozone, is not
unique to ozonation, but is associated with chlorination and other
oxidants as well.
2.7.2 Chloramine organic by-products
When Suwannee River (USA) fulvic acid was reacted with aqueous
solutions of 15N-labelled chloramine and 15N-labelled ammonia,
lyophilized products exhibiting nuclear magnetic resonances between 90
and 120 ppm were observed, denoting the formation of amides,
enaminones and aminoquinones (Ginwalla & Mikita, 1992). This
represents evidence for the formation of nitrogen-containing compounds
from the chloramination of NOM in natural waters.
Amino acids, peptides and amino sugars were chlorinated under
various chlorine/nitrogen ratios (Bruchet et al., 1992). Six natural
amino acids (alanine, methionine, valine, phenylalanine, leucine and
isoleucine) were shown to induce tastes and odours at concentrations
in the range of 10-20 µg/litre. Detectable odours were consistently
induced in a multicomponent mixture containing each of these amino
acids after a 2-h contact time with chlorine. Investigation of the
by-products indicated that the odours generated were systematically
linked to the aliphatic aldehydes formed. The peptides investigated
had varying degrees of odour formation potential, while the amino
sugars did not impart any odour. Chlorinous odours occasionally
detected during these experiments were found to be due to organic
chloramines and other oxidation by-products.
2.7.3 Chlorine dioxide organic by-products
Gilli (1990) showed the formation of carbonyl compounds
(34 µg/litre) such as n-valeraldehyde (7-15 µg/litre), formaldehyde
(3.4-9 µg/litre), acetaldehyde (4.5 µg/litre) and acetone (3.2
µg/litre) after using chlorine dioxide.
2.7.4 Ozone organic by-products
Ozone aliphatic oxidation products from organic impurities in
water are usually acids, ketones, aldehydes and alcohols. So-called
ultimate oxidation products of organic materials are carbon dioxide,
water, oxalic acid and acetic acid. However, ozonation conditions
generally employed in treating drinking-water are rarely sufficient to
form high concentrations of these ultimate products.
When source waters containing NOM and unsaturated organic
compounds are ozonated, ozonides, peroxides, diperoxides, triperoxides
and peroxy acids, for example, can be produced. The limited research
that has been conducted in aqueous solutions indicates that these
intermediates decompose readily in water to form products such as
aldehydes, ketones, carboxylic acids and ketoacids.
Coleman et al. (1992) identified numerous compounds in addition
to the following in ozonated humic samples: monocarboxylic acids up to
C-24, dicarboxylic acids up to C-10, ketoacids, furan carboxylic
acids, and benzene mono-, di- and tricarboxylic acids. Among the
various aldehydes, Paode et al. (1997) found four (formaldehyde,
acetaldehyde, glyoxal and methylglyoxal) to be dominant. Table 7
provides a range of concentrations for aldehydes from the ozonation of
a variety of source waters.
2.8 Influence of source water characteristics on the amount and type
of by-products produced
The extensive literature pertaining to DBP levels in disinfected
source waters and control of DBPs by various treatment processes
attests to the wide variety of factors influencing DBP formation and
the complex interrelationships between these factors. Variables
including the concentration and characteristics of precursor material,
pH, chlorine concentration, bromide level, presence of
chlorine-demanding substances such as ammonia, temperature and contact
time all play a role in DBP formation reactions.
2.8.1 Effect of natural organic matter and UV absorbance at 254 nm
NOM consists of a mixture of humic substances (humic and fulvic
acids) and non-humic (hydrophilic) material. Both the amount (as
indicated by TOC or UVA254) and the character (as described by UVA254)
of NOM can affect DBP formation. NOM provides the precursor material
from which organic DBPs are formed; consequently, increasing
concentrations of NOM lead to increasing concentrations of
by-products. This relationship has led to the use of TOC and UVA254
measurements as surrogate parameters for estimating the extent of DBP
formation.
The removal of NOM is strongly influenced by those properties
embodying the size, structure and functionality of this heterogeneous
mixture. The humic acids are more reactive than fulvic acids with
chlorine (Reckhow et al., 1990) and ozone, in terms of both
oxidant/disinfectant demand and DBP formation. Processes such as
coagulation, adsorption and membrane filtration are separation
processes that remove NOM intact, while ozonation transforms part of
the NOM into biodegradable organic matter, potentially removable by
biofiltration. Coagulation preferentially removes humic/higher
molecular weight NOM; the selectivity of membranes for NOM removal is
largely dictated by the molecular weight cutoff of the membrane; the
use of granular activated carbon (GAC) requires a significant empty
bed contact time; biofiltration can remove only the rapidly
biodegradable NOM fraction.
Table 7. Effect of ozone dose and TOC on non-halogenated organic by-products
Ozone dose TOC Formal Acetal Glyoxal Methyl-glyoxal Reference
(mg/litre) (mg/litre) (µg/litre) (µg/litre) (µg/litre) (µg/litre)
1.2-4.4 2.66 8-24 2-4 4-11 4-15 Miltner et al. (1992)
1.0-9.2 1.0-25.9 3-30 7-65 3-15 3-35 Weinberg et al. (1993)
5.5-28.5 5.4-17.4 58-567 6-28 15-166 17-54 Schechter & Singer (1995)
In an investigation of the nature of humic and fulvic acids
isolated from a variety of natural waters, Reckhow et al. (1990) found
that the fulvic fractions had a lower aromatic content and smaller
molecular size than the humic fractions. UV absorbance was
correspondingly higher for the humic fractions, owing to the higher
aromatic content and larger size. These researchers also found that
for all of the organic material investigated, the production of
chloroform, TCA, DCA and DCAN was higher upon chlorination of the
humic fractions than upon chlorination of the corresponding fulvic
fractions. These findings support the findings of other researchers
and show that the UV absorbance measurement is an indicator of the
nature of the precursor material present in a sample. This
measurement, in conjunction with the TOC (or DOC) measurement, can be
employed in the evaluation of data to provide an indication of the
reactivity of NOM towards forming DBPs.
The reaction of ozone with NOM can occur directly or by radical
processes. The disappearance of disinfecting chemical is influenced by
the type and concentration of NOM present in natural waters. Direct
consumption of these chemicals is greater when the UV absorbance (due
to electrophilic and nucleophilic sites of NOM) of the source water is
significant, resulting in decreased DBP formation potential.
It appears that the nature of the organic material in a source
water may have some impact on the relative concentrations of THMs and
HAAs formed upon chlorination. Treatment techniques that lower the
levels of DOC without affecting bromide levels have been implicated in
a shift from chlorinated to brominated THM compounds. This is of
concern because the theoretical risk to humans varies for the
individual THMs, with the brominated species generally being of more
concern (Bull & Kopfler, 1991).
2.8.2 Effect of pH
The impact of pH on THM concentrations has been reported by a
number of researchers since THMs in drinking-water first came to the
attention of the water industry (Stevens et al., 1976; Lange &
Kawczynski, 1978; Trussell & Umphres, 1978). More recently, the impact
of pH on a number of other chlorination by-products has been reported
(Miller & Uden, 1983; Reckhow & Singer, 1985). The rate of THM
formation increases with the pH (Stevens et al., 1976). Kavanaugh et
al. (1980) reported a 3-fold increase in the reaction rate per unit
pH.
In general, increasing pH has been associated with increasing
concentrations of THMs and decreasing concentrations of HAAs (pH
primarily impacting TCA), HANs and haloketones. The concentrations of
TCA tend to be higher in waters with pH levels less than 8.0 than in
waters with pH levels greater than 8.0; a less marked trend is
observed for DCA. Other researchers have reported similar findings
with respect to the pH dependency of HAA concentrations. For example,
Stevens et al. (1976) found that TCA concentrations were significantly
lower at a pH of 9.4 than at pH levels of approximately 5 and 7. TCA
was by far the most predominant of the measured HAA species at six of
the eight utilities surveyed. Carlson & Hardy (1998) reported that at
pH levels greater than 9.0, THM formation decreased with increasing
pH. It is possible that the shift in chlorine species from
hypochlorous acid to hypochlorite affects THM formation during short
reaction times.
AWWARF (1991) observed no relationship between pH and the
concentrations of THMs at eight utilities over time, suggesting that
although THM concentrations for a particular water are known to be pH
dependent, factors other than pH influence THM concentrations over a
variety of source waters. Nieminski et al. (1993) reported that
treatment plants with a pH of about 5.5 in finished water produced
equal amounts of THMs and HAAs, whereas plants with pHs greater than
7.0 in finished water produced higher amounts of THMs as compared with
HAAs.
No strong relationship has been observed between HAN
concentration and pH over time. Within the approximate pH range 7-8.5,
HAN concentrations increased slightly over time. In general, a trend
of decreasing HAN concentrations with increasing pH would be expected,
since these compounds are known to undergo base-catalysed hydrolysis
and have been identified as intermediates in the formation of
chloroform (Reckhow & Singer, 1985). Therefore, these compounds may be
unstable in the presence of free chlorine or under basic conditions.
In general, after an initial formation period, HAN and haloketone
concentrations level off or begin to decline over the remainder of the
reaction period. This indicates that base-catalysed hydrolysis may not
be a significant mechanism of reaction for the relatively low pH
sources.
Stevens et al. (1989) evaluated the effects of pH and reaction
time (4, 48 and 144 h) on the formation of chloral hydrate. Chloral
hydrate formation increased over time at pH 5 and 7, whereas chloral
hydrate that had formed within 4 h at pH 9.4 decayed over time at the
elevated pH.
The pH of the source water can also affect the formation of
by-products after chloramine addition. The disproportionation of
monochloramine, which is an important reaction leading to an oxidant
loss, has been shown by several researchers to be catalysed by
hydrogen ion, phosphate, carbonate and silicate (Valentine & Solomon,
1987).
Humic acids have shown reaction rates with chlorine dioxide that
increased by a factor of 3 per pH unit (pH 4-8) (Hoigne & Bader,
1994).
In addition to the impact of pH on THM and HAA formation noted
above, overall TOX formation decreases with increasing pH. Many of the
halogenated DBPs tend to hydrolyse at alkaline pH levels (>8.0)
(Singer, 1994a). This has significant implications, for example, for
precipitative softening facilities.
pH has a strong effect on aldehyde formation (Schechter & Singer,
1995). Higher ozonation pH values produced lower amounts of aldehydes,
supporting the theory that these DBPs are formed primarily through the
direct molecular ozone reaction pathway, as opposed to the radical
pathway. These results may also reflect greater destruction of
aldehydes by hydroxyl radicals at elevated pH levels.
2.8.3 Effect of bromide
The presence of bromide ion during water treatment disinfection
can lead to the formation of DBPs such as brominated organics and
bromate ion. Low but significant levels of bromide, the ultimate
precursor to bromate and other brominated compounds, may occur in
drinking-water sources as a result of pollution and saltwater
intrusion in addition to bromide from natural sources. An
understanding of the sources and levels of bromide ion in different
source waters is crucial for an understanding of the bromate ion
formation potential in drinking-waters. There are no known treatment
techniques available for economically removing bromide ion present in
source waters during drinking-water treatment.
The impact of bromide on the speciation of DBPs within a class of
compounds such as THMs or HAAs has been discussed by Cooper et al.
(1983, 1985) and Amy et al. (1998). Rook et al. (1978) reported that
bromine is more effective than chlorine in participating in
substitution reactions with organic molecules; furthermore, precursor
materials may differ in their susceptibility to bromination versus
chlorination reactions. Hypobromous acid formed from bromide may also
react with ammonia to form bromamines (Galal-Gorchev & Morris, 1965).
2.8.4 Effect of reaction rates
After chlorine addition, there is a period of rapid THM formation
for the initial few hours (e.g., 4 h), followed by a decline in the
rate of THM formation, suggesting fast and slow NOM reactive sites.
Many authors have indicated that the concentration of chloroform
appears to increase slowly even after 96 h, suggesting that as long as
low concentrations of free chlorine are present, chloroform continues
to form. Bromochlorinated THM species have been found to form more
rapidly than chloroform. Further data from many sources indicate that
bromoform formation slows at approximately 7-8 h and levels off almost
completely after 20 h (AWWARF, 1991; Koch et al., 1991). The same
general kinetic trend observed for THMs also appears to apply to HAAs.
A period of rapid formation occurs during the first 4-8 h, followed by
a reduction in the formation rate. In general, for most sources,
concentrations of chlorinated HAAs appear to slowly increase even
after 96 h, while the formation of DBA levels off after about 18-20 h.
Miller & Uden (1983) observed that nearly 90% of the final
concentrations of THMs, TCA and DCA form within the first 24 h of
chlorine addition to waters containing NOM. Reckhow et al. (1990)
found that although waters containing precursor materials isolated
from six different water sources differed in their yields of
chlorinated organic by-products, the formation curves for chloroform,
TCA and DCA had the same general shapes for all six precursor
materials. Some researchers have suggested that DCA may be an
intermediate in TCA formation; however, for all eight source waters
studied, both DCA and TCA concentrations increased or remained stable
throughout the 96-h reaction period, suggesting that DCA was an
end-product (AWWARF, 1991). Carlson & Hardy (1998) indicated that HAA
formation followed a pattern similar to that of THM formation. As with
the THMs, HAA formation rate appeared to be rapid for the first 30
min; after 30 min, HAAs formed at nearly a constant rate in four of
the source waters studied.
Different trends were observed in the HAN concentrations of
different source waters. For two source waters, HAN levels formed
rapidly for the first 8 h and continued to increase slowly or levelled
off after 96 h (AWWARF, 1991). DBAN levels remained relatively stable
over the 96 h, as did BCAN and DCAN levels. For other sources, levels
of HANs consisting mostly of DCAN increased rapidly up to 4-8 h and
began to decline by the end of the 96-h period. For these sources,
BCAN appeared to be slightly more stable than DCAN.
Very low levels of chloropicrin formation have been observed by
many researchers (AWWARF, 1991). The highest concentration observed
was 4.0 µg/litre. Chloropicrin appears to form slowly during the
incubation period, with concentrations tending to level off at
approximately 40 h.
2.8.5 Effect of temperature
The formation rates of THMs, HAAs, bromate ion and HANs have been
shown to increase with temperature (AWWARF, 1991; Siddiqui & Amy,
1993). Both haloketone and chloropicrin levels were found to be higher
at a lower temperature, while the concentrations of other DBP species
were similar or not significantly different. These results suggest
that a higher temperature allows for more rapid progression of the
transformation of haloketones to other by-products. In studies on the
effect of temperature on THMs, Peters et al. (1980) found an Arrhenius
dependency between the rate constant and temperature with an
activation energy of 10-20 kJ/mol.
The impact of temperature on THMs was strongest at longer contact
times (Carlson & Hardy, 1998). On a conceptual basis, it may be that
rapidly forming compounds are more reactive and form DBPs regardless
of temperature. On the other hand, slowly forming compounds require
higher activation energy, and an increase in the temperature supplies
the energy. In addition to reaction kinetics, the temperature of a
source water can also affect disinfection efficiency. The biocidal
effectiveness of monochloramine is significantly less than that of
free chlorine and is dependent on temperature, pH and residual
concentration.
2.8.6 Effect of alkalinity
Although pH is a very influential variable and alkalinity affects
pH, alkalinity itself does not appear to directly affect the formation
of THMs and HAAs (by chlorination) and has only a slight effect on
aldehydes and other organic by-products following ozonation (Andrews
et al., 1996). However, the majority of studies on the effect of
alkalinity on the formation of bromate during ozonation indicate that
increased alkalinity increases bromate formation (Siddiqui et al.,
1995). The quantity of aldehydes produced remains approximately
constant for similar changes in alkalinity and pH; however, deviation
from equivalent changes in pH and alkalinity results in increased
aldehyde concentrations. Therefore, conditions of high alkalinity and
low pH or low alkalinity and high pH produce greater quantities of
aldehydes than do intermediate values of these parameters (Andrews et
al., 1996).
2.9 Influence of water treatment variables on the amount and type of
by-products produced
Since DBPs are formed by all of the above chemical disinfectants,
the adoption of alternative disinfectants for DBP control often means
only a trade-off between one group of DBPs and another. The most
effective DBP control strategy is organic precursor (NOM) removal
through enhanced coagulation, biofiltration, GAC or membrane
filtration. There has been little success with bromide removal. Other
DBP control options include water quality modifications -- for
example, acid or ammonia addition for bromate minimization.
2.9.1 Effect of ammonia
The presence of ammonia in source waters during disinfection can
cause chlorine and ozone demand and participation in the formation of
by-products such as nitrate, cyanogen chloride and other nitrogenous
compounds.
Ammonia also does not consume chlorine dioxide. In contrast to
chlorine, chlorine dioxide can therefore be considered as a virucide
when ammonia is present. This might be one of the historical reasons
why chlorine dioxide has been adopted as a disinfectant by some
treatment plants using well oxidized waters but containing changing
ammonia concentrations. The addition of ammonia has been shown to
reduce the formation of bromate after ozonation (Siddiqui et al.,
1995), and the ammonia has been shown to participate in the formation
of HANs and cyanogen bromide (CNBr) (Siddiqui & Amy, 1993).
The growth of nitrifying bacteria is a potential problem in
chloraminated water supplies or chlorination of sources containing
nitrogen. In a study conducted by Cunliffe (1991), nitrifying bacteria
were detected in 64% of samples collected from five chloraminated
water supplies in South Australia and in 21% of samples that contained
more than 5 mg of monochloramine per litre. Increased numbers of the
bacteria were associated with monochloramine decay within the
distribution systems.
2.9.2 Effect of disinfectant dose
Chlorine dose is a factor affecting the type and concentration of
DBPs formed. The THM level rises with increasing chlorine dose
(Kavanaugh et al., 1980). However, there is some disagreement
regarding the quantitative relations between chlorine concentration
and THM levels (or the rate of THM production). Most investigators
found a linear relationship between chlorine consumption and THM
production, with an order of reaction greater than or equal to unity
(Trussell & Umphres, 1978; Kavanaugh et al., 1980). However, it is
also possible that the order of reaction changes during the course of
the reaction.
Reckhow & Singer (1985) found that the concentration of DBP
intermediates such as DCAN and 1,1,1-TCPN formed after 72 h of
reaction time was dependent on chlorine dose. DCAN, which was measured
at a concentration of approximately 5 µg/litre at a chlorine dose of
10 mg/litre, was not detected in samples dosed with 50 mg of chlorine
per litre. The concentration of chloroform was about 150 µg/litre in a
sample dosed with 10 mg of chlorine per litre but was approximately
200 µg/litre in a sample dosed with 20 mg of chlorine per litre. Thus,
it is imperative to have uniform chlorine doses for performing DBP
formation kinetic measurements.
Since chloramine residuals are longer-lasting than free chlorine
residuals, the doses for each set of chlorinated and chloraminated
samples will be different in order to achieve the prescribed target
residual. The disappearance of chloramines can be explained
approximately by a second-order reaction. However, as the chlorine
dose increased, the observed rate constant was found to decrease, then
increased after reaching a minimum value (Dlyamandoglu & Selleck,
1992). Below the chlorine dose at the minimum value of the observed
rate constant, the rate constant was proportional to the 1.4 power of
the chlorine dose, regardless of the ammonia concentration (Yamamoto
et al., 1988).
2.9.3 Effect of advanced oxidation processes
Water utilities can add treatment processes that remove DBP
precursors or DBPs. Many utilities will be using both approaches. The
hydrogen peroxide/UV process, an advanced oxidation process, offers
small water utilities a treatment process with the potential to
provide primary disinfection and a method of DBP control (Symons &
Worley, 1995). This process has been shown to oxidize dissolved
organic halogens and decrease TOC. TOC removal as a function of UV
dose has also been demonstrated by Worley (1994), with TOC removals of
between 0% and 80%. Andrews et al. (1996) evaluated the effect of the
hydrogen peroxide/UV process on THM formation and concluded that this
process is only slightly effective in reducing the formation of DBPs.
However, using hydrogen peroxide at 1 mg/litre in combination with UV
effectively reduced or prevented the formation of aldehydes. Other
advanced oxidation processes (e.g., hydrogen peroxide/ozone, ozone/UV)
involving hydroxyl (and hydroperoxyl) radical formation may provide
similar opportunities.
2.9.4 Effect of chemical coagulation
Enhanced coagulation and softening will remove TOC. Enhanced
coagulation is characterized by coagulant doses greater than those
required for optimum turbidity removal; as an alternative to higher
doses, a combination of acid (pH depression) and coagulant addition
can be practised.
All organic DBPs were reduced by the addition of commonly used
coagulants. Iron-based coagulants, such as ferric chloride, were
consistently more effective than alum in removing NOM (Crozes et al.,
1995). Alum coagulation removed all DBP precursors to a significant
extent. The percentage removals showed the same trends as, but were
not identical to, the percentage removals of TOC and UV absorbance. UV
absorbance was removed to a somewhat greater extent than TOC. Hence,
TOC and UV absorbance can serve as surrogate parameters for DBP
formation potential. A fairly good correlation was observed between
the ratio of HAAs to THMs and the ratio of UV absorbance to TOC,
indicating that the relative concentrations of HAAs and THMs do to
some extent depend on the nature of the precursor material. However,
more data from waters of different qualities would be required to
evaluate the validity of this relationship.
The effectiveness of coagulants in removing DBP precursors is
dependent upon the molecular size of the dissolved organic matter.
Normally, higher molecular weight fractions are effectively removed
through coagulation. In a study conducted by Teng & Veenstra (1995),
water containing dissolved organic matter with molecular weights in
the range 1000-10 000 daltons generally produced the largest amounts
of THMs and HAAs under conditions of free chlorination. Coagulation
and ozonation shift a proportionately greater amount of the THM and
HAA formation potential to the smallest molecular weight range (<1000
daltons).
Coagulation and filtration remove NOM but not bromide, hence
increasing the ratio of bromide to TOC. As a result, the subsequent
use of chlorine generally favours the formation of brominated organic
DBPs.
2.9.5 Effect of pre-ozonation
Several studies of ozone oxidation followed by chlorination
showed increased, rather than decreased, levels of THMs (Trussell &
Umphres, 1978). This is attributed, at least partially, to the
formation of aldehydes by ozonation. Another possibility is
hydroxylation of aromatic compounds to produce m-dihydroxy aromatic
derivatives, which are known THM precursors (Lykins & Clark, 1988).
Although the aldehydes produced contain polar groupings, they are
nevertheless not easily removed during the flocculation step by
complexation with aluminium or iron salts. A convenient and more
appropriate method for the removal of the aldehydes formed during
ozonation is the incorporation of a biological treatment step
(biofiltration) in the water treatment process following ozone
oxidation.
Pre-ozonation can have both positive and negative effects on DBP
formation. Pre-ozonation with typical water treatment dosages and
bicarbonate levels has been shown to remove TCA and DCAN precursors.
However, such treatment can result in no net change in the DCA
precursors and may lead to an increase in 1,1,1-TCPN precursors
(Reckhow & Singer, 1985). According to Teng & Veenstra (1995),
pre-ozonation resulted in enhanced formation of DCA during
chlorination and chloramination in the presence of precursors in the
<1000 dalton molecular weight range. They also indicated that
pre-ozonation plus chloramination controlled the overall production of
THMs and HAAs. However, the use of pre-ozonation coupled with free
chlorination increased the yield of DCA for both the hydrophilic and
hydrophobic fractions of NOM as compared with free chlorination alone.
With ozone-chlorine treatment, chloral hydrate formation can be
enhanced. This behaviour, which has also been observed for DCA,
suggests that the reaction that produces chloral hydrate is
accelerated under the conditions of ozonation in combination with
prechlorination and warm water temperatures (LeBel et al., 1995).
Ozonation in the presence of traces of hypochlorite ion can form
inorganic by-products such as chlorate. Siddiqui et al. (1996a) showed
that if there is any residual chlorine present, ozone can potentially
oxidize hypochlorite ion to chlorate.
Coleman et al. (1992) suggested that brominated MX analogues and
other mixed bromochlorinated by-products formed after ozonation and
chlorination can possibly increase mutagenic activity.
2.9.6 Effect of biofiltration
Biofiltration (ozone-sand filtration or ozone-GAC) can
potentially reduce TOC, organic by-products and the formation of
halogenated DBPs.
Passage of ozonated water samples through a rapid sand filter
reduced the concentration of aldehydes by 62% (Lykins & Clark, 1988).
Chlorinated samples experienced a 26% reduction in aldehyde
concentrations under the same conditions. These reductions in aldehyde
levels are attributable to biological activity in the sand filters. If
GAC filtration follows sand filtration, ozone oxidation can be
expected to promote more bioactivity in the GAC filter, because a
better colonization environment is provided for microorganisms on GAC
particles than on sand. Thus, the biological conversion of oxidized
water impurities to carbon dioxide and water will be greater during
passage through GAC media. Similar aldehyde removals have been
observed by several researchers (Van Hoof et al., 1985; Sketchell et
al., 1995).
Drinking-water treatment techniques that remove organic
contaminants without affecting bromide concentrations cause a shift in
the formation of DBPs towards brominated DBPs. Sketchell et al. (1995)
studied three sources containing three different DOC levels and
ambient bromides, which were filtered through biologically active GAC
filters. Analysis of treated waters showed no removal of bromide ion
and a shift towards more brominated organo-DBPs. THM levels after
treatment with GAC with no added ozone decreased from 900-1700 to
100-700 µg/litre. These water sources contained DOC levels ranging
from 10 to 25 mg/litre and high concentrations of biodegradable DOC
(DOC removals ranged from 60% to 80% after GAC treatment).
Table 8 summarizes the effects of ozonation and biofiltration on
the formation of DBPs from various sources.
2.10 Comparative assessment of disinfectants
A comparative assessment (Table 9) of various disinfecting
chemicals for pre-disinfection (or oxidation) and post-disinfection
and maintaining a residual for 5 days to simulate concentrations in
the distribution system showed that the use of free chlorine produces
the largest concentration of halogenated DBPs (Clark et al., 1994).
The concentration of DBPs may be reduced by adding ozone or chlorine
dioxide as a preoxidant, although enhanced formation has been
observed.
Table 10 summarizes the effects of water quality and treatment
variables on the formation of DBPs.
2.11 Alternative strategies for disinfectant by-product control
The concern about chlorite, bromate, chlorate and other DBPs in
drinking-water following treatment with disinfectants has stimulated
research into ways to eliminate the production or enhance the removal
of DBPs. Strategies for DBP control include source control, precursor
removal, use of alternative disinfectants and removal of DBPs by
technologies such as air stripping, activated carbon, UV light and
advanced oxidation technologies. For DBPs that can arise in
hypochlorite solutions (e.g., chlorate), the purity and storage
conditions of the solutions are important concerns.
2.11.1 Source control
Source control options involve controlling nutrient inputs to
waters (e.g., algae growth control) (Hoehn et al., 1990) that are used
as drinking-water sources, watershed management (e.g., constructing
stormwater detention basins), saltwater intrusion control (e.g.,
development of structural or hydrodynamic barriers to control TOC and
bromide), and using the concept known as aquifer storage and recovery
(e.g., drawing water during seasons when the quality of the water is
best) (Singer, 1994a).
2.11.2 Organohalogen by-products
Strategies for control of organohalogen by-products include
removal of DBPs that are formed using technologies such as oxidation,
aeration and carbon adsorption (Clark et al., 1994); and removal of
precursors using treatment techniques such as conventional treatment,
Table 8. Effects of ozonation and biofiltration on chlorine organic by-products
DBPs Ozonation Biofiltration Ozonation + biofiltration Reference
(% change) (% change) (% change)
THMs -20 -20 -40 (chlorine) Speitel et al. (1993)
HAAs -10 -13 -25 (chlorine) Speitel et al. (1993)
Chloropicrin +50 to +250 -50 to -100 (chlorine) Miltner et al. (1992)
Aldehydes +425 to +1300 -40 to -50 -92 to -98 Miltner et al. (1992)
TOX -30 -51 (chlorine) Miltner et al. (1992)
TOX -10 -38 -47 (chlorine) Shukairy & Summers (1992)
TOX +32 -69 -60 (monochloramine) Shukairy & Summers (1992)
Table 9. Comparative assessment of organic disinfectant by-products (µg/litre) in distribution systemsa,b
DBPs Sand-Cl2 Cl2-Sand-Cl2 O3-Sand-Cl2 NH2Cl-Sand-NH2Cl O3-Sand-NH2Cl ClO2-Sand-Cl2
THMs 236.0 225.0 154.0 9.0 3.2 138.0
HAAs 60.0 146.0 82.0 14.0 9.0 44.0
HANs 3.1 2.9 2.7 <0.1 <0.1 <0.1
Haloketones 2.1 2.6 2.6 <0.1 <0.1 4.2
Chloropicrin 1.3 1.3 7.7 <0.1 <0.1 1.4
Chloral hydrate 79.0 75.0 55.0 <0.1 <0.1 45.0
TOX 557.0 540.0 339.0 59.0 27.0 379.0
a Clark et al. (1994).
b TOC = 3.0 mg/litre; pH = 7.6.
Table 10. Summary of impact of water quality and treatment variables on disinfectant by-product formation
Variable Impact on THMs Impact on HAAs Impact on aldehydes Impact on Impact on bromate
chlorate/chlorite
Contact time Curvilinear increase Curvilinear increase Linear increase as long Linear increase in Curvilinear increase
with increasing with increasing as residual chemical bleach solutions with most bromate
contact time contact time present No discernible effects forming in <5 min
Rapid formation <5 h Rapid formation <5 h Secondary reactions in dilute solutions Formation is a function
90% formation in 24 h 90% formation in 24 h between disinfectants If oxidation of of ozone residual and
Levels off at 96 h Levels off at 150 h and aldehydes possible hypochlorite, contact bromide
time has a positive
effect
Disinfectant Rapid and curvilinear Curvilinear increase Curvilinear with Concentrations related Linear increase after
dose increase after TOC after TOC demand with increasing ozone dose to hypochlorite doses TOC demand and then
demand with dose, increasing dose, or chlorine dose applied levelling off after
levelling off at 2.0 levelling off at 2.0 No appreciable effect Ozone oxidation of ozone residual
mg/litre for TOC of mg/litre after ozone/DOC = 2 : 1 hypochlorite increases disappearance
2.0 mg/litre with dose
pH Curvilinear increase Mixed, possible pH Negative effect (forms Positive effect Strong linear positive
with increasing pH to maximum for DCAA mostly through molecular Decomposition of effect
pH 7.0 and possible and DBAA ozone) hypochlorite increases Hydroxyl radical
pH maximum TCAA decreases up 25% decrease for pH with pH generation efficiency
No positive effect at to pH > 9 7-8.5 Oxidation of increases
pH > 9.5 DCAA maximum at pH hypochlorite by ozone
7-7.5 increases
Temperature Linear increase with Linear increase with Terminal products such Positive effect Curvilinear increase
increasing temperature increasing temperature as carbon dioxide Decomposition of 20-30% increase for
(10-30 °C; 15-25% (10-30 °C; 20-30% increase and total hypochlorite increases 15-25 °C
increase) increase) aldehydes slightly
decrease
Table 10. (continued)
Variable Impact on THMs Impact on HAAs Impact on aldehydes Impact on Impact on bromate
chlorate/chlorite
TOC Increase with Increase with Positive effect Negative effect if Decreases with
increasing TOC; increasing TOC; (hydrophobic fraction ozone is used for increasing TOC;
precursor content precursor content mostly responsible) hypochlorite oxidation precursor content
important important Doubles for every Most likely no effect important
Humic acids more Humic acids more 2 mg/litre with hypochlorite Non-humic acid being
reactive than fulvic reactive than fulvic less reactive
acids acids with ozone
UVA254 Increase with Increase with Positive effect Negative effect if Decreases with
increasing UV increasing UV Ozone demand ozone is used for increasing UV
absorbance; precursor absorbance; increases with UV hypochlorite oxidation absorbance; precursor
content important precursor content (UV absorbance is Probable negative content important
Aromaticity of TOC important mostly due to effect with Humic acid being
being more important Aromaticity of TOC aromaticity and hypochlorous acid more reactive with
being more important hydrophobic fraction) ozone
Bromide Shift towards Shift towards Independent of bromide Shift towards more Bromide threshold
brominated species brominated species at <0.25 mg/litre toxic bromate in Curvilinear increase
At >0.25 mg/litre, hypochlorite solutions and dependent upon
aldehydes can decrease ozone residual
due to ozone-bromide
oxidation
Alkalinity No discernible effect No discernible effect Slight positive effect Unknown Positive effect
Minimization TOC removal, TOC removal, pH control, TOC Avoid hypochlorite pH depression,
strategies minimizing chlorine minimizing chlorine removal by coagulation, dosing solution ammonia addition,
residual, alternative residual, alternative GAC, optimizing doses, Minimize storage radical scavengers,
disinfectants, pH disinfectants, pH contact time Properly tune minimizing and
control, minimizing control, minimizing generators optimizing ozone
contact time contact time Use freshly made residual
solutions
Table 10. (continued)
Variable Impact on THMs Impact on HAAs Impact on aldehydes Impact on Impact on bromate
chlorate/chlorite
Removal GAC, electron beam, GAC, electron beam Biofiltration, advanced Ferrous sulfate, GAC, Ferrous sulfate, UV
strategies air stripping oxidation, GAC, electron beam, UV irradiation,
nanofilters irradiation, high-energy electron
nanofilters beam, GAC
oxidation, membrane processes, carbon adsorption and biological
degradation. For many organic compounds that are difficult to oxidize,
such as chloroform, the kinetics of ozone oxidation are generally very
slow but are faster if used in combination with UV irradiation. GAC
adsorption and membrane filtration are relatively expensive processes;
moreover, NOM removal by GAC cannot be accomplished to any significant
degree in a filter/adsorber (i.e., GAC filter cap) mode but requires a
separate post-filtration adsorber bed. The use of membranes requires
pretreatment to prevent fouling, as well as processing of waste brine.
The use of ozone in combination with biologically active GAC filters
is a promising alternative to reduce DBP precursors.
2.11.3 Inorganic by-products
Properly designed and operated chlorine and chlorine dioxide
generator systems can minimize some of the production of chlorate ion.
Removal of chlorite and chlorate has been reported using reduction by
Fe2+ or sulfite or by GAC (Voudrias et al., 1983; Lykins & Clark,
1988). GAC is seen as problematic because of chlorate production and a
short bed life. A chemical process using an appropriate agent such as
reduced iron (e.g., ferrous sulfate) appears to be a more promising
approach (Kraft & van Eldick, 1989; Gordon et al., 1990).
If bromate is present in treated water entering the coagulation
process (i.e., formed during pre-ozonation), several options exist for
its removal. An aqueous-phase reducing agent (e.g., Fe2+) can be
added at the rapid mix step. Powdered activated carbon can likewise be
added as a solid-phase reductant to remove bromate and DBP precursors.
Not all utilities contemplating ozone application intend to employ
pre-ozonation. Rather, they may use intermediate ozonation prior to
the filtration process; in this situation, removal of bromate by
activated carbon is possible. This approach has potential relevance to
integration of GAC columns into a process train or, more
realistically, to retrofitting of rapid sand filters with GAC filters.
For groundwaters that require no coagulation, bromate can be removed
after ozonation using a GAC filter, UV irradiation or high-energy
electron beam irradiation (Siddiqui et al., 1994, 1996a,b,c).
Brominated or bromochlorinated amines formed during the oxidation
step of the process train using chlorine can potentially be removed
using a suitable activated carbon before terminal chlorination.
However, carbon that has an accumulation of surface oxides, which
develop through reaction of amines, will have a diminished capacity to
reduce halogenated amines to nitrogen. Organic amines can potentially
be removed by activated carbon adsorption.
2.11.4 Organic by-products
There are some technologies for removing organic contaminants
formed after chlorination and chloramination, a less viable option
than minimizing their formation through DBP precursor removal or use
of alternative disinfectants. Studies of ozone oxidation have shown
that aromatic compounds, alkenes and certain pesticides (some of which
have structural similarities to certain organic DBPs) are removed well
by ozone treatment, but that alkanes are poorly removed. Also, removal
efficiency improves for the alkenes and aromatic compounds with
increasing ozone dosage and for some alkanes with increasing pH. For
most compounds, the efficacy of ozone is not affected by the
background water matrix if the ozone is used after coagulation.
Andrews et al. (1996) showed that using hydrogen peroxide at 1.0
mg/litre in combination with UV effectively reduced or prevented the
formation of aldehydes.
2.12 Models for predicting disinfectant by-product formation
The regulation of THMs and other halogenated DBPs has been
complicated by findings that alternative disinfectants to free
chlorine may also form by-products that are of potential health
concern. Additional complicating factors impacting the regulation of
DBPs have been the emergence of Giardia and Cryptosporidium as
major waterborne pathogens.
In view of the finding that water chlorination produces DBPs,
some of which are carcinogenic, mutagenic or possibly teratogenic,
several countries have recently laid down standards for various DBP
levels. This stimulated the search for mathematical models to describe
or predict DBP formation in disinfected water and to evaluate the
effectiveness of water treatment technologies designed to reduce DBP
levels so as to comply with the standards. Most of these models are
based on fitting mathematical equations to various empirical
observations, rather than mechanistic and kinetic considerations. This
is mainly due to the complexity of the reactions between organic
precursors and disinfecting chemicals, which usually involve several
parallel pathways leading to a great variety of products. The
complexity of the DBP formation reactions also makes it difficult to
develop universally applicable models for simulating DBP formation
potential associated with disinfection of a diverse array of natural
source waters. However, the analysis presented by many models suggests
that many waters exhibit comparable general responses to changes in a
given parameter (i.e., responses lending themselves to simulation by a
particular mathematical functionality), although specific responses
associated with individual waters may vary. The multiple regression
models developed by many researchers represent a rational framework
for modelling DBP formation in many sources. Another potential
application is the modelling of DBP mixtures, e.g., predicting HAA
levels from THM and water quality data.
2.12.1 Factors affecting disinfectant by-product formation and
variables of interest in disinfectant by-product modelling
The information on the factors controlling DBP formation, which
is available in the literature, is briefly summarized below.
The extent of formation of DBPs is dependent on several water
quality parameters, such as TOC concentration, UVA254, bromide
concentration and temperature. It is also dependent on chlorination
conditions, such as chlorine dose, pH, ammonia concentration and
contact time. After the various statistically significant factors were
identified, mathematical equations were developed to describe the
formation of various DBPs. A least squares method was used to
determine the optimum equation coefficients that best describe the
experimental data. The optimum coefficients have been defined as those
that produce a minimum residual error between the mathematical
predictions and the experimental data.
2.12.2 Empirical models for disinfectant by-product formation
Numerous models for predicting THM formation through chlorination
have been reported (Moore et al., 1979a; Kavanaugh et al., 1980;
Engerholm & Amy, 1983; Urano et al., 1983; Amy et al., 1987, 1998;
Morrow & Minear, 1987; AWWARF, 1991; Hutton & Chung, 1992). Of these,
models reported by AWWARF (1991) and Amy et al. (1998) are more recent
and were derived from a variety of natural source waters and more
realistic treatment conditions. Not much information has been reported
on the formation of other chlorination DBPs. Only Amy et al. (1998)
summarized empirical models for THMs, HAAs and chloral hydrate. These
chlorination by-product models can be used to assess both in-plant and
distribution system formation of THMs, HAAs and chloral hydrate. Water
quality conditions such as DOC, pH, temperature and bromide are needed
as inputs to the models; such data then allow assessment of
chlorination DBP formation as a function of reaction time:
DBP concentration (total THMs or THM species, total HAAs or HAA
species, or chloral hydrate) =
f(TOC, bromide, chlorine, pH, temperature, time)
Relatively little is known about the kinetics of the formation of
bromate and other DBPs during ozonation and the quantitative effects
of water quality factors (temperature, pH, etc.); such an
understanding is crucial for evaluating various bromate control
strategies. Siddiqui & Amy (1993) and Amy et al. (1998) developed
statistical relations to predict the concentrations of various ozone
DBPs, including bromate, as a function of water treatment variables.
Correlation matrix analysis has shown that ozone dose, dissolved ozone
concentration, bromide concentration, pH and reaction time all have a
positive influence on bromate formation. Von Gunten & Hoigne (1994)
developed kinetic models for bromate formation.
Ozone, as a result of its strong oxidizing power, produces a
variety of organic by-products, such as aldehydes and ketoacids, when
used to treat natural source waters. These by-products -- especially
aldehydes -- are highly biodegradable, and there is concern for
regrowth of microorganisms following ozone treatment. They are also
potentially hazardous and may produce increased amounts of chlorinated
by-products upon chlorination. Siddiqui et al. (1997) developed a
model to estimate the potential for total aldehyde formation in source
waters upon ozonation.
2.12.3 Models for predicting disinfectant by-product precursor
removal
It is recognized that chlorination will continue to be the most
common disinfection process; hence, enhanced removal of DBP precursors
present in raw sources represents a valuable option for reducing the
potential for by-product formation. The removal of NOM can be achieved
either by providing additional processes, such as GAC and
nanofiltration, or by enhancing the existing coagulation, flocculation
and sedimentation processes. Predictive models have been developed for
assessing coagulation efficiency in removing NOM and reducing DBP
precursor levels (AWWARF, 1991; Amy et al., 1998).
Coagulation can reduce DOC and DBP precursors but not bromide
levels; hence, a greater proportion of brominated DBP species can
potentially be produced in the finished water.
The effects of precursor removal by chemical coagulation can be
assessed through the use of treated water models. One can either
predict DBPs formed under a given degree of precursor removal or
define the degree of precursor removal required to meet DBP
regulations. The impact of bromide ion on meeting regulations can also
be assessed. If one makes the assumption that precursor reactivity
(i.e., DBP/DOC) does not change, one can also assess other precursor
removal processes, such as GAC or membrane processes, through use of
the raw/untreated water models. Care should be exercised when using
models to approximate post-chlorination DBPs following an ozonation
step.
2.13 Summary
* The primary and most important role of drinking-water treatment is
to remove or inactivate harmful microorganisms. Another role is to
minimize the concentrations of disinfectants and DBPs without
compromising in any way the removal or inactivation of pathogens.
* Drinking-water utility managers must be more knowledgeable about
options to meet regulations. It is often more practical to use
treatment methods that control the concentration of several
contaminants than to modify treatment practices for each new
standard that is promulgated.
* A thorough understanding of DBP formation would help the successful
balancing of appropriate microbial inactivation with the
minimization of DBPs. Water quality variables affect DBP formation
and must be considered when developing a strategy to control DBPs
with various disinfectants.
* The chemistry of chlorine and its by-products has been well
studied, and ozone and its by-products have recently received much
attention. Studies of chlorine dioxide and chloramines and their
by-products are relatively few, although more work in these areas
is now being undertaken.
* One of the simplest processes to minimize halogenated DBP formation
is limiting the free chlorine contact time by using monochloramine
to maintain a distribution system residual following primary
disinfection by chlorine or ozone. Chloramines are an effective
means of controlling DBPs. However, the growth of nitrifying
bacteria (and related production of nitrite) is a potential problem
in chloraminated water supplies.
* Various nitrogen-containing organic compounds may be present in
source waters after chlorination and chloramination. Because of
analytical complexities, very few detailed studies have been
undertaken to determine the individual compounds present and their
concentrations.
* Many factors between the source and the tap can influence the DBPs
to which consumers are exposed. Although THMs and HAAs continue to
form with increasing contact time, some other halogenated DBPs,
such as HANs and haloketones, form rapidly but then decay in the
distribution system as a result of hydrolysis. This has major
implications regarding exposure to these DBPs, depending upon their
proximity to the treatment plant. For treated source waters, median
levels of HAAs are often approximately one-half of the median THM
levels.
* For low-bromide source waters, chloroform is normally the dominant
THM species; DCA and TCA are the most prevalent HAA species; DCAN
is the most prevalent HAN species; and 1,1,1-TCPN is the most
prevalent of the two measured haloketones. Very low levels of
chloropicrin have been observed by various researchers; this
compound appears to form slowly during the incubation period, with
concentrations tending to level off at 40 h. For high-bromide
waters, increased levels of brominated DBPs are observed.
* Chlorine dioxide is a strong oxidant that under certain conditions
surpasses chlorine in its ability to destroy pathogenic organisms.
When chlorine dioxide is prepared and administered without excess
free chlorine, THMs and other chlorinated by-products are not
produced, but inorganic by-products are formed.
* TOC levels have been found to be correlated with halogenated DBP
formation. The nature of this relationship varies with the source.
TOC removal can be used as a surrogate for the reduction of DBP
formation.
* Although the presence of chloral hydrate and HANs in chlorinated
samples may be attributed to precursors other than amino acids, the
potential for amino acids to be present in natural sources is well
documented. Surface waters, but not groundwaters, tend to contain
amino acids. However, the removal of these precursors during
conventional water treatment is not well understood.
* The amount of chlorate that is present in delivered hypochlorite
solutions depends on many factors. Freshly made hypochlorite
solutions will contain less chlorate than hypochlorite that is
stored without concern for temperature and pH. If a utility is
using a single tank to store hypochlorite, it is likely that the
level of chlorate is increasing in the tank. Thus, storage tanks
should be periodically flushed and cleaned, and, if possible, the
storage time should be reduced.
* Models have been developed that can be used to simulate the fate
and movement of DBP precursors in distribution systems. The models
can be designed as a planning tool for evaluating the impacts of
source water management strategies and estimating DBP exposures.
Some limitations of existing models include calibration with a
limited database, application to only a specific water source or
group of related sources, lack of terms to simulate important
parameters, such as reaction time, and inadequate validation.
3. TOXICOLOGY OF DISINFECTANTS
In assessing the hazards associated with drinking-water
disinfection, it is important not to neglect the disinfectants
themselves. Adding disinfectant in excess of the demand has several
practical benefits. First, it ensures that reaction of the
disinfectant with DBP precursors (largely organic material and
ammonia) does not shorten contact time to the point of ineffective
disinfection. Second, residual disinfectant helps to prevent regrowth
of organisms in the remaining portions of the treatment and
distribution systems.
The result of this practice, however, is that one of the
chemicals that is present in the finished water at the highest
concentration is the disinfectant. In the present regulatory climate
in many countries, chemicals that are introduced as direct additives
to food would be subjected to a significant amount of toxicological
screening before they could be used. Since the major disinfectants
were introduced almost 100 years ago, they were subjected to much less
thorough toxicological evaluations than would be required today.
However, many of these data gaps have been addressed in the past
decade.
3.1 Chlorine and hypochlorite
3.1.1 General toxicological properties and information on
dose-response in animals
Chlorine gas has long been recognized as a lung irritant. This
topic will not be reviewed in the present document, as it appears to
be largely irrelevant to the small amounts of chlorine that are
volatilized from chlorinated water in showers or other points of use
in the household. In water treatment plants, however, there is a
possibility of occupational exposures that could have severe sequelae.
For information on these higher-level exposures, the interested reader
is referred to a recent review by Das & Blanc (1993). The effects of
chlorine gas that have been observed in humans will be discussed in
section 3.1.3.
Sodium hypochlorite (NaOCl) or calcium hypochlorite (Ca(OCl)2)
solutions have also been utilized extensively in the disinfection of
drinking-water. The stock solutions used for this purpose are highly
caustic and are a clear concern for occupational exposures. The
concentration required to produce irritation and decreased basal cell
viability in the skin of guinea-pigs after an application period of
2 weeks was 0.5% sodium hypochlorite (Cotter et al., 1985). Reducing
the concentration to 0.1% resulted in no effect on basal cell
viability relative to control animals. Yarington (1970) demonstrated
that instillation of bleach into the oesophagus of dogs produced
irritation. The minimal exposure that produced oesophageal burns was
10 ml of commercial bleach with a 5-min exposure. It should be noted
that the highly alkaline pH (about pH 11) of sodium hypochlorite is
not likely to be encountered in drinking-water.
There have been relatively few evaluations of the effects of
chlorine or hypochlorite in drinking-water. The present review will
focus on studies with treatment periods longer than 4 weeks where
drinking-water was the primary route of exposure. Reference to earlier
studies of shorter duration and less general applicability to a safety
evaluation can be found in previous reviews (Bull, 1980, 1982a,b,
1992; Bull & Kopfler, 1991).
Daniel et al. (1990a) evaluated the toxicity of solutions of
chlorine prepared by bubbling chlorine gas into distilled water and
adjusting the pH to 9.4. The nominal concentrations of chlorine used
were 0, 25, 100, 175 or 250 mg/litre in distilled water (approximately
0, 3, 10, 16 or 21 mg/kg of body weight per day). These solutions were
provided as drinking-water to both male and female Sprague-Dawley rats
(10 per sex per dose) for 90 days. No deaths occurred in any treatment
group. However, there were statistically significant decreases in
drinking-water consumption in females treated with 100 mg/litre and
higher, probably due to decreased palatability. There were no
consistent effects of chlorine treatment on organ to body weight
ratios or clinical chemistry parameters. A no-observed-effect level
(NOEL) of 10 mg/kg of body weight per day was identified by the
authors based on reduced body weight gain. However, since this was
associated with reduced palatability of the drinking-water, it is not
considered to be a true toxicological end-point.
The study in rats was followed up with another study in B6C3F1
mice (Daniel et al., 1991a). Male and female B6C3F1 mice (10 per sex
per group) were administered 12.5, 25, 50, 100 or 200 mg of chlorine
per litre of drinking-water for 90 days (calculated mean daily doses
were 2.7, 5.1, 10.3, 19.8 or 34.4 mg/kg of body weight in males and
2.8, 5.8, 11.7, 21.2 or 39.2 mg/kg of body weight in females). Spleen
and liver weights were depressed in males, but not in females, at the
highest dose rates (100 and 200 mg/litre). There were no other
consistent indications of target organ effects based on serum enzyme
concentrations. No gross or microscopic lesions could be related to
treatment with chlorine.
Several of the following studies utilized solutions of sodium
hypochlorite as the treatment chemical. It is now known that such
solutions can contain very high concentrations of chlorate within a
short time of their preparation (Bolyard et al., 1993). The extent of
this contamination has not been reported.
Hasegawa et al. (1986) examined the effects of much higher
concentrations (0.025, 0.05, 0.1, 0.2 or 0.4%) of sodium hypochlorite
(equivalent to 7, 14, 28, 55 and 111 mg/kg of body weight per day)
administered in drinking-water to male and female F344 rats for
13 weeks. Twenty rats of each sex were assigned to each experimental
group. Significant suppression of body weight (as a result of
decreased consumption of water and food) occurred at 0.2% and above.
The authors noted slight damage to the liver as indicated by increased
levels of serum enzymes (not specified) at 0.2% and 0.4% sodium
hypochlorite in both sexes. No evidence of treatment-related pathology
was observed in this study or in a 2-year study in which males were
subjected to 0.05% or 0.1% (13.5 or 27.7 mg/kg of body weight per day)
and females to 0.1% or 0.2% (34 or 63 mg/kg of body weight per day)
sodium hypochlorite. The extended exposures were conducted with
50 animals of each sex per treatment group. Analysis of dosing
solutions was not reported.
In a 2-year bioassay, the National Toxicology Program (NTP)
examined chlorine at 0, 70, 140 or 275 mg/litre (expressed as atomic
chlorine, Cl) in drinking-water of F344 rats and B6C3F1 mice (70 per
sex per group) (NTP, 1992). These solutions were prepared from gaseous
chlorine and neutralized to pH 9 by the addition of sodium hydroxide.
Stability studies indicated that 85% of the initial target
concentration remained after 3 days of preparation. Stock solutions
(concentrations not specified) were prepared once weekly, and
solutions for drinking were prepared 4 times weekly. Based on body
weight and water consumption, doses in these studies were
approximately 0, 4, 7 or 14 mg/kg of body weight per day for male
rats; 0, 4, 8 or 14 mg/kg of body weight per day for female rats; 0,
7, 14 or 24 mg/kg of body weight per day for male mice; and 0, 8, 14
or 24 mg/kg of body weight per day for female mice. The only
treatment-related non-tumour pathology was found to be a dilatation of
renal tubules in male mice receiving 275 mg/litre for more than 66
weeks. No non-neoplastic lesions were observed in either male or
female rats.
A number of immunological changes have been associated with the
treatment of rodents with sodium hypochlorite in drinking-water. Water
containing 25-30 mg of sodium hypochlorite per litre was found to
reduce the mean number of peritoneal exudate cells recovered from
female C57BL/6N mice after 2 weeks of treatment. This was, in turn,
associated with a significant decrease in macrophage-mediated
cytotoxicity to melanoma and fibrosarcoma cell lines (Fidler, 1977).
The treatment period was increased to 4 weeks in a subsequent study,
which demonstrated that 25 mg of sodium hypochlorite per litre
decreased the ability of peritoneal macrophages to phagocytose
51Cr-labelled sheep red blood cells. Macrophages obtained from the
mice treated with hypochlorite were found to be less effective in
destroying B16-BL6 melanoma cells in vitro. Mice so treated were
also found to have increased pulmonary metastasis of B16-BL6 cells
when they were introduced by subcutaneous injection (Fidler et al.,
1982).
Exon et al. (1987) examined the immunotoxicological effects of
sodium hypochlorite at 5, 15 or 30 mg/litre (0.7, 2.1 or 4.2 mg/kg of
body weight per day) in the drinking-water of male Sprague-Dawley rats
(12 per dose) for 9 weeks. Delayed hypersensitivity reaction to bovine
serum albumin was observed at the highest dose administered. Oxidative
metabolism by adherent resident peritoneal cells was decreased at 15
and 30 mg/litre, and the prostaglandin E2 levels of these cells were
found to be significantly elevated. No effects on natural killer cell
cytotoxicity, antibody responses, interleukin 2 production or
phagocytic activity were observed. The effects on macrophage activity
suggest that some impairment does occur at relatively low levels of
sodium hypochlorite. As pointed out by the authors, these were
relatively mild effects, the significance of which was unknown. It is
not clear that these effects would be translated into a significant
impairment of the immune response to a particular infectious agent.
However, modification of macrophage function appears to be one of the
most sensitive responses identified in studies of chlorine or
hypochlorite in experimental animals. A study in which female C57BL/6
mice were administered hypochlorite in their drinking-water (7.5, 15
or 30 mg of hypochlorite per litre) for 2 weeks showed no effects on
the immune system as measured by spleen and thymus weight,
plaque-forming cell response, haemagglutination titre and lymphocyte
proliferation (French et al., 1998).
Altered liver lipid composition has been observed as a result of
acute intragastric administration of sodium hypochlorite (5 ml of a 1%
solution) to rats (Chang et al., 1981). These data do not provide a
clear indication of whether these effects might give rise to
pathology. The concentrations of hypochlorite utilized were much
greater than those that would be encountered in drinking-water.
The effects of hypochlorous acid and hypochlorite on the skin
have received relatively little attention despite the current interest
in bathing as a significant source of chemical exposure from
drinking-water. Robinson et al. (1986) examined the effects of both
hypochlorite and hypochlorous acid solutions applied to the skin of
the entire body of female Sencar mice except for the head. Exposures
were to 1, 100, 300 or 1000 mg/litre as hypochlorous acid at pH 6.5
for 10 min on 4 consecutive days. Hypochlorite (formed by raising the
pH to 8.5) was studied only at 1000 mg/litre. Significant increases in
epidermal thickness and cell counts within the epidermal layer were
observed at concentrations of hypochlorous acid (pH 6.5) of 300
mg/litre and above, but the thickness of the skin was not
significantly different from that in animals at 100 mg/litre. The
increases in skin thickness were associated with an epidermis whose
thickness was increased to 4-6 cells as compared with the normal 1-2
cells seen in mice. The effects of hypochlorite were much less marked.
Following a single application, the increased thickness of the skin
observed in mice exposed to hypochlorous acid (i.e., pH 6.5) did not
appear until 4 days after the treatment. This differed from
hypochlorite, other disinfectants and the positive control,
12- O-tetradecanoylphorbol-13-acetate (TPA). In the latter cases, the
maximal response was observed within 24-48 h after treatment. The
hyperplastic response to hypochlorous acid required 12 days to return
to normal. This study suggests a considerable margin of safety between
the concentrations of chlorine required to produce hyperplasia and
those that are found in drinking-water.
The reactive nature of chlorine always raises questions of
whether it is chlorine or a by-product that is responsible for any
effect. Several studies have examined the formation of by-products in
the gastrointestinal tract following the administration of chlorine.
Invariably, these studies have involved the administration of chlorine
or hypochlorite by gavage at very high concentrations relative to the
amounts that would be encountered in chlorinated drinking-water. As a
consequence, the by-products formed following gavage dosing of high
concentrations may not be representative of the by-products that would
be seen following the consumption of modest to moderate levels of
chlorine in larger volumes of water. A particular issue is that the
high organic carbon concentration relative to chlorine that would be
encountered in the gastrointestinal tract when water is consumed at
low concentrations should dissipate disinfectant before sufficient
oxidative power would be present to break down substrates to small
molecules. Despite these design flaws, the data do indicate that
by-products are formed. The bulk of them remain as higher molecular
weight products, which may have little toxicological importance.
Vogt et al. (1979) reported that chloroform could be measured in
the blood, brain, liver, kidneys and fat of rats to which sodium
hypochlorite was administered by gavage at doses of 20, 50 or 80 mg in
5 ml of water. Thus, the by-product chloroform can be formed by the
reaction of chlorine with stomach contents.
Mink and co-workers (1983) pursued this observation and found
that other by-products could be detected in the stomach contents and
plasma of rats that had been administered sodium hypochlorite
solutions neutralized to pH 7.9. In addition to chloroform, DCAN, DCA
and TCA were detected in the stomach contents analysis. DCA and TCA
were also detected in blood plasma.
The third group of compounds identified as by-products of
chlorination in stomach contents of the rat are the organic
N-chloramines (Scully et al., 1990). N-Chloroglycine,
N-chloroleucine or N-chloroisoleucine and N-chlorophenylalanine
were confirmed products of reactions with normal amino acids that
would ordinarily be found in the gastrointestinal tract.
N-Chlorovaline and N-chloroserine were also tentatively
identified. Organic chloramines are reactive and could be responsible
for toxic effects that may be attributed to chlorine in toxicological
studies. The chlorine demand of free amino acids in stomach contents
was found to be only about 4% of the total. Consequently, this process
may be substrate-limited at concentrations of chlorine found as
residuals in drinking-water. However, use of higher concentrations of
chlorine would also lead to breakdown of proteins present in the
stomach fluid. Thus, as concentrations are increased to levels that
would be used in animal studies, these products would be formed at a
much higher concentration, similar to the phenomena noted with THM and
HAA by-products.
3.1.2 Reproductive and developmental toxicity
In general, animal studies have demonstrated no reproductive or
teratogenic effects of chlorine. Druckrey (1968) examined the effects
of water chlorinated to a level of 100 mg/litre (approximately 10
mg/kg of body weight per day) in BDII rats for seven generations. No
effects were observed on fertility, growth or survival.
A number of subsequent studies have studied the effects of
chlorine or hypochlorite on more specific aspects of reproduction or
development. Meier et al. (1985b) reported that oral administration of
sodium hypochlorite (pH 8.5) prepared from chlorine gas and
administered at 4 or 8 mg/kg of body weight per day for 5 weeks
increased the incidence of sperm head abnormalities in B6C3F1 mice
(10 animals per group). The effect was not observed when the solutions
were administered at pHs at which hypochlorous acid was the
predominant species (pH 6.5). However, other studies have not been
able to associate adverse reproductive outcomes with the
administration of chlorine or sodium hypochlorite.
Carlton et al. (1986) found no evidence of sperm head
abnormalities or adverse reproductive outcomes in Long-Evans rats.
Male rats were treated for 56 days prior to mating and female rats
from 14 days prior to mating through gestation. Each experimental
group consisted of 11-12 males and 23-24 females. Solutions of
chlorine were prepared at pH 8.5, so the study evaluated hypochlorite
as the dominant form in the drinking-water. Doses were as high as
5 mg/kg of body weight per day.
3.1.3 Toxicity in humans
There have been significant human exposures to chlorine and
hypochlorite solutions. Much of that experience is with inhalation of
chlorine gas, which is known to be a strong respiratory irritant.
Chlorine gas is also the largest single component involved in toxic
release incidents. A third major source of exposure is solutions of
sodium hypochlorite, usually marketed as bleach. Bleach is frequently
involved in human poisonings. These exposures are not particularly
relevant to exposures to chlorine or hypochlorite in drinking-water.
Therefore, only a few case reports are identified that illustrate the
types of problems that have been encountered. There was no attempt to
make this review comprehensive.
The irritating effects of chlorine gas have been well documented
because of its use as a chemical warfare agent during World War I (Das
& Blanc, 1993). In a follow-up of survivors of gassing, it was
concluded that there was no evidence of permanent lung damage;
however, these studies clearly indicated that survivors had breath
sounds that suggested bronchitis and limited chest and diaphragmatic
movement, even emphysema. Most studies suggested that there were high
incidences of acute respiratory disease and a lesser prevalence of
chronic sequelae. Similar sequelae have been identified following
exposure of humans to accidental releases of chlorine gas. In these
more modern characterizations, the acute signs and symptoms included a
high incidence of pulmonary oedema and severe bronchitis. These signs
and symptoms are of generally short duration and resolve themselves
over the course of about 1-4 weeks. However, chronic sequelae are
observed in some individuals, depending in part upon the severity of
the exposure. In such cases, a decrease in the forced expiratory
volume is the most consistently reported clinical sign.
Two recent reports suggest that chronic sequelae to acute
exposures to chlorine gas may be more prevalent than previously
appreciated. Moore & Sherman (1991) reported on an individual who was
previously asymptomatic and who developed chronic, recurrent asthma
after exposure to chlorine gas in an enclosed place. Schwartz et al.
(1990) followed 20 individuals who had been exposed to chlorine gas in
a 1975 incident. The prevalence of low residual lung volume was
increased during the follow-up period. Sixty-seven per cent of those
tested were found to have residual volumes below 80% of their
predicted values. Five of 13 subjects tested for airway reactivity to
methacholine were found to have a greater than 15% decline in forced
expiratory volume.
Controlled studies have been conducted in healthy, non-smoking
men exposed to chlorine gas at 1.5 and 2.9 mg/m3 (0.5 and 1.0 ppm)
for 4 or 8 h (reviewed in Das & Blanc, 1993). Four hours of exposure
to 2.9 mg/m3 (1.0 ppm) produced significant decreases in the forced
expiratory volume. One individual who was found to be experiencing
more difficulty than other subjects at this dose and who was later
exposed to 1.5 mg/m3 (0.5 ppm) experienced a significant decrease in
forced expiratory volume. While 1.5 mg/m3 (0.5 ppm) appears
protective for most people, some more sensitive individuals may in
fact have more significant responses to chlorine gas.
The effects of chronic exposure to chlorine gas have received
only limited study. In one study of paper mill workers, a more rapid
age-related decrease in lung volumes of workers exposed to chlorine
relative to those exposed to sulfur dioxide was noted, but the trend
was not statistically significant (Das & Blanc, 1993). Other studies
failed to identify chronic sequelae.
There are frequent reports of human poisonings from bleach. Most
often these exposures result from the mixing of bleach with acidic
products or ammonia. Acidification converts hypochlorite to
hypochlorous acid, which dissociates to chlorine gas, offgasses very
rapidly from the solutions and presents an inhalation exposure (MMWR,
1991). Mixing bleach with ammonia results in the formation of
monochloramine and dichloramine, both of which are effective
respiratory irritants (MMWR, 1991).
Any potential effects of chlorine or hypochlorite in
drinking-water are obscured by the fact that by-products inevitably
coexist with the residual chlorine. One series of studies in which
by-products formed were minimized by dissolving chlorine in distilled
water attempted to identify effects of chlorine in drinking-water on
humans. Chlorine in drinking-water was administered in a rising-dose
tolerance study beginning with 0.1 mg/litre in two 500-ml portions and
rising to a concentration of 24 mg/litre, equivalent to 0.34 mg/kg of
body weight per day (Lubbers & Bianchine, 1984). No clinically
important changes were observed. No findings of clinical importance
were identified in a follow-up treatment with repeated dosing with
500-ml portions of a solution containing 5 mg of chlorine per litre
for a 12-week period (Lubbers et al., 1984a).
Another study attempted to determine whether consumption of
chlorinated drinking-water affected blood cholesterol levels (Wones et
al., 1993a). The impetus for this study was a toxicological study in
pigeons that suggested that chlorine raised blood cholesterol levels
and modified serum thyroid levels (Revis et al., 1986a,b) and an
epidemiological study that associated small increases in cholesterol
of women with residence in communities having chlorinated water
(Zeighami et al., 1990a,b; described in detail in section 5.2.2). A
prior study (quoted in Wones et al., 1993a) was conducted that
examined men who consumed water containing 2, 5 or 10 mg of chlorine
per litre and found a small increase in serum cholesterol levels at
the highest dose group. However, no control group was studied, so the
changes could have been attributed to the change in diet imposed as
part of the study protocol (Wones & Glueck, 1986). The longer-term
study was composed of 30 men and 30 women who received a controlled
diet for the duration of the study. The first 4 weeks represented an
acclimatization period during which all subjects received distilled
water. Half the subjects were assigned to a group that consumed 1.5
litres of water containing 20 mg of chlorine per litre for the
following 4 weeks. At the end of each 4-week period, blood was
analysed for cholesterol, triglycerides, high-density lipoprotein
(HDL) cholesterol, low-density lipoprotein (LDL) cholesterol or
apolipoproteins A1, A2 and B. There were no significant effects. There
was a slight trend towards lower thyroid hormone levels in men
consuming chlorine, but this was not clinically significant (Wones et
al., 1993a). These data suggest that observations obtained previously
in pigeons could not be repeated under comparable conditions of
chlorine consumption. It is notable that the animals utilized in the
original pigeon study had consumed a modified diet (Revis et al.,
1986a) that was deficient in calcium and other trace metals. A
subsequent study failed to replicate the previous results in pigeons
(Penn et al., 1990).
3.1.4 Carcinogenicity and mutagenicity
The International Agency for Research on Cancer (IARC) has
evaluated the carcinogenicity of hypochlorite salts and concluded that
there were no data available from studies in humans on their
carcinogenicity and inadequate evidence for their carcinogenicity in
experimental animals. Hypochlorite salts were assigned to Group 3: the
compounds are not classifiable as to their carcinogenicity to humans
(IARC, 1991).
Several studies have shown that sodium hypochlorite produces
mutagenic responses in bacterial systems and mammalian cells
in vitro. However, there is no evidence of activity in mammalian
test systems in vivo. It is not clear to what extent this is
influenced by the formation of mutagenic by-products as a result of
reactions with components of the incubation media. Wlodkowski &
Rosenkranz (1975) used short-term exposures of Salmonella
typhimurium strain TA1530 followed by ascorbic acid-induced
decomposition to reduce the cytotoxic effects of hypochlorite. The
investigators applied 0.14 µmol per tube and added ascorbic acid after
intervals of 5, 10 and 15 min. At 5 min, a clear positive response was
observed with minimal cytotoxicity. Significant responses were also
observed in strain TA1535, but not in strain TA1538.
Rosenkranz (1973) and Rosenkranz et al. (1976) also demonstrated
a positive mutagenic response in DNA polymerase A deficient
Escherichia coli to 0.006 µmol of sodium hypochlorite. This response
was unaffected by the addition of catalase, suggesting that the
response was not related to the generation of hydrogen peroxide.
Matsuoka et al. (1979) reported that sodium hypochlorite at a
concentration of 6.7 mmol/litre (0.5 mg/ml) produced chromosomal
aberrations in Chinese hamster ovary (CHO) cells in the presence of S9
mix. This concentration was cytotoxic in the absence of S9. Some
concern must be expressed about whether responses observed with such
high and clearly cytotoxic concentrations in an in vitro system
represent specific clastogenic effects. The authors report only one
concentration tested with and without S9. It is probable that the
positive response in the presence of S9, if it is a specific response,
was a result of detoxifying hypochlorite. This protection could be
non-specific as well, in that it may not have depended upon any
catalytic activities present in the S9 fraction (i.e., the added
protein may have acted as a reactive sink to dissipate excess
hypochlorite). Consequently, it is difficult to use these data in
interpreting the effects of chlorine or hypochlorite in vivo.
Ishidate (1987) studied the induction of chromosomal aberrations
in cultures of Chinese hamster CHL cells at sodium hypochlorite
concentrations ranging from 125 to 500 µg/ml without exogenous
metabolic activation and from 31 to 125 µg/ml with and without rat
liver S9 mix. A clear increase in the number of cells with structural
chromosomal aberrations was observed at 500 µg/ml without S9 mix,
while the results obtained in the other series, showing weakly
positive responses, were considered inconclusive.
Meier et al. (1985b) evaluated the ability of hypochlorite and
hypochlorous acid to induce chromosomal damage or micronuclei in the
bone marrow of CD-1 mice. The samples to be tested were generated by
bubbling chlorine gas into water and then adjusting the pH to 6.5
(predominantly hypochlorous acid) or 8.5 (predominantly hypochlorite).
The doses administered were 1.6, 4 or 8 mg/kg of body weight for 5
consecutive days. There was no evidence of increased micronuclei or
chromosomal abnormalities in bone marrow cells. Significant positive
responses were observed with positive control chemicals in both
assays. As reported in section 3.1.2, these authors detected a
positive response in the sperm head abnormality assay in mice treated
at these same doses of hypochlorite in two separate experiments. This
assay is used primarily as a mutagenicity assay rather than as an
assay for reproductive toxicities. Hypochlorous acid had no effect in
the sperm head abnormality assay.
Tests of the ability of hypochlorite to induce cancer in rodents
were conducted in F344 rats by Hasegawa et al. (1986). Sodium
hypochlorite concentrations of 0, 500 or 1000 mg/litre (males) and 0,
1000 or 2000 mg/litre (females) were administered in the
drinking-water for 104 weeks (equivalent to 13.5 and 27.7 mg/kg of
body weight per day for males and 34.3 and 63.2 mg/kg of body weight
per day for females). There were 50 male and 50 female rats assigned
to each experimental group. No tumours could be attributed to sodium
hypochlorite administration.
NTP (1992) conducted a 2-year bioassay of chlorine in F344 rats
and B6C3F1 mice. The concentrations administered in drinking-water
were 0, 70, 140 or 275 mg/litre, and there were 70 animals of each sex
assigned to each group (approximately 0, 4, 8 or 14 mg/kg of body
weight per day for rats and 0, 7, 14 or 24 mg/kg of body weight per
day for mice). There was an apparent positive trend in the induction
of stromal polyps of the uterus of female mice treated with chlorine,
but this was considered unlikely to be treatment-related because the
incidence was below those observed in historical controls. In female
rats, there was an increase in mononuclear cell leukaemia at both 140
and 275 mg/litre (8 and 14 mg/kg of body weight per day). However, the
response was not considered treatment-related because it fell within
the range of historical controls, there was no apparent dose-response,
and there was no evidence for such an increase in male F344 rats.
A single study suggested that sodium hypochlorite could act as a
promoter of skin tumours following initiation with
4-nitroquinoline-1-oxide in female ddN mice (Hayatsu et al., 1971). A
solution of sodium hypochlorite that contained 10% effective
concentrations of chlorine was utilized. Skin tumours were produced in
9 of 32 mice given 45 applications of sodium hypochlorite following
initiation. Sodium hydroxide solutions were utilized as a control for
the alkaline pH of sodium hypochlorite and produced no tumours. No
tumours were observed with 60 applications of sodium hypochlorite
solution in non-initiated mice. Pfeiffer (1978) conducted a much
larger experiment that utilized 100 mice per group. This author found
that a 1% sodium hypochlorite solution applied alternately with
benzo [a]pyrene for 128 weeks was ineffective in producing skin
tumours in female NMRI mice above those that had been initiated with
benzo [a]pyrene alone at doses of 750 or 1500 µg. Pretreatment with
the sodium hypochlorite solution before application of the
benzo [a]pyrene actually reduced tumour yields at 128 weeks with
doses of either 750 or 1500 µg of benzo [a]pyrene. Sodium
hypochlorite used in a more traditional initiation/promotion study
(i.e., sodium hypochlorite treatment following initiation with
benzo [a]pyrene) produced a decrease in the tumour yield with the 750
µg dose of benzo [a]pyrene, but had no effect following 1500 µg.
Thus, the ability of sodium hypochlorite to act as a tumour promoter
may depend upon the initiator used, or the smaller experiment of
Hayatsu et al. (1971) may simply be a false result.
As pointed out in section 3.1.1, application of solutions of
hypochlorous acid to the skin of Sencar mice results in the
development of hyperplasia. The concentrations required are
considerably lower (300 mg/litre) (Robinson et al., 1986) than those
used in the studies of either Hayatsu et al. (1971) or Pfeiffer
(1978). Sodium hypochlorite was also effective at lower doses, but
less so than equivalent concentrations of hypochlorous acid. These
results suggest that these prior evaluations may have been conducted
at too high a dose. There appear to be no reports on the effectiveness
of hypochlorous acid as a tumour promoter, but the lack of activity at
doses of less than 300 mg/litre would suggest that this is of no
concern.
3.1.5 Comparative pharmacokinetics and metabolism
A series of pharmacokinetic studies using 36Cl-labelled
hypochlorous acid were conducted by Abdel-Rahman and co-workers
(1983). These studies are of limited value because the form of 36Cl
could not be determined in various body compartments.
3.1.6 Mode of action
There are no specific toxicities of chlorine for which a
mechanism needs to be proposed. It is a strong oxidizing agent, and it
must be presumed that damage induced at high doses by either gaseous
chlorine or solutions of hypochlorite is at least partially related to
this property. In studies in which sodium hypochlorite is used without
neutralization, a strong alkaline pH can also contribute to its
effects. There is always the possibility that chlorine is inducing
subtle effects by virtue of its reaction with organic compounds that
are found in the stomach. Such reactions have been demonstrated, but
there is no convincing evidence to date that any specific toxicity can
be attributed to these by-products.
3.2 Chloramine
3.2.1 General toxicological properties and information on
dose-response in animals
There have been relatively few evaluations of the toxic
properties of chloramine in experimental animals. In large part this
is because it is not marketed as a product but is created for
disinfection purposes on-site and in situ. Chloramine is primarily
used as a residual disinfectant in the distribution system. The final
solution consists of mostly monochloramine, with traces of other
chloramines, such as dichloramine. Chloramines, as a group, are
generally recognized as potent respiratory irritants, because the
formation of these compounds when household bleach and ammonia are
mixed results in a number of poisoning cases each year (MMWR, 1991).
In spite of this, there has been no attempt to quantify dose-response
relationships in animals.
Eaton et al. (1973) investigated concerns about
chloramine-induced methaemoglobin formation in kidney patients
dialysed with chloramine-containing water. This was done by examining
the ability of relatively large volumes of tapwater to oxidize
haemoglobin in dilute suspensions of red blood cells. This
circumstance is reflective of dialysis, but not of the concentrated
suspension of these cells in vivo. Nevertheless, the authors were
able to show that methaemoglobin formation occurred in a dose-related
manner when 1 volume of red blood cells (human) was suspended in 100
volumes of tapwater containing 1 mg of chloramine per litre or above.
This effect was not produced by comparable concentrations of sodium
hypochlorite. The ability to induce methaemoglobin formation was
eliminated by treating the water by reverse osmosis followed by carbon
filtration. Clearly, chloramine is capable of inducing
methaemoglobinaemia at low concentrations when there is a large
reservoir of chloramine. This is a decidedly different exposure
pattern from that of normal humans and other mammals, as they consume
small volumes of water relative to the volume of red blood cells that
are exposed.
Moore et al. (1980a) studied alterations of blood parameters in
male A/J mice treated with 0, 2.5, 25, 50, 100 or 200 mg of
monochloramine per litre in carbonate/bicarbonate-buffered (pH 8.9)
drinking-water. Twelve animals were assigned to each group, and
treatments were maintained over a 30-day period. Consistent with the
interpretation provided above, there were no treatment-related effects
on osmotic fragility, methaemoglobin levels, haemoglobin
concentrations, reticulocyte counts or a number of other derived
parameters. Haematocrits of mice treated with 50, 100 or 200 mg/litre
were actually higher than those observed in control mice. White blood
cell counts were not altered in these animals.
Daniel et al. (1990a) conducted a more traditional 90-day study
of monochloramine in Sprague-Dawley rats (10 animals per sex per
dose). Treatment concentrations were 0, 25, 50, 100 or 200 mg/litre,
corresponding to doses of 0, 1.8, 3.4, 5.8 or 9.0 mg/kg of body weight
per day in males and 0, 2.6, 4.3, 7.7 or 12.1 mg/kg of body weight per
day in females. Controls received carbonated, pH-adjusted
drinking-water. A large number of haematological and clinical
chemistry measures were included in the evaluation. Body weights were
significantly depressed in both sexes at treatment concentrations in
the 50-200 mg/litre range, but this appeared to be related to
depressed water and food consumption. There were minor changes in
organ to body weight ratios at the highest dose, but no evidence of
treatment-related pathology was observed. Male rats were found to have
decreased haematocrits at 100 mg/litre, and red blood cell counts were
slightly depressed at 100 and 200 mg/litre. The authors concluded that
monochloramine was more toxic than chlorine or chlorine dioxide.
However, it must be noted that the changes in blood parameters were
small and in themselves of no clinical significance. Other measures
were not related to specific toxic reactions. Based on the decrease in
organ and body weights observed in both sexes, the authors concluded
that the no-observed-adverse-effect level (NOAEL) was 100 mg/litre,
equivalent to 5.8 mg/kg of body weight per day.
The work in rats was followed up with a second study in B6C3F1
mice (Daniel et al., 1991a). Male and female B6C3F1 mice (10 per sex
per group) were administered 0, 12.5, 25, 50, 100 or 200 mg of
chloramine per litre of drinking-water for 90 days (calculated mean
daily dose was 0, 2.5, 5.0, 8.6, 11.1 or 15.6 mg/kg of body weight for
males and 0, 2.8, 5.3, 9.2, 12.9 or 15.8 mg/kg of body weight for
females). Water consumption significantly decreased at 100 and 200
mg/litre in males and at 25-200 mg/litre in females. Weight gain was
significantly depressed in both sexes at 100 and 200 mg/litre.
Neutrophil concentrations in blood were significantly depressed in
both male and female mice at the two highest doses, but other white
blood cell counts were unaltered. Absolute and relative spleen and
liver weights were depressed at both 100 and 200 mg/litre. No gross or
microscopic evidence of target organ toxicity was observed that could
be related to treatment. Based on decreased organ weights, weight
gain, and food and water consumption, the authors concluded that the
NOAEL was 50 mg/litre, equivalent to 8.6 mg/kg of body weight per day.
A 13-week study in which groups of 10 Sprague-Dawley rats were
given drinking-water containing 200 mg of monochloramine per litre or
buffered water as a control, ad libitum or restricted to the same
consumption as the monochloramine group, was designed to resolve some
of the outstanding toxicological questions. The results of this study
indicated that the reduced body weight gain and the minor biochemical,
haematological, immunological and histopathological changes associated
with exposure to 200 mg of monochloramine per litre (equivalent to
21.6 mg/kg of body weight per day) in drinking-water were largely
related to reduced water and food consumption (Poon et al., 1997).
In a 9-week study, Exon et al. (1987) examined the ability of
monochloramine to modify immunological parameters in male
Sprague-Dawley rats (12 per dose) exposed to concentrations of 0, 9,
19 or 38 mg of monochloramine per litre, equivalent to 0, 1.3, 2.6 or
5.3 mg/kg of body weight per day. At the middle and highest dose,
chloramine treatment was observed to increase prostaglandin E2
synthesis by adherent resident peritoneal cells (which include
macrophages) in response to lipopolysaccharide stimulation. No attempt
was made to relate this finding to other indices of modified
macrophage function. A small depression in spleen weights was observed
at the highest dose. The implications of these data for immune
function are not clear. Other measures of immune function did not
reveal statistically significant changes with treatment. These
included a decrease in antibody formation at the lowest and middle
doses in response to keyhole limpet haemocyanin injection or
delayed-type hypersensitivity reactions to bovine serum albumin
injected into the footpad.
The effect of monochloramine on skin irritation was tested by
immersing Sencar mice into water containing chloramine at
concentrations ranging from 1 to 1000 mg/litre for 10 min a day
(Robinson et al., 1986). Unlike hypochlorous acid (pH 6.5) or
hypochlorite (pH 8.5), chloramine did not produce hyperplasia of the
skin.
3.2.2 Reproductive and developmental toxicity
Studies in laboratory animals have indicated no reproductive or
developmental effects associated with chloramine. Abdel-Rahman et al.
(1982a) administered monochloramine to female Sprague-Dawley rats at
concentrations of 0, 1, 10 or 100 mg/litre (0, 0.15, 1.5 or 15 mg/kg
of body weight per day) for 2.5 months prior to breeding and through
gestation. Only six animals were assigned to each treatment group.
Reproductive performance was comparable between groups, and fetal
weights were not adversely affected by treatment. Between 50 and 60
fetuses were available (male and female combined) for evaluation.
There was no evidence of treatment-related skeletal or soft tissue
anomalies.
Carlton et al. (1986) examined the effects of monochloramine
administered by gavage at doses of 0, 2.5, 5 or 10 mg/kg of body
weight per day on the reproductive performance of Long-Evans rats.
Males (12 per group) were treated from 56 days prior to and through
mating, and females (24 per group) from 14 days prior to mating and
throughout the mating period. No statistically significant effects on
sperm morphology, concentration or motility were observed, nor were
there any effects on fertility, viability, litter size, pup weights,
day of eye opening or day of vaginal patency.
3.2.3 Toxicity in humans
The primary harmful effects of chloramine have been documented in
humans poisoned by chloramine formed when household bleach was mixed
with ammonia for use as a cleaning solution. Chloramine is a strong
respiratory irritant. These effects were discussed in section 3.1.3.
Forty-eight men completed an 8-week protocol during which diet
and other factors known to affect lipid metabolism were controlled.
During the first 4 weeks of the protocol, all subjects consumed
distilled water. During the second 4 weeks, one-third of the subjects
were assigned randomly to drink 1.5 litres of water containing 0, 2 or
15 mg of monochloramine per litre each day. At 2 mg/litre, no
significant changes were observed in total, HDL or LDL cholesterol,
triglycerides or apolipoproteins A1, A2 or B. Parameters of thyroid
function were unchanged. However, an increase in the level of
apolipoprotein B was observed at 15 mg/litre (Wones et al., 1993b).
3.2.4 Carcinogenicity and mutagenicity
Shih & Lederberg (1976) first demonstrated that monochloramine
induced mutation in a Bacillus subtilis reversion assay. The
concentration range studied extended from 18 to 74 µmol/litre. A
positive dose-response was observed through 56 µmol/litre, but 74
µmol/litre was cytotoxic. Repair-deficient mutants of B. subtilis,
rec3, recA and polyA5, were consistently more sensitive to the
cytotoxic effects of chloramine, while the uvr and recB mutants were
not. The sensitivity of the polyA5 mutants parallels the observations
of Rosenkranz (1973) with sodium hypochlorite and suggests that DNA
polymerase A is involved in the repair of DNA lesions produced by both
chemicals. Thus, it is possible that a common intermediate or
mechanism is involved in the mutagenic effects of hypochlorite and
chloramine.
A broader list of chloramines was tested by Thomas et al. (1987)
in Salmonella typhimurium tester strains TA97a, TA100 and TA102. The
chloramines tested included those that could be formed at low levels
from natural substrates in drinking-water or in the stomach. TA100 was
found to consistently be the most sensitive strain. The most potent
mutagens were the lipophilic dichloramines formed with histamine,
ethanolamine and putrescine. The corresponding monochloramines were
less potent. The more hydrophilic chloramines, such as
taurine-chloramine, had little activity. Monochloramine was active in
the 50 µmol/litre range, remarkably consistent with the data of Shih &
Lederberg (1976). Hypochlorous acid was inactive at all concentrations
that were tested, up to and including concentrations that induced
cytotoxicity.
Ashby and co-workers (1987) were unable to induce clastogenic
effects in the mouse bone marrow micronucleus assay when chloramine
was administered orally. They suggested that the in vitro
clastogenic effects were probably attributable to non-specific
cytotoxic effects that are secondary to the release of hypochlorite to
the media.
Meier et al. (1985b) found that intraperitoneal administration of
monochloramine to CD-1 mice at doses of up to 8 mg/kg of body weight
was without significant effect on either micronuclei or chromosomal
aberrations in the bone marrow. These data would appear to be
consistent with the findings of Ashby et al. (1987).
Studies on the carcinogenicity of chloramine are limited to a
single set of 2-year experiments conducted by the NTP (1992).
Drinking-water containing 0, 50, 100 or 200 mg of chloramine per litre
was provided to F344 rats and B6C3F1 mice. Seventy animals of each
species and of each sex within a species were assigned to each
experimental and control group. Doses in rats were 0, 2.1, 4.8 or
8.7 mg/kg of body weight per day in males and 0, 2.8, 5.3 or 9.5 mg/kg
of body weight per day in females; doses in mice were 0, 5.0, 8.9 or
15.9 mg/kg of body weight per day in males and 0, 4.9, 9.0 or
17.2 mg/kg of body weight per day in females. Of some interest was the
finding that two renal cell adenomas were found in male B6C3F1 mice
treated with the high dose of chloramine. In addition, one renal
adenoma was found in one male mouse treated with 100 mg/litre and in
one female mouse treated with 200 mg/litre. While this tumour site is
rare in both species, there was no real dose-response trend, nor were
the differences between the control and treatment groups statistically
significant. A second finding of some concern was an increase in the
incidence of mononuclear cell leukaemia in F344 rats. This pathology
was increased in rats treated with chloramine or hypochlorite,
although the effects were not clearly dose-dependent. The incidence of
mononuclear cell leukaemia was significantly greater than in
concurrent controls and was elevated above the historical incidence as
well. Nevertheless, these increases were not considered to be
treatment-related. In part, this conclusion arose from the lack of a
clear dose-response. It was also based on the fact that there was no
comparable trend in male rats.
3.2.5 Comparative pharmacokinetics and metabolism
The pharmacokinetics of 36Cl derived from monochloramine have
been examined in male Sprague-Dawley rats (Abdel-Rahman et al., 1983).
These data are difficult to interpret because the specific form of the
label is not known.
3.3 Chlorine dioxide
3.3.1 General toxicological properties and information on
dose-response in animals
Despite its use as a disinfectant, there have been very few
general toxicological evaluations of chlorine dioxide, because most
studies have focused on its major by-product, chlorite, which is
considered in section 4.6. The present review will first focus on the
limited characterization of chlorine dioxide's general toxicology,
then follow up with a discussion of its haematological and thyroid
effects.
Some very cursory investigations of chlorine dioxide's effects as
a respiratory irritant were published by Haller & Northgraves (1955)
in an article dealing with the general chemical properties of the
compound. In essence, these data suggested that exposure to chlorine
dioxide in air at a concentration of more than 420 mg/m3 (150 ppm)
for longer than 15 min was fatal to guinea-pigs. The total study
involved six guinea-pigs.
Rats (3-5 per group) were exposed to chlorine dioxide in various
concentrations (0.28-9520 mg/m3 [0.1-3400 ppm]) and for various
periods (3 min-10 weeks). All the rats exposed daily to chlorine
dioxide at 28 mg/m3 (10 ppm) died in less than 14 days. Purulent
bronchitis and disseminated bronchopneumonia were found at necroscopy.
No such changes were demonstrable in rats exposed to approximately
0.28 mg/m3 (0.1 ppm) for about 10 weeks (Dalhamn, 1957).
The LC50 of chlorine dioxide in rats (5 per sex per group)
exposed by inhalation for 4 h was 90 mg/m3 (32 ppm) (Ineris, 1996).
Robinson et al. (1986) studied the ability of chlorine and
alternative disinfectants to induce epidermal hyperplasia in the skin
of Sencar mice. The thickness of the interfollicular epidermis was
significantly increased by 10-min daily exposures to water containing
up to 1000 mg of chlorine dioxide per litre for 4 days. The thickness
of the epidermis was similar to that induced by an equivalent dose of
hypochlorous acid. Unlike hypochlorous acid, however, there was no
significant increase in skin thickness at concentrations of 300
mg/litre or less.
The study of Daniel et al. (1990a) was the first subchronic study
that adhered to modern expectations of toxicological studies. These
authors provided male and female Sprague-Dawley rats (10 per sex per
treatment group) with 0, 25, 50, 100 or 200 mg of chlorine dioxide per
litre of drinking-water for 90 days, equivalent to 0, 2, 4, 6 or 12
and 0, 2, 5, 8 or 15 mg/kg of body weight per day for males and
females, respectively. Conventional measures of body weight, organ
weights, a broad battery of clinical chemistry parameters and
histopathological examinations were all included in the study design.
Body and organ weights were significantly depressed at 200 mg/litre in
both sexes. This appeared to be secondary to depressed water
consumption, which is known to be tightly coupled to food consumption
in rats. The only significant histopathological damage found was
goblet cell hyperplasia and inflammation. This was observed at all
doses of chlorine dioxide in both male and female rats. Presumably
this inflammation occurs as a result of volatilization of chlorine
dioxide from the water bottle. The amount of chlorine dioxide actually
inhaled as a result of volatilization from the drinking-water
containing the lowest dose of chlorine dioxide (25 mg/litre) must be
extremely low. This suggests that there might be some concern for
sensitive individuals showering with water containing chlorine
dioxide.
The ability of chlorine dioxide to induce methaemoglobinaemia and
haemolytic anaemia has received extensive study. Abdel-Rahman et al.
(1980) found decreased red blood cell glutathione (GSH) concentrations
and decreased osmotic fragility in Sprague-Dawley rats and white
leghorn chickens given drinking-water containing chlorine dioxide
concentrations of 1, 10, 100 and 1000 mg/litre for up to 4 months, but
the changes were not consistently dose-related. However, the authors
found that the morphology of red blood cells was modified (codocytes
and echinocytes) in all dose groups, the severity increasing with
increased treatment concentration. Methaemoglobin was not detected
throughout these studies. However, there was no formal statistical
evaluation of these results, and only four rats were assigned to each
experimental group. Administration of acute doses of as little as 1
mg/kg of body weight by gavage decreased red blood cell GSH
concentrations. This response was not increased as dose was increased
to 4 mg/kg of body weight.
Abdel-Rahman and co-workers extended these observations to longer
treatment periods in a subsequent publication (Abdel-Rahman et al.,
1984a). Again, only four animals were assigned to each treatment
group. At 7 and 9 months of treatment, red blood cells appeared to
become resistant to osmotic shock at all treatment concentrations
(1-1000 mg/litre). These data did not display a clear dose-response
despite the large variation in the dose administered.
The study of Abdel-Rahman et al. (1984a) also reported changes in
the incorporation of 3H-thymidine into the DNA of various organs.
Incorporation was significantly inhibited in testes and apparently
increased in the intestinal mucosa. The effect on apparent DNA
synthesis was particularly marked in the testes at 100 mg/litre,
amounting to about 60% inhibition. These data are difficult to
interpret for several reasons. First, rats were sacrificed 8 h after
being injected with 3H-thymidine. Ordinarily, sacrifices are made
30-60 min after injection because the blood is essentially depleted of
3H-thymidine in an hour. Thus, it is not possible to determine if the
lowered amount of label is related to decreased synthesis or to
increased turnover of DNA. Second, the result was based on total
counts in DNA, which makes it impossible to determine what cell type
is affected or whether the change was associated with replicative or
repair synthesis. Third, only four animals were used per experimental
group.
In a rising-dose protocol study, Bercz et al. (1982) evaluated
the effects of chlorine dioxide on African green monkeys. These
animals were provided chlorine dioxide in drinking-water at
concentrations of 0, 30, 100 or 200 mg/litre, corresponding to doses
of 0, 3.5, 9.5 or 11 mg/kg of body weight per day. Each dose was
maintained for 30-60 days. Animals showed signs of dehydration at the
highest dose (11 mg/kg of body weight per day), so exposure at that
dose was discontinued. No effect was observed on any haematological
parameter, including methaemoglobinaemia. However, statistically
significant depressions in serum thyroxine levels were observed when
animals were dosed with chlorine dioxide at a concentration of
100 mg/litre. No effect had been observed in a prior exposure of the
same animals to 30 mg/litre for 30 days. The NOAEL in this study was
3.5 mg/kg of body weight per day.
The effects of chlorine dioxide on thyroid function were followed
up by Harrington et al. (1986). Thyroxine levels in African green
monkeys administered drinking-water containing 100 mg of chlorine
dioxide per litre (4.6 mg/kg of body weight per day) were again found
to be depressed at 4 weeks of treatment, but rebounded to above-normal
levels after 8 weeks of treatment. These investigators also found
significantly depressed thyroxine levels in rats treated with 100 or
200 mg of chlorine dioxide per litre in drinking-water (equivalent to
14 and 28 mg/kg of body weight per day) for 8 weeks. This change was
dose-related. Lower doses were not examined in the rat study. The
authors indicated that the results were based on 12 determinations; it
was not clear if these measurements were made on individual animals.
A set of in vivo and in vitro experiments was conducted in an
attempt to explain the effects of chlorine dioxide on serum thyroid
hormone concentrations. The authors demonstrated that chlorine dioxide
oxidizes iodide to reactive iodine species that would bind to the
stomach and oesophageal epithelium. Rat chow that was treated with
chlorine dioxide at approximately 80 mg/litre was found to increase
the binding of iodine to chow constituents. This activation of iodine
resulted in retention of labelled iodine in the ileum and colon and
reduced uptake by the thyroid gland. Previous work demonstrated that
chlorine dioxide was more effective than chlorine in activating iodide
to a form that would covalently bind with a variety of natural
foodstuffs (Bercz et al., 1986). Based on these observations, the
authors concluded that the effects of chlorine dioxide were probably
due to altered gastrointestinal absorption of iodide and reduced
uptake into the thyroid gland.
3.3.2 Reproductive and developmental toxicity
A number of reproductive effects have been reported in studies
with laboratory animals, but the relevance for humans of these
findings remains uncertain. The reproductive effects of chlorine
dioxide in Long-Evans rats were studied by Carlton et al. (1991).
Chlorine dioxide was administered by gavage at doses of 2.5, 5 or 10
mg/kg of body weight per day to male rats (12 per group) for 56 days
prior to and through mating and to female rats (24 per group) from 14
days prior to mating and through pregnancy. Fertility measures were
not significantly different among the dose groups. There were no
dose-related changes in sperm parameters (i.e., concentration,
motility, progressive movement or morphology). Thyroid hormone levels
were altered significantly, but not in a consistent pattern. The only
significant difference was significantly depressed vaginal weights in
female pups whose dams had been treated with 10 mg/kg of body weight
per day.
An evaluation of the effects of chlorine dioxide on the fetal
development of Sprague-Dawley rats was conducted by Suh et al. (1983).
Chlorine dioxide was administered at 0, 1, 10 or 100 mg/litre (0, 0.1,
1 or 10 mg/kg of body weight per day) for 2.5 months prior to mating
and throughout gestation. The total number of implants per dam was
significantly reduced at the highest concentration of chlorine
dioxide. The percentage of anomalous fetuses was increased in a
dose-related manner, but the response was not statistically
significant. These anomalies arose primarily as the percentage of
abnormal or incomplete sternebrae in treated rats relative to
controls. The lack of statistical significance was undoubtedly related
to the relatively few female rats that were included in the study (6-8
females per treatment group). As a consequence, the results of this
study must be considered inconclusive.
Orme et al. (1985) found that chlorine dioxide administered in
the drinking-water of female Sprague-Dawley rats (13-16 per dose) at
concentrations of 0, 2, 20 or 100 mg/litre (0, 1, 3 or 14 mg/kg of
body weight per day) throughout pregnancy and through weaning
decreased thyroxine levels in the serum of the pups at 100 mg/litre.
This was associated with delayed development of exploratory behaviour
in the pups away from their dams, and this, in turn, was probably due
to an indirect effect on iodine uptake. In a second experiment, pups
given 14 mg of chlorine dioxide per kg of body weight per day directly
by gavage on postnatal days 5-20 showed significantly depressed
activity and a decrease in serum thyroxine levels. Studies by the same
group (Taylor & Pfohl, 1985) indicated that cerebellar and forebrain
cell counts (based on DNA measurements) were depressed in 11-day-old
pups that had been treated with chlorine dioxide at 14 mg/kg of body
weight per day by gavage from 5 days of age. Cerebellar cell counts
remained depressed in rats at 21 days, but forebrain counts were
essentially the same as in controls. At 50-60 days of age, the
locomotor activity (measured by wheel-running) of these animals was
depressed relative to control animals.
The effects of chlorine dioxide on brain development were
examined further by Toth et al. (1990). These authors administered
chlorine dioxide by gavage at 14 mg/kg of body weight per day from
postnatal day 1 to 20. Body weight was reduced, but cerebellar weight
was unaltered at any age. Forebrain weight and protein content were
reduced on postnatal days 21 and 35. DNA content was depressed on
postnatal day 35, and the number of dendritic spines on cerebral
cortical pyramidal cells was significantly reduced. No
histopathological changes in the forebrain, cerebellum or brain stem
were observed. There were no consistent changes in serum thyroxine or
triiodothyronine levels in treated animals.
Collectively, these data suggest some effects of chlorine dioxide
on brain development. In most studies, there are suggestions of
modified thyroid function associated with these effects. It must be
pointed out that the changes in thyroid hormone levels are modest,
much less than are produced with classical antithyroid drugs such as
propylthiouracil (Toth et al., 1990).
3.3.3 Toxicity in humans
The effects of chlorine dioxide were assessed in a two-phase
study in 10 healthy male volunteers. The first study was a rising-dose
tolerance study (Lubbers & Bianchine, 1984) in which doses of chlorine
dioxide were increased from 0.1 to 24 mg/litre, administered in two
500-ml portions. The maximum dose for a 70-kg person was 0.34 mg/kg of
body weight. The details of this study were described in section
3.1.3. Some small changes in a variety of clinical chemistry
parameters were observed, but none was found to be outside the
accepted range of normal. The second phase of the experiment involved
the daily administration of a 500-ml portion of a solution containing
5 mg/litre to 10 healthy volunteers for a period of 12 weeks (Lubbers
et al., 1984a). Again, measurement of a large battery of clinical
chemistry parameters and routine physical examination failed to
identify any effects of chlorine dioxide that fell outside of the
normal range. Parameters yielding significant differences appeared to
be primarily a result of parallel drift of values with the control
group.
As with other disinfectants, it is important to recognize that
chlorine dioxide is a potent respiratory irritant. No quantitative
data can be used to construct a dose-response relationship for this
effect.
3.3.4 Carcinogenicity and mutagenicity
The mutagenic or clastogenic effects of chlorine dioxide have
received little attention. Ishidate et al. (1984) found chlorine
dioxide to be positive in Salmonella typhimurium tester strain
TA100. A linear dose-response was observed at concentrations between 2
and 20 µg per plate. Chlorine dioxide was ineffective as a clastogenic
agent in a CHO system.
Meier et al. (1985b) evaluated the ability of chlorine dioxide to
induce chromosomal aberrations and micronuclei in bone marrow of CD-1
mice or sperm head anomalies. Chlorine dioxide failed to produce such
damage following gavage doses of up to 16 mg/kg of body weight for 5
days.
With the exception of a 1949 study by Haag (cited in TERA, 1998),
which has serious limitations, no tests of the carcinogenic activity
of chlorine dioxide in experimental animals were identified in the
scientific literature.
3.3.5 Comparative pharmacokinetics and metabolism
There are significant differences in the pharmacokinetics of
36Cl obtained from different disinfectants. The absorption rate for
36Cl-labelled chlorine dioxide was at least 10 times that observed
with chlorine, chloramine or chloride. The relative amount of 36Cl
that is eliminated in the urine and faeces at 24 h has a distinct
pattern from that observed with other disinfectants and sodium
chloride. However, the terminal half-life of the 36Cl appears similar
for all disinfectants. These data suggest that the form of 36Cl that
is being absorbed differs chemically with the different disinfectants.
In the case of chlorine dioxide, this is supported by the observation
that measurable amounts of chlorite (about 3% of the original dose)
are eliminated in the urine during the first 24 h, and chlorite
comprises about 20% of the label present in blood 72 h after
administration of the test dose (Abdel-Rahman et al., 1980). However,
this higher absorption rate is not explained by the absorption rates
of chlorite and chlorate, which are about one-tenth as rapid
(Abdel-Rahman et al., 1982b). This suggests that some of the
absorption could be as chlorine dioxide itself. On the surface, this
hypothesis would seem to be incompatible with the high reactivity of
this disinfectant. As with other disinfectants, the terminal
elimination phases observed for 36Cl from chlorine dioxide seem
compatible with the hypothesis that the bulk of the elimination is as
chloride ion.
4. TOXICOLOGY OF DISINFECTANT BY-PRODUCTS
4.1 Trihalomethanes
As a class, the THMs are generally the most prevalent by-products
of drinking-water disinfection by chlorine. A variety of
non-neoplastic toxic effects have been associated with short-term and
long-term exposure of experimental animals to high doses of THMs, and
each of the four most common THMs -- chloroform, BDCM, DBCM and
bromoform -- has been shown to be carcinogenic to rodents in high-dose
chronic studies. Chloroform is generally the predominant THM in
chlorinated water and is also the most extensively studied chemical of
this class. Because the World Health Organization (WHO) recently
published an Environmental Health Criteria monograph on chloroform
(IPCS, 1994), this section will only update the information contained
in that publication. A thorough review of findings relevant to the
toxicology of the brominated THMs is included.
As with the other DBPs, the chemical and physical properties of
the THMs influence their potential routes of human exposure, their
pharmacokinetic behaviour, their toxicity and methods for conducting
toxicological studies with these compounds. The THMs are volatile
liquids at room temperature; therefore, as these chemicals vaporize
during water usage (e.g., showering), inhalation becomes an important
exposure route in addition to ingestion. Volatility decreases somewhat
with bromine substitution, but each of the brominated THMs evaporates
from drinking-water. Like that of other alkanes, the water solubility
of THMs is poor, although adequate to permit dissolution of the low
levels generated via water disinfection. When administered at higher
levels to animals in toxicity experiments, THMs are often either
emulsified in aqueous solutions or dissolved in oils. The use of oils
as vehicles of administration can significantly alter the
pharmacokinetics and toxicity of the THMs. Bromine substitution
enhances the lipid solubility of the halomethanes (and, consequently,
uptake into tissues) and generally increases their chemical reactivity
and the likelihood of biotransformation to a reactive intermediate.
Because toxicity is dependent upon a reactive compound actually
reaching a sensitive target site, greater bromine substitution may not
necessarily translate into greater in vivo toxicity (i.e., innocuous
reactions may occur, preventing arrival at target sites). Perhaps to a
greater extent than with other chemicals in this class, BDCM appears
to reach a variety of target tissues where it can be readily
metabolized to several intermediates, leading to adverse effects in
experimental animals.
4.1.1 Chloroform
In 1994, the WHO published an Environmental Health Criteria
monograph on chloroform (IPCS, 1994). The following sections will
update that document with the most recent findings from health-related
chloroform research. Another evaluation of chloroform was included in
the 1998 Addendum to the WHO Guidelines for drinking-water quality
(WHO, 1998).
4.1.1.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
Keegan et al. (1998) determined the
lowest-observed-adverse-effect level (LOAEL) and NOAEL for the
induction of acute hepatotoxicity following oral administration of
chloroform in an aqueous vehicle to male F344 rats. Based on
elevations of serum clinical chemistry indicators of liver damage, a
LOAEL of 0.5 mmol/kg of body weight (60 mg/kg of body weight) and a
NOAEL of 0.25 mmol/kg of body weight (30 mg/kg of body weight) were
established. In a corn oil gavage study of single-dose chloroform
effects, an increase in renal cell proliferation was observed at doses
as low as 10 mg/kg of body weight in male Osborne-Mendel rats and
90 mg/kg of body weight in male F344 rats (Templin et al., 1996a). The
only increase in the hepatic labelling index was in F344 rats given
477 mg/kg of body weight. Effects in the nasal passages of both rat
strains at 90 mg/kg of body weight and above included oedema and
periosteal hypercellularity. Gemma et al. (1996) dosed male B6C3F1
mice with chloroform (150 mg/kg of body weight) by gavage and observed
increases in cell proliferation in both the liver and kidneys. The
effect was more dramatic in the kidneys, where severe necrosis was
also noted.
Nephrotoxicity of chloroform was evaluated in male Sprague-Dawley
rats treated orally with single doses of chloroform using corn oil or
an aqueous preparation (5%) of Emulphor or Tween 85 as vehicle (10
ml/kg of body weight). Comparison between gavage vehicles indicated
clear trends for enhanced potency and severity of nephrotoxic effects
with corn oil administration of chloroform (Raymond & Plaa, 1997).
2) Short-term toxicity
Chloroform was administered by corn oil gavage to male B6C3F1
mice at doses of 0, 34, 90, 138 or 277 mg/kg of body weight for 4 days
or 3 weeks (5 days per week) (Larson et al., 1994a). Mild degenerative
changes in centrilobular hepatocytes were noted in mice given 34 and
90 mg/kg of body weight per day after 4 days of treatment, but these
effects were absent at 3 weeks. At 138 and 277 mg/kg of body weight
per day, centrilobular necrosis was observed at 4 days and with
increased severity at 3 weeks. Hepatic cell proliferation was
increased in a dose-dependent manner at all chloroform doses after 4
days, but only in the 277 mg/kg of body weight dose group at 3 weeks.
Renal tubular necrosis was observed in all dose groups after 4 days,
while 3 weeks of exposure produced severe nephropathy at the highest
dose and regenerating tubules at the lower doses. The nuclear
labelling index was increased in the proximal tubules at all doses
after 4 days of treatment, but was elevated only in the two highest
dose groups after 3 weeks.
In a similar study (Larson et al., 1994b), female B6C3F1 mice
were administered chloroform dissolved in corn oil by gavage at doses
of 0, 3, 10, 34, 238 or 477 mg/kg of body weight per day for 4 days or
3 weeks (5 days per week). Dose-dependent changes included
centrilobular necrosis and markedly elevated labelling index in mice
given 238 and 477 mg/kg of body weight per day. The NOAEL for
histopathological changes was 10 mg/kg of body weight per day, and for
induced cell proliferation, 34 mg/kg of body weight per day.
In an inhalation study, Templin et al. (1996b) exposed BDF1 mice
to chloroform vapour at concentrations of 0, 149 or 446 mg/m3 (0, 30
or 90 ppm) 6 h per day for 4 days or 2 weeks (5 days per week). In the
kidneys of male mice exposed to 149 and 446 mg/m3 (30 and 90 ppm),
degenerative lesions and 7- to 10-fold increases in cell proliferation
were observed. Liver damage and an increased hepatic labelling index
were noted in male mice exposed to 149 and 446 mg/m3 (30 and 90 ppm)
and in female mice exposed to 446 mg/m3 (90 ppm). Both doses were
lethal in groups exposed for 2 weeks (40% and 80% mortality at 149 and
446 mg/m3 [30 and 90 ppm], respectively).
Female F344 rats were given chloroform by corn oil gavage for 4
consecutive days or 5 days per week for 3 weeks (Larson et al.,
1995b). In the liver, mild degenerative centrilobular changes and
dose-dependent increases in hepatocyte proliferation were noted at
doses of 100, 200 and 400 mg/kg of body weight per day. At 200 and
400 mg/kg of body weight per day, degeneration and necrosis of the
renal cortical proximal tubules were observed. Increased regenerative
proliferation of epithelial cells lining proximal tubules was seen at
doses of 100 mg/kg of body weight per day or more. Lesions of the
olfactory mucosa lining the ethmoid region of the nose (new bone
formation, periosteal hypercellularity and increased cell replication)
were seen at all doses, including the lowest dose of 34 mg/kg of body
weight per day. Larson et al. (1995a) also administered chloroform to
male F344 rats by corn oil gavage (0, 10, 34, 90 or 180 mg/kg of body
weight per day) or in the drinking-water (0, 60, 200, 400, 900 or
1800 mg/litre) for 4 days or 3 weeks. Gavage of 90 or 180 mg/kg of
body weight per day for 4 days induced mild to moderate degeneration
of renal proximal tubules and centrilobular hepatocytes -- changes
that were no longer present after 3 weeks. Increased cell
proliferation in the kidney was noted only at the highest gavage dose
after 4 days. The labelling index was elevated in the livers of the
high-dose group at both time points. With drinking-water
administration, rats consuming the water containing 1800 mg/litre were
dosed at a rate of 106 mg/kg of body weight per day, but no increase
in renal or hepatic cell proliferation was observed at this or any
lower dose.
In a study carried out to evaluate whether exposure to chloroform
in drinking-water would interact with the activity of chloroform when
administered by gavage in corn oil, female B6C3F1 mice were exposed
to chloroform in drinking-water for 33 days at 0, 300 or 1800 mg/litre
or for 31 days at 0, 120, 240 or 480 mg/litre. Three days prior to
termination, mice also received a daily dose of 263 mg of chloroform
per kg of body weight per day, administered by gavage in corn oil.
Exposure to chloroform in drinking-water reduced both the
hepatotoxicity and the enhanced cell proliferation elicited in
response to chloroform administered by gavage in corn oil (Pereira &
Grothaus, 1997).
The cardiotoxicity of chloroform was examined in male Wistar rats
given daily doses of 37 mg/kg of body weight (0.31 mmol/kg) by gavage
in olive oil for 4 weeks (Muller et al., 1997). Chloroform caused
arrhythmogenic and negative chronotropic and dromotropic effects as
well as extension of the atrioventricular conduction time and
depressed myocardial contractility.
A 90-day chloroform inhalation study was conducted using male and
female B6C3F1 mice and exposure concentrations of 0, 1.5, 10, 50, 149
and 446 mg/m3 (0, 0.3, 2, 10, 30 and 90 ppm) for 6 h per day, 7 days
per week (Larson et al., 1996). Large, sustained increases in
hepatocyte proliferation were seen in the 446 mg/m3 (90 ppm) groups
at all time points (4 days and 3, 6 and 13 weeks). In the more
sensitive female mice, a NOAEL of 50 mg/m3 (10 ppm) for this effect
was established. Renal histopathology and regenerative hyperplasia
were noted in male mice at 50, 149 and 446 mg/m3 (10, 30 and 90 ppm).
In another 90-day inhalation study, F344 rats were exposed to
chloroform at concentrations of 0, 10, 50, 149, 446 or 1490 mg/m3 (0,
2, 10, 30, 90 or 300 ppm) for 6 h per day, 7 days per week. The 1490
mg/m3 (300 ppm) level was extremely toxic and deemed by the authors
to be inappropriate for chronic studies. Increases in renal epithelial
cell proliferation in cortical proximal tubules were observed at
concentrations of 149 mg/m3 (30 ppm) and above. Hepatic lesions and
increased proliferation were noted only at the highest exposure level.
In the ethmoid turbinates of the nose, enhanced bone growth and
hypercellularity in the lamina propria were observed at concentrations
of 50 mg/m3 (10 ppm) and above, and a generalized atrophy of the
turbinates was seen at all exposure levels after 90 days (Templin et
al., 1996c).
3) Reproductive and developmental toxicity
Rat embryo culture was used to assess the developmental effects
of chloroform (Brown-Woodman et al., 1998). The effect and
no-effect culture medium concentrations of chloroform were 2.06 and
1.05 µmol/ml. The authors estimated that fatal or near-fatal blood
levels would be required in the mother for the embryotoxic level to be
reached.
4.1.1.2 Toxicity in humans
Fatal acute chloroform intoxication via inhalation was reported
to cause cardiomyocyte fragmentation and waviness indicative of acute
heart failure possibly caused by arrhythmias or cardiac depression
(Harada et al., 1997). These observations are consistent with the
results of the short-term rat study (Templin et al., 1996b) described
above in section 4.1.1.1.
4.1.1.3 Carcinogenicity and mutagenicity
Jamison et al. (1996) reported that F344 rats exposed to a high
concentration of chloroform vapour (1490 mg/m3 [300 ppm]) for 90 days
developed atypical glandular structures lined by intestinal-like
epithelium and surrounded by dense connective tissue in their livers.
These lesions appeared to arise from a population of cells remote from
the bile ducts. The authors also observed a treatment-related increase
in transforming growth factor-alpha (TGF-alpha) immunoreactivity in
hepatocytes, bile duct epithelium, bile canaliculi and oval cells and
an increase in transforming growth factor-beta (TGF-ß)
immunoreactivity in hepatocytes, bile duct epithelium and intestinal
crypt-like ducts. The lesions occurred only in conjunction with
significant hepatocyte necrosis, regenerative cell proliferation and
increased growth factor expression or uptake.
Chloroform was tested for mutagenicity and clastogenicity by Le
Curieux et al. (1995) and was negative in the SOS chromotest, the Ames
fluctuation test and the newt micronucleus test. It appeared to these
authors that the presence of bromine substituents was needed for
genotoxic activity in the THM class. Pegram et al. (1997) examined
chloroform mutagenicity in a strain of Salmonella typhimurium TA1535
transfected with rat glutathione- S-transferase (GST) T1-1.
Chloroform was negative in this assay over a range of concentrations
(992-23 800 mg/m3 [200-4800 ppm]) that produced large dose-dependent
increases in revertants with BDCM. A doubling of revertants was
induced by chloroform in the GST-transfected strain only at the two
highest concentrations tested (95 200 and 127 000 mg/m3 [19 200 and
25 600 ppm]). Brennan & Schiestl (1998) found that chloroform induced
intrachromosomal recombination in the yeast strain Saccharomyces
cerevisiae at culture medium concentrations of 3-5.6 mg/ml.
In an in vivo study, Potter et al. (1996) found that chloroform
did not induce DNA strand breaks in the kidneys of male F344 rats
following seven daily doses of 1.5 mmol/kg of body weight. In
long-term mutagenicity studies with chloroform in female lacI
transgenic B6C3F1 mice conducted by Butterworth et al. (1998), the
mice were exposed daily by inhalation to chloroform concentrations of
0, 50, 149 or 446 mg/m3 (0, 10, 30 or 90 ppm) for 6 h per day, 7 days
per week, and lacI mutant frequency was determined after 10, 30, 90
and 180 days of exposure. No increase in lacI mutant frequency was
observed in the liver at any dose or time point with chloroform.
4.1.1.4 Comparative pharmacokinetics and metabolism
The percutaneous absorption of 14C-chloroform was examined
in vivo using human volunteers and in vitro using fresh, excised
human skin in a flow-through diffusion cell system (Dick et al.,
1995). Aqueous and ethanol solutions of chloroform were applied to the
forearm (16 and 81 g/cm) of volunteers, and absorption was determined
to be 7.8% from the water vehicle and 1.6% from ethanol. More than 95%
of the absorbed dose was excreted via the lungs (88% as carbon
dioxide), and maximum pulmonary excretion occurred between 15 min and
2 h after dosing. In vitro, 5.6% of a low dose and 7.1% of a high
dose were absorbed (skin plus perfusate).
The systemic uptake of chloroform during dermal exposure was also
studied in hairless rats (Islam et al., 1996). Animals were immersed
in water containing chloroform for 30 min, and the compound was
detected in blood as early as 4 min following exposure. About 10 mg of
chloroform were systemically absorbed after dermal exposure of a rat
to an aqueous solution of 0.44 mg/ml.
Absorption and tissue dosimetry of chloroform were evaluated
after gavage administration in various vehicles to male Fischer 344
rats and female B6C3F1 mice (Dix et al., 1997). Animals received a
single dose of chloroform (15-180 and 70-477 mg/kg of body weight for
rats and mice, respectively) in corn oil, water or aqueous 2% Emulphor
(dose volumes of 2 and 10 ml/kg of body weight for rats and mice,
respectively). Blood, liver and kidney chloroform concentration-time
courses were determined. Gavage vehicle had minimal effects on
chloroform dosimetry in rats. In mice, however, tissue chloroform
concentrations were consistently greater for aqueous versus corn oil
vehicle. At the low vehicle volume used for rats (2 ml/kg of body
weight), gavage vehicle may not play a significant role in chloroform
absorption and tissue dosimetry; at the higher vehicle volume used for
mice (10 ml/kg of body weight), however, vehicle may be a critical
factor.
Because chloroform metabolism was reviewed in detail in the
recent Environmental Health Criteria monograph (IPCS, 1994), the
primary discussion of the comparative metabolism of the THMs as a
class can be found in section 4.1.2.6 of the present report.
The contributions of cytochromes P450 (CYP) 2E1 and 2B1/2 to
chloroform hepatotoxicity were investigated in male Wistar rats
(Nakajima et al., 1995). The severity of toxicity observed in
differentially induced rats suggests that CYP2E1 is a low
Michaelis-Menten constant ( Km) isoform and CYP2B1/2 is a high
Km isoform for chloroform activation. A high dose of chloroform
(0.5 ml/kg of body weight) induced CYP2E1 but decreased CYP2B1/2.
Testai et al. (1996) generated similar results in a study examining
the involvement of these isozymes in in vitro chloroform metabolism.
At a low substrate concentration (0.1 mmol/litre), oxidative
metabolism by liver microsomes was dependent primarily on CYP2E1; at 5
mmol/litre, on the other hand, CYP2B1/2 was the major participant
responsible for chloroform activation, although CYP2E1 and CYP2C11
were also significantly involved. The reductive pathway was expressed
only at 5 mmol/litre and was not significantly increased by any CYP
inducer tested.
The reductive metabolism of chloroform by rat liver microsomes
was examined by Testai et al. (1995). In hypoxic (1% oxygen partial
pressure) and anoxic (0% oxygen partial pressure) incubations using
microsomes from phenobarbital-induced animals, no evidence of
formation of monochloromethyl carbene was found. Dichloromethane was
detected as a metabolite of chloroform under variable oxygenation
conditions using microsomes from phenobarbital-induced animals. With
uninduced microsomes, significant levels of dichloromethane were
formed only in hypoxic or anoxic incubations. In an in vivo study of
chloroform reductive metabolism, Knecht & Mason (1991) detected no
free radical adducts in the bile of rats treated with chloroform,
while radicals were detected from bromoform. Lipid adducts resulting
from the reductive metabolism of chloroform by hepatocytes appeared to
be generated by the unspecific attack of the radical on the
phospholipid fatty acyl chains (Guastadisegni et al., 1996). The
primary lipid adduct has now been identified as a modified
phosphatidylethanolamine, with the phosgene-derived carbonyl bound to
the amine of the head group (Guastadisegni et al., 1998). Waller &
McKinney (1993) found that chloroform had a lower theoretical
potential to undergo reductive metabolism than the brominated THMs.
Ade et al. (1994) reported that microsomes from the renal cortex of
DBA/2J mice can metabolize chloroform through the reductive and
oxidative pathways, as had been previously described using hepatic
microsomes. However, cytolethality of chloroform to freshly isolated
rodent hepatocytes was not increased under reduced oxygen tension,
indicating that reductive metabolism does not contribute to
chloroform-induced toxicity (Ammann et al., 1998).
The potential of chloroform to participate in the recently
discovered GSH conjugation pathway for brominated THMs has been
investigated (Pegram et al., 1997). The GST examined in this study has
a very low affinity for chloroform compared with the brominated THMs.
Chloroform conjugation with GSH occurred only at extremely high
substrate concentrations.
4.1.1.5 Mode of action
Direct DNA reactivity and mutagenicity cannot be considered to be
key factors in chloroform-induced carcinogenesis in experimental
animals. A substantial body of data demonstrates a lack of direct
in vivo or in vitro genotoxicity of chloroform. If THMs produce
their genotoxic effects primarily via the GSH conjugation mechanism,
the results of Pegram et al. (1997) indicate that chloroform would be
mutagenic in mammals only at lethal doses.
There is, however, compelling evidence to support a mode of
action for tumour induction based on metabolism of chloroform by the
target cell population, followed by cytotoxicity of oxidative
metabolites and regenerative cell proliferation. The evidence for the
link with cytotoxicity is strongest for hepatic and renal tumours in
the mouse and more limited for renal tumours in the rat (ILSI, 1997).
A number of recent studies support the hypothesis that chloroform acts
to produce cancer in rodents through a non-genotoxic/cytotoxic mode of
action, with carcinogenesis resulting from events secondary to
chloroform-induced cytolethality and regenerative cell proliferation
(Larson et al., 1994a,b, 1996; Pereira, 1994; Templin et al.,
1996a,b,c, 1998). These studies have shown that organ toxicity and
regenerative hyperplasia are associated with the tumorigenicity of
chloroform and are apparently the key steps in its carcinogenic mode
of action. Thus, sustained toxicity would result in tumour
development. Chloroform induces liver and kidney tumours in long-term
rodent cancer bioassays only at doses that induce frank cytotoxicity
in these target organs. Furthermore, there are no instances of
chloroform-induced tumours that are not preceded by this pattern of
dose-dependent toxic responses (Golden et al., 1997).
The organ toxicity and carcinogenicity of chloroform are
dependent on oxidative metabolism and levels of CYP2E1. Numerous
studies have also shown that oxidative metabolism by CYP2E1 generates
highly reactive metabolites (phosgene and hydrogen chloride), which
would lead to cytotoxicity and regenerative hyperplasia.
4.1.2 Bromodichloromethane
4.1.2.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
The acute oral lethality of the brominated THMs has been
determined in ICR Swiss mice (Bowman et al., 1978) and Sprague-Dawley
rats (Chu et al., 1980). The resulting LD50s for BDCM were 450 and
900 mg/kg of body weight for male and female mice, respectively, and
916 and 969 mg/kg of body weight for male and female rats,
respectively. Clinical observations of animals dosed with high levels
of BDCM in these studies and others (NTP, 1987) included ataxia,
sedation, laboured breathing and anaesthesia (500 mg/kg of body
weight), as well as gross evidence of liver and kidney damage. Hewitt
et al. (1983) gave single doses of BDCM to male Sprague-Dawley rats by
corn oil gavage and found doses of 1980 mg/kg of body weight and above
to be lethal. Little clinical evidence of hepatic or renal toxicity
was observed at doses below 1980 mg/kg of body weight.
Acute hepatotoxic and nephrotoxic responses to orally dosed BDCM
and various factors affecting these toxicities (e.g., gavage vehicle
and glutathione status) have been studied in male F344 rats. Lilly et
al. (1994, 1997a) examined the time course of toxicity and
dose-response relationships following oral administration of aqueous
solutions of BDCM. BDCM-induced liver toxicity was maximal at 24 h
after dosing with 1-3 mmol/kg of body weight (164-492 mg/kg of body
weight), as indicated by elevations in serum levels of aspartate
aminotransferase (ASAT), alanine aminotransferase (ALAT), sorbitol
dehydrogenase (SDH) and lactate dehydrogenase (LDH) and
histopathological observations of centrilobular vacuolar degeneration
and hepatocellular necrosis. Significant abatement of hepatic toxicity
was noted by 48 h post-dosing. The acute oral NOAEL and LOAEL for
liver toxicity following aqueous delivery of BDCM, based on elevations
in serum enzymes, were determined to be 0.25 and 0.5 mmol of BDCM per
kg of body weight (41 and 82 mg/kg of body weight), respectively
(Keegan et al., 1998). BDCM and chloroform appear to be equipotent
hepatotoxicants in rats at 24 h after exposure, but BDCM causes more
persistent damage to the liver, based on observations at 48 h
post-dosing (Lilly et al., 1997a; Keegan et al., 1998).
Kidney toxicity after corn oil or aqueous dosing of BDCM
(1.5-3 mol/kg of body weight) peaked between 24 and 48 h, as indicated
by elevations in kidney weight, urinary N-acetyl-ß-glucosaminidase,
ASAT, ALAT, LDH and protein, serum urea and creatinine, and
histopathological findings of renal tubule degeneration and necrosis
(Lilly et al., 1994, 1997a). The actual time of peak renal effects was
dose-dependent; in contrast to findings in the liver, toxicity was
increasingly prolonged in the kidney with increasing dose.
Nephrotoxicity has been noted in rats given single BDCM doses as low
as 200 mg or 1.2 mmol/kg of body weight (Lilly et al., 1994, 1997a),
and BDCM is a slightly more potent acute oral renal toxicant than
chloroform (based on the magnitude of the responses), especially at
lower doses (Lilly et al., 1997a). Kroll et al. (1994a,b) found that
among the THMs that occur in drinking-water, BDCM was the most potent
inducer of renal dysfunction in rats following intraperitoneal
injection of single 3 mmol/kg of body weight doses. Glomerular
filtration, renal concentrating ability, and proximal tubular
secretion and reabsorption were all more severely affected by BDCM
than by chloroform.
Several factors have been found to influence dose-response
relationships for BDCM toxicity. Acute hepatotoxicity and
nephrotoxicity were more severe after administration of 400 mg of BDCM
per kg of body weight in corn oil than when the same dose was given in
an aqueous vehicle (Lilly et al., 1994). However, vehicle differences
at a lower dose (200 mg/kg of body weight), although less pronounced,
were reversed: greater renal toxicity at this dose was associated with
the aqueous vehicle. The adverse renal and hepatic effects of BDCM
were also exacerbated in GSH-depleted rats (Gao et al., 1996) and in
rats that were dosed during the active period of their diurnal cycle
(Pegram et al., 1993). Induction of the cytochrome P-450 isozymes
CYP2E1 and CYP2B1/2 also potentiated acute liver toxicity, but not
renal toxicity, following dosing with BDCM (Thornton-Mannning et al.,
1994).
2) Short-term toxicity
Studies employing repeated daily BDCM dosing regimens have also
yielded results demonstrating liver and kidney toxicity.
Thornton-Manning et al. (1994) administered five consecutive daily
BDCM doses to female F344 rats and female C57BL/6J mice by aqueous
gavage and found that BDCM is both hepatotoxic and nephrotoxic to
female rats (150-300 mg/kg of body weight per day), but only
hepatotoxic to female mice (75-150 mg/kg of body weight per day).
Hepatic cytochrome P450 activities were decreased in rats, but not in
mice, in this study. Munson et al. (1982) administered BDCM (50, 125
or 250 mg/kg of body weight per day) to male and female CD-1 mice by
aqueous gavage for 14 days and reported evidence for hepatic and renal
toxicity as well as effects on the humoral immune system.
Nephrotoxicity, as reflected by significant elevations of blood urea
nitrogen (BUN), occurred only at the highest dose in both males and
females. Male mice appeared more sensitive than females to
BDCM-induced hepatotoxicity; 2- to 3-fold elevations in ASAT and ALAT
(although not significant, according to the authors' statistical
analysis) occurred at the lowest dose only in males. Based on the
degree of these elevations, BDCM was the most potent hepatotoxicant
compared with chloroform, DBCM and bromoform, which were also tested
in this study. Immunotoxic effects described in the study included
decreases in both antibody-forming cells and haemagglutination titres
at the 125 and 250 mg/kg of body weight per day doses, although a
recent investigation found no effects of BDCM on immune function
(French et al., 1999). Condie et al. (1983) conducted a similar 14-day
comparative dosing study with THMs and male CD-1 mice, but used corn
oil as the vehicle of administration for doses of BDCM of 37, 74 and
147 mg/kg of body weight per day. Evidence of renal damage was
observed at the mid and high doses, whereas liver toxicity occurred
only at the high dose. A 14-day corn oil gavage study by NTP (1987)
demonstrated the greater sensitivity of the B6C3F1 mouse to BDCM: all
male mice that received 150 or 300 mg/kg of body weight per day died
before the end of the study. Aida et al. (1992a) incorporated
microencapsulated BDCM into the diet of Wistar rats for 1 month, and a
LOAEL of 66 mg/kg of body weight per day and a NOAEL of 21 mg/kg of
body weight per day were determined based on histopathological
findings of hepatocellular vacuolization.
In a 13-week corn oil gavage study, NTP (1987) administered BDCM
doses of 0, 19, 38, 75, 150 or 300 mg/kg of body weight per day, 5
days per week, to F344/N rats (10 per sex per dose). The highest dose
was lethal to 50% of males and 20% of females, and body weight
depression was observed at the two highest doses. BDCM-induced lesions
were found only at 300 mg/kg of body weight per day; these included
hepatic centrilobular degeneration in both sexes and renal tubular
degeneration and necrosis in males. Additional findings included mild
bile duct hyperplasia and atrophy of the thymus, spleen and lymph
nodes in both sexes. B6C3F1 mice were also dosed with BDCM in this
study, and doses of 50 mg/kg of body weight per day and below produced
no compound-related effects. Degeneration and necrosis of the kidney
were observed in male mice at 100 mg/kg of body weight per day,
whereas centrilobular degeneration of the liver was noted in females
at 200 and 400 mg/kg of body weight per day.
3) Chronic toxicity
Moore et al. (1994) administered BDCM in drinking-water
(containing 0.25% Emulphor) to male F344 rats and B6C3F1 mice for
1 year and evaluated clinical indicators of kidney toxicity. Water
containing BDCM concentrations of 0.08, 0.4 and 0.8 g/litre for rats
and 0.06, 0.3 and 0.6 g/litre for mice resulted in average daily doses
of 4.4, 21 and 39 mg/kg of body weight for rats and 5.6, 24 and 49
mg/kg of body weight for mice. A urinary marker for renal proximal
tubule damage, N-acetyl-ß-glucosaminidase, was elevated above
controls in each dose group in rats and at the highest treatment level
in mice. Significant increases in urinary protein, indicative of
glomerular damage, were also noted in low- and mid-dose rats as well
as high-dose mice.
In an NTP (1987) study, BDCM was administered by corn oil gavage
for 102 weeks, 5 days per week, to F344/N rats (50 per sex per dose)
at doses of 0, 50 or 100 mg/kg of body weight per day and to B6C3F1
mice at doses of 0, 25 or 50 mg/kg of body weight per day (50 males
per group) and 0, 75 or 150 mg/kg of body weight per day (50 females
per group). In male rats, compound-related non-neoplastic lesions
included renal cytomegaly and tubular cell hyperplasia and hepatic
necrosis and fatty metamorphosis. Kidney tubule cell hyperplasia was
also observed in female rats, as well as eosinophilic cytoplasmic
change, clear cell change, focal cellular change and fatty
metamorphosis of the liver. Histopathological changes were noted at
both doses in rats. BDCM-induced non-neoplastic lesions in male mice
included hepatic fatty metamorphosis, renal cytomegaly and follicular
cell hyperplasia of the thyroid gland, all observed in both dose
groups. In female mice, hyperplasia of the thyroid gland was observed
at both doses.
Microencapsulated BDCM was fed in the diet to Wistar rats for 24
months, resulting in average daily doses of 6, 26 or 138 mg/kg of body
weight for males and 8, 32 or 168 mg/kg of body weight for females
(Aida et al., 1992b). Relative liver weight was increased in both
sexes of all dose groups, as was relative kidney weight in the
high-dose group. BDCM induced hepatic fatty degeneration and granuloma
in all dose groups and cholangiofibrosis in the high-dose groups.
Therefore, this study identified a LOAEL for chronic liver toxicity of
6 mg/kg of body weight per day.
4.1.2.2 Reproductive and developmental toxicity
Klinefelter et al. (1995) studied the potential of BDCM to alter
male reproductive function in F344 rats. BDCM was consumed in the
drinking-water for 52 weeks, resulting in average dose rates of 22 and
39 mg/kg of body weight per day. No gross lesions in the reproductive
organs were revealed by histological examination, but exposure to the
high BDCM dose significantly decreased the mean straight-line, average
path and curvilinear velocities of sperm recovered from the cauda
epididymis. These effects of BDCM on sperm motility occurred at a
lower exposure level than was observed for other DBPs that compromised
sperm motility.
A teratological assessment of BDCM was conducted in
Sprague-Dawley rats by administering the compound by gavage from day 6
to day 15 of gestation (Ruddick et al., 1983). Doses of 50, 100 and
200 mg of BDCM per kg of body weight per day did not produce any
teratogenic effects or dose-related histopathological changes in
either the dams or fetuses, but sternebra aberrations were observed
with a dose-dependent incidence in all dose groups. The increased
incidence of these variations appeared to be significant, but no
statistical analysis of the data was performed. Maternal weight gain
was depressed in the high-dose group, and maternal liver and kidney
weights were increased. Narotsky et al. (1997) employed a similar
experimental model to test BDCM in F344 rats using doses of 0, 25, 50
or 75 mg/kg of body weight per day in aqueous or oil gavage vehicles.
BDCM induced full-litter resorptions in the 50 and 75 mg/kg of body
weight per day dose groups with either vehicle of administration. For
dams receiving corn oil, full-litter resorptions were noted in 8% and
83% of the litters at 50 and 75 mg/kg of body weight per day,
respectively. With the aqueous vehicle, 17% and 21% of the litters
were fully resorbed at 50 and 75 mg/kg of body weight per day,
respectively. All vehicle control litters and litters from the group
given 25 mg/kg of body weight per day survived the experimental
period. BDCM had been shown to cause maternal toxicity at these doses
in a previous study (Narotsky et al., 1992).
4.1.2.3 Neurotoxicity
Neurotoxicological findings for the brominated THMs are limited
to various observations of anaesthesia associated with acute high-dose
exposures and results from a behavioural study conducted by Balster &
Borzelleca (1982). Adult male ICR mice were dosed by aqueous gavage
for up to 90 days. Treatments of 1.2 or 11.6 mg/kg of body weight per
day were without effect in various behavioural tests, and dosing for
30 days with 100 mg/kg of body weight per day did not affect passive
avoidance learning. Animals dosed with either 100 or 400 mg/kg of body
weight per day for 60 days exhibited decreased response rates in an
operant behaviour test; these effects were greatest early in the
regimen, with no evidence of progressive deterioration.
4.1.2.4 Toxicity in humans
Clinical case findings resulting from human exposure to BDCM have
not been reported.
4.1.2.5 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of BDCM and concluded that
there is sufficient evidence for its carcinogenicity in experimental
animals and inadequate evidence for its carcinogenicity in humans. On
this basis, BDCM was assigned to Group 2B: the agent is possibly
carcinogenic to humans (IARC, 1991, 1999).
Among the four THMs commonly found in drinking-water, BDCM
appears to be the most potent rodent carcinogen. BDCM caused cancer at
lower doses and at more target sites than for any of the other THMs.
In the NTP (1987) 2-year bioassay, a corn oil gavage study (50 animals
per sex per group, dosed 5 days per week), compound-related tumours
were found in the liver, kidneys and large intestine. Daily doses were
0, 50 or 100 mg/kg of body weight (male and female rats), 0, 25 or 50
mg/kg of body weight (male mice) and 0, 75 or 150 mg/kg of body weight
(female mice). NTP (1987) concluded that there was clear evidence of
carcinogenic activity for both sexes of F344 rats and B6C3F1 mice, as
shown by increased incidences of tubular cell adenomas and
adenocarcinomas in the kidney and adenocarcinomas and adenomatous
polyps in the large intestine of male and female rats, increased
incidences of tubular cell adenomas and adenocarcinomas in the kidney
of male mice, and increased incidences of hepatocellular adenomas and
carcinomas in female mice (Table 11).
Table 11. Tumour frequencies in rats and mice exposed to bromodichloromethane
in corn oil for 2 yearsa
Animal/tissue/tumour Tumour frequency at control, low and high
doses (mg/kg of body weight per day)
Male rat 0 50 100
Large intestineb
Adenomatous polyp 0/50 3/49 33/50
Adenocarcinoma 0/50 11/49 38/50
Combined 0/50 13/49 45/50
Kidneyb
Tubular cell adenoma 0/50 1/49 3/50
Tubular cell adenocarcinoma 0/50 0/49 10/50
Combined 0/50 1/49 13/50
Large intestine and/or kidney 0/50 13/49 46/50
combinedb
Female rat 0 50 100
Large intestinec
Adenomatous polyp 0/46 0/50 7/47
Adenocarcinoma 0/46 0/50 6/47
Combined 0/46 0/50 12/47
Kidney
Tubular cell adenoma 0/50 1/50 6/50
Tubular cell adenocarcinoma 0/50 0/50 9/50
Combined 0/50 1/50 15/50
Large intestine and/or kidney 0/46 1/50 24/48
combinedd
Male mouse 0 25 100
Kidneye
Tubular cell adenoma 1/46 2/49 6/50
Tubular cell adenocarcinoma 0/46 0/49 4/50
Combined 1/46 2/49 9/50
Female mouse 0 75 150
Liver
Hepatocellular adenoma 1/50 13/48 23/50
Hepatocellular carcinoma 2/50 5/48 10/50
Combined 3/50 18/48 29/50
Table 11. (continued)
a Adapted from NTP (1987).
b One rat died at week 33 in the low-dose group and was eliminated from
the cancer risk calculation.
c Intestine not examined in four rats from the control group and three
rats from the high-dose group.
d One rat in the high-dose group was not examined for intestinal tumours
and kidney tumours.
e In the control group, two mice died during the first week, one mouse
died during week 9 and one escaped in week 79. One mouse in the
low-dose group died in the first week. All of these mice were
eliminated from the cancer risk calculations.
Aida et al. (1992b) maintained Slc:Wistar rats on diets
containing microencapsulated BDCM for 24 months and examined the
animals for neoplastic lesions. The only significant finding was a
slight increase in the incidence of liver tumours in females receiving
the high dose (168 mg/kg of body weight per day). These included
cholangiocarcinomas and hepatocellular adenomas.
To date, effects following chronic BDCM administration via
drinking-water have not been described in the literature. However, two
separate drinking-water studies are currently being conducted by the
US Environmental Protection Agency (EPA) and the NTP.
Although BDCM has given mixed results in bacterial assays for
genotoxicity, the results have tended to be positive in tests
employing closed systems to overcome the problem of the compound's
volatility (IARC, 1991, 1999; Pegram et al., 1997). Pegram et al.
(1997) tested the THMs using a TA1535 strain transfected with rat GST
T1-1 and found that the mutagenicity of the brominated THMs, but not
chloroform, was greatly enhanced by the expression of the transferase.
BDCM was mutagenic in this assay at medium concentrations below 0.1
mmol/litre. Mutation spectra of the brominated THMs at the hisG46
allele were characterized by DeMarini et al. (1997) using revertants
induced in the GST-transfected strain. The overwhelming majority
(96-100%) of the mutations induced by the brominated THMs were GC to
AT transitions, and 87-100% of these were at the second position of
the CCC/GGG target. BDCM produced primary DNA damage in the SOS
chromotest (Escherichia coli PQ37), but was negative in the Ames
fluctuation test with Salmonella typhimurium TA100 (Le Curieux et
al., 1995). A mixture of BDCM and benzo [a]pyrene was tested in an
Ames mutagenicity test with S. typhimurium strains TA98 and TA100
plus S9 (Kevekordes et al., 1998). BDCM in combination with
benzo [a]pyrene caused a 25% increase in revertants in both strains
compared with benzo [a]pyrene alone.
BDCM was also positive in the majority of in vitro genotoxicity
tests employing eukaryotic systems, but the responses with and without
an exogenous metabolizing system are less consistent. This may be due
to the fact that the reactive intermediates suspected to be involved
in THM mutagenicity must be generated within the target cells (Thier
et al., 1993; Pegram et al., 1997). Moreover, extensive metabolism of
the THMs by the supplemental S9 outside of the cells would greatly
diminish the intracellular dose. Many of the positive studies are for
the induction of sister chromatid exchange (SCE) (IARC, 1991, 1999).
Morimoto & Koizumi (1983) found that BDCM induced SCEs in human
lymphocytes in vitro in the absence of S9 activation at
concentrations greater than or equal to 0.4 mmol/litre, and Fujie et
al. (1993) reported increased SCEs in rat erythroblastic leukaemia
cells under similar conditions. Metabolically activated BDCM also
increased SCEs in vitro in human lymphocytes (at 1 mmol/litre) and
in rat hepatocytes (at 100 mmol/litre) (Sobti, 1984).
In vivo, doses of 50 mg of BDCM per kg of body weight and above
produced SCEs in male CR/SJ mice (Morimoto & Koizumi, 1983). BDCM was
negative in in vivo clastogenicity tests (micronucleus formation) in
mice and rats (Ishidate et al., 1982; Hayashi et al., 1988). Fujie et
al. (1990) reported that BDCM induced bone marrow chromosomal
aberrations (primarily chromatid and chromosome breaks) in Long-Evans
rats following oral or intraperitoneal dosing at doses as low as 16.4
mg/kg of body weight. Potter et al. (1996) found that BDCM did not
induce DNA strand breaks in the kidney of male F344 rats following
seven daily doses of 1.5 mmol/kg of body weight (246 mg/kg of body
weight). Stocker et al. (1997) studied the effect of gastric
intubation of aqueous solutions of BDCM on unscheduled DNA synthesis
(UDS) in the liver of male rats. BDCM did not cause UDS in hepatocytes
isolated after administration of single doses of 135 or 450 mg/kg of
body weight. The in vivo mutagenicity studies of BDCM and the other
brominated THMs are summarized in Table 12.
In comparison with other chemicals known to produce mutations via
direct DNA reactivity such as aflatoxin B1 and ethylene dibromide,
BDCM is a relatively weak mutagen.
4.1.2.6 Comparative pharmacokinetics and metabolism
Bromine substitution would be expected to confer greater
lipophilicity on the brominated THMs compared with chloroform, which
would affect tissue solubility and other factors that can influence
pharmacokinetics. Because metabolism of each THM is qualitatively
similar (with one known exception), this section addresses key
features of the metabolism of all four THMs.
The absorption, distribution and elimination of BDCM have been
studied in rats and mice, and more recent work has led to the
development of a physiologically based pharmacokinetic (PBPK) model
for BDCM in rats. Mink et al. (1986) compared the pharmacokinetics of
orally administered 14C-BDCM in male B6C3F1 mice and Sprague-Dawley
rats. The animals received single doses of 100 mg/kg of body weight
(rats) or 150 mg/kg of body weight (mice) in corn oil by gavage, and
tissue levels of radioactivity were determined after 8 h. Absorption
of BDCM appeared to be rapid and fairly complete, as would be expected
for small halocarbons. This was especially true in the mouse, where
93% of the dose was recovered within 8 h as carbon dioxide (81%), as
Table 12. Dose information for selected in vivo mutagenicity studies of brominated trihalomethanes
End-point Assay system Dosea Result Reference
Sister chromatid exchange Male CR/SJ mice, gavage, 4 days 50 mg BDCM/kg bw per day positive Morimoto & Koizumi (1983)
Sister chromatid exchange Male CR/SJ mice, gavage, 4 days 25 mg DBCM/kg bw per day positive Morimoto & Koizumi (1983)
Sister chromatid exchange Male CR/SJ mice, gavage, 4 days 25 mg bromoform/kg bw positive Morimoto & Koizumi (1983)
per day
Sister chromatid exchange B6C3F1 mice, i.p.b 200 mg bromoform/kg bw positive NTP (1989a)
Micronucleus formation ddY mice, MS mice, Wistar rats, 500 mg BDCM/kg bw per day negative Ishidate et al. (1982)
i.p. in olive oil
Micronucleus formation ddY mice, MS mice, Wistar rats, 500 mg DBCM/kg bw per day negative Ishidate et al. (1982)
i.p. in olive oil
Micronucleus formation ddY mice, MS mice, Wistar rats, 500 mg bromoform/kg bw negative Ishidate et al. (1982)
i.p. in olive oil per day
Micronucleus formation ddY mice, i.p., single dose in 500 mg BDCM/kg bw negative Hayashi et al. (1988)
corn oil
Micronucleus formation ddY mice, i.p., single dose in 1000 mg DBCM/kg bw negative Hayashi et al. (1988)
corn oil
Micronucleus formation ddY mice, i.p., single dose in 1400 mg bromoform/kg bw negative Hayashi et al. (1988)
corn oil
Chromosomal aberrations Long-Evans rats, bone marrow, 16.4 mg BDCM/kg bw positive Fujie et al. (1990)
i.p., single dose
Chromosomal aberrations Long-Evans rats, bone marrow, 20.8 mg DBCM/kg bw positive Fujie et al. (1990)
i.p., single dose
Table 12. (continued)
End-point Assay system Dosea Result Reference
Chromosomal aberrations Long-Evans rats, bone marrow, 25.3 mg bromoform/kg bw positive Fujie et al. (1990)
i.p., single dose
Chromosomal aberrations Long-Evans rats, bone marrow 253 mg bromoform/kg bw positive Fujie et al. (1990)
per day
Unscheduled DNA synthesis Rat liver, gavage 450 mg BDCM/kg bw per negative Stocker et al. (1997)
day
Unscheduled DNA synthesis Rat liver, gavage 2000 mg DBCM/kg bw per negative Stocker et al. (1997)
day
Unscheduled DNA synthesis Rat liver, gavage 1080 mg bromoform/kg bw negative Stocker et al. (1997)
per day
Micronucleus formation Mouse, bone marrow, gavage, 1000 mg bromoform/kg bw negative Stocker et al. (1997)
single dose
DNA strand break Male F344 rats, kidney, gavage, 1.5 mmol BDCM/kg bw per negative Potter et al. (1996)
7 days day
DNA strand break Male F344 rats, kidney, gavage, 1.5 mmol DBCM/kg bw per negative Potter et al. (1996)
7 days day
DNA strand break Male F344 rats, kidney, gavage, 1.5 mmol bromoform/kg bw negative Potter et al. (1996)
7 days per day
Sex-linked recessive Drosophila 1000 ppm solution positive NTP (1989a)
mutation
a Doses listed are the lowest at which an effect was observed or, in the case of negative results, the highest dose tested.
bw = body weight.
b Intraperitoneal.
expired volatile organics assumed to be unmetabolized parent compound
(7.2%), in urine (2.2%) or in organs (3.2%). Much more of the 14C
dose was expired as the assumed parent compound by the rat (42%) than
by the mouse, but total recovery after 8 h was less (63%) because of
lower conversion to carbon dioxide (14%). The liver, stomach and
kidneys were the organs with the highest residual radioactivity
levels. The authors estimated BDCM half-lives of 1.5 and 2.5 h in the
rat and mouse, respectively.
Mathews et al. (1990) studied the disposition of 14C-BDCM in
male F344 rats after single oral (corn oil gavage) doses of 1, 10, 32
or 100 mg/kg of body weight and 10-day repeat oral dosing of 10 or
100 mg/kg of body weight per day. The doses of BDCM were well absorbed
from the gastrointestinal tract, as demonstrated by 24-h recoveries
exceeding 90% in non-faecal excreta samples and tissues. Persistence
of radiolabelled residues in tissues after 24 h was low (3-4% of
dose), with the most marked accumulation in the liver (1-3% of dose).
The kidneys, particularly cortical regions, also contained significant
concentrations of radiolabelled residues. Approximately 3-6% of the
dose was eliminated as volatile organics in the breath (primarily the
parent compound, presumably), much less than in Sprague-Dawley rats
(Mink et al., 1986). Urinary and faecal elimination were low at all
dose levels, accounting for 4% and 1-3% of the administered doses,
respectively. Repeated doses had no effect on the tissue distribution
of BDCM, and significant bioaccumulation was not observed (0.9-1.1%
total retention of the label).
Lilly et al. (1998) examined absorption and tissue dosimetry of
BDCM in male F344 rats after doses of 50 or 100 mg/kg of body weight
were given orally using either an aqueous emulsion or corn oil as the
vehicle of administration. After delivery in the aqueous vehicle,
concentrations of BDCM in venous blood peaked at about 6 min, with
maximum concentration ( Cmax) values of 16 and 26 mg/litre for the
low and high doses. With corn oil dosing, Cmax occurred at 15-30
min, with lower peak blood levels attained (5 and 9 mg/litre). The
time required for blood concentrations to decline to half- Cmax was
about 1 h with the 50 mg/kg of body weight dose and 1.5 h with the 100
mg/kg of body weight dose. Tissue partition coefficient determinations
confirmed the anticipated effect of bromine substitution on THM tissue
solubility. BDCM partition coefficients for fat and liver were 526 and
30.6 (Lilly et al., 1997b), compared with 203 and 21.1 for chloroform
(Corley et al., 1990). Lilly et al. (1998) found slightly higher
maximum concentrations of BDCM in the liver and kidneys after aqueous
administration compared with corn oil delivery. With the 100 mg/kg of
body weight aqueous dose, hepatic and renal levels peaked at about 15
mg/litre at 5 min after dosing in the liver and at 5-30 min in the
kidneys. At 6 h after dosing, concentrations of BDCM in the liver and
kidneys were less than 1 mg/litre. More of the parent compound was
eliminated unmetabolized via exhaled breath after aqueous dosing
(8.9%, low dose; 13.2%, high dose) than after corn oil gavage (5.3%,
low dose; 5.8%, high dose).
The elimination kinetics of BDCM have been studied in humans who
had swum in chlorinated pools (Lindstrom et al., 1997; Pleil &
Lindstrom, 1997). BDCM half-lives of 0.45-0.63 min for blood were
estimated using breath elimination data.
The deleterious effects of the THMs result from reactive
metabolites generated by biotransformation. Two isoenzyme groups of
cytochrome P450, CYP2E1 and CYP2B1/2, as well as a theta-class GST,
have been implicated in the metabolism of BDCM to toxic species in
rats (Thornton-Manning et al., 1993; Pegram et al., 1997). No specific
information is available regarding human metabolism of brominated
THMs. In rats, P450-mediated metabolism of BDCM (Figure 1) is believed
to proceed by the same two pathways established for chloroform:
oxidation with phosgene the proposed active metabolite, and reduction
with the dichloromethyl free radical proposed as the reactive product
(Tomasi et al., 1985; IPCS, 1994; Gao et al., 1996). Most
investigations of THM metabolism and reaction mechanisms have focused
on chloroform, and a detailed review of chloroform biotransformation
has been published recently (IPCS, 1994). Although the qualitative
aspects of the cytochrome P450-mediated metabolism of brominated THMs
are similar to those for chloroform, numerous studies have
demonstrated that brominated THMs are metabolized to a greater extent
and at faster rates than chloroform. There is evidence to suggest that
both CYP2E1 and CYP2B1/2 can catalyse the oxidative pathway and that
CYP2B1/2 catalyses the reductive metabolism of haloforms, but it has
also been postulated that either isoform can catalyse both routes
(Tomasi et al., 1985; Testai et al., 1996). CYP2E1 is clearly involved
in the hepatotoxicity induced by BDCM in rats, but its role in the
nephrotoxic response is less certain (Thornton-Manning et al., 1993).
Because CYP2E1 is highly conserved across mammalian species, it seems
likely that this isoform metabolizes BDCM in humans, although this has
not yet been demonstrated. CYP2B1/2 are not expressed in humans, and
there is no direct analogue for their catalytic activity; however,
based on substrate similarities with CYP2B1/2, human forms CYP2A6,
CYP2D6 and CYP3A4 appear to be possibilities.
Gao et al. (1996) demonstrated that GSH affords protection
against the toxicity and macromolecular binding of BDCM, indicating
that oxidative metabolism of BDCM, like that of chloroform, generates
phosgene, which can then react with GSH (Stevens & Anders, 1981).
In vitro binding of a BDCM-derived intermediate to microsomal
protein under aerobic conditions and the prevention of this binding by
GSH supplementation provide further evidence for the production of
phosgene from BDCM (Gao et al., 1996). The reaction of GSH with
phosgene forms S-(chlorocarbonyl)-GSH, which may react with a second
GSH molecule to produce diglutathionyl dithiocarbonate (Pohl et al.,
1981) or to give glutathione disulfide and carbon monoxide as minor
products (Stevens & Anders, 1981). Anders et al. (1978) and Mathews et
al. (1990) reported that carbon monoxide is a product of BDCM
metabolism. The most likely outcome for phosgene is hydrolysis to
carbon dioxide and hydrogen chloride (Brown et al., 1974), and, in
fact, 70-80% of 14C-BDCM doses administered to F344 rats or B6C3F1
mice appeared as expired 14C-labelled carbon dioxide (Mink et al.,
1986; Mathews et al., 1990), which also shows the predominance of
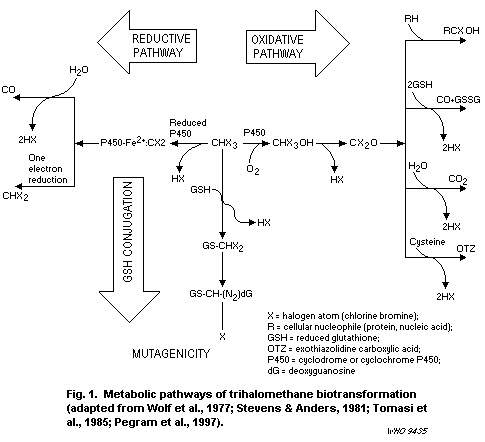 oxidative biotransformation as a metabolic route in these animals.
Sprague-Dawley rats were much less efficient metabolizers of THMs,
disposing of only 14% of a BDCM dose as carbon dioxide. In both rats
and mice, BDCM was more extensively metabolized to carbon dioxide than
was chloroform or bromoform (Mink et al., 1986). In vitro binding
assays with rat hepatic microsomes have also shown that BDCM has a
greater capacity than chloroform to be metabolized to intermediates
(presumably phosgene) that bind protein under aerobic conditions (Gao
& Pegram, 1992; Bull et al., 1995). Similar tests with kidney
microsomes have shown that GSH is much less effective in preventing
renal protein binding than in preventing liver protein binding,
suggesting that a significant portion of this binding in the kidney
may have resulted from generation of a reactive intermediate via a
different pathway (Gao et al., 1996).
Cytochrome P450-mediated reductive dehalogenation of BDCM to form
a dichloromethyl radical has been demonstrated in vivo in
phenobarbital-treated rats using an electron spin resonance (ESR)
spin-trapping technique (Tomasi et al., 1985). These authors reported
that more free radical was derived from BDCM than from chloroform and
that more radical was trapped from bromoform than from BDCM. Waller &
McKinney (1993) conducted a theoretical investigation into the
potential of halogenated methanes to undergo reductive metabolism
using density-functional theory-based computational chemistry. The
estimated reductive potentials for the THMs were in agreement with the
reaction order described in the ESR study. The dichloromethyl radical
reacts preferentially with the fatty acid skeleton of phospholipids to
give covalently bound adducts (De Biasi et al., 1992). In vitro
binding of metabolically activated THMs to hepatic microsomal lipids,
presumably by the radical, has also been investigated, and lipid
binding by BDCM was found to exceed that of chloroform by more than
300% (Gao & Pegram, 1992; Gao et al., 1996). Free radical generation
by this pathway may explain the loss of CYP2B1/2 in rats treated with
BDCM (Thornton-Manning et al., 1994). Finally, carbon monoxide has
also been postulated to be a product of the reductive pathway (Wolf et
al., 1977).
Although the reactions of GSH with phosgene are protective
against THM-induced hepatic and renal toxicity, direct conjugation of
the brominated THMs to GSH may lead to genotoxicity. A GST-mediated
mutagenic pathway of brominated THM metabolism has recently been
identified using a Salmonella strain transfected with rat GST T1-1
(Pegram et al., 1997). Base substitution revertants were produced in
this strain by BDCM (at medium concentrations of less than 0.1
mmol/litre) and the other brominated THMs, but not by chloroform (De
Marini et al., 1997; Pegram et al., 1997). The propensity for GSH
conjugation may therefore explain the different results noted in
mutagenicity tests with chloroform and the brominated THMs. Currently,
it is not known if GSH conjugation leading to genotoxicity occurs in
mammalian cells. However, it is likely that the analogous human GST
T1-1 will also activate the brominated THMs, because similar substrate
specificities have been demonstrated for the rat and human GST
isoforms (Thier et al., 1993, 1996). Human GST T1-1 is expressed
polymorphically and could therefore be a critical determinant of
susceptibility to the genotoxicity of the brominated THMs.
A PBPK model has recently been developed to describe the
absorption, distribution, tissue uptake and dosimetry, metabolism and
elimination of BDCM in rats (Lilly et al., 1997b, 1998). Metabolism
was characterized directly by measuring production of bromide ion,
which is liberated by all known biotransformation reactions of BDCM,
and indirectly by gas uptake techniques. Determinations of plasma
bromide concentrations after constant-concentration inhalation
exposures of rats to BDCM provided evidence for metabolic saturation
at concentrations of 1340 mg/m3 (200 ppm) and greater. Total
in vivo metabolism of BDCM was accurately described by the model as
a saturable process using metabolic rate constant values of 12.8 mg/h
for maximum rate of metabolism ( Vmax) and 0.5 mg/litre for Km.
This compares with a Vmax value for chloroform of 6.8 mg/h (Corley
et al., 1990), providing additional evidence for more rapid metabolism
and greater generation of reactive intermediates from BDCM than from
chloroform. Production of bromide from BDCM following treatment with
an inhibitor of CYP2E1, trans-dichloroethylene, increased the
apparent Km from 0.5 to 225 mg/litre, further demonstrating that
CYP2E1 is a major isoform involved in BDCM metabolism. The metabolism
model, derived from inhalation exposure data, was subsequently linked
to a multicompartment gastrointestinal tract submodel using estimates
of oral absorption rate constants determined by fitting blood and
exhaled breath chamber concentration-time curves obtained after gavage
of rats with BDCM. This model accurately predicted tissue dosimetry
and plasma bromide ion concentrations following oral exposure to BDCM
and can be utilized in estimating rates of formation of reactive
intermediates in target tissues.
4.1.2.7 Mode of action
As stated above, the metabolism of the THMs is believed to be a
prerequisite for the toxicity and carcinogenicity associated with
exposure to these DBPs. The primary target tissues for the THMs are
active sites of their metabolism, and treatments that increase or
decrease biotransformation also tend to cause parallel increases or
decreases in the toxicity induced by the THMs (Thornton-Manning et
al., 1993; US EPA, 1994b). The reactive intermediates generated from
the three brominated THM metabolic pathways react with macromolecules
to elicit both cytotoxic and genotoxic responses.
The cytotoxicity of the brominated THMs observed in the liver and
kidneys of exposed animals has been proposed to result from covalent
adducts formed between cellular proteins and lipids and
dihalocarbonyls or dihalomethyl free radicals. The adducts presumably
impair the function of these molecules and cause cell injury. BDCM
produced these adducts in in vitro incubations with hepatic and
renal microsomes to a significantly greater extent than did chloroform
(Gao & Pegram, 1992; Gao et al., 1996). Induction of lipid
peroxidation by free radical metabolites of reductive metabolism has
been proposed as another mechanism underlying THM cytotoxicity. Each
of the brominated THMs induced lipid peroxidation in rat liver
microsomes in vitro, which was maximal at low oxygen tensions (de
Groot & Noll, 1989).
As described above (section 4.1.2.6), it appears that direct
conjugation of BDCM and the other brominated THMs with GSH generates
mutagenic intermediates (Pegram et al., 1997), but this process has
not yet been demonstrated in mammals. However, the reaction was
dependent on a transfected rat enzyme (GST T1-1), and the human GST
T1-1 has been shown to catalyse GSH conjugation with the
dihalomethanes (Thier et al., 1996). The GC to AT transitions observed
in the Salmonella strain expressing GST T1-1 indicate that DNA
lesions on either guanine or cytosine occurred after exposure to
brominated THMs (De Marini et al., 1997). Based on similar reactions
by dihalomethanes, it can be proposed that S-(1,1-dihalomethyl)-GSH
is the product of GSH conjugation of brominated THMs, which then
reacts directly with guanine. Dechert & Dekant (1996) found that
S-(1-chloromethyl)-GSH reacts with deoxyguanosine to produce
S-[1-( N2-deoxyguanosinyl)-methyl]-GSH, and a methylguanosine
adduct was also formed from reactions of S-(1-acetoxymethyl)-GSH
with model nucleosides (Thier et al., 1993). In vivo covalent DNA
binding by 14C-BDCM has been observed in each of the cancer target
tissues for BDCM in the rat, but the nucleoside adducts have not yet
been identified (Bull et al., 1995).
An interplay of direct mutagenicity with cytotoxic responses
leading to regenerative hyperplasia may explain some, but not all, of
the carcinogenic effects of the brominated THMs. For example, renal
tubular cell hyperplasia coincident with tubular cell cancers was
observed in rats gavaged with BDCM for 2 years (NTP, 1987), but
necrosis and hyperplasia were not associated with the liver neoplasms
induced by BDCM in mice (NTP, 1987). Melnick et al. (1998)
recently revisited this issue and noted that high incidences of liver
tumours were observed with BDCM and DBCM at doses that had little or
no effect on hepatic regenerative hyperplasia. No evidence for
cytotoxic responses in the intestine was noted in the NTP study with
BDCM, but high incidences of intestinal carcinomas were reported (NTP,
1987). Therefore, while cytotoxic effects of BDCM may potentiate
tumorigenicity in certain rodent tissues at high dose levels, direct
induction of mutations by BDCM metabolites may also play a
carcinogenic role. The extent to which each of these processes
contributed to the induction of tumours observed in chronic animal
studies is at present unclear. Additional in vivo studies are
required to confirm the mechanism or mechanisms underlying brominated
THM-induced carcinogenesis.
4.1.3 Dibromochloromethane
4.1.3.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
Acute oral LD50s of 800 and 1200 mg of DBCM per kg of body
weight were reported by Bowman et al. (1978) for male and female ICR
Swiss mice, respectively, whereas Chu et al. (1980) found LD50s of
1186 and 848 mg/kg of body weight for male and female Sprague-Dawley
rats, respectively. A DBCM dose of 500 mg/kg of body weight produced
ataxia, sedation and anaesthesia in mice (Bowman et al., 1978). Hewitt
et al. (1983) dosed male Sprague-Dawley rats with DBCM by corn oil
gavage and found doses of 2450 mg/kg of body weight and above to be
lethal. No clinical evidence for significant liver or kidney toxicity
was found at sublethal doses.
Induction of acute renal toxicity by DBCM was studied by Kroll et
al. (1994a,b); a single intraperitoneal dose of 3 mmol/kg of body
weight resulted in elevated BUN, reductions in glomerular filtration
rate and renal concentrating ability, and interference with proximal
tubular secretion and reabsorption.
2) Short-term toxicity
Daily gavage of male and female CD-1 mice with DBCM in an aqueous
vehicle for 14 days produced hepatotoxicity in both sexes at the
highest dose of 250 mg/kg of body weight per day (Munson et al.,
1982). Depressed immune function was also observed in both sexes at
doses of 125 and 250 mg/kg of body weight per day, whereas the
50 mg/kg of body weight per day dose was without effect. Corn oil
gavage of DBCM to male CD-1 mice for 14 days (Condie et al., 1983) led
to observations of kidney and liver toxicity at a lower dose
(147 mg/kg of body weight per day) than had been observed with aqueous
delivery (Munson et al., 1982). In another 14-day corn oil gavage
study, NTP (1985) found that a dose of 500 mg/kg of body weight per
day was lethal to B6C3F1 mice, and doses of 500 and 1000 mg/kg of
body weight per day were lethal to F344/N rats. Dietary administration
of microencapsulated DBCM to Wistar rats for 1 month caused liver cell
vacuolization, with a LOAEL of 56 mg/kg of body weight per day and a
NOAEL of 18 mg/kg of body weight per day (Aida et al., 1992a).
DBCM-induced cardiotoxicity was reported in male Wistar rats
after short-term exposure (4 weeks of daily dosing with 0.4 mmol/kg of
body weight). Arrhythmogenic and negative chronotropic and dromotropic
effects were observed, as well as extension of atrioventricular
conduction times. Inhibitory actions of DBCM on calcium ion dynamics
in isolated cardiac myocytes were also noted.
In the NTP (1985) study, DBCM was administered by corn oil gavage
to F344/N rats and B6C3F1 mice (10 per sex per dose) for 13 weeks (5
days per week) at doses of 0, 15, 30, 60, 125 or 250 mg/kg of body
weight per day. The highest dose was lethal to 90% of the rats,
producing severe lesions and necrosis in kidney, liver and salivary
glands. Hepatocellular vacuolization indicative of fatty changes was
observed in male rats at doses of 60 mg/kg of body weight per day and
higher. In the mice, no DBCM-related effects were reported at doses of
125 mg/kg of body weight per day or lower. At the highest dose, fatty
liver and toxic nephropathy were noted in males, but not in females. A
NOAEL of 30 mg/kg of body weight per day can be derived from this
study.
A 90-day corn oil gavage study was conducted using Sprague-Dawley
rats and doses of 0, 50, 100 or 200 mg/kg of body weight per day
(Daniel et al., 1990b). Body weight gain was significantly depressed
in the high-dose groups to less than 50% and 70% of the controls in
males and females, respectively. Observations of liver damage included
elevated ALAT in mid- and high-dose males, centrilobular lipidosis
(vacuolization) in males at all doses and high-dose females, and
centrilobular necrosis in high-dose males and females. Kidney proximal
tubule cell degeneration was induced by DBCM in all high-dose rats and
to a lesser extent at 100 mg/kg of body weight per day in males and at
both 50 and 100 mg/kg of body weight per day in females.
3) Chronic toxicity
The chronic oral toxicity of DBCM was studied by NTP (1985) in
F344/N rats and B6C3F1 mice using corn oil gavage (5 days per week
for 104 weeks) and doses of 0, 40 or 80 mg/kg of body weight per day
for rats and 0, 50 or 100 mg/kg of body weight per day for mice. Liver
lesions, including fat accumulation, cytoplasmic changes and altered
basophilic staining, were observed in male and female rats at both
dose levels. The low-dose male mice in this study were lost as a
result of an overdosing accident. Compound-related hepatocytomegaly
and hepatic focal necrosis were observed in high-dose male mice, and
liver calcification (high dose) and fatty changes (both low and high
doses) were noted in female mice. Renal toxicity (nephrosis) was also
observed in male mice and female rats.
4.1.3.2 Reproductive and developmental toxicity
Borzelleca & Carchman (1982) conducted a two-generation
reproductive study of DBCM in ICR Swiss mice. Male and female mice at
9 weeks of age were maintained on drinking-water containing 0, 0.1,
1.0 or 4.0 mg of DBCM per ml, leading to average doses of 0, 17, 171
or 685 mg/kg of body weight per day. Fertility and gestational index
were reduced in the high-dose group for the F1 generations. Only
fertility was decreased (high-dose) in the F2 generation. At the mid
and high doses in both generations, litter size and the viability
index were decreased. Other effects included decreased lactation index
and reduced postnatal body weight. No dominant lethal or teratogenic
effects were observed in the F1 or F2 generations.
In a developmental study in rats conducted by Ruddick et al.
(1983), gavage of DBCM (0, 50, 100, or 200 mg/kg of body weight per
day) on gestational days 6-15 caused a depression of maternal weight
gain, but no fetal malformations.
4.1.3.3 Neurotoxicity
DBCM was tested for behavioural effects in male ICR mice by
Balster & Borzelleca (1982), who found no effect of treatments up to
100 mg/kg of body weight per day for 60 days. Dosing for 60 days with
400 mg/kg of body weight per day produced decreased response rates in
an operant behaviour test. Korz & Gattermann (1997) observed
DBCM-induced behavioural alterations in male golden hamsters exposed
either for 14 days to a dose of 5 mg/kg of body weight per day or
acutely to a single dose of 50 mg/kg of body weight. At the low dose,
subchronic treatment caused reduced aggressive behaviour during social
confrontation on day 14 as compared with vehicle-dosed controls.
Following the acute dose, increased locomotor activity on days 3-6 and
decreased wheel running on days 6-9 were observed, but no effects were
noted after day 9.
4.1.3.4 Toxicity in humans
Clinical case findings resulting from human exposure to DBCM have
not been reported.
4.1.3.5 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of DBCM and concluded that
there is inadequate evidence for its carcinogenicity in humans and
limited evidence for its carcinogenicity in experimental animals. The
compound was assigned to Group 3: DBCM is not classifiable as to its
carcinogenicity to humans (IARC 1991, 1999).
In a 104-week corn oil gavage study, DBCM was not carcinogenic in
F344 rats (50 per sex per dose) at doses of 0, 40 or 80 mg/kg of body
weight given 5 days per week (NTP, 1985). In female B6C3F1 mice,
however, DBCM significantly increased the incidence of hepatocellular
adenomas and the combined incidences of hepatocellular adenomas and
carcinomas at the high dose (100 mg/kg of body weight per day) (Table
13). The incidence of hepatocellular carcinomas was significantly
increased in male mice at the same dose. The low-dose (50 mg/kg of
body weight per day) male mice were lost midway through the study as a
result of an inadvertent overdose. NTP judged that these results
provided equivocal evidence of DBCM carcinogenicity in male mice and
some evidence of carcinogenicity in female mice.
Based on in vitro studies, mutagenic potency appears to
increase with the degree of bromine substitution in the THMs. DBCM is
mostly positive in tests employing closed systems to overcome the
problem of volatility (IARC, 1991, 1999; Pegram et al., 1997). In the
GST-transfected TA1535 strain (see section 4.1.1.3), DBCM is the most
potent THM, inducing greater than 10-fold more revertants per plate
than BDCM after exposure to THM vapour (3400 mg/m3 for DBCM; 2680
mg/m3 for BDCM [400 ppm]) (Pegram et al., 1996; DeMarini et al.,
1997). DBCM has given mostly positive results in eukaryotic test
systems (Loveday et al., 1990; IARC, 1991, 1999; McGregor et al.,
1991; Fujie et al., 1993), although there is less consistency in
results between the different assays when considered with or without
an exogenous metabolic system.
Data from in vivo studies are more equivocal. DBCM was positive
for SCE and chromosomal aberrations in mouse bone marrow (Morimoto &
Koizumi, 1983; Fujie et al., 1990) and in a newt micronucleus assay
(Le Curieux et al., 1995), but was negative for micronuclei and UDS in
the liver of rats (Ishidate et al., 1982; Hayashi et al., 1988;
Stocker et al., 1997). Potter et al. (1996) found that DBCM did not
Table 13. Frequencies of liver tumours in mice administered
dibromochloromethane in corn oil for 104 weeksa
Treatment (mg/kg Sex Adenoma Carcinoma Adenoma or
of body weight carcinoma
per day) (combined)
Vehicle control M 14/50 10/50 23/50
F 2/50 4/50 6/50
50 M -b - -
F 4/49 6/49 10/49
100 M 10/50 19/50c 27/50d
F 11/50c 8/50 19/50e
a Adapted from NTP (1985).
b Male low-dose group was inadequate for statistical analysis.
c P < 0.05 relative to controls.
d P < 0.01 (life table analysis); P = 0.065 (incidental tumour
tests) relative to controls.
e P < 0.01 relative to controls.
induce DNA strand breaks in the kidneys of male F344 rats following
seven daily doses of 1.5 mmol/kg of body weight.
These studies are summarized in Table 12.
4.1.3.6 Comparative pharmacokinetics and metabolism
The pharmacokinetics of DBCM have been studied the least among
the THMs. Mink et al. (1986) compared the absorption, distribution and
excretion of DBCM with those of the other THMs. A dose of 100 mg/kg of
body weight was administered orally in corn oil to male Sprague-Dawley
rats by gavage, and 150 mg/kg of body weight was administered
similarly to male B6C3F1 mice. The pattern of distribution and
elimination of DBCM was very similar to that observed with BDCM: rats
expired more of the dose than mice as a trapped organic component
presumed to be the parent compound (48% vs. 12%) and expired less as
carbon dioxide (18% vs. 72%) after 8 h. Total recovery from excised
organs was 1.4% in rats and 5.0% in mice, whereas less than 2% of the
dose was excreted in the urine in both species. Half-lives of DBCM in
rats and mice were estimated to be 1.2 h and 2.5 h, respectively.
The cytochrome P450-mediated metabolism of DBCM has not been
directly investigated. Presumably, metabolism proceeds via the same
routes of biotransformation as described for BDCM (section 4.1.2.6)
and chloroform (IPCS, 1994). Oxidative metabolism of DBCM would be
expected to yield a bromochlorocarbonyl rather than phosgene, and
reductive dehalogenation would produce a bromochloromethyl radical.
Anders et al. (1978) demonstrated in vivo carbon monoxide production
from DBCM in male Sprague-Dawley rats at a rate intermediate to those
of BDCM and bromoform. DBCM was very reactive in the Salmonella-GST
mutagenicity assay, indicating that it has greater potential for GSH
conjugation than BDCM (De Marini et al., 1997).
Pankow et al. (1997) reported the metabolism of DBCM to bromide
and carbon monoxide in rats after gavage of olive oil solutions
(0.4-3.1 mmol/kg of body weight). The DBCM concentrations in blood and
fat 6 h after the last of seven consecutive daily doses (0.8 mmol/kg
of body weight) were lower than at 6 h after a single gavage of this
dose, suggesting that more of the chemical was metabolized after the
seventh dose than after the single dose. The seven-dose regimen also
caused a 2-fold induction of a CYP2E1-specific activity. The
involvement of CYP2E1, CYP2B1/2 and GSH in DBCM metabolism was also
demonstrated.
4.1.3.7 Mode of action
Mechanistic issues for DBCM are similar to those addressed for
BDCM (sections 4.1.2.6 and 4.1.2.7). The greater propensity for the
metabolism of this compound and bromoform as compared with BDCM is
difficult to reconcile with its lower carcinogenicity in the NTP
(1985) bioassay. A possible explanation is less bioavailability
resulting from the greater lipophilicity of this compound and the use
of corn oil as the vehicle of administration. Greater lipophilicity
and reactivity of this compound or its metabolites (i.e., the
bromochlorocarbonyl metabolite) may also prevent it from reaching
critical target sites. It is also of note that DBCM did not induce
carcinomas of the large intestine in rats in the NTP studies using
corn oil vehicle, whereas both BDCM and bromoform did induce these
tumours.
4.1.4 Bromoform
4.1.4.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
Among the brominated THMs, bromoform is the least potent as a
lethal acute oral toxicant. The acute oral LD50s for bromoform were
1400 and 1550 mg/kg of body weight in male and female mice,
respectively (Bowman et al., 1978), and 1388 and 1147 mg/kg of body
weight in male and female rats, respectively (Chu et al., 1980).
Bromoform was also a less potent anaesthetic than BDCM and DBCM: a
1000 mg/kg of body weight dose was required to produce this effect in
mice. Intraperitoneal administration of bromoform in a corn oil
vehicle at doses of 25-300 µl/kg of body weight produced no
significant elevations of serum enzymes indicative of liver damage
(Agarwal & Mehendele, 1983).
Bromoform was included in the acute renal toxicity studies of
Kroll et al. (1994a,b) but was ranked as the least potent among the
THMs in general disruption of renal function. A single intraperitoneal
dose (3 mmol/kg of body weight) did, however, induce significant
decreases in glomerular filtration rate, renal concentrating ability,
and tubular secretion and reabsorption. Tubule function was affected
as early as 8 h after dosing, whereas the other effects were observed
at 24-48 h.
2) Short-term toxicity
Male and female CD-1 mice were gavaged daily with bromoform (50,
125 or 250 mg/kg of body weight) in an aqueous vehicle for 14 days,
leading to liver toxicity and a decrease in antibody-forming cells
only at the highest dose (Munson et al., 1982). The magnitude of the
liver effects was less than that observed with the other THMs, and BUN
was not elevated by bromoform. Condie et al. (1983) noted both liver
and kidney toxicity in CD-1 mice after dosing with 289 mg of bromoform
per kg of body weight per day in corn oil for 14 days, whereas no
significant effects were found at 72 and 145 mg/kg of body weight per
day. NTP (1989a) also conducted a 14-day corn oil gavage study with
bromoform and found that doses of 600 and 800 mg/kg of body weight per
day were lethal to both sexes of F344/N rats. Mice given doses of 400
mg/kg of body weight and higher had raised stomach nodules.
Microencapsulated bromoform was added to the diet of Wistar rats for 1
month, producing liver damage (Aida et al., 1992a).
Bromoform was given to F344/N rats (10 per sex per dose) and
B6C3F1 mice (10 per sex per dose) by corn oil gavage for 13 weeks
(5 days per week) at doses of 0, 25, 50, 100 or 200 mg/kg of body
weight per day, with an added dose of 400 mg/kg of body weight per day
for mice only (NTP, 1989a). The only significant finding, hepatic
vacuolation, was observed in male rats, but not in female rats, at
doses of 50 mg/kg of body weight per day and higher, and in male mice,
but not in female mice, at doses of 200 and 400 mg/kg of body weight
per day. The NOAEL in the rat study was 25 mg/kg of body weight per
day, whereas that in the study in mice was 100 mg/kg of body weight
per day.
3) Chronic toxicity
Bromoform was administered in drinking-water (containing 0.25%
Emulphor) to male F344 rats and B6C3F1 mice for 1 year, and clinical
indicators of kidney toxicity were examined (Moore et al., 1994).
Water containing bromoform concentrations of 0.12, 0.6 and 1.2 g/litre
for rats and 0.08, 0.4, and 0.8 g/litre for mice resulted in average
daily doses of 6.2, 29 or 57 mg/kg of body weight for rats and 8.3, 39
or 73 mg/kg of body weight for mice. Several indicators of tubular and
glomerular damage were elevated at each treatment level in mice, and
mice appeared more susceptible to the nephrotoxic effects of bromoform
than to those of BDCM (see section 4.1.2.1). As in mice, urinary
protein was increased in all rat dose groups, but little evidence of
loss of tubule function was observed in rats.
Two-year investigations of bromoform toxicity were conducted by
administering doses of 0, 100 or 200 mg/kg of body weight by corn oil
gavage, 5 days per week for 103 weeks, to F344/N rats (50 per sex per
dose) and female B6C3F1 mice (50 per dose) (NTP, 1989a). Male mice
(50 per dose) received doses of 0, 50 or 100 mg/kg of body weight per
day. Survival of high-dose male rats and both dose groups of female
mice was significantly lower than that of the vehicle controls. Focal
or diffuse fatty change of the liver was observed in a dose-dependent
fashion in both sexes of rats, and active chronic hepatic inflammation
was found in male and high-dose female rats. Minimal necrosis of the
liver was noted in high-dose male rats. In mice, the incidence of
fatty changes of the liver was increased in females, but not in males,
at both dose levels. Follicular cell hyperplasia of the thyroid gland
was observed in high-dose female mice. The different target organ
(liver) in this study as compared with the drinking-water study of
Moore et al. (1994), in which renal effects predominated, suggests
that the vehicle and mode of administration can affect which tissues
are affected by bromoform.
4.1.4.2 Reproductive and developmental toxicity
Ruddick et al. (1983) conducted a teratological investigation of
bromoform in Sprague-Dawley rats (15 per group). Bromoform was
administered by gavage at doses of 50, 100 or 200 mg/kg of body weight
per day on gestation days 6-15. Evidence of a fetotoxic response was
observed, but there were no fetal malformations. Interparietal
deviations were, however, noted in the mid- and high-dose groups. NTP
(1989b) utilized the same bromoform doses given by gavage for 105 days
to 20 male-female pairs of Swiss CD-1 mice to examine effects on
fertility and reproduction. There was no detectable effect of
bromoform on fertility, litters per pair, live pups per litter,
proportion of pups born alive, sex of live pups or pup body weights.
Bromoform was also found to induce full-litter resorptions in pregnant
F344 rats when administered orally on gestation days 6-15, but at
higher doses (150 and 200 mg/kg of body weight per day) than those
required to produce the same effect for BDCM (Narotsky et al., 1993).
4.1.4.3 Neurotoxicity
Bromoform was included in the mouse behavioural study of Balster
& Borzelleca (1982). Doses of 9.2 mg/kg of body weight per day for up
to 90 days had no effect on the outcome of several behavioural tests,
and 100 mg/kg of body weight per day for 30 days did not deter passive
avoidance learning. However, mice receiving either 100 or 400 mg/kg of
body weight per day for 60 days exhibited decreased response rates in
an operant behaviour test.
4.1.4.4 Toxicity in humans
Bromoform was used in the late 19th and early 20th centuries as a
sedative for children with whooping cough. Patients were typically
given doses of one drop (approximately 180 mg) 3-6 times per day
(Burton-Fanning, 1901), which usually resulted in mild sedation in the
children. A few rare instances of death or near-death were reported
but were believed to be due to accidental overdoses (Dwelle, 1903).
These clinical observations have been used to estimate a lethal dose
for a 10- to 20-kg child to be about 300 mg/kg of body weight and an
approximate minimal dose for sedation to be 50 mg/kg of body weight
per day (US EPA, 1994b).
4.1.4.5 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of bromoform and concluded
that there is inadequate evidence for its carcinogenicity in humans
and limited evidence for its carcinogenicity in experimental animals.
The compound was assigned to Group 3: bromoform is not classifiable as
to its carcinogenicity to humans (IARC, 1991, 1999).
Two-year studies of bromoform carcinogenicity were conducted by
administering doses of 0, 100 or 200 mg/kg of body weight in corn oil
by gavage, 5 days per week, to groups of F344/N rats (50 per sex per
group) and female B6C3F1 mice (50 per group) and doses of 0, 50 or
100 mg/kg of body weight to male mice (NTP, 1989a). No neoplastic
effects were associated with the exposure of mice to bromoform. In
rats, however, intestinal carcinomas were induced by bromoform, as had
also been observed with BDCM. The uncommon adenomatous polyps or
adenocarcinomas (combined) of the large intestine (colon or rectum)
were observed in 3 out of 50 high-dose male rats and in 8 out of 50
high-dose female rats as compared with 0 out of 50 rats in the male
controls and 0 out of 48 rats in the female controls (Table 14).
Because these tumours are very rare in the rat, these findings were
considered significant, and the NTP therefore concluded that there was
clear evidence for carcinogenic activity in female rats and some
evidence in male rats.
Bromoform, in common with the other brominated THMs, is largely
positive in bacterial assays of mutagenicity conducted in closed
systems (Zeiger, 1990; IARC, 1991, 1999). In the GST-transfected
Salmonella typhimurium TA1535 strain (see section 4.1.2.5),
bromoform produced about 5 times more revertants than BDCM at
comparable exposure levels (Pegram et al., 1997). As with BDCM, the
mutations were almost exclusively GC to AT transitions (DeMarini et
al., 1997). In prokaryotic tester strains, bromoform induced mutations
without metabolic activation in S. typhimurium strain TA100 (Simmon
& Tardiff, 1978; Ishidate et al., 1982; NTP, 1989a; Le Curieux et al.,
1995), with and without activation in TA98 (NTP, 1989a; Zeiger, 1990)
and with microsomal activation in TA97 (NTP, 1989a). In eukaryotic
test systems, bromoform is largely positive (IARC, 1991; Fujie et al.,
1993). As with bacterial assays, bromoform appeared more potent than
the other brominated THMs (Morimoto & Koizumi, 1983; Banerji &
Fernandes, 1996).
In vivo studies (summarized in Table 12) have given
contradictory results. Bromoform was positive and negative in
Drosophila (Woodruff et al., 1985). It was positive in the newt
micronucleus test (Le Curieux et al., 1995) and gave increased SCE and
chromosomal aberrations in mouse and rat bone marrow cells (Morimoto &
Koizumi, 1983; Fujie et al., 1990). It gave negative results in mouse
bone marrow (Hayashi et al., 1988; Stocker et al., 1997), in the rat
Table 14. Tumour frequencies in rats exposed to bromoform in corn oil
for 2 yearsa
Animal/tissue/tumour Tumour frequency
Control 100 mg/kg of 200 mg/kg of
body weight body weight
per day per day
Male rat
Large intestine
Adenocarcinoma 0/50 0/50 1/50
Polyp (adenomatous) 0/50 0/50 2/50
Female rat
Large intestine
Adenocarcinoma 0/48 0/50 2/50
Polyp (adenomatous) 0/48 1/50 6/50
a Adapted from NTP (1989a).
liver UDS assay (Pereira et al., 1982; Stocker et al., 1997) and in
the dominant lethal assay (Ishidate et al., 1982). In the studies
carried out by the NTP (1989a), it was positive for micronuclei and
SCE, but negative for chromosomal aberrations in mouse bone marrow.
Potter et al. (1996) found that bromoform did not induce DNA strand
breaks in the kidneys of male F344 rats following seven daily doses of
1.5 mmol/kg of body weight.
4.1.4.6 Comparative pharmacokinetics and metabolism
14C-Bromoform pharmacokinetics were examined in Sprague-Dawley
rats and B6C3F1 mice in a study that included all four of the common
THMs that occur as DBPs (Mink et al., 1986). Recoveries of 14C-label
8 h after gavage dosing were 79% in rats and 62% in mice. The
distribution and elimination of bromoform resembled those of
chloroform rather than those of the other brominated THMs. The
percentage of 14C-label recovered from excised organs and tissues was
2.1% in rats and 12.2% in mice. Tissue levels of 14C in mice were
substantially greater than those observed for the other brominated
THMs. Urinary excretion of label after 8 h (2.2-4.6%) was also greater
for bromoform than for BDCM and DBCM. Bromoform (and organic
metabolite) elimination via exhaled breath was greater than that for
all other THMs in the rat (67%), but less than that for all other THMs
in the mouse (6%). The estimated half-life of bromoform was 0.8 h in
rats and 8 h in mice.
The cytochrome P450-dependent metabolism of bromoform was studied
by Anders et al. (1978), who found that bromoform was converted to
carbon monoxide in vivo in male Sprague-Dawley rats at a rate
significantly greater than were the other three THMs. Evidence was
presented suggesting that some of the carbon monoxide arose from the
oxidative metabolism of bromoform to a dibromocarbonyl and subsequent
reactions with GSH. It is possible that some of the carbon monoxide
was also generated from reductive metabolism of bromoform, as
indicated by in vitro results of Wolf et al. (1977) from anaerobic
incubations using hepatic preparations derived from
phenobarbital-treated rats. Carbon monoxide was produced from
anaerobic bromoform metabolism in this study at much greater levels
than from chloroform metabolism. Among the four THMs tested by Mink et
al. (1986), bromoform exhibited the least metabolism to carbon dioxide
(4% in rats and 40% in mice in 8 h). Free radicals from the in vivo
reductive metabolism of bromoform were detected by ESR spin-trapping
after dosing phenobarbital-treated rats (Tomasi et al., 1985). More
radicals were generated from bromoform in vivo than from any other
bromochlorinated THM, which is consistent with computational chemistry
predictions that bromoform would have the greatest reductive potential
of the THMs (Waller & McKinney, 1993). Knecht & Mason (1991) also
detected the in vivo production of radicals from bromoform in the
bile of rats, but only after the induction of hypoxia.
Bromoform, like DBCM, has a much greater potential than BDCM to
be conjugated by GSH to form a mutagenic intermediate. Base pair
revertants were produced in a dose-dependent fashion in
GST-transfected Salmonella typhimurium strain TA1535 by bromoform
and BDCM, but not by chloroform (Pegram et al., 1996, 1997; De Marini
et al., 1997). At an exposure concentration of 33 100 mg/m3 for
bromoform and 22 800 mg/m3 for BDCM (3200 ppm), GST-dependent
revertants per plate induced by bromoform and BDCM averaged 373 and
1935, respectively, compared with a control rate of 23 per plate
(Pegram et al., 1996).
4.1.4.7 Mode of action
The basic mechanisms of action for bromoform are similar to those
described for BDCM (section 4.1.2.7). Although bromoform seems to have
a greater propensity for metabolism and is a more potent mutagen than
BDCM, it appears to be a less potent toxicant and carcinogen based on
the results of the NTP (1985, 1987) bioassays and numerous other
in vivo studies of toxicity. As with DBCM, a possible explanation is
less bioavailability resulting from the greater lipophilicity of this
compound and the use of corn oil as the vehicle of administration.
This concept may be supported by the occurrence of bromoform-induced
tumours in the intestinal tract, but not in the liver or kidneys.
Greater lipophilicity and reactivity of bromoform metabolites may also
prevent it from reaching critical target sites. Moreover, when
bromoform was injected intraperitoneally, its metabolism was greater
than that of the other THMs (Anders et al., 1978; Tomasi et al.,
1985); when administered by corn oil gavage, however, bromoform was
the least metabolized THM (Mink et al., 1986). Data from studies of
oral exposure to bromoform in aqueous solutions are therefore
required.
4.2 Haloacids
Like the THMs, the haloacids produced in the chlorination of
drinking-water consist of a series of chlorinated and brominated
forms. To date, the chlorinated acetic acids have been more thoroughly
characterized toxicologically than their brominated analogues. As
discussed in earlier chapters, the dihaloacetates and trihaloacetates
occur in significantly higher concentrations than the
monohaloacetates. The present review will emphasize the di- and
trihaloacetates as the dominant forms of the haloacids found in
drinking-water and the ones for which extensive toxicological data
have been developed. The probable existence of many longer-chain
halogenated acids in chlorinated drinking-water may be surmised from
studies of chlorinated humic acids (reviewed by Bull & Kopfler, 1991).
Few of these compounds have been extensively studied toxicologically.
However, brief reference will be made to those compounds that are
known to share some of the effects of the HAAs on intermediary
metabolism.
4.2.1 Dichloroacetic acid (dichloroacetate)
Before beginning discussions of available toxicological
evaluations of DCA, it is important to point out that this compound
exists in drinking-water as the salt, despite the fact that it is
widely referred to as dichloroacetic acid. DCA has a p Ka of 1.48 at
25°C (IARC, 1995). As a consequence, it occurs almost exclusively in
the ionized form at the pHs found in drinking-water (broadly speaking,
a pH range of 5-10). Failure to recognize this has resulted in a
number of studies that have employed the free acid in test systems. At
low doses, the buffering capacity of the physiological system can
neutralize acid, and the measured activity may in fact be
representative. However, most of the experimentation has been
conducted using doses ranging from 50 to 1000 mg/kg of body weight
in vivo or in the mmol/litre range in vitro. Therefore, the
applicability of the results of such studies to estimating human risks
will be uncertain because of the large pH artefacts that can be
expected when administering these quantities of a strong acid.
DCA has been shown to produce developmental, reproductive, neural
and hepatic effects in experimental animals. In general, these effects
occur when the compound has been administered at high dose rates, at
which there is evidence to indicate that the metabolic clearance of
DCA is substantially inhibited. This has important implications in
attempting to associate these toxicities with the low doses that are
obtained in drinking-water.
Because it was being developed as a potential therapeutic agent,
there is a toxicological literature that precedes DCA's discovery as a
by-product of chlorination. The present review discusses these early,
more general explorations of DCA's effects on intermediary metabolism
before proceeding to descriptions of studies designed to more
specifically study its toxicology.
4.2.1.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
DCA is not very toxic when administered acutely to rodents.
Woodard et al. (1941) reported LD50s of 4.5 and 5.5 g/kg of body
weight in rats and mice, respectively, for DCA administered as the
sodium salt. This is roughly in the same range as the LD50s for
acetic acid. There is reason to believe that other species, most
specifically the dog, may be more sensitive because of some
repeated-dose experiments discussed below; however, no specific data
seem to have been reported in the literature.
2) Short-term toxicity
Katz et al. (1981) published the first substantive evaluation of
DCA's subchronic toxicity in rats and dogs. DCA was administered by
gavage at 0, 125, 500 or 2000 mg/kg of body weight per day to rats
(10-15 per sex per group) and at 0, 50, 75 or 100 mg/kg of body weight
per day to dogs (3-4 per sex per group) for 3 months. One of three
female dogs died at 75 mg/kg of body weight per day, and one of four
male dogs died at 100 mg/kg of body weight per day. The most overt
toxicity in rats was hindlimb paralysis at the highest dose. Clinical
chemistry indicated significant increases in total and direct
bilirubin at 500 and 2000 mg/kg of body weight per day in rats, and
relative liver weights were significantly increased at all doses. In
dogs, an increase in the incidence of haemosiderin-laden Kupffer cells
was noted at all dose rates. Histopathological changes were observed
in the brain and testes of both species. In rats, oedematous brain
lesions were seen at 60% incidence (primarily in the cerebrum, but
also in the cerebellum) at the lowest dose and at 100% in the two
higher doses in both sexes. Slight to moderate vacuolation of
myelinated tracts was observed at all doses in both rats and dogs.
Testicular germinal epithelial degeneration was observed in rats at
doses of 500 mg/kg of body weight per day and above and at all doses
in dogs, with severity increasing with dose. In dogs, a high incidence
of ocular anomalies was observed, and lenticular opacities were found
to be irreversible upon suspension of treatment with DCA. In both rats
and dogs, glucose and lactate levels were suppressed in a dose-related
manner.
Bull et al. (1990) examined the effects of DCA on the liver of
B6C3F1 mice administered 1 or 2 g of DCA per litre (approximately 170
and 300 mg/kg of body weight per day) in their drinking-water, with
exposure lasting up to 1 year. As had been at least alluded to in
earlier studies, DCA was found to produce a severe hepatomegaly in
mice at concentrations in drinking-water of 1 g/litre and above. The
hepatomegaly could be largely accounted for by large increases in cell
size (cytomegaly). In shorter-term experiments, it was determined that
treatment with DCA produced only minor changes in the labelling index
of hepatocytes using a pulse dose of tritiated thymidine (Sanchez &
Bull, 1990). In general, increases in labelling indices were seen in
areas of acinar necrosis in the liver. Hepatocytes from these mice
stained very heavily for glycogen using periodic acid/Schiff's reagent
(PAS). The accumulation of glycogen began to occur with as little as
1-2 weeks of treatment (Sanchez & Bull, 1990), but became
progressively more severe with time (Bull et al., 1990). Sanchez &
Bull (1990) noted that these effects could not be replicated by
exposing mice to the metabolites of DCA -- glycolate, glyoxylate or
oxalate -- in the drinking-water.
Davis (1990) investigated the effects of DCA and TCA treatments
on male and female Sprague-Dawley rats for up to 14 days. The authors
reported decreased plasma glucose and lactic acid concentrations in
both plasma and liver. There were some inconsistencies in the
reporting of the doses in this study. However, the author has provided
the correct doses, which were 120 and 316 mg/kg of body weight per day
(M.E. Davis, personal communication, 1996). This places the results in
a context that is more consistent with those of other studies (e.g.,
Katz et al., 1981).
Mather et al. (1990) found increases in liver weight in rats
treated with DCA at 5 g/litre (350 mg/kg of body weight per day) in
their drinking-water for 90 days. Relative liver and kidney weights
were increased at concentrations of 0.5 g/litre (35 mg/kg of body
weight per day) and above. PAS staining of liver sections revealed
accumulation of glycogen in severely swollen hepatocytes, which was
quite marked with 5 g DCA/litre. The treatment caused small,
statistically significant increases in alkaline phosphatase (AP) and
ALAT in serum. However, these changes were too small to be of clinical
importance. DCA was found to approximately double the activity of
cyanide-insensitive acyl coenzyme A (CoA) activity in the liver at
5 g/litre, indicating some induction of peroxisome synthesis.
Cicmanec et al. (1991) examined the subchronic effects of DCA in
dogs. DCA was administered at doses of 0, 12.5, 39.5 or 72 mg/kg of
body weight per day for 90 days to groups of five males and five
females. Liver weights were significantly increased in a dose-related
manner, beginning with the lowest dose, and kidney weight was
increased at the highest dose. This was accompanied by
histopathological observation of vacuolar changes in the liver and
haemosiderosis. The pancreas displayed evidence of chronic
inflammation and acinar degeneration at the two highest doses.
Testicular degeneration was observed in virtually all the male dogs
administered DCA. This pathology was not observed in the control
animals. Vacuolization of white myelinated tracts in the cerebrum or
cerebellum was observed at all doses, and vacuolar changes were
observed in the medulla and spinal cord of male dogs. Although the
vacuolization was present in all dose groups, the authors described it
as being mild. These authors failed to find evidence of the lenticular
opacities that had been previously reported by Katz et al. (1981)
under very similar treatment conditions. They also found fairly
consistent decreases in erythrocyte counts and haemoglobin
concentrations at 72 mg/kg of body weight per day in both male and
female dogs. As found by others in rat studies, evidence of hindlimb
paralysis was reported, but the effect was expressed only sporadically
in dogs.
A number of other studies are consistent with the pattern of
toxicological effects produced by DCA described above. Included in
this group are the study of Bhat et al. (1991), which adds
observations of enlarged portal veins, deposition of collagen in the
area of the portal triads and some similar lesions in the vasculature
of the lung in rats. This study also noted atrophic testes and focal
vacuolation and gliosis in the brain. Only a single dose level of 10.5
g/litre of drinking-water (approximately 1100 mg/kg of body weight per
day) was utilized in this 90-day study, a level that substantially
exceeds concentrations reported to reduce water and food consumption
in other studies (Bull et al., 1990).
More recent studies have more closely examined the dose-response
relationships involved in the accumulation of glycogen in the liver of
male B6C3F1 mice treated with DCA in drinking-water at levels ranging
from 0.1 to 3 g/litre (approximately 20-600 mg/kg of body weight per
day) for up to 8 weeks (Kato-Weinstein et al., 1998). Significant
increases in the glycogen content of the liver of mice were seen with
concentrations as low as 0.5 g/litre (100 mg/kg of body weight per
day) in their drinking-water, with a small, but insignificant,
increase being observed at 0.2 g/litre (40 mg/kg of body weight per
day). Glycogen concentrations in the liver reached maximum levels
within 1 week of treatment with concentrations in drinking-water of
1 g/litre and above. At this early stage, the glycogen that
accumulates is subject to mobilization by fasting. With continued
treatment, the glycogen that is deposited becomes increasingly
resistant to mobilization until approximately 8 weeks of treatment,
when the glycogen contents of the livers of DCA-treated mice are not
different in fasted and non-fasted states.
The enzymatic basis of hepatic glycogen accumulation remains
unclear. DCA treatment has no effect on the total amount of either
form of glycogen synthase in the liver (e.g.,
glucose-6-phosphate-dependent vs. glucose-6-phosphate-independent
activity). The proportion of glycogen synthase in the active form was
significantly decreased in mice treated with DCA for as little as
1 week. The amount of phosphorylase in the active form appeared
unaltered by treatment. Such changes could indicate that a feedback
inhibition may have developed on the synthesis of glycogen as a result
of its accumulation in hepatocytes.
Carter et al. (1995) examined the time course of DCA's effects in
the liver of B6C3F1 mice at concentrations of 0, 0.5 or 5 g/litre in
drinking-water for up to 30 days. As reported in prior studies, the
high-dose group displayed severe liver hypertrophy. However, a
smaller, but consistent, increase in liver weight became evident with
as little as 10 days of treatment at 0.5 g/litre. Even at this
relatively low dose, some hepatocytes appeared to have lost nuclei or
possessed nuclei that had undergone some degree of karyolysis. These
experiments also appeared to rule out cytotoxicity and reparative
hyperplasia as consistent features of DCA's effects. The authors
suggested that this was in apparent contrast to the earlier
observations of Sanchez & Bull (1990). However, Sanchez & Bull (1990),
in a 14-day study in mice, noted that these effects were closely
associated with what appeared to be infarcted areas in the liver of
mice treated with high doses of DCA rather than cytotoxicity. These
infarcts are thought to be secondary to the severe swelling of
hepatocytes that results from DCA treatment. This interpretation is
supported by the apparent lack of cytotoxic effects of DCA in isolated
hepatocytes of both mice and rats at concentrations in the mmol/litre
range (Bruschi & Bull, 1993). Subsequently, these infarcted areas have
been identified as acinar necrosis (ILSI, 1997), which occurs in a
somewhat random fashion when high concentrations of DCA (> 2
g/litre) are administered for prolonged periods of time (Stauber &
Bull, 1997).
It is important to note that the dog appears to be very sensitive
to the effects of DCA on the liver; substantial increases in liver
weights are observed at daily doses as low as 12.5 mg/kg of body
weight per day for a 90-day period (Cicmanec et al., 1991). By
comparison, the lowest effect level noted in mice is 0.5 g/litre of
drinking-water, which approximates 70-100 mg/kg of body weight per day
(Carter et al. 1995), and the lowest effect level noted in rats is 125
mg/kg of body weight per day (Katz et al., 1981).
4.2.1.2 Reproductive effects
DCA produces testicular toxicity when administered at high doses
in drinking-water. These effects were first noted in studies of the
general toxicity of DCA (Katz et al., 1981) and were discussed above
(section 4.2.1.1). Cicmanec et al. (1991) followed up on these
original observations in dogs and detected degeneration of the
testicular epithelium and syncytial giant cell formation at doses as
low as 12.5 mg/kg of body weight.
Toth et al. (1992) examined DCA's ability to modify male
reproductive function in Long-Evans rats given 0, 31, 62 or 125 mg/kg
of body weight per day by gavage for 10 weeks. Reduced weights of
accessory organs (epididymis, cauda epididymis and preputial gland)
were observed at doses as low as 31 mg/kg of body weight per day.
Epididymal sperm counts were found to be depressed and sperm
morphology was increasingly abnormal at doses of 62 mg/kg of body
weight per day and above. These latter effects were accompanied by
changes in sperm motion. Fertility was tested in overnight matings and
was found to be depressed only at the highest dose evaluated,
125 mg/kg of body weight per day.
The testicular toxicity of DCA was evaluated in adult male rats
given both single and multiple (up to 14 days) oral doses. Delayed
spermiation and altered resorption of residual bodies were observed in
rats given single doses of 1500 or 3000 mg/kg of body weight; these
effects persisted to varying degrees on post-treatment days 2, 14 and
28. Delayed spermiation and formation of atypical residual bodies were
also observed on days 2, 5, 9 and 14 in rats dosed daily with 54, 160,
480 or 1440 mg/kg of body weight per day. Distorted sperm heads and
acrosomes were observed in step 15 spermatids after administration of
doses of 480 and 1440 mg/kg of body weight per day for 14 days.
Decreases in the percentage of motile sperm occurred after 9 days at
doses of 480 and 1440 mg/kg of body weight per day and after 14 days
at 160 mg/kg of body weight per day. Increased numbers of fused
epididymal sperm were observed on days 14, 9 and 5 in rats dosed with
160, 480 or 1440 mg/kg of body weight per day, respectively; other
morphological abnormalities occurred at 160 mg/kg of body weight per
day and higher. On day 14, a significant decrease in epididymis weight
was observed at 480 and 1440 mg/kg of body weight per day, and
epididymal sperm count was decreased at 160 mg/kg of body weight per
day and higher. These studies demonstrate that the testicular toxicity
induced by DCA is similar to that produced by the analogue DBA (see
section 4.2.3.2). However, the testicular toxicity of DCA is less
severe at equal mol/litre concentrations. Moreover, the DCA-induced
testicular lesions occur at lower doses as the duration of dosing
increases, indicating the importance of using low-dose subchronic
exposures to assess the health risk of prevalent DBPs (Linder et al.,
1997a).
DCA was found to be more potent than TCA in inhibiting
in vitro fertilization of B5D2F1 mouse gametes (Cosby & Dukelow,
1992). For DCA, the percentage of gametes fertilized dropped from
87.0% to 67.3% at the lowest concentration tested, 100 mg/litre. No
effects were noted for TCA at 100 mg/litre; at 1000 mg/litre, however,
71.8% of the gametes were fertilized. Both responses were
statistically different from those of their concurrent control groups.
4.2.1.3 Developmental effects
DCA has been shown to induce soft tissue abnormalities in fetal
rats when administered by gavage in a water vehicle to their dams
during gestation days 6-15 (Smith et al., 1992). These effects were
observed at doses of 140 mg/kg of body weight per day and above and
were not observed at 14 mg/kg of body weight per day. The heart was
the most common target organ. An interventricular septal defect
between the ascending aorta and the right ventricle was most commonly
observed. Urogenital defects (bilateral hydronephrosis and renal
papilla) and defects of the orbit were also observed. In a subsequent
publication, these authors identified the most sensitive period to be
days 12-15 of gestation (Epstein et al., 1992).
Some limited experimentation has been conducted to evaluate the
developmental effects of DCA in rat whole embryo culture (Saillenfait
et al., 1995). The applicability of these data to the in vivo
situation is difficult to evaluate based on the very high
concentrations that were utilized in these studies. At a concentration
of 1 mmol/litre, DCA retarded growth of the embryos by a variety of
measures. It required 2.5 mmol/litre to induce brain and eye defects
and 3.5 mmol/litre to produce abnormalities in other organ systems.
4.2.1.4 Neurotoxicity
Yount et al. (1982) demonstrated that DCA administered to rats in
their feed at doses of 2.5-4 mmol/kg of body weight per day (322-716
mg/kg of body weight per day) produced hindlimb weakness and abnormal
gait within 2-4 weeks of treatment. These effects were associated with
significant reductions in nerve conduction velocity and a decrease in
the cross-sectional area of the tibial nerve.
4.2.1.5 Toxicity in humans
DCA was first investigated as a potential orally effective
hypoglycaemic agent. Stacpoole et al. (1978) found that DCA,
administered in doses of 3-4 g of dichloroacetate as the sodium salt
per day for 6-7 days (43-57 mg/kg of body weight per day for a 70-kg
person), significantly reduced fasting hyperglycaemia in patients with
diabetes mellitus alone or in combination with hyperlipoproteinaemia
by 24%. In addition, plasma lactic acid concentrations dropped by 73%
and plasma alanine concentrations by 82%. Only mild sedation was
noticed by some of the patients, and there was no evidence of altered
blood counts or prothrombin time. There was a slower, but significant,
decline in plasma triglyceride levels and less consistent effects on
plasma cholesterol. ß-Hydroxybutyrate concentrations in plasma
increased significantly and continuously over the 6 days of
administration. Uric acid concentrations in serum increased and those
in urine decreased, reflecting a 50% decrease in urinary clearance.
Further details on the early investigations of DCA as an oral
antidiabetic agent have been reviewed extensively (Crabb et al., 1981;
Stacpoole, 1989; Stacpoole & Greene, 1992) and will not be dwelt on
extensively in the present review. The main reason that DCA was not
fully developed for this application was that longer-term
administration to patients induced a reversible polyneuropathy (Moore
et al., 1979b; Stacpoole, 1989). As discussed above, similar pathology
was reported in experimental animals by several authors, which added
significant weight to this observation. Subsequent work in rats
suggested that thiamine deficiency contributed to the development of
peripheral neuropathy (Stacpoole et al., 1990). However, a recent
report (Kurlemann et al., 1995) indicated that supplementing the diet
with 100 mg of thiamine daily did little to ameliorate the development
of polyneuropathy of a patient treated with DCA at a dose of 100 mg/kg
of body weight for 20 weeks.
Subsequently, DCA has been investigated extensively in the
treatment of congenital lactic acidosis (Coude et al., 1978; Stacpoole
& Greene, 1992; Toth et al., 1993), a disease that is frequently
fatal. More recently, DCA has been evaluated with success in the
treatment of lactic acidosis associated with severe malaria in
children (Krishna et al., 1995). It is also apparently effective in
treatment of lactic acidosis associated with liver transplantation
(Shangraw et al., 1994). DCA has been reported to improve psychiatric
symptoms, as well as to markedly decrease elevated lactic acid levels
in an individual with mitochondrial myopathy (Saijo et al., 1991).
Doses of up to 100 mg/kg of body weight per day were administered. In
a second study, DCA was shown to reverse lesions to the basal ganglia
in a patient with complex I deficiency as measured by computerized
tomography (CT) scan and magnetic resonance imaging (MRI) (Kimura et
al., 1995). However, a second patient with pyruvate dehydrogenase
complex deficiency displayed only transient improvement.
Based on work done in animals, several studies have examined the
effects of DCA on cardiovascular function under a variety of disease
conditions and altered physiological states. DCA was found to
stimulate myocardial lactate consumption and improve left ventricular
efficiency in 10 patients with congestive heart failure (Bersin et
al., 1994). On the other hand, although DCA decreased blood lactate
levels in patients with congestive heart failure, it had no effect on
exercise time, peak exercise oxygen consumption or flow to the
exercising leg (Wilson et al., 1988). In normal individuals, DCA
decreased blood lactate levels when exercising at less than 80% of
average maximal oxygen consumption, but it did not affect blood
lactate concentrations at exhaustion (Carraro et al., 1989).
These studies show that DCA has some beneficial effects in a
variety of metabolic diseases. Acutely, DCA produces little in the way
of risk, because short exposures are without apparent adverse effect.
However, despite the limited number of subjects that have been
studied, the results, when coupled with the results of animal
experiments, indicate relatively strongly that DCA is neurotoxic in
humans. The delayed induction of these toxicities may be attributable
in part to the fact that systemic concentrations of DCA can be
expected to sharply increase with prolonged treatment, at least at the
high doses used therapeutically, typically in the range 25-100 mg/kg
of body weight. There is no human evidence of adverse effects at lower
exposures to DCA (e.g., those that would be derived from drinking
chlorinated water).
4.2.1.6 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of DCA and, based on the
data available at the time, concluded that there is inadequate
evidence for its carcinogenicity in humans and limited evidence for
its carcinogenicity in experimental animals. The compound was assigned
to Group 3: not classifiable as to its carcinogenicity to humans
(IARC, 1995).
DCA is a very effective inducer of hepatic tumours in both mice
and rats at high doses. Several studies in male and female B6C3F1
mice found multiple tumours per animal with treatment concentrations
of 2 g/litre and above with as little as 1 year of treatment
(Herren-Freund et al., 1987; Bull et al., 1990; DeAngelo et al., 1991;
Daniel et al., 1992a; Pereira, 1996). These studies are summarized in
Table 15. Early in treatment (i.e., 52 weeks), the dose-response curve
is very steep, with essentially no response observed at concentrations
of 1 g/litre, but as many as four tumours per liver in mice treated
with 2 g/litre (Bull et al., 1990). However, concentrations as low as
0.5 g/litre will result in a hepatic tumour incidence of approximately
80% in a full 2-year study (Daniel et al., 1992a).
Hepatic tumours are also induced by DCA in male F344 rats
(Richmond et al., 1995). High doses of DCA given to rats also produce
overt signs of peripheral neuropathy. Nevertheless, increased
incidences of hyperplastic nodules, hepatocellular adenoma and
hepatocellular carcinoma were observed at 60 weeks of treatment at
Table 15. Carcinogenic effects of dichloroacetate in rodents
Species Dose Duration Tumour HN & HAa HCb Reference
(sex) (g/litre) (weeks) site
Incidence Tumour/n Incidence Tumour/n
(multiplicity) (multiplicity)
Mice
B6C3F1 (M) 0 61 Herren-Freund et al.
5 61 Liver 25/26 (4.6) 21/26 (1.7) (1987)
B6C3F1 (M) 1 52 Liver 2/11 0.3 - - Bull et al. (1990)
2 52 Liver 23/24 3.6 5/24 0.25
2 37 Liver 7/11 2.2 0/11 0
B6C3F1 (M) 0 60 Liver 0/10 0 0/10 0 DeAngelo et al. (1991)
0.5 60 Liver
3.5 60 Liver 12/12 2.3 8/12 1.7
5 60 Liver 27/30 2.3 25/30 2.2
0 75 Liver 2/28 0.07
0.05 75 Liver 4/29 0.31
0.5 75 Liver 3/27 0.11
0 104 Liver 1/20 0.05 2/20 0.1 Daniel et al. (1992a)
0.5 104 Liver 12/24 0.5 15/24 0.63
B6C3F1 (F) 0 52 Liver 1/40 0.03 0/40 0 Pereira (1996)
0.28 52 Liver 0/40 0 0/40 0
0.93 52 Liver 3/20 0.20 0/20 0
2.8 52 Liver 7/20 0.45 1/20 0.1
0 81 Liver 2/90 0.02 2/90 0.02
0.28 81 Liver 3/50 0.06 0/50 0
0.93 81 Liver 7/28 0.32 1/28 0.04
2.8 81 Liver 16/19 5.6 5/19 0.37
Table 15. (continued)
Species Dose Duration Tumour HN & HAa HCb Reference
(sex) (g/litre) (weeks) site
Incidence Tumour/n Incidence Tumour/n
(multiplicity) (multiplicity)
Rats
F344 (M) 0 60 Liver 0/7 0 0/7 0 Richmond et al. (1995)
0.05 60 Liver 0/7 0 0/7 0
0.5 60 Liver 0/7 0 0/7 0
2.4 60 Liver (26/27) 0.96 1/27 0.04
0 104 Liver 1/23 0.04 0/23 0
0.05 104 Liver 0/26 0 0/26 0
0.5 104 Liver (9/29) 0.31 3/29 0.1
2.4 104 Liver NRc NR NR NR
F344 (M) 0 104 Liver 1/33 0.03 1/33 0.03 DeAngelo et al. (1996)
0.05 104 Liver 0/26 0 0/26 0
0.5 104 Liver 5/29 0.17 3/29 0.10
1.6d 104 Liver 4/28 0.14 6/28 0.24
a Combined hepatocellular nodules and hepatocellular adenomas.
b Hepatocellular carcinoma.
c NR = not reported.
d Concentration was 2.6 g/litre of drinking-water for 18 weeks and then lowered to 1 g/litre to give a mean daily concentration
of 1.6 g/litre.
2.4 g/litre (Table 15). As in mice, if DCA treatment was extended to
104 weeks, the incidence of these lesions was 41% in a group of 29
rats at a treatment concentration of 0.5 g/litre. No tumours were
observed at 0.05 g/litre, and only one hepatic tumour was observed in
23 control rats.
Estimated doses of DCA in mg/kg of body weight per day for the
studies in Table 15 were as follows (ILSI, 1997; US EPA, 1998a):
Herren-Freund et al. (1997): 5 g/litre = 1000 mg/kg of body
weight per day
Bull et al. (1990): 1 or 2 g/litre = 140 or 300 mg/kg of body
weight per day
DeAngelo et al. (1991): 0.05, 0.5, 3.5 or 5 g/litre = 7.6, 77,
410 or 486 mg/kg of body weight per day
Daniel et al. (1992a): 0.5 g/litre = 95 mg/kg of body weight per
day
Pereira (1996): 0.26, 0.86 or 2.6 g/litre = 40, 120 or 330 mg/kg
of body weight per day
Richmond et al. (1995): 0.05, 0.5 or 2.4 g/litre = 4, 40 or
300 mg/kg of body weight per day
DeAngelo et al. (1996): 0.05, 0.5 or 1.6 g/litre = 4, 40 or
140 mg/kg of body weight per day.
Male F344 rats were exposed for 2 years to DCA in their
drinking-water at concentrations of 0.05, 0.5 or 1.6 g/litre. Based
upon the pathological examination, DCA induced observable signs of
toxicity in the nervous system, liver and myocardium. However,
treatment-related neoplastic lesions were observed only in the liver.
A statistically significant increase in carcinogenicity
(hepatocellular carcinoma) was noted at 1.6 g/litre. Exposure to 0.5
g/litre increased hepatocellular neoplasia (carcinoma and adenoma) at
100 weeks. Calculation of the time-weighted mean daily dose at which
50% of the animals exhibited liver neoplasia indicated that the F344
male rat (approximately 10 mg/kg of body weight per day) is 10 times
more sensitive than the B6C3F1 male mouse (approximately 100 mg/kg of
body weight per day) (DeAngelo et al., 1996).
The ability of DCA to induce damage to DNA that could give rise
to mutations or chromosomal damage has been studied both in vivo and
in vitro. Classical evaluations of DCA in Salmonella typhimurium
tester strains, both with and without metabolic activation, have been
largely negative if held to the standard of at least a 2-fold increase
in apparent mutation frequency (Waskell, 1978; Herbert et al., 1980).
However, a number of more recent studies have suggested some potential
for DCA-induced modifications in DNA. DeMarini et al. (1994) reported
that DCA induced prophage in Escherichia coli at a concentration of
0.26 mmol/litre and produced 2.7 and 4.2 revertants per ppm in
S. typhimurium strain TA100 with and without S9 addition,
respectively. There are some difficulties in interpreting this report,
as the authors introduced DCA as a vapour, and it is not clear whether
the concentrations reported (i.e., ppm) refer to air or medium
concentrations. Second, at least in the case of the Salmonella
assay, the DCA was introduced as the free acid and allowed to vaporize
and partition into the incubation medium. Because DCA is a strong
acid, and if sufficient time is allowed, such conditions could result
in near-quantitative transfer of DCA to the medium. The amount
volatilized in this case was approximately 60-600 mmol. Therefore, it
is likely that the pH of this small amount of medium (2.5 ml) was
substantially modified, even if only a fraction of this relatively
large amount of strong acid was indeed transferred to the medium. The
amount of DCA introduced into the prophage assay was unclear because
the method of addition was not described, although the introduction to
the journal article implied that it was again being tested as a
volatile.
Fox et al. (1996) recently published an evaluation of the
mutagenic effects of sodium dichloroacetate. These investigations
found no evidence of increased mutation rates in Salmonella
typhimurium tester strains TA98, TA100, TA1535 or TA1537;
Escherichia coli strain WP2urvA; or the mouse lymphoma forward
mutation assay, whether incubated in the presence or absence of rat
liver S9 fraction for metabolic activation. These authors found no
evidence that DCA was capable of inducing chromosomal aberrations in
CHO cells in vitro at doses of up to 1100 mg/kg of body weight for
3 days. These studies utilized neutralized DCA, supporting the
contention that positive results in prior studies may have been due to
artefactual results obtained by testing of the free acid or because
various sources of DCA have greater amounts of impurities.
Giller et al. (1997) examined the mutagenicity of DCA in the SOS
chromotest, the Ames fluctuation assay and the newt micronucleus
assay. DCA induced a positive response at 500 µg/ml (approximately 3.5
mmol/litre) in the SOS chromotest and at concentrations ranging from
100 to 1500 µg/ml (approximately 1-10 mmol/litre) in the Ames
fluctuation assay. The effects were observed at a lower concentration
in the absence of S9. The concentrations used in these studies exceed
the peak systemic concentrations of DCA that produce a high incidence
of liver tumours in mice by approximately 3 orders of magnitude
(Kato-Weinstein et al., 1998). Moreover, it appears that the authors
utilized the free acid in these experiments, raising the possibility
of a pH artefact. The newt micronucleus assay was found to be
negative.
DCA has been shown to produce a mutagenic and clastogenic
response in the in vitro mouse lymphoma assay, but only at doses at
or above 1 mmol/litre (Harrington-Brock et al., 1998).
Analogous difficulties have been encountered when attempting to
document the mutagenic effects of DCA in vivo. Nelson & Bull (1988)
and Nelson et al. (1989) reported that DCA induced single strand
breaks (SSB) in hepatic DNA when administered by gavage to both mice
and rats. Subsequent investigators were unable to replicate these
results in detail (Chang et al., 1992; Daniel et al., 1993a). However,
a small transitory increase in SSB was observed with doses of 5 and
10 mmol/kg of body weight in male B6C3F1 mice (Chang et al., 1992).
The bases of the discrepancies in these results are not clear, but
could, in part, be attributed to slightly different methods. As noted
in the subsequent section on TCA (section 4.2.2), the Nelson & Bull
(1988) results were not replicated by Styles et al. (1991), although
there was greater similarity in the methods used. More recently,
Austin et al. (1996) showed that acute doses of DCA oxidatively damage
nuclear DNA, measured as increases in the 8-hydroxy-2-deoxyguanosine
(8-OH-dG) relative to 2-deoxyguanosine content of the isolated DNA.
The time course of this damage is more consistent with the development
of SSB breaks reported by Chang et al. (1992) and could represent the
repair process that involves strand scission. There are two important
points that must be made: (i) the induction of SSB by Chang et al.
(1992) was very small relative to that seen with the positive
controls, diethylnitrosamine and methylmethane sulfonate; and (ii)
although increased 8-OH-dG was observed with acute treatments with
DCA, there was not a sustained elevation of this adduct in nuclear DNA
of mice when treatments were extended to 3 or 10 weeks in
drinking-water (Parrish et al., 1996).
Fuscoe et al. (1996) reported results obtained with the mouse
peripheral blood micronucleus assay. They found a small, but
statistically significant, increase in polychromatic erythrocytes
containing micronuclei in male B6C3F1 mice treated for 9 days with
3.5 g of DCA per litre of drinking-water. However, this response was
not maintained through 28 days of exposure. These investigators also
examined DNA migration in the single-cell gel assay. In this case, DCA
appeared to retard migration of DNA, suggesting the possibility of DNA
cross-linking after 28 days of treatment at 3.5 g/litre. Neither assay
revealed significant effects of DCA at concentrations of 2 g/litre or
below. DCA induces 3-4 tumours per animal within 1 year at 2 g/litre
in drinking-water (Bull et al., 1990). The higher dose adds little to
the tumorigenic response. More information with regard to the
possibility that DCA can cause mutations in liver cells is found in a
recent study using the lacI locus in the Big Blue(R) transgenic
mouse mutagenesis assay (Leavitt et al., 1997). These investigators
used a drinking-water route and the same doses of DCA as were used in
the rodent bioassay. After 10 and 60 weeks of DCA administration, an
increased frequency of mutants was observed at the high dose (3.5
g/litre). Mutational spectral analysis of these mutations revealed a
different spectrum in the mutants from DCA-treated animals than was
seen in the untreated animals. At this high dose of DCA, a large
portion of the liver can actually be tumour tissue. Because tumours
result from clonal expansion, the presence of tumour tissue in the
evaluated sample would give a falsely high mutation frequency if a
lacI mutation occurred in the rapidly expanding tumour clone. Thus,
these indications of genotoxic activity may have little to do with the
induction of hepatic cancer by DCA.
DCA appears to specifically stimulate outgrowth of hepatocellular
adenomas, rather than hepatocellular carcinomas. Pereira & Phelps
(1996) examined the role of DCA as a promoter of methylnitrosourea
(MNU)-initiated hepatic tumours in female B6C3F1 mice. These data are
provided in graphic form in Figure 2. At a concentration of
2.6 g/litre of drinking-water, DCA induced a very large increase in
the number of hepatocellular adenomas, but had no significant effect
on the induction of hepatocellular carcinomas. These data would appear
to be consistent with the stop experiments of Bull et al. (1990), who
found that suspension of treatment with DCA appeared to arrest
progression of liver tumours, but resulted in a yield of
hepatocellular adenomas and nodules that was proportional to the total
dose of DCA administered. In contrast, most of the tumours that
remained after the suspension of TCA treatment for 3 months were
hepatocellular carcinomas.
More recent studies on the effects of DCA on cell replication
within normal hepatocytes and hyperplastic nodules and tumours
(predominantly adenomas) indicate that DCA has selective effects.
Stauber & Bull (1997) found that DCA had a small, stimulatory effect
on the replication rate of normal hepatocytes over the first 14 days
of treatment. As treatment was extended to 28 days and beyond, these
effects became inhibitory at concentrations in drinking-water of
0.5 g/litre and above. In contrast, hepatocytes within nodules and
tumours appeared to be resistant to the inhibitory effects of DCA. At
a concentration of 2 g/litre, DCA doubled the rate at which c-Jun
immunoreactive hepatocytes replicated within hyperplastic nodules and
adenomas. This strong stimulation of tumour cell replication would
appear to be responsible for the very rapid induction of tumours in
mice treated with DCA in drinking-water at concentrations of 2 g/litre
and above. It would appear that the slower induction of liver tumours
at lower doses of DCA depends primarily on the selective suppression
of the replication of normal hepatocytes relative to that of initiated
cells.
The inhibitory effect of DCA on replication of normal hepatocytes
has been observed by a number of investigators (Carter et al., 1995).
The rate of replication is sharply inhibited within 5 days at
concentrations of DCA of 5 g/litre. At 0.5 g/litre, the replication
rate becomes inhibited to the same extent as observed with 5 g/litre
after 20 days of treatment. These decreases in replication were
accompanied by an increase in the percentage of the cells that were
mononucleated, which is probably associated with an increase in
tetraploid cells.
The suppression of cell replication by DCA in normal hepatocytes
of treated mice is accompanied by decreases in apoptosis (Snyder et
al., 1995). At concentrations of 5 g/litre, the frequency at which
apoptotic cells are observed drops by 60-75% with as few as 5 days of
treatment. At 0.5 g/litre, there is a downward trend that is observed
over the period from 5 to 30 days such that the frequency of apoptotic
bodies at this low dose approaches that observed at the highest dose
at 30 days. This result essentially parallels that described above for
suppression of the rates of cell replication. This raises a dilemma as
to whether the driver of the response is suppressed replication or
suppressed apoptosis. Whichever is the case, this has to translate
into suppressed turnover of normal hepatocytes. The question is
whether this suppressive effect on cell turnover increases the
probability of transformation of hepatocytes.
oxidative biotransformation as a metabolic route in these animals.
Sprague-Dawley rats were much less efficient metabolizers of THMs,
disposing of only 14% of a BDCM dose as carbon dioxide. In both rats
and mice, BDCM was more extensively metabolized to carbon dioxide than
was chloroform or bromoform (Mink et al., 1986). In vitro binding
assays with rat hepatic microsomes have also shown that BDCM has a
greater capacity than chloroform to be metabolized to intermediates
(presumably phosgene) that bind protein under aerobic conditions (Gao
& Pegram, 1992; Bull et al., 1995). Similar tests with kidney
microsomes have shown that GSH is much less effective in preventing
renal protein binding than in preventing liver protein binding,
suggesting that a significant portion of this binding in the kidney
may have resulted from generation of a reactive intermediate via a
different pathway (Gao et al., 1996).
Cytochrome P450-mediated reductive dehalogenation of BDCM to form
a dichloromethyl radical has been demonstrated in vivo in
phenobarbital-treated rats using an electron spin resonance (ESR)
spin-trapping technique (Tomasi et al., 1985). These authors reported
that more free radical was derived from BDCM than from chloroform and
that more radical was trapped from bromoform than from BDCM. Waller &
McKinney (1993) conducted a theoretical investigation into the
potential of halogenated methanes to undergo reductive metabolism
using density-functional theory-based computational chemistry. The
estimated reductive potentials for the THMs were in agreement with the
reaction order described in the ESR study. The dichloromethyl radical
reacts preferentially with the fatty acid skeleton of phospholipids to
give covalently bound adducts (De Biasi et al., 1992). In vitro
binding of metabolically activated THMs to hepatic microsomal lipids,
presumably by the radical, has also been investigated, and lipid
binding by BDCM was found to exceed that of chloroform by more than
300% (Gao & Pegram, 1992; Gao et al., 1996). Free radical generation
by this pathway may explain the loss of CYP2B1/2 in rats treated with
BDCM (Thornton-Manning et al., 1994). Finally, carbon monoxide has
also been postulated to be a product of the reductive pathway (Wolf et
al., 1977).
Although the reactions of GSH with phosgene are protective
against THM-induced hepatic and renal toxicity, direct conjugation of
the brominated THMs to GSH may lead to genotoxicity. A GST-mediated
mutagenic pathway of brominated THM metabolism has recently been
identified using a Salmonella strain transfected with rat GST T1-1
(Pegram et al., 1997). Base substitution revertants were produced in
this strain by BDCM (at medium concentrations of less than 0.1
mmol/litre) and the other brominated THMs, but not by chloroform (De
Marini et al., 1997; Pegram et al., 1997). The propensity for GSH
conjugation may therefore explain the different results noted in
mutagenicity tests with chloroform and the brominated THMs. Currently,
it is not known if GSH conjugation leading to genotoxicity occurs in
mammalian cells. However, it is likely that the analogous human GST
T1-1 will also activate the brominated THMs, because similar substrate
specificities have been demonstrated for the rat and human GST
isoforms (Thier et al., 1993, 1996). Human GST T1-1 is expressed
polymorphically and could therefore be a critical determinant of
susceptibility to the genotoxicity of the brominated THMs.
A PBPK model has recently been developed to describe the
absorption, distribution, tissue uptake and dosimetry, metabolism and
elimination of BDCM in rats (Lilly et al., 1997b, 1998). Metabolism
was characterized directly by measuring production of bromide ion,
which is liberated by all known biotransformation reactions of BDCM,
and indirectly by gas uptake techniques. Determinations of plasma
bromide concentrations after constant-concentration inhalation
exposures of rats to BDCM provided evidence for metabolic saturation
at concentrations of 1340 mg/m3 (200 ppm) and greater. Total
in vivo metabolism of BDCM was accurately described by the model as
a saturable process using metabolic rate constant values of 12.8 mg/h
for maximum rate of metabolism ( Vmax) and 0.5 mg/litre for Km.
This compares with a Vmax value for chloroform of 6.8 mg/h (Corley
et al., 1990), providing additional evidence for more rapid metabolism
and greater generation of reactive intermediates from BDCM than from
chloroform. Production of bromide from BDCM following treatment with
an inhibitor of CYP2E1, trans-dichloroethylene, increased the
apparent Km from 0.5 to 225 mg/litre, further demonstrating that
CYP2E1 is a major isoform involved in BDCM metabolism. The metabolism
model, derived from inhalation exposure data, was subsequently linked
to a multicompartment gastrointestinal tract submodel using estimates
of oral absorption rate constants determined by fitting blood and
exhaled breath chamber concentration-time curves obtained after gavage
of rats with BDCM. This model accurately predicted tissue dosimetry
and plasma bromide ion concentrations following oral exposure to BDCM
and can be utilized in estimating rates of formation of reactive
intermediates in target tissues.
4.1.2.7 Mode of action
As stated above, the metabolism of the THMs is believed to be a
prerequisite for the toxicity and carcinogenicity associated with
exposure to these DBPs. The primary target tissues for the THMs are
active sites of their metabolism, and treatments that increase or
decrease biotransformation also tend to cause parallel increases or
decreases in the toxicity induced by the THMs (Thornton-Manning et
al., 1993; US EPA, 1994b). The reactive intermediates generated from
the three brominated THM metabolic pathways react with macromolecules
to elicit both cytotoxic and genotoxic responses.
The cytotoxicity of the brominated THMs observed in the liver and
kidneys of exposed animals has been proposed to result from covalent
adducts formed between cellular proteins and lipids and
dihalocarbonyls or dihalomethyl free radicals. The adducts presumably
impair the function of these molecules and cause cell injury. BDCM
produced these adducts in in vitro incubations with hepatic and
renal microsomes to a significantly greater extent than did chloroform
(Gao & Pegram, 1992; Gao et al., 1996). Induction of lipid
peroxidation by free radical metabolites of reductive metabolism has
been proposed as another mechanism underlying THM cytotoxicity. Each
of the brominated THMs induced lipid peroxidation in rat liver
microsomes in vitro, which was maximal at low oxygen tensions (de
Groot & Noll, 1989).
As described above (section 4.1.2.6), it appears that direct
conjugation of BDCM and the other brominated THMs with GSH generates
mutagenic intermediates (Pegram et al., 1997), but this process has
not yet been demonstrated in mammals. However, the reaction was
dependent on a transfected rat enzyme (GST T1-1), and the human GST
T1-1 has been shown to catalyse GSH conjugation with the
dihalomethanes (Thier et al., 1996). The GC to AT transitions observed
in the Salmonella strain expressing GST T1-1 indicate that DNA
lesions on either guanine or cytosine occurred after exposure to
brominated THMs (De Marini et al., 1997). Based on similar reactions
by dihalomethanes, it can be proposed that S-(1,1-dihalomethyl)-GSH
is the product of GSH conjugation of brominated THMs, which then
reacts directly with guanine. Dechert & Dekant (1996) found that
S-(1-chloromethyl)-GSH reacts with deoxyguanosine to produce
S-[1-( N2-deoxyguanosinyl)-methyl]-GSH, and a methylguanosine
adduct was also formed from reactions of S-(1-acetoxymethyl)-GSH
with model nucleosides (Thier et al., 1993). In vivo covalent DNA
binding by 14C-BDCM has been observed in each of the cancer target
tissues for BDCM in the rat, but the nucleoside adducts have not yet
been identified (Bull et al., 1995).
An interplay of direct mutagenicity with cytotoxic responses
leading to regenerative hyperplasia may explain some, but not all, of
the carcinogenic effects of the brominated THMs. For example, renal
tubular cell hyperplasia coincident with tubular cell cancers was
observed in rats gavaged with BDCM for 2 years (NTP, 1987), but
necrosis and hyperplasia were not associated with the liver neoplasms
induced by BDCM in mice (NTP, 1987). Melnick et al. (1998)
recently revisited this issue and noted that high incidences of liver
tumours were observed with BDCM and DBCM at doses that had little or
no effect on hepatic regenerative hyperplasia. No evidence for
cytotoxic responses in the intestine was noted in the NTP study with
BDCM, but high incidences of intestinal carcinomas were reported (NTP,
1987). Therefore, while cytotoxic effects of BDCM may potentiate
tumorigenicity in certain rodent tissues at high dose levels, direct
induction of mutations by BDCM metabolites may also play a
carcinogenic role. The extent to which each of these processes
contributed to the induction of tumours observed in chronic animal
studies is at present unclear. Additional in vivo studies are
required to confirm the mechanism or mechanisms underlying brominated
THM-induced carcinogenesis.
4.1.3 Dibromochloromethane
4.1.3.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
Acute oral LD50s of 800 and 1200 mg of DBCM per kg of body
weight were reported by Bowman et al. (1978) for male and female ICR
Swiss mice, respectively, whereas Chu et al. (1980) found LD50s of
1186 and 848 mg/kg of body weight for male and female Sprague-Dawley
rats, respectively. A DBCM dose of 500 mg/kg of body weight produced
ataxia, sedation and anaesthesia in mice (Bowman et al., 1978). Hewitt
et al. (1983) dosed male Sprague-Dawley rats with DBCM by corn oil
gavage and found doses of 2450 mg/kg of body weight and above to be
lethal. No clinical evidence for significant liver or kidney toxicity
was found at sublethal doses.
Induction of acute renal toxicity by DBCM was studied by Kroll et
al. (1994a,b); a single intraperitoneal dose of 3 mmol/kg of body
weight resulted in elevated BUN, reductions in glomerular filtration
rate and renal concentrating ability, and interference with proximal
tubular secretion and reabsorption.
2) Short-term toxicity
Daily gavage of male and female CD-1 mice with DBCM in an aqueous
vehicle for 14 days produced hepatotoxicity in both sexes at the
highest dose of 250 mg/kg of body weight per day (Munson et al.,
1982). Depressed immune function was also observed in both sexes at
doses of 125 and 250 mg/kg of body weight per day, whereas the
50 mg/kg of body weight per day dose was without effect. Corn oil
gavage of DBCM to male CD-1 mice for 14 days (Condie et al., 1983) led
to observations of kidney and liver toxicity at a lower dose
(147 mg/kg of body weight per day) than had been observed with aqueous
delivery (Munson et al., 1982). In another 14-day corn oil gavage
study, NTP (1985) found that a dose of 500 mg/kg of body weight per
day was lethal to B6C3F1 mice, and doses of 500 and 1000 mg/kg of
body weight per day were lethal to F344/N rats. Dietary administration
of microencapsulated DBCM to Wistar rats for 1 month caused liver cell
vacuolization, with a LOAEL of 56 mg/kg of body weight per day and a
NOAEL of 18 mg/kg of body weight per day (Aida et al., 1992a).
DBCM-induced cardiotoxicity was reported in male Wistar rats
after short-term exposure (4 weeks of daily dosing with 0.4 mmol/kg of
body weight). Arrhythmogenic and negative chronotropic and dromotropic
effects were observed, as well as extension of atrioventricular
conduction times. Inhibitory actions of DBCM on calcium ion dynamics
in isolated cardiac myocytes were also noted.
In the NTP (1985) study, DBCM was administered by corn oil gavage
to F344/N rats and B6C3F1 mice (10 per sex per dose) for 13 weeks (5
days per week) at doses of 0, 15, 30, 60, 125 or 250 mg/kg of body
weight per day. The highest dose was lethal to 90% of the rats,
producing severe lesions and necrosis in kidney, liver and salivary
glands. Hepatocellular vacuolization indicative of fatty changes was
observed in male rats at doses of 60 mg/kg of body weight per day and
higher. In the mice, no DBCM-related effects were reported at doses of
125 mg/kg of body weight per day or lower. At the highest dose, fatty
liver and toxic nephropathy were noted in males, but not in females. A
NOAEL of 30 mg/kg of body weight per day can be derived from this
study.
A 90-day corn oil gavage study was conducted using Sprague-Dawley
rats and doses of 0, 50, 100 or 200 mg/kg of body weight per day
(Daniel et al., 1990b). Body weight gain was significantly depressed
in the high-dose groups to less than 50% and 70% of the controls in
males and females, respectively. Observations of liver damage included
elevated ALAT in mid- and high-dose males, centrilobular lipidosis
(vacuolization) in males at all doses and high-dose females, and
centrilobular necrosis in high-dose males and females. Kidney proximal
tubule cell degeneration was induced by DBCM in all high-dose rats and
to a lesser extent at 100 mg/kg of body weight per day in males and at
both 50 and 100 mg/kg of body weight per day in females.
3) Chronic toxicity
The chronic oral toxicity of DBCM was studied by NTP (1985) in
F344/N rats and B6C3F1 mice using corn oil gavage (5 days per week
for 104 weeks) and doses of 0, 40 or 80 mg/kg of body weight per day
for rats and 0, 50 or 100 mg/kg of body weight per day for mice. Liver
lesions, including fat accumulation, cytoplasmic changes and altered
basophilic staining, were observed in male and female rats at both
dose levels. The low-dose male mice in this study were lost as a
result of an overdosing accident. Compound-related hepatocytomegaly
and hepatic focal necrosis were observed in high-dose male mice, and
liver calcification (high dose) and fatty changes (both low and high
doses) were noted in female mice. Renal toxicity (nephrosis) was also
observed in male mice and female rats.
4.1.3.2 Reproductive and developmental toxicity
Borzelleca & Carchman (1982) conducted a two-generation
reproductive study of DBCM in ICR Swiss mice. Male and female mice at
9 weeks of age were maintained on drinking-water containing 0, 0.1,
1.0 or 4.0 mg of DBCM per ml, leading to average doses of 0, 17, 171
or 685 mg/kg of body weight per day. Fertility and gestational index
were reduced in the high-dose group for the F1 generations. Only
fertility was decreased (high-dose) in the F2 generation. At the mid
and high doses in both generations, litter size and the viability
index were decreased. Other effects included decreased lactation index
and reduced postnatal body weight. No dominant lethal or teratogenic
effects were observed in the F1 or F2 generations.
In a developmental study in rats conducted by Ruddick et al.
(1983), gavage of DBCM (0, 50, 100, or 200 mg/kg of body weight per
day) on gestational days 6-15 caused a depression of maternal weight
gain, but no fetal malformations.
4.1.3.3 Neurotoxicity
DBCM was tested for behavioural effects in male ICR mice by
Balster & Borzelleca (1982), who found no effect of treatments up to
100 mg/kg of body weight per day for 60 days. Dosing for 60 days with
400 mg/kg of body weight per day produced decreased response rates in
an operant behaviour test. Korz & Gattermann (1997) observed
DBCM-induced behavioural alterations in male golden hamsters exposed
either for 14 days to a dose of 5 mg/kg of body weight per day or
acutely to a single dose of 50 mg/kg of body weight. At the low dose,
subchronic treatment caused reduced aggressive behaviour during social
confrontation on day 14 as compared with vehicle-dosed controls.
Following the acute dose, increased locomotor activity on days 3-6 and
decreased wheel running on days 6-9 were observed, but no effects were
noted after day 9.
4.1.3.4 Toxicity in humans
Clinical case findings resulting from human exposure to DBCM have
not been reported.
4.1.3.5 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of DBCM and concluded that
there is inadequate evidence for its carcinogenicity in humans and
limited evidence for its carcinogenicity in experimental animals. The
compound was assigned to Group 3: DBCM is not classifiable as to its
carcinogenicity to humans (IARC 1991, 1999).
In a 104-week corn oil gavage study, DBCM was not carcinogenic in
F344 rats (50 per sex per dose) at doses of 0, 40 or 80 mg/kg of body
weight given 5 days per week (NTP, 1985). In female B6C3F1 mice,
however, DBCM significantly increased the incidence of hepatocellular
adenomas and the combined incidences of hepatocellular adenomas and
carcinomas at the high dose (100 mg/kg of body weight per day) (Table
13). The incidence of hepatocellular carcinomas was significantly
increased in male mice at the same dose. The low-dose (50 mg/kg of
body weight per day) male mice were lost midway through the study as a
result of an inadvertent overdose. NTP judged that these results
provided equivocal evidence of DBCM carcinogenicity in male mice and
some evidence of carcinogenicity in female mice.
Based on in vitro studies, mutagenic potency appears to
increase with the degree of bromine substitution in the THMs. DBCM is
mostly positive in tests employing closed systems to overcome the
problem of volatility (IARC, 1991, 1999; Pegram et al., 1997). In the
GST-transfected TA1535 strain (see section 4.1.1.3), DBCM is the most
potent THM, inducing greater than 10-fold more revertants per plate
than BDCM after exposure to THM vapour (3400 mg/m3 for DBCM; 2680
mg/m3 for BDCM [400 ppm]) (Pegram et al., 1996; DeMarini et al.,
1997). DBCM has given mostly positive results in eukaryotic test
systems (Loveday et al., 1990; IARC, 1991, 1999; McGregor et al.,
1991; Fujie et al., 1993), although there is less consistency in
results between the different assays when considered with or without
an exogenous metabolic system.
Data from in vivo studies are more equivocal. DBCM was positive
for SCE and chromosomal aberrations in mouse bone marrow (Morimoto &
Koizumi, 1983; Fujie et al., 1990) and in a newt micronucleus assay
(Le Curieux et al., 1995), but was negative for micronuclei and UDS in
the liver of rats (Ishidate et al., 1982; Hayashi et al., 1988;
Stocker et al., 1997). Potter et al. (1996) found that DBCM did not
Table 13. Frequencies of liver tumours in mice administered
dibromochloromethane in corn oil for 104 weeksa
Treatment (mg/kg Sex Adenoma Carcinoma Adenoma or
of body weight carcinoma
per day) (combined)
Vehicle control M 14/50 10/50 23/50
F 2/50 4/50 6/50
50 M -b - -
F 4/49 6/49 10/49
100 M 10/50 19/50c 27/50d
F 11/50c 8/50 19/50e
a Adapted from NTP (1985).
b Male low-dose group was inadequate for statistical analysis.
c P < 0.05 relative to controls.
d P < 0.01 (life table analysis); P = 0.065 (incidental tumour
tests) relative to controls.
e P < 0.01 relative to controls.
induce DNA strand breaks in the kidneys of male F344 rats following
seven daily doses of 1.5 mmol/kg of body weight.
These studies are summarized in Table 12.
4.1.3.6 Comparative pharmacokinetics and metabolism
The pharmacokinetics of DBCM have been studied the least among
the THMs. Mink et al. (1986) compared the absorption, distribution and
excretion of DBCM with those of the other THMs. A dose of 100 mg/kg of
body weight was administered orally in corn oil to male Sprague-Dawley
rats by gavage, and 150 mg/kg of body weight was administered
similarly to male B6C3F1 mice. The pattern of distribution and
elimination of DBCM was very similar to that observed with BDCM: rats
expired more of the dose than mice as a trapped organic component
presumed to be the parent compound (48% vs. 12%) and expired less as
carbon dioxide (18% vs. 72%) after 8 h. Total recovery from excised
organs was 1.4% in rats and 5.0% in mice, whereas less than 2% of the
dose was excreted in the urine in both species. Half-lives of DBCM in
rats and mice were estimated to be 1.2 h and 2.5 h, respectively.
The cytochrome P450-mediated metabolism of DBCM has not been
directly investigated. Presumably, metabolism proceeds via the same
routes of biotransformation as described for BDCM (section 4.1.2.6)
and chloroform (IPCS, 1994). Oxidative metabolism of DBCM would be
expected to yield a bromochlorocarbonyl rather than phosgene, and
reductive dehalogenation would produce a bromochloromethyl radical.
Anders et al. (1978) demonstrated in vivo carbon monoxide production
from DBCM in male Sprague-Dawley rats at a rate intermediate to those
of BDCM and bromoform. DBCM was very reactive in the Salmonella-GST
mutagenicity assay, indicating that it has greater potential for GSH
conjugation than BDCM (De Marini et al., 1997).
Pankow et al. (1997) reported the metabolism of DBCM to bromide
and carbon monoxide in rats after gavage of olive oil solutions
(0.4-3.1 mmol/kg of body weight). The DBCM concentrations in blood and
fat 6 h after the last of seven consecutive daily doses (0.8 mmol/kg
of body weight) were lower than at 6 h after a single gavage of this
dose, suggesting that more of the chemical was metabolized after the
seventh dose than after the single dose. The seven-dose regimen also
caused a 2-fold induction of a CYP2E1-specific activity. The
involvement of CYP2E1, CYP2B1/2 and GSH in DBCM metabolism was also
demonstrated.
4.1.3.7 Mode of action
Mechanistic issues for DBCM are similar to those addressed for
BDCM (sections 4.1.2.6 and 4.1.2.7). The greater propensity for the
metabolism of this compound and bromoform as compared with BDCM is
difficult to reconcile with its lower carcinogenicity in the NTP
(1985) bioassay. A possible explanation is less bioavailability
resulting from the greater lipophilicity of this compound and the use
of corn oil as the vehicle of administration. Greater lipophilicity
and reactivity of this compound or its metabolites (i.e., the
bromochlorocarbonyl metabolite) may also prevent it from reaching
critical target sites. It is also of note that DBCM did not induce
carcinomas of the large intestine in rats in the NTP studies using
corn oil vehicle, whereas both BDCM and bromoform did induce these
tumours.
4.1.4 Bromoform
4.1.4.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
Among the brominated THMs, bromoform is the least potent as a
lethal acute oral toxicant. The acute oral LD50s for bromoform were
1400 and 1550 mg/kg of body weight in male and female mice,
respectively (Bowman et al., 1978), and 1388 and 1147 mg/kg of body
weight in male and female rats, respectively (Chu et al., 1980).
Bromoform was also a less potent anaesthetic than BDCM and DBCM: a
1000 mg/kg of body weight dose was required to produce this effect in
mice. Intraperitoneal administration of bromoform in a corn oil
vehicle at doses of 25-300 µl/kg of body weight produced no
significant elevations of serum enzymes indicative of liver damage
(Agarwal & Mehendele, 1983).
Bromoform was included in the acute renal toxicity studies of
Kroll et al. (1994a,b) but was ranked as the least potent among the
THMs in general disruption of renal function. A single intraperitoneal
dose (3 mmol/kg of body weight) did, however, induce significant
decreases in glomerular filtration rate, renal concentrating ability,
and tubular secretion and reabsorption. Tubule function was affected
as early as 8 h after dosing, whereas the other effects were observed
at 24-48 h.
2) Short-term toxicity
Male and female CD-1 mice were gavaged daily with bromoform (50,
125 or 250 mg/kg of body weight) in an aqueous vehicle for 14 days,
leading to liver toxicity and a decrease in antibody-forming cells
only at the highest dose (Munson et al., 1982). The magnitude of the
liver effects was less than that observed with the other THMs, and BUN
was not elevated by bromoform. Condie et al. (1983) noted both liver
and kidney toxicity in CD-1 mice after dosing with 289 mg of bromoform
per kg of body weight per day in corn oil for 14 days, whereas no
significant effects were found at 72 and 145 mg/kg of body weight per
day. NTP (1989a) also conducted a 14-day corn oil gavage study with
bromoform and found that doses of 600 and 800 mg/kg of body weight per
day were lethal to both sexes of F344/N rats. Mice given doses of 400
mg/kg of body weight and higher had raised stomach nodules.
Microencapsulated bromoform was added to the diet of Wistar rats for 1
month, producing liver damage (Aida et al., 1992a).
Bromoform was given to F344/N rats (10 per sex per dose) and
B6C3F1 mice (10 per sex per dose) by corn oil gavage for 13 weeks
(5 days per week) at doses of 0, 25, 50, 100 or 200 mg/kg of body
weight per day, with an added dose of 400 mg/kg of body weight per day
for mice only (NTP, 1989a). The only significant finding, hepatic
vacuolation, was observed in male rats, but not in female rats, at
doses of 50 mg/kg of body weight per day and higher, and in male mice,
but not in female mice, at doses of 200 and 400 mg/kg of body weight
per day. The NOAEL in the rat study was 25 mg/kg of body weight per
day, whereas that in the study in mice was 100 mg/kg of body weight
per day.
3) Chronic toxicity
Bromoform was administered in drinking-water (containing 0.25%
Emulphor) to male F344 rats and B6C3F1 mice for 1 year, and clinical
indicators of kidney toxicity were examined (Moore et al., 1994).
Water containing bromoform concentrations of 0.12, 0.6 and 1.2 g/litre
for rats and 0.08, 0.4, and 0.8 g/litre for mice resulted in average
daily doses of 6.2, 29 or 57 mg/kg of body weight for rats and 8.3, 39
or 73 mg/kg of body weight for mice. Several indicators of tubular and
glomerular damage were elevated at each treatment level in mice, and
mice appeared more susceptible to the nephrotoxic effects of bromoform
than to those of BDCM (see section 4.1.2.1). As in mice, urinary
protein was increased in all rat dose groups, but little evidence of
loss of tubule function was observed in rats.
Two-year investigations of bromoform toxicity were conducted by
administering doses of 0, 100 or 200 mg/kg of body weight by corn oil
gavage, 5 days per week for 103 weeks, to F344/N rats (50 per sex per
dose) and female B6C3F1 mice (50 per dose) (NTP, 1989a). Male mice
(50 per dose) received doses of 0, 50 or 100 mg/kg of body weight per
day. Survival of high-dose male rats and both dose groups of female
mice was significantly lower than that of the vehicle controls. Focal
or diffuse fatty change of the liver was observed in a dose-dependent
fashion in both sexes of rats, and active chronic hepatic inflammation
was found in male and high-dose female rats. Minimal necrosis of the
liver was noted in high-dose male rats. In mice, the incidence of
fatty changes of the liver was increased in females, but not in males,
at both dose levels. Follicular cell hyperplasia of the thyroid gland
was observed in high-dose female mice. The different target organ
(liver) in this study as compared with the drinking-water study of
Moore et al. (1994), in which renal effects predominated, suggests
that the vehicle and mode of administration can affect which tissues
are affected by bromoform.
4.1.4.2 Reproductive and developmental toxicity
Ruddick et al. (1983) conducted a teratological investigation of
bromoform in Sprague-Dawley rats (15 per group). Bromoform was
administered by gavage at doses of 50, 100 or 200 mg/kg of body weight
per day on gestation days 6-15. Evidence of a fetotoxic response was
observed, but there were no fetal malformations. Interparietal
deviations were, however, noted in the mid- and high-dose groups. NTP
(1989b) utilized the same bromoform doses given by gavage for 105 days
to 20 male-female pairs of Swiss CD-1 mice to examine effects on
fertility and reproduction. There was no detectable effect of
bromoform on fertility, litters per pair, live pups per litter,
proportion of pups born alive, sex of live pups or pup body weights.
Bromoform was also found to induce full-litter resorptions in pregnant
F344 rats when administered orally on gestation days 6-15, but at
higher doses (150 and 200 mg/kg of body weight per day) than those
required to produce the same effect for BDCM (Narotsky et al., 1993).
4.1.4.3 Neurotoxicity
Bromoform was included in the mouse behavioural study of Balster
& Borzelleca (1982). Doses of 9.2 mg/kg of body weight per day for up
to 90 days had no effect on the outcome of several behavioural tests,
and 100 mg/kg of body weight per day for 30 days did not deter passive
avoidance learning. However, mice receiving either 100 or 400 mg/kg of
body weight per day for 60 days exhibited decreased response rates in
an operant behaviour test.
4.1.4.4 Toxicity in humans
Bromoform was used in the late 19th and early 20th centuries as a
sedative for children with whooping cough. Patients were typically
given doses of one drop (approximately 180 mg) 3-6 times per day
(Burton-Fanning, 1901), which usually resulted in mild sedation in the
children. A few rare instances of death or near-death were reported
but were believed to be due to accidental overdoses (Dwelle, 1903).
These clinical observations have been used to estimate a lethal dose
for a 10- to 20-kg child to be about 300 mg/kg of body weight and an
approximate minimal dose for sedation to be 50 mg/kg of body weight
per day (US EPA, 1994b).
4.1.4.5 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of bromoform and concluded
that there is inadequate evidence for its carcinogenicity in humans
and limited evidence for its carcinogenicity in experimental animals.
The compound was assigned to Group 3: bromoform is not classifiable as
to its carcinogenicity to humans (IARC, 1991, 1999).
Two-year studies of bromoform carcinogenicity were conducted by
administering doses of 0, 100 or 200 mg/kg of body weight in corn oil
by gavage, 5 days per week, to groups of F344/N rats (50 per sex per
group) and female B6C3F1 mice (50 per group) and doses of 0, 50 or
100 mg/kg of body weight to male mice (NTP, 1989a). No neoplastic
effects were associated with the exposure of mice to bromoform. In
rats, however, intestinal carcinomas were induced by bromoform, as had
also been observed with BDCM. The uncommon adenomatous polyps or
adenocarcinomas (combined) of the large intestine (colon or rectum)
were observed in 3 out of 50 high-dose male rats and in 8 out of 50
high-dose female rats as compared with 0 out of 50 rats in the male
controls and 0 out of 48 rats in the female controls (Table 14).
Because these tumours are very rare in the rat, these findings were
considered significant, and the NTP therefore concluded that there was
clear evidence for carcinogenic activity in female rats and some
evidence in male rats.
Bromoform, in common with the other brominated THMs, is largely
positive in bacterial assays of mutagenicity conducted in closed
systems (Zeiger, 1990; IARC, 1991, 1999). In the GST-transfected
Salmonella typhimurium TA1535 strain (see section 4.1.2.5),
bromoform produced about 5 times more revertants than BDCM at
comparable exposure levels (Pegram et al., 1997). As with BDCM, the
mutations were almost exclusively GC to AT transitions (DeMarini et
al., 1997). In prokaryotic tester strains, bromoform induced mutations
without metabolic activation in S. typhimurium strain TA100 (Simmon
& Tardiff, 1978; Ishidate et al., 1982; NTP, 1989a; Le Curieux et al.,
1995), with and without activation in TA98 (NTP, 1989a; Zeiger, 1990)
and with microsomal activation in TA97 (NTP, 1989a). In eukaryotic
test systems, bromoform is largely positive (IARC, 1991; Fujie et al.,
1993). As with bacterial assays, bromoform appeared more potent than
the other brominated THMs (Morimoto & Koizumi, 1983; Banerji &
Fernandes, 1996).
In vivo studies (summarized in Table 12) have given
contradictory results. Bromoform was positive and negative in
Drosophila (Woodruff et al., 1985). It was positive in the newt
micronucleus test (Le Curieux et al., 1995) and gave increased SCE and
chromosomal aberrations in mouse and rat bone marrow cells (Morimoto &
Koizumi, 1983; Fujie et al., 1990). It gave negative results in mouse
bone marrow (Hayashi et al., 1988; Stocker et al., 1997), in the rat
Table 14. Tumour frequencies in rats exposed to bromoform in corn oil
for 2 yearsa
Animal/tissue/tumour Tumour frequency
Control 100 mg/kg of 200 mg/kg of
body weight body weight
per day per day
Male rat
Large intestine
Adenocarcinoma 0/50 0/50 1/50
Polyp (adenomatous) 0/50 0/50 2/50
Female rat
Large intestine
Adenocarcinoma 0/48 0/50 2/50
Polyp (adenomatous) 0/48 1/50 6/50
a Adapted from NTP (1989a).
liver UDS assay (Pereira et al., 1982; Stocker et al., 1997) and in
the dominant lethal assay (Ishidate et al., 1982). In the studies
carried out by the NTP (1989a), it was positive for micronuclei and
SCE, but negative for chromosomal aberrations in mouse bone marrow.
Potter et al. (1996) found that bromoform did not induce DNA strand
breaks in the kidneys of male F344 rats following seven daily doses of
1.5 mmol/kg of body weight.
4.1.4.6 Comparative pharmacokinetics and metabolism
14C-Bromoform pharmacokinetics were examined in Sprague-Dawley
rats and B6C3F1 mice in a study that included all four of the common
THMs that occur as DBPs (Mink et al., 1986). Recoveries of 14C-label
8 h after gavage dosing were 79% in rats and 62% in mice. The
distribution and elimination of bromoform resembled those of
chloroform rather than those of the other brominated THMs. The
percentage of 14C-label recovered from excised organs and tissues was
2.1% in rats and 12.2% in mice. Tissue levels of 14C in mice were
substantially greater than those observed for the other brominated
THMs. Urinary excretion of label after 8 h (2.2-4.6%) was also greater
for bromoform than for BDCM and DBCM. Bromoform (and organic
metabolite) elimination via exhaled breath was greater than that for
all other THMs in the rat (67%), but less than that for all other THMs
in the mouse (6%). The estimated half-life of bromoform was 0.8 h in
rats and 8 h in mice.
The cytochrome P450-dependent metabolism of bromoform was studied
by Anders et al. (1978), who found that bromoform was converted to
carbon monoxide in vivo in male Sprague-Dawley rats at a rate
significantly greater than were the other three THMs. Evidence was
presented suggesting that some of the carbon monoxide arose from the
oxidative metabolism of bromoform to a dibromocarbonyl and subsequent
reactions with GSH. It is possible that some of the carbon monoxide
was also generated from reductive metabolism of bromoform, as
indicated by in vitro results of Wolf et al. (1977) from anaerobic
incubations using hepatic preparations derived from
phenobarbital-treated rats. Carbon monoxide was produced from
anaerobic bromoform metabolism in this study at much greater levels
than from chloroform metabolism. Among the four THMs tested by Mink et
al. (1986), bromoform exhibited the least metabolism to carbon dioxide
(4% in rats and 40% in mice in 8 h). Free radicals from the in vivo
reductive metabolism of bromoform were detected by ESR spin-trapping
after dosing phenobarbital-treated rats (Tomasi et al., 1985). More
radicals were generated from bromoform in vivo than from any other
bromochlorinated THM, which is consistent with computational chemistry
predictions that bromoform would have the greatest reductive potential
of the THMs (Waller & McKinney, 1993). Knecht & Mason (1991) also
detected the in vivo production of radicals from bromoform in the
bile of rats, but only after the induction of hypoxia.
Bromoform, like DBCM, has a much greater potential than BDCM to
be conjugated by GSH to form a mutagenic intermediate. Base pair
revertants were produced in a dose-dependent fashion in
GST-transfected Salmonella typhimurium strain TA1535 by bromoform
and BDCM, but not by chloroform (Pegram et al., 1996, 1997; De Marini
et al., 1997). At an exposure concentration of 33 100 mg/m3 for
bromoform and 22 800 mg/m3 for BDCM (3200 ppm), GST-dependent
revertants per plate induced by bromoform and BDCM averaged 373 and
1935, respectively, compared with a control rate of 23 per plate
(Pegram et al., 1996).
4.1.4.7 Mode of action
The basic mechanisms of action for bromoform are similar to those
described for BDCM (section 4.1.2.7). Although bromoform seems to have
a greater propensity for metabolism and is a more potent mutagen than
BDCM, it appears to be a less potent toxicant and carcinogen based on
the results of the NTP (1985, 1987) bioassays and numerous other
in vivo studies of toxicity. As with DBCM, a possible explanation is
less bioavailability resulting from the greater lipophilicity of this
compound and the use of corn oil as the vehicle of administration.
This concept may be supported by the occurrence of bromoform-induced
tumours in the intestinal tract, but not in the liver or kidneys.
Greater lipophilicity and reactivity of bromoform metabolites may also
prevent it from reaching critical target sites. Moreover, when
bromoform was injected intraperitoneally, its metabolism was greater
than that of the other THMs (Anders et al., 1978; Tomasi et al.,
1985); when administered by corn oil gavage, however, bromoform was
the least metabolized THM (Mink et al., 1986). Data from studies of
oral exposure to bromoform in aqueous solutions are therefore
required.
4.2 Haloacids
Like the THMs, the haloacids produced in the chlorination of
drinking-water consist of a series of chlorinated and brominated
forms. To date, the chlorinated acetic acids have been more thoroughly
characterized toxicologically than their brominated analogues. As
discussed in earlier chapters, the dihaloacetates and trihaloacetates
occur in significantly higher concentrations than the
monohaloacetates. The present review will emphasize the di- and
trihaloacetates as the dominant forms of the haloacids found in
drinking-water and the ones for which extensive toxicological data
have been developed. The probable existence of many longer-chain
halogenated acids in chlorinated drinking-water may be surmised from
studies of chlorinated humic acids (reviewed by Bull & Kopfler, 1991).
Few of these compounds have been extensively studied toxicologically.
However, brief reference will be made to those compounds that are
known to share some of the effects of the HAAs on intermediary
metabolism.
4.2.1 Dichloroacetic acid (dichloroacetate)
Before beginning discussions of available toxicological
evaluations of DCA, it is important to point out that this compound
exists in drinking-water as the salt, despite the fact that it is
widely referred to as dichloroacetic acid. DCA has a p Ka of 1.48 at
25°C (IARC, 1995). As a consequence, it occurs almost exclusively in
the ionized form at the pHs found in drinking-water (broadly speaking,
a pH range of 5-10). Failure to recognize this has resulted in a
number of studies that have employed the free acid in test systems. At
low doses, the buffering capacity of the physiological system can
neutralize acid, and the measured activity may in fact be
representative. However, most of the experimentation has been
conducted using doses ranging from 50 to 1000 mg/kg of body weight
in vivo or in the mmol/litre range in vitro. Therefore, the
applicability of the results of such studies to estimating human risks
will be uncertain because of the large pH artefacts that can be
expected when administering these quantities of a strong acid.
DCA has been shown to produce developmental, reproductive, neural
and hepatic effects in experimental animals. In general, these effects
occur when the compound has been administered at high dose rates, at
which there is evidence to indicate that the metabolic clearance of
DCA is substantially inhibited. This has important implications in
attempting to associate these toxicities with the low doses that are
obtained in drinking-water.
Because it was being developed as a potential therapeutic agent,
there is a toxicological literature that precedes DCA's discovery as a
by-product of chlorination. The present review discusses these early,
more general explorations of DCA's effects on intermediary metabolism
before proceeding to descriptions of studies designed to more
specifically study its toxicology.
4.2.1.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
DCA is not very toxic when administered acutely to rodents.
Woodard et al. (1941) reported LD50s of 4.5 and 5.5 g/kg of body
weight in rats and mice, respectively, for DCA administered as the
sodium salt. This is roughly in the same range as the LD50s for
acetic acid. There is reason to believe that other species, most
specifically the dog, may be more sensitive because of some
repeated-dose experiments discussed below; however, no specific data
seem to have been reported in the literature.
2) Short-term toxicity
Katz et al. (1981) published the first substantive evaluation of
DCA's subchronic toxicity in rats and dogs. DCA was administered by
gavage at 0, 125, 500 or 2000 mg/kg of body weight per day to rats
(10-15 per sex per group) and at 0, 50, 75 or 100 mg/kg of body weight
per day to dogs (3-4 per sex per group) for 3 months. One of three
female dogs died at 75 mg/kg of body weight per day, and one of four
male dogs died at 100 mg/kg of body weight per day. The most overt
toxicity in rats was hindlimb paralysis at the highest dose. Clinical
chemistry indicated significant increases in total and direct
bilirubin at 500 and 2000 mg/kg of body weight per day in rats, and
relative liver weights were significantly increased at all doses. In
dogs, an increase in the incidence of haemosiderin-laden Kupffer cells
was noted at all dose rates. Histopathological changes were observed
in the brain and testes of both species. In rats, oedematous brain
lesions were seen at 60% incidence (primarily in the cerebrum, but
also in the cerebellum) at the lowest dose and at 100% in the two
higher doses in both sexes. Slight to moderate vacuolation of
myelinated tracts was observed at all doses in both rats and dogs.
Testicular germinal epithelial degeneration was observed in rats at
doses of 500 mg/kg of body weight per day and above and at all doses
in dogs, with severity increasing with dose. In dogs, a high incidence
of ocular anomalies was observed, and lenticular opacities were found
to be irreversible upon suspension of treatment with DCA. In both rats
and dogs, glucose and lactate levels were suppressed in a dose-related
manner.
Bull et al. (1990) examined the effects of DCA on the liver of
B6C3F1 mice administered 1 or 2 g of DCA per litre (approximately 170
and 300 mg/kg of body weight per day) in their drinking-water, with
exposure lasting up to 1 year. As had been at least alluded to in
earlier studies, DCA was found to produce a severe hepatomegaly in
mice at concentrations in drinking-water of 1 g/litre and above. The
hepatomegaly could be largely accounted for by large increases in cell
size (cytomegaly). In shorter-term experiments, it was determined that
treatment with DCA produced only minor changes in the labelling index
of hepatocytes using a pulse dose of tritiated thymidine (Sanchez &
Bull, 1990). In general, increases in labelling indices were seen in
areas of acinar necrosis in the liver. Hepatocytes from these mice
stained very heavily for glycogen using periodic acid/Schiff's reagent
(PAS). The accumulation of glycogen began to occur with as little as
1-2 weeks of treatment (Sanchez & Bull, 1990), but became
progressively more severe with time (Bull et al., 1990). Sanchez &
Bull (1990) noted that these effects could not be replicated by
exposing mice to the metabolites of DCA -- glycolate, glyoxylate or
oxalate -- in the drinking-water.
Davis (1990) investigated the effects of DCA and TCA treatments
on male and female Sprague-Dawley rats for up to 14 days. The authors
reported decreased plasma glucose and lactic acid concentrations in
both plasma and liver. There were some inconsistencies in the
reporting of the doses in this study. However, the author has provided
the correct doses, which were 120 and 316 mg/kg of body weight per day
(M.E. Davis, personal communication, 1996). This places the results in
a context that is more consistent with those of other studies (e.g.,
Katz et al., 1981).
Mather et al. (1990) found increases in liver weight in rats
treated with DCA at 5 g/litre (350 mg/kg of body weight per day) in
their drinking-water for 90 days. Relative liver and kidney weights
were increased at concentrations of 0.5 g/litre (35 mg/kg of body
weight per day) and above. PAS staining of liver sections revealed
accumulation of glycogen in severely swollen hepatocytes, which was
quite marked with 5 g DCA/litre. The treatment caused small,
statistically significant increases in alkaline phosphatase (AP) and
ALAT in serum. However, these changes were too small to be of clinical
importance. DCA was found to approximately double the activity of
cyanide-insensitive acyl coenzyme A (CoA) activity in the liver at
5 g/litre, indicating some induction of peroxisome synthesis.
Cicmanec et al. (1991) examined the subchronic effects of DCA in
dogs. DCA was administered at doses of 0, 12.5, 39.5 or 72 mg/kg of
body weight per day for 90 days to groups of five males and five
females. Liver weights were significantly increased in a dose-related
manner, beginning with the lowest dose, and kidney weight was
increased at the highest dose. This was accompanied by
histopathological observation of vacuolar changes in the liver and
haemosiderosis. The pancreas displayed evidence of chronic
inflammation and acinar degeneration at the two highest doses.
Testicular degeneration was observed in virtually all the male dogs
administered DCA. This pathology was not observed in the control
animals. Vacuolization of white myelinated tracts in the cerebrum or
cerebellum was observed at all doses, and vacuolar changes were
observed in the medulla and spinal cord of male dogs. Although the
vacuolization was present in all dose groups, the authors described it
as being mild. These authors failed to find evidence of the lenticular
opacities that had been previously reported by Katz et al. (1981)
under very similar treatment conditions. They also found fairly
consistent decreases in erythrocyte counts and haemoglobin
concentrations at 72 mg/kg of body weight per day in both male and
female dogs. As found by others in rat studies, evidence of hindlimb
paralysis was reported, but the effect was expressed only sporadically
in dogs.
A number of other studies are consistent with the pattern of
toxicological effects produced by DCA described above. Included in
this group are the study of Bhat et al. (1991), which adds
observations of enlarged portal veins, deposition of collagen in the
area of the portal triads and some similar lesions in the vasculature
of the lung in rats. This study also noted atrophic testes and focal
vacuolation and gliosis in the brain. Only a single dose level of 10.5
g/litre of drinking-water (approximately 1100 mg/kg of body weight per
day) was utilized in this 90-day study, a level that substantially
exceeds concentrations reported to reduce water and food consumption
in other studies (Bull et al., 1990).
More recent studies have more closely examined the dose-response
relationships involved in the accumulation of glycogen in the liver of
male B6C3F1 mice treated with DCA in drinking-water at levels ranging
from 0.1 to 3 g/litre (approximately 20-600 mg/kg of body weight per
day) for up to 8 weeks (Kato-Weinstein et al., 1998). Significant
increases in the glycogen content of the liver of mice were seen with
concentrations as low as 0.5 g/litre (100 mg/kg of body weight per
day) in their drinking-water, with a small, but insignificant,
increase being observed at 0.2 g/litre (40 mg/kg of body weight per
day). Glycogen concentrations in the liver reached maximum levels
within 1 week of treatment with concentrations in drinking-water of
1 g/litre and above. At this early stage, the glycogen that
accumulates is subject to mobilization by fasting. With continued
treatment, the glycogen that is deposited becomes increasingly
resistant to mobilization until approximately 8 weeks of treatment,
when the glycogen contents of the livers of DCA-treated mice are not
different in fasted and non-fasted states.
The enzymatic basis of hepatic glycogen accumulation remains
unclear. DCA treatment has no effect on the total amount of either
form of glycogen synthase in the liver (e.g.,
glucose-6-phosphate-dependent vs. glucose-6-phosphate-independent
activity). The proportion of glycogen synthase in the active form was
significantly decreased in mice treated with DCA for as little as
1 week. The amount of phosphorylase in the active form appeared
unaltered by treatment. Such changes could indicate that a feedback
inhibition may have developed on the synthesis of glycogen as a result
of its accumulation in hepatocytes.
Carter et al. (1995) examined the time course of DCA's effects in
the liver of B6C3F1 mice at concentrations of 0, 0.5 or 5 g/litre in
drinking-water for up to 30 days. As reported in prior studies, the
high-dose group displayed severe liver hypertrophy. However, a
smaller, but consistent, increase in liver weight became evident with
as little as 10 days of treatment at 0.5 g/litre. Even at this
relatively low dose, some hepatocytes appeared to have lost nuclei or
possessed nuclei that had undergone some degree of karyolysis. These
experiments also appeared to rule out cytotoxicity and reparative
hyperplasia as consistent features of DCA's effects. The authors
suggested that this was in apparent contrast to the earlier
observations of Sanchez & Bull (1990). However, Sanchez & Bull (1990),
in a 14-day study in mice, noted that these effects were closely
associated with what appeared to be infarcted areas in the liver of
mice treated with high doses of DCA rather than cytotoxicity. These
infarcts are thought to be secondary to the severe swelling of
hepatocytes that results from DCA treatment. This interpretation is
supported by the apparent lack of cytotoxic effects of DCA in isolated
hepatocytes of both mice and rats at concentrations in the mmol/litre
range (Bruschi & Bull, 1993). Subsequently, these infarcted areas have
been identified as acinar necrosis (ILSI, 1997), which occurs in a
somewhat random fashion when high concentrations of DCA (> 2
g/litre) are administered for prolonged periods of time (Stauber &
Bull, 1997).
It is important to note that the dog appears to be very sensitive
to the effects of DCA on the liver; substantial increases in liver
weights are observed at daily doses as low as 12.5 mg/kg of body
weight per day for a 90-day period (Cicmanec et al., 1991). By
comparison, the lowest effect level noted in mice is 0.5 g/litre of
drinking-water, which approximates 70-100 mg/kg of body weight per day
(Carter et al. 1995), and the lowest effect level noted in rats is 125
mg/kg of body weight per day (Katz et al., 1981).
4.2.1.2 Reproductive effects
DCA produces testicular toxicity when administered at high doses
in drinking-water. These effects were first noted in studies of the
general toxicity of DCA (Katz et al., 1981) and were discussed above
(section 4.2.1.1). Cicmanec et al. (1991) followed up on these
original observations in dogs and detected degeneration of the
testicular epithelium and syncytial giant cell formation at doses as
low as 12.5 mg/kg of body weight.
Toth et al. (1992) examined DCA's ability to modify male
reproductive function in Long-Evans rats given 0, 31, 62 or 125 mg/kg
of body weight per day by gavage for 10 weeks. Reduced weights of
accessory organs (epididymis, cauda epididymis and preputial gland)
were observed at doses as low as 31 mg/kg of body weight per day.
Epididymal sperm counts were found to be depressed and sperm
morphology was increasingly abnormal at doses of 62 mg/kg of body
weight per day and above. These latter effects were accompanied by
changes in sperm motion. Fertility was tested in overnight matings and
was found to be depressed only at the highest dose evaluated,
125 mg/kg of body weight per day.
The testicular toxicity of DCA was evaluated in adult male rats
given both single and multiple (up to 14 days) oral doses. Delayed
spermiation and altered resorption of residual bodies were observed in
rats given single doses of 1500 or 3000 mg/kg of body weight; these
effects persisted to varying degrees on post-treatment days 2, 14 and
28. Delayed spermiation and formation of atypical residual bodies were
also observed on days 2, 5, 9 and 14 in rats dosed daily with 54, 160,
480 or 1440 mg/kg of body weight per day. Distorted sperm heads and
acrosomes were observed in step 15 spermatids after administration of
doses of 480 and 1440 mg/kg of body weight per day for 14 days.
Decreases in the percentage of motile sperm occurred after 9 days at
doses of 480 and 1440 mg/kg of body weight per day and after 14 days
at 160 mg/kg of body weight per day. Increased numbers of fused
epididymal sperm were observed on days 14, 9 and 5 in rats dosed with
160, 480 or 1440 mg/kg of body weight per day, respectively; other
morphological abnormalities occurred at 160 mg/kg of body weight per
day and higher. On day 14, a significant decrease in epididymis weight
was observed at 480 and 1440 mg/kg of body weight per day, and
epididymal sperm count was decreased at 160 mg/kg of body weight per
day and higher. These studies demonstrate that the testicular toxicity
induced by DCA is similar to that produced by the analogue DBA (see
section 4.2.3.2). However, the testicular toxicity of DCA is less
severe at equal mol/litre concentrations. Moreover, the DCA-induced
testicular lesions occur at lower doses as the duration of dosing
increases, indicating the importance of using low-dose subchronic
exposures to assess the health risk of prevalent DBPs (Linder et al.,
1997a).
DCA was found to be more potent than TCA in inhibiting
in vitro fertilization of B5D2F1 mouse gametes (Cosby & Dukelow,
1992). For DCA, the percentage of gametes fertilized dropped from
87.0% to 67.3% at the lowest concentration tested, 100 mg/litre. No
effects were noted for TCA at 100 mg/litre; at 1000 mg/litre, however,
71.8% of the gametes were fertilized. Both responses were
statistically different from those of their concurrent control groups.
4.2.1.3 Developmental effects
DCA has been shown to induce soft tissue abnormalities in fetal
rats when administered by gavage in a water vehicle to their dams
during gestation days 6-15 (Smith et al., 1992). These effects were
observed at doses of 140 mg/kg of body weight per day and above and
were not observed at 14 mg/kg of body weight per day. The heart was
the most common target organ. An interventricular septal defect
between the ascending aorta and the right ventricle was most commonly
observed. Urogenital defects (bilateral hydronephrosis and renal
papilla) and defects of the orbit were also observed. In a subsequent
publication, these authors identified the most sensitive period to be
days 12-15 of gestation (Epstein et al., 1992).
Some limited experimentation has been conducted to evaluate the
developmental effects of DCA in rat whole embryo culture (Saillenfait
et al., 1995). The applicability of these data to the in vivo
situation is difficult to evaluate based on the very high
concentrations that were utilized in these studies. At a concentration
of 1 mmol/litre, DCA retarded growth of the embryos by a variety of
measures. It required 2.5 mmol/litre to induce brain and eye defects
and 3.5 mmol/litre to produce abnormalities in other organ systems.
4.2.1.4 Neurotoxicity
Yount et al. (1982) demonstrated that DCA administered to rats in
their feed at doses of 2.5-4 mmol/kg of body weight per day (322-716
mg/kg of body weight per day) produced hindlimb weakness and abnormal
gait within 2-4 weeks of treatment. These effects were associated with
significant reductions in nerve conduction velocity and a decrease in
the cross-sectional area of the tibial nerve.
4.2.1.5 Toxicity in humans
DCA was first investigated as a potential orally effective
hypoglycaemic agent. Stacpoole et al. (1978) found that DCA,
administered in doses of 3-4 g of dichloroacetate as the sodium salt
per day for 6-7 days (43-57 mg/kg of body weight per day for a 70-kg
person), significantly reduced fasting hyperglycaemia in patients with
diabetes mellitus alone or in combination with hyperlipoproteinaemia
by 24%. In addition, plasma lactic acid concentrations dropped by 73%
and plasma alanine concentrations by 82%. Only mild sedation was
noticed by some of the patients, and there was no evidence of altered
blood counts or prothrombin time. There was a slower, but significant,
decline in plasma triglyceride levels and less consistent effects on
plasma cholesterol. ß-Hydroxybutyrate concentrations in plasma
increased significantly and continuously over the 6 days of
administration. Uric acid concentrations in serum increased and those
in urine decreased, reflecting a 50% decrease in urinary clearance.
Further details on the early investigations of DCA as an oral
antidiabetic agent have been reviewed extensively (Crabb et al., 1981;
Stacpoole, 1989; Stacpoole & Greene, 1992) and will not be dwelt on
extensively in the present review. The main reason that DCA was not
fully developed for this application was that longer-term
administration to patients induced a reversible polyneuropathy (Moore
et al., 1979b; Stacpoole, 1989). As discussed above, similar pathology
was reported in experimental animals by several authors, which added
significant weight to this observation. Subsequent work in rats
suggested that thiamine deficiency contributed to the development of
peripheral neuropathy (Stacpoole et al., 1990). However, a recent
report (Kurlemann et al., 1995) indicated that supplementing the diet
with 100 mg of thiamine daily did little to ameliorate the development
of polyneuropathy of a patient treated with DCA at a dose of 100 mg/kg
of body weight for 20 weeks.
Subsequently, DCA has been investigated extensively in the
treatment of congenital lactic acidosis (Coude et al., 1978; Stacpoole
& Greene, 1992; Toth et al., 1993), a disease that is frequently
fatal. More recently, DCA has been evaluated with success in the
treatment of lactic acidosis associated with severe malaria in
children (Krishna et al., 1995). It is also apparently effective in
treatment of lactic acidosis associated with liver transplantation
(Shangraw et al., 1994). DCA has been reported to improve psychiatric
symptoms, as well as to markedly decrease elevated lactic acid levels
in an individual with mitochondrial myopathy (Saijo et al., 1991).
Doses of up to 100 mg/kg of body weight per day were administered. In
a second study, DCA was shown to reverse lesions to the basal ganglia
in a patient with complex I deficiency as measured by computerized
tomography (CT) scan and magnetic resonance imaging (MRI) (Kimura et
al., 1995). However, a second patient with pyruvate dehydrogenase
complex deficiency displayed only transient improvement.
Based on work done in animals, several studies have examined the
effects of DCA on cardiovascular function under a variety of disease
conditions and altered physiological states. DCA was found to
stimulate myocardial lactate consumption and improve left ventricular
efficiency in 10 patients with congestive heart failure (Bersin et
al., 1994). On the other hand, although DCA decreased blood lactate
levels in patients with congestive heart failure, it had no effect on
exercise time, peak exercise oxygen consumption or flow to the
exercising leg (Wilson et al., 1988). In normal individuals, DCA
decreased blood lactate levels when exercising at less than 80% of
average maximal oxygen consumption, but it did not affect blood
lactate concentrations at exhaustion (Carraro et al., 1989).
These studies show that DCA has some beneficial effects in a
variety of metabolic diseases. Acutely, DCA produces little in the way
of risk, because short exposures are without apparent adverse effect.
However, despite the limited number of subjects that have been
studied, the results, when coupled with the results of animal
experiments, indicate relatively strongly that DCA is neurotoxic in
humans. The delayed induction of these toxicities may be attributable
in part to the fact that systemic concentrations of DCA can be
expected to sharply increase with prolonged treatment, at least at the
high doses used therapeutically, typically in the range 25-100 mg/kg
of body weight. There is no human evidence of adverse effects at lower
exposures to DCA (e.g., those that would be derived from drinking
chlorinated water).
4.2.1.6 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of DCA and, based on the
data available at the time, concluded that there is inadequate
evidence for its carcinogenicity in humans and limited evidence for
its carcinogenicity in experimental animals. The compound was assigned
to Group 3: not classifiable as to its carcinogenicity to humans
(IARC, 1995).
DCA is a very effective inducer of hepatic tumours in both mice
and rats at high doses. Several studies in male and female B6C3F1
mice found multiple tumours per animal with treatment concentrations
of 2 g/litre and above with as little as 1 year of treatment
(Herren-Freund et al., 1987; Bull et al., 1990; DeAngelo et al., 1991;
Daniel et al., 1992a; Pereira, 1996). These studies are summarized in
Table 15. Early in treatment (i.e., 52 weeks), the dose-response curve
is very steep, with essentially no response observed at concentrations
of 1 g/litre, but as many as four tumours per liver in mice treated
with 2 g/litre (Bull et al., 1990). However, concentrations as low as
0.5 g/litre will result in a hepatic tumour incidence of approximately
80% in a full 2-year study (Daniel et al., 1992a).
Hepatic tumours are also induced by DCA in male F344 rats
(Richmond et al., 1995). High doses of DCA given to rats also produce
overt signs of peripheral neuropathy. Nevertheless, increased
incidences of hyperplastic nodules, hepatocellular adenoma and
hepatocellular carcinoma were observed at 60 weeks of treatment at
Table 15. Carcinogenic effects of dichloroacetate in rodents
Species Dose Duration Tumour HN & HAa HCb Reference
(sex) (g/litre) (weeks) site
Incidence Tumour/n Incidence Tumour/n
(multiplicity) (multiplicity)
Mice
B6C3F1 (M) 0 61 Herren-Freund et al.
5 61 Liver 25/26 (4.6) 21/26 (1.7) (1987)
B6C3F1 (M) 1 52 Liver 2/11 0.3 - - Bull et al. (1990)
2 52 Liver 23/24 3.6 5/24 0.25
2 37 Liver 7/11 2.2 0/11 0
B6C3F1 (M) 0 60 Liver 0/10 0 0/10 0 DeAngelo et al. (1991)
0.5 60 Liver
3.5 60 Liver 12/12 2.3 8/12 1.7
5 60 Liver 27/30 2.3 25/30 2.2
0 75 Liver 2/28 0.07
0.05 75 Liver 4/29 0.31
0.5 75 Liver 3/27 0.11
0 104 Liver 1/20 0.05 2/20 0.1 Daniel et al. (1992a)
0.5 104 Liver 12/24 0.5 15/24 0.63
B6C3F1 (F) 0 52 Liver 1/40 0.03 0/40 0 Pereira (1996)
0.28 52 Liver 0/40 0 0/40 0
0.93 52 Liver 3/20 0.20 0/20 0
2.8 52 Liver 7/20 0.45 1/20 0.1
0 81 Liver 2/90 0.02 2/90 0.02
0.28 81 Liver 3/50 0.06 0/50 0
0.93 81 Liver 7/28 0.32 1/28 0.04
2.8 81 Liver 16/19 5.6 5/19 0.37
Table 15. (continued)
Species Dose Duration Tumour HN & HAa HCb Reference
(sex) (g/litre) (weeks) site
Incidence Tumour/n Incidence Tumour/n
(multiplicity) (multiplicity)
Rats
F344 (M) 0 60 Liver 0/7 0 0/7 0 Richmond et al. (1995)
0.05 60 Liver 0/7 0 0/7 0
0.5 60 Liver 0/7 0 0/7 0
2.4 60 Liver (26/27) 0.96 1/27 0.04
0 104 Liver 1/23 0.04 0/23 0
0.05 104 Liver 0/26 0 0/26 0
0.5 104 Liver (9/29) 0.31 3/29 0.1
2.4 104 Liver NRc NR NR NR
F344 (M) 0 104 Liver 1/33 0.03 1/33 0.03 DeAngelo et al. (1996)
0.05 104 Liver 0/26 0 0/26 0
0.5 104 Liver 5/29 0.17 3/29 0.10
1.6d 104 Liver 4/28 0.14 6/28 0.24
a Combined hepatocellular nodules and hepatocellular adenomas.
b Hepatocellular carcinoma.
c NR = not reported.
d Concentration was 2.6 g/litre of drinking-water for 18 weeks and then lowered to 1 g/litre to give a mean daily concentration
of 1.6 g/litre.
2.4 g/litre (Table 15). As in mice, if DCA treatment was extended to
104 weeks, the incidence of these lesions was 41% in a group of 29
rats at a treatment concentration of 0.5 g/litre. No tumours were
observed at 0.05 g/litre, and only one hepatic tumour was observed in
23 control rats.
Estimated doses of DCA in mg/kg of body weight per day for the
studies in Table 15 were as follows (ILSI, 1997; US EPA, 1998a):
Herren-Freund et al. (1997): 5 g/litre = 1000 mg/kg of body
weight per day
Bull et al. (1990): 1 or 2 g/litre = 140 or 300 mg/kg of body
weight per day
DeAngelo et al. (1991): 0.05, 0.5, 3.5 or 5 g/litre = 7.6, 77,
410 or 486 mg/kg of body weight per day
Daniel et al. (1992a): 0.5 g/litre = 95 mg/kg of body weight per
day
Pereira (1996): 0.26, 0.86 or 2.6 g/litre = 40, 120 or 330 mg/kg
of body weight per day
Richmond et al. (1995): 0.05, 0.5 or 2.4 g/litre = 4, 40 or
300 mg/kg of body weight per day
DeAngelo et al. (1996): 0.05, 0.5 or 1.6 g/litre = 4, 40 or
140 mg/kg of body weight per day.
Male F344 rats were exposed for 2 years to DCA in their
drinking-water at concentrations of 0.05, 0.5 or 1.6 g/litre. Based
upon the pathological examination, DCA induced observable signs of
toxicity in the nervous system, liver and myocardium. However,
treatment-related neoplastic lesions were observed only in the liver.
A statistically significant increase in carcinogenicity
(hepatocellular carcinoma) was noted at 1.6 g/litre. Exposure to 0.5
g/litre increased hepatocellular neoplasia (carcinoma and adenoma) at
100 weeks. Calculation of the time-weighted mean daily dose at which
50% of the animals exhibited liver neoplasia indicated that the F344
male rat (approximately 10 mg/kg of body weight per day) is 10 times
more sensitive than the B6C3F1 male mouse (approximately 100 mg/kg of
body weight per day) (DeAngelo et al., 1996).
The ability of DCA to induce damage to DNA that could give rise
to mutations or chromosomal damage has been studied both in vivo and
in vitro. Classical evaluations of DCA in Salmonella typhimurium
tester strains, both with and without metabolic activation, have been
largely negative if held to the standard of at least a 2-fold increase
in apparent mutation frequency (Waskell, 1978; Herbert et al., 1980).
However, a number of more recent studies have suggested some potential
for DCA-induced modifications in DNA. DeMarini et al. (1994) reported
that DCA induced prophage in Escherichia coli at a concentration of
0.26 mmol/litre and produced 2.7 and 4.2 revertants per ppm in
S. typhimurium strain TA100 with and without S9 addition,
respectively. There are some difficulties in interpreting this report,
as the authors introduced DCA as a vapour, and it is not clear whether
the concentrations reported (i.e., ppm) refer to air or medium
concentrations. Second, at least in the case of the Salmonella
assay, the DCA was introduced as the free acid and allowed to vaporize
and partition into the incubation medium. Because DCA is a strong
acid, and if sufficient time is allowed, such conditions could result
in near-quantitative transfer of DCA to the medium. The amount
volatilized in this case was approximately 60-600 mmol. Therefore, it
is likely that the pH of this small amount of medium (2.5 ml) was
substantially modified, even if only a fraction of this relatively
large amount of strong acid was indeed transferred to the medium. The
amount of DCA introduced into the prophage assay was unclear because
the method of addition was not described, although the introduction to
the journal article implied that it was again being tested as a
volatile.
Fox et al. (1996) recently published an evaluation of the
mutagenic effects of sodium dichloroacetate. These investigations
found no evidence of increased mutation rates in Salmonella
typhimurium tester strains TA98, TA100, TA1535 or TA1537;
Escherichia coli strain WP2urvA; or the mouse lymphoma forward
mutation assay, whether incubated in the presence or absence of rat
liver S9 fraction for metabolic activation. These authors found no
evidence that DCA was capable of inducing chromosomal aberrations in
CHO cells in vitro at doses of up to 1100 mg/kg of body weight for
3 days. These studies utilized neutralized DCA, supporting the
contention that positive results in prior studies may have been due to
artefactual results obtained by testing of the free acid or because
various sources of DCA have greater amounts of impurities.
Giller et al. (1997) examined the mutagenicity of DCA in the SOS
chromotest, the Ames fluctuation assay and the newt micronucleus
assay. DCA induced a positive response at 500 µg/ml (approximately 3.5
mmol/litre) in the SOS chromotest and at concentrations ranging from
100 to 1500 µg/ml (approximately 1-10 mmol/litre) in the Ames
fluctuation assay. The effects were observed at a lower concentration
in the absence of S9. The concentrations used in these studies exceed
the peak systemic concentrations of DCA that produce a high incidence
of liver tumours in mice by approximately 3 orders of magnitude
(Kato-Weinstein et al., 1998). Moreover, it appears that the authors
utilized the free acid in these experiments, raising the possibility
of a pH artefact. The newt micronucleus assay was found to be
negative.
DCA has been shown to produce a mutagenic and clastogenic
response in the in vitro mouse lymphoma assay, but only at doses at
or above 1 mmol/litre (Harrington-Brock et al., 1998).
Analogous difficulties have been encountered when attempting to
document the mutagenic effects of DCA in vivo. Nelson & Bull (1988)
and Nelson et al. (1989) reported that DCA induced single strand
breaks (SSB) in hepatic DNA when administered by gavage to both mice
and rats. Subsequent investigators were unable to replicate these
results in detail (Chang et al., 1992; Daniel et al., 1993a). However,
a small transitory increase in SSB was observed with doses of 5 and
10 mmol/kg of body weight in male B6C3F1 mice (Chang et al., 1992).
The bases of the discrepancies in these results are not clear, but
could, in part, be attributed to slightly different methods. As noted
in the subsequent section on TCA (section 4.2.2), the Nelson & Bull
(1988) results were not replicated by Styles et al. (1991), although
there was greater similarity in the methods used. More recently,
Austin et al. (1996) showed that acute doses of DCA oxidatively damage
nuclear DNA, measured as increases in the 8-hydroxy-2-deoxyguanosine
(8-OH-dG) relative to 2-deoxyguanosine content of the isolated DNA.
The time course of this damage is more consistent with the development
of SSB breaks reported by Chang et al. (1992) and could represent the
repair process that involves strand scission. There are two important
points that must be made: (i) the induction of SSB by Chang et al.
(1992) was very small relative to that seen with the positive
controls, diethylnitrosamine and methylmethane sulfonate; and (ii)
although increased 8-OH-dG was observed with acute treatments with
DCA, there was not a sustained elevation of this adduct in nuclear DNA
of mice when treatments were extended to 3 or 10 weeks in
drinking-water (Parrish et al., 1996).
Fuscoe et al. (1996) reported results obtained with the mouse
peripheral blood micronucleus assay. They found a small, but
statistically significant, increase in polychromatic erythrocytes
containing micronuclei in male B6C3F1 mice treated for 9 days with
3.5 g of DCA per litre of drinking-water. However, this response was
not maintained through 28 days of exposure. These investigators also
examined DNA migration in the single-cell gel assay. In this case, DCA
appeared to retard migration of DNA, suggesting the possibility of DNA
cross-linking after 28 days of treatment at 3.5 g/litre. Neither assay
revealed significant effects of DCA at concentrations of 2 g/litre or
below. DCA induces 3-4 tumours per animal within 1 year at 2 g/litre
in drinking-water (Bull et al., 1990). The higher dose adds little to
the tumorigenic response. More information with regard to the
possibility that DCA can cause mutations in liver cells is found in a
recent study using the lacI locus in the Big Blue(R) transgenic
mouse mutagenesis assay (Leavitt et al., 1997). These investigators
used a drinking-water route and the same doses of DCA as were used in
the rodent bioassay. After 10 and 60 weeks of DCA administration, an
increased frequency of mutants was observed at the high dose (3.5
g/litre). Mutational spectral analysis of these mutations revealed a
different spectrum in the mutants from DCA-treated animals than was
seen in the untreated animals. At this high dose of DCA, a large
portion of the liver can actually be tumour tissue. Because tumours
result from clonal expansion, the presence of tumour tissue in the
evaluated sample would give a falsely high mutation frequency if a
lacI mutation occurred in the rapidly expanding tumour clone. Thus,
these indications of genotoxic activity may have little to do with the
induction of hepatic cancer by DCA.
DCA appears to specifically stimulate outgrowth of hepatocellular
adenomas, rather than hepatocellular carcinomas. Pereira & Phelps
(1996) examined the role of DCA as a promoter of methylnitrosourea
(MNU)-initiated hepatic tumours in female B6C3F1 mice. These data are
provided in graphic form in Figure 2. At a concentration of
2.6 g/litre of drinking-water, DCA induced a very large increase in
the number of hepatocellular adenomas, but had no significant effect
on the induction of hepatocellular carcinomas. These data would appear
to be consistent with the stop experiments of Bull et al. (1990), who
found that suspension of treatment with DCA appeared to arrest
progression of liver tumours, but resulted in a yield of
hepatocellular adenomas and nodules that was proportional to the total
dose of DCA administered. In contrast, most of the tumours that
remained after the suspension of TCA treatment for 3 months were
hepatocellular carcinomas.
More recent studies on the effects of DCA on cell replication
within normal hepatocytes and hyperplastic nodules and tumours
(predominantly adenomas) indicate that DCA has selective effects.
Stauber & Bull (1997) found that DCA had a small, stimulatory effect
on the replication rate of normal hepatocytes over the first 14 days
of treatment. As treatment was extended to 28 days and beyond, these
effects became inhibitory at concentrations in drinking-water of
0.5 g/litre and above. In contrast, hepatocytes within nodules and
tumours appeared to be resistant to the inhibitory effects of DCA. At
a concentration of 2 g/litre, DCA doubled the rate at which c-Jun
immunoreactive hepatocytes replicated within hyperplastic nodules and
adenomas. This strong stimulation of tumour cell replication would
appear to be responsible for the very rapid induction of tumours in
mice treated with DCA in drinking-water at concentrations of 2 g/litre
and above. It would appear that the slower induction of liver tumours
at lower doses of DCA depends primarily on the selective suppression
of the replication of normal hepatocytes relative to that of initiated
cells.
The inhibitory effect of DCA on replication of normal hepatocytes
has been observed by a number of investigators (Carter et al., 1995).
The rate of replication is sharply inhibited within 5 days at
concentrations of DCA of 5 g/litre. At 0.5 g/litre, the replication
rate becomes inhibited to the same extent as observed with 5 g/litre
after 20 days of treatment. These decreases in replication were
accompanied by an increase in the percentage of the cells that were
mononucleated, which is probably associated with an increase in
tetraploid cells.
The suppression of cell replication by DCA in normal hepatocytes
of treated mice is accompanied by decreases in apoptosis (Snyder et
al., 1995). At concentrations of 5 g/litre, the frequency at which
apoptotic cells are observed drops by 60-75% with as few as 5 days of
treatment. At 0.5 g/litre, there is a downward trend that is observed
over the period from 5 to 30 days such that the frequency of apoptotic
bodies at this low dose approaches that observed at the highest dose
at 30 days. This result essentially parallels that described above for
suppression of the rates of cell replication. This raises a dilemma as
to whether the driver of the response is suppressed replication or
suppressed apoptosis. Whichever is the case, this has to translate
into suppressed turnover of normal hepatocytes. The question is
whether this suppressive effect on cell turnover increases the
probability of transformation of hepatocytes.
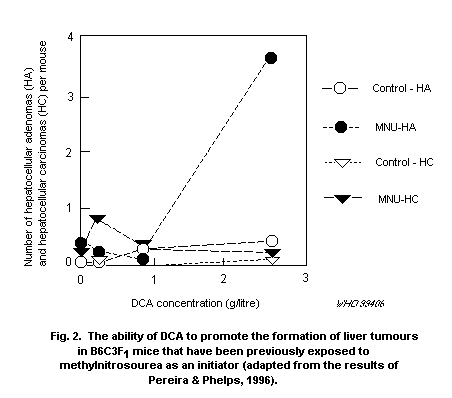 Small, but statistically significant, increases in the rate of
DNA synthesis in primary cultures of rat hepatocytes have been
reported at a concentration of 1 mmol of DCA per litre (Reddy et al.,
1992). This is indirect evidence of an effect on the rate of cell
division, because replication rates were not actually measured in this
study. Nevertheless, it seems probable that DCA does act as a weak
mitogen. Adaptation or down-regulation of this response has been
consistently observed in vivo as described above. The key
observation appears to be that the mitogenic response is not
down-regulated in hyperplastic nodules or tumours (Stauber & Bull,
1997).
Hyperplastic nodules and tumours induced by DCA have some common
characteristics that distinguish them from nodules and tumours that
are induced by TCA. In female mice, Pereira (1996) indicated that
liver tumours induced by DCA tended to be eosinophilic, whereas those
induced by TCA were basophilic. In male B6C3F1 mice treated with 2 g
of DCA per litre, a substantial fraction (66%) of the altered hepatic
foci found and nodules were reported to be eosinophilic. However, the
larger lesions tend to be basophilic (Stauber & Bull, 1997). These
larger lesions included hyperplastic nodules, adenomas and carcinomas.
These data suggest that there are some differences in tumour induction
by DCA based on sex. However, this difference appears to be important
primarily at high doses (>2 g/litre), where the rate of cell
replication is enhanced in a set of basophilic lesions. The
development of these lesions may account for the much shorter
latencies observed in male mice as compared with female mice at high
doses.
As pointed out by previous investigators examining responses in
male mice (Bull et al., 1990; DeAngelo et al., 1991), Pereira (1996)
found the dose-response curves describing the induction of total
lesions by DCA to be non-linear in female mice. Conversely, the
effects of TCA are essentially linear with dose.
Stauber & Bull (1997) found that DCA-induced liver tumours in
male mice were immunoreactive to c-Jun and c-Fos antibodies, whereas
TCA-induced liver tumours were not. This difference would appear
consistent with the observation that DCA-induced tumours in female
mice expressed the GST-pi at high levels, whereas TCA-induced tumours
were largely GST-pi negative. The expression of GST-pi is dependent on
AP-1 transcription factor binding sites in the promoter region of the
gene. Thus, elevations of c-Jun and c-Fos would be expected to
increase GST-pi expression (Angel & Karin, 1991). Conversely,
peroxisome proliferator activated receptor (PPAR)-alpha is known to
interfere with the c-Jun activity (Sakai et al., 1995). As a
consequence, GST-pi is generally not observed in tumours induced by
peroxisome proliferators.
Tao et al. (1996) reported a further differentiation of DCA- and
TCA-induced tumours. Non-neoplastic hepatocytes observed in mice
treated with DCA were found to have high levels of TGF-alpha, whereas
cells within the tumour expressed much lower levels. The opposite was
observed with TGF-ß expression, which was high in tumours and low in
normal tissues. This differential distribution of expression was not
observed in non-involved tissue and tumours from TCA-treated mice. The
precise involvement of these growth factors in the growth and
development of tumours cannot be stated. However, both TGF-alpha and
TGF-ß are known to be intimately involved with cell birth and cell
death processes. In liver tissue, TGF-ß expression is associated with
apoptosis (programmed cell death) and TGF-alpha expression is
associated with proliferative states. It should be noted that it is
not known whether these differences reflect characteristics of the
neoplastic cells or are actually responses induced by DCA.
Anna et al. (1994) and Ferreira-Gonzalez et al. (1995)
independently assessed the frequency and spectra of H- ras mutations
in DCA-induced tumours. These data and those of historical controls
for male B6C3F1 mice (Maronpot et al., 1995) specifically at codon-61
of H- ras are displayed in Table 16. The mutation frequency in
DCA-induced tumours does not differ significantly from that observed
in spontaneous tumours. However, there is an obvious change in the
mutation spectra in codon 61, involving a significant increase in the
H- ras-61(CTA) mutation largely at the expense of the H- ras-61(AAA)
lesion. A traditional interpretation of changes in mutation spectra
would be that this is evidence of mutation (Reynolds et al., 1987). As
pointed out by Anna et al. (1994), however, such an effect could be
accounted for if cells expressing a particular mutation were selected
for by treatment. Since the H- ras-61(CTA) mutation codes for
leucine, a neutral amino acid, whereas the H- ras-61(AAA) mutation
codes for lysine, a charged amino acid in this position, the
structures of these two mutant proteins are potentially quite
different in the Switch 2 region of the ras protein. Alterations in
structure within this region could significantly affect the affinity
of H- ras binding to raf-1 and other proteins involved in signal
transduction (Drugan et al., 1996).
4.2.1.7 Comparative pharmacokinetics and metabolism
The mammalian metabolism of DCA has received relatively little
study. However, there is sufficient information to show that its
metabolism is very dose-dependent and is dramatically affected by
prior exposure. While some significant differences in the details of
metabolism appear between species, these general statements hold for
both rodents and humans.
A proposed metabolic scheme for DCA, adapted from Larson & Bull
(1992), is provided in Figure 3. Oxalate, glyoxylate, MCA and carbon
dioxide have all been established as metabolites of DCA (Stacpoole,
1989; Larson & Bull, 1992; Lin et al., 1993; Gonzalez-Leon et al.,
1997). In addition to the metabolites depicted, thiodiacetate has been
observed in small amounts in the urine of mice and rats (Larson &
Bull, 1992). This may arise from the reaction of MCA with GSH (Yllner,
1971), but other mechanisms are also possible. The intermediates
indicated are hypothetical but reasonable in terms of the end-products
observed. The extent to which reductive dehalogenation and peroxy
radical formation play a role in the metabolism of DCA is unclear. As
discussed later, such reactions clearly play a role in the metabolism
Table 16. Mutation frequency and spectra with codon 61 of H- ras of B6C3F1 mice treated with dichloroacetate
and trichloroacetatea
Chemical No. of H-ras 61/ Fraction CAA AAA CGA CTA
no. of tumours
Spontaneous hepatocellular
carcinomasb 183/333 0.56 150 (0.45) 106 (0.32) 50 (0.15) 21 (0.06)
Dichloroacetateb,c,d 61/110 0.55 48 (0.44) 15 (0.14) 25 (0.22) 22 (0.20)
Trichloroacetatec 5/11 0.45 6 (0.55) 4 (0.36) 1 (0.09) 0 (0)
a Mutations at other codons are not included, although these tumours are kept as part of the denominator.
Therefore, all mutants and wild-type at codon 61 do not add up to the total number of tumours.
b Anna et al. (1994).
c Ferreira-Gonzalez et al. (1995).
d Maronpot et al. (1995).
of trihaloacetates, and they are included here for completeness. An
additional pathway to glycolate could also be rationalized by the
oxidative metabolism of MCA.
The human metabolism of DCA first came under study because of its
proposed use as an oral hypoglycaemic agent. Lukas et al. (1980)
studied intravenously infused (over a 20-min interval) doses of 10 and
20 mg/kg of body weight in two human volunteers at each dose. The low
and high doses led to mean half-lives of 0.34 and 0.51 h,
respectively. Wells et al. (1980) noted that the peak concentration
increased disproportionately when intravenous doses (30-min infusion)
of DCA increased from 1 to 50 mg/kg of body weight and departed from
linearity as doses approached 30 mg/kg of body weight. Whereas the
half-life of DCA in this study was seen to be approximately 20 min at
doses below 25 mg/kg of body weight, the half-life at higher doses was
closer to 40 min, and the mean half-life was 31.8 ± 10.9 (standard
deviation [SD]) for all 11 subjects. The authors also noted that the
effects of DCA on plasma lactate and alanine persisted several days
after cessation of repeated oral treatment with DCA, but ended within
12 h after administering a single intravenous dose. Curry et al.
(1985) found that the mean half-life of DCA increased from 63.3 min to
an average of 374 min following the fifth of a series of 50 mg/kg of
body weight doses of DCA administered intravenously at 2-h intervals.
These data suggest that high repeated doses of DCA appear to hinder
the metabolic clearance of DCA.
There are substantial species differences in the metabolism of
DCA. Dogs, in particular, clear DCA from blood at a very low rate.
Lukas et al. (1980) found that the half-life of DCA administered as a
single 100 mg/kg of body weight dose was between 17.1 and 24.6 h. In
contrast, the clearance of the same dose in rats occurred with a
half-life of 2.1-4.4 h. The very much lower metabolic clearance of DCA
in the dog is probably responsible for its much greater acute toxicity
in this species (Katz et al., 1981). However, the half-life of DCA in
humans is much closer to that in rats than to that in dogs (Curry et
al., 1991).
Larson & Bull (1992) studied the metabolism of DCA in mice and
rats. These authors estimated a half-life of 1.5 h in mice and 0.9 h
in rats following oral doses. The estimates of half-life in mice in
this study were problematic, because it was clear that there is a
tremendous first-pass effect on DCA's absorption from the
gastrointestinal tract, which was particularly marked in mice. These
authors also estimated a maximum concentration of DCA from oral doses
of 20 and 100 mg/kg of body weight. In mice, the Cmax was found to
be 4 and 20 nmol/ml, respectively. In rats, the Cmax was found to
be 15 and 380 at the same doses. These authors also found that
significant amounts of DCA were metabolized to carbon dioxide and the
non-halogenated acids -- glycolate, glyoxylate and oxalate. The
fraction of DCA that was metabolized to carbon dioxide was
substantially underestimated in these studies, as was shown in the
more recent study of Xu et al. (1995), in which approximately 45% of
an oral dose of DCA was metabolized to carbon dioxide in mice within
24 h. Lin et al. (1993) also provided more definitive analyses of the
Small, but statistically significant, increases in the rate of
DNA synthesis in primary cultures of rat hepatocytes have been
reported at a concentration of 1 mmol of DCA per litre (Reddy et al.,
1992). This is indirect evidence of an effect on the rate of cell
division, because replication rates were not actually measured in this
study. Nevertheless, it seems probable that DCA does act as a weak
mitogen. Adaptation or down-regulation of this response has been
consistently observed in vivo as described above. The key
observation appears to be that the mitogenic response is not
down-regulated in hyperplastic nodules or tumours (Stauber & Bull,
1997).
Hyperplastic nodules and tumours induced by DCA have some common
characteristics that distinguish them from nodules and tumours that
are induced by TCA. In female mice, Pereira (1996) indicated that
liver tumours induced by DCA tended to be eosinophilic, whereas those
induced by TCA were basophilic. In male B6C3F1 mice treated with 2 g
of DCA per litre, a substantial fraction (66%) of the altered hepatic
foci found and nodules were reported to be eosinophilic. However, the
larger lesions tend to be basophilic (Stauber & Bull, 1997). These
larger lesions included hyperplastic nodules, adenomas and carcinomas.
These data suggest that there are some differences in tumour induction
by DCA based on sex. However, this difference appears to be important
primarily at high doses (>2 g/litre), where the rate of cell
replication is enhanced in a set of basophilic lesions. The
development of these lesions may account for the much shorter
latencies observed in male mice as compared with female mice at high
doses.
As pointed out by previous investigators examining responses in
male mice (Bull et al., 1990; DeAngelo et al., 1991), Pereira (1996)
found the dose-response curves describing the induction of total
lesions by DCA to be non-linear in female mice. Conversely, the
effects of TCA are essentially linear with dose.
Stauber & Bull (1997) found that DCA-induced liver tumours in
male mice were immunoreactive to c-Jun and c-Fos antibodies, whereas
TCA-induced liver tumours were not. This difference would appear
consistent with the observation that DCA-induced tumours in female
mice expressed the GST-pi at high levels, whereas TCA-induced tumours
were largely GST-pi negative. The expression of GST-pi is dependent on
AP-1 transcription factor binding sites in the promoter region of the
gene. Thus, elevations of c-Jun and c-Fos would be expected to
increase GST-pi expression (Angel & Karin, 1991). Conversely,
peroxisome proliferator activated receptor (PPAR)-alpha is known to
interfere with the c-Jun activity (Sakai et al., 1995). As a
consequence, GST-pi is generally not observed in tumours induced by
peroxisome proliferators.
Tao et al. (1996) reported a further differentiation of DCA- and
TCA-induced tumours. Non-neoplastic hepatocytes observed in mice
treated with DCA were found to have high levels of TGF-alpha, whereas
cells within the tumour expressed much lower levels. The opposite was
observed with TGF-ß expression, which was high in tumours and low in
normal tissues. This differential distribution of expression was not
observed in non-involved tissue and tumours from TCA-treated mice. The
precise involvement of these growth factors in the growth and
development of tumours cannot be stated. However, both TGF-alpha and
TGF-ß are known to be intimately involved with cell birth and cell
death processes. In liver tissue, TGF-ß expression is associated with
apoptosis (programmed cell death) and TGF-alpha expression is
associated with proliferative states. It should be noted that it is
not known whether these differences reflect characteristics of the
neoplastic cells or are actually responses induced by DCA.
Anna et al. (1994) and Ferreira-Gonzalez et al. (1995)
independently assessed the frequency and spectra of H- ras mutations
in DCA-induced tumours. These data and those of historical controls
for male B6C3F1 mice (Maronpot et al., 1995) specifically at codon-61
of H- ras are displayed in Table 16. The mutation frequency in
DCA-induced tumours does not differ significantly from that observed
in spontaneous tumours. However, there is an obvious change in the
mutation spectra in codon 61, involving a significant increase in the
H- ras-61(CTA) mutation largely at the expense of the H- ras-61(AAA)
lesion. A traditional interpretation of changes in mutation spectra
would be that this is evidence of mutation (Reynolds et al., 1987). As
pointed out by Anna et al. (1994), however, such an effect could be
accounted for if cells expressing a particular mutation were selected
for by treatment. Since the H- ras-61(CTA) mutation codes for
leucine, a neutral amino acid, whereas the H- ras-61(AAA) mutation
codes for lysine, a charged amino acid in this position, the
structures of these two mutant proteins are potentially quite
different in the Switch 2 region of the ras protein. Alterations in
structure within this region could significantly affect the affinity
of H- ras binding to raf-1 and other proteins involved in signal
transduction (Drugan et al., 1996).
4.2.1.7 Comparative pharmacokinetics and metabolism
The mammalian metabolism of DCA has received relatively little
study. However, there is sufficient information to show that its
metabolism is very dose-dependent and is dramatically affected by
prior exposure. While some significant differences in the details of
metabolism appear between species, these general statements hold for
both rodents and humans.
A proposed metabolic scheme for DCA, adapted from Larson & Bull
(1992), is provided in Figure 3. Oxalate, glyoxylate, MCA and carbon
dioxide have all been established as metabolites of DCA (Stacpoole,
1989; Larson & Bull, 1992; Lin et al., 1993; Gonzalez-Leon et al.,
1997). In addition to the metabolites depicted, thiodiacetate has been
observed in small amounts in the urine of mice and rats (Larson &
Bull, 1992). This may arise from the reaction of MCA with GSH (Yllner,
1971), but other mechanisms are also possible. The intermediates
indicated are hypothetical but reasonable in terms of the end-products
observed. The extent to which reductive dehalogenation and peroxy
radical formation play a role in the metabolism of DCA is unclear. As
discussed later, such reactions clearly play a role in the metabolism
Table 16. Mutation frequency and spectra with codon 61 of H- ras of B6C3F1 mice treated with dichloroacetate
and trichloroacetatea
Chemical No. of H-ras 61/ Fraction CAA AAA CGA CTA
no. of tumours
Spontaneous hepatocellular
carcinomasb 183/333 0.56 150 (0.45) 106 (0.32) 50 (0.15) 21 (0.06)
Dichloroacetateb,c,d 61/110 0.55 48 (0.44) 15 (0.14) 25 (0.22) 22 (0.20)
Trichloroacetatec 5/11 0.45 6 (0.55) 4 (0.36) 1 (0.09) 0 (0)
a Mutations at other codons are not included, although these tumours are kept as part of the denominator.
Therefore, all mutants and wild-type at codon 61 do not add up to the total number of tumours.
b Anna et al. (1994).
c Ferreira-Gonzalez et al. (1995).
d Maronpot et al. (1995).
of trihaloacetates, and they are included here for completeness. An
additional pathway to glycolate could also be rationalized by the
oxidative metabolism of MCA.
The human metabolism of DCA first came under study because of its
proposed use as an oral hypoglycaemic agent. Lukas et al. (1980)
studied intravenously infused (over a 20-min interval) doses of 10 and
20 mg/kg of body weight in two human volunteers at each dose. The low
and high doses led to mean half-lives of 0.34 and 0.51 h,
respectively. Wells et al. (1980) noted that the peak concentration
increased disproportionately when intravenous doses (30-min infusion)
of DCA increased from 1 to 50 mg/kg of body weight and departed from
linearity as doses approached 30 mg/kg of body weight. Whereas the
half-life of DCA in this study was seen to be approximately 20 min at
doses below 25 mg/kg of body weight, the half-life at higher doses was
closer to 40 min, and the mean half-life was 31.8 ± 10.9 (standard
deviation [SD]) for all 11 subjects. The authors also noted that the
effects of DCA on plasma lactate and alanine persisted several days
after cessation of repeated oral treatment with DCA, but ended within
12 h after administering a single intravenous dose. Curry et al.
(1985) found that the mean half-life of DCA increased from 63.3 min to
an average of 374 min following the fifth of a series of 50 mg/kg of
body weight doses of DCA administered intravenously at 2-h intervals.
These data suggest that high repeated doses of DCA appear to hinder
the metabolic clearance of DCA.
There are substantial species differences in the metabolism of
DCA. Dogs, in particular, clear DCA from blood at a very low rate.
Lukas et al. (1980) found that the half-life of DCA administered as a
single 100 mg/kg of body weight dose was between 17.1 and 24.6 h. In
contrast, the clearance of the same dose in rats occurred with a
half-life of 2.1-4.4 h. The very much lower metabolic clearance of DCA
in the dog is probably responsible for its much greater acute toxicity
in this species (Katz et al., 1981). However, the half-life of DCA in
humans is much closer to that in rats than to that in dogs (Curry et
al., 1991).
Larson & Bull (1992) studied the metabolism of DCA in mice and
rats. These authors estimated a half-life of 1.5 h in mice and 0.9 h
in rats following oral doses. The estimates of half-life in mice in
this study were problematic, because it was clear that there is a
tremendous first-pass effect on DCA's absorption from the
gastrointestinal tract, which was particularly marked in mice. These
authors also estimated a maximum concentration of DCA from oral doses
of 20 and 100 mg/kg of body weight. In mice, the Cmax was found to
be 4 and 20 nmol/ml, respectively. In rats, the Cmax was found to
be 15 and 380 at the same doses. These authors also found that
significant amounts of DCA were metabolized to carbon dioxide and the
non-halogenated acids -- glycolate, glyoxylate and oxalate. The
fraction of DCA that was metabolized to carbon dioxide was
substantially underestimated in these studies, as was shown in the
more recent study of Xu et al. (1995), in which approximately 45% of
an oral dose of DCA was metabolized to carbon dioxide in mice within
24 h. Lin et al. (1993) also provided more definitive analyses of the
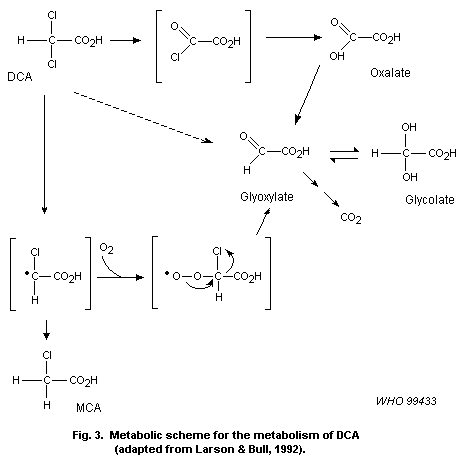 production of glycolate, glyoxylate and oxalate as major urinary
metabolites of DCA in the F344 rat. It is notable for later
discussions that these authors found a smaller percentage of the dose
of 1-14C-DCA ending up in glycolate than was observed with 2-14C-DCA.
This was offset by a somewhat greater yield (not statistically
significant) of carbon dioxide from 1-14C-DCA than from 2-14C-DCA.
These data would suggest that there are alternative pathways from DCA
to carbon dioxide in the rat besides the conversion to glyoxylate.
The decreases in DCA clearance with repeated doses appear to be
largely due to the inactivation of one enzyme involved in its
metabolism. There appear to be subtle differences in the metabolism of
mice and rats that lead to different manifestations of this
inhibition. In experiments that involved pretreatment of F344 rats at
a level of 0.2 or 2 g/litre in their drinking-water for a 14-day
period, it was found that the conversion of an oral dose of DCA to
carbon dioxide was substantially inhibited. Similar pretreatment of
male B6C3F1 mice did not affect carbon dioxide production from DCA.
However, in both cases, it is apparent that the metabolic clearance of
DCA from blood was affected by the DCA pretreatment. In rats, the
kinetics of DCA disappearance were studied with intravenous dosing,
which allows more precise definition of the kinetics. This
pretreatment led to an increase in the half-life of DCA in the blood
of rats from 2.4 ± 0.8 h to 10.8 ± 2.0 h when animals were
administered a dose of 100 mg/kg of body weight intravenously. Oral
dosing was used in mice, and the major impact of pretreatment was to
increase the Cmax from 2.6 ± 2.6 µg/ml in naive mice to 129.9 µg/ml
in mice that had DCA in their drinking-water at 2 g/litre until the
prior day (16 h before administration of the test dose). Therefore,
the phenomenon that DCA treatment inhibited its own metabolism, which
was originally observed in humans, could be replicated in both mice
and rats.
The mechanism by which this tremendous change in metabolism
occurs has not been established. It has been shown by Lipscomb et al.
(1995) that the bulk of the metabolism of DCA occurs in cytosolic
fractions. Very little DCA is metabolized in microsomes. It is
apparent that the metabolism of DCA in cytosolic preparations from
rodent liver is dependent upon nicotinamide cofactor and GSH. However,
the metabolism is not mediated by enzymes that can be recovered on a
GSH-sepharose column. Glutathione transferase activities towards
chlorodinitrobenzene were observed in the column. Subsequent work has
shown that it is the activity in the cytosol, however, that is
eliminated by DCA treatment (Gonzalez-Leon et al., 1997). Tong et al.
(1998) showed that a substantial fraction of DCA's metabolism is
mediated by a novel GST, GST-zeta. This enzyme appears to be subject
to autoinhibition by DCA.
4.2.1.8 Mode of action
Some indication of mechanisms by which DCA produces its effects
can be gleaned from studies cited in previous sections of this
document. While DCA produces many different types of toxicological
effects (i.e., neurotoxicity, reproductive and developmental
toxicities and carcinogenicity), there may be some features that are
common to the mechanisms that produce all these effects. The
particulars of those mechanisms remain to be established. What follows
is a brief outline of what is known and some speculation on how new
research may be applied to developing this information to better
assess the risks associated with the production of DCA as a by-product
of the chlorination of drinking-water.
Given the lack of evidence that induction of DNA damage by DCA is
involved in liver cancer induction, are there plausible alternative
mechanisms that may be invoked? The most common alternative mode of
action would be evidence that carcinogenic doses of DCA induce
cytotoxic damage in the target organ, which leads to reparative
hyperplasia. Although there is some evidence of single-cell necrosis
with chronic exposure (Stauber & Bull, 1997) and infarcts are
occasionally observed (Sanchez & Bull, 1990) in the livers of B6C3F1
mice, this mode of action plays a negligible role at the lowest doses
that induce liver cancer. Small and variable initial increases in
replication rates of hepatocytes in treated mice are reversed with
continued treatment, with the dominant effect becoming inhibition of
replication within a 4- to 8-week period (Carter et al., 1995; Stauber
& Bull, 1997). These observations indicate that necrosis followed by
reparative hyperplasia do not explain the rapid carcinogenic responses
to DCA.
Additional data add support to the hypothesis that DCA does in
fact act largely by a "non-genotoxic" mode of action. Early data
indicated that DCA was capable of inducing peroxisome proliferation
(Nelson & Bull, 1988; DeAngelo et al., 1989). While these effects have
been observed by others, they are clearly of short duration,
disappearing within a few months (DeAngelo et al., 1989). Moreover, it
has become apparent that DCA induces hepatic tumours at dose rates
that are significantly below those required to induce peroxisome
proliferation (cf. DeAngelo et al., 1989 and Daniel et al., 1992a).
New research has shown that DCA acts primarily to increase the
growth rate of pre-initiated cells in the liver. Stauber et al. (1998)
found that DCA induces growth of colonies on soft agar when cell
suspensions were obtained from the liver of neonatal mice. The most
impressive aspect of these studies was that the colonies expressed the
same phenotype (c-Jun+) as was found in DCA-induced tumours in
vivo. Similar experiments produced a c-Jun phenotype when TCA was
incorporated into the soft agar. A concentration of 0.5 mmol of DCA
per litre was required in the medium to produce a significant increase
in the number of colonies formed in a 10-day interval when the cells
were derived from naive mice. However, if mice were pretreated with
0.5 g of DCA per litre in their drinking-water prior to the isolation
of the cells, 0.02 mmol of DCA per litre was as effective as 0.5
mmol/litre. Moreover, the yield of colonies was increased by this
pretreatment. The increased sensitivity to DCA appears closely related
to the finding that pretreating animals with low levels of DCA
(<0.2 g/litre of drinking-water) reduces its metabolism by more than
90% (Gonzalez-Leon et al., 1997). The increased number of colonies
produced by pretreatment appears to be due to clonal expansion of
these cells. It appears that this activity accounts for the tumours
induced by DCA at higher doses (>2 g/litre) where blood
concentrations are found to be in the 100-500 µmol/litre range.
However, blood concentrations ranging from 1 to 7 µmol/litre produced
in mice treated with 0.5 g/litre (Kato-Weinstein et al., 1998) result
in an 80% incidence of liver tumours (Daniel et al., 1992a),
suggesting that a second mechanism may be involved at lower doses.
At all carcinogenic doses studied, DCA increases the deposition
of glycogen in the liver (Kato-Weinstein et al., 1998). This suggests
that DCA is modifying cell signalling pathways. Low intraperitoneal
doses of DCA produce increases in serum insulin concentrations in
response to glucose challenge (Kato-Weinstein et al., 1998). In
contrast, decreases in serum insulin concentrations have been observed
in mice chronically administered DCA at either 0.5 or 2 g/litre (Smith
et al., 1997). However, these measurements were made during the
daylight hours when both serum glucose and blood DCA concentrations
are low. Clearly, the involvement of insulin in DCA-induced liver
tumorigenesis needs to be studied further.
Recent studies have provided more substantive evidence that DCA
at very high levels possesses some ability to induce genotoxic
effects. Of particular note are the studies of Harrington-Brock et al.
(1998), who found significant increases in mutant frequencies in mouse
lymphoma cells in vitro at concentrations in excess of 1 mmol/litre.
Similar potency of DCA has been observed in the Ames fluctuation assay
by Giller et al. (1997). Previous studies were largely negative or
made use of the free acid form of DCA. Brusick (1986) had previously
documented that low pH produces increased evidence of genotoxic damage
in cultured mammalian cells.
Leavitt et al. (1997) reported increased recovery of mutant cells
from the lacI transgenic mouse with varying periods of treatment
with DCA in drinking-water. Significant increases were observed only
when mice had been treated with 3.5 g/litre for 60 weeks, but not for
shorter time intervals. No significant increases were noted at 1
g/litre. Although the authors took care to ensure that nodules and
tumours were excluded from the sampling, Stauber & Bull (1997)
demonstrated that there are numerous lesions that are smaller than
nodules in B6C3F1 mice maintained on 2 g of DCA per litre for only
40 weeks. It was inevitable that some of these microscopic lesions
were included within the tissue samples described. Given the marked
stimulation of cell replication that occurs within lesions in mice, it
is not possible to determine if the effect reported by Leavitt et al.
(1997) is due to mutagenic effects of DCA or its demonstrated ability
to selectively stimulate the growth of tumour phenotype.
Based on the available evidence, it is probable that genotoxic
effects of DCA play little, if any, role in the induction of liver
cancer in rodents at low doses. This conclusion is based on clear
evidence that DCA is capable of acting as a tumour promoter and
produces effects on cell replication or apoptosis at all carcinogenic
doses (Snyder et al., 1995; Pereira & Phelps, 1996; Stauber & Bull,
1997). The concentrations of DCA required to produce genotoxic effects
in vitro and the blood levels necessary to detect minimal genotoxic
effects in vivo are 3 orders of magnitude higher than those
necessary for induction of an 80% tumour incidence. The recent
evaluation by ILSI (1997) also concluded that the mechanism by which
DCA increased liver tumours was non-genotoxic. However, new data
indicate that the actual mechanism is by tumour promotion rather than
by cytotoxicity and reparative hyperplasia.
The metabolism of DCA is very dose-dependent, with metabolism and
clearance of the chemical being inhibited sharply with high dose
rates. Most data now suggest that it is the parent compound that is
responsible for the effects related to carcinogenicity. Thus, simply
on the basis of considerations of target organ dosimetry, the effects
of DCA would be predicted to increase sharply at chronic dosing levels
that approach or exceed 30 mg/kg of body weight per day rather than
being simple linear functions of dose. Unfortunately, the available
data do not allow the systemic dose versus external dose relationships
to be determined with any precision on the basis of current
information. In rats, the full inhibition was observed at
concentrations of DCA in drinking-water as low as 0.2 g/litre
(Gonzalez-Leon et al., 1997). The minimum treatment level for DCA's
inhibition of its own metabolism in mice has not been established.
However, blood concentrations of DCA increase from 2-4 µmol/litre when
the mice are treated with 0.5 g/litre in drinking-water to
approximately 300 µmol/litre when the treatment concentration is
2 g/litre (Kato-Weinstein et al., 1998). These are peak concentrations
measured during the night when mice are consuming the DCA-containing
water. This 100-fold increase in blood concentrations with a mere
4-fold increase in dose undoubtedly contributes to the highly
non-linear tumorigenic response for DCA reported previously (Bull et
al., 1990). The effect in humans has been documented to occur with
doses in a similar range, 30 mg/kg of body weight (Wells et al.,
1980). The animal data need to be extended to lower dosing rates or
the human treatments need to be extended in time to more precisely
define the level of chronic exposure that is required to produce this
phenomenon. Alternatively, a better understanding of the mechanism of
this inhibition should allow the critical question of whether the
inhibition is a function of cumulative dose or daily dose to be
answered.
The available data indicate that DCA differentially affects the
replication rates of normal hepatocytes and hepatocytes that have been
initiated. The dose-response relationships are complex, with DCA
initially stimulating division of normal hepatocytes. However, at the
lower chronic doses used in animal studies (but still very high
relative to those that would be derived from drinking-water), the
replication rate of normal hepatocytes is eventually sharply
inhibited. This indicates that normal hepatocytes eventually
down-regulate those pathways that are sensitive to stimulation by DCA.
However, altered cells, particularly those that express high amounts
of a protein that is immunoreactive to a c-Jun antibody, do not seem
to be able to down-regulate this response. Thus, the rates of
replication in the preneoplastic lesions with this phenotype are very
high at the doses that cause DCA tumours to develop with a very low
latency. Preliminary data suggest that this continued alteration in
cell birth and death rates is also necessary for the tumours to
progress to malignancy (Bull et al., 1990). This interpretation is
supported by studies that employ initiation/promotion designs as well
(Pereira, 1996).
On the basis of the above considerations, it is suggested that
the currently available cancer risk estimates for DCA should be
modified by incorporating newly developing information on its
comparative metabolism and modes of action to formulate a biologically
based dose-response model. These data are not available at the time of
this writing, but should become available within the next 2-3 years.
The dose-response data for effects other than cancer vary
significantly, with dogs being extraordinarily sensitive (Katz et al.,
1981). However, inhibition of the metabolism of DCA in chronically
treated rodents (Gonzalez-Leon et al., 1997) and humans (Wells et al.,
1980; Curry et al., 1985) may cause differences in sensitivities
between species to converge somewhat with repeated treatment or
exposure. However, a significant difference in species sensitivity
remains. Cicmanec et al. (1991) identified a LOAEL of 12.5 mg/kg of
body weight per day in dogs treated for 90 days. NOAELs for
hepatomegaly in mice appear to be in the neighbourhood of 0.2 g/litre,
which is approximately 40 mg/kg of body weight per day. On the basis
of the rates of metabolic clearance and the assumption that the
intrinsic sensitivities of different species are similar, the average
human would seem to more closely approximate rats than dogs. To obtain
more accurate pictures of human sensitivity at low doses, however, it
is clear that future work must focus more specifically on
toxicodynamic variables.
4.2.2 Trichloroacetic acid (trichloroacetate)
Like DCA, TCA exists almost exclusively in the salt form at pHs
found in drinking-water because of its very low p Ka of 0.70 (IARC,
1995).
4.2.2.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
Very little information exists on the mammalian toxicology of TCA
before it was discovered as a by-product of drinking-water
chlorination. Woodard et al. (1941) determined the oral LD50 of
trichloroacetate (i.e., neutralized to pH 6-7) to be 3.32 g/kg of body
weight in rats and 4.97 g/kg of body weight in albino mice when the
compound was administered in aqueous solution. These values were in
the same general range as those for neutralized acetic acid.
Davis (1990) examined the effects of TCA on blood glucose and
lactate levels following a dosing regimen of total doses of 0.92 or
2.45 mmol/kg of body weight administered 3 times in 1 day to
Sprague-Dawley rats by gavage. A typographical error in dosage was
suspected and confirmed with the author (M.E. Davis, personal
communication, 1996). Reductions in plasma glucose concentrations were
observed in females at the high dose, and lactic acid levels were
decreased at 0.92 and 2.45 mmol/kg of body weight doses in females,
but only at the high dose in males. The authors noted that these high
concentrations were neutralized with sodium hydroxide. However, these
are very low doses of TCA relative to those found to produce similar
effects in other studies. These initial experiments were followed by a
study of the effects of TCA administered in drinking-water for 14 days
at 0.04, 0.16, 0.63 or 2.38 g/litre. Effects on urine volume and
osmolality were reported to occur at the highest dose, but not at
0.63 g/litre. Effects on glucose and lactate were not reported.
2) Short-term toxicity
Mather et al. (1990) administered TCA to male Sprague-Dawley rats
in drinking-water at concentrations of 0, 50, 500 or 5000 mg/litre (0,
4.1, 36.5 or 355 mg/kg of body weight per day) for 90 days. Small, but
statistically insignificant, decreases in body weight were observed at
the highest dose. TCA produced a significant increase in the liver to
body weight ratio at this dose, but not at 500 mg/litre. This effect
was associated with a small, but statistically significant, increase
in cyanide-insensitive acyl CoA oxidase activity in the liver, an
indicator of peroxisome proliferation.
Unlike DCA and MCA, TCA does not appear to be a substrate for the
mitochondrial pyruvate carrier (Halestrap, 1975). TCA does appear to
inhibit the pig heart pyruvate dehydrogenase kinase at approximately
the same concentrations as for DCA (Whitehouse et al., 1974). However,
the authors noted that DCA influenced the proportion of active
pyruvate dehydrogenase in the perfused rat heart, but TCA was inactive
under these circumstances. Although these data were obtained from
mitochondria from different sources, they suggest that there may be
some differences in effects of DCA and TCA on intermediary metabolism
related to their transport into various cellular compartments.
Bhat et al. (1991) administered TCA to male Sprague-Dawley rats
at a concentration of 45.8 mmol/litre (7.5 g/litre) in their
drinking-water for 90 days to provide an approximate intake of 785
mg/kg of body weight per day. These levels produced minimal evidence
of liver toxicity by histopathological examination. However, lower
concentrations in drinking-water have been shown to seriously impair
water and food consumption in experimental animals (Bull et al., 1990;
Davis, 1990; Mather et al., 1990; DeAngelo et al., 1991).
Consequently, it is difficult to determine how these data relate to
the potential effects of the low concentrations of TCA that are found
in chlorinated drinking-water (e.g., in the 10-100 µg/litre range).
The most obvious target organ for TCA is the liver. This effect
is marked by a hepatomegaly (Goldsworthy & Popp, 1987; Bull et al.,
1990; Mather et al., 1990; Sanchez & Bull, 1990), which is presumably
related to the ability of TCA to act as a peroxisome proliferator
(DeAngelo et al., 1989), since that is a common finding with this
class of rodent carcinogens. It could also be related to the
metabolism of TCA to DCA (Larson & Bull, 1992). However, more recent
data suggest that the apparent conversion of TCA to DCA has largely
been the result of artefactual conversion when fresh oxygenated blood
is acidified prior to derivatization (Merdink et al., 1998). TCA is
clearly without substantive cytotoxic effects at doses of less than
300 mg/kg of body weight in vivo (Bull et al., 1990; Sanchez & Bull,
1990; Acharya et al., 1995) or concentrations of up to 5 mmol/litre
in vitro (Bruschi & Bull, 1993).
4.2.2.2 Reproductive effects
There have been limited studies of TCA's effects on reproductive
performance. TCA was found to inhibit in vitro fertilization of
gametes from B6D2F1 mice at a concentration of 1000 mg/litre, but was
without effect at 100 mg/litre (Cosby & Dukelow, 1992). It is unlikely
that these effects at very high doses relative to those that might be
expected from human consumption of TCA at concentrations less than
0.1% of the NOEL are of relevance in assessing risks from exposure to
TCA in drinking-water.
4.2.2.3 Developmental effects
Treatment of pregnant rats with TCA at 0, 330, 800, 1200 or
1800 mg/kg of body weight per day by gavage in a water vehicle on
gestation days 6-15 produced dose-dependent reductions in body weight
and length of rat pups from dams administered doses of 800 mg/kg of
body weight per day and above. No effects were observed at 330 mg/kg
of body weight per day. However, there was a significant and
dose-related increase in soft tissue malformations at all doses
studied. The mean frequency of soft tissue malformations was 3.5 ±
8.7% (SD), 9.06 ± 12.9%, 30.4 ± 28.1%, 55.4 ± 36.1% and 96.9 ± 8.8% at
0, 330, 800, 1200 and 1800 mg/kg of body weight, respectively. Most of
the increased soft tissue abnormalities were accounted for by defects
in the cardiovascular system. The major malformation seen was
laevocardia. However, at doses of 800 mg/kg of body weight and above,
a significant incidence of an interventricular septal defect was
observed (Smith et al., 1989a).
Saillenfait et al. (1995) found that TCA, administered to rats in
embryo culture, began to produce consistent increases in defects at
concentrations of 2.5 mmol/litre and above, with few or no effects
observed at 1 mmol/litre. These defects included brain and eye
defects, reduction in the branchial arch and otic system defects. At
concentrations of 3.5 mmol/litre, some evidence of skeletal defects
was observed (i.e., absence of hindlimb bud). As is discussed below,
these concentrations can be achieved in the blood of rats administered
high doses of TCA (>100 mg/kg of body weight) (Larson & Bull,
1992). However, there is considerable doubt about whether these
effects would be induced by the doses of less than 1 µg/kg of body
weight experienced by humans consuming chlorinated drinking-water.
4.2.2.4 Neurotoxicity
No reports of neurotoxic effects of TCA were located.
4.2.2.5 Toxicity in humans
TCA is a strong acid. It is widely recognized that contact of TCA
with the skin has the potential to produce acid burns, and ingestion
of TCA has the potential to damage tissues of the gastrointestinal
tract or produce systemic acidosis, even though specific studies of
these effects do not appear in the literature. Such effects would
occur from contact with the crystal or strong solutions of the free
acid. However, such effects have little relevance to the production of
low levels of TCA, as the salt, as a by-product of the chlorination of
drinking-water.
Indirectly, it may be presumed that TCA presents little overt
hazard to human health because it is a major metabolite of commonly
used solvents such as trichloroethylene and tetrachloroethylene.
Occupational exposures to these solvents have been quite high in the
past, but few, if any, effects of the solvents in humans have been
attributed to TCA. Therefore, one would surmise that TCA is relatively
non-toxic to humans under circumstances of low exposures such as those
encountered in chlorinated drinking-water. However, these largely
negative data do not insure against chronic hazards such as cancer or
adverse reproductive outcomes or teratogenicity. The only reasonable
evidence of carcinogenicity due to TCA in animals relates very
specifically to the induction of liver tumours. If TCA's apparent mode
of action is taken into consideration, it is difficult to identify
other tumours that would be attributable to TCA from animal studies.
Recent studies of workers in degreasing operations provide little
evidence of hepatocellular tumours (Spirtas et al., 1991; Weiss,
1996).
4.2.2.6 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of TCA and concluded that
there is inadequate evidence for its carcinogenicity in humans and
limited evidence for its carcinogenicity in experimental animals. The
compound was assigned to Group 3: not classifiable as to its
carcinogenicity to humans (IARC, 1995).
TCA induces hepatocellular carcinomas when administered in
drinking-water to male B6C3F1 mice (Herren-Freund et al., 1987; Bull
et al., 1990; Daniel et al., 1993a). Although some of the data in the
literature are of a preliminary nature, consistent results were
obtained in three independent studies (Table 17). In two of these
studies, dose-related increases in the incidence of malignant tumours
and precancerous lesions were obtained in B6C3F1 mice at
concentrations in water of between 1 and 5 g/litre and with as little
as 12 months of treatment (Bull et al., 1990; Daniel et al., 1993a).
Under similar conditions of treatment, TCA did not induce hepatic
tumours in F344 rats (DeAngelo et al., 1997).
In the study in mice by Pereira (1996) reported in Table 17, a
NOAEL of 0.35 g of TCA per litre can be identified. Drinking-water
consumption and body weight were reported only during the first
4 weeks of the study. These data were not used; instead, it was
Table 17. Carcinogenic effects of trichloroacetate in rodents
Species Dose Duration Tumour HN & HAa HCb Reference
(sex) (g/litre) (weeks) site
Incidence Tumour/n Incidence Tumour/n
(multiplicity) (multiplicity)
Mice
B6C3F1 (M) 0 61 Liver 2/22 (0.09) 0/22 (0) Herren-Freund et al.
5 61 Liver 8/22 (0.5) 7/22 (0.5) (1987)
B6C3F1 (M) 0 52 Liver 1/35 0.03 0/35 0 Bull et al. (1990)
1 52 Liver 5/11 0.45 2/11 0.18
2 52 Liver 15/24 0.63 4/24 0.17
2 37 Liver 2/11 0.18 3/11 0.27
B6C3F1 (M) 0 60-95 Liver NRc NR 6.7-10% 0.07-0.15 Daniel et al. (1993a)
0.05 60 Liver NR NR 22% 0.31
0.5 60 Liver NR NR 38% 0.55
4.5 95 Liver NR NR 87% 2.2
5 60 Liver NR NR 55% 0.97
B6C3F1 (F) 0 52 Liver 1/40 0.03 0/40 0 Pereira (1996)
0.35 52 Liver 6/40 0.15 0/40 0
1.2 52 Liver 3/19 0.16 0/19 0
3.5 52 Liver 2/20 0.10 5/20 0.25
0 81 Liver 2/90 0.02 2/90 0.02
0.35 81 Liver 14/53 0.26 0/53 0
1.2 81 Liver 12/27 0.44 5/27 0.19
3.5 81 Liver 18/18 1.0 5/18 0.28
Rats
F344 (M) 0 104 Liver 2/23 0.09 0/23 0 DeAngelo et al. (1997)
0.05 104 Liver 2/24 0.08 0/24 0
0.5 104 Liver 5/20 0.25 0/20 0
5.0 104 Liver 1/22 0.045 1/22 0.045
Table 17. (continued)
a Combined hyperplastic nodule and hepatocellular adenoma.
b Hepatocellular carcinoma.
c NR = not reported.
assumed that the average daily water consumption was about 10% of the
animal body weight. On this basis, 0.35 g of TCA per litre is
equivalent to approximately 40 mg/kg of body weight per day.
The available data suggest that TCA has some tumour-promoting
activity. Pereira (1995) and Pereira & Phelps (1996) reported that TCA
increased the yield of both hepatocellular adenomas and hepatocellular
carcinomas in MSU-initiated mice (Figure 4). This effect appears to be
evident at the lowest concentration tested, 0.35 g/litre of
drinking-water. Unlike the circumstance described above with DCA, TCA
significantly increased the yield of hepatocellular carcinomas as well
as hepatocellular adenomas after 362 days of treatment. An earlier
study by Parnell et al. (1986) suggested that TCA was capable of
promoting tumours initiated by diethylnitrosamine. However, this study
employed gamma-glutamyl transpeptidase (GGT) as a marker for
preneoplastic foci and was extended for only 6 months. As a
consequence, no increase in tumour yield was noted, although there was
a significant increase in GGT-positive foci that appeared
dose-related. GGT is a poor marker for foci induced by peroxisome
proliferators (Sakai et al., 1995). Therefore, this study may have
underestimated the promoting activity of TCA.
The mechanisms by which TCA induces tumours are not clear. TCA
induces peroxisome proliferation in male B6C3F1 mice over the same
dose range at which it induces hepatic tumours (DeAngelo et al.,
1989). Unlike the situation with DCA, the induction of peroxisome
synthesis by TCA appears to be sustained over time. Despite a large
number of data that strongly link peroxisome proliferation with
carcinogenesis, the actual mechanism by which such chemicals actually
produce cancer may be only loosely associated with peroxisome
proliferation per se (discussed further in section 4.2.2.8).
As noted in Table 16, the mutation spectra of mouse liver tumours
obtained from mice treated with TCA appear to be different from those
observed with DCA (Ferreria-Gonzalez et al., 1995). However, it is
important to note that this result was obtained from a very limited
number of animals (11), only five of which had hepatic tumours.
Although none of the tumours carried the mutation that is apparently
selected by DCA treatment, the sparseness of these data prevents a
clear conclusion.
Numerous mutagenicity tests have been conducted on TCA (IARC,
1995). TCA did not induce lambda prophage in Escherichia coli and
was not mutagenic to Salmonella typhimurium strains in the presence
or absence of metabolic activation. TCA, however, reacts with dimethyl
sulfoxide (a solvent used commonly in this assay) to form unstable
mutagenic substances, which have not been identified (Nestmann et al.,
1980). TCA did not induce DNA strand breaks in mammalian cells
in vitro. Chromosomal aberrations were not induced in human
lymphocytes exposed in vitro to TCA neutralized to avoid the effects
of low pH seen in cultured mammalian cells.
production of glycolate, glyoxylate and oxalate as major urinary
metabolites of DCA in the F344 rat. It is notable for later
discussions that these authors found a smaller percentage of the dose
of 1-14C-DCA ending up in glycolate than was observed with 2-14C-DCA.
This was offset by a somewhat greater yield (not statistically
significant) of carbon dioxide from 1-14C-DCA than from 2-14C-DCA.
These data would suggest that there are alternative pathways from DCA
to carbon dioxide in the rat besides the conversion to glyoxylate.
The decreases in DCA clearance with repeated doses appear to be
largely due to the inactivation of one enzyme involved in its
metabolism. There appear to be subtle differences in the metabolism of
mice and rats that lead to different manifestations of this
inhibition. In experiments that involved pretreatment of F344 rats at
a level of 0.2 or 2 g/litre in their drinking-water for a 14-day
period, it was found that the conversion of an oral dose of DCA to
carbon dioxide was substantially inhibited. Similar pretreatment of
male B6C3F1 mice did not affect carbon dioxide production from DCA.
However, in both cases, it is apparent that the metabolic clearance of
DCA from blood was affected by the DCA pretreatment. In rats, the
kinetics of DCA disappearance were studied with intravenous dosing,
which allows more precise definition of the kinetics. This
pretreatment led to an increase in the half-life of DCA in the blood
of rats from 2.4 ± 0.8 h to 10.8 ± 2.0 h when animals were
administered a dose of 100 mg/kg of body weight intravenously. Oral
dosing was used in mice, and the major impact of pretreatment was to
increase the Cmax from 2.6 ± 2.6 µg/ml in naive mice to 129.9 µg/ml
in mice that had DCA in their drinking-water at 2 g/litre until the
prior day (16 h before administration of the test dose). Therefore,
the phenomenon that DCA treatment inhibited its own metabolism, which
was originally observed in humans, could be replicated in both mice
and rats.
The mechanism by which this tremendous change in metabolism
occurs has not been established. It has been shown by Lipscomb et al.
(1995) that the bulk of the metabolism of DCA occurs in cytosolic
fractions. Very little DCA is metabolized in microsomes. It is
apparent that the metabolism of DCA in cytosolic preparations from
rodent liver is dependent upon nicotinamide cofactor and GSH. However,
the metabolism is not mediated by enzymes that can be recovered on a
GSH-sepharose column. Glutathione transferase activities towards
chlorodinitrobenzene were observed in the column. Subsequent work has
shown that it is the activity in the cytosol, however, that is
eliminated by DCA treatment (Gonzalez-Leon et al., 1997). Tong et al.
(1998) showed that a substantial fraction of DCA's metabolism is
mediated by a novel GST, GST-zeta. This enzyme appears to be subject
to autoinhibition by DCA.
4.2.1.8 Mode of action
Some indication of mechanisms by which DCA produces its effects
can be gleaned from studies cited in previous sections of this
document. While DCA produces many different types of toxicological
effects (i.e., neurotoxicity, reproductive and developmental
toxicities and carcinogenicity), there may be some features that are
common to the mechanisms that produce all these effects. The
particulars of those mechanisms remain to be established. What follows
is a brief outline of what is known and some speculation on how new
research may be applied to developing this information to better
assess the risks associated with the production of DCA as a by-product
of the chlorination of drinking-water.
Given the lack of evidence that induction of DNA damage by DCA is
involved in liver cancer induction, are there plausible alternative
mechanisms that may be invoked? The most common alternative mode of
action would be evidence that carcinogenic doses of DCA induce
cytotoxic damage in the target organ, which leads to reparative
hyperplasia. Although there is some evidence of single-cell necrosis
with chronic exposure (Stauber & Bull, 1997) and infarcts are
occasionally observed (Sanchez & Bull, 1990) in the livers of B6C3F1
mice, this mode of action plays a negligible role at the lowest doses
that induce liver cancer. Small and variable initial increases in
replication rates of hepatocytes in treated mice are reversed with
continued treatment, with the dominant effect becoming inhibition of
replication within a 4- to 8-week period (Carter et al., 1995; Stauber
& Bull, 1997). These observations indicate that necrosis followed by
reparative hyperplasia do not explain the rapid carcinogenic responses
to DCA.
Additional data add support to the hypothesis that DCA does in
fact act largely by a "non-genotoxic" mode of action. Early data
indicated that DCA was capable of inducing peroxisome proliferation
(Nelson & Bull, 1988; DeAngelo et al., 1989). While these effects have
been observed by others, they are clearly of short duration,
disappearing within a few months (DeAngelo et al., 1989). Moreover, it
has become apparent that DCA induces hepatic tumours at dose rates
that are significantly below those required to induce peroxisome
proliferation (cf. DeAngelo et al., 1989 and Daniel et al., 1992a).
New research has shown that DCA acts primarily to increase the
growth rate of pre-initiated cells in the liver. Stauber et al. (1998)
found that DCA induces growth of colonies on soft agar when cell
suspensions were obtained from the liver of neonatal mice. The most
impressive aspect of these studies was that the colonies expressed the
same phenotype (c-Jun+) as was found in DCA-induced tumours in
vivo. Similar experiments produced a c-Jun phenotype when TCA was
incorporated into the soft agar. A concentration of 0.5 mmol of DCA
per litre was required in the medium to produce a significant increase
in the number of colonies formed in a 10-day interval when the cells
were derived from naive mice. However, if mice were pretreated with
0.5 g of DCA per litre in their drinking-water prior to the isolation
of the cells, 0.02 mmol of DCA per litre was as effective as 0.5
mmol/litre. Moreover, the yield of colonies was increased by this
pretreatment. The increased sensitivity to DCA appears closely related
to the finding that pretreating animals with low levels of DCA
(<0.2 g/litre of drinking-water) reduces its metabolism by more than
90% (Gonzalez-Leon et al., 1997). The increased number of colonies
produced by pretreatment appears to be due to clonal expansion of
these cells. It appears that this activity accounts for the tumours
induced by DCA at higher doses (>2 g/litre) where blood
concentrations are found to be in the 100-500 µmol/litre range.
However, blood concentrations ranging from 1 to 7 µmol/litre produced
in mice treated with 0.5 g/litre (Kato-Weinstein et al., 1998) result
in an 80% incidence of liver tumours (Daniel et al., 1992a),
suggesting that a second mechanism may be involved at lower doses.
At all carcinogenic doses studied, DCA increases the deposition
of glycogen in the liver (Kato-Weinstein et al., 1998). This suggests
that DCA is modifying cell signalling pathways. Low intraperitoneal
doses of DCA produce increases in serum insulin concentrations in
response to glucose challenge (Kato-Weinstein et al., 1998). In
contrast, decreases in serum insulin concentrations have been observed
in mice chronically administered DCA at either 0.5 or 2 g/litre (Smith
et al., 1997). However, these measurements were made during the
daylight hours when both serum glucose and blood DCA concentrations
are low. Clearly, the involvement of insulin in DCA-induced liver
tumorigenesis needs to be studied further.
Recent studies have provided more substantive evidence that DCA
at very high levels possesses some ability to induce genotoxic
effects. Of particular note are the studies of Harrington-Brock et al.
(1998), who found significant increases in mutant frequencies in mouse
lymphoma cells in vitro at concentrations in excess of 1 mmol/litre.
Similar potency of DCA has been observed in the Ames fluctuation assay
by Giller et al. (1997). Previous studies were largely negative or
made use of the free acid form of DCA. Brusick (1986) had previously
documented that low pH produces increased evidence of genotoxic damage
in cultured mammalian cells.
Leavitt et al. (1997) reported increased recovery of mutant cells
from the lacI transgenic mouse with varying periods of treatment
with DCA in drinking-water. Significant increases were observed only
when mice had been treated with 3.5 g/litre for 60 weeks, but not for
shorter time intervals. No significant increases were noted at 1
g/litre. Although the authors took care to ensure that nodules and
tumours were excluded from the sampling, Stauber & Bull (1997)
demonstrated that there are numerous lesions that are smaller than
nodules in B6C3F1 mice maintained on 2 g of DCA per litre for only
40 weeks. It was inevitable that some of these microscopic lesions
were included within the tissue samples described. Given the marked
stimulation of cell replication that occurs within lesions in mice, it
is not possible to determine if the effect reported by Leavitt et al.
(1997) is due to mutagenic effects of DCA or its demonstrated ability
to selectively stimulate the growth of tumour phenotype.
Based on the available evidence, it is probable that genotoxic
effects of DCA play little, if any, role in the induction of liver
cancer in rodents at low doses. This conclusion is based on clear
evidence that DCA is capable of acting as a tumour promoter and
produces effects on cell replication or apoptosis at all carcinogenic
doses (Snyder et al., 1995; Pereira & Phelps, 1996; Stauber & Bull,
1997). The concentrations of DCA required to produce genotoxic effects
in vitro and the blood levels necessary to detect minimal genotoxic
effects in vivo are 3 orders of magnitude higher than those
necessary for induction of an 80% tumour incidence. The recent
evaluation by ILSI (1997) also concluded that the mechanism by which
DCA increased liver tumours was non-genotoxic. However, new data
indicate that the actual mechanism is by tumour promotion rather than
by cytotoxicity and reparative hyperplasia.
The metabolism of DCA is very dose-dependent, with metabolism and
clearance of the chemical being inhibited sharply with high dose
rates. Most data now suggest that it is the parent compound that is
responsible for the effects related to carcinogenicity. Thus, simply
on the basis of considerations of target organ dosimetry, the effects
of DCA would be predicted to increase sharply at chronic dosing levels
that approach or exceed 30 mg/kg of body weight per day rather than
being simple linear functions of dose. Unfortunately, the available
data do not allow the systemic dose versus external dose relationships
to be determined with any precision on the basis of current
information. In rats, the full inhibition was observed at
concentrations of DCA in drinking-water as low as 0.2 g/litre
(Gonzalez-Leon et al., 1997). The minimum treatment level for DCA's
inhibition of its own metabolism in mice has not been established.
However, blood concentrations of DCA increase from 2-4 µmol/litre when
the mice are treated with 0.5 g/litre in drinking-water to
approximately 300 µmol/litre when the treatment concentration is
2 g/litre (Kato-Weinstein et al., 1998). These are peak concentrations
measured during the night when mice are consuming the DCA-containing
water. This 100-fold increase in blood concentrations with a mere
4-fold increase in dose undoubtedly contributes to the highly
non-linear tumorigenic response for DCA reported previously (Bull et
al., 1990). The effect in humans has been documented to occur with
doses in a similar range, 30 mg/kg of body weight (Wells et al.,
1980). The animal data need to be extended to lower dosing rates or
the human treatments need to be extended in time to more precisely
define the level of chronic exposure that is required to produce this
phenomenon. Alternatively, a better understanding of the mechanism of
this inhibition should allow the critical question of whether the
inhibition is a function of cumulative dose or daily dose to be
answered.
The available data indicate that DCA differentially affects the
replication rates of normal hepatocytes and hepatocytes that have been
initiated. The dose-response relationships are complex, with DCA
initially stimulating division of normal hepatocytes. However, at the
lower chronic doses used in animal studies (but still very high
relative to those that would be derived from drinking-water), the
replication rate of normal hepatocytes is eventually sharply
inhibited. This indicates that normal hepatocytes eventually
down-regulate those pathways that are sensitive to stimulation by DCA.
However, altered cells, particularly those that express high amounts
of a protein that is immunoreactive to a c-Jun antibody, do not seem
to be able to down-regulate this response. Thus, the rates of
replication in the preneoplastic lesions with this phenotype are very
high at the doses that cause DCA tumours to develop with a very low
latency. Preliminary data suggest that this continued alteration in
cell birth and death rates is also necessary for the tumours to
progress to malignancy (Bull et al., 1990). This interpretation is
supported by studies that employ initiation/promotion designs as well
(Pereira, 1996).
On the basis of the above considerations, it is suggested that
the currently available cancer risk estimates for DCA should be
modified by incorporating newly developing information on its
comparative metabolism and modes of action to formulate a biologically
based dose-response model. These data are not available at the time of
this writing, but should become available within the next 2-3 years.
The dose-response data for effects other than cancer vary
significantly, with dogs being extraordinarily sensitive (Katz et al.,
1981). However, inhibition of the metabolism of DCA in chronically
treated rodents (Gonzalez-Leon et al., 1997) and humans (Wells et al.,
1980; Curry et al., 1985) may cause differences in sensitivities
between species to converge somewhat with repeated treatment or
exposure. However, a significant difference in species sensitivity
remains. Cicmanec et al. (1991) identified a LOAEL of 12.5 mg/kg of
body weight per day in dogs treated for 90 days. NOAELs for
hepatomegaly in mice appear to be in the neighbourhood of 0.2 g/litre,
which is approximately 40 mg/kg of body weight per day. On the basis
of the rates of metabolic clearance and the assumption that the
intrinsic sensitivities of different species are similar, the average
human would seem to more closely approximate rats than dogs. To obtain
more accurate pictures of human sensitivity at low doses, however, it
is clear that future work must focus more specifically on
toxicodynamic variables.
4.2.2 Trichloroacetic acid (trichloroacetate)
Like DCA, TCA exists almost exclusively in the salt form at pHs
found in drinking-water because of its very low p Ka of 0.70 (IARC,
1995).
4.2.2.1 General toxicological properties and information on
dose-response in animals
1) Acute toxicity
Very little information exists on the mammalian toxicology of TCA
before it was discovered as a by-product of drinking-water
chlorination. Woodard et al. (1941) determined the oral LD50 of
trichloroacetate (i.e., neutralized to pH 6-7) to be 3.32 g/kg of body
weight in rats and 4.97 g/kg of body weight in albino mice when the
compound was administered in aqueous solution. These values were in
the same general range as those for neutralized acetic acid.
Davis (1990) examined the effects of TCA on blood glucose and
lactate levels following a dosing regimen of total doses of 0.92 or
2.45 mmol/kg of body weight administered 3 times in 1 day to
Sprague-Dawley rats by gavage. A typographical error in dosage was
suspected and confirmed with the author (M.E. Davis, personal
communication, 1996). Reductions in plasma glucose concentrations were
observed in females at the high dose, and lactic acid levels were
decreased at 0.92 and 2.45 mmol/kg of body weight doses in females,
but only at the high dose in males. The authors noted that these high
concentrations were neutralized with sodium hydroxide. However, these
are very low doses of TCA relative to those found to produce similar
effects in other studies. These initial experiments were followed by a
study of the effects of TCA administered in drinking-water for 14 days
at 0.04, 0.16, 0.63 or 2.38 g/litre. Effects on urine volume and
osmolality were reported to occur at the highest dose, but not at
0.63 g/litre. Effects on glucose and lactate were not reported.
2) Short-term toxicity
Mather et al. (1990) administered TCA to male Sprague-Dawley rats
in drinking-water at concentrations of 0, 50, 500 or 5000 mg/litre (0,
4.1, 36.5 or 355 mg/kg of body weight per day) for 90 days. Small, but
statistically insignificant, decreases in body weight were observed at
the highest dose. TCA produced a significant increase in the liver to
body weight ratio at this dose, but not at 500 mg/litre. This effect
was associated with a small, but statistically significant, increase
in cyanide-insensitive acyl CoA oxidase activity in the liver, an
indicator of peroxisome proliferation.
Unlike DCA and MCA, TCA does not appear to be a substrate for the
mitochondrial pyruvate carrier (Halestrap, 1975). TCA does appear to
inhibit the pig heart pyruvate dehydrogenase kinase at approximately
the same concentrations as for DCA (Whitehouse et al., 1974). However,
the authors noted that DCA influenced the proportion of active
pyruvate dehydrogenase in the perfused rat heart, but TCA was inactive
under these circumstances. Although these data were obtained from
mitochondria from different sources, they suggest that there may be
some differences in effects of DCA and TCA on intermediary metabolism
related to their transport into various cellular compartments.
Bhat et al. (1991) administered TCA to male Sprague-Dawley rats
at a concentration of 45.8 mmol/litre (7.5 g/litre) in their
drinking-water for 90 days to provide an approximate intake of 785
mg/kg of body weight per day. These levels produced minimal evidence
of liver toxicity by histopathological examination. However, lower
concentrations in drinking-water have been shown to seriously impair
water and food consumption in experimental animals (Bull et al., 1990;
Davis, 1990; Mather et al., 1990; DeAngelo et al., 1991).
Consequently, it is difficult to determine how these data relate to
the potential effects of the low concentrations of TCA that are found
in chlorinated drinking-water (e.g., in the 10-100 µg/litre range).
The most obvious target organ for TCA is the liver. This effect
is marked by a hepatomegaly (Goldsworthy & Popp, 1987; Bull et al.,
1990; Mather et al., 1990; Sanchez & Bull, 1990), which is presumably
related to the ability of TCA to act as a peroxisome proliferator
(DeAngelo et al., 1989), since that is a common finding with this
class of rodent carcinogens. It could also be related to the
metabolism of TCA to DCA (Larson & Bull, 1992). However, more recent
data suggest that the apparent conversion of TCA to DCA has largely
been the result of artefactual conversion when fresh oxygenated blood
is acidified prior to derivatization (Merdink et al., 1998). TCA is
clearly without substantive cytotoxic effects at doses of less than
300 mg/kg of body weight in vivo (Bull et al., 1990; Sanchez & Bull,
1990; Acharya et al., 1995) or concentrations of up to 5 mmol/litre
in vitro (Bruschi & Bull, 1993).
4.2.2.2 Reproductive effects
There have been limited studies of TCA's effects on reproductive
performance. TCA was found to inhibit in vitro fertilization of
gametes from B6D2F1 mice at a concentration of 1000 mg/litre, but was
without effect at 100 mg/litre (Cosby & Dukelow, 1992). It is unlikely
that these effects at very high doses relative to those that might be
expected from human consumption of TCA at concentrations less than
0.1% of the NOEL are of relevance in assessing risks from exposure to
TCA in drinking-water.
4.2.2.3 Developmental effects
Treatment of pregnant rats with TCA at 0, 330, 800, 1200 or
1800 mg/kg of body weight per day by gavage in a water vehicle on
gestation days 6-15 produced dose-dependent reductions in body weight
and length of rat pups from dams administered doses of 800 mg/kg of
body weight per day and above. No effects were observed at 330 mg/kg
of body weight per day. However, there was a significant and
dose-related increase in soft tissue malformations at all doses
studied. The mean frequency of soft tissue malformations was 3.5 ±
8.7% (SD), 9.06 ± 12.9%, 30.4 ± 28.1%, 55.4 ± 36.1% and 96.9 ± 8.8% at
0, 330, 800, 1200 and 1800 mg/kg of body weight, respectively. Most of
the increased soft tissue abnormalities were accounted for by defects
in the cardiovascular system. The major malformation seen was
laevocardia. However, at doses of 800 mg/kg of body weight and above,
a significant incidence of an interventricular septal defect was
observed (Smith et al., 1989a).
Saillenfait et al. (1995) found that TCA, administered to rats in
embryo culture, began to produce consistent increases in defects at
concentrations of 2.5 mmol/litre and above, with few or no effects
observed at 1 mmol/litre. These defects included brain and eye
defects, reduction in the branchial arch and otic system defects. At
concentrations of 3.5 mmol/litre, some evidence of skeletal defects
was observed (i.e., absence of hindlimb bud). As is discussed below,
these concentrations can be achieved in the blood of rats administered
high doses of TCA (>100 mg/kg of body weight) (Larson & Bull,
1992). However, there is considerable doubt about whether these
effects would be induced by the doses of less than 1 µg/kg of body
weight experienced by humans consuming chlorinated drinking-water.
4.2.2.4 Neurotoxicity
No reports of neurotoxic effects of TCA were located.
4.2.2.5 Toxicity in humans
TCA is a strong acid. It is widely recognized that contact of TCA
with the skin has the potential to produce acid burns, and ingestion
of TCA has the potential to damage tissues of the gastrointestinal
tract or produce systemic acidosis, even though specific studies of
these effects do not appear in the literature. Such effects would
occur from contact with the crystal or strong solutions of the free
acid. However, such effects have little relevance to the production of
low levels of TCA, as the salt, as a by-product of the chlorination of
drinking-water.
Indirectly, it may be presumed that TCA presents little overt
hazard to human health because it is a major metabolite of commonly
used solvents such as trichloroethylene and tetrachloroethylene.
Occupational exposures to these solvents have been quite high in the
past, but few, if any, effects of the solvents in humans have been
attributed to TCA. Therefore, one would surmise that TCA is relatively
non-toxic to humans under circumstances of low exposures such as those
encountered in chlorinated drinking-water. However, these largely
negative data do not insure against chronic hazards such as cancer or
adverse reproductive outcomes or teratogenicity. The only reasonable
evidence of carcinogenicity due to TCA in animals relates very
specifically to the induction of liver tumours. If TCA's apparent mode
of action is taken into consideration, it is difficult to identify
other tumours that would be attributable to TCA from animal studies.
Recent studies of workers in degreasing operations provide little
evidence of hepatocellular tumours (Spirtas et al., 1991; Weiss,
1996).
4.2.2.6 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of TCA and concluded that
there is inadequate evidence for its carcinogenicity in humans and
limited evidence for its carcinogenicity in experimental animals. The
compound was assigned to Group 3: not classifiable as to its
carcinogenicity to humans (IARC, 1995).
TCA induces hepatocellular carcinomas when administered in
drinking-water to male B6C3F1 mice (Herren-Freund et al., 1987; Bull
et al., 1990; Daniel et al., 1993a). Although some of the data in the
literature are of a preliminary nature, consistent results were
obtained in three independent studies (Table 17). In two of these
studies, dose-related increases in the incidence of malignant tumours
and precancerous lesions were obtained in B6C3F1 mice at
concentrations in water of between 1 and 5 g/litre and with as little
as 12 months of treatment (Bull et al., 1990; Daniel et al., 1993a).
Under similar conditions of treatment, TCA did not induce hepatic
tumours in F344 rats (DeAngelo et al., 1997).
In the study in mice by Pereira (1996) reported in Table 17, a
NOAEL of 0.35 g of TCA per litre can be identified. Drinking-water
consumption and body weight were reported only during the first
4 weeks of the study. These data were not used; instead, it was
Table 17. Carcinogenic effects of trichloroacetate in rodents
Species Dose Duration Tumour HN & HAa HCb Reference
(sex) (g/litre) (weeks) site
Incidence Tumour/n Incidence Tumour/n
(multiplicity) (multiplicity)
Mice
B6C3F1 (M) 0 61 Liver 2/22 (0.09) 0/22 (0) Herren-Freund et al.
5 61 Liver 8/22 (0.5) 7/22 (0.5) (1987)
B6C3F1 (M) 0 52 Liver 1/35 0.03 0/35 0 Bull et al. (1990)
1 52 Liver 5/11 0.45 2/11 0.18
2 52 Liver 15/24 0.63 4/24 0.17
2 37 Liver 2/11 0.18 3/11 0.27
B6C3F1 (M) 0 60-95 Liver NRc NR 6.7-10% 0.07-0.15 Daniel et al. (1993a)
0.05 60 Liver NR NR 22% 0.31
0.5 60 Liver NR NR 38% 0.55
4.5 95 Liver NR NR 87% 2.2
5 60 Liver NR NR 55% 0.97
B6C3F1 (F) 0 52 Liver 1/40 0.03 0/40 0 Pereira (1996)
0.35 52 Liver 6/40 0.15 0/40 0
1.2 52 Liver 3/19 0.16 0/19 0
3.5 52 Liver 2/20 0.10 5/20 0.25
0 81 Liver 2/90 0.02 2/90 0.02
0.35 81 Liver 14/53 0.26 0/53 0
1.2 81 Liver 12/27 0.44 5/27 0.19
3.5 81 Liver 18/18 1.0 5/18 0.28
Rats
F344 (M) 0 104 Liver 2/23 0.09 0/23 0 DeAngelo et al. (1997)
0.05 104 Liver 2/24 0.08 0/24 0
0.5 104 Liver 5/20 0.25 0/20 0
5.0 104 Liver 1/22 0.045 1/22 0.045
Table 17. (continued)
a Combined hyperplastic nodule and hepatocellular adenoma.
b Hepatocellular carcinoma.
c NR = not reported.
assumed that the average daily water consumption was about 10% of the
animal body weight. On this basis, 0.35 g of TCA per litre is
equivalent to approximately 40 mg/kg of body weight per day.
The available data suggest that TCA has some tumour-promoting
activity. Pereira (1995) and Pereira & Phelps (1996) reported that TCA
increased the yield of both hepatocellular adenomas and hepatocellular
carcinomas in MSU-initiated mice (Figure 4). This effect appears to be
evident at the lowest concentration tested, 0.35 g/litre of
drinking-water. Unlike the circumstance described above with DCA, TCA
significantly increased the yield of hepatocellular carcinomas as well
as hepatocellular adenomas after 362 days of treatment. An earlier
study by Parnell et al. (1986) suggested that TCA was capable of
promoting tumours initiated by diethylnitrosamine. However, this study
employed gamma-glutamyl transpeptidase (GGT) as a marker for
preneoplastic foci and was extended for only 6 months. As a
consequence, no increase in tumour yield was noted, although there was
a significant increase in GGT-positive foci that appeared
dose-related. GGT is a poor marker for foci induced by peroxisome
proliferators (Sakai et al., 1995). Therefore, this study may have
underestimated the promoting activity of TCA.
The mechanisms by which TCA induces tumours are not clear. TCA
induces peroxisome proliferation in male B6C3F1 mice over the same
dose range at which it induces hepatic tumours (DeAngelo et al.,
1989). Unlike the situation with DCA, the induction of peroxisome
synthesis by TCA appears to be sustained over time. Despite a large
number of data that strongly link peroxisome proliferation with
carcinogenesis, the actual mechanism by which such chemicals actually
produce cancer may be only loosely associated with peroxisome
proliferation per se (discussed further in section 4.2.2.8).
As noted in Table 16, the mutation spectra of mouse liver tumours
obtained from mice treated with TCA appear to be different from those
observed with DCA (Ferreria-Gonzalez et al., 1995). However, it is
important to note that this result was obtained from a very limited
number of animals (11), only five of which had hepatic tumours.
Although none of the tumours carried the mutation that is apparently
selected by DCA treatment, the sparseness of these data prevents a
clear conclusion.
Numerous mutagenicity tests have been conducted on TCA (IARC,
1995). TCA did not induce lambda prophage in Escherichia coli and
was not mutagenic to Salmonella typhimurium strains in the presence
or absence of metabolic activation. TCA, however, reacts with dimethyl
sulfoxide (a solvent used commonly in this assay) to form unstable
mutagenic substances, which have not been identified (Nestmann et al.,
1980). TCA did not induce DNA strand breaks in mammalian cells
in vitro. Chromosomal aberrations were not induced in human
lymphocytes exposed in vitro to TCA neutralized to avoid the effects
of low pH seen in cultured mammalian cells.
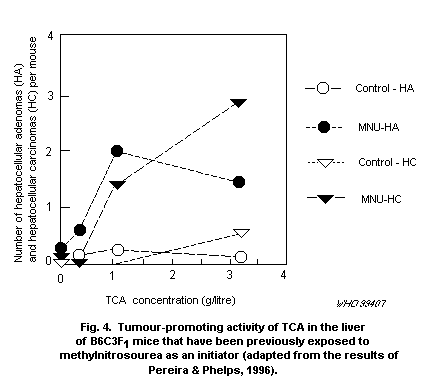 DNA strand breaks were reported in one laboratory in the livers
of mice and rats treated 4 h previously with TCA; none was observed
24 h after repeated daily dosing with 500 mg/kg of body weight (Nelson
& Bull, 1988; Nelson et al., 1989). Peroxisome proliferation, as
indicated by ß-oxidation of palmitoyl CoA, was observed only after
induction of DNA damage (Nelson et al., 1989). DNA strand breakage was
not observed in the livers of mice or rats (Chang et al., 1992). The
reasons for the contrasting results obtained using similar techniques
are unclear (IARC, 1995).
Giller et al. (1997) examined the effects of TCA in the SOS
chromotest in Escherichia coli Pq37, the Ames fluctuation assay and
the newt micronucleus assay. TCA was negative in the SOS chromotest
but exhibited weak activity in the Ames fluctuation assay. Effects
were observed at the lowest concentration, 1750 µg/ml, in the absence
of S9 fraction. This corresponds to TCA concentrations of
approximately 10 mmol/litre in the medium. Newt larvae were found to
have an increased frequency of micronuclei at TCA concentrations of
80 µg/ml. As with some previous studies, these tests appear to have
been conducted with the free acid, raising issues of potential
artefacts in the results.
Harrington-Brock et al. (1998) studied the mutagenic activity of
TCA in the mouse lymphoma system. A very weak positive result was
obtained at concentrations in excess of 20 mmol/litre. Concentrations
of TCA reached in the blood of mice treated with carcinogenic doses
can approach the mmol/litre range, so it is possible that these
results could be relevant to bioassay data. However, concentrations
anticipated in drinking-water would clearly be much lower (in the low
µmol/litre range).
In one study, TCA induced micronuclei and chromosomal aberrations
in bone marrow cells and abnormal sperm morphology after injection
into Swiss mice in vivo at doses of 125-500 mg/kg of body weight
(Bhunya & Behera, 1987). However, Mackay et al. (1995) could not
replicate this finding, even at doses 10-fold higher.
4.2.2.7 Comparative pharmacokinetics and metabolism
TCA is readily absorbed from the gastrointestinal tract in
experimental animals and humans (Muller et al., 1974; Larson & Bull,
1992). However, the major determinant of its blood concentrations at a
given dose is its relatively slow clearance from blood relative to
other HAAs. There are substantial differences in this clearance by
different species. The half-life is 5.8 h in mice (Larson & Bull,
1992), 9.3 h in rats (Merdink et al., 1999), 50 h in humans (Muller et
al., 1974) and approximately 200 h in dogs (Muller et al., 1974).
TCA is much less extensively metabolized than other HAAs found in
drinking-water. However, one metabolite is DCA (Larson & Bull, 1992),
which is subsequently converted to glyoxylate, glycolate and oxalate,
as outlined in Figure 5. This in turn explains the extensive
incorporation of radiolabel from 14C-TCA into blood (Stevens et al.,
DNA strand breaks were reported in one laboratory in the livers
of mice and rats treated 4 h previously with TCA; none was observed
24 h after repeated daily dosing with 500 mg/kg of body weight (Nelson
& Bull, 1988; Nelson et al., 1989). Peroxisome proliferation, as
indicated by ß-oxidation of palmitoyl CoA, was observed only after
induction of DNA damage (Nelson et al., 1989). DNA strand breakage was
not observed in the livers of mice or rats (Chang et al., 1992). The
reasons for the contrasting results obtained using similar techniques
are unclear (IARC, 1995).
Giller et al. (1997) examined the effects of TCA in the SOS
chromotest in Escherichia coli Pq37, the Ames fluctuation assay and
the newt micronucleus assay. TCA was negative in the SOS chromotest
but exhibited weak activity in the Ames fluctuation assay. Effects
were observed at the lowest concentration, 1750 µg/ml, in the absence
of S9 fraction. This corresponds to TCA concentrations of
approximately 10 mmol/litre in the medium. Newt larvae were found to
have an increased frequency of micronuclei at TCA concentrations of
80 µg/ml. As with some previous studies, these tests appear to have
been conducted with the free acid, raising issues of potential
artefacts in the results.
Harrington-Brock et al. (1998) studied the mutagenic activity of
TCA in the mouse lymphoma system. A very weak positive result was
obtained at concentrations in excess of 20 mmol/litre. Concentrations
of TCA reached in the blood of mice treated with carcinogenic doses
can approach the mmol/litre range, so it is possible that these
results could be relevant to bioassay data. However, concentrations
anticipated in drinking-water would clearly be much lower (in the low
µmol/litre range).
In one study, TCA induced micronuclei and chromosomal aberrations
in bone marrow cells and abnormal sperm morphology after injection
into Swiss mice in vivo at doses of 125-500 mg/kg of body weight
(Bhunya & Behera, 1987). However, Mackay et al. (1995) could not
replicate this finding, even at doses 10-fold higher.
4.2.2.7 Comparative pharmacokinetics and metabolism
TCA is readily absorbed from the gastrointestinal tract in
experimental animals and humans (Muller et al., 1974; Larson & Bull,
1992). However, the major determinant of its blood concentrations at a
given dose is its relatively slow clearance from blood relative to
other HAAs. There are substantial differences in this clearance by
different species. The half-life is 5.8 h in mice (Larson & Bull,
1992), 9.3 h in rats (Merdink et al., 1999), 50 h in humans (Muller et
al., 1974) and approximately 200 h in dogs (Muller et al., 1974).
TCA is much less extensively metabolized than other HAAs found in
drinking-water. However, one metabolite is DCA (Larson & Bull, 1992),
which is subsequently converted to glyoxylate, glycolate and oxalate,
as outlined in Figure 5. This in turn explains the extensive
incorporation of radiolabel from 14C-TCA into blood (Stevens et al.,
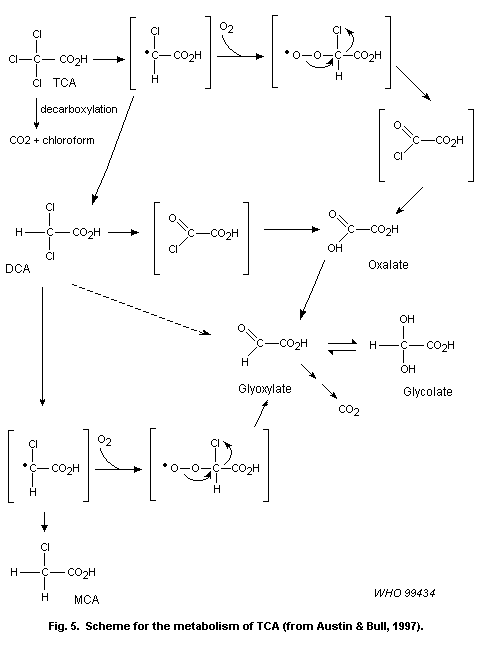 1992) and tissues (Eyre et al., 1995), as glyoxylate is rapidly
transaminated and converted to glycine.
In mice, some of TCA's metabolism is independent of the formation
of DCA as an intermediate. There is evidence that a significant amount
of oxalate is formed independently of DCA formation and that some
direct decarboxylation of trihaloacetates also occurs. The evidence
for these pathways comes largely from studies of the metabolism of
bromodichloroacetate (BDCA) (Xu et al., 1995). The conversion of TCA
is, however, much slower than that of BDCA. The basis for the overall
scheme is discussed more fully in the following section on brominated
HAAs. The reader is also referred to Figure 5 and the accompanying
text for explanation of the further metabolism of DCA.
4.2.2.8 Mode of action
The tumorigenic effects of TCA in the liver of B6C3F1 mice
appear to be closely related to its ability to induce synthesis of
peroxisomes and associated proteins (DeAngelo et al., 1989; Bull et
al., 1990; Pereira, 1996). Along with a number of other peroxisome
proliferators, TCA was shown to be capable of activating the PPAR
in vitro at concentrations consistent with the levels that are
achieved in vivo (Issemann & Green, 1990). The cause-and-effect
relationships between the activation of this receptor and the
induction of cancer are yet to be established. Based upon marked
increases in the numbers of peroxisomes that are observed in rodent
species that are susceptible to this class of carcinogen and the lack
of such responses in other mammalian species, it has been argued that
humans are minimally sensitive to the tumorigenic effects of these
compounds (Lake, 1995). Peroxisome proliferators are also known to
variably affect reproduction and development. Considering the types of
interactions in which the PPAR is involved, these observations are not
too surprising. However, it is not clear whether a consistent pattern
of developmental anomalies can be associated with activation of this
receptor.
A mouse that was genetically engineered with a targeted
disruption PPAR-alpha gene failed to respond to the pleiotropic
effects of peroxisome proliferators (Lee et al., 1995).
Non-rodent species, including humans, express the PPARs, and
peroxisome proliferator responsive elements (PPREs) have been
identified in the promoter regions of the genes that are analogous to
those that are induced in rodents (Varanasi et al., 1996). This
suggests that the responsiveness to peroxisome proliferators is in
some way modified by other factors. However, the expression of PPARs
in humans is low.
It is important to recognize that other mechanisms may be
involved in TCA-induced effects. Clearly, the production of DCA as a
metabolite could be involved in inducing effects associated with this
metabolite. The very high rate of DCA metabolism relative to the rate
of TCA metabolism has made it difficult to detect significant amounts
of DCA in the blood (Merdink et al., 1998). There is little doubt that
some DCA is formed in all species. In addition to DCA, there are a
number of reactive and stable metabolites that could contribute to
toxicity. At this time, there is no evidence that the acid chloride
intermediate postulated between DCA and oxalate contributes anything
to the toxic effects of TCA. Chronic TCA administration results in the
deposition of lipofuscin, a sign of increased oxidative stress.
Moreover, Austin et al. (1995, 1996) obtained evidence of increased
lipid peroxidation and increased levels of 8-OH-dG in nuclear DNA of
the liver of mice treated with single doses of TCA. Therefore, the
accumulation of lipofuscin could be associated with mechanisms
unrelated to peroxisome proliferation. The most likely
radical-generating sources for lipid peroxidation and oxidative damage
to DNA would be through the formation of the radical species
(carbon-centred radicals and the peroxy radicals that would be derived
from the reaction of molecular oxygen with the carbon-centred
radicals). Again, there is no direct evidence to indicate that these
processes are important to any toxicological response associated with
TCA treatment or exposure.
It is beyond the scope of the present review to resolve risk
assessment issues associated with peroxisome proliferators. However,
several points should be made specific to TCA. First, peroxisome
proliferative responses are not genotoxic responses. Second, TCA is
one of the weakest activators of the PPAR known (Issemann & Green,
1990). Finally, TCA appears to be only marginally active as a
peroxisome proliferator, even in rats (DeAngelo et al., 1989).
Furthermore, treatment of rats with high levels of TCA in
drinking-water does not induce liver tumours (DeAngelo et al., 1997).
These data strongly suggest that TCA presents little carcinogenic
hazard to humans at the low concentrations found in drinking-water.
The key question is whether the carcinogenic and teratogenic
effects of TCA observed at very high doses have any relevance at the
exposures that are obtained even under extreme conditions in
drinking-water. However, the application of conventional uncertainty
factors would suggest that TCA in drinking-water represents little
hazard to humans at the concentrations normally encountered in
chlorinated drinking-water.
4.2.3 Brominated haloacetic acids
Brominated HAAs are formed in waters that contain bromide, which
strong oxidants like chlorine and ozone are capable of oxidizing to
hypobromous acid. There are very limited data available on the
toxicity of these chemicals. Therefore, they will be considered as a
group. The discussion below focuses on similarities and differences
between brominated and chlorinated HAAs.
4.2.3.1 General toxicological properties and information on
dose-response in animals
Linder et al. (1994a) found that the oral LD50 for MBA was
177 mg/kg of body weight in adult male Sprague-Dawley rats. DBA was
much less toxic, with an LD50 of 1737 mg/kg of body weight. The acute
toxicities of the bromochloroacetates have not been determined.
No studies have been published on the brominated haloacetates.
Some of the information is available in abstract form and will be
touched on briefly.
Bull and co-workers (Bull & DeAngelo, 1995; Stauber et al., 1998)
conducted a series of experiments with DBA, bromochloroacetate (BCA)
and BDCA administered to male B6C3F1 mice at concentrations ranging
from 0.2 to 3 g/litre in their drinking-water. The data obtained from
these studies suggest that toxicological effects are observed in
approximately the same concentration ranges as for DCA- and
TCA-induced toxic effects.
The principal target organ in mice was identified as the liver
for all three brominated HAAs, but the nature of the effects on the
liver appears to be somewhat different for each compound. All the
brominated HAAs produce hepatomegaly, but glycogen accumulation and
cytomegaly are more prominent with BCA and BDCA than with DBA.
Conversely, DBA was reported to produce increases in
cyanide-insensitive acyl CoA activity in the liver. BCA and BDCA
produced only small and inconsistent effects on this marker of
peroxisome proliferation. Thus, it appears that DBA shares TCA's
ability to induce peroxisome proliferation, whereas BCA and BDCA
appear to produce effects much more like those induced by DCA.
Consistent with this, the severity of the hepatomegaly was BCA > BDCA
> DBA.
Administration of single doses of BDCA, BCA and DBA by gavage in
water as the vehicle induced increases in thiobarbituric acid reactive
substances (TBARS) and increased the 8-OH-dG content of nuclear DNA in
the liver of male B6C3F1 mice at doses as low as 30 mg/kg of body
weight (Austin et al., 1996). These effects were significantly greater
than those observed with TCA and DCA and tended to increase as the
bromine substitution increased within each series. The order of
potency was DBA = BCA > BDCA > DCA > TCA. Increases in 8-OH-dG
content were more rapid and more sustained with the brominated HAAs.
This indicates that brominated HAAs do induce oxidative stress.
Dose-related increases in the 8-OH-dG content of nuclear DNA of
the liver were observed when DBA and BCA were administered in
drinking-water to male B6C3F1 mice for periods from 3 to 10 weeks at
concentrations of 0.5 g/litre and above (Parrish et al., 1996). The
effect of BDCA in drinking-water was not evaluated. It is noted that
these effects were observed in the same range in which liver tumours
are induced in mice (discussed further below).
4.2.3.2 Reproductive effects
MBA and DBA have been examined for spermatotoxic effects in rats.
MBA did not affect parameters related to male reproductive function
(Linder et al., 1994a) when administered to rats as a single dose of
100 mg/kg of body weight or at 25 mg/kg of body weight per day
administered repeatedly for 14 consecutive days. In contrast, DBA
produced degenerating, misshapen epididymal sperm and abnormal
retention of step 19 spermatids following single doses of DBA in the
range 1000-2000 mg/kg of body weight. Caput sperm counts were
significantly reduced on the second day, and substantial reductions of
cauda sperm counts were observed at 14 and 28 days after treatment.
Serum testosterone levels were significantly depressed on day 2 but
returned to control levels by day 14. Sperm displayed defects in
development of the shape of sperm heads. Progressive motility of sperm
was significantly reduced at 14 and 28 days after treatment.
A subsequent paper by the same group described a study in which
doses of 0, 10, 30, 90 or 270 mg of DBA per kg of body weight were
administered to rats for 14 consecutive days (Linder et al., 1994b).
Marked effects on epididymal sperm counts and sperm morphology effects
were observed at the highest dose. Approximately 5% of the caput sperm
were fused. In contrast to the single-dose studies, serum testosterone
levels appeared to be unaffected. Spermiation also appeared to be
mildly affected, with step 19 spermatids being retained beyond stage
VIII in animals dosed with as little as 10 mg/kg of body weight per
day.
DBA was administered to rats for up to 79 days at 0, 2, 10, 50 or
250 mg/kg of body weight per day by gavage in water. Male fertility
was compromised during the second week of treatment at the high dose.
This effect appeared to result from behavioural changes, because
artificial insemination with sperm collected on day 9 of treatment
produced offspring. By day 15, however, no offspring were produced
with either biological or artificial insemination, indicating that
significant qualitative alterations had occurred in the sperm. Indeed,
the 50 mg/kg of body weight per day dose produced abnormal morphology,
decreased motility and decreases in epididymal sperm counts. However,
rats treated at this dose remained fertile. While no effects on sperm
quality were observed at lower doses, reproductive performance
appeared depressed at doses as low as 10 mg/kg of body weight per day,
suggesting that these lower doses modified behaviour (Linder et al.,
1995).
Histopathological changes were observed in the testis and
epididymis of rats gavaged daily for 2-79 days with DBA. On treatment
day 2, abnormal retention of step 19 spermatids was observed in
animals given the highest dose of 250 mg/kg of body weight per day.
Additional changes on day 5 included the fusion of mature spermatids
and the presence of atypical residual bodies (ARB) in the epithelium
and lumen of stage X-XII seminiferous tubules. By day 9, ARB were seen
in most stages of the seminiferous epithelial cycle and in the caput
epididymis. On day 16, distorted sperm heads were recognized in step
12 and older spermatids, and luminal cytoplasmic debris was found
throughout the epididymis. On day 31, there was vacuolation of the
Sertoli cell cytoplasm, extensive retention of step 19 spermatids near
the lumen of stage IX and X tubules, and vesiculation of the acrosomes
of late spermatids. Marked atrophy of the seminiferous tubules was
present 6 months after 42 doses of 250 mg/kg of body weight per day.
ARB and retention of step 19 spermatids were observed after 31 and 79
doses of 50 mg/kg of body weight per day, and increased retention of
step 19 spermatids was seen in several rats dosed with 10 mg/kg of
body weight per day. No abnormalities were detected at 2 mg/kg of body
weight per day. The changes suggest that the testicular effects of DBA
are sequelae to structural or functional changes in the Sertoli cell
(Linder et al., 1997b).
4.2.3.3 Neurotoxicity
Reference was made to neurotoxic effects in male Sprague-Dawley
rats treated with DBA in reproductive studies (Linder et al., 1994a,b,
1995). However, no specific investigations of the neurotoxic effects
of brominated HAAs appear to be available.
4.2.3.4 Toxicity in humans
There are no studies on the health effects of brominated HAAs in
humans.
4.2.3.5 Carcinogenicity and mutagenicity
No formal reports have been made of carcinogenic or mutagenic
effects of brominated HAAs. Indications that DBA, BCA and BDCA share
the hepatocarcinogenic effects of TCA and DCA in B6C3F1 mice have
been referred to in an abstract (Bull & DeAngelo, 1995).
Giller et al. (1997) examined the genotoxicity of MBA, DBA, and
tribromoacetic acid (TBA) in the SOS chromotest in Escherichia coli
PQ37, the Ames fluctuation assay utilizing Salmonella typhimurium
strain TA100 and the newt micronucleus assay in larvae at stage 53 of
the developmental table. MBA was negative in the SOS chromotest at
levels as high as 1000 µg/ml and in the newt micronucleus assay.
However, it was active at concentrations as low as 20 µg/ml in the
Ames fluctuation assay with S9 fraction added to the incubation
medium. DBA was positive with and without S9 fraction in the SOS
chromotest, requiring 100 µg/ml in the former case and 200 µg/ml in
the latter. Thus, it is about 5 times as potent as DCA in this test.
In the Ames fluctuation assay, DBA was active at 10 µg/ml without S9
and at 30 µg/ml with S9. TBA was active in the SOS chromotest at
100 µg/ml with S9 but required 2000 µg/ml for activity in the Ames
fluctuation assay without S9.
4.2.3.6 Comparative pharmacokinetics and metabolism
Metabolism of brominated HAAs has received little attention. Xu
et al. (1995) studied the metabolism of BDCA in male B6C3F1 mice. As
predicted, substitution of a bromine for a chlorine in TCA resulted in
a substantially greater extent of trihaloacetate metabolism. Whereas
45% of a 100 mg/kg of body weight dose of TCA was eliminated unchanged
in the urine of mice within 24 h, less than 4% of the same dose of
BDCA was found in the urine. At lower doses, only a fraction of a
percent of the BDCA was eliminated unchanged.
There is evidence for substantial conversion of BDCA to DCA in
both rats and mice (Xu et al., 1995; Schultz et al., 1999). Because of
the established ability of DCA to induce hepatic tumours in both mice
and rats (discussed above), this may have implications for assessing
the risk associated with this compound (see Figures 3 and 5 for a
general description of dihaloacetate and trihaloacetate metabolism,
respectively).
The metabolism of BDCA is differentially modified in mice and
rats as doses are increased. Xu et al. (1995) found that the kinetics
of carbon dioxide production from 1-14C-BDCA suggested an efficient
conversion of BDCA to carbon dioxide through DCA at low doses, but a
direct decarboxylation reaction became important as doses approached
100 mg/kg of body weight (Xu et al., 1995; Austin & Bull, 1997). This
complex activity was not observed in rats (Schultz et al., 1999), in
that a progressively smaller fraction of the dose is converted to
carbon dioxide as dose is increased. This suggests that direct
decarboxylation plays a less important role in the metabolism of BDCA
in rats than in mice.
The ratios of urinary metabolites produced by mice and rats
suggest that there are some substantive differences in the metabolism
of BDCA in the two species. Mice (Xu et al., 1995) produce much higher
amounts of oxalate (about 30% of the orally administered dose) than do
rats (about 20%) (Schultz et al., 1999). The much greater conversion
of BDCA to oxalate than for equivalent doses of DCA suggests that much
of the extra oxalate seen in mouse urine arises from reductive
dehalogenation of BDCA, followed by peroxy radical formation and
decomposition to oxalate (Xu et al., 1995).
As the BDCA dose is increased from 20 to 100 mg/kg of body weight
in the rat, the fraction of the dose that is eliminated in the urine
as DCA increases from about 2% to 13% (Schultz et al., 1999), whereas
in mice the increase is from 0.2% to approximately 3%. In the rat,
increasing the dose from 5 mg/kg of body weight to 20 or 100 mg/kg of
body weight was associated with a significantly extended half-life of
both BDCA and DCA (from 0.9 to 3.7 h). The increased half-life appears
to be attributed to saturation of the conversion of BDCA to DCA.
Despite this inhibition of DCA formation, the blood levels of DCA
observed are actually higher than those observed when equivalent doses
of DCA itself are administered. Thus, high doses of BDCA would appear
to inhibit the metabolic clearance of DCA as well. Consequently, the
toxicology of BDCA should share many of the attributes of DCA
toxicology in the rat. Comparable data do not exist on the blood
levels of DCA achieved by BDCA administration in mice, but the urinary
levels of DCA metabolites appear to be largely offset by the
conversion of BDCA to oxalate, suggesting that the blood levels of DCA
would be significantly lower in mice than in rats.
In animals previously treated with 1 g of DCA per litre in
drinking-water, metabolism of BDCA to carbon dioxide is significantly
increased in mice (Austin & Bull, 1997). This appears to be associated
with an increase in the capacity for the direct decarboxylation of
BDCA to form BDCM and carbon dioxide. Therefore, some effects of BDCA
in mice may be attributed to the formation of BDCM.
These data and data indicating that pretreatment with DCA also
affects its own metabolism suggest that there may be significant
interactions in the toxicity of these chemicals at high doses. It
remains to be seen if such interactions occur at treatment
concentrations that more closely approximate those observed in
drinking-water.
4.2.3.7 Mode of action
It is premature to attempt a definitive discussion of the
mechanisms by which brominated HAAs induce tumours. There are
suggestions of mechanisms that need to be pursued with this class, but
there are really no data that demonstrate how they contribute to the
induction of cancer, nor is there a firm basis for considering whether
these mechanisms are relevant at the low concentrations of these
chemicals found in drinking-water.
The mechanisms associated with the carcinogenic effects of HAAs
include those identified for DCA and TCA. It is apparent that more
than one mechanism is responsible for the effects of this class and
that the importance of these mechanisms to the activity of individual
members of the class varies. In part, these differences in mechanism
can be related to the differences in tumour phenotypes that are
induced. One phenotype seems to be associated with prior
characterizations of tumours induced by peroxisome proliferators and
is induced by TCA (DeAngelo et al., 1989; Stauber et al., 1998). The
second phenotype involves glycogen-poor tumours that stain heavily
with antibodies to c-Jun and c-Fos. This phenotype is produced by DCA.
These effects are probably produced by selection of lesions with
differing defects in cell signalling pathways that control the
processes of cell division and cell death.
Based upon the data of Giller at al. (1997), the brominated HAAs
are about 10-fold more potent than their chlorinated analogues in
their ability to induce point mutations. This does not establish that
they are inducing cancer by mutagenic mechanisms in vivo, but this
activity will have to be taken into account as data on their
carcinogenic activity become more complete.
The HAAs vary widely in their ability to induce oxidative stress
and to elevate the 8-OH-dG content of nuclear DNA of the liver. This
property becomes increasingly apparent with the brominated compounds
(Austin et al., 1996; Parrish et al., 1996). It is notable that the
brominated analogues are not more potent inducers of hepatic tumours
than the corresponding chlorinated HAAs. Therefore, it is doubtful
that this mechanism is the most important determinant of this effect.
No specific mechanisms have been associated with the effects of
DBA on male reproduction or as a developmental toxin. However, it
would be surprising if the effects on cell signalling systems that
appear to be involved in the carcinogenic responses do not also
contribute to these effects.
4.2.4 Higher molecular weight halogenated acids
Studies of by-product formation from humic and fulvic acids in
the 1970s and early 1980s demonstrated that there is a complex array
of halogenated carboxylic acids in addition to the HAAs (Christman et
al., 1983). Some of these acids have been identified in drinking-water
as well, but, as discussed elsewhere in this document, both the scope
and quantitative nature of the data that are available for
drinking-water itself are limited. Little attention has been paid to
these higher molecular weight acids in either the toxicological or
epidemiological literature. However, it is important to recognize that
they make up the bulk of the total organic halogen that is present in
drinking-water.
Beyond the HAAs, the most studied group of halogenated acids are
the chloropropionic acids. As with DCA, the major impetus for these
studies has been potential therapeutic applications rather than
concerns over exposure to the compounds as contaminants of
drinking-water. Therefore, there are few data that have been developed
for hazard identification purposes, and even less information is
available on dose-response on effects other than those being explored
for therapeutic purposes.
By far the most data on chloropropionic acids exist for
2-chloropropionate (2-CP). This compound shares the hypoglycaemic
effects of DCA. It was first used as an experimental tool to segregate
direct effects of DCA from those of its metabolites. Oxalate and
glyoxylate are two metabolites of DCA that seem to be responsible for
its effects on gluconeogenesis from lactate (Crabb & Harris, 1979).
Since 2-CP is not metabolized to these compounds and failed to inhibit
gluconeogenesis, these data effectively argue that this effect of DCA
is largely attributable to these two metabolites. In contrast, 2-CP
still decreased blood glucose in rats when infused intravenously at a
rate of 300 mg/kg of body weight per hour. As with DCA, 2-CP increased
concentrations of circulating ketone bodies but significantly reduced
blood concentrations of lactate. As had been previously demonstrated
with DCA, concentrations of 1 and 5 mmol of 2-CP per litre
significantly enhanced the activity of pyruvate dehydrogenase.
Yount et al. (1982) compared the effects of DCA and 2-CP in mice
and rats. They extended these investigations into the area of
comparative toxicities of the two compounds as well. The acute oral
LD50 for 2-CP in fasted ICR mice was 15.4 ± 0.1 mmol/kg of body
weight, making it approximately twice as toxic as DCA on a molar
basis. However, neither compound is really very toxic acutely (2-CP =
1671 mg/kg of body weight; DCA = 4100 mg/kg of body weight).
Male Wistar rats were administered the sodium salts of 2-CP and
DCA at 0.04 mol/kg of feed for 12 weeks. These concentrations
correspond to about 4.3 and 5.1 g of 2-CP and DCA per kg of feed,
respectively. Actual doses to the animals are difficult to calculate
accurately because the body weights were not provided and because
significant effects of both compounds on body weight gain were noted.
If an average weight of 300 g is assumed for rats for the duration of
the experiment (i.e., study was started with weanling rats), the doses
can be estimated to be roughly 300 mg/kg of body weight per day for
DCA and 250 mg/kg of body weight per day for 2-CP.
Both DCA and 2-CP decreased the growth rate and food consumption
of treated rats and caused neurotoxic effects (e.g., hind limb
weakness). 2-CP treatment caused testicular abnormalities and
significantly lowered plasma triacylglycerol levels compared with
control or DCA-treated rats. In mature rats, total serum ketone bodies
were increased by DCA but not by 2-CP (Yount et al., 1982).
2-CP, as either the L- or D-isomer, is rapidly and extensively
metabolized in the liver cytosol by a mechanism that depletes GSH
(Wyatt et al., 1996). Significant depletion of non-protein sulfhydryl
content (primarily GSH) was observed with single acute oral doses of
62.5 mg/kg of body weight and above in male Alderley Park
Wistar-derived rats. This effect was observed to be maximal at 4 h and
returned to control values in approximately 48 h. A slower depletion
of non-protein sulfhydryl content was observed in the cerebellum and
forebrain with doses of 750 mg/kg of body weight. The depletion was
observed to be maximal about 24 h after administration in the
cerebellum and between 12 and 24 h in the forebrain. The depletion in
the cerebellum occurred with doses that also resulted in the induction
of granule cell necrosis in the cerebellum. Although depletion of
non-protein sulfhydryl groups tended to recover in the liver, the
effect in the cerebellum appeared to be partially cumulative with each
daily dose.
As with DCA, the largest capacity for 2-CP metabolism appears to
be in the cytosolic fraction of the hepatocyte, whereas metabolism in
microsomes is quite small. L-CP appears to be slightly more active in
depleting cytosolic fractions of GSH. In the process, the metabolite
2- S-glutathionylpropanic acid is formed. On the basis of the
stoichiometry between GSH depletion and formation of this product and
prior observations of Polhuijs et al. (1989, 1991) with
2-bromocarboxylic acids, it was concluded that 2-CP was a substrate
for a theta-class GST.
4.3 Haloaldehydes and haloketones
A diverse set of halogenated aldehydes and ketones are formed in
the disinfection of drinking-water. The most important in terms of
having been identified in drinking-water is trichloroacetaldehyde or
chloral hydrate, which is discussed separately. The remainder of the
group is discussed collectively. From the perspective of
drinking-water problems, the class has received very little attention,
but its members have been identified as key metabolites of chemicals
such as trichloroethylene, vinyl chloride and dibromochloropropane
(Omichinski et al., 1988; Spengler & Singer, 1988). The principal
evidence that they are formed comes from studies of chlorinated humic
and fulvic acids (Meier et al., 1983, 1985a) and of kraft pulp
chlorination (Kringstad et al., 1981). However, when specifically
looked for in drinking-water using techniques with adequate analytical
sensitivity, selected members of the group have been found in
concentrations that are somewhere between those of the HANs and HAAs
(Coleman et al., 1984).
These two classes of chemicals played an important role in the
initial studies of chemicals formed in the chlorination of
drinking-water. The interest in them was sparked by the fact that they
were among the first by-products to be identified that contributed to
the mutagenic activity produced in chlorinated water (Cheh et al.,
1980). Attention faded from this group when MX was identified, because
this compound contributed the major proportion of the mutagenic
activity produced by chlorine (Meier, 1988). Because these chemicals
represent an additional group of mutagenic chemicals, some of which
are capable of initiating skin tumours, it is important to acknowledge
their presence. However, because of the lack of data that can be used
to assess the degree of hazard, the present review will not be
comprehensive.
4.3.1 Chloral hydrate (trichloroacetaldehyde, chloral)
Trichloroacetaldehyde (chloral) is hydrated in water and in the
body to form the well-known sedative-hypnotic, chloral hydrate. Most
toxicological and metabolic studies have been conducted with chloral
hydrate rather than trying to deal with maintaining the aldehyde in
the dehydrated state.
It should be recognized that where there are significant amounts
of bromide in treated drinking-water, the use of oxidants will also
produce brominated analogues of chloral hydrate. This has been poorly
documented with the trihaloacetaldehydes, but the brominated analogues
have been observed in a system where chlorination reactions with
fulvic acid are conducted in the presence of high bromide
concentrations (Xie & Reckhow, 1993). The lack of data from actual
water supplies is in large part due to the lack of appropriate
analytical standards. In a broad sense, however, the ratios of chloral
hydrate, bromodichloroacetaldehyde, dibromochloroacetaldehyde and
tribromoacetaldehyde should more or less parallel those seen with the
analogous trihaloacetates. A second problem is that there is not a
significant body of toxicological literature available for these
analogues because they are not utilized in commerce. This represents a
significant data gap because bromine substitution can be anticipated
to result in greater metabolism of the trihaloacetaldehydes, as has
been demonstrated for the trihaloacetates (Xu et al., 1995). As a
consequence, an exaggeration of those effects of chloral hydrate that
are secondary to metabolism through reactive intermediates might be
expected (Hoz et al., 1991). Owing to the lack of data, however, it is
inappropriate to speculate much further. Therefore, the remainder of
this section will specifically address the data that are available for
chloral hydrate.
4.3.1.1 General toxicological properties and information on
dose-response in animals
Chloral hydrate is primarily known for its depressant effects on
the central nervous system (Gilman et al., 1991). The usual doses
required to produce central nervous system depression in humans range
from about 500 to 2000 mg in adults. These effects do not appear to
have been extensively studied in experimental animals.
Chloral hydrate was administered for 90 days in drinking-water to
male and female Sprague-Dawley rats at concentrations of 300, 600,
1200 or 2400 mg/litre. Hepatocellular necrosis was observed in 2 of
10 male rats treated with concentrations of either 1200 or 2400 mg of
chloral hydrate per litre (Daniel et al., 1992b). No liver damage was
seen in female rats. The necrosis observed at the highest dose was
indicated as being more severe than that observed at 1200 mg/litre,
providing some indication of a dose-response. It is of interest that
there was no sign of hepatomegaly produced by chloral hydrate at
either this dose or any lower dose. If it is assumed that rats drink
about 10% of their body weight per day, the level of 1200 mg/litre
corresponds to approximately 120 mg/kg of body weight per day, or
about 8 g for a 70-kg human, close to the estimated doses in the study
by van Heijst et al. (1977).
In contrast to findings in rats, male CD-1 mice displayed
hepatomegaly when doses of 144 mg of chloral hydrate per kg of body
weight per day were administered by gavage for a period of 14 days
(Sanders et al., 1982). No effect was observed at 14.4 mg/kg of body
weight per day. Other organs remained normal at gross necropsy, and
there were no signs of altered serum enzyme levels (e.g., LDH or serum
glutamate-pyruvate transaminase [SGPT]) or altered BUN. These data
suggest that chloral hydrate is not cytotoxic at these doses. This
short-term experiment was followed by a second experiment in which
mice (140 per sex per group) were administered 70 or 700 mg of chloral
hydrate per litre in drinking-water for up to 90 days. These levels
were estimated to yield the same doses as those used in the 14-day
range-finding study and averaged 18 or 173 mg/kg of body weight per
day for female mice and 16 or 160 mg/kg of body weight per day for
male mice. Male mice displayed hepatomegaly after 90 days of treatment
at both the low and high doses (Sanders et al., 1982). Small, but
statistically significant, increases in LDH and serum
glutamate-oxaloacetate transaminase (SGOT)were observed at the high
dose, but not at 70 mg/litre. The female mice did not demonstrate the
hepatomegaly observed in males, but they did show alterations in
hepatic microsomal parameters. These data are of potential
significance in considering the greater sensitivity of mice (albeit of
another strain) to the hepatocarcinogenic effects of chloral hydrate
as compared with rats.
Chloral hydrate has the potential of interacting with other drugs
by direct and indirect means. Most commonly cited is a potentiation
with alcohol that has been associated with the so-called "Mickey Finn"
(Gilman et al., 1991). This is attributed to interactions with common
steps in the metabolism of both chloral hydrate and alcohol (Gessner &
Cabana, 1970; Sellers et al., 1972). Combined exposure appears to
interfere with glucuronidation of the metabolite trichloroethanol and
with its conversion to TCA (Kaplan et al., 1967; Sellers et al.,
1972). Moreover, trichloroethanol is probably the metabolite of
chloral hydrate that is primarily responsible for its central nervous
system depressant effects (Gilman et al., 1991); thus, there is
undoubtedly somewhat of a pharmacological interaction as well.
Increased sensitivity to the hypnotic activity of chloral hydrate in
paraoxon-treated animals has been suggested to result from an
increased sensitivity of the brain to hypoxia (Koepke et al., 1974).
The other major interaction associated with chloral hydrate is the
ability of its metabolites, particularly TCA, to compete for binding
sites on plasma proteins. This type of interaction has been held
responsible for increased pharmacological and toxicological reactions
to warfarin (Sellers & Koch-Weser, 1970; Koch-Weser et al., 1971) and
bis-hydroxycoumarin (Cucinell et al., 1966).
4.3.1.2 Toxicity in humans
The primary effect seen with ingestion of chloral hydrate is
central nervous system depression, the basis of its use in
therapeutics. The usual dose recommended for sedation is 250 mg 3
times daily. The hypnotic dose is generally given as 500-1000 mg, but
2000 mg is required to be effective in many adults (Gilman et al.,
1991). Neonates are frequently treated with doses of chloral hydrate
in the range of 30-40 mg/kg of body weight (Lambert et al., 1990). In
recent years, chloral hydrate has become popular for sedating
subjects, particularly children, to aid in performing diagnostic
procedures such as CT scans, electroencephalograms (EEG) and
electrocardiograms (ECG), where relatively higher doses have been
utilized (32-80 mg/kg of body weight, Silver & Steir, 1971; 100 mg/kg
of body weight, Farber & Abramow, 1985).
The most important acute toxic effect is the production of
cardiac arrhythmias. Where doses in adults have been estimated, they
are considerably above those commonly used for therapeutic purposes,
most often in excess of 8 g. Most of the available studies involved
poisoning or overdose situations. However, one study closely examined
the induction of arrhythmias in paediatric cases undergoing EEGs
(Silver & Steir, 1971). The dose range was 32-80 mg/kg of body weight,
and in only 2 of 12 subjects was a sinus arrhythmia associated with
the administration of chloral hydrate. This suggests that doses at the
lower end of this range may approximate a threshold. Doses in the
range of 96 mg/kg of body weight and above have consistently been
shown to produce arrhythmias in children (Nordenberg et al., 1971;
Farber & Abramow, 1985; Hirsch & Zauder, 1986). In a 70-kg adult, 32
mg/kg of body weight would equal a dose of 2240 mg.
Lower doses of chloral hydrate have been associated with some
adverse side-effects. A study of newborns administered chloral hydrate
indicated a high incidence of direct hyperbilirubinaemia (Lambert et
al., 1990). This effect was associated more with continuous use than
with an acute dose, however. The dose rate to affected (40 mg/kg of
body weight) and unaffected (33 mg/kg of body weight) neonates was not
significantly different. The total dose in affected children was
1035 mg relative to 135 mg in the unaffected children. Thus, a more
protracted use of chloral hydrate in the affected group was apparently
responsible for the hyperbilirubinaemia observed. These data suggest
that there is little concern over this effect with single doses of
chloral hydrate. Because neonates are generally thought to be more
sensitive to hyperbilirubinaemia, this effect is probably of less
concern for adults.
There have been occasional reports of liver damage induced by
high doses of chloral hydrate by ingestion (van Heijst et al., 1977;
Gilman et al., 1991). The short-term data from mice, discussed
previously, support the conclusion that an effect on the liver is
unlikely to be observed in humans until very high doses are reached.
Longer-term exposures (e.g., months) lead to some enlargement of the
liver, if humans are as sensitive as mice in this regard, but the
doses remain considerably above the fraction of a µg/kg of body weight
that would be expected from most chlorinated drinking-waters. The
clinical literature suggests that humans are closer to rats in the
sensitivity of their livers to chloral hydrate.
A single modern case of fixed cutaneous eruptions was noted in
the literature (Miller et al., 1966). This was associated with a
therapeutic dose of chloral hydrate (500 mg) in a 57-year-old man.
These lesions are termed "fixed" because they tend to occur at the
same locations on the body with repeated exposures. An earlier case
was reported in 1878. This seems to be a rare side-effect of chloral
hydrate that is completely reversible. There have been other reported
skin reactions to chloral hydrate ingestion, but these appear to be
relatively rare as well (Almeyda & Levantine, 1972).
4.3.1.3 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of chloral and chloral
hydrate and concluded that there is inadequate evidence for their
carcinogenicity in humans, limited evidence for the carcinogenicity of
chloral hydrate in experimental animals and inadequate evidence for
the carcinogenicity of chloral in experimental animals. Both chloral
and chloral hydrate were assigned to Group 3: the compounds are not
classifiable as to their carcinogenicity to humans (IARC, 1995).
A more recent concern with chloral hydrate has been findings that
it has some genotoxic and carcinogenic effects in animals and in
vitro test systems (Table 18). Interpretation of some of these data
is difficult, as many investigators failed to document the purity of
the chemical tested. Nevertheless, chloral hydrate tends to be
positive in Salmonella typhimurium strain TA100, but not in TA98
(Waskell, 1978) or TA1535 (Bignami et al., 1980). The activity towards
TA100 was very weak in one assay (Waskell, 1978) and substantially
greater in another (Bignami et al., 1980). It was notable that Waskell
(1978) recrystallized chloral hydrate from alcohol 6 times before
subjecting it to test. Bignami et al. (1980) also found that chloral
hydrate was capable of inducing point mutations in other test systems.
A third group found chloral hydrate to be negative in TA98, TA100,
Table 18. Results of genotoxicity assays of chloral hydrate
Dose Test system Result Reference
[conc.]a
10 mg/plate Salmonella Chloral hydrate purified before use Waskell (1978)
TA100 Positive, S9 enhanced slightly
TA98 Negative
1-5 mg/plate Salmonella Negative Bignami et al.
TA1535 Positive; decreased with S9 (1980)
TA100
2-10 mg/plate Streptomyces Positive Bignami et al.
coelicolor (1980)
1-10 mg/plate Aspergillus Positive Bignami et al.
nidulans (1980)
82.7-413.5 Mouse Produces non-disjunction Russo et al.
mg/kg bw, i.p. spermatocytes Increases hyperhaploidy at all (1984)
doses tested
[5-20 Saccharomyces Increased mitotic gene conversion Bronzetti et al.
mmol/litre] cerevisiae at trp+ locus in presence of S9; (1984)
ilv+ revertants not affected
[5-10 Aspergillus Increased mitotic segregation at Crebelli et al.
mmol/litre] nidulans both doses, purity stated 99% (1985)
[1-25 Saccharomyces Inhibits sporulation and increased Sora & Carbone
mmol/litre] cerevisiae diploid and disomic clones (1987)
[25-250 DNA-protein Negative Keller & Heck
mmol/litre] cross-links in (1988)
isolated rat
liver nuclei
[0.001-0.003%] Chinese hamster Increased number of aneuploid cells Furnus et al.
embryonic at all doses; other chromosomal (1990)
diploid cells aberrations produced at two higher
concentrations
0.008-5 Salmonella All negative, purity of compound Leuschner & Leuschner
mg/plate TA98 specified (1991)
TA100
TA1535
TA1537
TA1538
500 mg/kg Mouse Negative Leuschner & Leuschner
bw, i.p. micronucleus (1991)
Table 18. (continued)
Dose Test system Result Reference
[conc.]a
100-1000 Chromosome, Negative at 6 and 24 h Leuschner & Leuschner
mg/kg bw, rat bone (1991)
p.o. marrow
250-750 Human peripheral Increased hyperdiploid nuclei, and Vagnarelli et al.
µg/ml blood percentage of aneuploid mitosis; (1990)
lymphocytes purity specified at 99%
300 µg/ml Ames Metabolism-dependent positive Giller et al.
fluctuation response in TA100; negative at (1995)
assay 100 µg/ml
200 µg/ml Newt larvae Micronuclei in peripheral blood Giller et al.
micronucleus erythrocytes in vivo; negative at (1995)
assay 100 µg/ml
a Abbreviations used: conc. = concentration; bw = body weight; i.p. = intraperitoneal;
p.o. = per os.
TA1535, TA1537 and TA1538 (Leuschner & Leuschner, 1991), and the
purity of the chloral hydrate was specified. Keller & Heck (1988)
could find no evidence of DNA-protein cross-links with chloral hydrate
treatment of isolated rat liver nuclei. A number of laboratories have
shown that chloral hydrate is capable of producing chromosomal
aberrations in vitro (Bronzetti et al., 1984; Crebelli et al., 1985;
Sora & Carbone, 1987; Furnus et al., 1990; Vagnarelli et al., 1990),
including aneuploid cells. Chromosomal effects appear to be more
consistently observed, and, thus, these results are more convincing.
Even in these cases, however, the purity of the compound tested was
generally not determined.
Chloral hydrate has been extensively studied as a potentially
genotoxic agent. It has been evaluated in the recommended screening
battery and several other assays, including genetic alterations in
rodent germ cells. Chloral hydrate is positive in bacterial mutation
tests, indicating that it is capable of inducing point mutations
(Waskell, 1978; Haworth et al., 1983; Giller et al., 1995). It is
positive in the mouse lymphoma assay for mutations at the Tk locus
(Harrington-Brock et al., 1998). Chloral hydrate is also positive in
several other in vitro assays for genetic damage. It induces
anueploidy in Chinese hamster embryonic fibroblasts (Natarajan, 1993),
Chinese hamster pulmonary lines LUC2 and Don.Wq.3H (Warr et al., 1993)
and human peripheral blood lymphocytes (Sbrana et al., 1993). Positive
micronuclei induction was observed in Chinese hamster cells (Lynch &
Parry, 1993) and human peripheral blood lymphocytes (Ferguson et al.,
1993), and chromosomal aberrations were found in Chinese hamster
embryonic diploid cells (Furnus et al., 1990). It is not clear whether
chloral hydrate is capable of inducing genetic damage in vivo. There
is a mixture of positive and negative in vivo data. Russo & Levis
(1992) found chloral hydrate to be capable of inducing aneuploidy in
mouse spermatocytes. Two different groups observed an increase in
micronuclei in mouse spermatids when treatment involved exposure of
spermatogonia stem cells (Allen et al., 1994; Nutley et al., 1996).
Russo et al. (1992) found chloral hydrate to induce micronuclei in
mouse bone marrow erythrocytes. Other laboratories have found chloral
hydrate to be negative in in vivo experiments (Xu & Adler, 1990;
Adler, 1993). So far, chloral hydrate has been found to give negative
test results in studies with mouse oocytes (Mailhes et al., 1993).
Although chloral hydrate can induce a variety of genetic events
(mutation, aneuploidy, structural chromosomal aberrations), it does so
with a very low potency.
Chloral hydrate has been reported to produce hepatic tumours in
male B6C3F1 mice in two studies. One study administered by gavage a
single dose of 5 or 10 mg of chloral hydrate per kg of body weight to
groups of 25 and 20 male mice at 15 days of age (Rijhsinghani et al.,
1986). The response to the 10 mg/kg of body weight dose led to
statistically elevated levels of tumours between 48 and 92 weeks, but
the results are based on the appearance of three adenomas and three
carcinomas among eight animals. Moreover, the historical control
incidence of hepatic tumours in the male of this hybrid is generally
about 25% and has been reported to be in excess of 40% in individual
studies. Thus, the small numbers of animals make it difficult to give
much credence to the results of this study. A second study (Daniel et
al., 1992a), however, showed that chloral hydrate administered in
drinking-water to a group of 40 male B6C3F1 mice for 104 weeks at
1 g/litre (166 mg/kg of body weight per day) resulted in a 71%
incidence of hepatic tumours (combined adenomas and carcinomas). The
fact that much higher doses were required to induce tumours in the
same hybrid mouse in this study raises further questions about the
Rijhsinghani et al. (1986) study; however, mice of this age are known
to be very sensitive to tumour initiators (Vesselinovich et al.,
1974). However, the Daniel et al. (1992a) study does clearly indicate
that chloral hydrate is capable of inducing tumours in B6C3F1 mice
when the mice are subjected to a lifetime exposure.a
a After the Task Group meeting, three new studies in B6C3F1 mice
on the carcinogenicity of chloral hydrate appeared (NTP, 2000a,b;
George et al., in press). In the first (NTP, 2000a), no carcinogenic
effect was observed following the administration of a single dose of
chloral hydrate (the dose was up to 5 times higher than that used in
the study of Rijhsinghai et al., 1986), while in the two other
studies, males had an increased incidence of hepatic tumours after
life-time exposure (NTP, 2000b; George et al., in press). In the NTP
life-time study (NTP, 2000b), a slightly elevated incidence of
pituitary adenomas, of borderline statistical significance, was
observed in female mice.
The question of whether chloral hydrate itself contributes to its
carcinogenic effects is critical because at least two of its
metabolites, TCA and DCA, are comparatively potent inducers of hepatic
tumours in B6C3F1 mice. This question can be resolved only by
demonstrating (i) that the clastogenic effects of chloral hydrate play
a role in the development of tumours or (ii) that TCA, DCA or a
combination of both chemicals are produced in sufficient quantities to
completely account for the induction of liver tumours without
significant contribution from earlier, more reactive metabolites. This
question is more critically assessed in the next section.
4.3.1.4 Comparative metabolism and pharmacokinetics
The metabolism of chloral hydrate has received considerable
attention over the years because of its extensive use as a
sedative-hypnotic (Marshall & Owens, 1954; Kaplan et al., 1967;
Gessner & Cabana, 1970; Cabana & Gessner, 1970; Sellers et al., 1972;
Garrett & Lambert, 1973; Mayers et al., 1991). A series of older
studies provide a still valid general picture of the conversion of
chloral hydrate to its two major metabolites, trichloroethanol and
TCA. More recent studies have focused more specifically on species
differences in this metabolism and have begun to focus on minor
metabolic pathways.
Figure 6 provides a simplified scheme of chloral hydrate
metabolism. The reader is referred to the appropriate sections of this
document to evaluate the further metabolism of the products TCA and
DCA.
The major fate of chloral hydrate is to undergo reduction to
trichloroethanol, with a smaller, but significant, fraction being
oxidized to TCA. Initially, formation of trichloroethanol is favoured
because the redox potential within cells in vivo favours reduction
(Kawamoto et al., 1987). This initial tendency is accentuated by a
rapid glucuronidation of the trichloroethanol that is formed (Marshall
& Owens, 1954). As is pointed out below, this may be a key feature in
the interspecies differences in chloral hydrate metabolism. With time,
(continued)
References:
NTP (2000a) Toxicology and carcinogenesis studies of chloral hydrate
in B6C3F1 mice (gavage studies). Research Triangle Park, North
Carolina, US department of Health and Human Services, National
Toxicology Program (NTP-TR-502).
NTP (2000b) Toxicology and carcinogenesis studies of chloral hydrate
(ad libitum and dietary controlled) in male B6C3F1 mice (gavage
study). Research Triangle Park, North Carolina, US department of
Health and Human Services, National Toxicology Program (NTP-TR-503).
George MH, Kilburn S, Moore T & DeAngelo AB (in press) The
carcinogenicity of chloral hydrate administered in the drinking water
to the male B6C3F1 mouse and F344/N rat. Toxicol Pathol.
1992) and tissues (Eyre et al., 1995), as glyoxylate is rapidly
transaminated and converted to glycine.
In mice, some of TCA's metabolism is independent of the formation
of DCA as an intermediate. There is evidence that a significant amount
of oxalate is formed independently of DCA formation and that some
direct decarboxylation of trihaloacetates also occurs. The evidence
for these pathways comes largely from studies of the metabolism of
bromodichloroacetate (BDCA) (Xu et al., 1995). The conversion of TCA
is, however, much slower than that of BDCA. The basis for the overall
scheme is discussed more fully in the following section on brominated
HAAs. The reader is also referred to Figure 5 and the accompanying
text for explanation of the further metabolism of DCA.
4.2.2.8 Mode of action
The tumorigenic effects of TCA in the liver of B6C3F1 mice
appear to be closely related to its ability to induce synthesis of
peroxisomes and associated proteins (DeAngelo et al., 1989; Bull et
al., 1990; Pereira, 1996). Along with a number of other peroxisome
proliferators, TCA was shown to be capable of activating the PPAR
in vitro at concentrations consistent with the levels that are
achieved in vivo (Issemann & Green, 1990). The cause-and-effect
relationships between the activation of this receptor and the
induction of cancer are yet to be established. Based upon marked
increases in the numbers of peroxisomes that are observed in rodent
species that are susceptible to this class of carcinogen and the lack
of such responses in other mammalian species, it has been argued that
humans are minimally sensitive to the tumorigenic effects of these
compounds (Lake, 1995). Peroxisome proliferators are also known to
variably affect reproduction and development. Considering the types of
interactions in which the PPAR is involved, these observations are not
too surprising. However, it is not clear whether a consistent pattern
of developmental anomalies can be associated with activation of this
receptor.
A mouse that was genetically engineered with a targeted
disruption PPAR-alpha gene failed to respond to the pleiotropic
effects of peroxisome proliferators (Lee et al., 1995).
Non-rodent species, including humans, express the PPARs, and
peroxisome proliferator responsive elements (PPREs) have been
identified in the promoter regions of the genes that are analogous to
those that are induced in rodents (Varanasi et al., 1996). This
suggests that the responsiveness to peroxisome proliferators is in
some way modified by other factors. However, the expression of PPARs
in humans is low.
It is important to recognize that other mechanisms may be
involved in TCA-induced effects. Clearly, the production of DCA as a
metabolite could be involved in inducing effects associated with this
metabolite. The very high rate of DCA metabolism relative to the rate
of TCA metabolism has made it difficult to detect significant amounts
of DCA in the blood (Merdink et al., 1998). There is little doubt that
some DCA is formed in all species. In addition to DCA, there are a
number of reactive and stable metabolites that could contribute to
toxicity. At this time, there is no evidence that the acid chloride
intermediate postulated between DCA and oxalate contributes anything
to the toxic effects of TCA. Chronic TCA administration results in the
deposition of lipofuscin, a sign of increased oxidative stress.
Moreover, Austin et al. (1995, 1996) obtained evidence of increased
lipid peroxidation and increased levels of 8-OH-dG in nuclear DNA of
the liver of mice treated with single doses of TCA. Therefore, the
accumulation of lipofuscin could be associated with mechanisms
unrelated to peroxisome proliferation. The most likely
radical-generating sources for lipid peroxidation and oxidative damage
to DNA would be through the formation of the radical species
(carbon-centred radicals and the peroxy radicals that would be derived
from the reaction of molecular oxygen with the carbon-centred
radicals). Again, there is no direct evidence to indicate that these
processes are important to any toxicological response associated with
TCA treatment or exposure.
It is beyond the scope of the present review to resolve risk
assessment issues associated with peroxisome proliferators. However,
several points should be made specific to TCA. First, peroxisome
proliferative responses are not genotoxic responses. Second, TCA is
one of the weakest activators of the PPAR known (Issemann & Green,
1990). Finally, TCA appears to be only marginally active as a
peroxisome proliferator, even in rats (DeAngelo et al., 1989).
Furthermore, treatment of rats with high levels of TCA in
drinking-water does not induce liver tumours (DeAngelo et al., 1997).
These data strongly suggest that TCA presents little carcinogenic
hazard to humans at the low concentrations found in drinking-water.
The key question is whether the carcinogenic and teratogenic
effects of TCA observed at very high doses have any relevance at the
exposures that are obtained even under extreme conditions in
drinking-water. However, the application of conventional uncertainty
factors would suggest that TCA in drinking-water represents little
hazard to humans at the concentrations normally encountered in
chlorinated drinking-water.
4.2.3 Brominated haloacetic acids
Brominated HAAs are formed in waters that contain bromide, which
strong oxidants like chlorine and ozone are capable of oxidizing to
hypobromous acid. There are very limited data available on the
toxicity of these chemicals. Therefore, they will be considered as a
group. The discussion below focuses on similarities and differences
between brominated and chlorinated HAAs.
4.2.3.1 General toxicological properties and information on
dose-response in animals
Linder et al. (1994a) found that the oral LD50 for MBA was
177 mg/kg of body weight in adult male Sprague-Dawley rats. DBA was
much less toxic, with an LD50 of 1737 mg/kg of body weight. The acute
toxicities of the bromochloroacetates have not been determined.
No studies have been published on the brominated haloacetates.
Some of the information is available in abstract form and will be
touched on briefly.
Bull and co-workers (Bull & DeAngelo, 1995; Stauber et al., 1998)
conducted a series of experiments with DBA, bromochloroacetate (BCA)
and BDCA administered to male B6C3F1 mice at concentrations ranging
from 0.2 to 3 g/litre in their drinking-water. The data obtained from
these studies suggest that toxicological effects are observed in
approximately the same concentration ranges as for DCA- and
TCA-induced toxic effects.
The principal target organ in mice was identified as the liver
for all three brominated HAAs, but the nature of the effects on the
liver appears to be somewhat different for each compound. All the
brominated HAAs produce hepatomegaly, but glycogen accumulation and
cytomegaly are more prominent with BCA and BDCA than with DBA.
Conversely, DBA was reported to produce increases in
cyanide-insensitive acyl CoA activity in the liver. BCA and BDCA
produced only small and inconsistent effects on this marker of
peroxisome proliferation. Thus, it appears that DBA shares TCA's
ability to induce peroxisome proliferation, whereas BCA and BDCA
appear to produce effects much more like those induced by DCA.
Consistent with this, the severity of the hepatomegaly was BCA > BDCA
> DBA.
Administration of single doses of BDCA, BCA and DBA by gavage in
water as the vehicle induced increases in thiobarbituric acid reactive
substances (TBARS) and increased the 8-OH-dG content of nuclear DNA in
the liver of male B6C3F1 mice at doses as low as 30 mg/kg of body
weight (Austin et al., 1996). These effects were significantly greater
than those observed with TCA and DCA and tended to increase as the
bromine substitution increased within each series. The order of
potency was DBA = BCA > BDCA > DCA > TCA. Increases in 8-OH-dG
content were more rapid and more sustained with the brominated HAAs.
This indicates that brominated HAAs do induce oxidative stress.
Dose-related increases in the 8-OH-dG content of nuclear DNA of
the liver were observed when DBA and BCA were administered in
drinking-water to male B6C3F1 mice for periods from 3 to 10 weeks at
concentrations of 0.5 g/litre and above (Parrish et al., 1996). The
effect of BDCA in drinking-water was not evaluated. It is noted that
these effects were observed in the same range in which liver tumours
are induced in mice (discussed further below).
4.2.3.2 Reproductive effects
MBA and DBA have been examined for spermatotoxic effects in rats.
MBA did not affect parameters related to male reproductive function
(Linder et al., 1994a) when administered to rats as a single dose of
100 mg/kg of body weight or at 25 mg/kg of body weight per day
administered repeatedly for 14 consecutive days. In contrast, DBA
produced degenerating, misshapen epididymal sperm and abnormal
retention of step 19 spermatids following single doses of DBA in the
range 1000-2000 mg/kg of body weight. Caput sperm counts were
significantly reduced on the second day, and substantial reductions of
cauda sperm counts were observed at 14 and 28 days after treatment.
Serum testosterone levels were significantly depressed on day 2 but
returned to control levels by day 14. Sperm displayed defects in
development of the shape of sperm heads. Progressive motility of sperm
was significantly reduced at 14 and 28 days after treatment.
A subsequent paper by the same group described a study in which
doses of 0, 10, 30, 90 or 270 mg of DBA per kg of body weight were
administered to rats for 14 consecutive days (Linder et al., 1994b).
Marked effects on epididymal sperm counts and sperm morphology effects
were observed at the highest dose. Approximately 5% of the caput sperm
were fused. In contrast to the single-dose studies, serum testosterone
levels appeared to be unaffected. Spermiation also appeared to be
mildly affected, with step 19 spermatids being retained beyond stage
VIII in animals dosed with as little as 10 mg/kg of body weight per
day.
DBA was administered to rats for up to 79 days at 0, 2, 10, 50 or
250 mg/kg of body weight per day by gavage in water. Male fertility
was compromised during the second week of treatment at the high dose.
This effect appeared to result from behavioural changes, because
artificial insemination with sperm collected on day 9 of treatment
produced offspring. By day 15, however, no offspring were produced
with either biological or artificial insemination, indicating that
significant qualitative alterations had occurred in the sperm. Indeed,
the 50 mg/kg of body weight per day dose produced abnormal morphology,
decreased motility and decreases in epididymal sperm counts. However,
rats treated at this dose remained fertile. While no effects on sperm
quality were observed at lower doses, reproductive performance
appeared depressed at doses as low as 10 mg/kg of body weight per day,
suggesting that these lower doses modified behaviour (Linder et al.,
1995).
Histopathological changes were observed in the testis and
epididymis of rats gavaged daily for 2-79 days with DBA. On treatment
day 2, abnormal retention of step 19 spermatids was observed in
animals given the highest dose of 250 mg/kg of body weight per day.
Additional changes on day 5 included the fusion of mature spermatids
and the presence of atypical residual bodies (ARB) in the epithelium
and lumen of stage X-XII seminiferous tubules. By day 9, ARB were seen
in most stages of the seminiferous epithelial cycle and in the caput
epididymis. On day 16, distorted sperm heads were recognized in step
12 and older spermatids, and luminal cytoplasmic debris was found
throughout the epididymis. On day 31, there was vacuolation of the
Sertoli cell cytoplasm, extensive retention of step 19 spermatids near
the lumen of stage IX and X tubules, and vesiculation of the acrosomes
of late spermatids. Marked atrophy of the seminiferous tubules was
present 6 months after 42 doses of 250 mg/kg of body weight per day.
ARB and retention of step 19 spermatids were observed after 31 and 79
doses of 50 mg/kg of body weight per day, and increased retention of
step 19 spermatids was seen in several rats dosed with 10 mg/kg of
body weight per day. No abnormalities were detected at 2 mg/kg of body
weight per day. The changes suggest that the testicular effects of DBA
are sequelae to structural or functional changes in the Sertoli cell
(Linder et al., 1997b).
4.2.3.3 Neurotoxicity
Reference was made to neurotoxic effects in male Sprague-Dawley
rats treated with DBA in reproductive studies (Linder et al., 1994a,b,
1995). However, no specific investigations of the neurotoxic effects
of brominated HAAs appear to be available.
4.2.3.4 Toxicity in humans
There are no studies on the health effects of brominated HAAs in
humans.
4.2.3.5 Carcinogenicity and mutagenicity
No formal reports have been made of carcinogenic or mutagenic
effects of brominated HAAs. Indications that DBA, BCA and BDCA share
the hepatocarcinogenic effects of TCA and DCA in B6C3F1 mice have
been referred to in an abstract (Bull & DeAngelo, 1995).
Giller et al. (1997) examined the genotoxicity of MBA, DBA, and
tribromoacetic acid (TBA) in the SOS chromotest in Escherichia coli
PQ37, the Ames fluctuation assay utilizing Salmonella typhimurium
strain TA100 and the newt micronucleus assay in larvae at stage 53 of
the developmental table. MBA was negative in the SOS chromotest at
levels as high as 1000 µg/ml and in the newt micronucleus assay.
However, it was active at concentrations as low as 20 µg/ml in the
Ames fluctuation assay with S9 fraction added to the incubation
medium. DBA was positive with and without S9 fraction in the SOS
chromotest, requiring 100 µg/ml in the former case and 200 µg/ml in
the latter. Thus, it is about 5 times as potent as DCA in this test.
In the Ames fluctuation assay, DBA was active at 10 µg/ml without S9
and at 30 µg/ml with S9. TBA was active in the SOS chromotest at
100 µg/ml with S9 but required 2000 µg/ml for activity in the Ames
fluctuation assay without S9.
4.2.3.6 Comparative pharmacokinetics and metabolism
Metabolism of brominated HAAs has received little attention. Xu
et al. (1995) studied the metabolism of BDCA in male B6C3F1 mice. As
predicted, substitution of a bromine for a chlorine in TCA resulted in
a substantially greater extent of trihaloacetate metabolism. Whereas
45% of a 100 mg/kg of body weight dose of TCA was eliminated unchanged
in the urine of mice within 24 h, less than 4% of the same dose of
BDCA was found in the urine. At lower doses, only a fraction of a
percent of the BDCA was eliminated unchanged.
There is evidence for substantial conversion of BDCA to DCA in
both rats and mice (Xu et al., 1995; Schultz et al., 1999). Because of
the established ability of DCA to induce hepatic tumours in both mice
and rats (discussed above), this may have implications for assessing
the risk associated with this compound (see Figures 3 and 5 for a
general description of dihaloacetate and trihaloacetate metabolism,
respectively).
The metabolism of BDCA is differentially modified in mice and
rats as doses are increased. Xu et al. (1995) found that the kinetics
of carbon dioxide production from 1-14C-BDCA suggested an efficient
conversion of BDCA to carbon dioxide through DCA at low doses, but a
direct decarboxylation reaction became important as doses approached
100 mg/kg of body weight (Xu et al., 1995; Austin & Bull, 1997). This
complex activity was not observed in rats (Schultz et al., 1999), in
that a progressively smaller fraction of the dose is converted to
carbon dioxide as dose is increased. This suggests that direct
decarboxylation plays a less important role in the metabolism of BDCA
in rats than in mice.
The ratios of urinary metabolites produced by mice and rats
suggest that there are some substantive differences in the metabolism
of BDCA in the two species. Mice (Xu et al., 1995) produce much higher
amounts of oxalate (about 30% of the orally administered dose) than do
rats (about 20%) (Schultz et al., 1999). The much greater conversion
of BDCA to oxalate than for equivalent doses of DCA suggests that much
of the extra oxalate seen in mouse urine arises from reductive
dehalogenation of BDCA, followed by peroxy radical formation and
decomposition to oxalate (Xu et al., 1995).
As the BDCA dose is increased from 20 to 100 mg/kg of body weight
in the rat, the fraction of the dose that is eliminated in the urine
as DCA increases from about 2% to 13% (Schultz et al., 1999), whereas
in mice the increase is from 0.2% to approximately 3%. In the rat,
increasing the dose from 5 mg/kg of body weight to 20 or 100 mg/kg of
body weight was associated with a significantly extended half-life of
both BDCA and DCA (from 0.9 to 3.7 h). The increased half-life appears
to be attributed to saturation of the conversion of BDCA to DCA.
Despite this inhibition of DCA formation, the blood levels of DCA
observed are actually higher than those observed when equivalent doses
of DCA itself are administered. Thus, high doses of BDCA would appear
to inhibit the metabolic clearance of DCA as well. Consequently, the
toxicology of BDCA should share many of the attributes of DCA
toxicology in the rat. Comparable data do not exist on the blood
levels of DCA achieved by BDCA administration in mice, but the urinary
levels of DCA metabolites appear to be largely offset by the
conversion of BDCA to oxalate, suggesting that the blood levels of DCA
would be significantly lower in mice than in rats.
In animals previously treated with 1 g of DCA per litre in
drinking-water, metabolism of BDCA to carbon dioxide is significantly
increased in mice (Austin & Bull, 1997). This appears to be associated
with an increase in the capacity for the direct decarboxylation of
BDCA to form BDCM and carbon dioxide. Therefore, some effects of BDCA
in mice may be attributed to the formation of BDCM.
These data and data indicating that pretreatment with DCA also
affects its own metabolism suggest that there may be significant
interactions in the toxicity of these chemicals at high doses. It
remains to be seen if such interactions occur at treatment
concentrations that more closely approximate those observed in
drinking-water.
4.2.3.7 Mode of action
It is premature to attempt a definitive discussion of the
mechanisms by which brominated HAAs induce tumours. There are
suggestions of mechanisms that need to be pursued with this class, but
there are really no data that demonstrate how they contribute to the
induction of cancer, nor is there a firm basis for considering whether
these mechanisms are relevant at the low concentrations of these
chemicals found in drinking-water.
The mechanisms associated with the carcinogenic effects of HAAs
include those identified for DCA and TCA. It is apparent that more
than one mechanism is responsible for the effects of this class and
that the importance of these mechanisms to the activity of individual
members of the class varies. In part, these differences in mechanism
can be related to the differences in tumour phenotypes that are
induced. One phenotype seems to be associated with prior
characterizations of tumours induced by peroxisome proliferators and
is induced by TCA (DeAngelo et al., 1989; Stauber et al., 1998). The
second phenotype involves glycogen-poor tumours that stain heavily
with antibodies to c-Jun and c-Fos. This phenotype is produced by DCA.
These effects are probably produced by selection of lesions with
differing defects in cell signalling pathways that control the
processes of cell division and cell death.
Based upon the data of Giller at al. (1997), the brominated HAAs
are about 10-fold more potent than their chlorinated analogues in
their ability to induce point mutations. This does not establish that
they are inducing cancer by mutagenic mechanisms in vivo, but this
activity will have to be taken into account as data on their
carcinogenic activity become more complete.
The HAAs vary widely in their ability to induce oxidative stress
and to elevate the 8-OH-dG content of nuclear DNA of the liver. This
property becomes increasingly apparent with the brominated compounds
(Austin et al., 1996; Parrish et al., 1996). It is notable that the
brominated analogues are not more potent inducers of hepatic tumours
than the corresponding chlorinated HAAs. Therefore, it is doubtful
that this mechanism is the most important determinant of this effect.
No specific mechanisms have been associated with the effects of
DBA on male reproduction or as a developmental toxin. However, it
would be surprising if the effects on cell signalling systems that
appear to be involved in the carcinogenic responses do not also
contribute to these effects.
4.2.4 Higher molecular weight halogenated acids
Studies of by-product formation from humic and fulvic acids in
the 1970s and early 1980s demonstrated that there is a complex array
of halogenated carboxylic acids in addition to the HAAs (Christman et
al., 1983). Some of these acids have been identified in drinking-water
as well, but, as discussed elsewhere in this document, both the scope
and quantitative nature of the data that are available for
drinking-water itself are limited. Little attention has been paid to
these higher molecular weight acids in either the toxicological or
epidemiological literature. However, it is important to recognize that
they make up the bulk of the total organic halogen that is present in
drinking-water.
Beyond the HAAs, the most studied group of halogenated acids are
the chloropropionic acids. As with DCA, the major impetus for these
studies has been potential therapeutic applications rather than
concerns over exposure to the compounds as contaminants of
drinking-water. Therefore, there are few data that have been developed
for hazard identification purposes, and even less information is
available on dose-response on effects other than those being explored
for therapeutic purposes.
By far the most data on chloropropionic acids exist for
2-chloropropionate (2-CP). This compound shares the hypoglycaemic
effects of DCA. It was first used as an experimental tool to segregate
direct effects of DCA from those of its metabolites. Oxalate and
glyoxylate are two metabolites of DCA that seem to be responsible for
its effects on gluconeogenesis from lactate (Crabb & Harris, 1979).
Since 2-CP is not metabolized to these compounds and failed to inhibit
gluconeogenesis, these data effectively argue that this effect of DCA
is largely attributable to these two metabolites. In contrast, 2-CP
still decreased blood glucose in rats when infused intravenously at a
rate of 300 mg/kg of body weight per hour. As with DCA, 2-CP increased
concentrations of circulating ketone bodies but significantly reduced
blood concentrations of lactate. As had been previously demonstrated
with DCA, concentrations of 1 and 5 mmol of 2-CP per litre
significantly enhanced the activity of pyruvate dehydrogenase.
Yount et al. (1982) compared the effects of DCA and 2-CP in mice
and rats. They extended these investigations into the area of
comparative toxicities of the two compounds as well. The acute oral
LD50 for 2-CP in fasted ICR mice was 15.4 ± 0.1 mmol/kg of body
weight, making it approximately twice as toxic as DCA on a molar
basis. However, neither compound is really very toxic acutely (2-CP =
1671 mg/kg of body weight; DCA = 4100 mg/kg of body weight).
Male Wistar rats were administered the sodium salts of 2-CP and
DCA at 0.04 mol/kg of feed for 12 weeks. These concentrations
correspond to about 4.3 and 5.1 g of 2-CP and DCA per kg of feed,
respectively. Actual doses to the animals are difficult to calculate
accurately because the body weights were not provided and because
significant effects of both compounds on body weight gain were noted.
If an average weight of 300 g is assumed for rats for the duration of
the experiment (i.e., study was started with weanling rats), the doses
can be estimated to be roughly 300 mg/kg of body weight per day for
DCA and 250 mg/kg of body weight per day for 2-CP.
Both DCA and 2-CP decreased the growth rate and food consumption
of treated rats and caused neurotoxic effects (e.g., hind limb
weakness). 2-CP treatment caused testicular abnormalities and
significantly lowered plasma triacylglycerol levels compared with
control or DCA-treated rats. In mature rats, total serum ketone bodies
were increased by DCA but not by 2-CP (Yount et al., 1982).
2-CP, as either the L- or D-isomer, is rapidly and extensively
metabolized in the liver cytosol by a mechanism that depletes GSH
(Wyatt et al., 1996). Significant depletion of non-protein sulfhydryl
content (primarily GSH) was observed with single acute oral doses of
62.5 mg/kg of body weight and above in male Alderley Park
Wistar-derived rats. This effect was observed to be maximal at 4 h and
returned to control values in approximately 48 h. A slower depletion
of non-protein sulfhydryl content was observed in the cerebellum and
forebrain with doses of 750 mg/kg of body weight. The depletion was
observed to be maximal about 24 h after administration in the
cerebellum and between 12 and 24 h in the forebrain. The depletion in
the cerebellum occurred with doses that also resulted in the induction
of granule cell necrosis in the cerebellum. Although depletion of
non-protein sulfhydryl groups tended to recover in the liver, the
effect in the cerebellum appeared to be partially cumulative with each
daily dose.
As with DCA, the largest capacity for 2-CP metabolism appears to
be in the cytosolic fraction of the hepatocyte, whereas metabolism in
microsomes is quite small. L-CP appears to be slightly more active in
depleting cytosolic fractions of GSH. In the process, the metabolite
2- S-glutathionylpropanic acid is formed. On the basis of the
stoichiometry between GSH depletion and formation of this product and
prior observations of Polhuijs et al. (1989, 1991) with
2-bromocarboxylic acids, it was concluded that 2-CP was a substrate
for a theta-class GST.
4.3 Haloaldehydes and haloketones
A diverse set of halogenated aldehydes and ketones are formed in
the disinfection of drinking-water. The most important in terms of
having been identified in drinking-water is trichloroacetaldehyde or
chloral hydrate, which is discussed separately. The remainder of the
group is discussed collectively. From the perspective of
drinking-water problems, the class has received very little attention,
but its members have been identified as key metabolites of chemicals
such as trichloroethylene, vinyl chloride and dibromochloropropane
(Omichinski et al., 1988; Spengler & Singer, 1988). The principal
evidence that they are formed comes from studies of chlorinated humic
and fulvic acids (Meier et al., 1983, 1985a) and of kraft pulp
chlorination (Kringstad et al., 1981). However, when specifically
looked for in drinking-water using techniques with adequate analytical
sensitivity, selected members of the group have been found in
concentrations that are somewhere between those of the HANs and HAAs
(Coleman et al., 1984).
These two classes of chemicals played an important role in the
initial studies of chemicals formed in the chlorination of
drinking-water. The interest in them was sparked by the fact that they
were among the first by-products to be identified that contributed to
the mutagenic activity produced in chlorinated water (Cheh et al.,
1980). Attention faded from this group when MX was identified, because
this compound contributed the major proportion of the mutagenic
activity produced by chlorine (Meier, 1988). Because these chemicals
represent an additional group of mutagenic chemicals, some of which
are capable of initiating skin tumours, it is important to acknowledge
their presence. However, because of the lack of data that can be used
to assess the degree of hazard, the present review will not be
comprehensive.
4.3.1 Chloral hydrate (trichloroacetaldehyde, chloral)
Trichloroacetaldehyde (chloral) is hydrated in water and in the
body to form the well-known sedative-hypnotic, chloral hydrate. Most
toxicological and metabolic studies have been conducted with chloral
hydrate rather than trying to deal with maintaining the aldehyde in
the dehydrated state.
It should be recognized that where there are significant amounts
of bromide in treated drinking-water, the use of oxidants will also
produce brominated analogues of chloral hydrate. This has been poorly
documented with the trihaloacetaldehydes, but the brominated analogues
have been observed in a system where chlorination reactions with
fulvic acid are conducted in the presence of high bromide
concentrations (Xie & Reckhow, 1993). The lack of data from actual
water supplies is in large part due to the lack of appropriate
analytical standards. In a broad sense, however, the ratios of chloral
hydrate, bromodichloroacetaldehyde, dibromochloroacetaldehyde and
tribromoacetaldehyde should more or less parallel those seen with the
analogous trihaloacetates. A second problem is that there is not a
significant body of toxicological literature available for these
analogues because they are not utilized in commerce. This represents a
significant data gap because bromine substitution can be anticipated
to result in greater metabolism of the trihaloacetaldehydes, as has
been demonstrated for the trihaloacetates (Xu et al., 1995). As a
consequence, an exaggeration of those effects of chloral hydrate that
are secondary to metabolism through reactive intermediates might be
expected (Hoz et al., 1991). Owing to the lack of data, however, it is
inappropriate to speculate much further. Therefore, the remainder of
this section will specifically address the data that are available for
chloral hydrate.
4.3.1.1 General toxicological properties and information on
dose-response in animals
Chloral hydrate is primarily known for its depressant effects on
the central nervous system (Gilman et al., 1991). The usual doses
required to produce central nervous system depression in humans range
from about 500 to 2000 mg in adults. These effects do not appear to
have been extensively studied in experimental animals.
Chloral hydrate was administered for 90 days in drinking-water to
male and female Sprague-Dawley rats at concentrations of 300, 600,
1200 or 2400 mg/litre. Hepatocellular necrosis was observed in 2 of
10 male rats treated with concentrations of either 1200 or 2400 mg of
chloral hydrate per litre (Daniel et al., 1992b). No liver damage was
seen in female rats. The necrosis observed at the highest dose was
indicated as being more severe than that observed at 1200 mg/litre,
providing some indication of a dose-response. It is of interest that
there was no sign of hepatomegaly produced by chloral hydrate at
either this dose or any lower dose. If it is assumed that rats drink
about 10% of their body weight per day, the level of 1200 mg/litre
corresponds to approximately 120 mg/kg of body weight per day, or
about 8 g for a 70-kg human, close to the estimated doses in the study
by van Heijst et al. (1977).
In contrast to findings in rats, male CD-1 mice displayed
hepatomegaly when doses of 144 mg of chloral hydrate per kg of body
weight per day were administered by gavage for a period of 14 days
(Sanders et al., 1982). No effect was observed at 14.4 mg/kg of body
weight per day. Other organs remained normal at gross necropsy, and
there were no signs of altered serum enzyme levels (e.g., LDH or serum
glutamate-pyruvate transaminase [SGPT]) or altered BUN. These data
suggest that chloral hydrate is not cytotoxic at these doses. This
short-term experiment was followed by a second experiment in which
mice (140 per sex per group) were administered 70 or 700 mg of chloral
hydrate per litre in drinking-water for up to 90 days. These levels
were estimated to yield the same doses as those used in the 14-day
range-finding study and averaged 18 or 173 mg/kg of body weight per
day for female mice and 16 or 160 mg/kg of body weight per day for
male mice. Male mice displayed hepatomegaly after 90 days of treatment
at both the low and high doses (Sanders et al., 1982). Small, but
statistically significant, increases in LDH and serum
glutamate-oxaloacetate transaminase (SGOT)were observed at the high
dose, but not at 70 mg/litre. The female mice did not demonstrate the
hepatomegaly observed in males, but they did show alterations in
hepatic microsomal parameters. These data are of potential
significance in considering the greater sensitivity of mice (albeit of
another strain) to the hepatocarcinogenic effects of chloral hydrate
as compared with rats.
Chloral hydrate has the potential of interacting with other drugs
by direct and indirect means. Most commonly cited is a potentiation
with alcohol that has been associated with the so-called "Mickey Finn"
(Gilman et al., 1991). This is attributed to interactions with common
steps in the metabolism of both chloral hydrate and alcohol (Gessner &
Cabana, 1970; Sellers et al., 1972). Combined exposure appears to
interfere with glucuronidation of the metabolite trichloroethanol and
with its conversion to TCA (Kaplan et al., 1967; Sellers et al.,
1972). Moreover, trichloroethanol is probably the metabolite of
chloral hydrate that is primarily responsible for its central nervous
system depressant effects (Gilman et al., 1991); thus, there is
undoubtedly somewhat of a pharmacological interaction as well.
Increased sensitivity to the hypnotic activity of chloral hydrate in
paraoxon-treated animals has been suggested to result from an
increased sensitivity of the brain to hypoxia (Koepke et al., 1974).
The other major interaction associated with chloral hydrate is the
ability of its metabolites, particularly TCA, to compete for binding
sites on plasma proteins. This type of interaction has been held
responsible for increased pharmacological and toxicological reactions
to warfarin (Sellers & Koch-Weser, 1970; Koch-Weser et al., 1971) and
bis-hydroxycoumarin (Cucinell et al., 1966).
4.3.1.2 Toxicity in humans
The primary effect seen with ingestion of chloral hydrate is
central nervous system depression, the basis of its use in
therapeutics. The usual dose recommended for sedation is 250 mg 3
times daily. The hypnotic dose is generally given as 500-1000 mg, but
2000 mg is required to be effective in many adults (Gilman et al.,
1991). Neonates are frequently treated with doses of chloral hydrate
in the range of 30-40 mg/kg of body weight (Lambert et al., 1990). In
recent years, chloral hydrate has become popular for sedating
subjects, particularly children, to aid in performing diagnostic
procedures such as CT scans, electroencephalograms (EEG) and
electrocardiograms (ECG), where relatively higher doses have been
utilized (32-80 mg/kg of body weight, Silver & Steir, 1971; 100 mg/kg
of body weight, Farber & Abramow, 1985).
The most important acute toxic effect is the production of
cardiac arrhythmias. Where doses in adults have been estimated, they
are considerably above those commonly used for therapeutic purposes,
most often in excess of 8 g. Most of the available studies involved
poisoning or overdose situations. However, one study closely examined
the induction of arrhythmias in paediatric cases undergoing EEGs
(Silver & Steir, 1971). The dose range was 32-80 mg/kg of body weight,
and in only 2 of 12 subjects was a sinus arrhythmia associated with
the administration of chloral hydrate. This suggests that doses at the
lower end of this range may approximate a threshold. Doses in the
range of 96 mg/kg of body weight and above have consistently been
shown to produce arrhythmias in children (Nordenberg et al., 1971;
Farber & Abramow, 1985; Hirsch & Zauder, 1986). In a 70-kg adult, 32
mg/kg of body weight would equal a dose of 2240 mg.
Lower doses of chloral hydrate have been associated with some
adverse side-effects. A study of newborns administered chloral hydrate
indicated a high incidence of direct hyperbilirubinaemia (Lambert et
al., 1990). This effect was associated more with continuous use than
with an acute dose, however. The dose rate to affected (40 mg/kg of
body weight) and unaffected (33 mg/kg of body weight) neonates was not
significantly different. The total dose in affected children was
1035 mg relative to 135 mg in the unaffected children. Thus, a more
protracted use of chloral hydrate in the affected group was apparently
responsible for the hyperbilirubinaemia observed. These data suggest
that there is little concern over this effect with single doses of
chloral hydrate. Because neonates are generally thought to be more
sensitive to hyperbilirubinaemia, this effect is probably of less
concern for adults.
There have been occasional reports of liver damage induced by
high doses of chloral hydrate by ingestion (van Heijst et al., 1977;
Gilman et al., 1991). The short-term data from mice, discussed
previously, support the conclusion that an effect on the liver is
unlikely to be observed in humans until very high doses are reached.
Longer-term exposures (e.g., months) lead to some enlargement of the
liver, if humans are as sensitive as mice in this regard, but the
doses remain considerably above the fraction of a µg/kg of body weight
that would be expected from most chlorinated drinking-waters. The
clinical literature suggests that humans are closer to rats in the
sensitivity of their livers to chloral hydrate.
A single modern case of fixed cutaneous eruptions was noted in
the literature (Miller et al., 1966). This was associated with a
therapeutic dose of chloral hydrate (500 mg) in a 57-year-old man.
These lesions are termed "fixed" because they tend to occur at the
same locations on the body with repeated exposures. An earlier case
was reported in 1878. This seems to be a rare side-effect of chloral
hydrate that is completely reversible. There have been other reported
skin reactions to chloral hydrate ingestion, but these appear to be
relatively rare as well (Almeyda & Levantine, 1972).
4.3.1.3 Carcinogenicity and mutagenicity
IARC has evaluated the carcinogenicity of chloral and chloral
hydrate and concluded that there is inadequate evidence for their
carcinogenicity in humans, limited evidence for the carcinogenicity of
chloral hydrate in experimental animals and inadequate evidence for
the carcinogenicity of chloral in experimental animals. Both chloral
and chloral hydrate were assigned to Group 3: the compounds are not
classifiable as to their carcinogenicity to humans (IARC, 1995).
A more recent concern with chloral hydrate has been findings that
it has some genotoxic and carcinogenic effects in animals and in
vitro test systems (Table 18). Interpretation of some of these data
is difficult, as many investigators failed to document the purity of
the chemical tested. Nevertheless, chloral hydrate tends to be
positive in Salmonella typhimurium strain TA100, but not in TA98
(Waskell, 1978) or TA1535 (Bignami et al., 1980). The activity towards
TA100 was very weak in one assay (Waskell, 1978) and substantially
greater in another (Bignami et al., 1980). It was notable that Waskell
(1978) recrystallized chloral hydrate from alcohol 6 times before
subjecting it to test. Bignami et al. (1980) also found that chloral
hydrate was capable of inducing point mutations in other test systems.
A third group found chloral hydrate to be negative in TA98, TA100,
Table 18. Results of genotoxicity assays of chloral hydrate
Dose Test system Result Reference
[conc.]a
10 mg/plate Salmonella Chloral hydrate purified before use Waskell (1978)
TA100 Positive, S9 enhanced slightly
TA98 Negative
1-5 mg/plate Salmonella Negative Bignami et al.
TA1535 Positive; decreased with S9 (1980)
TA100
2-10 mg/plate Streptomyces Positive Bignami et al.
coelicolor (1980)
1-10 mg/plate Aspergillus Positive Bignami et al.
nidulans (1980)
82.7-413.5 Mouse Produces non-disjunction Russo et al.
mg/kg bw, i.p. spermatocytes Increases hyperhaploidy at all (1984)
doses tested
[5-20 Saccharomyces Increased mitotic gene conversion Bronzetti et al.
mmol/litre] cerevisiae at trp+ locus in presence of S9; (1984)
ilv+ revertants not affected
[5-10 Aspergillus Increased mitotic segregation at Crebelli et al.
mmol/litre] nidulans both doses, purity stated 99% (1985)
[1-25 Saccharomyces Inhibits sporulation and increased Sora & Carbone
mmol/litre] cerevisiae diploid and disomic clones (1987)
[25-250 DNA-protein Negative Keller & Heck
mmol/litre] cross-links in (1988)
isolated rat
liver nuclei
[0.001-0.003%] Chinese hamster Increased number of aneuploid cells Furnus et al.
embryonic at all doses; other chromosomal (1990)
diploid cells aberrations produced at two higher
concentrations
0.008-5 Salmonella All negative, purity of compound Leuschner & Leuschner
mg/plate TA98 specified (1991)
TA100
TA1535
TA1537
TA1538
500 mg/kg Mouse Negative Leuschner & Leuschner
bw, i.p. micronucleus (1991)
Table 18. (continued)
Dose Test system Result Reference
[conc.]a
100-1000 Chromosome, Negative at 6 and 24 h Leuschner & Leuschner
mg/kg bw, rat bone (1991)
p.o. marrow
250-750 Human peripheral Increased hyperdiploid nuclei, and Vagnarelli et al.
µg/ml blood percentage of aneuploid mitosis; (1990)
lymphocytes purity specified at 99%
300 µg/ml Ames Metabolism-dependent positive Giller et al.
fluctuation response in TA100; negative at (1995)
assay 100 µg/ml
200 µg/ml Newt larvae Micronuclei in peripheral blood Giller et al.
micronucleus erythrocytes in vivo; negative at (1995)
assay 100 µg/ml
a Abbreviations used: conc. = concentration; bw = body weight; i.p. = intraperitoneal;
p.o. = per os.
TA1535, TA1537 and TA1538 (Leuschner & Leuschner, 1991), and the
purity of the chloral hydrate was specified. Keller & Heck (1988)
could find no evidence of DNA-protein cross-links with chloral hydrate
treatment of isolated rat liver nuclei. A number of laboratories have
shown that chloral hydrate is capable of producing chromosomal
aberrations in vitro (Bronzetti et al., 1984; Crebelli et al., 1985;
Sora & Carbone, 1987; Furnus et al., 1990; Vagnarelli et al., 1990),
including aneuploid cells. Chromosomal effects appear to be more
consistently observed, and, thus, these results are more convincing.
Even in these cases, however, the purity of the compound tested was
generally not determined.
Chloral hydrate has been extensively studied as a potentially
genotoxic agent. It has been evaluated in the recommended screening
battery and several other assays, including genetic alterations in
rodent germ cells. Chloral hydrate is positive in bacterial mutation
tests, indicating that it is capable of inducing point mutations
(Waskell, 1978; Haworth et al., 1983; Giller et al., 1995). It is
positive in the mouse lymphoma assay for mutations at the Tk locus
(Harrington-Brock et al., 1998). Chloral hydrate is also positive in
several other in vitro assays for genetic damage. It induces
anueploidy in Chinese hamster embryonic fibroblasts (Natarajan, 1993),
Chinese hamster pulmonary lines LUC2 and Don.Wq.3H (Warr et al., 1993)
and human peripheral blood lymphocytes (Sbrana et al., 1993). Positive
micronuclei induction was observed in Chinese hamster cells (Lynch &
Parry, 1993) and human peripheral blood lymphocytes (Ferguson et al.,
1993), and chromosomal aberrations were found in Chinese hamster
embryonic diploid cells (Furnus et al., 1990). It is not clear whether
chloral hydrate is capable of inducing genetic damage in vivo. There
is a mixture of positive and negative in vivo data. Russo & Levis
(1992) found chloral hydrate to be capable of inducing aneuploidy in
mouse spermatocytes. Two different groups observed an increase in
micronuclei in mouse spermatids when treatment involved exposure of
spermatogonia stem cells (Allen et al., 1994; Nutley et al., 1996).
Russo et al. (1992) found chloral hydrate to induce micronuclei in
mouse bone marrow erythrocytes. Other laboratories have found chloral
hydrate to be negative in in vivo experiments (Xu & Adler, 1990;
Adler, 1993). So far, chloral hydrate has been found to give negative
test results in studies with mouse oocytes (Mailhes et al., 1993).
Although chloral hydrate can induce a variety of genetic events
(mutation, aneuploidy, structural chromosomal aberrations), it does so
with a very low potency.
Chloral hydrate has been reported to produce hepatic tumours in
male B6C3F1 mice in two studies. One study administered by gavage a
single dose of 5 or 10 mg of chloral hydrate per kg of body weight to
groups of 25 and 20 male mice at 15 days of age (Rijhsinghani et al.,
1986). The response to the 10 mg/kg of body weight dose led to
statistically elevated levels of tumours between 48 and 92 weeks, but
the results are based on the appearance of three adenomas and three
carcinomas among eight animals. Moreover, the historical control
incidence of hepatic tumours in the male of this hybrid is generally
about 25% and has been reported to be in excess of 40% in individual
studies. Thus, the small numbers of animals make it difficult to give
much credence to the results of this study. A second study (Daniel et
al., 1992a), however, showed that chloral hydrate administered in
drinking-water to a group of 40 male B6C3F1 mice for 104 weeks at
1 g/litre (166 mg/kg of body weight per day) resulted in a 71%
incidence of hepatic tumours (combined adenomas and carcinomas). The
fact that much higher doses were required to induce tumours in the
same hybrid mouse in this study raises further questions about the
Rijhsinghani et al. (1986) study; however, mice of this age are known
to be very sensitive to tumour initiators (Vesselinovich et al.,
1974). However, the Daniel et al. (1992a) study does clearly indicate
that chloral hydrate is capable of inducing tumours in B6C3F1 mice
when the mice are subjected to a lifetime exposure.a
a After the Task Group meeting, three new studies in B6C3F1 mice
on the carcinogenicity of chloral hydrate appeared (NTP, 2000a,b;
George et al., in press). In the first (NTP, 2000a), no carcinogenic
effect was observed following the administration of a single dose of
chloral hydrate (the dose was up to 5 times higher than that used in
the study of Rijhsinghai et al., 1986), while in the two other
studies, males had an increased incidence of hepatic tumours after
life-time exposure (NTP, 2000b; George et al., in press). In the NTP
life-time study (NTP, 2000b), a slightly elevated incidence of
pituitary adenomas, of borderline statistical significance, was
observed in female mice.
The question of whether chloral hydrate itself contributes to its
carcinogenic effects is critical because at least two of its
metabolites, TCA and DCA, are comparatively potent inducers of hepatic
tumours in B6C3F1 mice. This question can be resolved only by
demonstrating (i) that the clastogenic effects of chloral hydrate play
a role in the development of tumours or (ii) that TCA, DCA or a
combination of both chemicals are produced in sufficient quantities to
completely account for the induction of liver tumours without
significant contribution from earlier, more reactive metabolites. This
question is more critically assessed in the next section.
4.3.1.4 Comparative metabolism and pharmacokinetics
The metabolism of chloral hydrate has received considerable
attention over the years because of its extensive use as a
sedative-hypnotic (Marshall & Owens, 1954; Kaplan et al., 1967;
Gessner & Cabana, 1970; Cabana & Gessner, 1970; Sellers et al., 1972;
Garrett & Lambert, 1973; Mayers et al., 1991). A series of older
studies provide a still valid general picture of the conversion of
chloral hydrate to its two major metabolites, trichloroethanol and
TCA. More recent studies have focused more specifically on species
differences in this metabolism and have begun to focus on minor
metabolic pathways.
Figure 6 provides a simplified scheme of chloral hydrate
metabolism. The reader is referred to the appropriate sections of this
document to evaluate the further metabolism of the products TCA and
DCA.
The major fate of chloral hydrate is to undergo reduction to
trichloroethanol, with a smaller, but significant, fraction being
oxidized to TCA. Initially, formation of trichloroethanol is favoured
because the redox potential within cells in vivo favours reduction
(Kawamoto et al., 1987). This initial tendency is accentuated by a
rapid glucuronidation of the trichloroethanol that is formed (Marshall
& Owens, 1954). As is pointed out below, this may be a key feature in
the interspecies differences in chloral hydrate metabolism. With time,
(continued)
References:
NTP (2000a) Toxicology and carcinogenesis studies of chloral hydrate
in B6C3F1 mice (gavage studies). Research Triangle Park, North
Carolina, US department of Health and Human Services, National
Toxicology Program (NTP-TR-502).
NTP (2000b) Toxicology and carcinogenesis studies of chloral hydrate
(ad libitum and dietary controlled) in male B6C3F1 mice (gavage
study). Research Triangle Park, North Carolina, US department of
Health and Human Services, National Toxicology Program (NTP-TR-503).
George MH, Kilburn S, Moore T & DeAngelo AB (in press) The
carcinogenicity of chloral hydrate administered in the drinking water
to the male B6C3F1 mouse and F344/N rat. Toxicol Pathol.
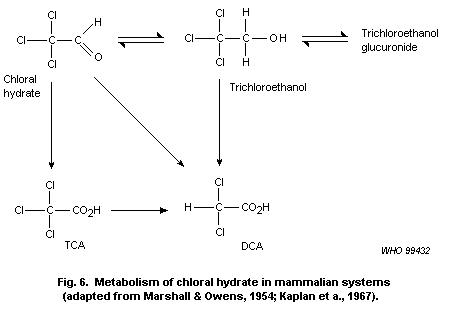 however, more TCA is formed as a result of enterohepatic circulation
of the trichloroethanol glucuronide (Stenner et al., 1996). The
glucuronide is hydrolysed to trichloroethanol, which can be oxidized
to TCA, with chloral hydrate as an intermediate. Under physiological
conditions, the formation of TCA is for all practical purposes
irreversible, and the net conversion of chloral hydrate to TCA will
continue as long as there are significant amounts of trichloroethanol
entrained in the enterohepatic circulation.
The production of these major products of chloral hydrate
metabolism is relatively well understood. However, the exact source of
a number of minor metabolites is less well understood. The reaction
rates and mechanisms involved are just beginning to be studied.
Understanding these mechanisms will be key to understanding whether
the effects of chloral hydrate can be attributed primarily to its
conversion to chemicals of established toxicological properties, such
as trichloroethanol, DCA and TCA, or whether the activities of
reactive intermediates must also be considered. Formation of DCA
probably requires radical formation, but it is not clear whether the
radical would be formed from trichloroethanol, chloral hydrate or TCA.
If DCA is derived from the first two compounds, the
dichloroacetaldehyde that would result as an intermediate could pose
some toxicological problems as well. Moreover, Ni et al. (1996)
suggested from their ESR data that a trichloromethyl radical is formed
from chloral hydrate. Such an intermediate could contribute to the
toxicological effects of chloral hydrate as well.
Available pharmacokinetic data have focused upon the relative
role of trichloroethanol as the metabolite of chloral hydrate that is
responsible for its central nervous system-depressant activity
(Butler, 1948, 1949; Marshall & Owens, 1954; Garrett & Lambert, 1973).
Conversely, a variety of metabolites have been suggested as
responsible for the toxic effects of chloral hydrate and its ability
to induce liver cancer in mice, in particular (Daniel et al., 1992a).
The critical question is whether an early reactive metabolite -- e.g.,
the free radicals identified by Ni et al. (1996) -- induces
clastogenic effects that are important to tumour development. The
competing, but not necessarily exclusive, hypothesis is that the
clearly hepatocarcinogenic metabolites TCA and DCA are produced in
sufficient quantity to account for some or all of the liver cancer
that results from chloral hydrate treatment.
While TCA and trichloroethanol are well established metabolites
of chloral hydrate, DCA has been only recently recognized as a
potentially important metabolite with respect to liver tumour
induction. Part of the difficulty is that DCA is much more rapidly
metabolized than either TCA or trichloroethanol, and peak
concentrations would be expected to be significantly lower. This first
became apparent when DCA appeared to be produced in significant
quantities from trichloroethylene administered to B6C3F1 mice
(Templin et al., 1993). Chloral hydrate is the first stable metabolite
of trichloroethylene metabolism (Cole et al., 1975); thus, these data
prompt an examination of the role that chloral hydrate plays in the
formation of DCA. In contrast with these results in mice, DCA was not
detectable in rats and dogs administered similar doses (Templin et
al., 1995). It became apparent that some of the DCA that was measured
in these studies may have arisen artefactually through dehalogenation
of TCA under acid conditions in fresh blood (Ketcha et al., 1996). As
a consequence of this series of findings, newer studies have examined
whether DCA is produced from chloral hydrate in a number of species.
Recent results of Abbas et al. (1996) showed that doses of 10 and
100 mg of chloral hydrate per kg of body weight result in
approximately 2.4 and 10 µg of DCA per ml of blood. However, it must
be noted that a variety of artefacts of DCA formation from TCA in
blood suggest that these results need to be viewed with caution
(Ketcha et al., 1996). Indeed, Merdink et al. (1998) found that DCA
was not measurable in mice dosed with 50 mg of chloral hydrate per kg
of body weight.
4.3.1.5 Mode of action
The mechanism of action involved in chloral hydrate-induced liver
tumours in mice remains to be established. Clearly, chloral hydrate is
converted to at least one metabolite, TCA, that appears to act as a
peroxisome proliferator. There is some possibility that it is
converted to DCA, a compound that acts primarily as a tumour promoter
(Stauber & Bull, 1997). On the other hand, chloral hydrate is distinct
from these other two compounds in that it appears to be clastogenic
in vivo, but at very high doses. The question is, does this
clastogenic activity play a role in tumorigenesis? The fact that
chloral hydrate appears to produce only hepatic tumours in mice
parallels the species specificity of TCA and suggests that other
activities are perhaps not involved.
4.3.2 Halogenated aldehydes and ketones other than chloral hydrate
4.3.2.1 General toxicological properties and information on
dose-response in animals
1) Haloaldehydes
Relatively few data are available to describe acute, short-term
or chronic toxicities for the haloacetaldehydes. In general, aldehydes
are irritant chemicals, and substitution of chlorine generally
increases this irritancy. However, it is unlikely that irritant
effects will occur at concentrations that are encountered in
drinking-water. Chloroacetaldehyde is an example of a haloaldehyde for
which some data exist, although it has not been commonly found in
drinking-water. Concentrations of chloroacetaldehyde as low as 0.02%
produced intradermal irritation, 7.5% produced rather severe dermal
irritation and 0.03% irritated the eyes of rabbits (Lawrence et al.,
1972).
When administered systemically, halogenated aldehydes are quite
toxic. Again because of a lack of appropriate data, chloroacetaldehyde
will be used for illustrative purposes. Lawrence et al. (1972)
reported the oral LD50 in Sprague-Dawley rats to be 89 and 103 mg/kg
of body weight in males and females, respectively. In male ICR mice,
the oral LD50 was found to be 82 mg/kg of body weight. In longer-term
investigations, Lawrence and co-workers (1972) utilized
intraperitoneal injections or inhalation as the method of
administration. Intraperitoneal injections of 2.2 or 4.4 mg/kg of body
weight per day for 30 days in male Sprague-Dawley rats produced
significant decreases in haemoglobin, haematocrit and erythrocytes at
the highest dose. This was consistent with its ability to induce
haemolysis of erythrocytes at concentrations of 0.2 mol of
chloroacetaldehyde per litre and above. However, the validity of this
in vitro observation as a dependable indicator of in vivo effects
is suspect because of the extremely high concentrations that were
utilized. The 4.4 mg/kg of body weight dose produced reductions in
body weight and induced significant increases in the organ to body
weight ratios for the brain, gonads, heart, kidneys, lungs and spleen.
These effects appeared to be largely the result of reduced body weight
rather than changes in absolute organ weights.
Lawrence et al. (1972) pointed out the importance of route of
administration to the toxicity of chloroacetaldehyde. If administered
intraperitoneally, it is 10-30 times as potent as its metabolite
2-chloroethanol. However, the toxicity of the two compounds is more or
less equivalent when they are administered orally, and 2-chloroethanol
is about 4 times as toxic as chloroacetaldehyde when applied
topically. These differences are most likely attributed to relative
rates of absorption versus metabolic conversion and the many
non-specific reactions in which chloroacetaldehyde would be expected
to be involved in the gastrointestinal tract and on the skin. This
issue should be reflected in the systemic toxicities of halogenated
aldehydes, in general.
A 104-week study of chloroacetaldehyde utilizing drinking-water
as the mode of administration was conducted in male B6C3F1 mice
(Daniel et al., 1992a). Only a single concentration was used
(100 mg/litre), which yielded an average dose of 17 mg/kg of body
weight (i.e., larger than administered to rats intraperitoneally).
This dose did not lead to excessive mortality or depress body weight
gains. It did not affect weights of the liver, kidneys, testes or
spleen. There was no remarkable non-tumour pathology in 40 tissues
that were sampled and examined microscopically in five of the animals
that were serially sacrificed during the course of the experiment. An
apparent increase in the incidence of liver histopathological change
was described as hepatocellular necrosis, hyperplasia and cytomegaly.
However, these effects were very mild and of doubtful significance as
compared with the same types of pathology in control mice.
Sood & O'Brien (1993) examined the effects of chloroacetaldehyde
in isolated rat hepatocytes. A concentration of 0.5 mmol/litre was
found to be cytotoxic, whereas 0.2 mmol/litre was without apparent
effect. The cytotoxicity could be essentially abolished by
dithiothreitol in the incubation media. The requirement for high
concentrations in this in vitro experiment and the apparent lack of
effect (albeit administered to a different species) at relatively high
concentrations in the animals' drinking-water (100 mg/litre) suggest
that there is little concern about hepatotoxicity at the very low
concentrations that might be expected to be found in chlorinated
drinking-water.
2) Haloketones
Toxicological data in experimental animals for the haloketones
are extremely limited. The halopropanones are the most commonly
studied group of this class, but most of the work has been directed at
mutagenic effects of the chemicals.
Laurie et al. (1986) studied the effects of 1,1- and 1,3-DCPN in
CD-1 mice. 1,1-DCPN was administered in paraffin, and 1,3-DCPN was
administered as an aqueous solution. 1,1-DCPN significantly increased
the levels of ASAT, ALAT and LDH at doses of 325 mg/kg of body weight.
1,3-DCPN was evaluated to a maximum dose of 20 mg/kg of body weight
and appeared to be without effect on the serum enzymes. However, the
LD50 for 1,3-DCPN was stated to be 25 mg/kg of body weight,
indicating that the liver was probably not the critical target organ
for this compound, at least with acute treatment. At doses of 130
mg/kg of body weight and greater, 1,1-DCPN significantly depressed
hepatic GSH levels. Again, 1,3-DCPN was without effect at the dose of
20 mg/kg of body weight. Most of the decrease in GSH produced by
1,1-DCPN was observed in the post-mitochondrial cellular fraction as
opposed to the mitochondrial fraction.
A major thrust of the Laurie et al. (1986) paper was to determine
the extent to which 1,1- and 1,3-DCPN modified the toxicity of carbon
tetrachloride. Carbon tetrachloride was administered at doses ranging
from 0.02 to 1.0 ml/kg of body weight. The 0.02 ml/kg of body weight
dose produced an elevation of serum enzymes on its own. However, when
carbon tetrachloride was administered 4 h after 1,1-DCPN, the dose of
1,1-DCPN that was required to significantly increase serum enzyme
levels was decreased from 325 to 130 mg/kg of body weight. Since a
doubling of the dose of 1,1-DCPN would have produced a similar
response and the dose of carbon tetrachloride was above a threshold
response level, the interaction between carbon tetrachloride and
1,1-DCPN would seem to be no more than additive. Inhibition of the
toxicological effects of carbon tetrachloride in a dose-related manner
was observed when 1,3-DCPN was administered prior to carbon
tetrachloride.
Merrick et al. (1987) studied the cytotoxic effects of
chloropropanone (CPN), 1,1-DCPN and 1,3-DCPN on isolated hepatocytes
of male Sprague-Dawley rats. The chloropropanones were all shown to
react with GSH in solution. At concentrations of 10 mmol/litre, the
rate of GSH reaction was most rapid with 1,3-DCPN, followed by CPN and
then 1,1-DCPN. This reactivity paralleled the ability of the compounds
to induce cytotoxicity in isolated hepatocytes. Significant increases
in ASAT release were observed with 1,3-DCPN at 0.5 mmol/litre, CPN at
1 mmol/litre and 1,1-DCPN at 5 mmol/litre. As would be expected, GSH
depletion was observed at concentrations of 1,3-DCPN as low as 0.1
mmol/litre. The other two compounds were significantly less active in
depleting GSH, but were of approximately equivalent potency with one
another.
A study of 1,1,1-TCPN was conducted in Sprague-Dawley rats by
Daniel et al. (1993b). Acute, 10-day and 90-day experiments were
performed. 1,1,1-TCPN was administered in corn oil by gavage. Doses in
the 10-day study were 0, 16, 48, 161 or 483 mg/kg of body weight per
day. In the 90-day study, doses of 30, 90 or 270 mg/kg of body weight
per day were administered. In the 10-day study, 8 out of 10 male and 7
out of 10 female rats died at 483 mg/kg of body weight per day before
the conclusion of the treatments. Two male rats also died at 161 mg/kg
of body weight per day. Although there was not a significant effect on
body weight in the survivors, there was a 10% increase in liver to
body weight ratios at 161 mg/kg of body weight per day in both male
and female rats. Evidence of hyperkeratosis was found in the
forestomach of both male and female rats treated at doses of 48 mg/kg
of body weight per day and greater. No adverse effect was observed at
16 mg/kg of body weight per day.
Increases in relative liver weight were observed in the 90-day
experiment at 270 mg/kg of body weight per day in male rats, but not
at 90 mg/kg of body weight per day. Ataxia was reported in both sexes
at the 270 mg/kg of body weight per day dose level. Increases in the
incidence of forestomach lesions were observed in both sexes at 90 and
270 mg/kg of body weight per day, with the most frequent observation
being hyperkeratosis followed by acanthosis. The overall NOAEL in the
10-day and 90-day studies is 30 mg/kg of body weight per day.
In summary, the toxicological effects of the halopropanones
provide evidence that some of the representatives of this class are
highly toxic, with acute lethal doses being as low as 25 mg/kg of body
weight. The gastrointestinal tract and liver appear to be key target
organs for some members of the class. However, no target organ was
identified for the most acutely toxic of the group, 1,3-DCPN.
4.3.2.2 Toxicity in humans
No data on the effects of either halogenated aldehydes or
halogenated ketones on human subjects were identified.
4.3.2.3 Carcinogenicity and mutagenicity
There are considerable data on the mutagenic properties of
various halogenated aldehydes and ketones. A comparison of the
mutagenic activity, measured in Salmonella typhimurium tester
strains, of the halogenated aldehydes found in drinking-water with
that of chloroacetaldehyde is summarized (not comprehensively) in
Table 19. Table 20 provides a similar summary of results with
halogenated ketones. These data are presented to demonstrate that the
activity of these compounds is not dissimilar from that observed with
chloroacetaldehyde, a metabolite of a large number of carcinogenic
chemicals, including vinyl chloride. Therefore, it will be used as a
prototype for the class. This is not to suggest that this is an
adequate substitute for appropriate data for the other compounds. It
must be recognized that the data available for this class are
completely inadequate for making substantive estimates of the impact
of these chemicals on human health; in particular, mutagenicity data
in bacterial systems do not necessarily reflect activity in vivo.
1) Haloaldehydes
Chloroacetaldehyde has been extensively studied with respect to
the types of interactions that it has with DNA. The adducts formed in
animals treated with vinyl chloride are the same as those produced
with chloroacetaldehyde (Green & Hathway, 1978). The cyclic etheno
adducts formed with cytosine and adenine seem particularly important
in mutagenic responses observed with chloroacetaldehyde (Spengler &
Singer, 1988; Jacobsen et al., 1989).
There are a limited number of studies that have examined the
carcinogenic properties of chloroacetaldehyde in rather specialized
test systems. Van Duuren et al. (1979) examined the carcinogenic
activity of chloroacetaldehyde in mouse skin initiation/promotion
studies, with subcutaneous injection and by stomach tube. In the
initiation/promotion assay, chloroacetaldehyde was applied to the skin
of 30 female Ha:ICR Swiss mice, at a dose of 1.0 mg per application
per mouse, 3 times weekly for up to 581 days, or as a single dose
followed by 2.5 µg of TPA 3 times weekly for 576 days. No evidence of
increased skin tumour yield was found. The intragastric treatments
involved administration of 0.25 mg per mouse per week (1.8 mg/kg of
body weight per day if a 20-g body weight is assumed). In this case,
sections of lung, liver and stomach were taken for histopathological
examination. No signs of increased tumour incidence were found. A
fourth experiment involved the subcutaneous injection in 30 mice of
0.25 mg per mouse (1.8 mg/kg of body weight per day, assuming a 20-g
body weight for the mouse). Microscopic examination of sections of the
liver and injection sites revealed no evidence of increased tumour
yield.
A second group of mouse skin initiation/promotion experiments
with chloroacetaldehyde were conducted by Zajdela et al. (1980).
Single doses of 0.05, 0.1, 1.0 or 2.5 mg of chloroacetaldehyde
dissolved in acetone were applied to male and female XVIInc/Z mice
(20-28 per group). This was followed by application of TPA at 2 µg, 3
times weekly for 42 weeks. There was no significant difference between
mice receiving TPA alone and those that had been initiated by
chloroacetaldehyde.
There appears to be only one study that examined the carcinogenic
activity of halogenated aldehydes administered in drinking-water over
a lifetime (Daniel et al., 1992a). Male B6C3F1 mice treated with 0.1
g of chloroacetaldehyde per litre of drinking-water for 104 weeks were
found to have an incidence of eight hepatocellular carcinomas in
26 mice examined (31%). In addition, 8% of these mice were diagnosed
as having hepatocellular adenoma, and another 8% were found to have
hyperplastic nodules. This compared with two carcinomas in 20 control
mice examined (10%), one with adenoma (5%) and no hyperplastic nodules
(0%). In a comparison with previous studies, the experiment utilized a
Table 19. Mutagenic activity of halogenated aldehydes produced by chlorination in
the Salmonella/microsome assay
Compound Strain Net revertants/plate Reference
-S9 +S9
Chloroacetaldehyde TA100 440 ~330 Bignami et al. (1980)
2-Chloropropenal TA100 135 49 Segall et al. (1985)
2-Bromopropenal TA100 1140 108 Segall et al. (1985)
2-Bromopropenal TA100 400 Gordon et al. (1985)
2,3-Dibromopropanal TA100 300 Gordon et al. (1985)
2,3-Dichloropropenal TA100 91 5 Segall et al. (1985)
3,3-Dichloropropenal TA100 0.7 Meier et al. (1985a,b)
2,3,3-Trichloropropenal TA100 224 Rosen et al. (1980)
3-Chloro-2-butenal TA100 68 42 Segall et al. (1985)
3-Bromo-2-butenal TA100 108 39 Segall et al. (1985)
2-Bromo-3-methyl-2-butenal TA100 <0.5 <0.5 Segall et al. (1985)
Table 20. Mutagenic activity of halogenated ketones produced by chlorination in the
Salmonella/microsome assay
Compound Strain Net revertants/plate Reference
-S9 +S9
CPN TA100 NMa NM Merrick et al. (1987)
1,1-DCPN TA100 0.04 Meier et al. (1985a)
1,3-DCPN TA100 25.2 Meier et al. (1985a)
1,1,1-TCPN TA100 0.12 Meier et al. (1985a)
1,1,3-TCPN TA100 3.9 Douglas et al. (1985)
1,1,3,3-Tetrachloropropanone TA100 1.5 Meier et al. (1985a)
Pentachloropropanone TA100 0.86 Meier et al. (1985a)
a NM = non-mutagenic.
significantly higher dose (17 mg/kg of body weight per day) as well as
a continuous treatment. It is possible that these tumours were induced
by the genotoxic properties of the chemical.
Robinson et al. (1989) examined the ability of four other
halogenated aldehydes to act as tumour initiators in the skin of
Sencar mice: 2-chloropropenal, 2-bromopropenal, 3,3-dichloropropenal
and 2,3,3-trichloropropenal. The compounds were administered topically
in six divided doses over a 2-week period (total doses were
600-2400 mg/kg of body weight). Two weeks after the final initiating
dose, TPA was applied at a dose of 1 µg, 3 times weekly for 20 weeks.
Both 2-chloropropenal and 2-bromopropenal significantly increased
tumour yield at 24 weeks and significantly increased the yield of
squamous cell carcinomas at 52 weeks at total topical doses of
1200 mg/kg of body weight and above. Both the benign tumour and
malignant tumour yields were greater with 2-bromopropenal than with
2-chloropropenal. An experiment utilizing oral administration of these
compounds during the initiation period was included in this study.
Oral administration of 2-chloropropenal did not produce consistent,
dose-related responses. However, there appeared to be a substantial
increase in skin tumour yield at an oral dose of 300 mg of
2-bromopropenal per kg of body weight (19 tumours in 38 mice [50%] vs.
20 tumours in 110 control mice [18%]).
On the basis of these studies, it must be concluded that there is
a potential carcinogenic hazard associated with the halogenated
aldehydes. Only a single compound, chloroacetaldehyde, was evaluated
as a carcinogen in a lifetime study, and only one dose level was
studied. It appears to be more potent as a carcinogen than the
corresponding THM and HAA by-products. Many members of the class are
mutagenic, and chloroacetaldehyde, at least, appears to produce
tumours in the liver at less than cytotoxic doses. Based upon the
comparison between 2-chloropropenal and 2-bromopropenal, there is some
reason to believe that the brominated by-products are more potent than
the corresponding chlorinated by-products. Therefore, concern must be
expressed over disinfection processes that activate bromide, as well
as those that simply chlorinate. However, the currently available data
are not sufficient to allow the hazards associated with these
compounds to be estimated.
2) Haloketones
A number of halopropanones have been tested in mutagenesis
assays. To facilitate comparison of their relative potencies, selected
results from assays that were conducted in Salmonella typhimurium
tester strain TA100 were incorporated into Table 20. For the most
part, the data selected for this table were abstracted from papers in
which more than one haloketone was evaluated, rather than being
selected because they were identified as the best value for each
individual chemical that exists in the literature. Some of the
compounds have been shown to be active in other Salmonella tester
strains and other mutagenesis and clastogenesis assays. There was
little to be gained from an exhaustive review of this literature, so
further consideration of the mutagenic activity will be limited to
those systems that extended evaluations to other end-points in
vitro or attempted to confirm in vitro observations in vivo.
1,3-DCPN was found to induce SCEs in V79 cells at concentrations
as low as 0.002 mmol/litre (von der Hude et al., 1987). Blazak et al.
(1988) found that 1,1,1-TCPN and 1,1,3-TCPN were able to act as
clastogens in CHO cells in vitro. Structural aberrations were
produced at a 1,1,3-TCPN concentration of 1.5 g/ml, whereas a
concentration of 23 µg/ml was required for a similar response to
1,1,1-TCPN. However, the dose-response for 1,1,3-TCPN was limited by
cytotoxicity. Experiments were also conducted focusing on the ability
of 1,1,1-TCPN and 1,1,3-TCPN to induce micronuclei in polychromatic
erythrocytes and to induce sperm head abnormalities in mice in
vivo. 1,1,1-TCPN was found to be negative in both assays in the dose
range 75-300 mg/kg of body weight, whereas 1,1,3-TCPN was negative in
the range 3-12 mg/kg of body weight.
Robinson et al. (1989) tested CPN, 1,1-DCPN, 1,3-DCPN, 1,1,1-TCPN
and 1,1,3-TCPN as initiators in the skin of Sencar mice. The compounds
were administered by topical application in acetone with total doses
that ranged from 37.5 to 4800 mg/kg of body weight (doses of 600 mg/kg
of body weight and above were administered in six equal doses over a
2-week period to avoid cytotoxic or lethal effects of the compounds).
Other groups of animals treated with the chemicals were also
administered similar doses by intragastric intubation. The initiating
treatments were followed by a promotion schedule that involved the
topical application of 1 µg of TPA 3 times weekly for 20 weeks. Tumour
counts were reported at 24 weeks; if the incidence was elevated within
this time period, the mice were held until 52 weeks on study prior to
sacrifice, and histological evaluations of the tumours were made.
Among the haloketones, only 1,3-DCPN was found to produce a
dose-related increase in tumour incidence. A single topical dose of
37.5 mg/kg of body weight was sufficient to initiate skin tumours, and
the response increased progressively as doses were increased to 150
mg/kg of body weight. At higher doses, the response decreased in
magnitude. Splitting the 300 mg/kg of body weight dose into six equal
doses over a 2-week period increased the tumorigenic response relative
to a single dose of 300 mg/kg of body weight. However, this response
was also attenuated, as the multiple-dose schedule utilized higher
doses. This attenuation of the response was particularly marked in
total tumour yields, which included many benign tumours. It was less
effective in limiting the yield of squamous cell carcinomas.
In conclusion, the carcinogenic activity of the DBPs in the
haloaldehyde and haloketone classes, with the exception of
chloroacetaldehyde, has not been evaluated in lifetime studies in
experimental animals. However, other tests confirm that they have
carcinogenic properties. 1,3-DCPN was the most potent tumour initiator
in both classes of DBPs. A single dose of 75 mg/kg of body weight
produces a total tumour yield equivalent to that produced by 1200 mg
of 2-bromopropenal, the most potent of the haloaldehydes, per kg of
body weight. 2-Bromopropenal is about 40 times as potent as 1,3-DCPN
as a mutagen. The other halopropanones do not appear to be capable of
acting as tumour initiators in the mouse skin.
4.3.2.4 Comparative pharmacokinetics and metabolism
No information was identified in the available literature.
4.3.2.5 Mode of action
The data available indicate that these two groups of chemicals
contain compounds that possess mutagenic activity. As these effects
have been identified in in vitro or bacterial test systems, there is
no assurance that this is the manner in which they contribute to
toxicity or carcinogenicity. A few chemicals have been shown to be
initiators in the mouse skin, but it is not clear whether that would
be a target organ as a result of chronic ingestion of these chemicals.
Other chemicals appear to have activities that could contribute less
directly to the induction of cancer, particularly as cytotoxic
compounds. It is clear from the limited data available that it would
be inappropriate to try to generalize data from only a few examples to
these two larger classes of DBPs.
4.4 Haloacetonitriles
4.4.1 General toxicological properties and information on
dose-response in animals and humans
The HANs are discussed in a single section of this document
because the toxicological data on them are quite limited. The
dihaloacetonitriles (DHAN) -- DCAN, BCAN and DBAN -- are the most
important in terms of concentrations found in chlorinated
drinking-water. However, there are limited data on bromoacetonitrile
(BAN), chloroacetonitrile (CAN) and trichloroacetonitrile (TCAN) that
are included for completeness.
Hayes et al. (1986) examined the general toxicological effects of
DCAN and DBAN in male and female ICR mice and CD rats. In mice, the
acute oral (by gavage in corn oil) LD50 was reported to be 270
(males) and 279 (females) mg/kg of body weight for DCAN and 289
(males) and 303 (females) mg/kg of body weight for DBAN. In rats, the
LD50 was found to be 339 (males) and 330 (females) mg/kg of body
weight for DCAN and 245 (males) and 361 (females) mg/kg of body weight
for DBAN. Hussein & Ahmed (1987) found somewhat lower oral LD50s in
rats: BAN, 25.8 mg/kg of body weight; DBAN, 98.9 mg/kg of body weight;
CAN, 152.8 mg/kg of body weight; and DCAN, 202.4 mg/kg of body weight.
These latter data were reported only in abstract form, and the vehicle
used was not indicated. As discussed below, some of the toxicological
responses to chemicals in this class appear to depend on the nature of
the vehicle in which they were administered.
DCAN and DBAN were also studied over 14- and 90-day treatment
intervals (Hayes et al., 1986). DCAN dissolved in corn oil was
administered to male and female CD rats by gavage at 12, 23, 45 or 90
mg/kg of body weight per day for 14 days and at 8, 33 or 65 mg/kg of
body weight per day for 90 days. DBAN was administered to male and
female CD rats at daily doses of 23, 45, 90 or 180 mg/kg of body
weight per day for 14 days and at 6, 23 or 45 mg/kg of body weight per
day for 90 days. Increased mortality was produced at 33 mg of DCAN per
kg of body weight per day and at 45 mg of DBAN per kg of body weight
per day in the 90-day studies. Body weight was decreased and lower
weights and organ to body weight ratios were observed for spleen and
gonads with doses of 65 mg of DCAN per kg of body weight per day and
above. The NOAELs for DCAN were 45 mg/kg of body weight per day for 14
days and 8 mg/kg of body weight per day for 90 days of exposure. The
NOAELs for DBAN were 23 mg/kg of body weight per day at 90 days and 45
mg/kg of body weight per day at 14 days. No serum chemistry changes
indicative of adverse effects were seen with either compound at
sublethal doses.
4.4.2 Reproductive and developmental toxicity
Smith et al. (1987) examined the effect of CAN, DCAN, TCAN, BCAN
and DBAN on female reproduction in an in vivo teratology screening
test in Long-Evans hooded rats. DCAN and TCAN at doses of 55 mg/kg of
body weight administered in tricaprylin by gavage from day 7 to day 21
of gestation significantly reduced the percentage of females
delivering viable litters, increased resorption rates and reduced
maternal weight gain. BCAN and DBAN at the same dose were without
effect. All of the HANs reduced the mean birth weight of pups, and the
DHANs reduced the postnatal weight gain till the fourth day after
birth. Postnatal survival was reduced with DCAN and TCAN but not with
BCAN or DBAN. These pups continued to display reduced body weights
into puberty. BCAN also resulted in significantly depressed weights at
puberty, although the effect was smaller than that observed with DCAN
or TCAN.
The hydra assay system for developmental toxicity has also been
used to screen some of the HANs (Fu et al., 1990). Both DBAN and TCAN
were found to be of the same general order of toxicity to adult and
embryonic animals. Based on these findings, the authors predicted that
DBAN and TCAN would not be teratogenic at non-maternally toxic doses.
The developmental toxicity of DCAN was followed up in full-scale
teratology studies (Smith et al., 1989b). In this case, DCAN dissolved
in tricaprylin was administered to Long-Evans rats at doses of 0, 5,
15, 25 or 45 mg/kg of body weight per day from day 6 to day 18 of
gestation. Embryolethality and fetal resorptions were statistically
significant at 25 and 45 mg/kg of body weight per day. The highest
dose was also maternally toxic. Soft tissue anomalies, including an
intraventricular septal defect in the heart, hydronephrosis, fused
ureters and cryptorchidism, were observed at this dose. Skeletal
abnormalities (fused and cervical ribs) were produced in a
dose-dependent manner and were significantly increased at 45 mg/kg of
body weight per day. A NOAEL was found to be 15 mg/kg of body weight
per day.
TCAN was evaluated in two teratology studies by the same
laboratory (Smith et al., 1988; Christ et al., 1996). The first of
these studies utilized tricaprylin as the vehicle, whereas the second
utilized corn oil. In the first study, embryolethality was observed at
doses as low as 7.5 mg/kg of body weight per day. Doses of 15 mg/kg of
body weight per day and above produced soft tissue abnormalities,
including fetal cardiovascular anomalies (Smith et al., 1988). TCAN
administered in a corn oil vehicle produced cardiovascular defects at
55 mg/kg of body weight per day (Christ et al., 1996). The effects
observed at this dose were found at significantly lower incidence than
observed in the 15 mg/kg of body weight per day dose of the previous
study. The abnormalities were milder, being simply positional
(laevocardia), instead of the interventricular septal defect and a
defect between the ascending aorta and right ventricle that were
observed with the tricaprylin vehicle. On the other hand, more
skeletal defects were observed with the corn oil vehicle. The authors
attributed these differences to an interaction between the tricaprylin
vehicle and TCAN. However, tricaprylin and corn oil are not
representative of drinking-water exposure. It is not possible to
determine whether the results obtained with tricaprylin or the results
obtained with corn oil provide the most valid test. The results could
be just as readily ascribed to an inhibition of the effects of the
corn oil vehicle. As a consequence, these data present somewhat of a
difficulty in interpreting results for all of the HANs that have been
tested, because the only vehicle in which most have been evaluated was
tricaprylin.
4.4.3 Carcinogenicity and mutagenicity
IARC has evaluated BCAN, CAN, DBAN, DCAN and TCAN and concluded
that there is inadequate evidence for their carcinogenicity in
experimental animals. No data were available on their carcinogenicity
in humans. Consequently, these HANs were assigned to Group 3: the
agent is not classifiable as to its carcinogenicity to humans (IARC,
1991, 1999).
Bull et al. (1985) tested the ability of CAN, DCAN, TCAN, BCAN
and DBAN to induce point mutations in the Salmonella/microsome
assay, to induce SCEs in CHO cells in vitro, to produce micronuclei
in polychromatic erythrocytes in CD-1 mice and to act as tumour
initiators in the skin of Sencar mice.
DCAN produced a clear increase in mutagenic activity in
Salmonella typhimurium strains TA1535 and TA100. This response was
not altered by the inclusion of the S9 system to metabolically
activate the compound, if needed. BCAN also produced a positive
response at low doses, but the dose-response curve was interrupted at
high doses by cytotoxicity. In this case, the inclusion of the S9
fraction appeared to simply allow the bacteria to survive cytotoxic
effects. This perhaps arose through a non-specific inactivation of the
electrophilic character of the compound. BCAN produced a similar, but
less marked, trend in strain TA100. The other HANs were negative in
the Salmonella/microsome assay.
All of the HANs tested increased the frequency of SCEs in CHO
cells. The potencies in the absence of S9 were DBAN > BCAN > DCAN
approx. or equiv. TCAN > CAN. As in the Salmonella/microsome assay,
the addition of S9 allowed higher doses to be tested rather than
modifying the response to a given concentration. In contrast, none of
the HANs was found to induce micronuclei in CD-1 mice in vivo.
The HANs were tested for their ability to initiate tumours in the
skin of Sencar mice (Bull et al., 1985). In this experiment, the HANs
were administered topically to the shaved backs of the mice in six
doses over a 2-week period. Two weeks following the last initiation
treatment, TPA dissolved in acetone was applied topically at a dose of
1 µg per mouse, 3 times weekly for 20 weeks. The total initiating
doses were 1200, 2400 and 4800 mg/kg of body weight. Significant
increases in skin tumours were observed with CAN, TCAN, BCAN and DBAN.
DBAN produced the greatest response at a dose of 2400 mg/kg of body
weight, but the response decreased in magnitude as the dose was
increased to 4800 mg/kg of body weight. The shape of this
dose-response curve was checked in a repetition of the experiment, and
the results were virtually identical. It was postulated that the
attenuated response at the higher dose was caused by the cytotoxicity
to initiated cells. BCAN also increased the incidence of skin tumours,
but no significant increase in tumour incidence was induced by DCAN.
The carcinogenicity of the DHANs in mouse skin was seen to
progressively increase as bromine was substituted for chlorine in the
compound. On the other hand, CAN appeared to be among the more potent
of the HANs in initiating skin tumours. TCAN gave inconsistent
results.
CAN, TCAN and BCAN produced small, but significant, increases in
the incidence of lung tumours in female A/J mice (40 mice per chemical
tested) when administered by gavage at doses of 10 mg/kg of body
weight, 3 times weekly for 8 weeks (Bull & Robinson, 1985). Mice were
started on treatment at 10 weeks of age and sacrificed at 9 months of
age. No significant effects were observed with DBAN and DCAN. The
differences in tumorigenic response are too small for meaningful
rankings of the compounds for potency in this lung tumour-susceptible
strain.
DCAN was found to induce aneuploidy in Drosophila (Osgood &
Sterling, 1991) at a concentration of 8.6 mg/litre. On the other hand,
DBAN produced inconsistent results but was tested at much lower
concentrations (0.3 mg/litre) because of its higher degree of
toxicity. Low levels of sodium cyanide (0.2 mg/litre) were also found
to be active in this test system. Since DCAN is metabolized to
cyanide, the authors suggested that the cyanide ion (CN-) was
responsible for the response. DCAN is somewhat more efficiently
converted to cyanide in rats than is DBAN (Pereira et al., 1984). This
difference would be multiplied by the much higher concentration of
DCAN that was tested.
Daniel et al. (1986) found that the HANs were direct-acting
electrophiles with the following decreasing order of reactivity: DBAN
>> BCAN > CAN >> DCAN >> TCAN. The ability to induce DNA strand
breakage in human CCRF-CEM cells was found to follow a considerably
different order: TCAN >> BCAN > DBAN > DCAN > CAN. It is of
interest that tumour-initiating activity paralleled alkylation
potential in a cell-free system rather than an ability to induce
strand breaks in DNA in intact cells or to induce mutation in
Salmonella (BCAN > DCAN >> DBAN > TCAN = CAN = 0). If CAN is
omitted from the group, mutagenicity parallels the extent to which the
HAN is converted to cyanide in vivo (Daniel et al., 1986). This
discordant set of parallels suggests that some property may be
affecting the ability of the test systems to measure the response or
that the responses are not mediated through a common mechanism.
Clearly, cytotoxic effects limited the responses of Salmonella to
the brominated HANs. Similar activity appeared to be affecting the
mouse skin initiation/promotion studies, but only after a fairly
robust response was observed. Some of the other effects may be only
loosely associated with a health effect. For example, the induction of
SSBs in DNA can arise from cytolethal effects. Moreover, such breaks
reflect DNA repair processes as well as damage. Therefore, it is
suggested that the carcinogenic potency of this class best parallels
alkylation potential.
TCAN was found to covalently bind with macromolecules in liver,
kidney and stomach of the F344 rat (Lin et al, 1992). The covalent
binding index was found to be the highest in the DNA of the stomach,
followed by the liver, and was lowest in the kidney. The binding of
14C was significantly higher when it was in the C2 position rather
than in C1, indicating that the nitrile carbon is lost. The adducts
formed were labile, and no specific adducts were identified. Adducts
to blood proteins were also observed with TCAN. Covalent binding of
DBAN or DCAN to DNA could not be demonstrated (Lin et al., 1986), but
binding to proteins was apparently not investigated with these HANs.
In conclusion, the HANs do possess carcinogenic and mutagenic
properties in short-term tests. However, without appropriate long-term
animal studies, the carcinogenic risk from HANs cannot be estimated.
4.4.4 Comparative pharmacokinetics and metabolism
The metabolism of the HANs has received some preliminary study,
but little information exists on the pharmacokinetics of the parent
compounds or their products. It is also important to note that some of
the HANs inhibit enzymes that are important in the metabolism of other
chemicals that are foreign to the body.
Pereira et al. (1984) found the following percentage of the
original doses of the HANs eliminated in the urine within 24 h as
thiocyanate: CAN, 14%; BCAN, 12.8%; DCAN, 9.3%; DBAN, 7.7%; and TCAN,
2.3%. This was compared with 42% of a dose of propionitrile. On the
basis of this limited information and a general scheme for the
elimination of cyanide from nitriles, published by Silver et al.
(1982), Pereira et al. (1984) proposed that additional products of HAN
metabolism would be as follows: CAN, formaldehyde; DHANs, formyl
cyanide or formyl halide; and TCAN, phosgene or cyanoformyl chloride.
These products would be direct-acting alkylating agents.
Roby et al. (1986) studied the metabolism and excretion of DCAN
labelled with 14C in either the C1 or C2 position in both male F344
rats and B6C3F1 mice. The metabolic fate of the two carbons was
significantly different in both mice and rats: C2 is metabolized much
more efficiently to carbon dioxide, whereas a very much higher
proportion of C1 is found as urinary metabolites, at least in mice.
These results are consistent with the proposal of Pereira et al.
(1984) suggesting metabolites that would be converted efficiently to
carbon dioxide from C2.
The HANs inhibit enzymes in the liver of the rat that are
traditionally associated with the metabolism of foreign compounds.
Pereira et al. (1984) demonstrated the inhibition of
dimethylnitrosamine demethylase activity. This activity has been
traditionally associated with the cytochrome P450 isoform 2E1,
although there was no direct confirmation of this in the study.
However, two forms of the enzyme activity were identified, one with a
Km of 2 × 10-5 and the other with a Km of 7 × 10-2. Based on a
plot presented in the paper, DBAN appears to be inhibiting the
high-affinity enzyme by either a non-competitive or uncompetitive
mechanism. The kinetics of inhibition were examined in vitro and the
enzyme : inhibitor dissociation constants ( Kis) reported to be as
follows: DBAN, 3 × 10-5; BCAN, 4 × 10-5; DCAN or TCAN, 2 × 10-4; and
CAN, 9 × 10-2. Although not commented upon by the authors, the
kinetics of inhibition by TCAN were clearly different from those of
the DHANs, suggesting some differences in the mechanism or the form of
the enzyme that might be affected. The authors examined the effects of
DBAN or TCAN administered orally to rats at doses of 0.75 mmol/kg of
body weight on the dimethylnitrosamine demethylase activity in the
liver at 3 and 10 h after administration. TCAN significantly reduced
the activity of the enzyme by about 30% at both time intervals, but
DBAN did not, despite the fact that it was the more potent inhibitor
in vitro. This could represent a difference in the extent to which
the two compounds are absorbed systemically, or it could be related to
the nature of the inhibition (i.e., reversible vs. irreversible).
Ahmed et al. (1989) demonstrated inhibition of cytosolic GSTs by
HANs in vitro. Doses (mmol/litre) at which 50% inhibition of the
activity of the enzyme GST occurred were as follows: DCAN, 2.49; TCAN,
0.34; DBAN, 0.82; CAN, >10; and BAN, >10. This latter observation
has not been established to occur in animals (Gao et al., 1996).
Activation and inactivation of various DBPs are catalysed by various
isoforms of GST (Pegram et al., 1997; Tong et al., 1998). In this
case, toxicity is reduced by inhibition of GST; however, mutagenic
activity of brominated THMs appears to depend upon a GSH pathway
(Pegram et al., 1997). Similarly, GSTs appear to play an important
role in the metabolism of HAAs (Tong et al., 1998).
While these effects on the metabolism of other DBPs could be of
importance, there are no data with which to relate these effects to
concentrations that would be encountered in drinking-water.
4.4.5 Mode of action
The induction of skin tumours appears closely correlated with the
alkylating potential within this class of chlorination by-products
(Daniel et al., 1986). This suggests that the carcinogenic activity of
the HANs may be related to mutagenic effects, despite the fact that
cytotoxicity limits the capability of test systems to detect such
activity. Cytotoxicity actually appears to inhibit the
tumour-initiating activity responses rather than to amplify them, as
has been seen with other DBPs. This suggests that the HANs retain some
specificity for inducing cytotoxic responses in initiated cells.
Another concern in this class is the ability of certain members
to induce developmental delays. At present, interpretation of these
results is somewhat clouded by the issues of interactions in the
toxicity of the test compound with its vehicles, as discussed in the
recent publication of Christ et al. (1996). No satisfactory
explanation has been offered for these results. Such effects may be
the result of fairly subtle changes in the pharmacokinetics and
metabolism of the compound, or, as Christ et al. (1996) suggest, TCAN
may have acted synergistically with a subthreshold effect of
tricaprylin on developmental processes. This is suggested by a
minimal, but consistent, response in treatment groups that received
tricaprylin only relative to a group of naive controls.
The potential importance of the HANs as cyanogens has not been
extensively explored. As noted above, Pereira et al. (1984) indicated
that significant portions of the dose of the HANs are eliminated as
thiocyanate in the urine. Thus, cyanide release could be contributing
significantly to the effects of these chemicals at high doses.
4.5 Halogenated hydroxyfuranone derivatives
The halogenated hydroxyfuranones were first identified in the
bleaching of pulp (Holmbom et al., 1984). The first member of this
class, MX, was found because of its high mutagenic activity in
Salmonella tester strains. In the same time frame, mutagenic
activity had been associated with the chlorination of drinking-water
(Meier, 1988). The high specific mutagenic activity of MX prompted
examination of the possibility that it could contribute to the
mutagenic activity that had been identified in chlorinated
drinking-water. Subsequent experimentation confirmed this hypothesis.
Estimates of the contribution of MX to the mutagenic activity in
drinking-water ranged from 3% to 57% (Hemming et al., 1986; Meier et
al., 1987a; Kronberg & Vartiainen, 1988). The lower estimates should
probably be discounted, because the methods for recovering MX and
other mutagens from water have varied. Moreover, it is important to
recognize that these estimates apply only to MX itself. A variety of
related compounds are produced that are also mutagens (Daniel et al.,
1991b; DeMarini et al., 1995; Suzuki & Nakanishi, 1995), but which
have received very little toxicological study. The contribution of
these related compounds has not been estimated.
The present section will focus on the research subsequent to that
which identified MX as an important chlorination by-product. This
review will confine itself to newer data that provide some perspective
on possible mechanisms of action in vivo. To avoid unnecessary
redundancy, other furanones will be discussed only as they have been
investigated to aid in an understanding of responses to MX.
4.5.1 General toxicological properties and information on
dose-response in animals
The lack of a commercial source of MX has limited research in
experimental animals. However, a number of in vivo studies and a
carcinogenesis study of MX have been published (Bull et al., 1995;
Komulainen et al., 1997). Reported epidemiological associations of
drinking-water mutagenicity with cancer of the gastrointestinal and
urinary tracts (Koivusalo et al., 1994b, 1996, 1997) provided
additional impetus for investigating the compound.
Three studies explicitly examined the acute toxicity of MX. The
oral LD50 for MX in Swiss-Webster mice was determined to be 128 mg/kg
of body weight when MX was administered for 2 consecutive days by
gavage (Meier et al., 1987b). Doses of 70% of the LD50 and less
(<90 mg/kg of body weight) had no significant effect on body weight
of the mice, nor were they lethal. Most mice died within 24 h of
receiving the first dose. Mice that died were found to have enlarged
stomachs with moderate haemorrhagic areas in the forestomach. Very
limited mortality was observed in weanling CD-1 mice administered a
single dose of 144 mg/kg of body weight (Mullins & Proudlock, 1990).
In this study, focal epithelial hyperplasia was observed in the
stomach, and some vacuolation of the superficial villus epithelium was
observed in the duodenum and jejunum. Evidence of increased numbers of
mitotic figures was observed in the liver, and the possibility of some
cytotoxicity was identified in the urinary bladder. Komulainen et al.
(1994) administered MX in distilled water to male Wistar rats. Rats
tolerated doses of 200 mg/kg of body weight administered by gavage
but displayed severe symptoms including dyspnoea, laborious breathing,
depressed motor activity and cyanosis at higher doses (Komulainen et
al 1994). At necropsy, gastrointestinal inflammation was observed,
and oedema was noted in the lungs and kidneys. An LD50 of 230 mg/kg
in 48 hours was identified in this study.
The study of Meier et al. (1996) examined the effects of a 14-day
course of MX treatment by gavage at a dose of 64 mg/kg of body weight
per day on a number of enzyme activities in the liver of rats. MX
treatment reduced hepatic levels of catalase, cytochrome P450
reductase, aminopyrine demethylase and aromatic hydrocarbon
hydroxylase. It did not affect fatty acyl CoA oxidase,
glutamylcysteine synthetase, GST or glutathione peroxidase. The main
result of such effects would be potential modifications of metabolism
of various xenobiotics and endogenous biochemicals.
A more extensive study of the effect of MX on enzyme activities
in various tissues was conducted by Heiskanen et al. (1995) in Wistar
rats. This study employed a constant daily dose of 30 mg/kg of body
weight administered by gavage for 18 weeks as the low dose, whereas
the higher dose was achieved by initiating treatment at 45 mg/kg of
body weight (7 weeks) and raising it to 60 mg/kg of body weight
(2 weeks) and to 75 mg/kg of body weight (5 weeks). A dose-related
decrease in ethoxyresorufin- O-deethylase activity was observed in
liver and kidney. MX appears to inhibit this enzyme's activity
directly based upon in vitro experiments conducted by the authors.
However, the high concentrations required (0.9 mmol/litre) are
unlikely to be approached systemically from the very low levels found
in chlorinated drinking-water. The treatment also increased the
activities of two phase 2 enzymes, uridine
diphosphate-glucuronosyltransferase and GST, in the kidneys in a
dose-dependent manner, but only in female rats. The health
consequences of such modifications are uncertain, but could reflect an
effect on physiological mechanisms associated with differences in sex.
Again, it is important to recognize that these effects were produced
at doses that were chronically, as well as acutely, toxic and unlikely
to be remotely approached at the low concentrations found in
chlorinated drinking-water.
In a subchronic (14-18 weeks) toxicity study, Wistar rats (15 per
sex per group) were given MX by gavage, 5 days per week, at doses of 0
or 30 mg/kg of body weight (low dose) for 18 weeks or, in the
high-dose group, at doses increasing from 45 to 75 mg/kg of body
weight over 14 weeks. The high dose was finally lethal (two males and
one female died) and caused hypersalivation, wheezing respiration,
emaciation and tangled fur in animals. Increased water consumption,
decreased body weights and food consumption, elevated plasma
cholesterol and triglycerides, and increased urine excretion were
noted in high-dose male rats. Urine specific gravity was decreased and
the relative weights of the liver and kidneys were increased in both
sexes at both doses in comparison with the controls. At both doses,
duodenal hyperplasia occurred in males and females, and slight focal
epithelial hyperplasia in the forestomach was observed in males.
Splenic atrophy and haemosiderosis were seen in two high-dose females,
and epithelial cell atypia was seen in the urinary bladder of one
high-dose male and female. The frequency of bone marrow polychromatic
erythrocytes with micronuclei was slightly increased only in low-dose
male rats (Vaittinen et al., 1995).
4.5.2 Toxicity in humans
There have been no studies of the effects of these compounds in
humans.
4.5.3 Carcinogenicity and mutagenicity
4.5.3.1 Studies in bacteria and mammalian cells in vitro
There have been extensive studies of the mutagenic activity of MX
and related chemicals. In vitro studies have extended knowledge
beyond the initial characterization of simple mutagenic responses to
(i) demonstrate effects in higher test systems, (ii) extend the data
to other genotoxic end-points, (ii) characterize the mutagenic lesions
produced in DNA and (iv) develop structural correlates. At higher
levels of biological organization, a limited number of studies have
been conducted to document that the genotoxic form of MX reaches the
systemic circulation and to measure mutagenic effects in particular
cell types in vivo.
Meier et al. (1987b) demonstrated that MX induced chromosomal
aberrations in CHO cells at concentrations as low as 4 µg/ml in
vitro. However, these authors were unable to demonstrate an
increased frequency of micronuclei in the bone marrow of Swiss-Webster
mice following two consecutive daily doses administered by gavage at
70% of the LD50 (90 mg/kg of body weight × 2). Treatment with
30-300 µmol of MX per litre (1 h) induced DNA damage in a
concentration-dependent manner in suspensions of rat hepatocytes. DNA
damage was induced in V79 Chinese hamster cells and in isolated rat
testicular cells at the same concentrations as in hepatocytes. V79
cells exposed to 2-5 µmol of MX per litre (2 h) showed an increased
frequency of SCE, whereas no significant effect on
hypoxanthine-guanine phosphoribosyltransferase mutation induction was
observed (Brunborg et al., 1991).
Watanabe et al. (1994) found that the mutagenic activity of MX
was effectively inhibited by sulfhydryl compounds such as cysteine,
cysteamine, GSH, dithiothreitol and 2-mercaptoethanol. Pre-incubation
of 0.5 µg of MX with 15 µg of cysteine in a phosphate buffer at 37°C
for 15 min prior to exposure of bacterial cells depleted the mutagenic
activity of MX. Together with the result showing a change in the UV
spectra, the authors suggested that sulfhydryl compounds inactivate MX
by direct chemical interaction before MX induces DNA damage. On the
other hand, a variety of antioxidants other than the sulfhydryl
compounds showed no inhibitory effects. Investigation using structural
analogues of cysteine revealed that the thiol moiety was indispensable
for antimutagenic activity, and the amino moiety appeared to enhance
the MX-inactivating reaction of the sulfhydryl group.
Incubation of both rat and mouse hepatocytes with MX in vitro
resulted in a dose-dependent increase in UDS at subcytotoxic
concentrations (1-10 µmol of MX per litre; 20-h incubation). Depletion
of GSH stores by pretreatment of rat hepatocytes with buthionine
sulfoximine did not result in a significant increase in UDS produced
by MX. In contrast, MX did not induce UDS in mouse hepatocytes ex
vivo either 3 or 16 h following administration of a single oral dose
of 100 mg of MX per kg of body weight. Despite the ability of MX to
produce repairable DNA damage, restricted access of MX to the liver
may prevent a measurable UDS response in vivo (Nunn et al., 1997).
Jansson et al. (1995) examined MX and a related compound,
3,4-(dichloro)-5-hydroxy-2(5H)-furanone (MA, also known as mucochloric
acid), in the CHO hypoxanthine phosphoribosyl transferase (hprt)
locus assay system where 6-thioguanine resistance (TGr) is the
parameter measured. Both MX and MA induced TGr mutants. Indirect
evidence was provided to suggest that the difference in sensitivity in
the bacterial systems was related to differential ability to repair
bulky adducts hypothesized to be induced by MX versus smaller adducts
suggested to occur as a result of MA treatment. These results are
interesting, considering the fact that Daniel et al. (1991b) found
that MX and MA were of approximately equivalent potencies in inducing
nuclear anomalies in gastrointestinal cells of the B6C3F1 mouse.
Harrington-Brock et al. (1995) recently examined MX in the
L5178/TK+1C3.7.2C mouse lymphoma system. A mutant frequency of 1027
per 106 surviving cells was found. There was, however, a predominance
of small-colony mutants. Small colonies more commonly arise from
clastogenic effects than from point mutations. In parallel
experiments, MX was found to have clastogenic effects (chromatid
breaks and rearrangements). Point mutations and clastogenic effects
were both observed at a medium concentration of 0.75 µg/ml.
MX and MA also induced micronuclei when applied to inflorescences
of pollen mother cells of Tradescantia (Helma et al., 1995). MX was
approximately 5 times as potent as MA in this assay system.
DeMarini et al. (1995) compared the mutation spectra induced by
MX and extracts of water treated with chlorine, chloramine, ozone or
ozone followed by chlorine or chloramine in Salmonella typhimurium
strains TA98 and TA100. The mutation spectra induced in the hisG46
codon displayed a predominance of the GAC mutation with MX, but there
were also significant increases in the CTC and to a lesser extent ACC
mutations. These latter mutations are not typical of MX. Since MX has
never been identified as a by-product of ozonation, it is somewhat
surprising that extracts of ozonated water produced a similar
spectrum, even though these extracts are much less potent than those
obtained from chlorinated or chloraminated water (i.e., they produced
net increases in mutant colonies that are only about twice the
spontaneous rate). The mutation spectra induced in the TA98
hisD3052 allele more clearly differentiated between ozone and
chlorinated or chloraminated water. In this case, virtually all of the
frameshift mutations induced by raw and ozonated water extracts
involved hotspot mutations, whereas only 30-50% of those induced by MX
or extracts from water that had been treated with chlorine or
chloramine involved the hotspot. Thus, the TA98 mutations are
consistent with the hypothesis that the chemicals responsible for
mutagenic effects of ozonated water are distinct from those induced by
chlorination by-products.
Hyttinen et al. (1996) found that MX and MA induce different
mutation spectra in the DNA of Salmonella typhimurium hisG46 codons
(target codon sequence is CCC). The predominant mutation was GAC
followed by ACC in MX-treated colonies, whereas CTC dominated the
spectra produced by MA. MX primarily induced G:C -> T:A
transversions, whereas MA produced G:C -> A:T transitions in the
second base of the codon. The G:C -> T:A transversion in
Salmonella was also observed in the hprt gene of CHO cells
(Hyttinen et al., 1996). Knasmuller et al. (1996) found the same
difference in the mutation spectra of MX and MA in S. typhimurium
mutants. These latter authors further found that
3-chloro-4-(chloromethyl)-5-hydroxy-2(5H)-furanone (CMCF) produced the
same transversion as MX, while chloromalonaldehyde produced the same
transition found with MA.
Some complex structure-activity relationships appear to occur
with these two types of halofuranones. Kronberg et al. (1993) found
that MA forms ethenocarbaldehyde derivatives with adenosine and
cytidine, with chloroacetaldehyde being an intermediate. As pointed
out by Knasmuller et al. (1996), ethenocytosine adducts formed by
vinyl chloride cause G:C -> A:T transitions as reported for MA. The
mutations induced by MX are similar to those produced by carcinogens
that form bulky adducts, such as benzo [a]pyrene and 4-aminobiphenyl.
Bulky adducts block replication leading to base substitutions
according to the "adenine rule" (Strauss, 1991). As a consequence,
there may be some significant differences in the health impact (e.g.,
tumour site or character if they are found to be carcinogenic) of the
mutagenic activities of these two chlorohydroxyfuranone derivatives.
Ishiguro et al. (1988) examined structure-activity relationships
for MX and related compounds. Their studies identified the chlorine
substitution on C3 as being very important to the mutagenic activity
of MX. Association of similar losses in mutagenic activity by removing
the analogous chlorine from an open-ring structure compound,
3-(dichloromethyl)-4,4-dichloro-2-chlorobutenoic acid, strongly
supported this hypothesis.
LaLonde and co-workers (LaLonde et al., 1991a,b, 1992; LaLonde &
Xie, 1992, 1993) conducted a series of experimental and computational
studies to relate the electronic structure of MX and related compounds
with mutagenic activity within the class. Substitutions for the
hydroxyl group led to reduction of mutagenic activity by a factor of
100. Removal of the C3 or C6 chlorines from the structure reduced
mutagenic activity by a factor of 10. Removal of the second chlorine
at C3 caused a very large further reduction of mutagenic activity (by
a factor of 1000) (LaLonde et al., 1991b). The mutagenicity appears to
depend on the electron density at C2, C3 or C4 based upon 13C
chemical shifts observed by nuclear magnetic resonance. Expansion of
these studies supported the hypothesis that the mutagenic properties
of the class paralleled the electrophilic character of chemicals
within the class and the ability to stabilize a radical anion
following acceptance of a single electron.
Another difference in the chemistry of MA and MX, which might
have biological implications, is that GSH readily displaces the
chlorine on C4 of MA, greatly reducing its electrophilicity. On the
other hand, GSH or N-acetylcysteine reacts with MX to produce
mixtures that are intractable to analysis, with the release of
hydrogen sulfide (LaLonde & Xie, 1993).
4.5.3.2 Studies in experimental animals
In vivo studies are of two types: those that use bacterial
systems to document absorption of MX (or mutagenic metabolite), and
those in which effects on the test animal are directly measured.
Fekadu et al. (1994) injected mixtures of repair-competent and
repair-deficient Escherichia coli K-12 cells intravenously into mice
as test cells, and the animals were subsequently treated with 200 mg
of test chemical per kg of body weight. Two hours later, the mice were
sacrificed and cells recovered from various organs. MX, CMCF and MA
were the test chemicals. The differential survival of the DNA
repair-deficient strain versus a repair-competent variant is used to
detect mutagenic activity. All three compounds significantly reduced
recovery of the repair-deficient strain in the stomach, lung,
intestine, liver, kidney and spleen. In a further experiment, the
effects of lower doses of MX (4.3, 13 and 40 mg/kg of body weight)
were investigated. Significantly depressed recovery was seen with MX
doses as low as 4.3 mg/kg of body weight. MA did not modify recovery
of the repair-deficient strain at doses less than or equal to 40 mg/kg
of body weight. These data suggest that significant amounts of MX or a
mutagenic metabolite reach the systematic circulation and at least
reach the extracellular water. They do not clearly demonstrate effects
in the target tissue of the experimental animal.
Meier et al. (1996) found that only 0.3% of the original dose was
excreted in a genotoxically active form in the urine of rats
administered MX at a dose of 64 mg/kg of body weight for 14 days by
gavage. No evidence of micronuclei induction was detected in
peripheral blood erythrocytes in mice treated with a similar protocol.
Whereas mutagenic activity was observed in urine at doses of 64 mg/kg
of body weight, no significant mutagenic activity was observed at
doses of 32 mg/kg of body weight and below.
Brunborg et al. (1990, 1991) studied DNA damage induced by MX and
other compounds in organs of rats using the alkaline elution assay (to
detect strand breaks). While clear evidence of strand breaks was
obtained with dibromochloropropane and
2-amino-3,4-dimethylimidazo[4,5- f]quinoline, no significant effects
were observed with MX after an intraperitoneal dose of 18 mg/kg of
body weight or at oral doses of up to 125 mg/kg of body weight. The
organs examined included the small and large intestine, stomach,
liver, kidney, lung, bone marrow, urinary bladder and testis.
Nishikawa et al. (1994) investigated cell proliferation and lipid
peroxidation in the glandular stomach mucosa in Wistar rats given 0,
6.25, 12.5, 25 or 50 mg of MX per litre in their drinking-water for
5 weeks. Statistically significant cell proliferation increased in a
dose-dependent manner up to 25 mg/litre. The MX treatment was also
associated with increased lipid peroxidation levels in the gastric
mucosa as well as in the urine, with loose dose dependence, although
not at 50 mg/litre. Histopathologically, gastric erosion was noted in
rats receiving 25 mg of MX per litre or more. These results suggest
that MX may exert a gastric tumour-promoting action in rats, even at
low doses that do not give rise to toxic effects, because of the clear
dose-response relationship evident at low levels.
The peripheral lymphocytes of male and female Han:Wistar rats
exposed to MX at 30 or 45-75 mg/kg of body weight per day by gavage, 5
days a week for 14-18 weeks, showed significant dose-related increases
in SCEs at both levels of exposure in both sexes (Jansson et al.,
1993).
The peripheral lymphocytes of male Han:Wistar rats exposed to MX
(25-150 mg/kg of body weight) by gavage on 3 consecutive days showed a
significant dose-related increase in chromosomal damage measured as
micronuclei, in addition to SCEs. Moreover, MX produced a significant
dose-related increase in SCEs in the kidney cells of the exposed rats.
However, the magnitude of the genotoxic responses observed was
relatively weak (Maki-Paakkanen & Jansson, 1995).
Daniel et al. (1991b) found that MX and MA induced nuclear
anomalies in the epithelial cells of the gastrointestinal tract of
B6C3F1 mice. Doses of 0.37 mmol/kg of body weight (approximately 80
mg/kg of body weight) produced a modest increase in nuclear anomalies
in the duodenum. There was no effect at 0.28 mmol/kg of body weight.
Mullins & Proudlock (1990) and Proudlock & Crouch (1990) also found
insignificant increases in nuclear anomalies in the non-glandular
stomach, urinary bladder, jejunum and ileum. These latter authors
noted that at the top dose at which nuclear anomalies were observed
(144 mg/kg of body weight), there was significant irritation,
inflammation and evidence of apoptotic cells in the gastrointestinal
tract. These changes render the significance of the observed nuclear
anomalies uncertain.
MX was administered to Wistar rats (50 per sex per group) in
drinking-water for 104 weeks at 0, 0.4, 1.3 or 5.0 mg/kg of body
weight per day for males and 0, 0.6, 1.9 or 6.6 mg/kg of body weight
per day for females. Dose-dependent increases in the incidence of some
tumours were observed in rats, while the same MX doses had no obvious
toxic effects on animals. Increases in tumours of the lung, mammary
gland, haematopoietic system, liver, pancreas, adrenal gland and
thyroid were observed, but few showed a clear dose-response (Table 21)
(Komulainen et al., 1997).
4.5.4 Comparative pharmacokinetics and metabolism
There are very few data on the metabolism and pharmacokinetics of
MX or related compounds. Ringhand et al. (1989) examined the
distribution of radioactivity derived from 3-14C-MX in male F344
rats. Approximately 35% of the radiolabel was eliminated in the urine
and 47% in the faeces, with about 6% remaining in the body after 48 h.
Neither the parent compound nor any specific metabolites were
identified in any body compartment or fluid.
Horth et al. (1991) studied the disposition of 3-14C-MX in male
CD-1 mice. 14C was rapidly absorbed, reaching peak values in blood
within 15 min of its administration. There was some evidence for
binding to protein and retention of label within tissues, but no
attempt was made to identify the chemical form in which the 14C was
bound, so it was not clear whether MX was binding by virtue of its
electrophilic character or whether this represented metabolic
incorporation of metabolites of MX. Approximately 57% of the
radioactivity was eliminated in the urine and 28% in the faeces. Less
than 1% of the initial dose was retained in the carcass 120 h after
administration, but most of this was associated with the stomach. It
was stated that the urinary metabolites were polar, but no specific
identifications were made.
Komulainen et al. (1992) evaluated the pharmacokinetics of MX
after a single oral or intravenous administration in Han:Wistar rats
using 14C-labelled compound. Approximately 20-35% of the dose was
absorbed into circulation from the gastrointestinal tract. The mean
elimination half-life of the radioactivity in blood was 3.8 h. Traces
of radioactivity remained in the blood for several days. The tissues
lining the gastrointestinal and urinary tracts, kidney, stomach, small
intestine and urinary bladder contained the highest radioactivity. The
activity declined most slowly in the kidneys. Urine was the main
excretion route, with 77% of the total radioactivity appearing in
urine in 12 h and 90% in 24 h. No radioactivity was exhaled in air.
After an intravenous administration of 14C-MX, the mean elimination
half-life was much longer, 22.9 h, and the total elimination half-life
was 42.1 h. Results indicate that MX is absorbed from the
gastrointestinal tract to a considerable degree and is excreted in
urine very rapidly. A fraction of MX or its metabolites is retained in
blood for a longer period of time.
No data are available in the scientific literature on the
metabolism of MX or related compounds in humans.
In conclusion, there are data to suggest that MX or a
mutagenically active metabolite reaches the systemic circulation in
experimental animals. Mutagenic activity has been detected in various
organs and tissues using doses as low as 4.3 mg/kg of body weight
(Fekadu et al., 1994). If these data are to have application to
estimation of the hazards that MX presents to humans consuming
chlorinated drinking-water, it is essential to understand whether MX
or a metabolite reaches critical targets in the human body. An
essential component of the information required would be an
understanding of the metabolism and pharmacokinetics of MX and those
of critical metabolites. The available data are too limited to provide
much more than very general guidance in this area.
4.6 Chlorite
4.6.1 General toxicological properties and information on
dose-response in animals
Concerns over chlorite in drinking-water first arose as chlorine
dioxide began to play a role in the primary disinfection of
drinking-water. Chlorite is the principal by-product of oxidative
reactions of chlorine dioxide, but acidification of chlorite solutions
is one method for generating chlorine dioxide for purposes of water
disinfection (Aieta & Berg, 1986).
Unless otherwise noted, references to chlorite will generally be
to the sodium salt. This is the form most frequently studied. The term
chlorite will be used if the authors expressed their doses in terms of
chlorite; if dose levels were expressed as sodium chlorite, they will
be identified as such. There is no reason to suspect that other salts
of chlorite would exert inherently different toxicological effects, so
this convention should not lead to confusion. A preparation of sodium
chlorite with lactic acid is specifically excluded from consideration
here because the actual composition of the product has not been
specified (Scatina et al., 1984). Consequently, it is not clear that
the data on this product have any relevance to chlorite or vice versa.
Table 21. Summary of primary tumours observed in selected tissues in male rats after exposure to
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX) in drinking-water for 104 weeksa
Tissue Control MX (mg/kg of body weight per day) Pb
0.4 1.3 5.0
Integumentary system
Skin, subcutaneous tissuec 50 50 50 50
Basal cell tumourd 1 (2%) 1 (2%) 3 (6%) 0.0314
Mammary glandsc 50 50 50 49
Adenocarcinomad 1 (2%) 0.3162
Fibroadenomad 1 (2%) 3 (6%) 1 (2%) 0.2996
Fibromad 1 (2%) 0.4749
Respiratory system
Lungsc 50 50 50 50
Alveolar & bronchiolar carcinomasd 1 (2%)
Alveolar & bronchiolar adenomasd 2 (4%) 1 (2%) 1 (2%) 7 (14%) 0.0015
Haematopoietic system
Multiple tissuesc 50 50 50 50
Lymphoma & leukaemiad 3 (6%) 4 (8%) 3 (6%) 0.1527
Digestive system
Liverc 50 50 50 50
Carcinomad 2 (4%) 1 (2%) 0.1605
Hepatocholangiocarcinomad 1 (2%) 0.4897
Cholangiomad 1 (2%) 4 (8%) 0.0009
Adenomad 1 (2%) 2 (4%) 4 (8%) 0.0142
Pancreasc 50 50 50 50
Langerhans' cell carcinomad 4 (8%) 3 (6%) 5 (10%) 4 (8%) 0.3769
Langerhans' cell adenomad 5 (10%) 8 (16%) 8 (16%) 12 (24%) 0.0116
Acinar cell adenomad 2 (4%) 3 (6%) 2 (4%) 4 (8%) 0.1243
Table 21. (continued)
Tissue Control MX (mg/kg of body weight per day) Pb
0.4 1.3 5.0
Endocrine system
Adrenal glandsc 50 50 50 50
Pheochromocytoma, malignantd 1 (2%) 0.3213
Pheochromocytoma, benignd 5 (10%) 2 (4%) 9 (18%) 3 (6%) 0.4830
Cortical carcinomad 2 (4%) 1 (2%) 0.9447
Cortical adenomad 5 (10%) 2 (4%) 7 (14%) 14 (28%) 0.0001
Thyroid glandsc 49 50 50 50
Follicular carcinomad 1 (2%) 9 (18%) 27 (55%) 0.0000
Follicular adenomad 2 (4%) 20 (40%) 34 (68%) 21 (43%) 0.0045
C-cell carcinomad 2 (4%) 0.4478
C-cell adenomad 11 (22%) 7 (14%) 10 (20%) 11 (22%) 0.2459
a From Komulainen et al. (1997).
b P value from the one-sided trend test. A statistically positive trend at P < 0.05 or lower.
c Values (reading across) = number of animals analysed.
d Values (reading across) = number of animals with one or more indicated tumours (frequency of animals
with tumour as percentage of examined animals.
Early investigations of chlorite's toxic properties focused
almost entirely on it potential ability to produce methaemoglobin and
haemolysis. Heffernan et al. (1979a) examined the ability of sodium
chlorite to induce methaemoglobinaemia in cats and Sprague-Dawley
rats. When administered to cats as an oral bolus, as little as 20 mg
of sodium chlorite per kg of body weight resulted in formation of
significant amounts of methaemoglobin. Intraperitoneal doses of
20 mg/kg of body weight in rats also induced methaemoglobin formation.
However, when administered in drinking-water, no significant elevation
in methaemoglobin was observed in cats (up to 1000 mg/litre as sodium
chlorite) or rats (up to 500 mg/litre). Thus, chlorite must enter into
the systemic circulation at a rapid rate, i.e., as a bolus dose, to
induce methaemoglobin formation.
Short-term toxic effects of chlorite were more systematically
assessed in rats using gavage doses ranging from 25 to 200 mg/kg of
body weight (Harrington et al., 1995a). Minor effects were observed at
25 and 50 mg/kg of body weight. At 100 mg/kg of body weight and above,
signs of haemolytic anaemia became apparent, with decreases in red
blood cell count, haemoglobin concentration and haematocrit. These
data support the idea that bolus doses were necessary to determine
substantial effects on oxidative stress.
Treatment of both cats and rats with sodium chlorite in
drinking-water for extended periods (up to 90 days) resulted in
decreases in red blood cell counts, haemoglobin concentrations and
packed cell volume. These effects were observed with 500 mg of sodium
chlorite per litre in cats (equivalent to 7 mg/kg of body weight per
day) and with as little as 100 mg/litre in rats (equivalent to 10
mg/kg of body weight per day) (Heffernan et al., 1979a). The changes
in these blood parameters appeared to generally decrease in severity
as the treatment was extended from 30 to 90 days, suggesting that
adaptation to the treatment was occurring. In rats, however, red blood
cell glutathione concentrations remained significantly depressed and
2,3-diphosphoglycerate levels elevated through 90 days of treatment
with concentrations of sodium chlorite as low as 50 mg/litre (5 mg/kg
of body weight per day). In the cat, increased turnover of
erythrocytes was detectable at concentrations of 100 mg/litre, with no
significant effect being observed at 10 mg/litre. This latter
concentration resulted in a daily dose of 0.6 mg/kg of body weight per
day. Red blood cells drawn from rats treated with 100 mg/litre had
significantly less ability to detoxify hydrogen peroxide that was
generated by the addition of chlorite in vitro. These data show that
while the anaemia caused by haemolysis was largely compensated for in
subchronic treatment of healthy rats, there was still evidence of
oxidative stress being exerted by the chlorite treatment. Depletion of
this reserve capacity could be of importance in individuals who are in
a more compromised state (e.g., glucose-6-phosphate dehydrogenase
deficiency) or who might be exposed to other haemolytic agents. The
lowest concentration at which GSH was depleted significantly from
control levels in rats was 50 mg/litre, and no effect was observed at
10 mg/litre (equivalent to 1 mg/kg of body weight per day).
The results of Heffernan et al. (1979a) have been generally
confirmed by subsequent studies in a variety of species. Abdel-Rahman
et al. (1980) and Couri & Abdel-Rahman (1980) obtained very similar
effects in rats treated for up to 11 months. Moore & Calabrese (1980,
1982) produced similar results in mice, and Bercz et al. (1982)
demonstrated reduced red blood cell counts and decreased haemoglobin
levels at similar doses in African green monkeys. These studies tended
to identify altered forms of erythrocytes that are commonly associated
with oxidative damage at treatment doses below those that produced
actual anaemia (most consistently at a concentration of 100 mg/litre,
with hints of such effects at lower doses).
A more recent study employed doses of sodium chlorite
administered by gavage to male and female Crl: CD (SD) BR rats (15 per
sex per group) (Harrington et al., 1995a). Doses of 0, 10, 25 or 80 mg
of sodium chlorite per kg of body weight per day were administered
daily by gavage for 13 weeks (equivalent to 0, 7.4, 18.6 or 59.7 mg of
chlorite per kg of body weight per day). This study is important
because it included many of the standard parameters of subchronic
toxicological studies, whereas previous studies had focused almost
entirely on blood parameters. A gavage dose of 80 mg/kg of body weight
per day produced death in a number of animals. It also resulted in
morphological changes in erythrocytes and significant decreases in
haemoglobin concentrations. Red blood cell counts were reduced
slightly, but not significantly, at doses of 10 mg/kg of body weight
per day in male rats, with further decreases being observed at 80
mg/kg of body weight per day. Red blood cell counts were significantly
depressed in female rats at doses of 25 mg/kg of body weight per day
and above. As would be expected where haemolysis is occurring, splenic
weights were increased. Adrenal weights were increased in females at
25 and 80 mg/kg of body weight per day, whereas statistically
significant changes were observed only at 80 mg/kg of body weight per
day in males. Histopathological examination of necropsied tissues
revealed squamous cell epithelial hyperplasia, hyperkeratosis,
ulceration, chronic inflammation and oedema in the stomach of 7 out of
15 males and 8 out of 15 females given 80 mg/kg of body weight per day
doses. This effect was observed in only 2 out of 15 animals at the 25
mg/kg of body weight per day dose and was not observed at all at 10
mg/kg of body weight per day. Microscopic evaluations were made in 40
additional tissues, and no treatment-related abnormalities were found.
The Harrington et al. (1995a) study confirms the essential
findings of previous studies and, in retrospect, justifies their focus
on the blood cells as the critical target for chlorite toxicity. It
also confirmed negative results of other studies that failed to
identify significant effects in investigations of particular target
organs (Moore et al., 1984; Connor et al., 1985).
4.6.2 Reproductive and developmental toxicity
Sodium chlorite did not exert any spermatotoxic effects in
short-duration (1-5 days) tests (Linder et al., 1992).
Moore et al. (1980b) reported that sodium chlorite administered
at a concentration of 100 mg/litre throughout gestation and through
28 days of lactation reduced the conception rate and the number of
pups alive at weaning in A/J mice. A significantly reduced pup weight
at weaning was interpreted as indicating that chlorite retarded growth
rate.
Groups of 4-13 Sprague-Dawley rats were treated on gestation days
8-15 with sodium chlorite at concentrations of 0, 100, 500 or 2000
mg/litre in drinking-water, by injection of 10, 20 or 50 mg/kg of body
weight per day intraperitoneally or by gavaging with 200 mg/kg of body
weight per day. Calculated daily doses of sodium chlorite administered
to pregnant rats in drinking-water were 0, 34, 163 or 212 mg. Rats
body weights were approximately 0.3 kg, giving estimated doses of 0,
110, 540 or 710 mg/kg of body weight per day. Sodium chlorite at 20 or
50 mg/kg of body weight per day intraperitoneally or at 200 mg/kg of
body weight per day by gavage caused vaginal and urethral bleeding.
Doses of 10, 20 and 50 mg/kg of body weight per day intraperitoneally
caused 0%, 50% and 100% mortality of dams, respectively. No deaths
were caused by sodium chlorite in the drinking-water, but the body
weight and food consumption of the dams were decreased at 500 and 2000
mg/litre. Blood smears from the dams injected intraperitoneally with
all doses or drinking water containing 2000 mg of sodium chlorite per
litre showed irregular, bizarre and ruptured erythrocytes. Injection
of 10 or 20 mg/kg of body weight per day or drinking a solution
containing 2000 mg/litre resulted in a decrease in litter size and an
increase in stillbirths and resorption sites. Drinking 100 or 500 mg
of sodium chlorite per litre did not produce any significant
embryotoxicity. With all treatments, no significant gross soft tissue
or skeletal malformations were observed. Postnatal growth of the pups
was not affected by any treatment of the dams during the gestation
period (Couri et al., 1982a,b).
The effects of chlorite at 1 or 10 mg/litre in drinking-water for
2.5 months prior to mating and throughout gestation were studied in
Sprague-Dawley rats (Suh et al., 1983). This study indicated an
increase in the incidence of anomalies in fetuses at both
concentrations in two separate experiments; however, because the
treatment groups were small (6-9 pregnant females per group), the
effects were not considered statistically significant. Moreover, there
were no consistent differences in either skeletal or soft tissue
anomalies.
Male and female Long-Evans rats were given 0, 1, 10 or 100 mg of
sodium chlorite per litre of drinking-water. Males were exposed for 56
days before mating and during 10 days of mating; females were treated
for 14 days prior to mating, throughout the 10-day breeding period and
gestation and through to day 21 of lactation. Males were evaluated for
sperm parameters and reproductive tract histopathology following the
breeding period. Dams and pups were necropsied at weaning. There was
no effect on fertility, litter size or survival of neonates or on the
weight of the testis, epididymis or cauda epididymis when males
treated as described above were mated with these females. Decreases in
the concentrations of triiodothyronine and thyroxine in blood were
observed on postnatal days 21 and 40 in male and female pups exposed
to 100 mg/litre. There were no effects at lower doses. Additionally,
groups of males were exposed to 0, 10, 100 or 500 mg of sodium
chlorite per litre for 72-76 days to confirm subtle observed changes
in sperm count, morphology and movement. A significant increase in the
percentage of abnormal sperm morphology and a decrease in the
progressive sperm motility were observed for adult males at 100 and
500 mg/litre (Carlton et al., 1987).
Mobley et al. (1990) exposed groups of female Sprague-Dawley rats
(12 per group) for 9 weeks to drinking-water containing 0, 20 or 40 mg
of sodium chlorite per litre (0, 3 or 6 mg of chlorite per kg of body
weight per day) beginning 10 days prior to breeding with untreated
males and until the pups were sacrificed at 35-42 days
post-conception. Animals exposed to a dose of 6 mg/kg of body weight
per day exhibited a consistent and significant depression in
exploratory behaviour on post-conception days 36-39. Exploratory
activity was comparable between treated and control groups after
post-conception day 39.
In a two-generation study conducted by CMA (1997) and described
in TERA (1998), Sprague-Dawley rats (30 per sex per dose) received
drinking-water containing 0, 35, 70 or 300 mg of sodium chlorite per
litre for 10 weeks and were then paired for mating. Males were exposed
through mating, then sacrificed. Exposure for the females continued
through mating, pregnancy, lactation and until necropsy following
weaning of their litters. Twenty-five males and females from each of
the first 25 litters to be weaned in a treatment group were chosen to
produce the F1 generation. The F1 pups were continued on the same
treatment regimen as their parents. At approximately 14 weeks of age,
they were mated to produce the F2a generation. Because of a reduced
number of litters in the 70 mg/litre F1-F2a generation, the F1
animals were remated following weaning of the F2a generation to
produce the F2b generation. Doses for the F0 animals were 0, 3.0,
5.6 or 20.0 mg of chlorite per kg of body weight per day for males and
0, 3.8, 7.5 or 28.6 mg of chlorite per kg of body weight per day for
females. For the F1 animals, doses were 0, 2.9, 5.9 or 22.7 mg of
chlorite per kg of body weight per day for males and 0, 3.8, 7.9 or
28.6 mg of chlorite per kg of body weight per day for females. There
were reductions in water consumption, food consumption and body weight
gain in both sexes in all generations at various times throughout the
experiment, primarily in the 70 and 300 mg/litre groups; these were
attributed to a lack of palatability of the water. At 300 mg/litre,
reduced pup survival, reduced body weight at birth and throughout
lactation in F1 and F2, lower thymus and spleen weights in both
generations, lowered incidence of pups exhibiting a normal righting
reflex, delays in sexual development in males and females in F1 and
F2, and lower red blood cell parameters in F1 were noted.
Significant reductions in absolute and relative liver weights in F0
females and F1 males and females, reduced absolute brain weights in
F1 and F2, and a decrease in the maximum response to an auditory
startle stimulus on postnatal day 24 but not at postnatal day 60 were
noted in the 300 and 70 mg/litre groups. Minor changes in red blood
cell parameters in the F1 generation were seen at 35 and 70 mg/litre,
but these appear to be within normal ranges based on historical data.
The NOAEL in this study was 35 mg/litre (2.9 mg/kg of body weight per
day), based on lower auditory startle amplitude, decreased absolute
brain weight in the F1 and F2 generations, and altered liver weights
in two generations.
Harrington et al. (1995b) examined the developmental toxicity of
chlorite in New Zealand white rabbits. The rabbits (16 per group) were
treated with 0, 200, 600 or 1200 mg of sodium chlorite per litre in
their drinking-water (equal to 0, 10, 26 or 40 mg of chlorite per kg
of body weight per day) from day 7 to day 19 of pregnancy. The animals
were necropsied on day 28. There were no dose-related increases in
defects identified. Minor skeletal anomalies were observed as the
concentration of chlorite in water was increased and food consumption
was depressed.
4.6.3 Toxicity in humans
The effects of chlorite have received some attention in
toxicological and epidemiological investigations in human subjects.
All of these studies were conducted at doses within an order of
magnitude of concentrations of chlorite that might be expected in
water supplies disinfected with chlorine dioxide. None pushed the
limit of tolerance such that clear effects were observed. As a
consequence, they are not informative for establishing a margin of
safety.
An experimental epidemiological study was conducted in the USA in
a small city that had been using chlorine dioxide for some time in the
summer months (April to October) to avoid taste and odour problems
associated with the use of chlorine (Michael et al., 1981). Chlorine
dioxide was generated from sodium chlorite that was mixed with
chlorine gas and metered into the water. During the active use of
chlorine dioxide, the chlorite concentrations in the water averaged
5.2 mg/litre (range about 3-7 mg/litre). Subjects were monitored for
11 parameters: haematocrit, haemoglobin, red cell count, white cell
count, mean corpuscular volume, methaemoglobin, BUN, serum creatinine,
total bilirubin, reticulocyte count and osmotic fragility of red blood
cells. No effects could be associated with the switch of treatment
from chlorine to chlorine dioxide disinfection. A total of 197 people
were monitored in the exposed population, and there were 112
non-exposed individuals. Each person served as his/her own control.
Chlorine dioxide, free chlorine, chloramine and chlorate
concentrations were also measured and were found to be 0.3-1.1,
0.5-0.9, 0.9-1.8 and 0.3-1.8 mg/litre, respectively. The sampling for
clinical measurements was done 1 week before chlorine dioxide
disinfection began and 10 weeks into the cycle. Water samples taken
during weeks 10-13 had chlorite levels that were systematically
somewhat below those observed in the prior 9 weeks of sampling, and
the same general trend was observable in other measures of chlorine
dioxide and chlorate. This was not observed with chlorine or
chloramines, suggesting that some change in water treatment had
occurred. The authors provided no explanation for this change in water
quality, but, since clinical samples were taken in week 10, this
change in water quality could have resulted in lower exposure to
chlorite and chlorate.
The second set of evaluations of chlorite in humans involved
direct administration of sodium chlorite in a rising-dose tolerance
study and a follow-up study in which volunteers were treated for 12
weeks, which were reported on in several publications. Lubbers et al.
(1981, 1982) provided an overview of the studies. The detailed results
of the rising-dose tolerance study were reported in Lubbers &
Bianchine (1984), and those of the repeated-dose study in Lubbers et
al. (1982, 1984a). A fourth paper (Lubbers et al., 1984b) reported
results for three male volunteers that had glucose-6-phosphate
dehydrogenase deficiency.
The rising-dose tolerance study (Lubbers & Bianchine, 1984)
involved administration of progressively increasing single doses of
chlorite (0.01, 0.1, 0.5, 1.0, 1.8 or 2.4 mg/litre) in two 500-ml
portions to a group of 10 healthy adult male volunteers. Doses were
administered on days 1, 4, 7, 10, 13 and 16. In the interval between
doses, clinical evaluations of the subjects were performed and a
battery of clinical chemistry tests was performed on blood and urine
samples. These latter tests were primarily directed at potential
haematological effects of chlorite, but serum thyroxine and uptake of
triiodothyronine were also determined. In addition, blood pressure,
ECGs and other physiological parameters were monitored. No
treatment-related effects were observed.
In the repeated-dose study (Lubbers et al., 1984a), 10 male
volunteers were administered 5 mg of chlorite per litre in a 500-ml
portion for 12 weeks (0.036 mg/kg of body weight per day). Physical
examinations and blood and urine analyses were conducted throughout
the duration of treatment and for 8 weeks following the last dose of
the solutions. None of the parameters investigated was found to fall
outside the normal range; although there were some consistent changes
in values with time, none of these appeared to be related to chlorite
treatment.
Three individuals with glucose-6-phosphate dehydrogenase
deficiency were identified in the course of the study. This genetic
disorder makes individuals more sensitive to oxidative damage, which
is frequently manifested as increased methaemoglobin production and
haemolysis when the individuals are exposed to oxidative chemicals in
sufficient doses. All three individuals were treated with chlorite in
the same concentrations and in the same manner as described for the
study of normal individuals (Lubbers et al., 1984b). No clinically
significant changes were found in these individuals.
A study (Ames & Stratton, 1987) was conducted of renal dialysis
patients in California (USA) after a water district introduced
chlorine dioxide as a drinking-water disinfectant but failed to inform
the clinic for 12 months. Water treatment at the clinic consisted of
ion exchange, GAC, 5-µm filtration and reverse osmosis. Chlorite
levels measured after this treatment were 0.02-0.08 mg/litre, but
there were periods during which no chlorine dioxide was added, and
exposures to the patients may have been lower. Measures for 28 serum
and haematological parameters were available for 17 renal dialysis
patients for a period of 3 months before and 1 month after exposure.
Methaemoglobin measures were not available. Only one measure was
statistically associated with the use of water disinfected by chlorine
dioxide: serum uric acid declined by 10% after exposure to disinfected
water, a change that was not considered clinically important. The
study found no evidence of anaemia or other adverse effects of
chlorine dioxide-disinfected water for these renal dialysis patients,
but the interpretation of these results is severely limited because of
the small sample size and apparently very low exposures.
Collectively, these studies suggest that humans are probably not
sensitive to the concentrations of chlorite that are likely to be
found in water disinfected with chlorine dioxide. Some safety factor
is present in these data, because it is unlikely that concentrations
of chlorite would exceed 1 mg/litre with new methods of application.
However, these studies provide little information relative to the
actual margin of safety that exists between those concentrations seen
or administered and concentrations that would lead to clear adverse
effects. Consequently, these studies do not imply that the
concentrations of chlorite in drinking-water should be without limits.
4.6.4 Carcinogenicity and mutagenicity
Sodium chlorite was reported to produce a concentration-dependent
increase in revertants in Salmonella typhimurium strain TA100 in
both the presence and absence of rat liver S9 fraction (Ishidate et
al., 1984). A linear dose-response curve was observed, and the net
number of revertants produced at 0.3 mg per plate was 88. The S9 mix
used for metabolic activation was from the liver of F344 rats
pretreated for 5 days with polychlorinated biphenyls at 500 mg/kg of
body weight.
Meier et al. (1985b) evaluated chlorite in the mouse micronucleus
assay, the mouse bone marrow cytogenetics assay and the mouse sperm
head abnormality assay. The doses administered were 0.2, 0.5 or 1 mg
per mouse or approximately 40 mg/kg of body weight at the highest
dose. No statistically significant results were found in any of the
tests. In a later reference, it was indicated that chlorite also
induced chromosomal aberrations (Kurokawa et al., 1986b), but the data
were not provided. Hayashi et al. (1988, 1989) found an increase in
micronuclei in the bone marrow of mice given 0, 7.5, 15, 30 or 60
mg/kg of body weight by intraperitoneal injection at doses of 15 and
30 mg/kg of body weight. In a repeat study in which mice were given 0
or 15 mg/kg of body weight on 4 successive days, no increase in
micronuclei was observed. In a study using the oral route with doses
of 0, 37.5, 75, 150 or 300 mg/kg of body weight, a significant
increase in micronuclei was observed only at 150 mg/kg of body weight.
In a carcinogenicity study, sodium chlorite was administered to
F344 rats (50 per sex per dose) at concentrations of 0, 300 or
600 mg/litre of drinking-water (equivalent to 0, 18 or 32 and 0, 28 or
41 mg of chlorite per kg of body weight per day in males and females,
respectively) and to B6C3F1 mice (50 per sex per dose) at
concentrations of 250 or 500 mg/litre (equivalent to 0, 36 or 71 mg/kg
of body weight per day) for 85 weeks (Kurokawa et al., 1986b). The
rats became infected with a Sendai virus in all groups, which resulted
in the termination of the study after only 85 weeks. There was a
statistically significant increase in the incidence of hyperplastic
nodules in male mice treated with 250 mg/litre, but not in females.
The incidence of these lesions did not increase when the dose of
chlorite was increased to 500 mg/litre. Hepatocellular carcinomas were
too few for their observation to add anything substantive to the
evaluation. There were no other treatment-related changes in the
incidence of other tumours in either male or female mice.
Groups of 50 male and female B6C3F1 mice were given 0, 250 or
500 mg of sodium chlorite per litre in the drinking-water for 80 weeks
(0, 36 or 71 mg of chlorite per kg of body weight per day). A small,
but statistically significant ( p < 0.05), increase in the incidence
of lung adenomas was observed at 500 mg/litre. The authors noted that
this was not accompanied by the appearance of lung adenocarcinomas and
that the incidence was within the range of historical controls; thus,
it was not possible to conclude from these data that chlorite induced
lung tumours (Yokose et al., 1987).
In an associated experiment, Kurokawa et al. (1984) assessed the
ability of sodium chlorite to promote skin tumours in a group of
20 female Sencar mice. These mice were initiated with a single topical
application of 20 nmol (5.1 µg) of dimethylbenzanthracene in acetone
followed by 0.2-ml applications of sodium chlorite at 20 mg/ml in
acetone twice weekly for 51 weeks. A group of 15 female mice given a
single application of dimethylbenzanthracene followed by applications
of acetone were used as controls. This treatment resulted in 5 of 25
mice having squamous cell carcinomas at 52 weeks. No tumours were
found in the corresponding initiated control mice. Both TPA and
benzoyl peroxide produced increased tumour incidence in
dimethylbenzanthracene-initiated mice. These data indicate the
potential for a weak tumour-promoting activity for sodium chlorite.
However, no dose-response information has been forthcoming in the
literature.
4.6.5 Comparative pharmacokinetics and metabolism
Some limited data on the absorption, distribution and excretion
of chlorite have been developed in rats using 36Cl-labelled chlorite.
The label was absorbed with a half-life of about 3.5 min and
eliminated with a terminal half-life of 35.2 h (Abdel-Rahman et al.,
1982b, 1984b). In 72 h, approximately 35% of the label was recovered
in the urine and another 5% in the faeces. In the urine, 32% of the
administered dose was determined to be chloride, whereas 6% was found
to be chlorite, utilizing a fractionation procedure developed in a
prior study (Abdel-Rahman et al., 1980). While these studies did not
determine the form of the radiolabel found in blood, plasma and
tissues, it was clear that there were significant differences in the
behaviour of the label derived from chlorite and chlorate. However,
the efforts in this area have been seriously hampered by the lack of
an analytical method to discriminate between chlorine dioxide,
chlorite, chlorate and chloride in vivo.
4.6.6 Mode of action
The adverse effects of chlorite appear to be mediated through its
activity as an oxidant. However, this question has received very
limited attention, except for the involvement of oxidation in its
haematological effects. Heffernan et al. (1979b) demonstrated that
chlorite was consumed during the oxidation of haemoglobin to
methaemoglobin in vitro. It was also observed that, unlike
methaemoglobin induction by nitrite, the action of chlorite also
depleted the red blood cells of GSH, and that this could be partially
counteracted by including glucose in the incubation medium. The
oxidative action of chlorite could be associated with the production
of hydrogen peroxide as measured by the formation of complex I with
catalase. This production of hydrogen peroxide was associated with
oxidative damage by demonstrating that it could also be attenuated by
the inclusion of glucose in the medium. In a dose-response comparison,
it could be demonstrated that the loss of GSH and the loss of catalase
activity paralleled one another and occurred at concentrations an
order of magnitude lower than those required for methaemoglobin
formation. This is consistent with the behaviour of other oxidants
that produce haemolytic anaemia. These observations also appear to
explain why destruction of the red blood cell (measured as decreased
haematocrit, decreased haemoglobin concentrations and increased red
blood cell turnover) is a much more sensitive and important measure of
chlorite toxicity than methaemoglobin formation.
4.7 Chlorate
4.7.1 General toxicological properties and information on
dose-response in animals
Toxicological data on chlorate in the open scientific literature
are limited to two short-term studies in dogs (Sheahan et al., 1971;
Heywood et al., 1972) and a series of studies that focused primarily
on its ability to induce oxidative damage in the blood of rats and
chickens (Abdel-Rahman et al., 1980; Couri & Abdel-Rahman, 1980) and
African green monkeys (Bercz et al., 1982). Two short-term studies,
one in dogs and one in rats, carried out by Bio/Dynamics Inc. in 1987,
were reviewed by WHO (1996). In the dog study, a NOAEL of 360 mg/kg of
body weight was identified based on no significant effects on any
measured parameter. In the rat study, a NOAEL of 100 mg/kg of body
weight was identified based on haematological effects at the highest
dose (1000 mg/kg of body weight). There is also a single subchronic
study of toxicity conducted in rats in which histopathological
examination of tissues was performed (McCauley et al., 1995). This
limited data set has hindered attempts to establish a guideline value
for chlorate in drinking-water (WHO, 1993).
The studies in dogs documented the fact that high acute doses of
1 or 2 g/kg of body weight induce methaemoglobinaemia (Sheahan et al.,
1971; Heywood et al., 1972). In addition, Heywood et al. (1972)
administered lower doses (200-300 mg/kg of body weight) for 5 days and
observed no clinical signs at doses lower than 300 mg/kg of body
weight per day. Those doses that produced some evidence of
methaemoglobinaemia were also found to have produced some
discoloration of the kidneys and haematogenous cases in renal tubules
at necropsy.
No consistent effects were observed when chlorate was
administered to rats at concentrations of 10 or 100 mg/litre
(equivalent to 1 or 10 mg/kg of body weight per day) for 12 months
(Couri & Abdel-Rahman, 1980). These authors documented some loss of
the normal sensitivity of erythrocytes to osmotic shock at these
doses, however.
Bercz et al. (1982) administered drinking-water containing sodium
chlorate to African green monkeys for a total of 8 weeks in a
rising-dose experiment. Drinking-water concentrations were 25, 50,
100, 200 or 400 mg/litre, equivalent to 4, 7.5, 15, 30 or 58.4 mg/kg
of body weight per day. Chlorate was found to be without significant
effect on a number of serum parameters related to oxidative damage and
thyroid hormone levels at concentrations of up to 400 mg/litre.
In the subchronic study (McCauley et al., 1995), concentrations
of chlorate of 3, 12 or 48 mmol/litre in drinking-water were provided
to both male and female Sprague-Dawley rats for 90 days. These
concentrations correspond to 250, 1000 and 4000 mg of chlorate per
litre, equal to 30, 100 or 510 mg/kg of body weight per day in males
and 42, 164 or 800 mg/kg of body weight per day in females, based on
measured water consumption of each group. Body weight gain was sharply
curtailed in both sexes at the highest concentration. These effects
were generally paralleled by smaller organ weights (except for brain
and testes). Some decreases in haemoglobin, haematocrit and red blood
cell counts were observed at this same dose. Pituitary lesions
(vacuolization in the cytoplasm of the pars distalis) and thyroid
gland colloid depletion were observed in both the mid- and high-dose
groups of both sexes. The NOAEL in this study was 30 mg/kg of body
weight per day.
4.7.2 Reproductive and developmental toxicity
Suh et al. (1983) examined the effects on fetal development of
chlorate at 0, 1 or 10 mg/litre administered to rats for 2.5 months
prior to mating and throughout gestation. This was a very limited
study, involving only six female rats per treatment group. Therefore,
the apparent increase of anomalous fetuses from 30.7% in the control
group to 52% and 55.2% in the groups receiving 1 and 10 mg/litre,
respectively, was not statistically significant. The abnormalities
were limited to relatively mild skeletal defects (missing sternebra
and rudimentary ribs).
A teratogenicity study carried out by Bio/Dynamics Inc. in 1987
was reviewed by WHO (1996). In this study, rats given 0, 10, 100 or
1000 mg of chlorate per kg of body weight per day on days 6-15 of
gestation showed no effects on maternal or fetal health.
4.7.3 Toxicity in humans
There have been sporadic reports of poisoning with sodium or
potassium salts of chlorate (Temperman & Maes, 1966; Mengele et al.,
1969; Yoshida et al., 1977; Bloxham et al., 1979; Helliwell & Nunn,
1979; Steffen & Seitz, 1981). Most of these cases involved ingestion
of preparations of sodium chlorate used for pesticidal purposes. The
symptomatology observed is consistent with that observed in the acute
studies in dogs identified above. There was generally evidence of
oxidative damage to erythrocytes, methaemoglobin formation and the
renal complications of haemolytic anaemia. The lethal dose to humans
has been estimated to be in the range 20-30 g.
A study in 10 male human volunteers was conducted by
administering solutions of 0.01-2.4 mg of chlorate per litre in two
500-ml portions (highest dose 0.034 mg/kg of body weight per day) in a
6-day rising-dose tolerance design (Lubbers & Bianchine, 1984). No
adverse effects were noted. This test of acute studies was followed by
an experiment (Lubbers et al., 1981) that provided 500 ml of water
containing 5 mg of chlorate per litre per day to 10 subjects for
12 weeks (average dose 0.036 mg/kg of body weight per day). Volunteers
in both studies were monitored using a battery of clinical and
physiological parameters and routine physical examinations throughout
the course of the study and for 8 weeks following termination of
treatments. Again, no adverse effects were observed.
4.7.4 Carcinogenicity and mutagenicity
There are no published studies of the carcinogenic potential of
chlorate administered alone. Sodium and potassium chlorate were
evaluated as promoters of renal tumours in
N-ethyl- N-hydroxyethylnitrosamine (EHEN)-initiated F344 rats.
Sodium chlorate and potassium chlorate were administered in the
drinking-water for 28 weeks. There was an increased incidence of renal
cell tumours (7/15 rats) in the EHEN-initiated group treated with
sodium chlorate, but no effect was observed with potassium chlorate
(1/5) relative to control rats (2/15). The small numbers of animals
used in this study make the treatment groups indistinguishable from
one another statistically (Kurokawa et al., 1985b).
Chlorate has long been known to select nitrate
reductase-deficient mutants of Aspergillus nidulans (Cove, 1976).
However, Prieto & Fernandez (1993) demonstrated that there is also a
mutagenic effect of chlorate in Chlamydomonas reinhardtii and
Rhodobacter capsulatus. Chlorate failed to induce mutations in the
BA-13 strain of Salmonella typhimurium. The positive mutagenic
effects were separated from simple selection of nitrate reductase
mutants by incubating cells in nitrogen-free media. Lack of nitrogen
prevents cell division during the treatment period. In the case of
C. reinhardtii, significant increases in mutants were observed at
concentration of 4-5 mmol/litre and above.
Meier et al. (1985b) examined chlorate in assays for micronuclei
and chromosomal aberrations in bone marrow and sperm head anomalies,
but all findings were negative.
4.7.5 Mode of action
Some research has been directed towards establishing the
mechanisms by which chlorate oxidatively damages erythrocytes and
their contents. There is a characteristic delay in the production of
methaemoglobin by chlorate when erythrocytes are incubated in the
presence of chlorate (Singelmann et al., 1984). It has been suggested
that this delay was due to the conversion of chlorate to chlorite
(Heubner & Jung, 1941; Koransky, 1952), but there is no direct
evidence to support this view. A competing hypothesis suggested that
chlorate formed a complex with methaemoglobin, which autocatalytically
increased methaemoglobin formation. This suggestion is supported by
experiments demonstrating that the formation of methaemoglobin
accelerates the further formation of methaemoglobin in the presence of
chlorate (Huebner & Jung, 1941; Jung, 1947, 1965). It is further
supported by the observation that compounds that compete for binding
of chlorate to methaemoglobin (e.g., azide or cyanide) block the
effect.
The properties of the erythrocyte membrane are also modified by
chlorate. Increased resistance to haemolysis is the most readily
observed effect (Singelmann et al., 1984). These effects appear to be
related to the formation of high molecular weight complexes of
erythrocytic proteins (Singelmann et al., 1984). These changes could
not be reversed by disulfide reduction. The formation of these
complexes was associated with the loss of activity of several enzymes,
the most sensitive being glucose-6-phosphate dehydrogenase (Singelmann
et al., 1984; Steffen & Wetzel, 1993). The inactivation of this enzyme
accounts for the insensitivity of chlorate-induced methaemoglobinaemia
to treatment with methylene blue. Reduction of nicotinamide adenine
dinucleotide phosphate (NADP) by the pentose pathway is necessary for
methylene blue to be effective. The cross-linking of protein is not
limited to cytosolic proteins and methaemoglobin, because
cross-linking of membrane proteins has also been demonstrated (Steffen
& Wetzel, 1993). The cross-linking requires the presence of
haemoglobin. Similar changes are induced by hypochlorite, but in this
case haemoglobin is not necessary.
The oxidative damage to the erythrocyte appears to be the basis
of chlorate's renal toxicity. This hypothesis is supported primarily
by the observation that species less sensitive to methaemoglobin
formation are also resistant to the nephrotoxic effects of chlorate
(Steffen & Wetzel, 1993). The observation is consistent with the
finding of haematogenous casts in kidney tubules of dogs treated with
doses of chlorate that induce methaemoglobin and their absence in dogs
treated with slightly lower doses that did not produce
methaemoglobinaemia (Heywood et al., 1972).
4.8 Bromate
4.8.1 General toxicological properties and information on
dose-response in animals
The acute toxic effects of bromate (administered as either the
potassium or sodium salt) have been studied in F344 rats, B6C3F1 mice
and Syrian golden hamsters (Kurokawa et al., 1990). The mean LD50
values in these species ranged from 280 to 495 mg/kg of body weight,
with slightly but consistently lower values found in males than in
females of each species. Mice appear to be somewhat more sensitive
than the other two species, but the lethal doses are remarkably
similar across species. Toxic signs and symptoms at lethal doses
included suppressed locomotor activity, ataxia, tachypnoea,
hypothermia, diarrhoea, lacrimation and piloerection. Hyperaemia of
the stomach and congestion of lungs were observed at autopsy. Damage
to renal tubules was seen microscopically, including necrosis in the
proximal tubular epithelium. Regenerative changes were observed from
48 h to 2 weeks after treatment. These effects were less marked in
mice and hamsters. No glomerular lesions were observed in any species.
Treatment of rats for 10 weeks with potassium bromate
concentrations of 250, 500, 1000, 2000 or 4000 mg/litre of
drinking-water established a maximally tolerated concentration of less
than 1000 mg/litre. As treatments were extended to 13 weeks, elevated
levels of glutamate-oxalate transaminase (GOT), glutamate-pyruvate
transaminase (GPT), LDH, AP and BUN were observed in blood samples
(Onodera et al., 1986; Kurokawa et al., 1990).
Eosinophilic droplets were observed in the cytoplasm of the
proximal renal tubule cells in male F344 rats receiving 600 mg of
potassium bromate per litre for 12 weeks (Onodera et al., 1986;
Kurokawa et al., 1990). These droplets were determined to be
eosinophilic bodies rather than hyaline droplets. Lipofuscin pigments
were also observed in the proximal tubular epithelium.
Dogs, rats and monkeys were fed bread or flour treated with up to
200 mg of potassium bromate per kg of body weight for up to 17 months
(FAO/WHO, 1989). These studies revealed no adverse effects, but, as
pointed out by Kurokawa et al. (1990), substantial portions of bromate
are presumed to be converted to bromide during the dough-making
process. It is also important to note that the numbers of animals
included in these studies were quite limited. Subsequent studies
conducted for longer periods of time indicated an increase
periarteritis in male rats and pathology to the adrenal glands in
female rats (Fisher et al., 1979).
Lifetime studies in female rats administered potassium bromate in
drinking-water found significant increases in GPT, albumin/globulin
ratios, serum potassium ion and cholinesterase activity at
concentrations of 500 mg/litre. Slight increases in BUN were also
observed at this dose (Kurokawa et al., 1990).
4.8.2 Toxicity in humans
Human poisonings have been associated with the ingestion of
sodium bromate and potassium bromate. Many of these poisonings result
from accidental or deliberate ingestion of preparations used as
neutralizers in permanent wave kits (Warshaw et al., 1985; Lue et al.,
1988). Clinical signs of bromate poisoning include anaemia and
haemolysis, renal failure and hearing loss. Loss of hearing appears to
be more common in adults than in children (Lichtenberg et al., 1989).
The hearing loss and renal failure can have a prolonged course in
some, but not all, people poisoned by bromate (Kuwahara et al., 1984).
Poisoning with bromate is frequently fatal when doses exceed 6 g
(Kurokawa et al., 1990).
4.8.3 Carcinogenicity and mutagenicity
IARC evaluated potassium bromate in 1986 and concluded that there
is sufficient evidence for its carcinogenicity in experimental
animals, whereas no data were available on its carcinogenicity to
humans. On this basis, potassium bromate was assigned to Group 2B: the
agent is possibly carcinogenic to humans (IARC, 1986, 1987).
In 1992, the Joint FAO/WHO Expert Committee on Food Additives
(JECFA) evaluated potassium bromate and concluded that it was
genotoxic and carcinogenic. On this basis, JECFA concluded that the
use of potassium bromate as a flour treatment agent was not
appropriate (FAO/WHO, 1993).
Potassium bromate was found to be weakly mutagenic in
Salmonella typhimurium strain TA100 when incubated with rat S9
fraction for metabolic activation (Kawachi et al., 1980; Ishidate et
al., 1984). Negative results were found in strains TA98, TA1535,
TA1537 and TA1538 (Kurokawa et al., 1990). Potassium bromate was also
inactive in Escherichia coli Wptry- and E. coli WP2try- his-
(Ishidate et al., 1984). Bromate was later found to be active in
S. typhimurium strains TA102 and TA104, which were developed to
detect compounds that generate oxygen radicals (Kurokawa et al.,
1990).
Potassium bromate also induced chromosomal aberrations in a
Chinese hamster fibroblasts cell line. The concentrations required
were, however, very high (>30 mmol/litre) (Ishidate et al., 1984).
Such high doses may induce changes by indirect mechanisms.
Bromate appears capable of inducing micronuclei in vivo.
Significant increases in the frequency of micronuclei were observed in
polychromatic erythrocytes when potassium bromate was administered by
either the oral or intraperitoneal route (Hayashi et al., 1988;
Nakajima et al., 1989). Positive results were obtained at doses of
24 mg/kg of body weight when administered intraperitoneally. Oral
doses of less than 100 mg/kg of body weight were negative. Ms/Ae and
CD-1 mice were found to be equally sensitive to these effects.
Several reports of bromate-induced cancer in experimental animals
are available. The clearest evidence comes from studies in F344 rats
(Kurokawa et al., 1983, 1986a, 1987a; DeAngelo et al., 1998). The
dose-response curves for the principal target organs, the kidney and
peritoneum, are provided in Figure 7. Tumours found in the kidney were
of tubular origin, with significantly increased numbers of both
adenomas and adenocarcinomas being observed in both males and females
(Kurokawa et al., 1983). The peritoneal tumours were mesotheliomas,
but treatment-related increases were observed only in male rats. In
the Kurokawa et al. (1983) study, concentrations of potassium bromate
of 0, 250 or 500 mg/litre correspond to doses of 0, 9.6 or 21.3 mg of
bromate per kg of body weight per day in males and 0, 9.6 or 19.6
mg/kg of body weight per day in females (as cited in IARC, 1986 and
WHO, 1996). The Kurokawa et al. (1986a) study represented, in part, a
repeat of the earlier study, except that more doses were included and
only male rats were studied. Concentrations in this study were 0, 15,
30, 60, 125, 250 or 500 mg of potassium bromate per litre,
corresponding to 0, 0.7, 1.3, 2.5, 5.6, 12 or 33 mg of bromate per kg
of body weight per day (as cited in WHO, 1996). The study provided
verification of the ability of potassium bromate to induce both renal
cell tumours and mesothelioma. An increased incidence of renal tumours
was observed at 125 mg of potassium bromate per litre of
drinking-water. Results with mesothelioma were not observed at a dose
of 250 mg/litre, but this is partially attributable to the smaller
number of animals per treatment group in this experiment. However, the
incidences of mesothelioma were very similar at 500 mg/litre in the
two studies. Significant increases in the occurrence of dysplastic
foci of the kidney (considered to be preneoplastic lesions) were found
in groups at doses higher than 30 mg/litre.
The time course of renal tumour development was examined in a
third study (dose-response provided in panel C of Figure 7) by
Kurokawa et al. (1987a). These experiments were designed to include
sacrifices of animals at 13, 26, 39 or 52 weeks as well as the
104-week period examined in prior studies. Additional groups were
included, however, that involved treatment for the above periods, but
the animals were maintained on bromate-free water until 104 weeks
before they were sacrificed. The concentration of potassium bromate
was 500 mg/litre during active treatment periods (average dose 32.3 mg
of bromate per kg of body weight per day). If animals were held for
104 weeks, 13 weeks of treatment was sufficient to produce the same
tumour incidence as was produced by longer treatment periods.
Moreover, the same incidence of renal cell tumours was observed in
animals that had been treated for 52 weeks and sacrificed at 52 weeks.
These data indicate that the tumour yield is not dependent upon the
total dose administered, but rather that sufficient time simply had to
be provided for the tumours to become evident.
The carcinogenicity of bromate has also been studied in three
hybrid strains of mice (B6C3F1, BDF1 and CDF1). Treatments of
female mice were conducted at concentrations of 0, 500 or
1000 mg/litre for 78 weeks (average dose 0, 43.5 or 91.6 mg of bromate
per kg of body weight per day). No treatment-related increases in
tumour incidence were observed (Kurokawa et al., 1986b). Groups of 27
however, more TCA is formed as a result of enterohepatic circulation
of the trichloroethanol glucuronide (Stenner et al., 1996). The
glucuronide is hydrolysed to trichloroethanol, which can be oxidized
to TCA, with chloral hydrate as an intermediate. Under physiological
conditions, the formation of TCA is for all practical purposes
irreversible, and the net conversion of chloral hydrate to TCA will
continue as long as there are significant amounts of trichloroethanol
entrained in the enterohepatic circulation.
The production of these major products of chloral hydrate
metabolism is relatively well understood. However, the exact source of
a number of minor metabolites is less well understood. The reaction
rates and mechanisms involved are just beginning to be studied.
Understanding these mechanisms will be key to understanding whether
the effects of chloral hydrate can be attributed primarily to its
conversion to chemicals of established toxicological properties, such
as trichloroethanol, DCA and TCA, or whether the activities of
reactive intermediates must also be considered. Formation of DCA
probably requires radical formation, but it is not clear whether the
radical would be formed from trichloroethanol, chloral hydrate or TCA.
If DCA is derived from the first two compounds, the
dichloroacetaldehyde that would result as an intermediate could pose
some toxicological problems as well. Moreover, Ni et al. (1996)
suggested from their ESR data that a trichloromethyl radical is formed
from chloral hydrate. Such an intermediate could contribute to the
toxicological effects of chloral hydrate as well.
Available pharmacokinetic data have focused upon the relative
role of trichloroethanol as the metabolite of chloral hydrate that is
responsible for its central nervous system-depressant activity
(Butler, 1948, 1949; Marshall & Owens, 1954; Garrett & Lambert, 1973).
Conversely, a variety of metabolites have been suggested as
responsible for the toxic effects of chloral hydrate and its ability
to induce liver cancer in mice, in particular (Daniel et al., 1992a).
The critical question is whether an early reactive metabolite -- e.g.,
the free radicals identified by Ni et al. (1996) -- induces
clastogenic effects that are important to tumour development. The
competing, but not necessarily exclusive, hypothesis is that the
clearly hepatocarcinogenic metabolites TCA and DCA are produced in
sufficient quantity to account for some or all of the liver cancer
that results from chloral hydrate treatment.
While TCA and trichloroethanol are well established metabolites
of chloral hydrate, DCA has been only recently recognized as a
potentially important metabolite with respect to liver tumour
induction. Part of the difficulty is that DCA is much more rapidly
metabolized than either TCA or trichloroethanol, and peak
concentrations would be expected to be significantly lower. This first
became apparent when DCA appeared to be produced in significant
quantities from trichloroethylene administered to B6C3F1 mice
(Templin et al., 1993). Chloral hydrate is the first stable metabolite
of trichloroethylene metabolism (Cole et al., 1975); thus, these data
prompt an examination of the role that chloral hydrate plays in the
formation of DCA. In contrast with these results in mice, DCA was not
detectable in rats and dogs administered similar doses (Templin et
al., 1995). It became apparent that some of the DCA that was measured
in these studies may have arisen artefactually through dehalogenation
of TCA under acid conditions in fresh blood (Ketcha et al., 1996). As
a consequence of this series of findings, newer studies have examined
whether DCA is produced from chloral hydrate in a number of species.
Recent results of Abbas et al. (1996) showed that doses of 10 and
100 mg of chloral hydrate per kg of body weight result in
approximately 2.4 and 10 µg of DCA per ml of blood. However, it must
be noted that a variety of artefacts of DCA formation from TCA in
blood suggest that these results need to be viewed with caution
(Ketcha et al., 1996). Indeed, Merdink et al. (1998) found that DCA
was not measurable in mice dosed with 50 mg of chloral hydrate per kg
of body weight.
4.3.1.5 Mode of action
The mechanism of action involved in chloral hydrate-induced liver
tumours in mice remains to be established. Clearly, chloral hydrate is
converted to at least one metabolite, TCA, that appears to act as a
peroxisome proliferator. There is some possibility that it is
converted to DCA, a compound that acts primarily as a tumour promoter
(Stauber & Bull, 1997). On the other hand, chloral hydrate is distinct
from these other two compounds in that it appears to be clastogenic
in vivo, but at very high doses. The question is, does this
clastogenic activity play a role in tumorigenesis? The fact that
chloral hydrate appears to produce only hepatic tumours in mice
parallels the species specificity of TCA and suggests that other
activities are perhaps not involved.
4.3.2 Halogenated aldehydes and ketones other than chloral hydrate
4.3.2.1 General toxicological properties and information on
dose-response in animals
1) Haloaldehydes
Relatively few data are available to describe acute, short-term
or chronic toxicities for the haloacetaldehydes. In general, aldehydes
are irritant chemicals, and substitution of chlorine generally
increases this irritancy. However, it is unlikely that irritant
effects will occur at concentrations that are encountered in
drinking-water. Chloroacetaldehyde is an example of a haloaldehyde for
which some data exist, although it has not been commonly found in
drinking-water. Concentrations of chloroacetaldehyde as low as 0.02%
produced intradermal irritation, 7.5% produced rather severe dermal
irritation and 0.03% irritated the eyes of rabbits (Lawrence et al.,
1972).
When administered systemically, halogenated aldehydes are quite
toxic. Again because of a lack of appropriate data, chloroacetaldehyde
will be used for illustrative purposes. Lawrence et al. (1972)
reported the oral LD50 in Sprague-Dawley rats to be 89 and 103 mg/kg
of body weight in males and females, respectively. In male ICR mice,
the oral LD50 was found to be 82 mg/kg of body weight. In longer-term
investigations, Lawrence and co-workers (1972) utilized
intraperitoneal injections or inhalation as the method of
administration. Intraperitoneal injections of 2.2 or 4.4 mg/kg of body
weight per day for 30 days in male Sprague-Dawley rats produced
significant decreases in haemoglobin, haematocrit and erythrocytes at
the highest dose. This was consistent with its ability to induce
haemolysis of erythrocytes at concentrations of 0.2 mol of
chloroacetaldehyde per litre and above. However, the validity of this
in vitro observation as a dependable indicator of in vivo effects
is suspect because of the extremely high concentrations that were
utilized. The 4.4 mg/kg of body weight dose produced reductions in
body weight and induced significant increases in the organ to body
weight ratios for the brain, gonads, heart, kidneys, lungs and spleen.
These effects appeared to be largely the result of reduced body weight
rather than changes in absolute organ weights.
Lawrence et al. (1972) pointed out the importance of route of
administration to the toxicity of chloroacetaldehyde. If administered
intraperitoneally, it is 10-30 times as potent as its metabolite
2-chloroethanol. However, the toxicity of the two compounds is more or
less equivalent when they are administered orally, and 2-chloroethanol
is about 4 times as toxic as chloroacetaldehyde when applied
topically. These differences are most likely attributed to relative
rates of absorption versus metabolic conversion and the many
non-specific reactions in which chloroacetaldehyde would be expected
to be involved in the gastrointestinal tract and on the skin. This
issue should be reflected in the systemic toxicities of halogenated
aldehydes, in general.
A 104-week study of chloroacetaldehyde utilizing drinking-water
as the mode of administration was conducted in male B6C3F1 mice
(Daniel et al., 1992a). Only a single concentration was used
(100 mg/litre), which yielded an average dose of 17 mg/kg of body
weight (i.e., larger than administered to rats intraperitoneally).
This dose did not lead to excessive mortality or depress body weight
gains. It did not affect weights of the liver, kidneys, testes or
spleen. There was no remarkable non-tumour pathology in 40 tissues
that were sampled and examined microscopically in five of the animals
that were serially sacrificed during the course of the experiment. An
apparent increase in the incidence of liver histopathological change
was described as hepatocellular necrosis, hyperplasia and cytomegaly.
However, these effects were very mild and of doubtful significance as
compared with the same types of pathology in control mice.
Sood & O'Brien (1993) examined the effects of chloroacetaldehyde
in isolated rat hepatocytes. A concentration of 0.5 mmol/litre was
found to be cytotoxic, whereas 0.2 mmol/litre was without apparent
effect. The cytotoxicity could be essentially abolished by
dithiothreitol in the incubation media. The requirement for high
concentrations in this in vitro experiment and the apparent lack of
effect (albeit administered to a different species) at relatively high
concentrations in the animals' drinking-water (100 mg/litre) suggest
that there is little concern about hepatotoxicity at the very low
concentrations that might be expected to be found in chlorinated
drinking-water.
2) Haloketones
Toxicological data in experimental animals for the haloketones
are extremely limited. The halopropanones are the most commonly
studied group of this class, but most of the work has been directed at
mutagenic effects of the chemicals.
Laurie et al. (1986) studied the effects of 1,1- and 1,3-DCPN in
CD-1 mice. 1,1-DCPN was administered in paraffin, and 1,3-DCPN was
administered as an aqueous solution. 1,1-DCPN significantly increased
the levels of ASAT, ALAT and LDH at doses of 325 mg/kg of body weight.
1,3-DCPN was evaluated to a maximum dose of 20 mg/kg of body weight
and appeared to be without effect on the serum enzymes. However, the
LD50 for 1,3-DCPN was stated to be 25 mg/kg of body weight,
indicating that the liver was probably not the critical target organ
for this compound, at least with acute treatment. At doses of 130
mg/kg of body weight and greater, 1,1-DCPN significantly depressed
hepatic GSH levels. Again, 1,3-DCPN was without effect at the dose of
20 mg/kg of body weight. Most of the decrease in GSH produced by
1,1-DCPN was observed in the post-mitochondrial cellular fraction as
opposed to the mitochondrial fraction.
A major thrust of the Laurie et al. (1986) paper was to determine
the extent to which 1,1- and 1,3-DCPN modified the toxicity of carbon
tetrachloride. Carbon tetrachloride was administered at doses ranging
from 0.02 to 1.0 ml/kg of body weight. The 0.02 ml/kg of body weight
dose produced an elevation of serum enzymes on its own. However, when
carbon tetrachloride was administered 4 h after 1,1-DCPN, the dose of
1,1-DCPN that was required to significantly increase serum enzyme
levels was decreased from 325 to 130 mg/kg of body weight. Since a
doubling of the dose of 1,1-DCPN would have produced a similar
response and the dose of carbon tetrachloride was above a threshold
response level, the interaction between carbon tetrachloride and
1,1-DCPN would seem to be no more than additive. Inhibition of the
toxicological effects of carbon tetrachloride in a dose-related manner
was observed when 1,3-DCPN was administered prior to carbon
tetrachloride.
Merrick et al. (1987) studied the cytotoxic effects of
chloropropanone (CPN), 1,1-DCPN and 1,3-DCPN on isolated hepatocytes
of male Sprague-Dawley rats. The chloropropanones were all shown to
react with GSH in solution. At concentrations of 10 mmol/litre, the
rate of GSH reaction was most rapid with 1,3-DCPN, followed by CPN and
then 1,1-DCPN. This reactivity paralleled the ability of the compounds
to induce cytotoxicity in isolated hepatocytes. Significant increases
in ASAT release were observed with 1,3-DCPN at 0.5 mmol/litre, CPN at
1 mmol/litre and 1,1-DCPN at 5 mmol/litre. As would be expected, GSH
depletion was observed at concentrations of 1,3-DCPN as low as 0.1
mmol/litre. The other two compounds were significantly less active in
depleting GSH, but were of approximately equivalent potency with one
another.
A study of 1,1,1-TCPN was conducted in Sprague-Dawley rats by
Daniel et al. (1993b). Acute, 10-day and 90-day experiments were
performed. 1,1,1-TCPN was administered in corn oil by gavage. Doses in
the 10-day study were 0, 16, 48, 161 or 483 mg/kg of body weight per
day. In the 90-day study, doses of 30, 90 or 270 mg/kg of body weight
per day were administered. In the 10-day study, 8 out of 10 male and 7
out of 10 female rats died at 483 mg/kg of body weight per day before
the conclusion of the treatments. Two male rats also died at 161 mg/kg
of body weight per day. Although there was not a significant effect on
body weight in the survivors, there was a 10% increase in liver to
body weight ratios at 161 mg/kg of body weight per day in both male
and female rats. Evidence of hyperkeratosis was found in the
forestomach of both male and female rats treated at doses of 48 mg/kg
of body weight per day and greater. No adverse effect was observed at
16 mg/kg of body weight per day.
Increases in relative liver weight were observed in the 90-day
experiment at 270 mg/kg of body weight per day in male rats, but not
at 90 mg/kg of body weight per day. Ataxia was reported in both sexes
at the 270 mg/kg of body weight per day dose level. Increases in the
incidence of forestomach lesions were observed in both sexes at 90 and
270 mg/kg of body weight per day, with the most frequent observation
being hyperkeratosis followed by acanthosis. The overall NOAEL in the
10-day and 90-day studies is 30 mg/kg of body weight per day.
In summary, the toxicological effects of the halopropanones
provide evidence that some of the representatives of this class are
highly toxic, with acute lethal doses being as low as 25 mg/kg of body
weight. The gastrointestinal tract and liver appear to be key target
organs for some members of the class. However, no target organ was
identified for the most acutely toxic of the group, 1,3-DCPN.
4.3.2.2 Toxicity in humans
No data on the effects of either halogenated aldehydes or
halogenated ketones on human subjects were identified.
4.3.2.3 Carcinogenicity and mutagenicity
There are considerable data on the mutagenic properties of
various halogenated aldehydes and ketones. A comparison of the
mutagenic activity, measured in Salmonella typhimurium tester
strains, of the halogenated aldehydes found in drinking-water with
that of chloroacetaldehyde is summarized (not comprehensively) in
Table 19. Table 20 provides a similar summary of results with
halogenated ketones. These data are presented to demonstrate that the
activity of these compounds is not dissimilar from that observed with
chloroacetaldehyde, a metabolite of a large number of carcinogenic
chemicals, including vinyl chloride. Therefore, it will be used as a
prototype for the class. This is not to suggest that this is an
adequate substitute for appropriate data for the other compounds. It
must be recognized that the data available for this class are
completely inadequate for making substantive estimates of the impact
of these chemicals on human health; in particular, mutagenicity data
in bacterial systems do not necessarily reflect activity in vivo.
1) Haloaldehydes
Chloroacetaldehyde has been extensively studied with respect to
the types of interactions that it has with DNA. The adducts formed in
animals treated with vinyl chloride are the same as those produced
with chloroacetaldehyde (Green & Hathway, 1978). The cyclic etheno
adducts formed with cytosine and adenine seem particularly important
in mutagenic responses observed with chloroacetaldehyde (Spengler &
Singer, 1988; Jacobsen et al., 1989).
There are a limited number of studies that have examined the
carcinogenic properties of chloroacetaldehyde in rather specialized
test systems. Van Duuren et al. (1979) examined the carcinogenic
activity of chloroacetaldehyde in mouse skin initiation/promotion
studies, with subcutaneous injection and by stomach tube. In the
initiation/promotion assay, chloroacetaldehyde was applied to the skin
of 30 female Ha:ICR Swiss mice, at a dose of 1.0 mg per application
per mouse, 3 times weekly for up to 581 days, or as a single dose
followed by 2.5 µg of TPA 3 times weekly for 576 days. No evidence of
increased skin tumour yield was found. The intragastric treatments
involved administration of 0.25 mg per mouse per week (1.8 mg/kg of
body weight per day if a 20-g body weight is assumed). In this case,
sections of lung, liver and stomach were taken for histopathological
examination. No signs of increased tumour incidence were found. A
fourth experiment involved the subcutaneous injection in 30 mice of
0.25 mg per mouse (1.8 mg/kg of body weight per day, assuming a 20-g
body weight for the mouse). Microscopic examination of sections of the
liver and injection sites revealed no evidence of increased tumour
yield.
A second group of mouse skin initiation/promotion experiments
with chloroacetaldehyde were conducted by Zajdela et al. (1980).
Single doses of 0.05, 0.1, 1.0 or 2.5 mg of chloroacetaldehyde
dissolved in acetone were applied to male and female XVIInc/Z mice
(20-28 per group). This was followed by application of TPA at 2 µg, 3
times weekly for 42 weeks. There was no significant difference between
mice receiving TPA alone and those that had been initiated by
chloroacetaldehyde.
There appears to be only one study that examined the carcinogenic
activity of halogenated aldehydes administered in drinking-water over
a lifetime (Daniel et al., 1992a). Male B6C3F1 mice treated with 0.1
g of chloroacetaldehyde per litre of drinking-water for 104 weeks were
found to have an incidence of eight hepatocellular carcinomas in
26 mice examined (31%). In addition, 8% of these mice were diagnosed
as having hepatocellular adenoma, and another 8% were found to have
hyperplastic nodules. This compared with two carcinomas in 20 control
mice examined (10%), one with adenoma (5%) and no hyperplastic nodules
(0%). In a comparison with previous studies, the experiment utilized a
Table 19. Mutagenic activity of halogenated aldehydes produced by chlorination in
the Salmonella/microsome assay
Compound Strain Net revertants/plate Reference
-S9 +S9
Chloroacetaldehyde TA100 440 ~330 Bignami et al. (1980)
2-Chloropropenal TA100 135 49 Segall et al. (1985)
2-Bromopropenal TA100 1140 108 Segall et al. (1985)
2-Bromopropenal TA100 400 Gordon et al. (1985)
2,3-Dibromopropanal TA100 300 Gordon et al. (1985)
2,3-Dichloropropenal TA100 91 5 Segall et al. (1985)
3,3-Dichloropropenal TA100 0.7 Meier et al. (1985a,b)
2,3,3-Trichloropropenal TA100 224 Rosen et al. (1980)
3-Chloro-2-butenal TA100 68 42 Segall et al. (1985)
3-Bromo-2-butenal TA100 108 39 Segall et al. (1985)
2-Bromo-3-methyl-2-butenal TA100 <0.5 <0.5 Segall et al. (1985)
Table 20. Mutagenic activity of halogenated ketones produced by chlorination in the
Salmonella/microsome assay
Compound Strain Net revertants/plate Reference
-S9 +S9
CPN TA100 NMa NM Merrick et al. (1987)
1,1-DCPN TA100 0.04 Meier et al. (1985a)
1,3-DCPN TA100 25.2 Meier et al. (1985a)
1,1,1-TCPN TA100 0.12 Meier et al. (1985a)
1,1,3-TCPN TA100 3.9 Douglas et al. (1985)
1,1,3,3-Tetrachloropropanone TA100 1.5 Meier et al. (1985a)
Pentachloropropanone TA100 0.86 Meier et al. (1985a)
a NM = non-mutagenic.
significantly higher dose (17 mg/kg of body weight per day) as well as
a continuous treatment. It is possible that these tumours were induced
by the genotoxic properties of the chemical.
Robinson et al. (1989) examined the ability of four other
halogenated aldehydes to act as tumour initiators in the skin of
Sencar mice: 2-chloropropenal, 2-bromopropenal, 3,3-dichloropropenal
and 2,3,3-trichloropropenal. The compounds were administered topically
in six divided doses over a 2-week period (total doses were
600-2400 mg/kg of body weight). Two weeks after the final initiating
dose, TPA was applied at a dose of 1 µg, 3 times weekly for 20 weeks.
Both 2-chloropropenal and 2-bromopropenal significantly increased
tumour yield at 24 weeks and significantly increased the yield of
squamous cell carcinomas at 52 weeks at total topical doses of
1200 mg/kg of body weight and above. Both the benign tumour and
malignant tumour yields were greater with 2-bromopropenal than with
2-chloropropenal. An experiment utilizing oral administration of these
compounds during the initiation period was included in this study.
Oral administration of 2-chloropropenal did not produce consistent,
dose-related responses. However, there appeared to be a substantial
increase in skin tumour yield at an oral dose of 300 mg of
2-bromopropenal per kg of body weight (19 tumours in 38 mice [50%] vs.
20 tumours in 110 control mice [18%]).
On the basis of these studies, it must be concluded that there is
a potential carcinogenic hazard associated with the halogenated
aldehydes. Only a single compound, chloroacetaldehyde, was evaluated
as a carcinogen in a lifetime study, and only one dose level was
studied. It appears to be more potent as a carcinogen than the
corresponding THM and HAA by-products. Many members of the class are
mutagenic, and chloroacetaldehyde, at least, appears to produce
tumours in the liver at less than cytotoxic doses. Based upon the
comparison between 2-chloropropenal and 2-bromopropenal, there is some
reason to believe that the brominated by-products are more potent than
the corresponding chlorinated by-products. Therefore, concern must be
expressed over disinfection processes that activate bromide, as well
as those that simply chlorinate. However, the currently available data
are not sufficient to allow the hazards associated with these
compounds to be estimated.
2) Haloketones
A number of halopropanones have been tested in mutagenesis
assays. To facilitate comparison of their relative potencies, selected
results from assays that were conducted in Salmonella typhimurium
tester strain TA100 were incorporated into Table 20. For the most
part, the data selected for this table were abstracted from papers in
which more than one haloketone was evaluated, rather than being
selected because they were identified as the best value for each
individual chemical that exists in the literature. Some of the
compounds have been shown to be active in other Salmonella tester
strains and other mutagenesis and clastogenesis assays. There was
little to be gained from an exhaustive review of this literature, so
further consideration of the mutagenic activity will be limited to
those systems that extended evaluations to other end-points in
vitro or attempted to confirm in vitro observations in vivo.
1,3-DCPN was found to induce SCEs in V79 cells at concentrations
as low as 0.002 mmol/litre (von der Hude et al., 1987). Blazak et al.
(1988) found that 1,1,1-TCPN and 1,1,3-TCPN were able to act as
clastogens in CHO cells in vitro. Structural aberrations were
produced at a 1,1,3-TCPN concentration of 1.5 g/ml, whereas a
concentration of 23 µg/ml was required for a similar response to
1,1,1-TCPN. However, the dose-response for 1,1,3-TCPN was limited by
cytotoxicity. Experiments were also conducted focusing on the ability
of 1,1,1-TCPN and 1,1,3-TCPN to induce micronuclei in polychromatic
erythrocytes and to induce sperm head abnormalities in mice in
vivo. 1,1,1-TCPN was found to be negative in both assays in the dose
range 75-300 mg/kg of body weight, whereas 1,1,3-TCPN was negative in
the range 3-12 mg/kg of body weight.
Robinson et al. (1989) tested CPN, 1,1-DCPN, 1,3-DCPN, 1,1,1-TCPN
and 1,1,3-TCPN as initiators in the skin of Sencar mice. The compounds
were administered by topical application in acetone with total doses
that ranged from 37.5 to 4800 mg/kg of body weight (doses of 600 mg/kg
of body weight and above were administered in six equal doses over a
2-week period to avoid cytotoxic or lethal effects of the compounds).
Other groups of animals treated with the chemicals were also
administered similar doses by intragastric intubation. The initiating
treatments were followed by a promotion schedule that involved the
topical application of 1 µg of TPA 3 times weekly for 20 weeks. Tumour
counts were reported at 24 weeks; if the incidence was elevated within
this time period, the mice were held until 52 weeks on study prior to
sacrifice, and histological evaluations of the tumours were made.
Among the haloketones, only 1,3-DCPN was found to produce a
dose-related increase in tumour incidence. A single topical dose of
37.5 mg/kg of body weight was sufficient to initiate skin tumours, and
the response increased progressively as doses were increased to 150
mg/kg of body weight. At higher doses, the response decreased in
magnitude. Splitting the 300 mg/kg of body weight dose into six equal
doses over a 2-week period increased the tumorigenic response relative
to a single dose of 300 mg/kg of body weight. However, this response
was also attenuated, as the multiple-dose schedule utilized higher
doses. This attenuation of the response was particularly marked in
total tumour yields, which included many benign tumours. It was less
effective in limiting the yield of squamous cell carcinomas.
In conclusion, the carcinogenic activity of the DBPs in the
haloaldehyde and haloketone classes, with the exception of
chloroacetaldehyde, has not been evaluated in lifetime studies in
experimental animals. However, other tests confirm that they have
carcinogenic properties. 1,3-DCPN was the most potent tumour initiator
in both classes of DBPs. A single dose of 75 mg/kg of body weight
produces a total tumour yield equivalent to that produced by 1200 mg
of 2-bromopropenal, the most potent of the haloaldehydes, per kg of
body weight. 2-Bromopropenal is about 40 times as potent as 1,3-DCPN
as a mutagen. The other halopropanones do not appear to be capable of
acting as tumour initiators in the mouse skin.
4.3.2.4 Comparative pharmacokinetics and metabolism
No information was identified in the available literature.
4.3.2.5 Mode of action
The data available indicate that these two groups of chemicals
contain compounds that possess mutagenic activity. As these effects
have been identified in in vitro or bacterial test systems, there is
no assurance that this is the manner in which they contribute to
toxicity or carcinogenicity. A few chemicals have been shown to be
initiators in the mouse skin, but it is not clear whether that would
be a target organ as a result of chronic ingestion of these chemicals.
Other chemicals appear to have activities that could contribute less
directly to the induction of cancer, particularly as cytotoxic
compounds. It is clear from the limited data available that it would
be inappropriate to try to generalize data from only a few examples to
these two larger classes of DBPs.
4.4 Haloacetonitriles
4.4.1 General toxicological properties and information on
dose-response in animals and humans
The HANs are discussed in a single section of this document
because the toxicological data on them are quite limited. The
dihaloacetonitriles (DHAN) -- DCAN, BCAN and DBAN -- are the most
important in terms of concentrations found in chlorinated
drinking-water. However, there are limited data on bromoacetonitrile
(BAN), chloroacetonitrile (CAN) and trichloroacetonitrile (TCAN) that
are included for completeness.
Hayes et al. (1986) examined the general toxicological effects of
DCAN and DBAN in male and female ICR mice and CD rats. In mice, the
acute oral (by gavage in corn oil) LD50 was reported to be 270
(males) and 279 (females) mg/kg of body weight for DCAN and 289
(males) and 303 (females) mg/kg of body weight for DBAN. In rats, the
LD50 was found to be 339 (males) and 330 (females) mg/kg of body
weight for DCAN and 245 (males) and 361 (females) mg/kg of body weight
for DBAN. Hussein & Ahmed (1987) found somewhat lower oral LD50s in
rats: BAN, 25.8 mg/kg of body weight; DBAN, 98.9 mg/kg of body weight;
CAN, 152.8 mg/kg of body weight; and DCAN, 202.4 mg/kg of body weight.
These latter data were reported only in abstract form, and the vehicle
used was not indicated. As discussed below, some of the toxicological
responses to chemicals in this class appear to depend on the nature of
the vehicle in which they were administered.
DCAN and DBAN were also studied over 14- and 90-day treatment
intervals (Hayes et al., 1986). DCAN dissolved in corn oil was
administered to male and female CD rats by gavage at 12, 23, 45 or 90
mg/kg of body weight per day for 14 days and at 8, 33 or 65 mg/kg of
body weight per day for 90 days. DBAN was administered to male and
female CD rats at daily doses of 23, 45, 90 or 180 mg/kg of body
weight per day for 14 days and at 6, 23 or 45 mg/kg of body weight per
day for 90 days. Increased mortality was produced at 33 mg of DCAN per
kg of body weight per day and at 45 mg of DBAN per kg of body weight
per day in the 90-day studies. Body weight was decreased and lower
weights and organ to body weight ratios were observed for spleen and
gonads with doses of 65 mg of DCAN per kg of body weight per day and
above. The NOAELs for DCAN were 45 mg/kg of body weight per day for 14
days and 8 mg/kg of body weight per day for 90 days of exposure. The
NOAELs for DBAN were 23 mg/kg of body weight per day at 90 days and 45
mg/kg of body weight per day at 14 days. No serum chemistry changes
indicative of adverse effects were seen with either compound at
sublethal doses.
4.4.2 Reproductive and developmental toxicity
Smith et al. (1987) examined the effect of CAN, DCAN, TCAN, BCAN
and DBAN on female reproduction in an in vivo teratology screening
test in Long-Evans hooded rats. DCAN and TCAN at doses of 55 mg/kg of
body weight administered in tricaprylin by gavage from day 7 to day 21
of gestation significantly reduced the percentage of females
delivering viable litters, increased resorption rates and reduced
maternal weight gain. BCAN and DBAN at the same dose were without
effect. All of the HANs reduced the mean birth weight of pups, and the
DHANs reduced the postnatal weight gain till the fourth day after
birth. Postnatal survival was reduced with DCAN and TCAN but not with
BCAN or DBAN. These pups continued to display reduced body weights
into puberty. BCAN also resulted in significantly depressed weights at
puberty, although the effect was smaller than that observed with DCAN
or TCAN.
The hydra assay system for developmental toxicity has also been
used to screen some of the HANs (Fu et al., 1990). Both DBAN and TCAN
were found to be of the same general order of toxicity to adult and
embryonic animals. Based on these findings, the authors predicted that
DBAN and TCAN would not be teratogenic at non-maternally toxic doses.
The developmental toxicity of DCAN was followed up in full-scale
teratology studies (Smith et al., 1989b). In this case, DCAN dissolved
in tricaprylin was administered to Long-Evans rats at doses of 0, 5,
15, 25 or 45 mg/kg of body weight per day from day 6 to day 18 of
gestation. Embryolethality and fetal resorptions were statistically
significant at 25 and 45 mg/kg of body weight per day. The highest
dose was also maternally toxic. Soft tissue anomalies, including an
intraventricular septal defect in the heart, hydronephrosis, fused
ureters and cryptorchidism, were observed at this dose. Skeletal
abnormalities (fused and cervical ribs) were produced in a
dose-dependent manner and were significantly increased at 45 mg/kg of
body weight per day. A NOAEL was found to be 15 mg/kg of body weight
per day.
TCAN was evaluated in two teratology studies by the same
laboratory (Smith et al., 1988; Christ et al., 1996). The first of
these studies utilized tricaprylin as the vehicle, whereas the second
utilized corn oil. In the first study, embryolethality was observed at
doses as low as 7.5 mg/kg of body weight per day. Doses of 15 mg/kg of
body weight per day and above produced soft tissue abnormalities,
including fetal cardiovascular anomalies (Smith et al., 1988). TCAN
administered in a corn oil vehicle produced cardiovascular defects at
55 mg/kg of body weight per day (Christ et al., 1996). The effects
observed at this dose were found at significantly lower incidence than
observed in the 15 mg/kg of body weight per day dose of the previous
study. The abnormalities were milder, being simply positional
(laevocardia), instead of the interventricular septal defect and a
defect between the ascending aorta and right ventricle that were
observed with the tricaprylin vehicle. On the other hand, more
skeletal defects were observed with the corn oil vehicle. The authors
attributed these differences to an interaction between the tricaprylin
vehicle and TCAN. However, tricaprylin and corn oil are not
representative of drinking-water exposure. It is not possible to
determine whether the results obtained with tricaprylin or the results
obtained with corn oil provide the most valid test. The results could
be just as readily ascribed to an inhibition of the effects of the
corn oil vehicle. As a consequence, these data present somewhat of a
difficulty in interpreting results for all of the HANs that have been
tested, because the only vehicle in which most have been evaluated was
tricaprylin.
4.4.3 Carcinogenicity and mutagenicity
IARC has evaluated BCAN, CAN, DBAN, DCAN and TCAN and concluded
that there is inadequate evidence for their carcinogenicity in
experimental animals. No data were available on their carcinogenicity
in humans. Consequently, these HANs were assigned to Group 3: the
agent is not classifiable as to its carcinogenicity to humans (IARC,
1991, 1999).
Bull et al. (1985) tested the ability of CAN, DCAN, TCAN, BCAN
and DBAN to induce point mutations in the Salmonella/microsome
assay, to induce SCEs in CHO cells in vitro, to produce micronuclei
in polychromatic erythrocytes in CD-1 mice and to act as tumour
initiators in the skin of Sencar mice.
DCAN produced a clear increase in mutagenic activity in
Salmonella typhimurium strains TA1535 and TA100. This response was
not altered by the inclusion of the S9 system to metabolically
activate the compound, if needed. BCAN also produced a positive
response at low doses, but the dose-response curve was interrupted at
high doses by cytotoxicity. In this case, the inclusion of the S9
fraction appeared to simply allow the bacteria to survive cytotoxic
effects. This perhaps arose through a non-specific inactivation of the
electrophilic character of the compound. BCAN produced a similar, but
less marked, trend in strain TA100. The other HANs were negative in
the Salmonella/microsome assay.
All of the HANs tested increased the frequency of SCEs in CHO
cells. The potencies in the absence of S9 were DBAN > BCAN > DCAN
approx. or equiv. TCAN > CAN. As in the Salmonella/microsome assay,
the addition of S9 allowed higher doses to be tested rather than
modifying the response to a given concentration. In contrast, none of
the HANs was found to induce micronuclei in CD-1 mice in vivo.
The HANs were tested for their ability to initiate tumours in the
skin of Sencar mice (Bull et al., 1985). In this experiment, the HANs
were administered topically to the shaved backs of the mice in six
doses over a 2-week period. Two weeks following the last initiation
treatment, TPA dissolved in acetone was applied topically at a dose of
1 µg per mouse, 3 times weekly for 20 weeks. The total initiating
doses were 1200, 2400 and 4800 mg/kg of body weight. Significant
increases in skin tumours were observed with CAN, TCAN, BCAN and DBAN.
DBAN produced the greatest response at a dose of 2400 mg/kg of body
weight, but the response decreased in magnitude as the dose was
increased to 4800 mg/kg of body weight. The shape of this
dose-response curve was checked in a repetition of the experiment, and
the results were virtually identical. It was postulated that the
attenuated response at the higher dose was caused by the cytotoxicity
to initiated cells. BCAN also increased the incidence of skin tumours,
but no significant increase in tumour incidence was induced by DCAN.
The carcinogenicity of the DHANs in mouse skin was seen to
progressively increase as bromine was substituted for chlorine in the
compound. On the other hand, CAN appeared to be among the more potent
of the HANs in initiating skin tumours. TCAN gave inconsistent
results.
CAN, TCAN and BCAN produced small, but significant, increases in
the incidence of lung tumours in female A/J mice (40 mice per chemical
tested) when administered by gavage at doses of 10 mg/kg of body
weight, 3 times weekly for 8 weeks (Bull & Robinson, 1985). Mice were
started on treatment at 10 weeks of age and sacrificed at 9 months of
age. No significant effects were observed with DBAN and DCAN. The
differences in tumorigenic response are too small for meaningful
rankings of the compounds for potency in this lung tumour-susceptible
strain.
DCAN was found to induce aneuploidy in Drosophila (Osgood &
Sterling, 1991) at a concentration of 8.6 mg/litre. On the other hand,
DBAN produced inconsistent results but was tested at much lower
concentrations (0.3 mg/litre) because of its higher degree of
toxicity. Low levels of sodium cyanide (0.2 mg/litre) were also found
to be active in this test system. Since DCAN is metabolized to
cyanide, the authors suggested that the cyanide ion (CN-) was
responsible for the response. DCAN is somewhat more efficiently
converted to cyanide in rats than is DBAN (Pereira et al., 1984). This
difference would be multiplied by the much higher concentration of
DCAN that was tested.
Daniel et al. (1986) found that the HANs were direct-acting
electrophiles with the following decreasing order of reactivity: DBAN
>> BCAN > CAN >> DCAN >> TCAN. The ability to induce DNA strand
breakage in human CCRF-CEM cells was found to follow a considerably
different order: TCAN >> BCAN > DBAN > DCAN > CAN. It is of
interest that tumour-initiating activity paralleled alkylation
potential in a cell-free system rather than an ability to induce
strand breaks in DNA in intact cells or to induce mutation in
Salmonella (BCAN > DCAN >> DBAN > TCAN = CAN = 0). If CAN is
omitted from the group, mutagenicity parallels the extent to which the
HAN is converted to cyanide in vivo (Daniel et al., 1986). This
discordant set of parallels suggests that some property may be
affecting the ability of the test systems to measure the response or
that the responses are not mediated through a common mechanism.
Clearly, cytotoxic effects limited the responses of Salmonella to
the brominated HANs. Similar activity appeared to be affecting the
mouse skin initiation/promotion studies, but only after a fairly
robust response was observed. Some of the other effects may be only
loosely associated with a health effect. For example, the induction of
SSBs in DNA can arise from cytolethal effects. Moreover, such breaks
reflect DNA repair processes as well as damage. Therefore, it is
suggested that the carcinogenic potency of this class best parallels
alkylation potential.
TCAN was found to covalently bind with macromolecules in liver,
kidney and stomach of the F344 rat (Lin et al, 1992). The covalent
binding index was found to be the highest in the DNA of the stomach,
followed by the liver, and was lowest in the kidney. The binding of
14C was significantly higher when it was in the C2 position rather
than in C1, indicating that the nitrile carbon is lost. The adducts
formed were labile, and no specific adducts were identified. Adducts
to blood proteins were also observed with TCAN. Covalent binding of
DBAN or DCAN to DNA could not be demonstrated (Lin et al., 1986), but
binding to proteins was apparently not investigated with these HANs.
In conclusion, the HANs do possess carcinogenic and mutagenic
properties in short-term tests. However, without appropriate long-term
animal studies, the carcinogenic risk from HANs cannot be estimated.
4.4.4 Comparative pharmacokinetics and metabolism
The metabolism of the HANs has received some preliminary study,
but little information exists on the pharmacokinetics of the parent
compounds or their products. It is also important to note that some of
the HANs inhibit enzymes that are important in the metabolism of other
chemicals that are foreign to the body.
Pereira et al. (1984) found the following percentage of the
original doses of the HANs eliminated in the urine within 24 h as
thiocyanate: CAN, 14%; BCAN, 12.8%; DCAN, 9.3%; DBAN, 7.7%; and TCAN,
2.3%. This was compared with 42% of a dose of propionitrile. On the
basis of this limited information and a general scheme for the
elimination of cyanide from nitriles, published by Silver et al.
(1982), Pereira et al. (1984) proposed that additional products of HAN
metabolism would be as follows: CAN, formaldehyde; DHANs, formyl
cyanide or formyl halide; and TCAN, phosgene or cyanoformyl chloride.
These products would be direct-acting alkylating agents.
Roby et al. (1986) studied the metabolism and excretion of DCAN
labelled with 14C in either the C1 or C2 position in both male F344
rats and B6C3F1 mice. The metabolic fate of the two carbons was
significantly different in both mice and rats: C2 is metabolized much
more efficiently to carbon dioxide, whereas a very much higher
proportion of C1 is found as urinary metabolites, at least in mice.
These results are consistent with the proposal of Pereira et al.
(1984) suggesting metabolites that would be converted efficiently to
carbon dioxide from C2.
The HANs inhibit enzymes in the liver of the rat that are
traditionally associated with the metabolism of foreign compounds.
Pereira et al. (1984) demonstrated the inhibition of
dimethylnitrosamine demethylase activity. This activity has been
traditionally associated with the cytochrome P450 isoform 2E1,
although there was no direct confirmation of this in the study.
However, two forms of the enzyme activity were identified, one with a
Km of 2 × 10-5 and the other with a Km of 7 × 10-2. Based on a
plot presented in the paper, DBAN appears to be inhibiting the
high-affinity enzyme by either a non-competitive or uncompetitive
mechanism. The kinetics of inhibition were examined in vitro and the
enzyme : inhibitor dissociation constants ( Kis) reported to be as
follows: DBAN, 3 × 10-5; BCAN, 4 × 10-5; DCAN or TCAN, 2 × 10-4; and
CAN, 9 × 10-2. Although not commented upon by the authors, the
kinetics of inhibition by TCAN were clearly different from those of
the DHANs, suggesting some differences in the mechanism or the form of
the enzyme that might be affected. The authors examined the effects of
DBAN or TCAN administered orally to rats at doses of 0.75 mmol/kg of
body weight on the dimethylnitrosamine demethylase activity in the
liver at 3 and 10 h after administration. TCAN significantly reduced
the activity of the enzyme by about 30% at both time intervals, but
DBAN did not, despite the fact that it was the more potent inhibitor
in vitro. This could represent a difference in the extent to which
the two compounds are absorbed systemically, or it could be related to
the nature of the inhibition (i.e., reversible vs. irreversible).
Ahmed et al. (1989) demonstrated inhibition of cytosolic GSTs by
HANs in vitro. Doses (mmol/litre) at which 50% inhibition of the
activity of the enzyme GST occurred were as follows: DCAN, 2.49; TCAN,
0.34; DBAN, 0.82; CAN, >10; and BAN, >10. This latter observation
has not been established to occur in animals (Gao et al., 1996).
Activation and inactivation of various DBPs are catalysed by various
isoforms of GST (Pegram et al., 1997; Tong et al., 1998). In this
case, toxicity is reduced by inhibition of GST; however, mutagenic
activity of brominated THMs appears to depend upon a GSH pathway
(Pegram et al., 1997). Similarly, GSTs appear to play an important
role in the metabolism of HAAs (Tong et al., 1998).
While these effects on the metabolism of other DBPs could be of
importance, there are no data with which to relate these effects to
concentrations that would be encountered in drinking-water.
4.4.5 Mode of action
The induction of skin tumours appears closely correlated with the
alkylating potential within this class of chlorination by-products
(Daniel et al., 1986). This suggests that the carcinogenic activity of
the HANs may be related to mutagenic effects, despite the fact that
cytotoxicity limits the capability of test systems to detect such
activity. Cytotoxicity actually appears to inhibit the
tumour-initiating activity responses rather than to amplify them, as
has been seen with other DBPs. This suggests that the HANs retain some
specificity for inducing cytotoxic responses in initiated cells.
Another concern in this class is the ability of certain members
to induce developmental delays. At present, interpretation of these
results is somewhat clouded by the issues of interactions in the
toxicity of the test compound with its vehicles, as discussed in the
recent publication of Christ et al. (1996). No satisfactory
explanation has been offered for these results. Such effects may be
the result of fairly subtle changes in the pharmacokinetics and
metabolism of the compound, or, as Christ et al. (1996) suggest, TCAN
may have acted synergistically with a subthreshold effect of
tricaprylin on developmental processes. This is suggested by a
minimal, but consistent, response in treatment groups that received
tricaprylin only relative to a group of naive controls.
The potential importance of the HANs as cyanogens has not been
extensively explored. As noted above, Pereira et al. (1984) indicated
that significant portions of the dose of the HANs are eliminated as
thiocyanate in the urine. Thus, cyanide release could be contributing
significantly to the effects of these chemicals at high doses.
4.5 Halogenated hydroxyfuranone derivatives
The halogenated hydroxyfuranones were first identified in the
bleaching of pulp (Holmbom et al., 1984). The first member of this
class, MX, was found because of its high mutagenic activity in
Salmonella tester strains. In the same time frame, mutagenic
activity had been associated with the chlorination of drinking-water
(Meier, 1988). The high specific mutagenic activity of MX prompted
examination of the possibility that it could contribute to the
mutagenic activity that had been identified in chlorinated
drinking-water. Subsequent experimentation confirmed this hypothesis.
Estimates of the contribution of MX to the mutagenic activity in
drinking-water ranged from 3% to 57% (Hemming et al., 1986; Meier et
al., 1987a; Kronberg & Vartiainen, 1988). The lower estimates should
probably be discounted, because the methods for recovering MX and
other mutagens from water have varied. Moreover, it is important to
recognize that these estimates apply only to MX itself. A variety of
related compounds are produced that are also mutagens (Daniel et al.,
1991b; DeMarini et al., 1995; Suzuki & Nakanishi, 1995), but which
have received very little toxicological study. The contribution of
these related compounds has not been estimated.
The present section will focus on the research subsequent to that
which identified MX as an important chlorination by-product. This
review will confine itself to newer data that provide some perspective
on possible mechanisms of action in vivo. To avoid unnecessary
redundancy, other furanones will be discussed only as they have been
investigated to aid in an understanding of responses to MX.
4.5.1 General toxicological properties and information on
dose-response in animals
The lack of a commercial source of MX has limited research in
experimental animals. However, a number of in vivo studies and a
carcinogenesis study of MX have been published (Bull et al., 1995;
Komulainen et al., 1997). Reported epidemiological associations of
drinking-water mutagenicity with cancer of the gastrointestinal and
urinary tracts (Koivusalo et al., 1994b, 1996, 1997) provided
additional impetus for investigating the compound.
Three studies explicitly examined the acute toxicity of MX. The
oral LD50 for MX in Swiss-Webster mice was determined to be 128 mg/kg
of body weight when MX was administered for 2 consecutive days by
gavage (Meier et al., 1987b). Doses of 70% of the LD50 and less
(<90 mg/kg of body weight) had no significant effect on body weight
of the mice, nor were they lethal. Most mice died within 24 h of
receiving the first dose. Mice that died were found to have enlarged
stomachs with moderate haemorrhagic areas in the forestomach. Very
limited mortality was observed in weanling CD-1 mice administered a
single dose of 144 mg/kg of body weight (Mullins & Proudlock, 1990).
In this study, focal epithelial hyperplasia was observed in the
stomach, and some vacuolation of the superficial villus epithelium was
observed in the duodenum and jejunum. Evidence of increased numbers of
mitotic figures was observed in the liver, and the possibility of some
cytotoxicity was identified in the urinary bladder. Komulainen et al.
(1994) administered MX in distilled water to male Wistar rats. Rats
tolerated doses of 200 mg/kg of body weight administered by gavage
but displayed severe symptoms including dyspnoea, laborious breathing,
depressed motor activity and cyanosis at higher doses (Komulainen et
al 1994). At necropsy, gastrointestinal inflammation was observed,
and oedema was noted in the lungs and kidneys. An LD50 of 230 mg/kg
in 48 hours was identified in this study.
The study of Meier et al. (1996) examined the effects of a 14-day
course of MX treatment by gavage at a dose of 64 mg/kg of body weight
per day on a number of enzyme activities in the liver of rats. MX
treatment reduced hepatic levels of catalase, cytochrome P450
reductase, aminopyrine demethylase and aromatic hydrocarbon
hydroxylase. It did not affect fatty acyl CoA oxidase,
glutamylcysteine synthetase, GST or glutathione peroxidase. The main
result of such effects would be potential modifications of metabolism
of various xenobiotics and endogenous biochemicals.
A more extensive study of the effect of MX on enzyme activities
in various tissues was conducted by Heiskanen et al. (1995) in Wistar
rats. This study employed a constant daily dose of 30 mg/kg of body
weight administered by gavage for 18 weeks as the low dose, whereas
the higher dose was achieved by initiating treatment at 45 mg/kg of
body weight (7 weeks) and raising it to 60 mg/kg of body weight
(2 weeks) and to 75 mg/kg of body weight (5 weeks). A dose-related
decrease in ethoxyresorufin- O-deethylase activity was observed in
liver and kidney. MX appears to inhibit this enzyme's activity
directly based upon in vitro experiments conducted by the authors.
However, the high concentrations required (0.9 mmol/litre) are
unlikely to be approached systemically from the very low levels found
in chlorinated drinking-water. The treatment also increased the
activities of two phase 2 enzymes, uridine
diphosphate-glucuronosyltransferase and GST, in the kidneys in a
dose-dependent manner, but only in female rats. The health
consequences of such modifications are uncertain, but could reflect an
effect on physiological mechanisms associated with differences in sex.
Again, it is important to recognize that these effects were produced
at doses that were chronically, as well as acutely, toxic and unlikely
to be remotely approached at the low concentrations found in
chlorinated drinking-water.
In a subchronic (14-18 weeks) toxicity study, Wistar rats (15 per
sex per group) were given MX by gavage, 5 days per week, at doses of 0
or 30 mg/kg of body weight (low dose) for 18 weeks or, in the
high-dose group, at doses increasing from 45 to 75 mg/kg of body
weight over 14 weeks. The high dose was finally lethal (two males and
one female died) and caused hypersalivation, wheezing respiration,
emaciation and tangled fur in animals. Increased water consumption,
decreased body weights and food consumption, elevated plasma
cholesterol and triglycerides, and increased urine excretion were
noted in high-dose male rats. Urine specific gravity was decreased and
the relative weights of the liver and kidneys were increased in both
sexes at both doses in comparison with the controls. At both doses,
duodenal hyperplasia occurred in males and females, and slight focal
epithelial hyperplasia in the forestomach was observed in males.
Splenic atrophy and haemosiderosis were seen in two high-dose females,
and epithelial cell atypia was seen in the urinary bladder of one
high-dose male and female. The frequency of bone marrow polychromatic
erythrocytes with micronuclei was slightly increased only in low-dose
male rats (Vaittinen et al., 1995).
4.5.2 Toxicity in humans
There have been no studies of the effects of these compounds in
humans.
4.5.3 Carcinogenicity and mutagenicity
4.5.3.1 Studies in bacteria and mammalian cells in vitro
There have been extensive studies of the mutagenic activity of MX
and related chemicals. In vitro studies have extended knowledge
beyond the initial characterization of simple mutagenic responses to
(i) demonstrate effects in higher test systems, (ii) extend the data
to other genotoxic end-points, (ii) characterize the mutagenic lesions
produced in DNA and (iv) develop structural correlates. At higher
levels of biological organization, a limited number of studies have
been conducted to document that the genotoxic form of MX reaches the
systemic circulation and to measure mutagenic effects in particular
cell types in vivo.
Meier et al. (1987b) demonstrated that MX induced chromosomal
aberrations in CHO cells at concentrations as low as 4 µg/ml in
vitro. However, these authors were unable to demonstrate an
increased frequency of micronuclei in the bone marrow of Swiss-Webster
mice following two consecutive daily doses administered by gavage at
70% of the LD50 (90 mg/kg of body weight × 2). Treatment with
30-300 µmol of MX per litre (1 h) induced DNA damage in a
concentration-dependent manner in suspensions of rat hepatocytes. DNA
damage was induced in V79 Chinese hamster cells and in isolated rat
testicular cells at the same concentrations as in hepatocytes. V79
cells exposed to 2-5 µmol of MX per litre (2 h) showed an increased
frequency of SCE, whereas no significant effect on
hypoxanthine-guanine phosphoribosyltransferase mutation induction was
observed (Brunborg et al., 1991).
Watanabe et al. (1994) found that the mutagenic activity of MX
was effectively inhibited by sulfhydryl compounds such as cysteine,
cysteamine, GSH, dithiothreitol and 2-mercaptoethanol. Pre-incubation
of 0.5 µg of MX with 15 µg of cysteine in a phosphate buffer at 37°C
for 15 min prior to exposure of bacterial cells depleted the mutagenic
activity of MX. Together with the result showing a change in the UV
spectra, the authors suggested that sulfhydryl compounds inactivate MX
by direct chemical interaction before MX induces DNA damage. On the
other hand, a variety of antioxidants other than the sulfhydryl
compounds showed no inhibitory effects. Investigation using structural
analogues of cysteine revealed that the thiol moiety was indispensable
for antimutagenic activity, and the amino moiety appeared to enhance
the MX-inactivating reaction of the sulfhydryl group.
Incubation of both rat and mouse hepatocytes with MX in vitro
resulted in a dose-dependent increase in UDS at subcytotoxic
concentrations (1-10 µmol of MX per litre; 20-h incubation). Depletion
of GSH stores by pretreatment of rat hepatocytes with buthionine
sulfoximine did not result in a significant increase in UDS produced
by MX. In contrast, MX did not induce UDS in mouse hepatocytes ex
vivo either 3 or 16 h following administration of a single oral dose
of 100 mg of MX per kg of body weight. Despite the ability of MX to
produce repairable DNA damage, restricted access of MX to the liver
may prevent a measurable UDS response in vivo (Nunn et al., 1997).
Jansson et al. (1995) examined MX and a related compound,
3,4-(dichloro)-5-hydroxy-2(5H)-furanone (MA, also known as mucochloric
acid), in the CHO hypoxanthine phosphoribosyl transferase (hprt)
locus assay system where 6-thioguanine resistance (TGr) is the
parameter measured. Both MX and MA induced TGr mutants. Indirect
evidence was provided to suggest that the difference in sensitivity in
the bacterial systems was related to differential ability to repair
bulky adducts hypothesized to be induced by MX versus smaller adducts
suggested to occur as a result of MA treatment. These results are
interesting, considering the fact that Daniel et al. (1991b) found
that MX and MA were of approximately equivalent potencies in inducing
nuclear anomalies in gastrointestinal cells of the B6C3F1 mouse.
Harrington-Brock et al. (1995) recently examined MX in the
L5178/TK+1C3.7.2C mouse lymphoma system. A mutant frequency of 1027
per 106 surviving cells was found. There was, however, a predominance
of small-colony mutants. Small colonies more commonly arise from
clastogenic effects than from point mutations. In parallel
experiments, MX was found to have clastogenic effects (chromatid
breaks and rearrangements). Point mutations and clastogenic effects
were both observed at a medium concentration of 0.75 µg/ml.
MX and MA also induced micronuclei when applied to inflorescences
of pollen mother cells of Tradescantia (Helma et al., 1995). MX was
approximately 5 times as potent as MA in this assay system.
DeMarini et al. (1995) compared the mutation spectra induced by
MX and extracts of water treated with chlorine, chloramine, ozone or
ozone followed by chlorine or chloramine in Salmonella typhimurium
strains TA98 and TA100. The mutation spectra induced in the hisG46
codon displayed a predominance of the GAC mutation with MX, but there
were also significant increases in the CTC and to a lesser extent ACC
mutations. These latter mutations are not typical of MX. Since MX has
never been identified as a by-product of ozonation, it is somewhat
surprising that extracts of ozonated water produced a similar
spectrum, even though these extracts are much less potent than those
obtained from chlorinated or chloraminated water (i.e., they produced
net increases in mutant colonies that are only about twice the
spontaneous rate). The mutation spectra induced in the TA98
hisD3052 allele more clearly differentiated between ozone and
chlorinated or chloraminated water. In this case, virtually all of the
frameshift mutations induced by raw and ozonated water extracts
involved hotspot mutations, whereas only 30-50% of those induced by MX
or extracts from water that had been treated with chlorine or
chloramine involved the hotspot. Thus, the TA98 mutations are
consistent with the hypothesis that the chemicals responsible for
mutagenic effects of ozonated water are distinct from those induced by
chlorination by-products.
Hyttinen et al. (1996) found that MX and MA induce different
mutation spectra in the DNA of Salmonella typhimurium hisG46 codons
(target codon sequence is CCC). The predominant mutation was GAC
followed by ACC in MX-treated colonies, whereas CTC dominated the
spectra produced by MA. MX primarily induced G:C -> T:A
transversions, whereas MA produced G:C -> A:T transitions in the
second base of the codon. The G:C -> T:A transversion in
Salmonella was also observed in the hprt gene of CHO cells
(Hyttinen et al., 1996). Knasmuller et al. (1996) found the same
difference in the mutation spectra of MX and MA in S. typhimurium
mutants. These latter authors further found that
3-chloro-4-(chloromethyl)-5-hydroxy-2(5H)-furanone (CMCF) produced the
same transversion as MX, while chloromalonaldehyde produced the same
transition found with MA.
Some complex structure-activity relationships appear to occur
with these two types of halofuranones. Kronberg et al. (1993) found
that MA forms ethenocarbaldehyde derivatives with adenosine and
cytidine, with chloroacetaldehyde being an intermediate. As pointed
out by Knasmuller et al. (1996), ethenocytosine adducts formed by
vinyl chloride cause G:C -> A:T transitions as reported for MA. The
mutations induced by MX are similar to those produced by carcinogens
that form bulky adducts, such as benzo [a]pyrene and 4-aminobiphenyl.
Bulky adducts block replication leading to base substitutions
according to the "adenine rule" (Strauss, 1991). As a consequence,
there may be some significant differences in the health impact (e.g.,
tumour site or character if they are found to be carcinogenic) of the
mutagenic activities of these two chlorohydroxyfuranone derivatives.
Ishiguro et al. (1988) examined structure-activity relationships
for MX and related compounds. Their studies identified the chlorine
substitution on C3 as being very important to the mutagenic activity
of MX. Association of similar losses in mutagenic activity by removing
the analogous chlorine from an open-ring structure compound,
3-(dichloromethyl)-4,4-dichloro-2-chlorobutenoic acid, strongly
supported this hypothesis.
LaLonde and co-workers (LaLonde et al., 1991a,b, 1992; LaLonde &
Xie, 1992, 1993) conducted a series of experimental and computational
studies to relate the electronic structure of MX and related compounds
with mutagenic activity within the class. Substitutions for the
hydroxyl group led to reduction of mutagenic activity by a factor of
100. Removal of the C3 or C6 chlorines from the structure reduced
mutagenic activity by a factor of 10. Removal of the second chlorine
at C3 caused a very large further reduction of mutagenic activity (by
a factor of 1000) (LaLonde et al., 1991b). The mutagenicity appears to
depend on the electron density at C2, C3 or C4 based upon 13C
chemical shifts observed by nuclear magnetic resonance. Expansion of
these studies supported the hypothesis that the mutagenic properties
of the class paralleled the electrophilic character of chemicals
within the class and the ability to stabilize a radical anion
following acceptance of a single electron.
Another difference in the chemistry of MA and MX, which might
have biological implications, is that GSH readily displaces the
chlorine on C4 of MA, greatly reducing its electrophilicity. On the
other hand, GSH or N-acetylcysteine reacts with MX to produce
mixtures that are intractable to analysis, with the release of
hydrogen sulfide (LaLonde & Xie, 1993).
4.5.3.2 Studies in experimental animals
In vivo studies are of two types: those that use bacterial
systems to document absorption of MX (or mutagenic metabolite), and
those in which effects on the test animal are directly measured.
Fekadu et al. (1994) injected mixtures of repair-competent and
repair-deficient Escherichia coli K-12 cells intravenously into mice
as test cells, and the animals were subsequently treated with 200 mg
of test chemical per kg of body weight. Two hours later, the mice were
sacrificed and cells recovered from various organs. MX, CMCF and MA
were the test chemicals. The differential survival of the DNA
repair-deficient strain versus a repair-competent variant is used to
detect mutagenic activity. All three compounds significantly reduced
recovery of the repair-deficient strain in the stomach, lung,
intestine, liver, kidney and spleen. In a further experiment, the
effects of lower doses of MX (4.3, 13 and 40 mg/kg of body weight)
were investigated. Significantly depressed recovery was seen with MX
doses as low as 4.3 mg/kg of body weight. MA did not modify recovery
of the repair-deficient strain at doses less than or equal to 40 mg/kg
of body weight. These data suggest that significant amounts of MX or a
mutagenic metabolite reach the systematic circulation and at least
reach the extracellular water. They do not clearly demonstrate effects
in the target tissue of the experimental animal.
Meier et al. (1996) found that only 0.3% of the original dose was
excreted in a genotoxically active form in the urine of rats
administered MX at a dose of 64 mg/kg of body weight for 14 days by
gavage. No evidence of micronuclei induction was detected in
peripheral blood erythrocytes in mice treated with a similar protocol.
Whereas mutagenic activity was observed in urine at doses of 64 mg/kg
of body weight, no significant mutagenic activity was observed at
doses of 32 mg/kg of body weight and below.
Brunborg et al. (1990, 1991) studied DNA damage induced by MX and
other compounds in organs of rats using the alkaline elution assay (to
detect strand breaks). While clear evidence of strand breaks was
obtained with dibromochloropropane and
2-amino-3,4-dimethylimidazo[4,5- f]quinoline, no significant effects
were observed with MX after an intraperitoneal dose of 18 mg/kg of
body weight or at oral doses of up to 125 mg/kg of body weight. The
organs examined included the small and large intestine, stomach,
liver, kidney, lung, bone marrow, urinary bladder and testis.
Nishikawa et al. (1994) investigated cell proliferation and lipid
peroxidation in the glandular stomach mucosa in Wistar rats given 0,
6.25, 12.5, 25 or 50 mg of MX per litre in their drinking-water for
5 weeks. Statistically significant cell proliferation increased in a
dose-dependent manner up to 25 mg/litre. The MX treatment was also
associated with increased lipid peroxidation levels in the gastric
mucosa as well as in the urine, with loose dose dependence, although
not at 50 mg/litre. Histopathologically, gastric erosion was noted in
rats receiving 25 mg of MX per litre or more. These results suggest
that MX may exert a gastric tumour-promoting action in rats, even at
low doses that do not give rise to toxic effects, because of the clear
dose-response relationship evident at low levels.
The peripheral lymphocytes of male and female Han:Wistar rats
exposed to MX at 30 or 45-75 mg/kg of body weight per day by gavage, 5
days a week for 14-18 weeks, showed significant dose-related increases
in SCEs at both levels of exposure in both sexes (Jansson et al.,
1993).
The peripheral lymphocytes of male Han:Wistar rats exposed to MX
(25-150 mg/kg of body weight) by gavage on 3 consecutive days showed a
significant dose-related increase in chromosomal damage measured as
micronuclei, in addition to SCEs. Moreover, MX produced a significant
dose-related increase in SCEs in the kidney cells of the exposed rats.
However, the magnitude of the genotoxic responses observed was
relatively weak (Maki-Paakkanen & Jansson, 1995).
Daniel et al. (1991b) found that MX and MA induced nuclear
anomalies in the epithelial cells of the gastrointestinal tract of
B6C3F1 mice. Doses of 0.37 mmol/kg of body weight (approximately 80
mg/kg of body weight) produced a modest increase in nuclear anomalies
in the duodenum. There was no effect at 0.28 mmol/kg of body weight.
Mullins & Proudlock (1990) and Proudlock & Crouch (1990) also found
insignificant increases in nuclear anomalies in the non-glandular
stomach, urinary bladder, jejunum and ileum. These latter authors
noted that at the top dose at which nuclear anomalies were observed
(144 mg/kg of body weight), there was significant irritation,
inflammation and evidence of apoptotic cells in the gastrointestinal
tract. These changes render the significance of the observed nuclear
anomalies uncertain.
MX was administered to Wistar rats (50 per sex per group) in
drinking-water for 104 weeks at 0, 0.4, 1.3 or 5.0 mg/kg of body
weight per day for males and 0, 0.6, 1.9 or 6.6 mg/kg of body weight
per day for females. Dose-dependent increases in the incidence of some
tumours were observed in rats, while the same MX doses had no obvious
toxic effects on animals. Increases in tumours of the lung, mammary
gland, haematopoietic system, liver, pancreas, adrenal gland and
thyroid were observed, but few showed a clear dose-response (Table 21)
(Komulainen et al., 1997).
4.5.4 Comparative pharmacokinetics and metabolism
There are very few data on the metabolism and pharmacokinetics of
MX or related compounds. Ringhand et al. (1989) examined the
distribution of radioactivity derived from 3-14C-MX in male F344
rats. Approximately 35% of the radiolabel was eliminated in the urine
and 47% in the faeces, with about 6% remaining in the body after 48 h.
Neither the parent compound nor any specific metabolites were
identified in any body compartment or fluid.
Horth et al. (1991) studied the disposition of 3-14C-MX in male
CD-1 mice. 14C was rapidly absorbed, reaching peak values in blood
within 15 min of its administration. There was some evidence for
binding to protein and retention of label within tissues, but no
attempt was made to identify the chemical form in which the 14C was
bound, so it was not clear whether MX was binding by virtue of its
electrophilic character or whether this represented metabolic
incorporation of metabolites of MX. Approximately 57% of the
radioactivity was eliminated in the urine and 28% in the faeces. Less
than 1% of the initial dose was retained in the carcass 120 h after
administration, but most of this was associated with the stomach. It
was stated that the urinary metabolites were polar, but no specific
identifications were made.
Komulainen et al. (1992) evaluated the pharmacokinetics of MX
after a single oral or intravenous administration in Han:Wistar rats
using 14C-labelled compound. Approximately 20-35% of the dose was
absorbed into circulation from the gastrointestinal tract. The mean
elimination half-life of the radioactivity in blood was 3.8 h. Traces
of radioactivity remained in the blood for several days. The tissues
lining the gastrointestinal and urinary tracts, kidney, stomach, small
intestine and urinary bladder contained the highest radioactivity. The
activity declined most slowly in the kidneys. Urine was the main
excretion route, with 77% of the total radioactivity appearing in
urine in 12 h and 90% in 24 h. No radioactivity was exhaled in air.
After an intravenous administration of 14C-MX, the mean elimination
half-life was much longer, 22.9 h, and the total elimination half-life
was 42.1 h. Results indicate that MX is absorbed from the
gastrointestinal tract to a considerable degree and is excreted in
urine very rapidly. A fraction of MX or its metabolites is retained in
blood for a longer period of time.
No data are available in the scientific literature on the
metabolism of MX or related compounds in humans.
In conclusion, there are data to suggest that MX or a
mutagenically active metabolite reaches the systemic circulation in
experimental animals. Mutagenic activity has been detected in various
organs and tissues using doses as low as 4.3 mg/kg of body weight
(Fekadu et al., 1994). If these data are to have application to
estimation of the hazards that MX presents to humans consuming
chlorinated drinking-water, it is essential to understand whether MX
or a metabolite reaches critical targets in the human body. An
essential component of the information required would be an
understanding of the metabolism and pharmacokinetics of MX and those
of critical metabolites. The available data are too limited to provide
much more than very general guidance in this area.
4.6 Chlorite
4.6.1 General toxicological properties and information on
dose-response in animals
Concerns over chlorite in drinking-water first arose as chlorine
dioxide began to play a role in the primary disinfection of
drinking-water. Chlorite is the principal by-product of oxidative
reactions of chlorine dioxide, but acidification of chlorite solutions
is one method for generating chlorine dioxide for purposes of water
disinfection (Aieta & Berg, 1986).
Unless otherwise noted, references to chlorite will generally be
to the sodium salt. This is the form most frequently studied. The term
chlorite will be used if the authors expressed their doses in terms of
chlorite; if dose levels were expressed as sodium chlorite, they will
be identified as such. There is no reason to suspect that other salts
of chlorite would exert inherently different toxicological effects, so
this convention should not lead to confusion. A preparation of sodium
chlorite with lactic acid is specifically excluded from consideration
here because the actual composition of the product has not been
specified (Scatina et al., 1984). Consequently, it is not clear that
the data on this product have any relevance to chlorite or vice versa.
Table 21. Summary of primary tumours observed in selected tissues in male rats after exposure to
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX) in drinking-water for 104 weeksa
Tissue Control MX (mg/kg of body weight per day) Pb
0.4 1.3 5.0
Integumentary system
Skin, subcutaneous tissuec 50 50 50 50
Basal cell tumourd 1 (2%) 1 (2%) 3 (6%) 0.0314
Mammary glandsc 50 50 50 49
Adenocarcinomad 1 (2%) 0.3162
Fibroadenomad 1 (2%) 3 (6%) 1 (2%) 0.2996
Fibromad 1 (2%) 0.4749
Respiratory system
Lungsc 50 50 50 50
Alveolar & bronchiolar carcinomasd 1 (2%)
Alveolar & bronchiolar adenomasd 2 (4%) 1 (2%) 1 (2%) 7 (14%) 0.0015
Haematopoietic system
Multiple tissuesc 50 50 50 50
Lymphoma & leukaemiad 3 (6%) 4 (8%) 3 (6%) 0.1527
Digestive system
Liverc 50 50 50 50
Carcinomad 2 (4%) 1 (2%) 0.1605
Hepatocholangiocarcinomad 1 (2%) 0.4897
Cholangiomad 1 (2%) 4 (8%) 0.0009
Adenomad 1 (2%) 2 (4%) 4 (8%) 0.0142
Pancreasc 50 50 50 50
Langerhans' cell carcinomad 4 (8%) 3 (6%) 5 (10%) 4 (8%) 0.3769
Langerhans' cell adenomad 5 (10%) 8 (16%) 8 (16%) 12 (24%) 0.0116
Acinar cell adenomad 2 (4%) 3 (6%) 2 (4%) 4 (8%) 0.1243
Table 21. (continued)
Tissue Control MX (mg/kg of body weight per day) Pb
0.4 1.3 5.0
Endocrine system
Adrenal glandsc 50 50 50 50
Pheochromocytoma, malignantd 1 (2%) 0.3213
Pheochromocytoma, benignd 5 (10%) 2 (4%) 9 (18%) 3 (6%) 0.4830
Cortical carcinomad 2 (4%) 1 (2%) 0.9447
Cortical adenomad 5 (10%) 2 (4%) 7 (14%) 14 (28%) 0.0001
Thyroid glandsc 49 50 50 50
Follicular carcinomad 1 (2%) 9 (18%) 27 (55%) 0.0000
Follicular adenomad 2 (4%) 20 (40%) 34 (68%) 21 (43%) 0.0045
C-cell carcinomad 2 (4%) 0.4478
C-cell adenomad 11 (22%) 7 (14%) 10 (20%) 11 (22%) 0.2459
a From Komulainen et al. (1997).
b P value from the one-sided trend test. A statistically positive trend at P < 0.05 or lower.
c Values (reading across) = number of animals analysed.
d Values (reading across) = number of animals with one or more indicated tumours (frequency of animals
with tumour as percentage of examined animals.
Early investigations of chlorite's toxic properties focused
almost entirely on it potential ability to produce methaemoglobin and
haemolysis. Heffernan et al. (1979a) examined the ability of sodium
chlorite to induce methaemoglobinaemia in cats and Sprague-Dawley
rats. When administered to cats as an oral bolus, as little as 20 mg
of sodium chlorite per kg of body weight resulted in formation of
significant amounts of methaemoglobin. Intraperitoneal doses of
20 mg/kg of body weight in rats also induced methaemoglobin formation.
However, when administered in drinking-water, no significant elevation
in methaemoglobin was observed in cats (up to 1000 mg/litre as sodium
chlorite) or rats (up to 500 mg/litre). Thus, chlorite must enter into
the systemic circulation at a rapid rate, i.e., as a bolus dose, to
induce methaemoglobin formation.
Short-term toxic effects of chlorite were more systematically
assessed in rats using gavage doses ranging from 25 to 200 mg/kg of
body weight (Harrington et al., 1995a). Minor effects were observed at
25 and 50 mg/kg of body weight. At 100 mg/kg of body weight and above,
signs of haemolytic anaemia became apparent, with decreases in red
blood cell count, haemoglobin concentration and haematocrit. These
data support the idea that bolus doses were necessary to determine
substantial effects on oxidative stress.
Treatment of both cats and rats with sodium chlorite in
drinking-water for extended periods (up to 90 days) resulted in
decreases in red blood cell counts, haemoglobin concentrations and
packed cell volume. These effects were observed with 500 mg of sodium
chlorite per litre in cats (equivalent to 7 mg/kg of body weight per
day) and with as little as 100 mg/litre in rats (equivalent to 10
mg/kg of body weight per day) (Heffernan et al., 1979a). The changes
in these blood parameters appeared to generally decrease in severity
as the treatment was extended from 30 to 90 days, suggesting that
adaptation to the treatment was occurring. In rats, however, red blood
cell glutathione concentrations remained significantly depressed and
2,3-diphosphoglycerate levels elevated through 90 days of treatment
with concentrations of sodium chlorite as low as 50 mg/litre (5 mg/kg
of body weight per day). In the cat, increased turnover of
erythrocytes was detectable at concentrations of 100 mg/litre, with no
significant effect being observed at 10 mg/litre. This latter
concentration resulted in a daily dose of 0.6 mg/kg of body weight per
day. Red blood cells drawn from rats treated with 100 mg/litre had
significantly less ability to detoxify hydrogen peroxide that was
generated by the addition of chlorite in vitro. These data show that
while the anaemia caused by haemolysis was largely compensated for in
subchronic treatment of healthy rats, there was still evidence of
oxidative stress being exerted by the chlorite treatment. Depletion of
this reserve capacity could be of importance in individuals who are in
a more compromised state (e.g., glucose-6-phosphate dehydrogenase
deficiency) or who might be exposed to other haemolytic agents. The
lowest concentration at which GSH was depleted significantly from
control levels in rats was 50 mg/litre, and no effect was observed at
10 mg/litre (equivalent to 1 mg/kg of body weight per day).
The results of Heffernan et al. (1979a) have been generally
confirmed by subsequent studies in a variety of species. Abdel-Rahman
et al. (1980) and Couri & Abdel-Rahman (1980) obtained very similar
effects in rats treated for up to 11 months. Moore & Calabrese (1980,
1982) produced similar results in mice, and Bercz et al. (1982)
demonstrated reduced red blood cell counts and decreased haemoglobin
levels at similar doses in African green monkeys. These studies tended
to identify altered forms of erythrocytes that are commonly associated
with oxidative damage at treatment doses below those that produced
actual anaemia (most consistently at a concentration of 100 mg/litre,
with hints of such effects at lower doses).
A more recent study employed doses of sodium chlorite
administered by gavage to male and female Crl: CD (SD) BR rats (15 per
sex per group) (Harrington et al., 1995a). Doses of 0, 10, 25 or 80 mg
of sodium chlorite per kg of body weight per day were administered
daily by gavage for 13 weeks (equivalent to 0, 7.4, 18.6 or 59.7 mg of
chlorite per kg of body weight per day). This study is important
because it included many of the standard parameters of subchronic
toxicological studies, whereas previous studies had focused almost
entirely on blood parameters. A gavage dose of 80 mg/kg of body weight
per day produced death in a number of animals. It also resulted in
morphological changes in erythrocytes and significant decreases in
haemoglobin concentrations. Red blood cell counts were reduced
slightly, but not significantly, at doses of 10 mg/kg of body weight
per day in male rats, with further decreases being observed at 80
mg/kg of body weight per day. Red blood cell counts were significantly
depressed in female rats at doses of 25 mg/kg of body weight per day
and above. As would be expected where haemolysis is occurring, splenic
weights were increased. Adrenal weights were increased in females at
25 and 80 mg/kg of body weight per day, whereas statistically
significant changes were observed only at 80 mg/kg of body weight per
day in males. Histopathological examination of necropsied tissues
revealed squamous cell epithelial hyperplasia, hyperkeratosis,
ulceration, chronic inflammation and oedema in the stomach of 7 out of
15 males and 8 out of 15 females given 80 mg/kg of body weight per day
doses. This effect was observed in only 2 out of 15 animals at the 25
mg/kg of body weight per day dose and was not observed at all at 10
mg/kg of body weight per day. Microscopic evaluations were made in 40
additional tissues, and no treatment-related abnormalities were found.
The Harrington et al. (1995a) study confirms the essential
findings of previous studies and, in retrospect, justifies their focus
on the blood cells as the critical target for chlorite toxicity. It
also confirmed negative results of other studies that failed to
identify significant effects in investigations of particular target
organs (Moore et al., 1984; Connor et al., 1985).
4.6.2 Reproductive and developmental toxicity
Sodium chlorite did not exert any spermatotoxic effects in
short-duration (1-5 days) tests (Linder et al., 1992).
Moore et al. (1980b) reported that sodium chlorite administered
at a concentration of 100 mg/litre throughout gestation and through
28 days of lactation reduced the conception rate and the number of
pups alive at weaning in A/J mice. A significantly reduced pup weight
at weaning was interpreted as indicating that chlorite retarded growth
rate.
Groups of 4-13 Sprague-Dawley rats were treated on gestation days
8-15 with sodium chlorite at concentrations of 0, 100, 500 or 2000
mg/litre in drinking-water, by injection of 10, 20 or 50 mg/kg of body
weight per day intraperitoneally or by gavaging with 200 mg/kg of body
weight per day. Calculated daily doses of sodium chlorite administered
to pregnant rats in drinking-water were 0, 34, 163 or 212 mg. Rats
body weights were approximately 0.3 kg, giving estimated doses of 0,
110, 540 or 710 mg/kg of body weight per day. Sodium chlorite at 20 or
50 mg/kg of body weight per day intraperitoneally or at 200 mg/kg of
body weight per day by gavage caused vaginal and urethral bleeding.
Doses of 10, 20 and 50 mg/kg of body weight per day intraperitoneally
caused 0%, 50% and 100% mortality of dams, respectively. No deaths
were caused by sodium chlorite in the drinking-water, but the body
weight and food consumption of the dams were decreased at 500 and 2000
mg/litre. Blood smears from the dams injected intraperitoneally with
all doses or drinking water containing 2000 mg of sodium chlorite per
litre showed irregular, bizarre and ruptured erythrocytes. Injection
of 10 or 20 mg/kg of body weight per day or drinking a solution
containing 2000 mg/litre resulted in a decrease in litter size and an
increase in stillbirths and resorption sites. Drinking 100 or 500 mg
of sodium chlorite per litre did not produce any significant
embryotoxicity. With all treatments, no significant gross soft tissue
or skeletal malformations were observed. Postnatal growth of the pups
was not affected by any treatment of the dams during the gestation
period (Couri et al., 1982a,b).
The effects of chlorite at 1 or 10 mg/litre in drinking-water for
2.5 months prior to mating and throughout gestation were studied in
Sprague-Dawley rats (Suh et al., 1983). This study indicated an
increase in the incidence of anomalies in fetuses at both
concentrations in two separate experiments; however, because the
treatment groups were small (6-9 pregnant females per group), the
effects were not considered statistically significant. Moreover, there
were no consistent differences in either skeletal or soft tissue
anomalies.
Male and female Long-Evans rats were given 0, 1, 10 or 100 mg of
sodium chlorite per litre of drinking-water. Males were exposed for 56
days before mating and during 10 days of mating; females were treated
for 14 days prior to mating, throughout the 10-day breeding period and
gestation and through to day 21 of lactation. Males were evaluated for
sperm parameters and reproductive tract histopathology following the
breeding period. Dams and pups were necropsied at weaning. There was
no effect on fertility, litter size or survival of neonates or on the
weight of the testis, epididymis or cauda epididymis when males
treated as described above were mated with these females. Decreases in
the concentrations of triiodothyronine and thyroxine in blood were
observed on postnatal days 21 and 40 in male and female pups exposed
to 100 mg/litre. There were no effects at lower doses. Additionally,
groups of males were exposed to 0, 10, 100 or 500 mg of sodium
chlorite per litre for 72-76 days to confirm subtle observed changes
in sperm count, morphology and movement. A significant increase in the
percentage of abnormal sperm morphology and a decrease in the
progressive sperm motility were observed for adult males at 100 and
500 mg/litre (Carlton et al., 1987).
Mobley et al. (1990) exposed groups of female Sprague-Dawley rats
(12 per group) for 9 weeks to drinking-water containing 0, 20 or 40 mg
of sodium chlorite per litre (0, 3 or 6 mg of chlorite per kg of body
weight per day) beginning 10 days prior to breeding with untreated
males and until the pups were sacrificed at 35-42 days
post-conception. Animals exposed to a dose of 6 mg/kg of body weight
per day exhibited a consistent and significant depression in
exploratory behaviour on post-conception days 36-39. Exploratory
activity was comparable between treated and control groups after
post-conception day 39.
In a two-generation study conducted by CMA (1997) and described
in TERA (1998), Sprague-Dawley rats (30 per sex per dose) received
drinking-water containing 0, 35, 70 or 300 mg of sodium chlorite per
litre for 10 weeks and were then paired for mating. Males were exposed
through mating, then sacrificed. Exposure for the females continued
through mating, pregnancy, lactation and until necropsy following
weaning of their litters. Twenty-five males and females from each of
the first 25 litters to be weaned in a treatment group were chosen to
produce the F1 generation. The F1 pups were continued on the same
treatment regimen as their parents. At approximately 14 weeks of age,
they were mated to produce the F2a generation. Because of a reduced
number of litters in the 70 mg/litre F1-F2a generation, the F1
animals were remated following weaning of the F2a generation to
produce the F2b generation. Doses for the F0 animals were 0, 3.0,
5.6 or 20.0 mg of chlorite per kg of body weight per day for males and
0, 3.8, 7.5 or 28.6 mg of chlorite per kg of body weight per day for
females. For the F1 animals, doses were 0, 2.9, 5.9 or 22.7 mg of
chlorite per kg of body weight per day for males and 0, 3.8, 7.9 or
28.6 mg of chlorite per kg of body weight per day for females. There
were reductions in water consumption, food consumption and body weight
gain in both sexes in all generations at various times throughout the
experiment, primarily in the 70 and 300 mg/litre groups; these were
attributed to a lack of palatability of the water. At 300 mg/litre,
reduced pup survival, reduced body weight at birth and throughout
lactation in F1 and F2, lower thymus and spleen weights in both
generations, lowered incidence of pups exhibiting a normal righting
reflex, delays in sexual development in males and females in F1 and
F2, and lower red blood cell parameters in F1 were noted.
Significant reductions in absolute and relative liver weights in F0
females and F1 males and females, reduced absolute brain weights in
F1 and F2, and a decrease in the maximum response to an auditory
startle stimulus on postnatal day 24 but not at postnatal day 60 were
noted in the 300 and 70 mg/litre groups. Minor changes in red blood
cell parameters in the F1 generation were seen at 35 and 70 mg/litre,
but these appear to be within normal ranges based on historical data.
The NOAEL in this study was 35 mg/litre (2.9 mg/kg of body weight per
day), based on lower auditory startle amplitude, decreased absolute
brain weight in the F1 and F2 generations, and altered liver weights
in two generations.
Harrington et al. (1995b) examined the developmental toxicity of
chlorite in New Zealand white rabbits. The rabbits (16 per group) were
treated with 0, 200, 600 or 1200 mg of sodium chlorite per litre in
their drinking-water (equal to 0, 10, 26 or 40 mg of chlorite per kg
of body weight per day) from day 7 to day 19 of pregnancy. The animals
were necropsied on day 28. There were no dose-related increases in
defects identified. Minor skeletal anomalies were observed as the
concentration of chlorite in water was increased and food consumption
was depressed.
4.6.3 Toxicity in humans
The effects of chlorite have received some attention in
toxicological and epidemiological investigations in human subjects.
All of these studies were conducted at doses within an order of
magnitude of concentrations of chlorite that might be expected in
water supplies disinfected with chlorine dioxide. None pushed the
limit of tolerance such that clear effects were observed. As a
consequence, they are not informative for establishing a margin of
safety.
An experimental epidemiological study was conducted in the USA in
a small city that had been using chlorine dioxide for some time in the
summer months (April to October) to avoid taste and odour problems
associated with the use of chlorine (Michael et al., 1981). Chlorine
dioxide was generated from sodium chlorite that was mixed with
chlorine gas and metered into the water. During the active use of
chlorine dioxide, the chlorite concentrations in the water averaged
5.2 mg/litre (range about 3-7 mg/litre). Subjects were monitored for
11 parameters: haematocrit, haemoglobin, red cell count, white cell
count, mean corpuscular volume, methaemoglobin, BUN, serum creatinine,
total bilirubin, reticulocyte count and osmotic fragility of red blood
cells. No effects could be associated with the switch of treatment
from chlorine to chlorine dioxide disinfection. A total of 197 people
were monitored in the exposed population, and there were 112
non-exposed individuals. Each person served as his/her own control.
Chlorine dioxide, free chlorine, chloramine and chlorate
concentrations were also measured and were found to be 0.3-1.1,
0.5-0.9, 0.9-1.8 and 0.3-1.8 mg/litre, respectively. The sampling for
clinical measurements was done 1 week before chlorine dioxide
disinfection began and 10 weeks into the cycle. Water samples taken
during weeks 10-13 had chlorite levels that were systematically
somewhat below those observed in the prior 9 weeks of sampling, and
the same general trend was observable in other measures of chlorine
dioxide and chlorate. This was not observed with chlorine or
chloramines, suggesting that some change in water treatment had
occurred. The authors provided no explanation for this change in water
quality, but, since clinical samples were taken in week 10, this
change in water quality could have resulted in lower exposure to
chlorite and chlorate.
The second set of evaluations of chlorite in humans involved
direct administration of sodium chlorite in a rising-dose tolerance
study and a follow-up study in which volunteers were treated for 12
weeks, which were reported on in several publications. Lubbers et al.
(1981, 1982) provided an overview of the studies. The detailed results
of the rising-dose tolerance study were reported in Lubbers &
Bianchine (1984), and those of the repeated-dose study in Lubbers et
al. (1982, 1984a). A fourth paper (Lubbers et al., 1984b) reported
results for three male volunteers that had glucose-6-phosphate
dehydrogenase deficiency.
The rising-dose tolerance study (Lubbers & Bianchine, 1984)
involved administration of progressively increasing single doses of
chlorite (0.01, 0.1, 0.5, 1.0, 1.8 or 2.4 mg/litre) in two 500-ml
portions to a group of 10 healthy adult male volunteers. Doses were
administered on days 1, 4, 7, 10, 13 and 16. In the interval between
doses, clinical evaluations of the subjects were performed and a
battery of clinical chemistry tests was performed on blood and urine
samples. These latter tests were primarily directed at potential
haematological effects of chlorite, but serum thyroxine and uptake of
triiodothyronine were also determined. In addition, blood pressure,
ECGs and other physiological parameters were monitored. No
treatment-related effects were observed.
In the repeated-dose study (Lubbers et al., 1984a), 10 male
volunteers were administered 5 mg of chlorite per litre in a 500-ml
portion for 12 weeks (0.036 mg/kg of body weight per day). Physical
examinations and blood and urine analyses were conducted throughout
the duration of treatment and for 8 weeks following the last dose of
the solutions. None of the parameters investigated was found to fall
outside the normal range; although there were some consistent changes
in values with time, none of these appeared to be related to chlorite
treatment.
Three individuals with glucose-6-phosphate dehydrogenase
deficiency were identified in the course of the study. This genetic
disorder makes individuals more sensitive to oxidative damage, which
is frequently manifested as increased methaemoglobin production and
haemolysis when the individuals are exposed to oxidative chemicals in
sufficient doses. All three individuals were treated with chlorite in
the same concentrations and in the same manner as described for the
study of normal individuals (Lubbers et al., 1984b). No clinically
significant changes were found in these individuals.
A study (Ames & Stratton, 1987) was conducted of renal dialysis
patients in California (USA) after a water district introduced
chlorine dioxide as a drinking-water disinfectant but failed to inform
the clinic for 12 months. Water treatment at the clinic consisted of
ion exchange, GAC, 5-µm filtration and reverse osmosis. Chlorite
levels measured after this treatment were 0.02-0.08 mg/litre, but
there were periods during which no chlorine dioxide was added, and
exposures to the patients may have been lower. Measures for 28 serum
and haematological parameters were available for 17 renal dialysis
patients for a period of 3 months before and 1 month after exposure.
Methaemoglobin measures were not available. Only one measure was
statistically associated with the use of water disinfected by chlorine
dioxide: serum uric acid declined by 10% after exposure to disinfected
water, a change that was not considered clinically important. The
study found no evidence of anaemia or other adverse effects of
chlorine dioxide-disinfected water for these renal dialysis patients,
but the interpretation of these results is severely limited because of
the small sample size and apparently very low exposures.
Collectively, these studies suggest that humans are probably not
sensitive to the concentrations of chlorite that are likely to be
found in water disinfected with chlorine dioxide. Some safety factor
is present in these data, because it is unlikely that concentrations
of chlorite would exceed 1 mg/litre with new methods of application.
However, these studies provide little information relative to the
actual margin of safety that exists between those concentrations seen
or administered and concentrations that would lead to clear adverse
effects. Consequently, these studies do not imply that the
concentrations of chlorite in drinking-water should be without limits.
4.6.4 Carcinogenicity and mutagenicity
Sodium chlorite was reported to produce a concentration-dependent
increase in revertants in Salmonella typhimurium strain TA100 in
both the presence and absence of rat liver S9 fraction (Ishidate et
al., 1984). A linear dose-response curve was observed, and the net
number of revertants produced at 0.3 mg per plate was 88. The S9 mix
used for metabolic activation was from the liver of F344 rats
pretreated for 5 days with polychlorinated biphenyls at 500 mg/kg of
body weight.
Meier et al. (1985b) evaluated chlorite in the mouse micronucleus
assay, the mouse bone marrow cytogenetics assay and the mouse sperm
head abnormality assay. The doses administered were 0.2, 0.5 or 1 mg
per mouse or approximately 40 mg/kg of body weight at the highest
dose. No statistically significant results were found in any of the
tests. In a later reference, it was indicated that chlorite also
induced chromosomal aberrations (Kurokawa et al., 1986b), but the data
were not provided. Hayashi et al. (1988, 1989) found an increase in
micronuclei in the bone marrow of mice given 0, 7.5, 15, 30 or 60
mg/kg of body weight by intraperitoneal injection at doses of 15 and
30 mg/kg of body weight. In a repeat study in which mice were given 0
or 15 mg/kg of body weight on 4 successive days, no increase in
micronuclei was observed. In a study using the oral route with doses
of 0, 37.5, 75, 150 or 300 mg/kg of body weight, a significant
increase in micronuclei was observed only at 150 mg/kg of body weight.
In a carcinogenicity study, sodium chlorite was administered to
F344 rats (50 per sex per dose) at concentrations of 0, 300 or
600 mg/litre of drinking-water (equivalent to 0, 18 or 32 and 0, 28 or
41 mg of chlorite per kg of body weight per day in males and females,
respectively) and to B6C3F1 mice (50 per sex per dose) at
concentrations of 250 or 500 mg/litre (equivalent to 0, 36 or 71 mg/kg
of body weight per day) for 85 weeks (Kurokawa et al., 1986b). The
rats became infected with a Sendai virus in all groups, which resulted
in the termination of the study after only 85 weeks. There was a
statistically significant increase in the incidence of hyperplastic
nodules in male mice treated with 250 mg/litre, but not in females.
The incidence of these lesions did not increase when the dose of
chlorite was increased to 500 mg/litre. Hepatocellular carcinomas were
too few for their observation to add anything substantive to the
evaluation. There were no other treatment-related changes in the
incidence of other tumours in either male or female mice.
Groups of 50 male and female B6C3F1 mice were given 0, 250 or
500 mg of sodium chlorite per litre in the drinking-water for 80 weeks
(0, 36 or 71 mg of chlorite per kg of body weight per day). A small,
but statistically significant ( p < 0.05), increase in the incidence
of lung adenomas was observed at 500 mg/litre. The authors noted that
this was not accompanied by the appearance of lung adenocarcinomas and
that the incidence was within the range of historical controls; thus,
it was not possible to conclude from these data that chlorite induced
lung tumours (Yokose et al., 1987).
In an associated experiment, Kurokawa et al. (1984) assessed the
ability of sodium chlorite to promote skin tumours in a group of
20 female Sencar mice. These mice were initiated with a single topical
application of 20 nmol (5.1 µg) of dimethylbenzanthracene in acetone
followed by 0.2-ml applications of sodium chlorite at 20 mg/ml in
acetone twice weekly for 51 weeks. A group of 15 female mice given a
single application of dimethylbenzanthracene followed by applications
of acetone were used as controls. This treatment resulted in 5 of 25
mice having squamous cell carcinomas at 52 weeks. No tumours were
found in the corresponding initiated control mice. Both TPA and
benzoyl peroxide produced increased tumour incidence in
dimethylbenzanthracene-initiated mice. These data indicate the
potential for a weak tumour-promoting activity for sodium chlorite.
However, no dose-response information has been forthcoming in the
literature.
4.6.5 Comparative pharmacokinetics and metabolism
Some limited data on the absorption, distribution and excretion
of chlorite have been developed in rats using 36Cl-labelled chlorite.
The label was absorbed with a half-life of about 3.5 min and
eliminated with a terminal half-life of 35.2 h (Abdel-Rahman et al.,
1982b, 1984b). In 72 h, approximately 35% of the label was recovered
in the urine and another 5% in the faeces. In the urine, 32% of the
administered dose was determined to be chloride, whereas 6% was found
to be chlorite, utilizing a fractionation procedure developed in a
prior study (Abdel-Rahman et al., 1980). While these studies did not
determine the form of the radiolabel found in blood, plasma and
tissues, it was clear that there were significant differences in the
behaviour of the label derived from chlorite and chlorate. However,
the efforts in this area have been seriously hampered by the lack of
an analytical method to discriminate between chlorine dioxide,
chlorite, chlorate and chloride in vivo.
4.6.6 Mode of action
The adverse effects of chlorite appear to be mediated through its
activity as an oxidant. However, this question has received very
limited attention, except for the involvement of oxidation in its
haematological effects. Heffernan et al. (1979b) demonstrated that
chlorite was consumed during the oxidation of haemoglobin to
methaemoglobin in vitro. It was also observed that, unlike
methaemoglobin induction by nitrite, the action of chlorite also
depleted the red blood cells of GSH, and that this could be partially
counteracted by including glucose in the incubation medium. The
oxidative action of chlorite could be associated with the production
of hydrogen peroxide as measured by the formation of complex I with
catalase. This production of hydrogen peroxide was associated with
oxidative damage by demonstrating that it could also be attenuated by
the inclusion of glucose in the medium. In a dose-response comparison,
it could be demonstrated that the loss of GSH and the loss of catalase
activity paralleled one another and occurred at concentrations an
order of magnitude lower than those required for methaemoglobin
formation. This is consistent with the behaviour of other oxidants
that produce haemolytic anaemia. These observations also appear to
explain why destruction of the red blood cell (measured as decreased
haematocrit, decreased haemoglobin concentrations and increased red
blood cell turnover) is a much more sensitive and important measure of
chlorite toxicity than methaemoglobin formation.
4.7 Chlorate
4.7.1 General toxicological properties and information on
dose-response in animals
Toxicological data on chlorate in the open scientific literature
are limited to two short-term studies in dogs (Sheahan et al., 1971;
Heywood et al., 1972) and a series of studies that focused primarily
on its ability to induce oxidative damage in the blood of rats and
chickens (Abdel-Rahman et al., 1980; Couri & Abdel-Rahman, 1980) and
African green monkeys (Bercz et al., 1982). Two short-term studies,
one in dogs and one in rats, carried out by Bio/Dynamics Inc. in 1987,
were reviewed by WHO (1996). In the dog study, a NOAEL of 360 mg/kg of
body weight was identified based on no significant effects on any
measured parameter. In the rat study, a NOAEL of 100 mg/kg of body
weight was identified based on haematological effects at the highest
dose (1000 mg/kg of body weight). There is also a single subchronic
study of toxicity conducted in rats in which histopathological
examination of tissues was performed (McCauley et al., 1995). This
limited data set has hindered attempts to establish a guideline value
for chlorate in drinking-water (WHO, 1993).
The studies in dogs documented the fact that high acute doses of
1 or 2 g/kg of body weight induce methaemoglobinaemia (Sheahan et al.,
1971; Heywood et al., 1972). In addition, Heywood et al. (1972)
administered lower doses (200-300 mg/kg of body weight) for 5 days and
observed no clinical signs at doses lower than 300 mg/kg of body
weight per day. Those doses that produced some evidence of
methaemoglobinaemia were also found to have produced some
discoloration of the kidneys and haematogenous cases in renal tubules
at necropsy.
No consistent effects were observed when chlorate was
administered to rats at concentrations of 10 or 100 mg/litre
(equivalent to 1 or 10 mg/kg of body weight per day) for 12 months
(Couri & Abdel-Rahman, 1980). These authors documented some loss of
the normal sensitivity of erythrocytes to osmotic shock at these
doses, however.
Bercz et al. (1982) administered drinking-water containing sodium
chlorate to African green monkeys for a total of 8 weeks in a
rising-dose experiment. Drinking-water concentrations were 25, 50,
100, 200 or 400 mg/litre, equivalent to 4, 7.5, 15, 30 or 58.4 mg/kg
of body weight per day. Chlorate was found to be without significant
effect on a number of serum parameters related to oxidative damage and
thyroid hormone levels at concentrations of up to 400 mg/litre.
In the subchronic study (McCauley et al., 1995), concentrations
of chlorate of 3, 12 or 48 mmol/litre in drinking-water were provided
to both male and female Sprague-Dawley rats for 90 days. These
concentrations correspond to 250, 1000 and 4000 mg of chlorate per
litre, equal to 30, 100 or 510 mg/kg of body weight per day in males
and 42, 164 or 800 mg/kg of body weight per day in females, based on
measured water consumption of each group. Body weight gain was sharply
curtailed in both sexes at the highest concentration. These effects
were generally paralleled by smaller organ weights (except for brain
and testes). Some decreases in haemoglobin, haematocrit and red blood
cell counts were observed at this same dose. Pituitary lesions
(vacuolization in the cytoplasm of the pars distalis) and thyroid
gland colloid depletion were observed in both the mid- and high-dose
groups of both sexes. The NOAEL in this study was 30 mg/kg of body
weight per day.
4.7.2 Reproductive and developmental toxicity
Suh et al. (1983) examined the effects on fetal development of
chlorate at 0, 1 or 10 mg/litre administered to rats for 2.5 months
prior to mating and throughout gestation. This was a very limited
study, involving only six female rats per treatment group. Therefore,
the apparent increase of anomalous fetuses from 30.7% in the control
group to 52% and 55.2% in the groups receiving 1 and 10 mg/litre,
respectively, was not statistically significant. The abnormalities
were limited to relatively mild skeletal defects (missing sternebra
and rudimentary ribs).
A teratogenicity study carried out by Bio/Dynamics Inc. in 1987
was reviewed by WHO (1996). In this study, rats given 0, 10, 100 or
1000 mg of chlorate per kg of body weight per day on days 6-15 of
gestation showed no effects on maternal or fetal health.
4.7.3 Toxicity in humans
There have been sporadic reports of poisoning with sodium or
potassium salts of chlorate (Temperman & Maes, 1966; Mengele et al.,
1969; Yoshida et al., 1977; Bloxham et al., 1979; Helliwell & Nunn,
1979; Steffen & Seitz, 1981). Most of these cases involved ingestion
of preparations of sodium chlorate used for pesticidal purposes. The
symptomatology observed is consistent with that observed in the acute
studies in dogs identified above. There was generally evidence of
oxidative damage to erythrocytes, methaemoglobin formation and the
renal complications of haemolytic anaemia. The lethal dose to humans
has been estimated to be in the range 20-30 g.
A study in 10 male human volunteers was conducted by
administering solutions of 0.01-2.4 mg of chlorate per litre in two
500-ml portions (highest dose 0.034 mg/kg of body weight per day) in a
6-day rising-dose tolerance design (Lubbers & Bianchine, 1984). No
adverse effects were noted. This test of acute studies was followed by
an experiment (Lubbers et al., 1981) that provided 500 ml of water
containing 5 mg of chlorate per litre per day to 10 subjects for
12 weeks (average dose 0.036 mg/kg of body weight per day). Volunteers
in both studies were monitored using a battery of clinical and
physiological parameters and routine physical examinations throughout
the course of the study and for 8 weeks following termination of
treatments. Again, no adverse effects were observed.
4.7.4 Carcinogenicity and mutagenicity
There are no published studies of the carcinogenic potential of
chlorate administered alone. Sodium and potassium chlorate were
evaluated as promoters of renal tumours in
N-ethyl- N-hydroxyethylnitrosamine (EHEN)-initiated F344 rats.
Sodium chlorate and potassium chlorate were administered in the
drinking-water for 28 weeks. There was an increased incidence of renal
cell tumours (7/15 rats) in the EHEN-initiated group treated with
sodium chlorate, but no effect was observed with potassium chlorate
(1/5) relative to control rats (2/15). The small numbers of animals
used in this study make the treatment groups indistinguishable from
one another statistically (Kurokawa et al., 1985b).
Chlorate has long been known to select nitrate
reductase-deficient mutants of Aspergillus nidulans (Cove, 1976).
However, Prieto & Fernandez (1993) demonstrated that there is also a
mutagenic effect of chlorate in Chlamydomonas reinhardtii and
Rhodobacter capsulatus. Chlorate failed to induce mutations in the
BA-13 strain of Salmonella typhimurium. The positive mutagenic
effects were separated from simple selection of nitrate reductase
mutants by incubating cells in nitrogen-free media. Lack of nitrogen
prevents cell division during the treatment period. In the case of
C. reinhardtii, significant increases in mutants were observed at
concentration of 4-5 mmol/litre and above.
Meier et al. (1985b) examined chlorate in assays for micronuclei
and chromosomal aberrations in bone marrow and sperm head anomalies,
but all findings were negative.
4.7.5 Mode of action
Some research has been directed towards establishing the
mechanisms by which chlorate oxidatively damages erythrocytes and
their contents. There is a characteristic delay in the production of
methaemoglobin by chlorate when erythrocytes are incubated in the
presence of chlorate (Singelmann et al., 1984). It has been suggested
that this delay was due to the conversion of chlorate to chlorite
(Heubner & Jung, 1941; Koransky, 1952), but there is no direct
evidence to support this view. A competing hypothesis suggested that
chlorate formed a complex with methaemoglobin, which autocatalytically
increased methaemoglobin formation. This suggestion is supported by
experiments demonstrating that the formation of methaemoglobin
accelerates the further formation of methaemoglobin in the presence of
chlorate (Huebner & Jung, 1941; Jung, 1947, 1965). It is further
supported by the observation that compounds that compete for binding
of chlorate to methaemoglobin (e.g., azide or cyanide) block the
effect.
The properties of the erythrocyte membrane are also modified by
chlorate. Increased resistance to haemolysis is the most readily
observed effect (Singelmann et al., 1984). These effects appear to be
related to the formation of high molecular weight complexes of
erythrocytic proteins (Singelmann et al., 1984). These changes could
not be reversed by disulfide reduction. The formation of these
complexes was associated with the loss of activity of several enzymes,
the most sensitive being glucose-6-phosphate dehydrogenase (Singelmann
et al., 1984; Steffen & Wetzel, 1993). The inactivation of this enzyme
accounts for the insensitivity of chlorate-induced methaemoglobinaemia
to treatment with methylene blue. Reduction of nicotinamide adenine
dinucleotide phosphate (NADP) by the pentose pathway is necessary for
methylene blue to be effective. The cross-linking of protein is not
limited to cytosolic proteins and methaemoglobin, because
cross-linking of membrane proteins has also been demonstrated (Steffen
& Wetzel, 1993). The cross-linking requires the presence of
haemoglobin. Similar changes are induced by hypochlorite, but in this
case haemoglobin is not necessary.
The oxidative damage to the erythrocyte appears to be the basis
of chlorate's renal toxicity. This hypothesis is supported primarily
by the observation that species less sensitive to methaemoglobin
formation are also resistant to the nephrotoxic effects of chlorate
(Steffen & Wetzel, 1993). The observation is consistent with the
finding of haematogenous casts in kidney tubules of dogs treated with
doses of chlorate that induce methaemoglobin and their absence in dogs
treated with slightly lower doses that did not produce
methaemoglobinaemia (Heywood et al., 1972).
4.8 Bromate
4.8.1 General toxicological properties and information on
dose-response in animals
The acute toxic effects of bromate (administered as either the
potassium or sodium salt) have been studied in F344 rats, B6C3F1 mice
and Syrian golden hamsters (Kurokawa et al., 1990). The mean LD50
values in these species ranged from 280 to 495 mg/kg of body weight,
with slightly but consistently lower values found in males than in
females of each species. Mice appear to be somewhat more sensitive
than the other two species, but the lethal doses are remarkably
similar across species. Toxic signs and symptoms at lethal doses
included suppressed locomotor activity, ataxia, tachypnoea,
hypothermia, diarrhoea, lacrimation and piloerection. Hyperaemia of
the stomach and congestion of lungs were observed at autopsy. Damage
to renal tubules was seen microscopically, including necrosis in the
proximal tubular epithelium. Regenerative changes were observed from
48 h to 2 weeks after treatment. These effects were less marked in
mice and hamsters. No glomerular lesions were observed in any species.
Treatment of rats for 10 weeks with potassium bromate
concentrations of 250, 500, 1000, 2000 or 4000 mg/litre of
drinking-water established a maximally tolerated concentration of less
than 1000 mg/litre. As treatments were extended to 13 weeks, elevated
levels of glutamate-oxalate transaminase (GOT), glutamate-pyruvate
transaminase (GPT), LDH, AP and BUN were observed in blood samples
(Onodera et al., 1986; Kurokawa et al., 1990).
Eosinophilic droplets were observed in the cytoplasm of the
proximal renal tubule cells in male F344 rats receiving 600 mg of
potassium bromate per litre for 12 weeks (Onodera et al., 1986;
Kurokawa et al., 1990). These droplets were determined to be
eosinophilic bodies rather than hyaline droplets. Lipofuscin pigments
were also observed in the proximal tubular epithelium.
Dogs, rats and monkeys were fed bread or flour treated with up to
200 mg of potassium bromate per kg of body weight for up to 17 months
(FAO/WHO, 1989). These studies revealed no adverse effects, but, as
pointed out by Kurokawa et al. (1990), substantial portions of bromate
are presumed to be converted to bromide during the dough-making
process. It is also important to note that the numbers of animals
included in these studies were quite limited. Subsequent studies
conducted for longer periods of time indicated an increase
periarteritis in male rats and pathology to the adrenal glands in
female rats (Fisher et al., 1979).
Lifetime studies in female rats administered potassium bromate in
drinking-water found significant increases in GPT, albumin/globulin
ratios, serum potassium ion and cholinesterase activity at
concentrations of 500 mg/litre. Slight increases in BUN were also
observed at this dose (Kurokawa et al., 1990).
4.8.2 Toxicity in humans
Human poisonings have been associated with the ingestion of
sodium bromate and potassium bromate. Many of these poisonings result
from accidental or deliberate ingestion of preparations used as
neutralizers in permanent wave kits (Warshaw et al., 1985; Lue et al.,
1988). Clinical signs of bromate poisoning include anaemia and
haemolysis, renal failure and hearing loss. Loss of hearing appears to
be more common in adults than in children (Lichtenberg et al., 1989).
The hearing loss and renal failure can have a prolonged course in
some, but not all, people poisoned by bromate (Kuwahara et al., 1984).
Poisoning with bromate is frequently fatal when doses exceed 6 g
(Kurokawa et al., 1990).
4.8.3 Carcinogenicity and mutagenicity
IARC evaluated potassium bromate in 1986 and concluded that there
is sufficient evidence for its carcinogenicity in experimental
animals, whereas no data were available on its carcinogenicity to
humans. On this basis, potassium bromate was assigned to Group 2B: the
agent is possibly carcinogenic to humans (IARC, 1986, 1987).
In 1992, the Joint FAO/WHO Expert Committee on Food Additives
(JECFA) evaluated potassium bromate and concluded that it was
genotoxic and carcinogenic. On this basis, JECFA concluded that the
use of potassium bromate as a flour treatment agent was not
appropriate (FAO/WHO, 1993).
Potassium bromate was found to be weakly mutagenic in
Salmonella typhimurium strain TA100 when incubated with rat S9
fraction for metabolic activation (Kawachi et al., 1980; Ishidate et
al., 1984). Negative results were found in strains TA98, TA1535,
TA1537 and TA1538 (Kurokawa et al., 1990). Potassium bromate was also
inactive in Escherichia coli Wptry- and E. coli WP2try- his-
(Ishidate et al., 1984). Bromate was later found to be active in
S. typhimurium strains TA102 and TA104, which were developed to
detect compounds that generate oxygen radicals (Kurokawa et al.,
1990).
Potassium bromate also induced chromosomal aberrations in a
Chinese hamster fibroblasts cell line. The concentrations required
were, however, very high (>30 mmol/litre) (Ishidate et al., 1984).
Such high doses may induce changes by indirect mechanisms.
Bromate appears capable of inducing micronuclei in vivo.
Significant increases in the frequency of micronuclei were observed in
polychromatic erythrocytes when potassium bromate was administered by
either the oral or intraperitoneal route (Hayashi et al., 1988;
Nakajima et al., 1989). Positive results were obtained at doses of
24 mg/kg of body weight when administered intraperitoneally. Oral
doses of less than 100 mg/kg of body weight were negative. Ms/Ae and
CD-1 mice were found to be equally sensitive to these effects.
Several reports of bromate-induced cancer in experimental animals
are available. The clearest evidence comes from studies in F344 rats
(Kurokawa et al., 1983, 1986a, 1987a; DeAngelo et al., 1998). The
dose-response curves for the principal target organs, the kidney and
peritoneum, are provided in Figure 7. Tumours found in the kidney were
of tubular origin, with significantly increased numbers of both
adenomas and adenocarcinomas being observed in both males and females
(Kurokawa et al., 1983). The peritoneal tumours were mesotheliomas,
but treatment-related increases were observed only in male rats. In
the Kurokawa et al. (1983) study, concentrations of potassium bromate
of 0, 250 or 500 mg/litre correspond to doses of 0, 9.6 or 21.3 mg of
bromate per kg of body weight per day in males and 0, 9.6 or 19.6
mg/kg of body weight per day in females (as cited in IARC, 1986 and
WHO, 1996). The Kurokawa et al. (1986a) study represented, in part, a
repeat of the earlier study, except that more doses were included and
only male rats were studied. Concentrations in this study were 0, 15,
30, 60, 125, 250 or 500 mg of potassium bromate per litre,
corresponding to 0, 0.7, 1.3, 2.5, 5.6, 12 or 33 mg of bromate per kg
of body weight per day (as cited in WHO, 1996). The study provided
verification of the ability of potassium bromate to induce both renal
cell tumours and mesothelioma. An increased incidence of renal tumours
was observed at 125 mg of potassium bromate per litre of
drinking-water. Results with mesothelioma were not observed at a dose
of 250 mg/litre, but this is partially attributable to the smaller
number of animals per treatment group in this experiment. However, the
incidences of mesothelioma were very similar at 500 mg/litre in the
two studies. Significant increases in the occurrence of dysplastic
foci of the kidney (considered to be preneoplastic lesions) were found
in groups at doses higher than 30 mg/litre.
The time course of renal tumour development was examined in a
third study (dose-response provided in panel C of Figure 7) by
Kurokawa et al. (1987a). These experiments were designed to include
sacrifices of animals at 13, 26, 39 or 52 weeks as well as the
104-week period examined in prior studies. Additional groups were
included, however, that involved treatment for the above periods, but
the animals were maintained on bromate-free water until 104 weeks
before they were sacrificed. The concentration of potassium bromate
was 500 mg/litre during active treatment periods (average dose 32.3 mg
of bromate per kg of body weight per day). If animals were held for
104 weeks, 13 weeks of treatment was sufficient to produce the same
tumour incidence as was produced by longer treatment periods.
Moreover, the same incidence of renal cell tumours was observed in
animals that had been treated for 52 weeks and sacrificed at 52 weeks.
These data indicate that the tumour yield is not dependent upon the
total dose administered, but rather that sufficient time simply had to
be provided for the tumours to become evident.
The carcinogenicity of bromate has also been studied in three
hybrid strains of mice (B6C3F1, BDF1 and CDF1). Treatments of
female mice were conducted at concentrations of 0, 500 or
1000 mg/litre for 78 weeks (average dose 0, 43.5 or 91.6 mg of bromate
per kg of body weight per day). No treatment-related increases in
tumour incidence were observed (Kurokawa et al., 1986b). Groups of 27
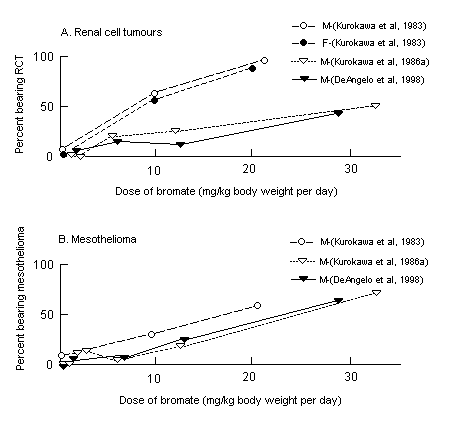
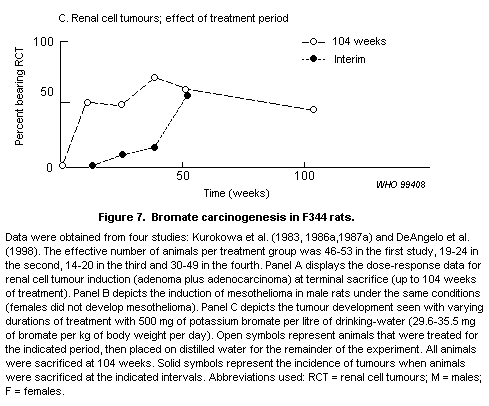 male mice of the same strains were provided 750 mg of potassium
bromate per litre (approximately 60-90 mg/kg of body weight per day,
as cited in FAO/WHO, 1993) for 88 weeks. A control group of 15 males
per strain was used. Increased numbers of renal cell tumours were not
observed in any of the strains. There was, however, an increased
frequency of adenomas (14/27 mice) relative to control mice (1/15) of
the CDF1 hybrid (Kurokawa et al., 1990).
In a separate study, male Syrian golden hamsters were treated
with potassium bromate at concentrations of 0, 125, 250, 500 or 2000
mg/litre of drinking-water for 89 weeks (Takamura et al., 1985). No
renal cell tumours were observed in control (0/20) or 125 mg/litre
(0/19) groups. At the higher concentrations, the incidences were 1/17
at 250, 4/20 at 500 and 2/19 at 2000 mg/litre.
DeAngelo et al. (1998) administered potassium bromate to male
F344 rats and male B6C3F1 mice (78 per group) in drinking-water at
concentrations of 0, 20, 100, 200 or 400 mg/litre or 0, 80, 400 or
800 mg/litre, respectively, for 100 weeks. Time-weighted mean daily
doses were calculated by the authors from mean daily water consumption
and the measured concentrations of potassium bromate. For rats, six
animals per group were included for interim sacrifices, which occurred
at 12, 26, 52 and 77 weeks. Statistically significant, dose-dependent
increases in tumour incidence were observed in the kidney (adenomas
and carcinomas combined and carcinomas alone), thyroid (adenomas and
carcinomas combined and carcinomas alone) and tunical vaginalis testis
(mesotheliomas). Historical control incidences for these tumour sites
in male F344 rats are as follows: renal cell tumours, 0.6%; thyroid
follicular cell adenomas and carcinomas, 2.1%; and mesotheliomas,
1.5%. The dose-response information for the renal cell tumours and
mesotheliomas is provided in Figure 7 to facilitate comparisons with
the Kurokawa studies. The combined thyroid cell tumour incidences were
0/36 (0%), 4/39 (10%), 1/43 (2%), 4/35 (11%) and 14/30 (47%) in the 0,
0.1, 6.1, 12.9 and 28.7 mg of bromate per kg of body weight per day
dose groups, respectively. Thyroid tumours were not reported in the
Kurokawa studies discussed above.
As seen in Figure 7, there is remarkable correspondence in the
dose-response relationships for renal cell tumour induction by bromate
observed in the DeAngelo et al. (1998) and the Kurokawa et al. (1986a)
studies. There was no indication of a positive trend in the
dose-response curve at the lowest doses. The DeAngelo et al. (1998)
study produced a positive trend in mesothelioma induction at the lower
dose, but this response was not significant until the dose was raised
to 6.1 mg/kg of body weight per day, with a strong positive trend in
tumour incidence with additional doses. This is different from the
Kurokawa studies, in that a significant background incidence of
mesotheliomas was observed in one study (Kurokawa et al., 1983),
whereas there was no indication of spontaneous lesions in a second
study (Kurokawa et al., 1986a). When the studies are combined, a clear
positive response is observed at concentrations of 200 mg/litre or
more in drinking-water (>12.9 mg/kg of body weight per day).
Tumour responses in male B6C3F1 mice were confined to kidney
tumours, but the incidence was not clearly dose-dependent. The tumour
incidence at terminal sacrifice was 0/40 (0%), 5/38 (13%), 3/41 (7%)
and 1/44 (2%) in mice treated with the equivalent of 0, 6.9, 33 or
60 mg/kg of body weight per day (DeAngelo et al., 1998).
A series of additional studies have evaluated the ability of
bromate to act as a tumour promoter in a variety of animal models.
Bromate was found to be inactive when applied to the skin of
dimethylbenzanthracene-initiated female Sencar mice in 0.2 ml of
acetone at 40 mg of potassium bromate per ml twice per week for 51
weeks (Kurokawa et al., 1984). Potassium bromate was administered for
24 weeks at concentrations of 15, 30, 60, 125, 250 or 500 mg/litre to
male F344 rats initiated by EHEN given in the drinking-water for the
first 2 weeks at a concentration of 500 mg/litre (Kurokawa et al.,
1985b). A dose-related increase in renal cell tumours was observed in
rats treated with more than 30 mg of potassium bromate per litre.
These changes were apparently not analysed for dose-relatedness, but
individual groups were compared using the Student t-test and
considered statistically insignificant ( p = 0.05). There were,
however, statistically significant (again compared using the Student
t-test) increases in the mean number of dysplastic foci at doses of
30 mg/litre and above ( p < 0.01) and in the mean number of renal
cell tumours observed per cm2 at 500 mg/litre. These data provide
evidence that at least some of the carcinogenic activity of bromate
can be attributed to its activity as a promoter.
4.8.4 Comparative pharmacokinetics and metabolism
The absorption and distribution of bromate and bromide were
studied following administration of a single dose of 50 mg of bromate
per kg of body weight (Kurokawa et al., 1990). Concentrations of
bromate of approximately 4 µg/ml were seen in plasma 15 min after
administration. This concentration was quickly reduced to
approximately 1 µg/ml within another 15 min and was not detectable in
plasma at 2 h. Concentrations of bromate in urine peaked at
approximately 1 h after administration. No bromate was detected in
urine until doses reached 5 mg/kg of body weight. About 3-6% of the
higher doses were recovered in the urine.
These observations are generally consistent with the observed
elimination of bromate in human urine following poisonings
(Lichtenberg et al., 1989). However, the data available in humans are
quite limited and generally involved much higher doses than would be
encountered in drinking-water.
4.8.5 Mode of action
Attempts have been made to link the toxic effects of bromate with
its oxidant properties. The administration of potassium bromate
induces TBARS as a measure of lipid peroxidation (Kurokawa et al.,
1987b). Single doses of 77 mg/kg of body weight and higher
significantly increased TBARS in the kidney of F344 rats. Mice
displayed smaller and less consistent responses, with statistically
significant responses being seen in male CDF1 mice, but not B6C3F1
or BDF1 mice. Lipid peroxidation was not observed in male Syrian
golden hamsters. Treatment of rats with antioxidants (GSH or cysteine)
decreased the lethality of potassium bromate. Clinical indicators of
kidney damage (non-protein nitrogen, BUN and creatinine) were
consistently reduced by GSH or cysteine and increased by
co-administration of diethylmaleate, a depletor of GSH. These
treatments modified the histopathological changes in renal tubules
that were consistent with the clinical findings.
In vitro studies have shown that hydroxyl radical is produced
in renal homogenates or kidney cells treated with potassium bromate
(Sai et al., 1992a,b). Incubation of hepatocytes or liver homogenates
with bromate did not produce evidence of oxygen radicals.
Several studies have demonstrated the formation of 8-OH-dG in the
DNA of the kidneys of rats treated with bromate (Kasai et al., 1987;
Sai et al., 1991; Cho et al., 1993). Such modifications in DNA can be
produced by oxygen radicals. The formation of 8-OH-dG was not observed
in liver DNA of the same animals, paralleling the target organ
specificity of the compound. Increases in 8-OH-dG were blocked by
parallel treatment with GSH, cysteine or vitamin C (Sai et al.,
1992c). Superoxide dismutase and vitamin E were ineffective in
modifying DNA damage produced by potassium bromate.
GSH and cysteine administered 30 min before or after bromate
significantly inhibited the induction of micronuclei in rat peripheral
blood reticulocytes (Sai et al., 1992c). Treatment of superoxide
dismutase had no effect on the induction of micronucleated
reticulocytes. This suggests that the mechanisms involved in
micronuclei formation parallel those involved in production of lipid
peroxidation and DNA damage via the production of oxygen radicals.
Chipman et al. (1998) studied the production of DNA oxidation
with bromate in vitro and with intraperitoneal doses of potassium
bromate. Studies with isolated calf thymus DNA demonstrated a
GSH-dependent oxidation of guanosine bases. In vivo studies
indicated that this mechanism was apparently not active. High doses of
potassium bromate (100 mg/kg of body weight) administered
intraperitoneally induced a statistically significant increase in
8-OH-dG adducts in total cellular DNA. A trend towards an increase was
observed at 20 mg/kg of body weight per day.
Studies demonstrating that clastogenic effects of bromate can be
suppressed by antioxidant treatments provide evidence that effects in
the bone marrow can be attributed to generation of oxygen radicals and
the Fenton chemistry that subsequently occurs. The acute doses
required to produce evidence of damage to DNA mediated by oxygen
radicals generated by bromate have been considerably higher than the
daily oral doses required to induce a significant tumour response in
the rat kidney (6.1 mg/kg of body weight per day). Consequently, more
precise dose metrics and better dose-response information will be
necessary to demonstrate the relationship of these effects of bromate
with tumour induction. The possibility that increased rates of cell
replication could contribute to the renal cancer induced by bromate
was investigated by Umemura et al. (1993). Both sodium and potassium
salts were utilized and were found to induce significant increases in
the cumulative replication fraction of cells in the proximal
convoluted tubules of male F344 rats. The effect was significantly
smaller in proximal straight or distal tubules. However, female rats
did not display elevated rates of replication (Umemura et al., 1993).
Thus, these effects in male rats appear to be associated with the
formation of bodies that are stained with Mallory-Heidenham stain,
used to detect hyaline droplets. These bodies were not seen in female
rats. The absence of these responses in female rats, despite a very
similar incidence of renal tumours in lifetime exposures (Figure 7),
renders an argument of alpha-2u globulin in the target tissue
immaterial. While such pathology could contribute to the tumorigenic
response in the male, it is clearly unnecessary for the response in
females.
4.9 Other DBPs
Many other DBPs can be found in drinking-water, as indicated in
chapter 2. Most of these are present at very low concentrations.
Several were considered by WHO (1993, 1996, 1998) in the Guidelines
for drinking-water quality. Those considered in the Guidelines but
not in this document include formaldehyde, chlorophenols, chloropicrin
and cyanogen chloride. There are no new data that materially change
those evaluations.
male mice of the same strains were provided 750 mg of potassium
bromate per litre (approximately 60-90 mg/kg of body weight per day,
as cited in FAO/WHO, 1993) for 88 weeks. A control group of 15 males
per strain was used. Increased numbers of renal cell tumours were not
observed in any of the strains. There was, however, an increased
frequency of adenomas (14/27 mice) relative to control mice (1/15) of
the CDF1 hybrid (Kurokawa et al., 1990).
In a separate study, male Syrian golden hamsters were treated
with potassium bromate at concentrations of 0, 125, 250, 500 or 2000
mg/litre of drinking-water for 89 weeks (Takamura et al., 1985). No
renal cell tumours were observed in control (0/20) or 125 mg/litre
(0/19) groups. At the higher concentrations, the incidences were 1/17
at 250, 4/20 at 500 and 2/19 at 2000 mg/litre.
DeAngelo et al. (1998) administered potassium bromate to male
F344 rats and male B6C3F1 mice (78 per group) in drinking-water at
concentrations of 0, 20, 100, 200 or 400 mg/litre or 0, 80, 400 or
800 mg/litre, respectively, for 100 weeks. Time-weighted mean daily
doses were calculated by the authors from mean daily water consumption
and the measured concentrations of potassium bromate. For rats, six
animals per group were included for interim sacrifices, which occurred
at 12, 26, 52 and 77 weeks. Statistically significant, dose-dependent
increases in tumour incidence were observed in the kidney (adenomas
and carcinomas combined and carcinomas alone), thyroid (adenomas and
carcinomas combined and carcinomas alone) and tunical vaginalis testis
(mesotheliomas). Historical control incidences for these tumour sites
in male F344 rats are as follows: renal cell tumours, 0.6%; thyroid
follicular cell adenomas and carcinomas, 2.1%; and mesotheliomas,
1.5%. The dose-response information for the renal cell tumours and
mesotheliomas is provided in Figure 7 to facilitate comparisons with
the Kurokawa studies. The combined thyroid cell tumour incidences were
0/36 (0%), 4/39 (10%), 1/43 (2%), 4/35 (11%) and 14/30 (47%) in the 0,
0.1, 6.1, 12.9 and 28.7 mg of bromate per kg of body weight per day
dose groups, respectively. Thyroid tumours were not reported in the
Kurokawa studies discussed above.
As seen in Figure 7, there is remarkable correspondence in the
dose-response relationships for renal cell tumour induction by bromate
observed in the DeAngelo et al. (1998) and the Kurokawa et al. (1986a)
studies. There was no indication of a positive trend in the
dose-response curve at the lowest doses. The DeAngelo et al. (1998)
study produced a positive trend in mesothelioma induction at the lower
dose, but this response was not significant until the dose was raised
to 6.1 mg/kg of body weight per day, with a strong positive trend in
tumour incidence with additional doses. This is different from the
Kurokawa studies, in that a significant background incidence of
mesotheliomas was observed in one study (Kurokawa et al., 1983),
whereas there was no indication of spontaneous lesions in a second
study (Kurokawa et al., 1986a). When the studies are combined, a clear
positive response is observed at concentrations of 200 mg/litre or
more in drinking-water (>12.9 mg/kg of body weight per day).
Tumour responses in male B6C3F1 mice were confined to kidney
tumours, but the incidence was not clearly dose-dependent. The tumour
incidence at terminal sacrifice was 0/40 (0%), 5/38 (13%), 3/41 (7%)
and 1/44 (2%) in mice treated with the equivalent of 0, 6.9, 33 or
60 mg/kg of body weight per day (DeAngelo et al., 1998).
A series of additional studies have evaluated the ability of
bromate to act as a tumour promoter in a variety of animal models.
Bromate was found to be inactive when applied to the skin of
dimethylbenzanthracene-initiated female Sencar mice in 0.2 ml of
acetone at 40 mg of potassium bromate per ml twice per week for 51
weeks (Kurokawa et al., 1984). Potassium bromate was administered for
24 weeks at concentrations of 15, 30, 60, 125, 250 or 500 mg/litre to
male F344 rats initiated by EHEN given in the drinking-water for the
first 2 weeks at a concentration of 500 mg/litre (Kurokawa et al.,
1985b). A dose-related increase in renal cell tumours was observed in
rats treated with more than 30 mg of potassium bromate per litre.
These changes were apparently not analysed for dose-relatedness, but
individual groups were compared using the Student t-test and
considered statistically insignificant ( p = 0.05). There were,
however, statistically significant (again compared using the Student
t-test) increases in the mean number of dysplastic foci at doses of
30 mg/litre and above ( p < 0.01) and in the mean number of renal
cell tumours observed per cm2 at 500 mg/litre. These data provide
evidence that at least some of the carcinogenic activity of bromate
can be attributed to its activity as a promoter.
4.8.4 Comparative pharmacokinetics and metabolism
The absorption and distribution of bromate and bromide were
studied following administration of a single dose of 50 mg of bromate
per kg of body weight (Kurokawa et al., 1990). Concentrations of
bromate of approximately 4 µg/ml were seen in plasma 15 min after
administration. This concentration was quickly reduced to
approximately 1 µg/ml within another 15 min and was not detectable in
plasma at 2 h. Concentrations of bromate in urine peaked at
approximately 1 h after administration. No bromate was detected in
urine until doses reached 5 mg/kg of body weight. About 3-6% of the
higher doses were recovered in the urine.
These observations are generally consistent with the observed
elimination of bromate in human urine following poisonings
(Lichtenberg et al., 1989). However, the data available in humans are
quite limited and generally involved much higher doses than would be
encountered in drinking-water.
4.8.5 Mode of action
Attempts have been made to link the toxic effects of bromate with
its oxidant properties. The administration of potassium bromate
induces TBARS as a measure of lipid peroxidation (Kurokawa et al.,
1987b). Single doses of 77 mg/kg of body weight and higher
significantly increased TBARS in the kidney of F344 rats. Mice
displayed smaller and less consistent responses, with statistically
significant responses being seen in male CDF1 mice, but not B6C3F1
or BDF1 mice. Lipid peroxidation was not observed in male Syrian
golden hamsters. Treatment of rats with antioxidants (GSH or cysteine)
decreased the lethality of potassium bromate. Clinical indicators of
kidney damage (non-protein nitrogen, BUN and creatinine) were
consistently reduced by GSH or cysteine and increased by
co-administration of diethylmaleate, a depletor of GSH. These
treatments modified the histopathological changes in renal tubules
that were consistent with the clinical findings.
In vitro studies have shown that hydroxyl radical is produced
in renal homogenates or kidney cells treated with potassium bromate
(Sai et al., 1992a,b). Incubation of hepatocytes or liver homogenates
with bromate did not produce evidence of oxygen radicals.
Several studies have demonstrated the formation of 8-OH-dG in the
DNA of the kidneys of rats treated with bromate (Kasai et al., 1987;
Sai et al., 1991; Cho et al., 1993). Such modifications in DNA can be
produced by oxygen radicals. The formation of 8-OH-dG was not observed
in liver DNA of the same animals, paralleling the target organ
specificity of the compound. Increases in 8-OH-dG were blocked by
parallel treatment with GSH, cysteine or vitamin C (Sai et al.,
1992c). Superoxide dismutase and vitamin E were ineffective in
modifying DNA damage produced by potassium bromate.
GSH and cysteine administered 30 min before or after bromate
significantly inhibited the induction of micronuclei in rat peripheral
blood reticulocytes (Sai et al., 1992c). Treatment of superoxide
dismutase had no effect on the induction of micronucleated
reticulocytes. This suggests that the mechanisms involved in
micronuclei formation parallel those involved in production of lipid
peroxidation and DNA damage via the production of oxygen radicals.
Chipman et al. (1998) studied the production of DNA oxidation
with bromate in vitro and with intraperitoneal doses of potassium
bromate. Studies with isolated calf thymus DNA demonstrated a
GSH-dependent oxidation of guanosine bases. In vivo studies
indicated that this mechanism was apparently not active. High doses of
potassium bromate (100 mg/kg of body weight) administered
intraperitoneally induced a statistically significant increase in
8-OH-dG adducts in total cellular DNA. A trend towards an increase was
observed at 20 mg/kg of body weight per day.
Studies demonstrating that clastogenic effects of bromate can be
suppressed by antioxidant treatments provide evidence that effects in
the bone marrow can be attributed to generation of oxygen radicals and
the Fenton chemistry that subsequently occurs. The acute doses
required to produce evidence of damage to DNA mediated by oxygen
radicals generated by bromate have been considerably higher than the
daily oral doses required to induce a significant tumour response in
the rat kidney (6.1 mg/kg of body weight per day). Consequently, more
precise dose metrics and better dose-response information will be
necessary to demonstrate the relationship of these effects of bromate
with tumour induction. The possibility that increased rates of cell
replication could contribute to the renal cancer induced by bromate
was investigated by Umemura et al. (1993). Both sodium and potassium
salts were utilized and were found to induce significant increases in
the cumulative replication fraction of cells in the proximal
convoluted tubules of male F344 rats. The effect was significantly
smaller in proximal straight or distal tubules. However, female rats
did not display elevated rates of replication (Umemura et al., 1993).
Thus, these effects in male rats appear to be associated with the
formation of bodies that are stained with Mallory-Heidenham stain,
used to detect hyaline droplets. These bodies were not seen in female
rats. The absence of these responses in female rats, despite a very
similar incidence of renal tumours in lifetime exposures (Figure 7),
renders an argument of alpha-2u globulin in the target tissue
immaterial. While such pathology could contribute to the tumorigenic
response in the male, it is clearly unnecessary for the response in
females.
4.9 Other DBPs
Many other DBPs can be found in drinking-water, as indicated in
chapter 2. Most of these are present at very low concentrations.
Several were considered by WHO (1993, 1996, 1998) in the Guidelines
for drinking-water quality. Those considered in the Guidelines but
not in this document include formaldehyde, chlorophenols, chloropicrin
and cyanogen chloride. There are no new data that materially change
those evaluations.
Please Note: This is a corrigendum after the task group
Note: After the printing of the document, Dr James Huff kindly
brought to the attention of the Secretariat that a study on the
carcinogenicity of sodium hypochlorite, and another on the
carcinogenicity of bromodichloromethane, chlorodibromomethane, bromoform,
chlorine, and chloramine, were not cited in the document. The authors'
abstracts of these studies are given below.
Soffritti M, Belpoggi F, Lenzi A, Maltoni C (1997) Results of long-term
carcinogenicity studies of chlorine in rats. Ann NY Acad Sci,
837: 189-208.
Four groups, each of 50 male and 50 female Sprague-Dawley rats, of the
colony used in the Cancer Research Center of Bentivoglio of the Ramazzini
Foundation, 12 weeks old at the start of the study, received drinking
water containing sodium hypochlorite, resulting in concentrations of active
chlorine of 750, 500, and 100 mg/l (treated groups), and tap water (active
chlorine < 0.2 mg/l) (control group), respectively, for 104 weeks. Among
the female rats of the treated groups, an increased incidence of lymphomas
and leukemias has been observed, although this is not clearly dose related.
Moreover, sporadic cases of some tumors, the occurrence of which is extremely
unusual among the untreated rats of the colony used (historical controls),
were detected in chlorine-exposed animals. The results of this study confirm
the results of the experiment of the United States National Toxicology
Program (1991), which showed an increase of leukemia among female Fischer
344/N rats following the administration of chlorine (in the form of sodium
hypochlorite and chloramine) in their drinking water. The data here presented
call for further research aimed at quantifying the oncogenic risks related to
the chlorination of drinking water, to be used as a basis for consequent
public health measures.
Dunnick JK, Melnick RL (1993) Assessment of the carcinogenic potential of
chlorinated water: experimental studies of chlorine, chloramine, and
trihalomethanes. J Natl Cancer Inst, 85: 817-822.
BACKGROUND: Water chlorination has been one of the major disease prevention
treatments of this century. While epidemiologic studies suggest an association
between cancer in humans and consumption of chlorination byproducts in
drinking water, these studies have not been adequate to draw definite
conclusions about the carcinogenic potential of the individual byproducts
PURPOSE: The purpose of this study was to investigate the carcinogenic
potential of chlorinated or chloraminated drinking water and of four organic
trihalomethane byproducts of chlorination (chloroform, bromodichloromethane,
chlorodibromomethane, and bromoform) in rats and mice.
METHODS: Bromodichloromethane, chlorodibromomethane, bromoform, chlorine, or
chloramine was administered to both sexes of F344/N rats and (C57BL/6 x C3H)F1
mice (hereafter called B6C3F1 mice). Chloroform was given to both sexes of
Osborne-Mendel rats and B6C3F1 mice. Chlorine or chloramine was administered
daily in the drinking water for 2 years at doses ranging from 0.05 to 0.3
mmol/kg per day. The trihalomethanes were administered by gavage in corn oil
at doses ranging from 0.15 to 4.0 mmol/kg per day for 2 years, with the
exception of chloroform, which was given for 78 weeks.
RESULTS: The trihalomethanes were carcinogenic in the liver, kidney, and/or
intestine of rodents. There was equivocal evidence for carcinogenicity in
female rats that received chlorinated or chloraminated drinking water; this
evidence was based on a marginal increase in the incidence of mononuclear
cell leukemia. Rodents were generally exposed to lower doses of chlorine and
chloramine than to the trihalomethanes, but the doses in these studies were
the maximum that the animals would consume in the drinking water. The highest
doses used in the chlorine and chloramine studies were equivalent to a daily
gavage dose of bromodichloromethane that induced neoplasms of the large
intestine in rats. In contrast to the results with the trihalomethanes,
administration of chlorine or chloramine did not cause a clear carcinogenic
response in rats or mice after long-term exposure.
CONCLUSION: These results suggest that organic byproducts of chlorination are
the chemicals of greatest concern in assessment of the carcinogenic potential
of chlorinated drinking water.
5. EPIDEMIOLOGICAL STUDIES
This chapter reviews observational and experimental
epidemiological studies that have been conducted to determine
associations between disinfected drinking-water and adverse health
outcomes. Disinfection practices vary throughout the world. Applied
and residual concentrations have varied over the years and from
country to country.
Epidemiological study designs, sources of systematic and random
error (bias), and guidelines for assessing the causality of
associations are discussed in section 5.1. Epidemiological studies of
exposures to disinfected drinking-water and to specific DBPs are
evaluated in sections 5.2 and 5.3, respectively.
Observational epidemiological studies have been conducted to
determine possible associations between adverse health-related
outcomes and drinking-water disinfected with chlorine and chloramine.
Chlorinated drinking-water was studied most often, and studies
primarily compared health risks associated with chlorinated
drinking-water from surface water sources with those associated with
unchlorinated drinking-water from groundwater sources. Also studied
were specific DBPs, including chloroform and other THMs. Only one
study considered DBPs other than THMs. Two studies considered risks
that may be associated with chloraminated water and chlorine dioxide.
The mutagenic activity of drinking-water, which may represent exposure
to the non-volatile, acid/neutral fraction of chlorinated organic
material in water, was also considered. Health effects studied
included cancer, cardiovascular disease and adverse reproductive and
developmental outcomes. Most of the studies focused on bladder cancer
risks. Also studied were risks of colon, rectal and other cancers.
5.1 Epidemiological study designs and causality of epidemiological
associations
Both observational and experimental epidemiological studies have
been conducted to assess the health risks associated with
drinking-water disinfection (Table 22).
5.1.1 Experimental studies
Results of experimental epidemiological studies, which include
clinical trials, are reported in chapter 4 as appropriate under
toxicity in humans. These studies consider the effect of varying some
characteristic or exposure that is under the investigator's control,
much like in a toxicological study. Comparable individuals are
randomly assigned to a treatment or intervention group and observed
for a specific health-related outcome. Ethical concerns must be fully
addressed. Several clinical trials have evaluated changes in lipid,
thyroid and haematological parameters that may be affected by
consumption of disinfected drinking-water.
Table 22. Types of epidemiological studiesa
I. Experimental
A. Clinical
B. Population
II. Observational
A. Descriptive
1. Disease surveillance and surveys
2. Ecological
B. Analytical
1. Longitudinal
a. Cohort (follow-up)
b. Case-control (case-comparison)
2. Cross-sectional
a Adapted from Monson (1990).
5.1.2 Observational studies
Two basic kinds of observational epidemiological studies have
been conducted to determine risks associated with disinfection of
drinking-water: ecological and analytical. These two study approaches
differ primarily in the supportive evidence they can provide about a
possible causal association. Unlike the analytical study, an
ecological study does not link individual outcome events to individual
exposure or confounding characteristics, and it does not link
individual exposure and confounding characteristics to one another. In
an ecological study, information about exposure and disease is
available only for groups of people, and critical information can be
lost in the process of aggregating these data (Piantadosi, 1994).
Results from ecological studies are difficult to interpret, and
serious errors can result when it is assumed that inferences from an
ecological analysis pertain either to the individuals within the group
or to individuals across the groups (Connor & Gillings, 1974;
Piantadosi et al., 1988). Theoretical and empirical analyses have
offered no consistent guidelines for the interpretation of ecological
associations (Greenland & Robins, 1994a,b; Piantadosi, 1994).
Investigators (Greenland & Robins, 1994a,b; Piantadosi, 1994; Poole,
1994; Susser, 1994a,b) have examined the limitations of ecological
studies and determined when and under what assumptions this type of
study may be appropriate.
Analytical studies can provide the necessary information to help
evaluate the causality of an association and estimate the magnitude of
risk. For each person included in the study, information is obtained
about their disease status, their exposure to various contaminants and
confounding characteristics. Analytical studies are either
longitudinal or cross-sectional. In a longitudinal study, the time
sequence can be inferred between exposure and disease; in other words,
exposure precedes disease. In a cross-sectional study, exposure and
disease information relate to the same time period; in these studies,
it may not always be correct to presume that exposure preceded
disease. The cross-sectional study design was used to investigate
possible risks of cardiovascular disease and reproductive and
developmental risks.
Longitudinal studies are of two opposite approaches: the cohort
study and the case-control study. The cohort study begins with the
identification of individuals having an exposure of interest and a
non-exposed population for comparison; disease consequences or other
health-related outcomes are then determined for each group. In a
case-control study, the investigator identifies individuals having a
disease or health outcome of interest and a control or comparison
group of individuals without the disease of interest; exposures and
risk factors are evaluated in these persons. In a case-control study,
a variety of exposures can be studied, whereas in a cohort study, a
variety of diseases can be studied.
The cohort or follow-up study can be either retrospective or
prospective, and sometimes a combination retrospective-prospective
approach is used. Two or more groups of people are assembled for study
strictly according to their exposure status. Incidence or mortality
rates for the disease of interest are compared between exposed and
unexposed groups. Multiple disease end-points can be evaluated, but a
disadvantage is that large numbers of people must be studied,
especially for environmental exposures. Because of the lengthy latent
period for cardiovascular disease and cancer, a long follow-up period
is required for a prospective cohort, and this is usually not
feasible. The retrospective cohort study design was used to evaluate
the possible association of chlorinated drinking-water with cancer and
cardiovascular disease risks.
In a case-control study, persons with the disease of interest
(the cases) and persons without this disease (the controls or
comparison group) are sampled from either the general population or a
special population (e.g., hospitals or a select group) within a
specified geographic area. Exposures among the cases are compared with
exposures among non-diseased persons. Multiple exposures can be
evaluated, and a relatively small number of study participants is
needed to obtain reasonably precise estimates of risk associated with
environmental exposures. Retrospective exposures must be considered,
and, because of the lengthy latency period for cancer and
cardiovascular disease, exposures to water sources and contaminants
must be assessed over the previous 20-30 years, or perhaps even a
person's lifetime. It may be difficult to assess these exposures
accurately. Two types of case-control studies have been conducted to
investigate associations between disinfected drinking-water and
cancer:
* Decedent cases and controls without interviewing next-of-kin or
survivors for information about residential histories, risk factors
and possible confounding characteristics.
* Decedent and incident cases and controls using interviews or other
methods to obtain information about possible confounding
characteristics and document long-term mobility and changes in
residences to allow documentation of lifetime exposure to
disinfected water. In several studies, a person's intake of
tapwater and historical exposures to chlorinated water, chloroform
or other THMs were assessed.
5.1.3 Random and systematic error
Biases that occur during the design and conduct of a study can
lead to a false or spurious association or a measure of risk that
departs systematically from the true value. All reported
epidemiological associations require evaluation of random and
systematic error so that results can be interpreted properly.
Systematic error (bias) affects the validity of a study's observed
association; random error affects the precision of the estimated
magnitude of the risk. Random error is governed by chance and is
influenced by the size of the study. The likelihood that a positive
association is due to random error can be assessed by calculating the
level of statistical significance ( "P" value) or confidence interval
(CI). A small P value or a CI that does not include unity (1.0)
suggests that chance may be an unlikely explanation for an observed
association, but the association may, nevertheless, be spurious
because of systematic error. Statistical significance does not imply
causality or biological significance, nor does it mean that random
error or chance can ever be completely ruled out as a possible
explanation for the observed association. Many epidemiologists believe
that strict reliance on statistical significance testing is not
appropriate (US EPA, 1994a).
Potential sources of systematic error include observation,
selection, misclassification and confounding biases. When information
on exposure and disease is collected by methods that are not
comparable for each participant (e.g., selective recall), an incorrect
association will be due to observation bias. When the criteria used to
enrol individuals in the study are not comparable, the observed
association between exposure and disease will be due to selection
bias. A wrong diagnosis of disease or assessment of exposure can
result in misclassification bias. This type of bias may be randomly
distributed (non-differential misclassification bias), which almost
always biases study results towards the direction of not observing an
effect (or observing a smaller change in risk than may actually be
present), or it may be non-random (differential misclassification
bias), which can result in either higher or lower estimates of risk,
depending on how the misclassification is distributed. Lynch et al.
(1989) examined the effects of misclassification of exposure using
empirical data from an interview-based case-control study of bladder
cancer in Iowa (USA). Bladder cancer risk estimates were found to be
higher when more information was known about the study participants'
residential history and their possible exposure to chlorinated water
sources. This suggests that misclassification bias in epidemiological
studies of chlorinated water may be primarily non-differential,
underestimating the risk; however, in study areas where residential
mobility is different from that in Iowa, the magnitude of risk may be
overestimated rather than underestimated.
Confounding bias may convey the appearance of an association;
that is, a confounding characteristic rather than the putative cause
or exposure may be responsible for all or much of the observed
association. Although negative confounding bias may occur, concern is
usually with positive effects of confounding bias -- is confounding
bias responsible for the observed association? Confounding bias is
potentially present in all epidemiological studies and should always
be evaluated as a possible explanation for an association. Information
on known or suspected confounding characteristics is collected to
evaluate and control confounding during the analysis. In the design of
case-control studies, matching is a technique that is used to prevent
confounding bias. For example, if smoking is thought to be a possible
confounding characteristic, an equal number or proportion of smoking
cases and controls can be selected for study in order to avoid
confounding bias by this exposure. Techniques are also available to
assess and control confounding during the data analysis. In an
experimental epidemiological study, randomization is possible; that
is, each individual in the study has an equal or random chance of
being assigned to an exposed or unexposed group. Because of this
random assignment of exposure, all characteristics, confounding or
not, tend to be distributed equally between the selected study groups
of different exposure.
Procedures in the study's design and conduct are used to prevent
or reduce possible bias. If bias has been identified in a study, the
direction of the bias can often be determined, but its effect on the
magnitude of the association may not. For example, information may be
available to determine whether the bias was responsible for an
increased or decreased likelihood of observing an association, but its
magnitude usually cannot be estimated.
Two basic measures of an association between exposure and disease
in analytical studies are the rate ratio or relative risk (RR) and
exposure odds ratio (OR). A mortality odds ratio (MOR) is reported
when mortality is studied. An RR or OR of 1.0 indicates no
association; any other ratio signifies either a positive or negative
association. For example, an RR or OR of 1.8 indicates an 80%
increased risk among the exposed. Decreased risk and protective
effects are indicated by an RR or OR that is less than 1.0. The size
of the relative risk and odds ratio is also used to help assess if an
observed association may be spurious (Table 23). Based on Monson's
(1990) experience, an RR or OR of 0.9-1.2 indicates essentially no
association. Associations in this range are generally considered too
weak to be detected by epidemiological methods. It is difficult to
interpret a weak association, i.e., an RR or OR of 1.2-1.5. One or
more confounding characteristics can easily lead to a weak association
between exposure and disease, and it is usually not possible to
identify, measure or control weak confounding bias. On the other hand,
a large relative risk is unlikely to be completely explained by some
uncontrolled or unidentified confounding characteristic. When the
study has a reasonably large number of participants and the relative
risk or odds ratio is large, random variability and confounding bias
are much less likely to be responsible for an observed association.
Table 23. Guide to the strength of an epidemiological
associationa
Relative risk Strength of association
1.0 None
>1.0-<1.5 Weak
1.5-3.0 Moderate
3.1-10.0 Strong
>10.0 Infinite
a Adapted from Monson (1990).
Another measure of effect is the standardized mortality ratio
(SMR). An SMR of 100 indicates no association; an SMR of 150 indicates
a 50% increased risk.
5.1.4 Causality of an epidemiological association
Epidemiological associations may be causal; however, before
causality can be assessed, each study must be evaluated to determine
whether its design is appropriate, the study size is adequate and
systematic bias has not influenced the observed association. In
addition, the association should be consistent with prior hypotheses
and previous study results, and its magnitude should be moderately
large. Causality requires sufficient evidence from several well
designed and well conducted epidemiological studies in various
geographic areas. Supporting toxicological and pharmacological data
are also important. Guidelines are available to help epidemiologists
assess the possible causality of associations observed in well
designed and well conducted studies. Epidemiological data should be
interpreted with caution and in the context of other available
scientific information. Epidemiologists apply the following guidelines
to assess evidence about causality (Hill, 1965; Rothman, 1986):
* Biological plausibility. When the association is supported by
evidence from clinical research or toxicology about biological
behaviour or mechanisms, an inference of causality is strengthened.
* Temporal association. Exposure must precede the disease, and in
most epidemiological studies this can be inferred. When exposure
and disease are measured simultaneously, it is possible that
exposure has been modified by the presence of disease.
* Study precision and validity. Individual studies that provide
evidence of an association are well designed with an adequate
number of study participants (good precision) and well conducted
with valid results (i.e., the association is not likely due to
systematic bias).
* Strength of association. The larger the relative risk or odds
ratio, the less likely the association is to be spurious or due to
unidentified confounding. However, a causal association cannot be
ruled out simply because a weak association is observed.
* Consistency. Repeated observation of an association under
different study conditions supports an inference of causality, but
the absence of consistency does not rule out causality.
* Specificity. A putative cause or exposure leads to a specific
effect. The presence of specificity argues for causality, but its
absence does not rule it out.
* Dose-response relationship. A causal interpretation is more
plausible when an epidemiological gradient is found (e.g., higher
risk is associated with larger exposures).
* Reversibility or preventability. An observed association leads to
some preventive action, and removal of the possible cause leads to
a reduction of disease or risk of disease.
5.2 Epidemiological associations between disinfectant use and adverse
health outcomes
Studies of water disinfected with chlorine and chloramine are
reviewed in this section. Chlorinated drinking-water was studied most
often. Studies primarily compared health risks associated with
ingestion of chlorinated drinking-water from surface water sources
with those associated with ingestion of unchlorinated drinking-water
from groundwater sources, but risks were also compared among
populations using chloraminated and chlorinated surface water
supplies. Studies that considered exposures to specific DBPs are
reviewed in section 5.3. Studies that considered exposures to both
disinfected drinking-water and specific by-products are described in
either section 5.2 or 5.3.
One study assessed the effects on haematological and serum
chemical parameters that may be associated with the use of chlorine
dioxide. Because of their relevance to other experimental studies,
results are reported in section 4.6.3.
5.2.1 Epidemiological studies of cancer and disinfected
drinking-water
Since 1974, numerous epidemiological studies have attempted to
assess the association between cancer and the long-term consumption of
disinfected drinking-water. Studies were conducted in various
geographic locations with different types of water sources, chemical
quality and levels and types of DBPs. Ecological, cohort and
case-control studies of incident and decedent cases were conducted.
The quality of information about water disinfection exposures and
potential confounding characteristics differs dramatically between
these studies. In many of the case-control studies, interviews or
other methods were used to obtain information about various risk
factors, confounding characteristics and residential histories, to
determine long-term exposures to disinfected drinking-water supplies;
in several studies, individual tapwater consumption was estimated.
However, in several other case-control studies, limited information
about exposure and confounding factors was obtained only from the
death certificates.
In most studies, disease incidence or mortality was compared
between populations supplied with chlorinated surface water and those
supplied with unchlorinated groundwater. The chemical quality of
drinking-water for a number of chemical constituents, including DBPs,
differs between surface water and groundwater and also among the
various surface waters in the different geographic locations studied.
Surface water sources may also be contaminated with non-volatile
synthetic organic compounds from industrial, agricultural and
residential runoff. Groundwater may be contaminated with volatile
synthetic organic compounds and inorganic constituents, such as
arsenic and nitrate. It is not feasible to consider epidemiological
studies of cancer in populations consuming unchlorinated
drinking-water from surface water sources because so few people
consume such drinking-water, but exposures to water contaminants in
addition to DBPs must be considered. Since the quality of water
sources may also affect the concentration and type of DBPs, even when
the same disinfectant is used, it is important to assess specific
by-products in water systems included in epidemiological studies.
5.2.1.1 Cancer associations in ecological studies
Early epidemiological studies analysed group or aggregate data
available on drinking-water exposures and cancer. Usually the
variables selected for analysis were readily available in published
census, vital statistics or public records. Cancer mortality rates,
usually obtained for census tracts, counties or other geographic
regions, were compared for areas with different water sources and
disinfection practices. Differing source waters and their disinfection
were usually used as surrogates for drinking-water exposures of the
populations. For example, the drinking-water for the area was
categorized as being primarily from a surface water or from a
groundwater source and as chlorinated or not chlorinated. In some
instances, exposure variables included estimates of the proportion of
the area's population that received drinking-water from chlorinated
surface water or unchlorinated groundwater. In several ecological
analyses, the investigators studied the association between cancer
mortality and an estimate of population exposures to levels of
chlorination by-products based on the measure of THMs or chloroform
levels from a limited number of water samples from monitoring studies
(see section 5.3.1.1).
The first ecological studies reported higher mortality from
several cancers in Louisiana (USA) parishes where the majority of the
population used the lower Mississippi River as a source of
drinking-water. Lower mortality was reported in parishes where the
majority of the population used groundwater sources (Harris, 1974;
Page et al., 1976). Additional ecological studies for different
geographic areas of the USA, including Louisiana, Ohio, Missouri,
Kentucky, New York, Massachusetts and Iowa, reported increased cancer
mortality or incidence in areas using chlorinated surface water
(Craun, 1991). A wide range of cancer sites was found to be
statistically associated with the use of chlorinated surface water.
These cancer sites included gall bladder, oesophagus, kidney, breast,
liver, pancreas, prostate, stomach, bladder, colon and rectum. These
studies have been extensively reviewed (NAS, 1980, 1986; Crump &
Guess, 1982; Shy, 1985; Craun, 1991; Murphy & Craun, 1990). The NAS
(1980) noted the limitations of these studies and recommended that
analytical studies be conducted to further assess the possible
association of chlorinated drinking-water and cancer. It was
recommended that studies focus on cancers of the bladder, stomach,
colon and rectum because they had been most often associated with
chlorinated water in ecological studies.
As noted in section 5.1.2, ecological analyses have theoretical
deficiencies, and the interpretation of results from these studies is
difficult. Associations reported from ecological analyses cannot be
evaluated for causality, nor do they provide an estimate of the
magnitude of risk. Yet these studies continue to be conducted. An
ecological study in Norway reported weak associations between
chlorinated drinking-water supplies and cancer of the colon and rectum
in men and women (Flaten, 1992). No associations between chlorinated
surface water supplies and bladder or stomach cancer were found in
Valencia province, Spain (Suarez-Varela et al., 1994). A study in New
Jersey found no association between DBPs and either bladder or rectal
cancer (Savin & Cohn, 1996). A study in Taiwan, China, reported
associations between the use of chlorinated drinking-water and cancer
of the rectum, lung, bladder and kidney (Yang et al., 1998). THMs were
not found to be associated with breast cancer risk in an ecological
study in North Carolina (Marcus et al., 1998).
5.2.1.2 Cancer associations in analytical studies
More case-control studies have been conducted than cohort
studies. Case-control studies have included the traditional
interview-based study and those where information was obtained only
from death certificates or other readily available sources.
1) Cohort studies
Wilkins & Comstock (1981) found no statistically significant
associations between the incidence of cancer mortality in Washington
County, Maryland, USA, and residence in an area supplied with
chlorinated surface water. Information was available for individuals
in a well-defined homogeneous cohort that allowed disease rates to be
computed by presumed degree of exposure to chlorination by-products.
The cohort was established from a private census during the summer of
1963 and followed for 12 years. The source of drinking-water at home
was ascertained, and personal and socioeconomic data were collected
for each county resident, including age, education, smoking history
and number of years lived at the 1963 address.
Potential cases of cancer were obtained from death certificate
records, the county's cancer registry and medical records of the
county hospital and a regional medical centre. Census data were used
to compute age-gender-site-specific cancer mortality incidence rates
for 27 causes of death, including 16 cancer sites, cardiovascular
disease, vehicular accidents, all causes of death and pneumonia. Three
exposure categories were examined: a high-exposure group of residents
served by chlorinated surface water, a low-exposure group served by
unchlorinated deep wells and a third group served by a combination of
chlorinated surface water and groundwater. The average chloroform
level from an extensive analysis of chlorinated surface water samples
was 107 µg/litre. The third group, which represented an intermediate
exposure, was not used in detailed analyses. Confounding bias was
controlled and incidence rates were adjusted by multiple regression
analysis for age, marital status, education, smoking history,
frequency of church attendance, adequacy of housing and number of
persons per room. Selected cancer mortality rates for males and
females are reported in Table 24. Small increased risks of bladder and
liver cancer were reported; these risk estimates are not statistically
stable. Although the study was of high quality and well conducted, the
associations reported are subject to random error (i.e., all relative
risks had a confidence interval that included 1.0 and thus were not
statistically significant). Even though over 31 000 people were
included in the cohort, estimates of specific cancer risks were based
on relatively few deaths.
History of length of residence was used to estimate a person's
duration of exposure to chlorinated and unchlorinated water. For
bladder and liver cancer in females and bladder cancer in males, the
association was stronger for persons who had lived in their 1963
domicile for 12 or more years than for those who had lived in it
3 years or less. Among men who had at least 24 years' exposure to
Table 24. Cohort study: Selected cancer mortality, water source and disinfection, Washington
County, Maryland (USA)a
Cause of death Chlorinated surface water Unchlorinated groundwater Risk
Deaths Incidence rateb Deaths Incidence rateb RR 95% CI
Females
Liver cancer 31 19.9 2 11.0 1.8 0.6-6.8
Kidney cancer 11 7.2 2 7.1 1.0 0.3-6.0
Bladder cancer 27 16.6 2 10.4 1.6 0.5-6.3
Males
Liver cancer 9 6.4 2 9.0 0.7 0.2-3.5
Kidney cancer 15 10.6 3 13.6 0.8 0.3-2.7
Bladder cancer 46 34.6 5 19.2 1.8 0.8-4.8
a From Wilkins & Comstock (1981).
b Adjusted incidence rate per 100 000 person-years.
chlorinated surface water, the bladder cancer risk was high (RR = 6.5;
95% CI = 1.0->100) but very imprecise and statistically unstable (the
CI is high). Additional follow-up of the cohort for several more years
can possibly provide more meaningful associations. Freedman et al.
(1997) and Ijsselmuiden et al. (1992) conducted case-control studies
in Washington County, Maryland (USA) to evaluate risks of cancer of
the bladder and pancreas. The results of these two studies are given
below.
Zierler et al. (1986) examined mortality patterns of
Massachusetts (USA) residents at least 45 years of age who died
between 1969 and 1983 and whose last residence was in a community
where drinking-water was disinfected with either chlorine or
chloramine. A standardized mortality study found little differences in
the patterns of 51 645 deaths due to cancer in 43 Massachusetts
communities with water supplies disinfected with either chlorine or
chloramine as compared with all cancer deaths reported in
Massachusetts. The mortality rate of residents from these selected
communities with chlorinated drinking-water was slightly higher than
expected for stomach cancer (SMR = 109; 95% CI = 104-114) and lung
cancer (SMR = 105; 95% CI = 103-107). Mortality rates in selected
communities served by chloraminated drinking-water was slightly less
than expected for bladder cancer (SMR = 93; 95% CI = 88-98) but
greater than expected for lung cancer (SMR = 104; 95% CI = 102-106).
Because residence at death was used to assign the exposure status of
persons to either chlorinated or chloraminated drinking-water, there
is a serious potential for exposure misclassification bias. In
addition, errors in death certificate classification of the cause of
death may also affect the interpretation of these findings.
Bean et al. (1982) conducted a cohort study of municipalities in
Iowa (USA) that were classified into groups based on the source of
their drinking-water -- surface water or groundwater of various
depths. Municipalities included were those with a 1970 population of
more than 999 and a public water supply that used exclusively either
surface water or groundwater sources that had remained stable for at
least 14 years. All of the surface water sources were chlorinated;
groundwater was less frequently chlorinated, especially as the depth
of the well increased. In the regression analysis, each group was
considered a single population, and age-adjusted, sex-specific cancer
incidence rates were determined for the years 1969-1978. Indicators of
socioeconomic status were obtained for the municipalities to determine
if they could explain any observed differences in cancer incidence,
and detailed information about residential mobility, water usage and
smoking, collected from a sample of non-cancer controls in a large
bladder cancer case-control study, was used to assess exposure
misclassification and confounding. Municipalities supplied by
chlorinated surface water had higher lung and rectal cancer incidence
rates than those using groundwater sources for all population groups
(<10 000; 10 000-50 000; >50 000); no differences were found for
colon and bladder cancer incidence between surface water and
groundwater sources. Using information reported by Bean et al. (1982),
Poole (1997) estimated a single relative risk for lung cancer
(RR = 1.1; 95% CI = 1.1-1.2) and rectal cancer (RR = 1.2; 95%
CI = 1.1-1.3).
Selected results from a prospective cohort study of 41 836
post-menopausal women in Iowa are reported in Table 25. The study
compared risks among users of groundwater and surface water sources;
an assessment of exposure to chloroform and other THMs was also
included (Doyle et al., 1997). In 1989, the participants were asked
about their source of drinking-water and the length of time this
source had been used. Information on potential confounding
characteristics was obtained from the baseline questionnaire. Analyses
were limited to those who reported drinking municipal or private
well-water for more than the past 10 years ( n = 28 237). Historical
water treatment and water quality data were used to ascertain exposure
to THMs (see section 5.3.1.2). No increased risks were found for women
who used private wells. In comparison with women who used 100%
municipal groundwater sources, women who used 100% municipal surface
water sources were at a statistically increased risk of all cancers
combined (RR = 1.3; 95% CI = 1.1-1.6), colon cancer and breast cancer.
No increased risk was observed for bladder cancer or cancer of the
rectum and anus; however, only two and six cases of cancer were
observed in the cohort. No increased risks were found for cancer of
the kidney and six other sites. Reported relative risks were adjusted
for age, education, smoking status, physical activity, fruit and
vegetable intake, total energy intake, body mass index and waist to
hip ratio. Limitations of this study include the relatively short
period (8 years) of follow-up of the cohort and possible
misclassification of exposure because the source of drinking-water was
assessed at only one point in time.
2) Decedent case-control studies without interviews or residential
histories
Six case-control studies where no information was obtained from
next-of-kin interviews (Alavanja et al., 1978; Struba, 1979; Brenniman
et al., 1980; Gottlieb et al., 1981, 1982; Young et al., 1981; Zierler
et al., 1986) considered only information that was routinely recorded
on death certificates or readily available from vital statistics, such
as occupation, race, age and gender. Deceased cases of cancer of
interest were identified, and controls were non-cancer deaths from the
same geographic area. In four of these studies, controls were matched
for certain characteristics, including age, race, gender and year of
death, to prevent possible confounding bias by these characteristics.
In studies where matching was not employed, these characteristics were
controlled in the analysis. Other possible confounders considered
included occupation listed on the death certificate and a measure of
urbanization of the person's residence. Information about important
potential confounders, such as diet and smoking, was not available and
could not be assessed. In one study, smoking was indirectly controlled
by assessing lung cancer risks among those exposed to chlorinated
water.
Table 25. Cohort study: Selected incidence of cancer in post-menopausal women and water source
in Iowa (USA)a
Cancer 100% municipal Mixed municipal surface water 100% municipal surface water
groundwater and groundwater
Cases RRb Cases RRb 95% CI Cases RRb 95% CI
Bladder 23 1.0 16 2.4 1.3-4.6 2 0.7 0.2-2.9
Colon 106 1.0 47 1.5 1.1-2.2 23 1.7 1.1-2.7
Rectal 53 1.0 20 1.3 0.8-2.2 6 0.9 0.4-2.1
Breast 381 1.0 106 0.9 0.8-1.2 65 1.3 1.03-1.8
Kidney 21 1.0 5 0.8 0.3-2.2 3 1.1 0.3-3.8
Lung 95 1.0 30 1.1 0.7-1.1 17 1.4 0.9-2.4
Melanoma 29 1.0 12 1.4 1.7-2.8 4 1.1 0.4-3.2
All cancers 631 1.0 223 1.2 1.04-1.4 112 1.3 1.1-1.6
a From Doyle et al. (1997).
b Reported relative risks were adjusted for age, education, smoking status, physical activity,
fruit and vegetable intake, total energy intake, body mass index and waist to hip ratio.
A single address (address at death or usual address) or
combination of addresses (address at birth and death) was used to
determine place of residence for assessing exposure to disinfected
water. This place of residence was linked to public records of water
source and treatment practices to classify the type of water source
and drinking-water disinfection. Exposures considered were surface
water or groundwater sources and chlorinated or unchlorinated water
sources at the residence (Shy, 1985); in one study, chlorinated and
chloraminated water were evaluated (Zierler et al., 1986). In three
studies, a statistically significant increased risk of bladder cancer
was found to be associated with chlorinated surface water; an
increased risk of colon cancer was found in three studies, and an
increased risk of rectal cancer was found in four studies (Shy, 1985;
Zierler et al., 1986).
Alavanja et al. (1978) studied 3446 deaths due to total urinary
tract and total gastrointestinal cancers in seven New York (USA)
counties during 1968-1970. The comparison group was non-cancer deaths
from the same period matched on age, race, gender, county of birth and
county of residence; occupation and the urban nature of the county
were considered as potential confounders. Statistically significant
increased risks of colon (OR = 2.0) and bladder (OR = 2.0) cancer
mortality were found to be higher in men but not women who resided in
communities with chlorinated surface water. Other statistically
significant increased risks in men who resided in communities with
chlorinated surface water were for liver and kidney (OR = 2.8),
oesophagus (OR = 2.4) and pancreas (OR = 2.2) cancer mortality;
stomach cancer risks were increased for both men (OR = 2.4) and women
(OR = 2.2). A study by Struba (1979) of bladder, colon and rectal
cancer deaths (700-1500 cases per site) and a comparison group of
deaths matched on age, race, gender and region of residence in North
Carolina (USA) during 1975-1978 also found statistically significant
increased risks for bladder (OR = 1.5), rectal (OR = 1.5) and colon
(OR = 1.3) cancer mortality associated with chlorinated water. In
studies of similar design, Brenniman et al. (1980), Young et al.
(1981) and Gottlieb et al. (1981, 1982) did not find significant
increased risks of bladder or colon cancer mortality associated with
chlorinated water.
Gottlieb et al. (1981, 1982) found increased risks for rectal
(OR = 1.7) and breast (OR = 1.6) cancer mortality associated with
chlorinated water in Louisiana (USA). In Illinois (USA), Brenniman et
al. (1980) found an increased risk of rectal cancer mortality, but
only for females (OR = 1.4). Young et al. (1981) reported an
association between colon cancer mortality in Wisconsin (USA) and the
average daily chlorine dosage of drinking-water over a 20-year period.
The study included 8029 cancer deaths and 8029 non-cancer deaths in
white women matched on county of residence, age and year of death.
Urbanization, marital status and occupation were considered as
potential confounders. Logistic regression analysis was used to
evaluate risks for a number of site-specific cancer deaths associated
with drinking-water classified by investigators as high, medium and
low chlorine-dosed water. Only cancer of the colon was found to be
statistically associated with the use of chlorine, but the risk did
not increase with higher chlorine dosages. Young et al. (1981) found
no association between chlorinated drinking-water and mortality from
cancer of the bladder, liver, kidney, oesophagus, stomach, pancreas,
lung, brain or breast.
Zierler et al. (1986) evaluated exposures to surface water
supplies disinfected with either chlorine or chloramine among 51 645
persons aged 45 years and older who died from cancer and 214 988
controls who died from cardiovascular, cerebrovascular or pulmonary
disease or from lymphatic cancer in 43 Massachusetts (USA)
communities. Using lymphatic cancer deaths as the comparison group,
bladder cancer mortality was found to be moderately increased
(OR = 1.7; 95% CI = 1.3-2.2) among residents who died in communities
with chlorinated drinking-water as compared with those who died in
communities with chloraminated water. This analysis controlled for the
potential effects of differences in population density, poverty, age
at death and year of death between the communities treated with
different disinfectants. Because smoking is a known risk factor for
bladder cancer, a special comparison group of lung cancer deaths was
enrolled to evaluate this potential cause of confounding bias. Results
suggested that smoking did not explain all of the excess mortality
from bladder cancer; thus, chlorinated drinking-water may present some
risk. However, because residence at death is a poor measure of
long-term exposure to disinfected water, an interview-based
case-control study was designed to further evaluate the possible
association between chlorinated water and bladder cancer (see below).
Although not subject to all of the design limitations of
ecological studies, these decedent case-control studies are,
nevertheless, still limited in their ability to provide information
about the causal nature of the associations observed. Interpretation
of results reported by these studies is limited because of likely
systematic bias due to misclassification and uncontrolled confounding.
Use of decedent instead of incident cancer cases means that
differential survival among cases may also influence the observed
association. Selection bias is also likely in the control group.
Insufficient information was available in these studies to adequately
assess historical, long-term exposures to chlorinated water, and use
of a single residential address can result in exposure
misclassification bias, which may lead to risk being underestimated or
overestimated. While the magnitude of risk may be underestimated, as
was the case in Iowa (Lynch et al., 1989), risk may also be
overestimated as a result of possible different residential mobility
patterns. Thus, these studies can provide very limited information
about causality and the magnitude of cancer risks of chlorinated
water. The findings from these studies should be interpreted with
caution because of study design limitations.
3) Case-control studies with interviews or residential histories
Two additional decedent case-control studies included more
complete information about residential histories for a better
assessment of exposure to disinfected water. Lawrence et al. (1984)
studied the relationship of THMs and colo-rectal cancer among a cohort
of white female teachers in New York State (USA) who died of either
colo-rectal cancer or non-cancer causes. A case-control study (Zierler
et al., 1990) of individuals who had died of primary bladder cancer
and other causes was conducted in selected Massachusetts (USA)
communities that obtained drinking-water from surface water sources
disinfected by either chlorine or chloramine. Eleven case-control
studies assessed risks of cancer incidence and included interviews
with study participants or a surrogate to obtain information about
complete residential histories, long-term drinking-water exposures to
chlorinated or chloraminated water and potential confounding
characteristics (Cantor et al., 1985, 1987, 1990, 1995, 1997, 1998;
Cragle et al., 1985; Young et al., 1987, 1990; Lynch et al., 1990;
Ijsselmuiden et al., 1992; McGeehin et al., 1993; Vena et al., 1993;
King & Marrett, 1996; Freedman et al., 1997; Hildesheim et al., 1998).
In seven of these studies (Cantor et al., 1987, 1990, 1995, 1997,
1998; McGeehin et al., 1993; Vena et al., 1993; King & Marrett, 1996;
Hildesheim et al., 1998), information about tapwater consumption was
also obtained. In six studies (Young et al., 1987, 1990; McGeehin et
al., 1993; Cantor et al., 1995, 1997, 1998; Hildesheim et al., 1998;
King & Marrett, 1996), THM exposure was evaluated (see also section
5.3.1.2).
Bladder cancer risk
Chlorinated water studies. Cantor et al. (1985) reported results
from a national study of water chlorination and bladder cancer in the
USA, the largest case-control study of water chlorination risks
reported to date. The study included 2982 people between the ages of
21 and 84 diagnosed with bladder cancer in 1978 and residing in five
states and five metropolitan areas of the USA and 5782
population-based comparison subjects, randomly selected and frequency
matched on gender, age and study area. The primary purpose of the
study was to evaluate the possible association of bladder cancer and
artificial sweeteners; however, because of its case-control design, it
was possible to include an assessment of drinking-water exposures.
Participants were interviewed for information about past residences,
smoking, occupation, artificial sweetener use, coffee and tea
consumption and use of hair dyes. A lifetime residence history
categorized individuals according to water sources and chlorination
status on a year-by-year basis. Information was obtained on use of
bottled water and fluid consumption. Logistic regression analysis was
used to control for potential confounding bias. Overall, no
association was found between bladder cancer risk and duration of
exposure to chlorinated water. In all study areas combined, no
increased risk was found among participants who lived in areas with
chlorinated water supplies for <20, 20-39, 40-59 and 60 or more years
(Table 26A). However, an increased bladder cancer risk was found among
persons who never smoked, were never employed in a high-risk
occupation and resided in areas served by chlorinated surface sources
(Table 27A). There was little evidence of an exposure-response
relationship among persons who had never smoked. Only in the low-risk
group of non-smokers who had resided 60 or more years in an area
served by chlorinated water was the increased bladder cancer risk
statistically significant (RR = 2.3; 95% CI = 1.3-4.2). Although the
study included a large number of cases and controls, this subgroup
analysis included relatively few study participants (Table 27A). Only
46 cases and 77 controls were included in the analysis, which found a
doubling of risk among non-smokers who had resided 60 or more years in
an area served by chlorinated water.
Lynch et al. (1990) conducted a separate analysis of the Iowa
(USA) portion of the national bladder cancer study. Included were
294 primary, histologically confirmed cases of bladder cancer in
whites and 686 comparison subjects, all of whom had spent more than
50% of their lifetimes on primary water sources with known
chlorination exposure. Study participants exposed to chlorinated water
sources for more than 40 years were found to have twice the risk of
bladder cancer (OR = 2.0; 95% CI = 1.3-3.1) compared with participants
exposed to unchlorinated groundwater sources. Unadjusted risks were
higher (OR = 9.9; 95% CI = 2.6-38.0) when the analysis was restricted
to study participants who used, for more than 40 years, water sources
that had been chlorinated prior to filtration, a water treatment
practice known to result in higher THM levels. However, for both of
these chlorination practices, no statistically significant risks were
found to be associated with the use of chlorinated water for 40 or
fewer years. The risk of bladder cancer was also increased for
cigarette smokers with longer duration of exposure to chlorinated
drinking-water. Unadjusted risks were 4 times as great in heavy
smokers (OR = 9.9; 95% CI = 2.6-38.0) as in non-smokers (OR = 2.7; 95%
CI = 1.2-5.8) who were exposed to chlorinated water for more than 30
years. A greater risk of bladder cancer was also seen in heavy smokers
(OR = 4.0; 95% CI = 2.0-8.4) who were exposed to chlorinated water for
30 or fewer years.
King & Marrett (1996) conducted a population-based case-control
study in Ontario (Canada). Cases were residents between 25 and 74
years of age with a histologically confirmed diagnosis of primary
cancer or carcinoma in situ of the bladder, diagnosed between
1 September 1992 and 1 May 1994. Of 1694 eligible cases, 250 patients
were not included in the study because an appropriate physician could
not be identified or the patients were recently deceased or too ill to
be contacted. Of the remaining 1444 patients, consent was received to
contact 1262 (87%). The remaining 13% of cases did not participate
because the physician refused to participate, did not provide consent
or did not respond. Controls were an age-sex frequency matched sample
of the general population from households randomly selected from a
computerized database of residential telephone listings in the same
area. Since the control subjects were also used to study cancers of
the colon and rectum with respect to the same exposures (results not
yet published), they were selected to have the expected age-sex
Table 26. Bladder cancer risks and duration of exposure to chlorinated surface water in
five interview-based incident case-control studies
Years of Odds 95% CI Comments Reference
exposure to ratio
chlorinated
surface water
A. Ten areas of the USA Whites only; adjusted Cantor et al. (1985)
0 1.0 for age, gender, smoking,
1-19 1.1 0.8-1.4 usual employment as a
20-39 1.0 0.8-1.3 farmer, study area
40-59 1.0 0.8-1.3
>60 1.1 0.8-1.5
B. Ontario, Canada Adjusted for age, gender, King & Marrett (1996)
0-9 1.0 smoking, education, calorie
10-19 1.0 0.7-1.5 intake
20-34 1.2 0.9-1.5
>35 1.4 1.1-1.8
C. Colorado Whites only; adjusted for McGeehin et al. (1993)
0 1.0 gender, smoking, coffee
1-10 0.7 0.4-1.3 consumption, tapwater
11-20 1.4 0.8-2.5 intake, family history
21-30 1.5 0.8-2.9 of bladder cancer, medical
>30 1.8 1.1-2.9 history of bladder infection
or kidney stone
D. Iowa Adjusted for age, gender, Cantor et al. (1998)
0 1.0 smoking, education, high-risk
<19 1.0 0.8-1.2 employment, study area
20-39 1.1 0.8-1.4
40-59 1.2 0.8-1.7
>60 1.5 0.9-2.6
E. Washington County, Maryland Adjusted for age, gender, Freedman et al. (1997)
0 1.0 smoking, urbanicity
1-10 1.0 0.6-1.5
11-20 1.0 0.6-1.6
21-30 1.1 0.6-1.8
31-40 1.1 0.6-2.2
>40 1.4 0.7-2.9
Table 27. Bladder cancer risks for smokers and non-smokers exposed to chlorinated water
Years at a residence Cases Controls Odds ratio Cases Controls Odds ratio Reference/comments
served by chlorinated (95% CI) (95% CI)
water
A. Males and females, Never Current Cantor et al. (1985)
10 areas of the USA smoked smoker
0 61 268 1.0 87 109 1.0 Whites, adjusted for study
1-19 29 110 1.3 (0.7-2.2) 63 71 0.9 (0.6-1.5) area, gender, age, usual
20-39 73 236 1.5 (0.9-2.4) 136 186 0.7 (0.4-1.1) employment as a farmer,
40-59 108 348 1.4 (0.9-2.3) 166 211 0.7 (0.5-1.2) smoking
>60 46 77 2.3 (1.3-4.2) 27 37 0.6 (0.3-1.2)
B. Males and females, Non-smokers Smokers McGeehin et al. (1993)
Colorado
0 19 45 1 85 57 1 Whites only; adjusted for
1-11 7 21 0.8 (0.2-2.4) 36 33 0.8 (0.4-1.4) gender, smoking, coffee
12-34 11 34 0.9 (0.3-2.3) 70 34 1.4 (0.8-2.4) consumption, tapwater intake,
>34 21 25 2.9 (1.2-7.4) 77 25 2.1 (1.1-3.8) family history of bladder
cancer, medical history of
bladder infection or kidney stone
C. Males and females, Never Smoker (Freedman et al., 1997)
Washington County, smoked (past/present)
Maryland
0 32 331 1.0 47 391 1.3 (0.8-2.2) Adjusted for age, gender, and
1-10 21 232 0.8 (0.4-1.6) 70 469 1.4 (0.8-2.5) urbanicity
11-20 15 147 0.9 (0.5-1.9) 41 285 1.4 (0.8-2.5)
21-30 6 89 0.6 (0.2-1.5) 32 177 1.7 (0.9-3.2)
31-40 3 53 0.5 (0.1-1.5) 13 54 2.2 (1.0-4.7)
>40 5 51 0.9 (0.3-2.3) 8 27 2.8 (1.0-6.9)
Table 27. (continued)
Years at a residence Cases Controls Odds ratio Cases Controls Odds ratio Reference/comments
served by chlorinated (95% CI) (95% CI)
water
D. Males, Iowa Never Current Cantor et al. (1998)
smoked smoker
0 112 332 1.0 188 156 3.5 (2.5-4.7) Adjusted for age, study period,
1-19 27 75 1.0 (0.6-1.6) 73 62 3.5 (2.3-5.3) education, high-risk population
20-39 6 22 0.8 (0.3-2.0) 37 20 5.7 (3.1-10.4)
>40 5 26 0.7 (0.3-1.9) 29 16 5.8 (3.0-11.3)
distribution of these three cancer sites combined. In over 90% of the
2768 households with an eligible resident, the person selected for the
study agreed to participate. The overall response rate considering
actual participation was 73% for cases and 72% for controls.
Relevant exposure and confounding variables were collected using
a mailed questionnaire in combination with a computer-assisted
telephone interview. Interviewers were blinded as to the case or
control status of the subject. Questions were included on demographics
(e.g., gender, date of birth and education), other potentially
important confounding variables (e.g., smoking history and usual diet
prior to diagnosis) and information pertaining to the primary
exposures of interest (e.g., residence and water source history and
usual water consumption prior to diagnosis). Persons reported their
drinking-water source and other household water sources at each
residence as municipal, household well, bottled water or other. Volume
of tapwater was calculated from the reported daily frequency of
consuming beverages containing water during the 2-year period before
the interview and usual source of water (tap or bottled) used to make
hot and cold beverages. The water supply for each participant was
characterized by source (surface water vs. groundwater) and
chlorination status (chlorinated vs. unchlorinated).
The analysis considered the 696 cases (75%) and 1545 controls
(73%) for whom water source characteristics were available for at
least 30 of the 40 years ending 2 years before the person's interview.
To reduce possible misclassification of exposure, only persons with 30
or more years of known water history were included in a separate
analysis. Logistic regression was used to estimate bladder cancer
risks. Potentially important confounders considered were age, gender,
smoking, education, consumption of alcoholic beverages, coffee
consumption, total fluid consumption and dietary intake of energy
(total calories), protein, fat, cholesterol, fibre and vitamin A.
The study found a pattern of increasing bladder cancer risk with
increasing number of years exposed to chlorinated surface water, but a
statistically significant association was found only for lengthy
exposures (Table 26B). For persons exposed to a chlorinated surface
water source for 35 years or more, the bladder cancer relative risk
was increased by 40% in comparison with those exposed for less than
10 years. An analysis restricted to those who had relatively
homogenous water exposures found that exposure to chlorinated surface
water for 30 or more years was associated with a weak increased risk
(OR = 1.4; 95% CI = 1.1-1.8) compared with exposure to a groundwater
source. King & Marrett (1996) found higher relative risk estimates for
non-smokers associated with many years of exposure to chlorinated
drinking-water from surface water sources, but the difference in risk
compared with smokers was not statistically significant, nor was the
pattern of higher risks in non-smokers observed consistently.
McGeehin et al. (1993) conducted a case-control study to assess
the relationship between chlorinated or chloraminated drinking-water
and bladder cancer in Colorado. Study participants were identified
from a population-based cancer registry; 327 histologically confirmed
incident bladder cancer cases and 261 randomly selected controls with
other cancers, except colo-rectal and lung, were interviewed by a
single blinded interviewer about demographic data, drinking-water
source, fluid consumption and personal habits. Persons with
colo-rectal and lung cancers were excluded from the control group
because of a possible association with chlorinated surface water
reported in earlier studies. Of the originally identified cases, 38%
could not be interviewed because permission was denied by the
physician, cases were dead or persons refused to participate. For
controls, 47% were not interviewed for these reasons. Because of the
large number of excluded study participants, selection bias is a
concern. Bias could be present if the excluded persons were different
from participating persons in ways that are related to the exposure
and outcome.
Information was collected from individuals on tapwater
consumption and from local water utilities regarding water source and
method of disinfection in order to construct individual lifetime
profiles of exposure to disinfected drinking-water. Although 91% of
person-years of community exposure to specific water sources could be
determined, only 81% of disinfection methods, 60% of THM levels and
55% of residual chlorine levels could be determined for use in
assessing exposures.
Logistic regression analysis, adjusting for coffee consumption,
smoking, tapwater intake, family history of bladder cancer, sex and
medical history of bladder infection or kidney stone, found no
statistically significant increased risk of bladder cancer for persons
exposed to chlorinated drinking-water for 1-10, 11-20 or 21-30 years
compared with those with no exposure to chlorinated drinking-water
(Table 26C). The increased relative risk (OR = 1.8; 95% CI = 1.1-2.9)
of bladder cancer for persons exposed to chlorinated drinking-water
for more than 30 years was statistically significant. A trend of
increasing risk with increasing duration of exposure was reported
(trend P < 0.01), but no association was found between THM levels
and bladder cancer (also see section 5.3.1.2). The authors reported
that these results support the hypothesis that prolonged (i.e., more
than 30 years) exposure to chlorinated drinking-water from surface
water sources is associated with an increased risk of bladder cancer.
However, incomplete characterization of the exposure profiles for
water sources and disinfection, potential bias from non-participation
and potential confounding bias from undetermined confounders tend to
limit this interpretation. An increased risk of bladder cancer was
seen among both smokers and non-smokers with more than 34 years of
exposure to chlorinated water (Table 27B).
Freedman et al. (1997) conducted a population-based case-control
study in Washington County, Maryland (USA) to evaluate the association
between the incidence of bladder cancer and use of chlorinated surface
water (Table 26E). The study included 294 bladder cancer cases in
white residents enumerated in a 1975 county census and reported to the
county cancer registry between 1975 and 1992; 2326 white controls,
frequency matched by age and gender, were randomly selected from the
census. Duration of exposure to chlorinated surface water was based on
length of residence in the census household before 1975, and relative
risks were calculated using logistic regression methods adjusting for
age, gender, tobacco use and urbanicity. Nearly all municipal sources
in 1975 were supplied by surface waters that had been chlorinated for
more than 30 years. Bladder cancer risk was found to be weakly
associated with municipal water and duration of exposure to municipal
water (for exposure to municipal water for more than 40 years,
OR = 1.4; 95% CI = 0.7-2.9). The association was limited, however, to
those who smoked cigarettes (Table 27C), primarily male smokers. Male
smokers who had resided more than 40 years in an area with municipal
water supplies had a 3-fold greater risk than male smokers who had not
resided in an area with municipal water supplies or had resided in
such an area for 20 years or less (OR = 3.2; 95% CI = 1.1-8.6). No
increased relative risk was found for female smokers; in fact,
relative risks for exposure to municipal water for 1-30 years were
less than those for no exposure to municipal water (OR ranged from 0.4
to 0.6; 95% CI ranged from 0.1 to 2.9). A limitation of this study is
that duration of exposure was based solely on place of residence in
1975. Also, there was no information on prior or subsequent domiciles,
but the authors felt that because the population was relatively
stable, residential mobility was not a major concern. No water
consumption data were available, nor were data on DBPs available. As
previously reported, however, chloroform analyses in 1975 found an
average chloroform concentration of 107 µg/litre in chlorinated
surface waters used by the cohort (Wilkins & Comstock, 1981).
Cantor et al. (1998) and Hildesheim et al. (1998) conducted a
population-based case-control study in Iowa (USA) in 1986-1989 to
evaluate cancer risks that may be associated with chlorinated water;
results have been reported for bladder, colon and rectal cancer.
Information about residential history, drinking-water source, beverage
intake and other factors was combined with historical data from water
utilities and measured THM levels to create indices of past exposure
to chlorinated by-products. The bladder cancer study was composed of
1123 incident cases who were residents of Iowa, aged 40-85 years and
diagnosed with histologically confirmed bladder cancer in 1986-1989
and 1983 randomly selected controls from driver's licence records and
US Health Care Financing Administration listings for whom data
relating to at least 70% of their lifetime drinking-water source were
available. Of 1716 eligible bladder cancer cases, 85% participated.
Adjusted odds ratios were determined using unconditional logistic
regression analysis. Where appropriate, risks were adjusted for
gender, age, cigarette smoking, years of education and employment in
an occupation with elevated bladder cancer risk, including in the
analysis persons with at least 70% of lifetime years with available
information on drinking-water source. No increased risk was observed
for men and women combined (Table 26D). Risk increased among men, with
duration of chlorinated surface water exposure, duration of
chlorinated groundwater exposure and duration of exposure to any
chlorinated water source. For women, risks did not increase, and a
protective effect for duration of exposure to chlorinated surface
water was suggested, with OR values of less than unity; OR values
ranged from 0.7 to 0.9, and 95% CIs ranged from 0.2 to 2.4. The
increased relative risks in men were restricted to current smokers
(Table 27D) and past smokers. No increased risk was found for men who
had never smoked. Among non-smoking men and women, regardless of their
previous smoking habit, there was no association between duration of
exposure to chlorinated water and bladder cancer risk. Little or no
association of risk was found for either total daily tapwater intake
or intake of all beverages for men or women (Table 28A).
Table 28. Bladder cancer risks associated with daily tapwater consumption
Tapwater Odds 95% CI Comments Reference
consumption ratio
(litres/day)
A. Males and females, Iowa Beverages from Cantor et al.
<1.58 1.0 tapwater; adjusted for (1998)
1.58-<2.13 1.2 0.9-1.5 gender, age, study
2.13-2.85 1.3 1.0-1.6 period, education,
>2.85 1.2 0.9-1.5 high-risk occupation,
smoking
B. Males and females, 10 areas Adjusted for age, Cantor et al.
of the USA gender, smoking, (1985, 1987,
<0.80 1.0 high-risk occupation, 1990)
0.81-1.12 1.1 0.9-1.3 population size,
1.13-1.44 1.1 1.0-1.3 usual residence
1.45-1.95 1.3 1.1-1.5
>1.95 1.4 1.2-1.7
Chloraminated water studies. In Massachusetts (USA), a number
of towns have used surface water disinfected only with chlorine or
chloramine since 1938, providing an opportunity to compare cancer
risks between these two disinfectants. In a previous study, Zierler et
al. (1986) found bladder cancer mortality to be weakly associated with
residence at death in Massachusetts communities using chlorine for
water disinfection; however, because of the concern that residence at
time of death is a poor measure of historical exposure to a water
disinfectant and likely resulted in misclassification bias, an
analytical study (Zierler et al., 1988, 1990) was also conducted.
Eligible for the study were all persons who were over 44 years of age
at death and who died during 1978-1984 from bladder cancer, lung
cancer, lymphoma, cardiovascular disease, cerebrovascular disease or
chronic obstructive pulmonary disease while residing in 43 selected
communities. Included were 614 persons who died of primary bladder
cancer and 1074 individuals who died of other causes. Possible
confounding bias by age, gender, smoking, occupation and socioeconomic
status was controlled by multiple logistic regression. Detailed
information on each person's residential history and possible
confounding characteristics was obtained from survivors and census
records. Analyses included a person's usual exposure (at least 50% of
their residence since 1938 was in a community where surface water was
disinfected by only one of the two disinfectants, either chlorine or
chloramine) or lifetime exposure to water disinfected with only one of
the two disinfectants.
An association was found between bladder cancer mortality and
both usual and lifetime exposure to chlorinated drinking-water. The
bladder cancer mortality risk was higher for lifetime exposure than
for usual exposure; a 60% increased risk of bladder cancer mortality
(MOR = 1.6; 95% CI = 1.2-2.1) was found among lifetime residents of
communities where only chlorinated surface water was used. The
association is statistically significant, and the estimate of risk is
precise (i.e., the CI is small). Bias is not likely present, but the
magnitude of the association is not large and may be subject to
residual confounding by unknown, unmeasured characteristics. The
magnitude of the association found by Zierler et al. (1988, 1990),
however, may be even larger than a 60% increased risk. Using only
lymphatic cancers as the comparison group, the risk of bladder cancer
mortality among lifetime consumers of chlorinated water was 3 times
the risk for consumers of chloraminated drinking-water (MOR = 2.7; 95%
CI = 1.7-4.3). This suggests that one or more causes of death in the
comparison group may also be associated with water chlorination.
McGeehin et al. (1993) also found that the risk of bladder cancer
decreased with increasing duration of exposure to chloraminated
surface water (trend P < 0.01). Persons who consumed chloraminated
water for 21-40 years had a decreased, but not statistically
significant, bladder cancer risk (OR = 0.7; 95% CI = 0.1-1.1). Those
who consumed chloraminated water for more than 40 years also had a
slightly decreased risk of cancer (OR = 0.6; 95% CI = 0.4-1.0).
However, neither of these risks was statistically significant.
McGeehin et al. (1993) reported that their results do not imply that
chloraminated water conveys a protective effect, because there is no
plausible biological explanation for suggesting that choramination
inhibits neoplastic transformation of the bladder epithelium. Zierler
et al. (1988, 1990) found that lifetime users of chloraminated surface
water had a lower bladder cancer mortality risk than lifetime users of
chlorinated surface water, and the findings of McGeehin et al. (1993),
if real and not due to bias, provide additional evidence to support
the conclusion that bladder cancer risks may be lower in persons using
chloraminated drinking-water for long periods of time.
Water and fluid consumption studies. Studies have reported both
increased (Claude et al., 1986; Jensen et al., 1986) and decreased
(Slattery et al., 1988) risk of bladder cancer associated with a high
total fluid intake. Vena et al. (1993) conducted a case-control study
that investigated the relationship between the incidence of bladder
cancer and fluid intake and consumption of drinking-water. The study
included 351 white males with histologically confirmed transitional
cell carcinoma of the bladder and 855 white male controls with cancer
of one of six other sites (oral cavity, oesophagus, stomach, colon,
rectum and larynx).
Study subjects were interviewed about diet and their total daily
fluid intake of alcoholic beverages, bottled beverages, soda, milk,
coffee, tea, all juices and glasses of tapwater. A dose-response
relationship was found between daily intake of total liquids and risk
of bladder cancer when a number of potential confounding factors were
controlled (trend P < 0.001). For persons under the age of 65, up
to a 6-fold increased risk of bladder cancer was found for those who
drank more than 7 cups of fluids compared with those who drank 2-7
cups of total fluids daily (the lowest quartile studied). The OR
ranged from 2.6 (95% CI = 1.2-5.7) for those who drank 8-10 cups of
total fluids per day to 3.7 (95% CI = 1.7-8.2) for those who drank
11-13 cups and 6.3 (95% CI = 2.8-14.1) for those who drank 14-49 cups
of total fluids daily. These ORs are adjusted for age, education,
cigarette smoking, coffee, carotene and sodium by logistic regression.
Statistically significant but slightly smaller risks were observed for
those 65 and older. Total fluid consumption was divided into tapwater
and non-tapwater consumption. Tapwater beverages included coffee, tea
(hot and iced), 75% reconstituted orange juice, all other juices and
glasses of water taken directly from the tap. A dose-response
relationship was observed between increased tapwater consumption and
bladder cancer (trend P < 0.001), but no statistically significant
increased risk of bladder cancer, regardless of age, was found for
persons consuming either 6-7 or 8-9 cups of tapwater per day compared
with persons consuming 0-5 cups. Only in the highest group (10-39 cups
of tapwater per day) was the association between tapwater consumption
and bladder cancer statistically significant (OR = 2.6, 95% CI =
1.5-4.5, for those under the age of 65; OR = 3.0, 95% CI = 1.8-5.0,
for those 65 and older). Increased bladder cancer risk was also found
among persons under the age of 65 with the highest quartile of
non-tapwater intake.
The results provide additional hypotheses for further study, but
limitations of this study preclude definitive conclusions regarding a
potential water chlorination-cancer association. Over a third of
potential cases were excluded for reasons including death (3%),
refusal to be interviewed (24%) and extreme illness (11%). Selection
bias would be present if the excluded cases were different from the
participating cases in ways that are related to the exposure and
outcome. Recall bias may also have been present, as bladder cancer
cases were asked to recall usual dietary habits for the year before
onset of cancer symptoms, whereas the controls, other cancer patients,
were asked to recall dietary habits for the year before their
interview for the study. Assessing fluid consumption in the year
before onset of symptoms may not reflect historical or lifetime
patterns, and increased consumption may occur during the early stages
of bladder cancer.
Since increased risks were seen primarily in total fluid
consumption, it is also possible that total fluid consumption may be
relevant in the pathogenesis of bladder cancer. Total daily fluid
intake may be a marker for some unmeasured risk factor, and it is
important to determine the biological relevance of increased fluid
intake, independent of chlorinated tapwater, in the pathogenesis of
bladder cancer (US EPA, 1994a).
Since over 70% of the study population spent more than 90% of
their lives using chlorinated surface water from public water supplies
in western New York State, it is possible that the observed increased
risks for bladder cancer may be associated with a high consumption of
chlorinated tapwater. However, limited information was available with
which to determine a study participant's duration of exposure to
specific municipal water systems, and risks were not compared for
populations using chlorinated surface water supplies.
Cantor et al. (1987, 1990) conducted a further analysis of the
national bladder cancer study to include beverage consumption
information that was available for 5793 men and 1983 women. After
correcting for age, smoking and other potential confounding
characteristics, it was found that people who drank the most
chlorinated tapwater had a bladder cancer relative risk about 40%
higher than people who drank the least (Table 28B). When tapwater
consumption was analysed separately for men and women, however, the
association between water consumption and bladder cancer risk was
statistically significant only among males. Bladder cancer risk was
also evaluated for the combined effects of tapwater consumption and
duration of chlorinated surface water use. No increased risk was
associated with a high consumption of chlorinated drinking-water from
surface water sources for less than 40 years. Increased bladder cancer
risk (OR = 3.2; 95% CI = 1.2-8.7) was seen primarily in populations
who had resided 60 or more years in areas served by chlorinated
surface water and whose tapwater consumption was above the median of
greater than 1.4 litres per day. Among non-smoking men, a risk
gradient was apparent for those who consumed more than the population
median of tapwater (Table 29), but a higher risk was also seen for
non-smoking women who consumed less than the median amount. The
increasingly smaller numbers of participants available for these
subgroup analyses generally lead to statistically unstable estimates,
making it difficult to evaluate trends in these data.
In Colorado (USA) (McGeehin et al., 1993), the risk of bladder
cancer (OR = 2.0; 95% CI = 1.1-2.8) was found to be elevated among
persons who consumed more than five glasses of tapwater per day, and a
dose-response trend was found ( P < 0.01). However, tapwater
consumption appeared to be an independent risk factor for bladder
cancer. There was no evidence of an increased relative risk of bladder
cancer when both the volume of water consumed and years of exposure to
chlorinated water were considered in the analysis. For example,
similar estimates of risk were found for those who consumed more than
five glasses of chlorinated water for fewer than 12 years (OR = 2.0;
95% CI = 0.8-4.7) and those who consumed more than five glasses of
Table 29. Bladder cancer risks among non-smokers according to daily tapwater consumption and exposure
to chlorinated surface water in 10 areas of the USAa
Years at Odds ratio (95% CI)b
residence with
chlorinated water Tapwater consumption below 1.4 litres Tapwater consumption above 1.4 litres
Males Females Males Females
0 1.0 1.0 1.0 1.0
1-19 1.6 (0.7-4.0) 1.2 (0.4-3.7) 0.8 (0.3-2.5) 1.7 (0.5-5.4)
20-39 1.0 (0.4-2.6) 1.5 (0.5-4.4) 2.1 (0.9-5.2) 1.8 (0.6-5.4)
40-59 0.7 (0.3-1.9) 2.1 (0.8-5.9) 2.5 (0.9-6.6) 1.8 (0.6-5.9)
>60 1.3 (0.4-4.4) 4.3 (1.3-14.5) 3.7 (1.1-12.0) 3.6 (0.8-15.1)
a From Cantor et al. (1987, 1990).
b Adjusted for age, gender, smoking, high-risk occupation, population size, usual residence.
chlorinated water for 12 years or more (OR = 2.4; 95% CI = 1.0-5.9).
Similar to the study in western New York (Vena et al., 1993), tapwater
consumption in Colorado was assessed for the year prior to diagnosis
for cancer and controls and thus may not reflect historical patterns.
King & Marrett (1996) also evaluated the combined effects of
tapwater consumption and duration of exposure to chlorinated water
(see also section 5.3.1.2). Overall, the pattern of risk estimates did
not provide evidence for the interdependence of water consumption and
years of exposure to THMs at levels above 49 µg/litre to increase
bladder cancer risks ( P for interaction = 0.775). Similar low, but
not statistically significant, estimated relative risks are observed
for those with less than 19 years' exposure to high THM levels,
regardless of the volume of water they consumed. For persons with
20-34 years of exposure to THMs at levels above 49 µg/litre, the
estimated relative risk of bladder cancer was also similar for those
who consumed less than 1.54 litres per day (OR = 1.7; 95%
CI = 1.1-2.7) and those who consumed more than 2.08 litres per day
(OR = 1.7; 95% CI = 1.1-2.7). For persons with 35 or more years of
exposure to high THM levels, statistically significant risk estimates
representing more than a doubling of estimated relative risk are
observed for those who consumed between 1.54 and 2.08 litres of
tapwater per day (OR = 2.6; 95% CI = 1.3-5.2) or more than 2.08 litres
per day (OR = 2.3; 95% CI = 1.1-4.7). Cantor et al. (1998) studied
tapwater consumption in Iowa (USA) and found little or no association
of risk for either total daily tapwater intake or intake of all
beverages for men or women (Table 28A).
Colon cancer risk
Cragle et al. (1985) investigated the relationship between water
chlorination and colon cancer using 200 incident cases from seven
hospitals and 407 hospital-based comparison subjects without evidence
of cancer who had been North Carolina (USA) residents for at least 10
years. Comparison subjects were matched on age, race, gender, vital
status and hospital to prevent potential confounding by these
characteristics. Additional information on potential confounders,
including alcohol consumption, genetic risk (number of first-degree
relatives with cancer), diet, geographic region, urbanicity, education
and number of pregnancies, was obtained by either mailed questionnaire
or telephone interview. Water exposures were verified for each address
and categorized as chlorinated or unchlorinated. Logistic regression
analysis showed genetic risk, a combination of alcohol consumption and
high-fat diet, and an interaction between age and chlorination to be
positively associated with colo-rectal cancer. Risks for people who
drank chlorinated water at their residences for 16 or more years were
consistently higher than risks for those exposed to chlorinated water
for less than 16 years, but a statistically significant association
between water chlorination and colo-rectal cancer, controlling for
possible confounding bias, was found only for those above age 60
(Table 30). For example, 70- to 79-year-old participants who drank
chlorinated water for more than 15 years had twice the relative risk
of colo-rectal cancer, but for 70- to 79-year old participants who
drank chlorinated water for less than 16 years, the risk of colon
cancer was only about 50% higher. Confusing the interpretation of the
results is an apparent protective effect of chlorinated water for
colo-rectal cancer found in age groups under age 50 (Table 30). For
example, 40- to 49-year-old persons who drank chlorinated water for
less than 16 years had about half the relative risk, and 20- to
29-year-old persons who drank chlorinated water for less than 16 years
had about one-quarter the risk. These apparent protective effects may
indicate lack of control for an important confounding characteristic.
Table 30. Colon cancer risks associated with exposure to
chlorinated water supplies in North Carolinaa
Age Odds ratio (95% CI)b
1-15 years exposure >15 years exposure
20-29 0.2 (0.1-0.5) 0.5 (0.2-1.0)
30-39 0.4 (0.2-0.7) 0.6 (0.3-1.1)
40-49 0.6 (0.4-0.9) 0.8 (0.5-1.2)
50-59 0.9 (0.8-1.1) 0.9 (0.7-1.3)
60-69 1.2 (0.9-1.5) 1.4 (1.1-1.7)
70-79 1.5 (1.2-1.8) 2.2 (1.7-2.7)
80-89 1.8 (1.3-2.5) 3.4 (2.4-4.6)
a From Cragle et al. (1985).
b Adjusted for various confounders, including gender,
race, diet, alcohol consumption, education, region and
medical history of intestinal disorder.
The colon cancer association in Wisconsin (USA) was further
pursued in an interview-based study (Young et al., 1987, 1990) of
incident cases of colon cancer and population-based comparison
subjects where historical exposures to THMs were estimated. When water
disinfection within only the most recent 10-year exposure period was
considered, colon cancer cases were more likely supplied with
chlorinated rather than with unchlorinated water (OR = 1.6; 95%
CI = 1.0-2.4) and used municipal groundwater rather than private
groundwater (OR = 1.7; 95% CI = 1.1-2.4). Increased risks were not
found for use of chlorinated water or municipal groundwater for 20 or
30 years prior to diagnosis of cancer. THMs are not usually found in
chlorinated municipal groundwaters in Wisconsin, but contaminants such
as tetrachloroethylene, trichloroethylene and 1,1,1-trichloroethane
have been found. Lawrence et al. (1984) studied the relationship of
THMs and colo-rectal cancer in New York (USA) where THM exposure was
higher than in Wisconsin (see section 5.3.1.2 for details about these
studies).
Hildesheim et al. (1998) conducted a population-based
case-control study in Iowa (USA) in 1986-1989 to evaluate cancer risks
that may be associated with chlorinated water; results have been
reported for colon and rectal cancer. Information about residential
history, drinking-water source, beverage intake and other factors was
combined with historical data from water utilities and measured THM
levels to create indices of past exposure to chlorinated by-products.
The colon cancer study was composed of 560 incident cases who were
residents of Iowa during March-December 1987, aged 40-85 years and
with histological confirmation and 2434 age and gender frequency
matched controls for whom water exposure information was available for
at least 70% of their lifetime. Of the 801 eligible colon cancer
cases, 685 (86%) participated; of these cases, 560 (82%) had
sufficient information about water exposures. Unconditional multiple
logistic regression analysis was used to estimate odds ratios for
risks associated with chlorinated surface water and groundwater, THM
exposure and tapwater consumption, while adjusting for potentially
confounding factors. For colon cancer and subsites, no increase in
risk was associated with duration of exposure to chlorinated surface
water or chlorinated groundwater (Table 31A). A slight decrease in
colon cancer risk was found with increased tapwater consumption. Those
who drank 2.9 litres or more of tapwater daily had a 25% reduced risk
of colon cancer compared with those who drank less than 1.5 litres per
day.
Rectal cancer risk
Hildesheim et al. (1998) conducted a population-based
case-control study in Iowa (USA) in 1986-1989 to evaluate cancer risks
that may be associated with chlorinated water; results have been
reported for colon and rectal cancer. Information about residential
history, drinking-water source, beverage intake and other factors was
combined with historical data from water utilities and measured THM
levels to create indices of past exposure to chlorinated by-products.
The study was composed of 537 incident rectal cancer cases aged 40-85
years who were residents of Iowa from January 1986 to December 1988
and whose cancer was confirmed histologically and 2434 age and gender
frequency matched controls for whom water exposure information was
available for at least 70% of their lifetime. Of the 761 eligible
rectal cancer cases, 655 (86%) participated; sufficient information
about water exposures was available for 537 (82%) of these 655 cases.
Unconditional multiple logistic regression analysis was used to
estimate odds ratios for risks associated with chlorinated surface
water, chlorinated groundwater, THM exposure and tapwater consumption,
while adjusting for potentially confounding factors. An increasing
rectal cancer risk was associated with both increasing cumulative THM
Table 31. Colo-rectal cancer risks associated with exposures to
chlorinated water supplies in Iowa (USA)a
Years of exposure Odds 95% CI Comments
to chlorinated ratio
surface water
A. Colon cancer risks Adjusted for age,
0 1.0 gender
1-19 1.0 0.8-1.3
20-39 1.0 0.7-1.5
40-59 1.2 0.8-1.8
>60 0.8 0.4-1.7
B. Rectal cancer risks Adjusted for age,
0 1.0 gender
1-19 1.1 0.8-1.4
20-39 1.6 1.1-2.2
40-59 1.6 1.0-2.6
>60 2.6 1.4-5.0
a From Hildesheim et al. (1998).
exposure and duration of exposure to chlorinated surface water (Table
31B). However, the amount of tapwater consumed did not confound the
risk, as the authors reported that little association (no data
provided) was found between water consumption and rectal cancer after
adjustment for age and gender. Larger relative risks for rectal cancer
were found among persons with low dietary fibre intake and
longer-duration exposure to chlorinated surface water source compared
with persons with high-fibre diets and no exposure to chlorinated
surface water.
Pancreatic cancer risk
A population-based case-control study (Ijsselmuiden et al., 1992)
was conducted in Washington County, Maryland (USA), the same area in
which an earlier cohort study had been conducted by Wilkins & Comstock
(1981). Included in the study were 101 residents who were identified
by the county cancer registry with a diagnosis of pancreatic cancer
from July 1975 to December 1989 and 206 controls randomly chosen from
the county population defined by a specially conducted census in 1975.
Drinking-water source obtained from the 1975 census was used to assess
exposure. Multivariate analysis found an increased risk of pancreatic
cancer (RR = 2.2; 95% CI = 1.2-4.1) associated with chlorinated
municipal surface water after adjusting for cigarette smoking;
however, the authors recommended caution in the interpretation of this
finding because of limitations in the assessment of exposure for study
participants. Exposure was assessed at only one point in time, 1975,
and this may not accurately reflect long-term exposure to chlorinated
water. No increased risk of pancreatic cancer (RR = 0.8; 95% CI =
0.4-1.5) was found by Wilkins & Comstock (1981) in an earlier cohort
study in the same county. Ijsselmuiden et al. (1992) did not report
the reason for studying pancreatic cancer rather than evaluating
further the previous findings of Wilkins & Comstock (1981) of
increased, but not statistically significant, risks for bladder, liver
or kidney cancer.
Brain cancer risk
In an abstract, Cantor et al. (1996) described the results of a
population-based case-control study of 375 incident brain cancer
patients, diagnosed in 1984-1987, and 2434 controls in Iowa. After
controlling for age, farm occupation and other potential confounding
characteristics, brain cancer risk among men, but not women, was
associated with increased duration of exposure to chlorinated surface
water. The risk was greatest for over 40 years of exposure (OR = 2.4;
no CI reported) compared with 20-39 years of exposure (OR = 1.8) and
1-19 years of exposure (OR = 1.3). Historical exposures to
chlorination by-products in drinking-water were also estimated from
recent measures of THMs, other water quality data and information from
study participants, but results of these analyses were not presented.
5.2.1.3 Meta-analysis of cancer studies
Meta-analysis is the application of quantitative techniques to
literature reviewing. Two complementary approaches may be taken (US
EPA, 1994a). One is an aggregative approach to summarize the compiled
research on a given topic. This summary typically provides a measure
of overall statistical significance or a consolidated estimate of
effect, such as a relative risk. In practice, "when the world's
literature on a topic is declared to be statistically significant,"
results of this type of meta-analysis are usually interpreted as an
indication that research should cease and action should begin (US EPA,
1994a).
The other approach to meta-analysis is an analytical or
explanatory approach (Greenland, 1994), in which the goal is to see
whether differences among the studies can explain differences among
their results. A formal analysis of the explanatory type might take
the form of a meta-regression (Greenland, 1987), in which the
dependent variable is the measure of effect, as estimated by each
study, and the independent variables are the potentially explanatory
factors that might yield higher or lower estimates of effect. The
strength of explanatory meta-analysis is that it produces an enhanced
understanding and appreciation of the strengths and weaknesses of the
studies that have been conducted on the selected topic (US EPA,
1994a).
In the case of chlorinated drinking-water and cancer, a
meta-analysis that used the aggregative approach was published by
Morris et al. (1992). The authors conducted significance tests for
several cancers and reported two, bladder cancer and rectal cancer, as
statistically significant. Emphasized were the summary estimates of
effect: pooled RRs of 1.2 (95% CI = 1.1-1.2) for bladder cancer and
1.4 (95% CI = 1.1-1.9) for rectal cancer. In conjunction with crude
data on population exposure to chlorinated surface water, an
attributable risk was estimated by the authors, suggesting that about
4200 cases (9%) of bladder cancer per year and 6500 cases (18%) of
rectal cancer per year may be associated with consumption of
chlorinated surface water in the USA. The meta-analysis and its
quantitative estimate of risk have caused considerable controversy
because there are significant differences in the design of the 10
individual epidemiological studies included (IARC, 1991; Risch et al.,
1992; Craun et al., 1993; Murphy, 1993; Bailar, 1995). Reported
quality scores for the individual studies were low (43-78 of a
possible 100), and their study populations, research methods and
results are not homogeneous. Several of the included studies were well
designed and attempted to assess historical exposures to chlorinated
water and possible confounding characteristics, but most did not
adequately assess historical exposures and confounding bias. Only
three studies adjusted or matched for smoking (one bladder cancer
study, one colon cancer study and a cohort study of bladder, colon and
rectal cancer), and only one study considered diet as a potential
confounder. In four studies, a single reported address on a death
certificate was used to assess the study participants' exposure to
chlorinated water.
It is important to ask whether the meta-analysis should have
restricted the analysis to those studies that were more homogeneous or
those with adequate exposure assessments and control of confounding
bias. Bailar (1995) questioned the summary findings for bladder cancer
based on his assessment of the weaknesses of the individual studies
and concluded that "bias could well explain the whole of the
apparently positive findings." A recently completed formal evaluation
of the meta-analysis by Poole (1997) concluded that studies should not
have been combined into aggregate estimates of relative risk or used
as the basis for national attributable risk estimates.
5.2.1.4 Summary of results of cancer studies
Various types of epidemiological studies, primarily in the USA,
have attempted to assess the cancer risks associated with chlorinated
water systems. Water disinfected with ozone and chlorine dioxide has
not been studied for cancer associations, but chloraminated
drinking-water was considered in two studies. Many, but not all,
ecological studies reported associations between chlorinated water and
cancer incidence or mortality and helped develop hypotheses for
further study.
Analytical studies reported small relative risks for colon and
bladder cancer incidence for populations consuming chlorinated
drinking-water for long periods of time. Because of probable bias,
interpretation of observed associations is severely limited in
case-control studies where information was not obtained from
interviews and residence histories. Interview-based case-control
epidemiological studies provide a basis for evaluating the potential
cancer risk that may be associated with chlorinated drinking-water.
Based on Monson's (1980) guide to interpreting the strength of an
association, a weak to moderate epidemiological association was found
between water chlorination and colon cancer incidence among an elderly
population in North Carolina (USA). However, a moderate to strong
protective effect was also found among persons 20-49 years of age,
confusing the interpretation of these results. The higher risks for
those above age 70 in North Carolina suggest that the association may
be evident only after a very long duration of exposure to chlorinated
surface water. Colon cancer incidence was weakly associated with the
use of chlorinated drinking-water for the most recent 10-year period
in Wisconsin (USA), but no association was found when 20 or 30 years
of exposure were considered. In Iowa (USA), no association was found
between colon cancer or any subsites and duration of exposure to
chlorinated surface water.
In a national study of 8764 persons in the USA, no overall
association was found between chlorinated drinking-water and bladder
cancer risk. A moderate association between chlorinated surface water
and bladder cancer incidence was observed among an otherwise low-risk
population of non-smokers that had received chlorinated surface water
for 60 or more years, but this analysis included only 123 persons. In
the Iowa portion of this study, moderate to strong associations were
found for smokers and non-smokers with at least 30 years of exposure
to chlorinated water. Interview case-control studies found a moderate
risk of bladder cancer incidence associated with more than 30 years of
exposure to chlorinated surface water in Colorado (USA), a weak to
moderate risk of bladder cancer incidence associated with more than 35
years of exposure to chlorinated surface water in Ontario (Canada), a
weak to moderate risk associated with more than 59 years of exposure
to chlorinated surface water in Iowa (USA), and a weak association
with more than 40 years of exposure to chlorinated surface water in
Washington County, Maryland (USA). Inconsistencies, however, were
observed in risks for non-smokers and smokers and in risks for women
and men.
In New York State (USA), a dose-response relationship was
observed between daily intake of total liquids and risk of bladder
cancer and between increased tapwater consumption and risk of bladder
cancer. Since increased risks were seen primarily in total fluid
consumption, it is possible that total fluid consumption may be
relevant in the pathogenesis of bladder cancer. Total daily fluid
intake may be a marker for some unmeasured risk factor, and it is
important to determine the biological relevance of increased fluid
intake, independent of chlorinated tapwater, in the pathogenesis of
bladder cancer. Several studies found the risk of bladder cancer to be
elevated among persons who consumed more tapwater per day, but
increased tapwater consumption appears to be an independent risk
factor for bladder cancer. There was no evidence of an increased
relative risk of bladder cancer when both the volume of water consumed
and duration of exposure to chlorinated water or THMs were considered
in the analysis.
In Massachusetts (USA), an increased risk of bladder cancer
mortality was observed in a population receiving chlorinated surface
water compared with a population receiving chloraminated surface
water. These associations are weak to moderate in strength, depending
upon the diseases used for comparison, and also considered long
exposures, over 40 years' duration. A decreased risk of bladder cancer
incidence was also associated with a similar duration of exposure to
chloraminated surface water in Colorado (USA), but investigators felt
that the results do not imply that chloraminated water conveys a
protective effect because there is no plausible biological explanation
for suggesting that choramination inhibits neoplastic transformation
of the bladder epithelium.
A case-control study reported a large increased risk of rectal
cancer among those with long duration of exposure to chlorinated
water, but two cohort studies did not find an increased relative risk.
A single case-control study reported a moderate to strong risk of
pancreatic cancer associated with chlorinated surface water in
Washington County, Maryland (USA), but the interpretation of this
study is hampered because exposure was assessed at only one point in
time, and this may not accurately reflect long-term exposure to
chlorinated water. Preliminary results from a study in Iowa (USA)
found that a moderate risk of brain cancer among men, but not women,
was associated with increased duration of exposure to chlorinated
surface water, especially for over 40 years of exposure. Additional
details of this study are required to allow these conclusions to be
evaluated. A weak increased risk of lung cancer incidence was also
seen for users of chlorinated surface water in Iowa.
A controversial meta-analysis study, which statistically combined
the results of 10 previously published epidemiological studies,
reported a small pooled increased relative risk for bladder and rectal
cancer but not for colon cancer. This meta-analysis, however, included
a number of low-quality studies with likely bias.
Current evidence from epidemiological studies is insufficient to
allow a causal relationship between the use of chlorinated
drinking-water and the incidence of bladder cancer to be established.
Several studies reported weak to moderate associations of
long-duration exposure to chlorinated water and bladder cancer, but
risks have differed between smokers and non-smokers in several
studies. Inconsistent risks have also been seen when gender and water
consumption were considered.
For colon cancer, the epidemiological data appear to be equivocal
and inconclusive. For rectal cancer, insufficient data are available
with which to evaluate the moderate associations observed in one
study. Similarly, single studies of reported associations for
pancreatic, lung, brain and breast provide insufficient data.
5.2.2 Epidemiological studies of cardiovascular disease and
disinfected drinking-water
Several epidemiological studies have evaluated possible risks of
cardiovascular disease associated with the chlorination of
drinking-water. The cohort study (Wilkins & Comstock, 1981) of
31 000 residents of Washington County, Maryland (USA) found a slightly
increased, but not statistically significant, risk (RR = 1.1; 95%
CI = 1.0-1.3) of death due to arteriosclerotic heart disease in
residents exposed to chlorinated drinking-water from a surface water
and springs (average chloroform level was reported as 107 µg/litre)
compared with residents of towns where unchlorinated well-water was
used. A standardized mortality study (Zierler et al., 1986) found
little difference in the patterns of 35 539 and 166 433 deaths (age of
death was at least 45 years) due to cerebrovascular and cardiovascular
disease, respectively, during 1969-1983 in 43 Massachusetts (USA)
communities with water supplies disinfected with either chlorine or
chloramine as compared with all deaths reported in Massachusetts due
to these causes. The mortality rate in these selected communities with
chlorinated drinking-water was slightly higher than expected for
cerebrovascular disease (SMR = 108; 95% CI = 106-109) and
cardiovascular disease (SMR = 104; 95% CI = 104-105). The mortality
rate in selected communities with chloraminated drinking-water was
slightly lower than expected for cerebrovascular disease (SMR = 86;
95% CI = 85-88) and about the same as expected for cardiovascular
disease (SMR = 101; 95% CI = 100-101). As noted in section 5.2.1.2,
the serious potential for exposure and disease misclassification bias
in the Massachusetts study limits the interpretation of these
findings.
A cross-sectional study (Zeighami et al., 1990a,b) of 1520 adult
residents, aged 40-70 years, in 46 Wisconsin (USA) communities was
conducted to determine whether the hardness or chlorination of
drinking-water affects serum lipids. The water for the communities
contained total hardness of less than or equal to either 80 or 200 mg
of calcium carbonate per litre; 858 participants (59% female) resided
in 24 communities that provided chlorinated water, and 662 (55%
female) resided in 22 communities that did not disinfect water. An
age-gender-stratified sampling technique was used to choose a single
participant from each eligible household, and a questionnaire was
administered to obtain data on occupation, health history,
medications, dietary history, water use, water supply and other basic
demographic information. Among women who resided in communities with
chlorinated, hard drinking-water, mean serum cholesterol levels were
found to be higher (249.6 mg/dl, standard error [SE] = 6.4) than for
women who resided in unchlorinated, hard-water communities (235.3
mg/dl, SE = 6.4). Among women who resided in communities with
chlorinated, soft drinking-water, mean serum cholesterol levels were
also found to be higher (248.0 mg/dl, SE = 6.2) than in women residing
in unchlorinated, soft-water communities (239.7 mg/dl, SE = 6.3). Mean
serum cholesterol levels were also higher for men in chlorinated
communities, but the differences in mean cholesterol levels between
chlorinated and unchlorinated communities were smaller and not
statistically significant. The age-specific and overall risks of
elevated serum cholesterol levels (>270 mg/dl) in communities with
chlorinated drinking-water were also evaluated for men and women.
Statistically significant increased risks of elevated serum
cholesterol levels were found among women but not men and primarily in
women aged 50-59 (OR = 3.1; 95% CI = 1.5-6.7).
Mean levels of LDL cholesterol in men and women had a similar
pattern to total cholesterol. However, mean levels of HDL cholesterol
were nearly identical in the chlorinated and unchlorinated communities
for each gender, and the implication for increased cardiovascular
disease risk in communities with chlorinated water remains unclear.
Caution is urged in the interpretation of the results of this study.
It is possible that an undetermined confounding characteristic due to
lifestyle differences may be responsible for the observed association
in the chlorinated communities.
The relationship between consumption of chlorinated
drinking-water in the home and serum lipids was also evaluated in a
cohort of 2070 white women, aged 65-93, participating in a study of
osteoporotic fractures in western Pennsylvania (USA) (Riley et al.,
1995). Mean serum cholesterol levels (247 mg/dl, SD = 41.3) in 1869
women using chlorinated water sources were similar to levels (246
mg/dl, SD = 41.9) in 201 women using unchlorinated water sources.
Women with the largest cumulative exposure to chlorinated water had
higher serum cholesterol levels (247 mg/dl, SD = 41.3) than women with
unchlorinated water (241 mg/dl, SD = 0.7), but the difference was not
statistically significant. There was no evidence that increasing the
duration of exposure to chlorinated water influenced LDL or HDL
cholesterol, triglycerides or apolipoproteins. In this cohort, women
exposed to chlorinated water sources tended to smoke and drink more
than women not exposed to chlorinated water sources, suggesting that
the reported association in Wisconsin may be due primarily to
inadequate control of lifestyle characteristics differentially
distributed across chlorinated exposure groups.
5.2.2.1 Summary of results of cardiovascular studies
A mortality study showed little difference between patterns of
death due to cardiovascular and cerebrovascular disease in the general
population of Massachusetts (USA) and among residents of communities
using either chlorine or chloramine . A cohort study in Washington
County, Maryland (USA) found a slightly increased, but not
statistically significant, risk of death due to arteriosclerotic heart
disease in residents exposed to chlorinated drinking-water. A
cross-sectional epidemiological study in Wisconsin (USA) found higher
serum cholesterol in those exposed to chlorinated drinking-water than
in those not exposed, but the association was confined to women aged
50-59. Mean serum lipids and lipoproteins were found to be similar in
elderly white women exposed to chlorinated and unchlorinated
drinking-water in western Pennsylvania (USA).
There is inadequate evidence from epidemiological studies that
chlorinated or chloraminated drinking-water increases cardiovascular
disease risks.
5.2.3 Epidemiological studies of adverse reproductive/developmental
outcomes and disinfected drinking-water
After adjustment for potential confounders, Aschengrau et al.
(1989) found a statistically significant increase in the frequency of
spontaneous abortion in Massachusetts (USA) communities that used
surface water sources compared with those that used groundwater
sources. No measures of DBPs were available for the communities. When
surface water sources are chlorinated, DBPs are typically higher than
when groundwater sources are chlorinated, but levels of other water
parameters also differ between surface water and groundwater sources.
Aschengrau et al. (1993) also conducted a case-control study of late
adverse pregnancy outcomes and water quality in Massachusetts
community water systems. Among women who delivered infants during
August 1977 and March 1980 at Brigham and Women's Hospital, various
water quality indices were compared for 1039 congenital anomaly cases,
77 stillbirth cases, 55 neonatal deaths and 1177 controls. Risk of
neonatal death or all congenital anomalies was not found to be
increased in women exposed to chlorinated surface water supplies
compared with chloraminated water supplies. Stillbirth risk was
associated with chlorinated water supplies; however, after adjustment
for appropriate confounding characteristics, the risk was not
statistically significant (OR = 2.6; 95% CI = 0.9-7.5). Water quality
measures were available for trace metals, but no water quality
measures were available with which to assess risks associated with
THMs or other DBPs.
A population-based case-control study of miscarriage, preterm
delivery and low birth weight was conducted in three counties in
central North Carolina (USA) (Savitz et al., 1995). Preterm deliveries
(<37 weeks completed gestation) and low birth weight infants
(<2500 g) were identified at hospitals with virtually all births to
residents of Orange and Durham counties from September 1988 to August
1989 and Alamance County from September 1988 to April 1991. About 50%
of eligible live births were both preterm and low birth weight. All
medically treated miscarriages among women in Alamance County from
September 1988 to August 1991 were also identified for study.
Full-term, normal-weight births immediately following a preterm or low
birth weight delivery were selected as controls. Race and hospital
were controlled for in the analysis. Telephone interviews were used to
obtain information on a number of potential risk factors, including
age, race, education, marital status, income, pregnancy history,
tobacco and alcohol use, prenatal care, employment and psychological
stress. Questions about drinking-water sources at home, including
bottled water, and amount of water consumed around the time of
pregnancy were also asked. Only 62-71% participated; the lowest
response rate was for miscarriage cases. The analysis, which
considered water consumption and THMs, was further restricted to women
served by public water sources who reported drinking one or more
glasses of water per day, and this limited the analysis to 70% of
those participating. Selection bias is of concern in these studies, as
is recall bias, where cases are more likely to more accurately recall
previous exposures. It was found that water source was not related to
miscarriage, preterm delivery or low birth weight. Risk of miscarriage
was slightly increased among women who used bottled water compared
with those who used private wells, but the risk was not statistically
significant (OR = 1.6; 95% CI = 0.6-4.3).
A cross-sectional study (Kanitz et al., 1996) was conducted of
548 births at Galliera Hospital in Genoa and 128 births at Chiavari
Hospital in Chiavari (Italy) during 1988-1989 to mothers residing in
each city. Women in Genoa were exposed to filtered water disinfected
with chlorine dioxide (Brugneto River wells, reservoir and surface
water) and/or chlorine (Val Noci reservoir). Women residing in
Chiavari used untreated well-water. Assignment to a water source and
type of disinfectant was based on the mother's address (undisinfected
well-water, chlorine, chlorine dioxide or both). Municipal records
were used to determine family income, and hospital records were used
to obtain information about mother's age, smoking, alcohol consumption
and education level and birth outcomes -- low birth weight (<2500
g), preterm delivery (<37 weeks), body length (<49.5 cm),
cranial circumference (<35 cm) and neonatal jaundice. Neonatal
jaundice was almost twice as likely (OR = 1.7; 95% CL = 1.1-3.1) in
infants whose mothers resided in the area where drinking-water from
surface water sources was disinfected with chlorine dioxide as in
infants whose mothers used undisinfected well-water. No increased risk
for neonatal jaundice was found for infants whose mothers lived in an
area using chlorinated surface water. Large increased risks of smaller
cranial circumference and body length were associated with
drinking-water from surface water sources disinfected with chlorine or
chlorine dioxide. Infants born to mothers residing in areas where
surface water was disinfected with chlorine or chlorine dioxide had
smaller cranial circumference (OR = 3.5; 95% CI = 2.1-8.5 for chlorine
vs. untreated well-water; OR = 2.2; 95% CI = 1.4-3.9 for chlorine
dioxide vs. untreated well water) and smaller body length (OR = 2.0;
95% CI = 1.2-3.3 for chlorine dioxide vs. untreated well-water; OR =
2.3; 95% CI = 1.3-4.2 for chlorine vs. untreated well-water). Risks of
low birth weight infants were also increased for mothers residing in
areas using water disinfected with chlorine and chlorine dioxide, but
these associations were not statistically significant. For preterm
delivery, small and not statistically significant increased risks were
found among mothers residing in the area using chlorine dioxide. This
study suggests possible risks associated with surface water
disinfected with chlorine or chlorine dioxide, but the results should
be interpreted very cautiously (US EPA, 1997). THM levels were low in
both chlorinated water (8-16 µg/litre) and chlorine
dioxide-disinfected water (1-3 µg/litre). No information was collected
to assess the mothers' water consumption or nutritional habits, and
the age distribution of the mothers was not considered. It is
important to determine whether municipal or bottled water was consumed
by the mothers and how much water was consumed. If mothers routinely
consumed untreated municipal well-water but did not consume
disinfected municipal surface water, drinking bottled water instead,
the observed relative risks would not be associated with the
disinfection of surface water. In addition, there are concerns about
incomplete ascertainment of births and whether the population may be
different in respects other than the studied water system differences.
On the other hand, if the observed associations with water source and
disinfection are not spurious, a question is raised about what water
contaminants may be responsible. Exposures to surface water and
groundwater sources are compared in this study, and no information is
presented about other possible water quality differences.
Preliminary analyses were available for a cross-sectional study
(Nuckols et al., 1995) of births to women in Northglenn, Colorado
(USA) who were exposed to chlorinated water from Stanley Lake with
high THM levels (32-72 µg/litre) and to women in Westminster who were
exposed to chloraminated water from Stanley Lake with low THM levels
(<20 µg/litre). Lower, but not statistically significant, relative
risks for low infant birth weight and preterm delivery were found in
the water district using chlorine. Further analysis in the chlorinated
water system found increased, but not statistically significant,
relative risks for low birth weight infants and preterm delivery in
areas where the THMs were higher. These results must be interpreted
with caution because very limited information was provided about the
epidemiological methods. Instead, the article focused on water
distribution system quality modelling and the use of geographic
information systems.
5.2.3.1 Summary of results of reproductive/developmental studies
Results of several exploratory epidemiological studies of adverse
reproductive effects/developmental outcomes and chlorinated water
should be cautiously interpreted because of limitations of study
design and likely bias.
5.3 Epidemiological associations between disinfectant by-products
and adverse health outcomes
In this section, studies of specific DBPs are reviewed. In some
studies, both water source and disinfectant type were considered in
addition to the specific by-products; if this is the case, the details
of the study design and any limitations are reported in section 5.2.
5.3.1 Epidemiological studies of cancer and disinfectant by-products
5.3.1.1 Cancer associations in ecological studies
1) Volatile by-products
Cantor et al. (1978) studied the relationship between THM levels
and age-standardized cancer mortality for 1968-1971 of white men and
women in urban counties in the USA. THM concentrations in
drinking-water were estimated at the county level from data obtained
in two national surveys of water supplies conducted by the US EPA. The
analysis took into account the median number of school years completed
by county inhabitants over age 25, the foreign-born and native
population of the county, the change in county population from 1950 to
1970, the percentage of each county that is considered urban, the
percentage of the county work force engaged in all manufacturing
industries, and the geographic region of the USA. Multivariate
regression analysis found the variability among these counties for
gender-specific and cancer-site-specific mortality rates, and the
residual mortality rates were then correlated directly with estimated
THM exposure for those 76 counties in which 50% or more of the
population was served by the sampled water supplies. Among both men
and women, a statistically significant positive correlation was found
between non-chloroform THM levels and bladder cancer mortality. No
association between THM levels and colon cancer mortality rates was
observed after controlling for the ethnicity of the population. Hogan
et al. (1979) studied county cancer mortality data for an earlier
period and county chloroform levels estimated from the same EPA
surveys. Multivariate regression analysis of the county cancer
mortality rates included, as independent variables, estimated exposure
to chloroform concentrations, 1960 county population, county
population density, percentage of county that is urban, percentage of
county population that is non-white, percentage of county population
that is foreign-born, median number of school years completed by
county residents over age 25, median family income of county and
percentage of county work force engaged in manufacturing. The results
suggested that county cancer mortality rates for the rectum, bladder
and possibly large intestine increased with increased levels of
chloroform in drinking-water supplies.
McCabe (1975) found that age-adjusted total cancer mortality
rates correlated positively with estimated chloroform concentrations
in 80 US cities but included no attempt to control for potential
confounding bias on a group or aggregate level. Carlo & Mettlin (1980)
studied 4255 incident cases of oesophageal, stomach, colon, rectal,
bladder and pancreatic cancers reported through the New York State
Tumor Registry for Erie County, New York (USA) between 1973 and 1976.
Age-adjusted incidence rates were calculated by census tract, and
levels of THMs were estimated from a single water survey in July 1978.
Statistically significant positive associations were found between
consumption of chlorinated drinking-water from surface water sources
and oesophageal and pancreatic cancer and between THM levels and
pancreatic cancer in white males. The authors placed little credence
on these findings, noting that the pancreatic cancer-THMs relationship
was found only in one gender-race subgroup, the range of THM
concentrations in the study was narrow (the largest variation was only
71 µg/litre) and no data were available with which to estimate
historical trends in THM levels.
Tuthill & Moore (1980) studied cancer mortality rates from 1969
to 1976 in Massachusetts (USA) communities supplied by surface water.
Chlorination exposure data were assessed by considering past chlorine
dosage, recent THM levels and recent chlorine dosage. Stomach and
rectal cancer mortality rates were associated with recent THM levels
and recent chlorine dosage, but not with past chlorine dosage in the
communities. However, when regression models included migration
patterns and ethnicity, none of the associations was statistically
significant.
Wigle et al. (1986) studied selected contaminants in
drinking-water and cancer risks in Canadian cities with populations of
at least 10 000. Water quality data from three national surveys of
urban drinking-water supplies, demographic data and age-standardized
cancer mortality rates for 1973-1979 were analysed by multivariate
regression techniques. No statistically significant associations were
found between chlorine dosage and risk of death with any disease
category. When chlorine dosage was replaced in the model by TOC, a
statistically significant association was found between this variable
and cancer of the large intestine among males but not females. There
were no statistically significant associations when chlorine dosage
was replaced by THM, chloroform or non-chloroform THM levels. A recent
ecological analysis of cancer cases reported to the New Jersey (USA)
cancer registry in 1979-1990 found no association between bladder and
rectal cancer incidence and THM or BDCM levels in public
drinking-water systems (Savin & Cohn, 1996). Exposure was based on
address at the time of cancer diagnosis and the water quality of the
public water system from monitoring conducted during the 1980s.
These ecological studies used limited information about current
THM levels in drinking-water to estimate exposures for a census tract,
county or community, and these exposures may not represent long-term
exposures or be relevant for the population whose cancer statistics
were studied. Available demographic characteristics were included as
group variables to assess or control for confounding bias, but these
likely have limited value in controlling for such bias. Because of the
ecological design of the study, these results cannot be easily
interpreted. Even if the exposure information were accurately
assessed, it cannot be determined whether the observed associations
were a result of exposure to chloroform, THM levels or other DBPs or
were confounded by characteristics that were not assessed (e.g.,
cigarette smoking) or incorrectly assessed by using available
demographic data. Ecological studies where no associations were found
suffer from similar limitations.
2) Mutagenicity
Several ecological studies have considered the mutagenic activity
of drinking-water, as determined by the Salmonella/microsome assay.
These mutagenicity tests assess the non-volatile, acid/neutral
fraction of chlorinated organic material in a concentrated water
sample. A mutagen of particular concern is MX, a potent mutagen, as
measured by strain S. typhimurium TA100, which may be responsible
for up to 57% of the mutagenicity in chlorinated drinking-water (Meier
et al., 1986, 1987b).
High levels of mutagenic activity have been observed in Finnish
chlorinated drinking-water, and Koivusalo et al. (1994a,b, 1995)
investigated the relationship between mutagenic activity in
drinking-water and gastrointestinal, urinary tract and other cancers
in 56 Finnish municipalities. Included in the analysis were cases of
bladder, kidney, stomach, colon, rectum, liver, pancreas and soft
tissue cancer, leukaemia (acute, chronic myeloid and chronic
lymphatic), Hodgkin's disease and non-Hodgkin's lymphoma obtained from
the population-based Finnish Cancer Registry for two periods,
1966-1976 and 1977-1989. The Cancer Registry includes virtually all
cancer cases in the country. On an ecological level, information was
also obtained for several potential confounding characteristics --
social class, urban living, time period, migration and exposures from
the chemical industry.
Koivusalo et al. (1994b) discussed the methodology used to
estimate the mutagenicity of drinking-water and assess past exposures
for the epidemiological studies. Exposure was assessed at the
ecological, not the individual, level and was based on the proportion
of population served by municipal water and the estimated mutagenicity
of the water. Previous drinking-water mutagenic activity was estimated
for each community on the basis of an equation derived by Vartiainen
et al. (1988), yielding an estimated drinking-water mutagenicity level
in net revertants per litre for each community for each of two
specific periods, 1955 and 1970. The equation used available
historical information on raw water quality (TOC, ammonia,
permanganate test for oxidizable organic matter, etc.), pre- and
post-chlorine dosages, and water treatment practices obtained by a
questionnaire sent to the municipalities. Vartiainen et al. (1988) and
Koivusalo et al. (1994b) reported a high correlation between estimated
and measured drinking-water mutagenicity after comparing the results
of assays of drinking-water mutagenicity in 1985 and 1987 with
estimated mutagenicity from their equation.
Observed and expected numbers of cancer cases in each
municipality were compared by sex, broad age group and time period,
and cancer risk was adjusted for social class as a surrogate for
lifestyle and smoking habits. For all 56 municipalities, no
statistically significant increases in risks of cancer of the kidney,
stomach, colon, rectum, liver, pancreas and soft tissue, leukaemia
(acute, chronic myeloid and chronic lymphatic) and non-Hodgkin's
lymphoma were found among those exposed to a typically mutagenic
drinking-water (3000 net revertants per litre) when adjusted for age,
sex, period, main cities and social class. The risks of bladder cancer
(RR = 1.2; 95% CI = 1.1-1.3) and Hodgkin's disease (RR = 1.2; 95%
CI = 1.0-1.4) were small but statistically significant. When this
analysis was restricted to those 34 municipalities with mutagenic
drinking-water, risks of non-Hodgkin's lymphoma, Hodgkin's disease and
cancer of the liver, pancreas, kidney, stomach and bladder were all
found to be slightly increased (RRs ranged from 1.1 to 1.3). As this
is an ecological study, the small increased relative risks that were
observed preclude definitive conclusions.
5.3.1.2 Cancer associations in analytical studies
1) Bladder cancer risk
In Colorado (USA), where an increased risk of bladder cancer was
found for persons exposed to chlorinated drinking-water for more than
30 years (OR = 1.8; 95% CI = 1.1-2.9), no association was found
between THM levels and bladder cancer (McGeehin et al., 1993). The
mean 1989 levels for THMs and residual chlorine for each water system
were multiplied by the number of years each study participant was
exposed to that system and summed to compute lifetime exposure indices
for each water quality indicator. Higher risks were found when the
cumulative exposure index for THMs was less than 200 or greater than
600 µg/litre-years, but not when it was 201-600 µg/litre-years. No
statistically significant trends were found; risks did not increase
with increased cumulative THM exposure. In logistic regression models
that included water source and disinfection variables and controlled
for years of exposure to chlorinated water, THMs assessed by this
exposure index were not associated with bladder cancer. Interpretation
of these results is limited, however, because only 61% of THM levels
and 55% of residual chlorine levels could be determined for use in
assessing exposures.
In Ontario (Canada), individual information about water
consumption and exposure to chlorinated water was supplemented by
water treatment data (e.g., area served, water source and
characteristics, and treatment practices for years of operation
between 1950 and 1990) collected from a survey of historical treatment
practices at water supplies in the study area (King & Marrett, 1996).
For each treatment facility, water treatment information was obtained
for an average day in August in 5-year intervals, and that observation
was used to represent water characteristics for the years surrounding
that date. The water supply for each participant was characterized by
the estimated annual maximum THM levels for each water facility.
Historical THM levels were estimated using a model developed to
predict the THM level in treated water from characteristics of the
treatment process. The model was developed from water quality
measurements recorded by the Ontario Drinking Water Surveillance
Program between 1988 and 1992 for 114 water treatment facilities.
Predicted THM levels were compared with those observed in 1986-1987,
and the correlation between observed and predicted values was 0.76.
When observed and predicted THM levels were considered as either above
or below 50 µg/litre, the model predicted values with a sensitivity of
84% and a specificity of 76%.
King & Marrett (1996) found an increased bladder cancer risk with
increasing duration of exposure to THMs, but the association was
statistically significant and of higher magnitude only after 35 or
more years of exposure (Table 32). A statistically significant
increased bladder cancer risk was also found for the highest quartile
of cumulative exposure to THMs (Table 33). The risk of bladder cancer
incidence was about 40% higher among persons exposed to greater than
1956 µg of THMs per litre-year in water compared with those exposed to
less than 584 µg/litre-year. No association between cumulative
exposure to THMs and increased bladder cancer risk was found in
Colorado (USA) (McGeehin et al., 1993), but the highest cumulative THM
levels in Colorado were much lower than those found in Ontario (Table
34).
A population-based case-control study conducted in 1986-1989 in
Iowa (USA) (Cantor et al., 1998) found associations of increased
bladder cancer risk with total and average lifetime exposure to THMs
(Table 35), but increased relative risks were restricted to men who
had ever smoked (see also section 5.2.1.2). These findings were
similar to associations found with duration of exposure to chlorinated
surface water. Relative risks for women suggested a protective effect
for increased lifetime average THM levels; ORs ranged from 0.6 to 0.9,
and 95% CIs ranged from 0.3 to 1.3. Past exposure to chlorination
by-products was estimated by combining information about water
sources, historical chlorine use, lifetime residential history, fluid
consumption, THM levels and other water quality data, including
volatile organic compounds, TOC, TOX, pesticides, total dissolved
solids and nitrates (Lynch et al., 1990; Neutra & Ostro, 1992). Data
reported thus far (Cantor et al., 1998) have been restricted to THMs.
Table 32. Bladder cancer risks and estimated maximum annual
exposure to trihalomethanes in water in Ontario (Canada)a
Years of Odds ratio (95% CI)b
exposure
THMs >24 µg/litre THMs >49 µg/litre THMs >74 µg/litre
0-9 1.0 1.0 1.0
10-19 1.2 (0.9-1.7) 1.1 (0.9-1.4) 1.1 (0.9-1.4)
20-34 1.2 (0.9-1.5) 1.4 (1.1-1.8) 1.3 (0.9-1.8)
>34 1.6 (1.2-2.1) 1.6 (1.1-2.5) 1.7 (1.1-2.7)
a From King & Marrett (1996).
b Adjusted for age, gender, smoking, education and calorie intake.
Table 33. Bladder cancer risks and estimated
cumulative exposure to trihalomethanes
in Ontario (Canada)a
THMs-years of exposure Odds ratio (95% CI)b
(µg/litre-year)
0-583 1.0
584-1505 1.2 (0.9-1.6)
1506-1956 1.1 (0.8-1.4)
1957-6425 1.4 (1.1-1.9)
a From King & Marrett (1996).
b Adjusted for age, gender, smoking, education
and calorie intake.
Table 34. Bladder cancer risks and estimated
cumulative exposure to trihalomethanes
in Colorado (USA)a
THMs-years of exposure Odds ratiob
(µg/litre-year)
0 1.0
<200 1.8
201-600 1.1
>600 1.8
a From McGeehin et al. (1993).
b 95% CI not available; P for trend = 0.16.
Table 35. Bladder cancer risks and estimated cumulative
exposure to trihalomethanes in Iowa (USA)a
Lifetime THM exposure Odds ratio (95% CI)b
Total lifetime exposure (g)
<0.04 1.0
0.05-0.12 1.3 (1.0-1.6)
0.13-0.34 1.1 (0.9-1.4)
0.35-1.48 1.1 (0.9-1.4)
1.49-2.41 1.2 (0.8-1.7)
>2.42 1.3 (0.9-2.0)
Lifetime average exposure (µg/litre)
<0.8 1.0
0.8-2.2 1.2 (1.0-1.5)
2.3-8.0 1.1 (0.8-1.4)
8.1-32.5 1.1 (0.8-1.4)
32.6-46.3 1.3 (0.9-1.8)
>46.3 1.2 (0.8-1.8)
a From Cantor et al. (1998).
b Adjusted for age, gender, study period, smoking,
education and high-risk occupation.
2) Colon cancer risk
A colon cancer association was studied in Wisconsin (USA) in an
interview-based study (Young et al., 1987, 1990) of 347 incident cases
of colon cancer, 611 population-based controls and 639 controls with
cancer of other sites. White males and females between the ages of 35
and 90 were eligible for selection in the study. Lifetime residential
and water source histories and information on water consumption
habits, diet, demographic information, medical and occupational
histories, lifestyle and other factors was obtained by a
self-administered questionnaire and augmented with information from
medical records. Historical exposures to THMs were estimated using a
predictive statistical model based on current THM levels and water
supply operation records. Multivariate logistic regression analysis
was used to estimate the risk of colon cancer adjusted for age, gender
and urbanization. Individuals exposed to drinking-water containing
more than 40 µg of THMs per litre, the highest exposure category, were
found to be at no greater risk of colon cancer than individuals
exposed to water with no or trace levels of THMs. Nor did cumulative
exposure to THMs present a colon cancer risk (Table 36). Although this
analysis suggests that the presence of THMs in Wisconsin
Table 36. Colon cancer risks and estimated cumulative exposure
to trihalomethanes in Wisconsin (USA)a
Lifetime THM exposure Odds ratiob 95% CI
(µg/litre-year)
<137 1.0
137-410 1.1 0.7-1.8
>410 0.7 0.4-1.2
a From Young et al. (1987).
b Adjusted for age, gender and population of place of
residence.
drinking-water is not associated with a colon cancer risk, THM levels
in Wisconsin are low: 98% of water samples had concentrations less
than 100 µg/litre.
Lawrence et al. (1984) studied the relationship of THMs and
colo-rectal cancer in New York State (USA) where THM exposure was
higher than in Wisconsin and found no association. Included in the
study were 395 white female teachers in New York State who died from
colo-rectal cancer and an equal number of teachers who died from
non-cancer causes. Cumulative chloroform exposure was estimated by a
statistical model that considered water treatment operational records
during the 20 years prior to death. The distribution of chloroform
exposure was not significantly different between cases and controls,
and no effect of cumulative exposure was found in a logistic analysis
controlling for average source type, population density, marital
status, age and year of death. The risk of cancer was not found to be
higher among those using a surface water source containing THMs (OR =
1.1; 90% CI = 0.8-1.4). Mean levels of cumulative THM exposure were
similar among cases (635 µg/litre-years) and controls
(623 µg/litre-years).
The population-based case-control study conducted from 1986 to
1989 in Iowa (USA) (Hildesheim et al., 1998) found no increased risk
of colon cancer or any subsites associated with estimates of exposure
to THMs, either total (g) or average lifetime (µg/litre) (Table 37).
A prospective cohort study of 41 836 post-menopausal women in
Iowa (USA) (Doyle et al., 1997) included an analysis of women who
reported drinking municipal or private well-water for more than the
past 10 years ( n = 28 237). Historical water treatment and water
quality data were used to ascertain exposure to THMs. The primary
source of information on THMs was a 1986-1987 water survey. All women
who lived in the same community were assigned the same level of
Table 37. Colon cancer rsks and estimated cumulative exposure
to trihalomethanes in Iowa (USA)a
Lifetime THM exposure Odds ratio (95% CI)b
Total lifetime exposure (g)
<0.04 1.0
0.05-0.12 1.0 (0.7-1.2)
0.13-0.34 0.9 (0.6-1.2)
0.35-1.48 1.2 (0.9-1.6)
1.49-2.41 0.5 (0.3-0.9)
>2.42 1.1 (0.7-1.8)
Lifetime average exposure (µg/litre)
<0.8 1.0
0.8-2.2 1.0 (0.8-1.3)
2.3-8.0 0.9 (0.7-1.3)
8.1-32.5 1.1 (0.8-1.4)
32.6-46.3 0.9 (0.6-1.5)
>46.3 1.1 (0.7-1.6)
a From Hildesheim et al. (1998).
b Adjusted for age and gender.
exposure to THMs. A dose-response relationship (Table 38) was found
with increasing chloroform levels in municipal drinking-water for all
cancers combined, colon cancer, lung cancer and melanoma (test for
trend P < 0.05). The highest exposure covered a wide range of
values, and selection of the exposure categories for chloroform is
unusual and does not reflect normal high exposures to chloroform.
Additional analyses after exclusion of women who reported a history of
colo-rectal polyps and adjustment for additional risk or protective
factors did not change the dose-response relationship and slightly
increased the relative risk estimates. No associations were observed
between colon cancer and BDCM, DBCM or bromoform, but levels were low,
and many water systems had no detectable levels of these THMs. For
example, the geometric mean levels of BDCM and DBCM in surface water
were 8.7 and 0.4 µg/litre, respectively. Because water quality data
were also available from a 1979 water survey for a national bladder
cancer study, analyses were also conducted using these data. Fewer
municipalities were sampled for THMs in 1979, and only 16 461
participants were included. In this analysis, colon cancer was found
to be associated with increasing exposure to chloroform levels.
Table 38. Risks of cancer incidence associated with chloroform levels in post-menopausal women, Iowa (USA)a
Cancer site Chloroform concentration (µg/litre)
1-2 3-13 14-287
Cases RRb 95% CI Cases RRb 95% CI Cases RRb 95% CI
Bladder 11 0.9 0.4-2.1 12 1.3 0.6-2.8 7 0.7 0.3-1.7
Colon 41 1.1 0.7-1.7 42 1.4 0.9-2.2 57 1.7 1.1-2.6
Rectal 19 0.8 0.4-1.5 14 0.8 0.4-1.5 22 1.1 0.6-2.0
Breast 151 1.1 0.9-1.3 131 1.2 0.9-1.5 136 1.1 0.9-1.4
Kidney 5 0.6 0.2-1.7 9 1.3 0.5-3.2 7 0.9 0.3-2.4
Lung 35 1.4 0.8-2.3 40 2.0 1.2-3.2 42 1.9 1.1-3.0
Melanoma 15 2.5 1.0-6.5 6 1.3 0.4-3.9 17 3.2 1.3-8.2
All cancers 253 1.1 0.9-1.3 220 1.3 1.05-1.5 268 1.3 1.1-1.5
a From Doyle et al. (1997).
b Reported relative risks were adjusted for age, education, smoking status, physical activity, fruit and
vegetable intake, total energy intake, body mass index and waist to hip ratio.
3) Rectal cancer risk
The population-based case-control study conducted in Iowa (USA)
(Hildesheim et al., 1998) found an increased risk of rectal cancer
associated with estimates of exposure to THMs after controlling for
age, gender and average population size for men and women. For total
lifetime exposure to THMs of greater than 1.48 g, odds ratios were
almost double (OR = 1.9; 95% CI = 1.2-3.0) those for total lifetime
exposure to THMs of less than 0.05 g. For lifetime average THM
concentrations of 32.6-46.3 µg/litre and greater than 46.4 µg/litre,
odds ratios (OR = 1.7; 95% CI = 1.1-2.6) were almost 70% greater than
those for average exposures of less than 0.8 µg/litre (Table 39). This
is the only study to report increased rectal cancer risks associated
with THM exposure.
Table 39. Rectal cancer risks and estimated cumulative
exposure to trihalomethanesa
Lifetime THM exposure Odds ratio (95% CI)b
Total lifetime exposure (g)
<0.04 1.0
0.05-0.12 1.3 (1.0-1.6)
0.13-0.34 1.3 (0.9-1.8)
0.35-1.48 1.5 (1.1-2.1)
1.49-2.41 1.9 (1.2-3.0)
>2.42 1.6 (1.0-2.6)
Lifetime average exposure (µg/litre)
<0.8 1.0
0.8-2.2 1.0 (0.8-1.4)
2.3-8.0 1.2 (0.9-1.7)
8.1-32.5 1.2 (0.9-1.7)
32.6-46.3 1.7 (1.1-2.6)
>46.3 1.7 (1.1-2.6)
a From Hildesheim et al. (1998).
b Adjusted for age and gender.
4) Mutagenicity
A cohort study of populations exposed to various levels of
mutagenicity in drinking-water and a case-control study of kidney and
bladder cancers are currently being conducted in Finland (Tuomisto et
al., 1995). No results have yet been reported for the case-control
study. The cohort study (Koivusalo et al., 1996, 1997) included
621 431 persons living in the same town in which they were born and
having a water connection in 1970. Cancer incidence in the cohort was
compared with national cancer incidence stratified by gender, time
period and age group. Cases were derived from the population-based
Finnish Cancer Registry, and follow-up of the cohort started in 1970.
Past exposure to drinking-water mutagenicity and THMs was assessed
using historical water quality information. The quantity of
mutagenicity was estimated for each 5-year period from 1955 to 1970
using an empirical equation relating mutagenicity and raw water pH,
potassium permanganate oxidation and chlorine dose. A good correlation
was found between estimated and measured values for the period 1986-
1987 when measures of mutagenicity were available. The quantity of
mutagenicity is minor in raw waters and predominantly results from the
chlorination process. The Salmonella/microsome assay is used to
assess the mutagenicity of the non-volatile, acid/neutral fraction of
chlorinated organic material in water. A mutagen of particular concern
is MX, a potent mutagen, as measured by strain S. typhimurium TA100,
which may be responsible for up to 57% of the mutagenicity in
chlorinated drinking-water (Meier et al., 1986, 1987). After adjusting
for age, time period, urbanization and social status, an average
exposure to mutagenicity in chlorinated water of 3000 net revertants
per litre was found to be associated with a statistically significant
increased risk in women for cancers of the bladder (RR = 1.5; 95% CI =
1.0-2.2), rectum (RR = 1.4; 95% CI = 1.0-1.9), oesophagus (RR = 1.9;
95% CI = 1.0-3.5) and breast (RR = 1.1; 95% CI = 1.0-1.2). Past
exposure to THMs, one group of volatile by-products of chlorination,
did not result in statistically significant excess risks (Koivusalo et
al., 1996). Although this study found a moderate association between
cancers of the bladder, rectum and oesophagus in women and high levels
of mutagenicity in drinking-water, the results should be interpreted
with caution, as this is the only analytical epidemiological study of
water mutagenicity. Significantly increased relative risks were found
only for women, and the magnitude of the risks suggests that results
may be due to residual uncontrolled confounding.
5.3.1.3 Summary of results of cancer studies
Cumulative exposure to THMs was slightly higher in New York (USA)
than in Wisconsin (USA), but no increased colon cancer risk associated
with THM exposure was observed in either study. Data reported thus far
from a study in Iowa (USA) show that colon cancer risk was not
associated with estimates of past exposure to THMs, but rectal cancer
risk was associated with increasing amounts of lifetime exposure to
THMs. Risks were not reported for THM levels found in drinking-water.
A cohort study in Iowa found moderately increased risks associated
with a wide reported range of chloroform concentrations (14-287
µg/litre).
In Ontario (Canada), the risk of bladder cancer incidence was
about 40% higher among persons exposed to greater than 1956 µg of THMs
per litre-year in water compared with those exposed to less than 584
µg/litre-year. No association between exposure to THM levels and
increased bladder cancer risk was found in Colorado (USA). Data
reported thus far from a study in Iowa (USA) show that risk of bladder
cancer was not associated with estimates of past exposure to
chlorination by-products except among men and smokers, where bladder
cancer risk increased with duration of exposure after control for
cigarette smoking.
In Finland, an average exposure to mutagenicity in chlorinated
water of 3000 net revertants per litre was found to be associated with
an increased risk in women for cancers of the bladder, rectum,
oesophagus and breast; however, past exposure to THMs did not result
in statistically significant excess cancer risks. THM levels in
drinking-water were not reported for Finland, and these results were
reported only in an abstract.
No increased risk of bladder cancer was associated with THM
exposure in Colorado (USA); cumulative exposure to THMs in Colorado
was similar to those in New York (USA) and Wisconsin (USA), where no
increased risk was found for colon cancer. In Ontario (Canada), THM
exposure was much higher, and a moderate increased risk of bladder
cancer was found.
At this time, the evidence for an association between THM
exposure in drinking-water and colon cancer must be considered
inconclusive. No evidence is available from epidemiological studies to
suggest an increased risk of colon cancer, but studies have been
conducted in areas where cumulative exposures were generally low. The
evidence for an association between chlorinated water or THM exposure
in drinking-water and bladder cancer is limited. No association was
found in Colorado, but cumulative exposures were low. In Canada, where
cumulative THM exposure was much higher, a moderate increased risk of
bladder cancer was found.
It is possible that other unmeasured by-products may be
associated with bladder cancer risks, as several studies have found an
association between chlorinated surface water and bladder cancer but
not between THMs and bladder cancer. It is possible that another DBP
or a water contaminant other than a DBP is responsible. A plausible
alternative explanation for the observed results is that residence in
an area served by a chlorinated surface water supply is simply a
surrogate for some other unidentified risk factor or characteristic of
urban populations that may be associated with an increased risk of
cancer. Other potential DBPs may include other volatile organic
contaminants, but studies have considered ingestion, not inhalation,
exposures, and very few studies have attempted to assess both
long-term exposures to chlorinated water and historical water
consumption patterns. Accurate long-term exposure assessment is
difficult. Exposures to other chlorinated or brominated compounds or
HAAs have also not been considered. Additional studies should continue
to assess the risk of the non-volatile fraction of organic
by-products.
Several studies found an elevated bladder cancer risk among
persons who consumed more tapwater per day, but increased tapwater
consumption appeared to be an independent risk factor for bladder
cancer. The association of water and other fluid consumption and
bladder cancer also requires additional study.
Weak associations reported between cumulative THM exposure and
bladder cancer risks do not provide adequate evidence that THMs cause
bladder cancer. In two studies, no association was found. A moderate
association was reported for rectal cancer and cumulative THMs, but
only in a single study. There is no evidence for an association
between the other cancer sites studied and THM exposure.
5.3.2 Epidemiological studies of cardiovascular disease and
disinfectant by-products
A cohort study of 31 000 residents of Washington County, Maryland
(USA) found a slightly increased, but not statistically significant,
risk (RR = 1.1; 95% CI = 1.0-1.3) of death due to arteriosclerotic
heart disease in residents exposed to chlorinated surface water and
springs compared with residents of towns where unchlorinated
well-water was used. Water sampling during the study found average
chloroform levels of 107 µg/litre in the Hagerstown water system, but
it is not known if these levels accurately represent long-term
exposures. Other observational (Zierler et al., 1988; Zeighami et al.,
1990a,b; Riley et al., 1995) epidemiological studies of disinfected
water evaluated the possible adverse cardiovascular effects of
chlorinated or chloraminated drinking-water, but no by-products were
measured.
5.3.2.1 Summary of results of cardiovascular studies
Epidemiological studies have not evaluated associations between
specific DBPs and cardiovascular disease, but there is no evidence of
an increased risk caused by chlorinated or chloraminated
drinking-water.
5.3.3 Epidemiological studies of adverse reproductive/developmental
outcomes and disinfectant by-products
In the Savitz et al. (1995) study, dates of pregnancy were used
to assign THM levels from the appropriate water supply and for the
periods in the pregnancy in which exposures might cause any adverse
effect. No associations were reported between THM levels in North
Carolina (USA) and estimated dose of THMs with miscarriage, preterm
delivery or low birth weight. Savitz et al. (1995) found no increased
risk of low infant birth weight or preterm delivery associated with
exposure to THM levels of 63-69 µg/litre or a computed dose of THMs of
170-1171 µg/litre-glasses per day. No increased risk of miscarriage
was associated with either (i) THM levels of 60-81.0 or 81.1-168.8
µg/litre or (ii) a computed dose of THMs of 140.0-275.0 or
275.1-1171.0 µg/litre-glasses per day. Although no increased
miscarriage risk was found in this categorical analysis, an analysis
using a continuous measure for THMs predicted an association (1.7 per
50 µg of THMs per litre increment; 95% CI = 1.1-2.7). This
association, however, was not part of an overall dose-response
gradient and may be a spurious finding. Another categorical analysis
using sextiles of THM exposures showed a much higher miscarriage risk
(adjusted OR = 2.8; 95% CI = 1.2-6.1) in the highest sextile but a
very low risk or even a possibly protective effect in the next highest
sextile (adjusted OR = 0.2; 95% CI = 0.0-0.5).
A population-based case-control study in Iowa (USA) used
information from birth certificates from January 1989 to June 1990 and
a water supply survey conducted in 1987 to study the association of
waterborne chloroform and other DBPs with low birth weight (<2500 g),
prematurity (<37 weeks' gestation) and intrauterine growth
retardation (Kramer et al., 1992). Cases were not mutually exclusive,
but each outcome was analysed separately. Controls were randomly
selected from the same birth certificates. The study included 159 low
birth weight and 795 normal birth weight infants, 342 premature
infants and 1710 controls, and 187 intrauterine growth-retarded
infants and 935 controls. Mothers were not interviewed to obtain
information about their residential history during pregnancy or
possible risk factors, and other exposures were not noted on the
certificate, which might potentially confound any observed association
or modify its effect. Information on maternal age, parity, adequacy of
prenatal care, marital status, education and maternal smoking was
available from the birth certificate. Residence of the mother at the
time of birth determined which water system was used to assign levels
of exposure to THM and TOX measured in a previous municipal water
survey. The only statistically significant finding was a moderately
increased risk (OR = 1.8; 95% CI = 1.1-2.9) of intrauterine growth
retardation associated with chloroform levels of greater than 9
µg/litre in water after controlling for confounding characteristics
from the certificate. Prematurity (OR = 1.1; 95% CI = 0.7-1.6) and low
birth weight (OR = 1.3; 95% CI = 0.8-2.2) were not found to be
associated with chloroform levels. No statistically significant
associations were seen with any of these developmental outcomes and
BDCM, DBCM, bromoform or organic halides. Interpretation of the
results of this study, however, is limited because the study design
was more ecological than analytical. The authors considered the
results to be preliminary because of possible bias. The ascertainment
and classification of exposure to the water contaminants were
imprecise and may have resulted in misclassification bias. Municipal
measures of by-products assigned to the residences for exposure
purposes may have been either higher or lower than actual exposures.
Characteristics that were not identified (e.g., alcohol consumption)
could be responsible for confounding bias. A cross-sectional
epidemiological study (Bove et al., 1992a, 1995) was conducted in four
northern New Jersey (USA) counties to explore possible associations
between THM levels and 13 developmental and adverse reproductive
outcomes: low birth weight (<2500 g), prematurity (<37 weeks), small
for gestational age, very low birth weight (<1500 g), stillbirths,
surveillance malformations for 33 selected categories, central nervous
system defects and subgroups, oral cleft defects and subgroups, and
cardiac defects and subgroups. A total of 143 hypotheses were formally
evaluated; as the stated objective of the study was to identify
promising leads for further research rather than for
decision-analytical purposes, each finding was reported as if it were
the sole focus of the study without statistical adjustment for
multiple comparisons.
Reproductive outcomes over a 4-year period, from January 1985 to
December 1988, were obtained from a population-based birth defects
registry and vital records, birth certificates and death certificates.
A total of 80 938 live births and 594 fetal deaths were studied in 75
towns selected because residents were mostly served by public water
systems. All information about reproductive outcomes, potential
confounding characteristics and risk factors, such as maternal age,
race, education, primipara, previous stillbirth or miscarriage, sex
and adequacy of prenatal care, was obtained from vital records.
Information on other potential important confounders -- maternal
occupation, drug use during pregnancy, smoking and alcohol consumption
-- was not available for analysis. Mothers were not interviewed for
information on individual exposures and potentially confounding
characteristics.
Information about water quality for the 75 towns was obtained
from existing records. Monthly estimates of each water contaminant in
a town's water system were used to assign exposure for each
gestational month of each live birth and fetal death. A mother's
residence at birth was assumed to be her residence throughout her
pregnancy. All water systems in the study were chlorinated, but
sufficient groundwater sources were available to ensure inclusion of
areas with very low levels of THMs. The drinking-water contaminants
studied were THMs, trichloroethylene, tetrachloroethylene,
dichloroethylenes, 1,1,1-trichloroethane, carbon tetrachloride,
1,2-dichloroethane, benzene and nitrate. Type of water source was also
considered. For evaluation of risk with different levels of exposure,
THMs were categorized into six different levels of potential exposure:
<20, >20-40, >40-60, >60-80, >80-100 and >100 µg/litre.
Reported associations with levels of THMs greater than 100
µg/litre included small for gestation age (OR = 1.5; 90% CI = 1.2-1.9)
and oral cleft defects (OR = 3.2; 90% CI = 1.2-7.3). Reported
associations with levels of THMs above 80 µg/litre included all
surveillance defects (OR = 1.6; 90% CI = 1.2-2.0), central nervous
system defects (OR = 2.6; 90% CI = 1.5-4.3), neural tube defects
(OR = 3.0; 90% CI = 1.3-6.6) and major cardiac defects (OR = 1.8; 90%
CI = 1.0-3.3). Moderate to strong associations were found for central
nervous system, oral cleft and neural tube defects, but only a small
number of cases were studied. The study included 4082 small for
gestational age infants, but fewer numbers with birth defects: 56
infants with neural tube defects, 83 with oral cleft defects, 108 with
major cardiac defects and 118 with central nervous system defects. The
observed increased risk for other reproductive outcomes was smaller,
and these weak associations could be due to unidentified confounding
bias.
As in the Iowa (USA) study, the ecological study design for
assessment of individual exposure and confounding bias limits the
interpretation of the results. The investigators noted "the difficulty
of interpreting the available water contamination data and the
numerous assumptions needed in order to estimate contaminant levels"
as a limitation (Bove et al., 1992a). "By itself, this study cannot
resolve whether the drinking-water contaminants caused the adverse
birth outcomes" (Bove et al., 1995).
Also reported were results of a population-based case-control
study in New Jersey (USA) to determine risks of cardiac defects,
neural tube defects, oral clefts, very low birth weight and low birth
weight associated with exposure to different THM levels (Bove et al.,
1992b). A total of 563 mothers of cases and controls were interviewed
by telephone some 6-54 months after giving birth. The study included
185 infants with birth defects, 37 of whom had neural tube defects,
97 infants with very low birth weights, 113 infants with low birth
weights and 138 infants of normal weight without birth defects.
Information was obtained for a potential exposure period 3 months
prior to conception through the end of pregnancy and included
residences of the mother, sources of drinking-water, tapwater
consumption, showering and smoking habits, alcohol consumption,
exposures in and around the home, prescription drugs, medical history
and previous adverse reproductive outcomes. For neural tube defects, a
4-fold increased risk was found to be associated with THM levels
greater than 80 µg/litre (OR = 4.25; 95% CI = 1.0-17.7); however, this
estimate is based on only 7 cases and 14 controls. The sample size for
the case-control study was small, resulting in low statistical power
and a lack of precision in estimating the risk; most importantly, a
majority (53.3%) of the mothers of the cases and controls could not be
located for interviews, causing possible selection bias. Once contact
was established with the mother of the case or control, only 78%
agreed to be interviewed. In addition, because of the long period
between pregnancy and the interview, inaccurate recall of the mothers
may be responsible for incorrect or biased information about both
potential exposures and confounding characteristics. An assessment of
selection bias by the investigators showed that risk had likely been
overestimated for neural tube defects. After correcting for selection
bias, the risk associated with THM levels greater than 80 µg/litre was
reduced from a 4-fold to a 50% increased risk. The observed risk now
represents a weak association where unknown confounding bias might be
responsible.
A second population-based case-control study was recently
reported in a published abstract (Klotz et al., 1996). Cases were
births or fetal deaths after 20 weeks' gestation in 1993 and 1994 with
anencephaly, spina bifida or encephalocele reported to the New Jersey
Birth Defects Registry or Vital Statistics. Controls were randomly
selected by month of birth from birth certificates during the same
period as cases. In home interviews, information was obtained on
ingestion and non-ingestion exposures, other environmental and
occupational exposures, and pregnancy characteristics. Exposures were
estimated from water quality monitoring data for the appropriate
public water system selected to approximate the critical time for
neural closure (fourth week of gestation), similar water quality data
from the same source 1 year later, and analysis of residential
tapwater collected about 1 year after the critical time. To prevent
misclassification of exposure, biological monitoring of urine and
exhaled breath from a sample of participants was also conducted.
Increased relative risks for neural tube defects were associated with
THMs; ORs were generally between 1.5 and 2.1 (Klotz & Pyrch, 1998).
The only statistically significant results were observed in infants
born with neural tube defects (and no other malformations) and whose
mothers' residence in early pregnancy was in an area where the THM
levels were greater than 40 µg/litre (OR = 2.1; 95% CI = 1.1-4.0). No
association was observed between HAAs, HANs or nitrates in
drinking-water and risk of neural tube defects.
A cohort study (Waller et al., 1998; Swan, 1998) of women members
of Kaiser Permanente Medical Care Program in California (USA)
evaluated associations of THMs and spontaneous abortion, low birth
weight, preterm delivery and intrauterine growth retardation. Results
of the spontaneous abortion analysis were recently reported.
Information about water consumption, water source and THM levels was
collected for the participating cohort members. Data for THM levels in
tapwater were obtained from each public water supply for a 3-year
period when distribution taps were monitored at least quarterly. A
woman's exposure to THMs was estimated using the average level of THMs
for each water supply of samples collected within the woman's first
trimester (77% of the cohort) or within 30 days of the subject's first
trimester (4%) or the annual average from the utility's annual water
quality report (9%). High first-trimester levels of THMs were based on
levels corresponding to the 75th percentile, i.e., >75 µg of total
THMs per litre, >16 µg of chloroform per litre, >15 µg of
bromoform per litre, >17 µg of BDCM per litre and >31 µg of DBCM
per litre. Pregnancy outcomes were obtained from hospital records, the
California Birth Registry and follow-up interviews. Interviews were
also used to obtain information about possible confounding
characteristics. The study found that an increased risk of miscarriage
was associated with a high consumption of water (five or more glasses
of cold tapwater per day) containing high levels of total THMs
(>75 µg/litre), especially for waters high in BDCM (>17
µg/litre). In the women with high exposures to total THMs (THM levels
>75 µg/litre and five or more glasses of water consumed per day),
the relative risk of a miscarriage was almost twice that of the
low-exposure groups (total THM levels below 75 µg/litre and fewer than
five glasses of water per day). Of the four THMs, only high exposure
to BDCM (>17 µg/litre and five or more glasses of water per day)
was associated with a 3-fold increased relative risk of miscarriage.
The risk for unemployed women was also found to be greater than that
for employed women, suggesting that women with a greater opportunity
to consume home tapwater with a high level of THMs may be at greater
risk. However, it is not certain that other characteristics of
employed women were adequately assessed and controlled for (e.g., a
healthy worker effect).
Preliminary analyses were available for a cross-sectional study
(Nuckols et al., 1995) of births to women in Northglenn, Colorado
(USA) exposed to chlorinated water from Stanley Lake with high THM
levels (32-72 µg/litre) and women in Westminster exposed to
chloraminated water from Stanley Lake with low THM levels
(<20 µg/litre). Gallagher et al. (1997)1 used a geographic
information system and water quality modelling as described by Nuckols
et al. (1995) to conduct a retrospective cohort study in the same
water districts. Information about developmental outcomes and possible
confounding characteristics was obtained from birth certificates, and
exposures to THMs were modelled based on hydraulic characteristics of
the water system and THM levels obtained from a monitoring program.
However, sufficient information was available to estimate THM
exposures for mothers in only 28 census blocks, 11 of 26 census blocks
from Northglenn and 17 of 60 blocks from Westminster. The exclusion of
such a large number of births from the study seriously limits the
interpretation of the observed results. Women with high or low THM
exposure may have been selectively excluded, but this could not be
determined with the information reported. After excluding births to
mothers in blocks with no THM data and births less than 400 g, only
25% of births remained for analysis, and the reported epidemiological
associations must be considered inconclusive.
5.3.3.1 Summary of results of reproductive/developmental studies
No associations were reported in North Carolina (USA) for THM
levels and estimated dose of THMs, but associations between chloroform
or THM levels in water and adverse reproductive or developmental
outcomes were reported in studies conducted in California, Iowa and
New Jersey (USA).
A recently completed case-control study (but not yet reported in
the peer review literature) in New Jersey (USA) reported increased
relative risks for infants born with neural tube defects (and no other
malformations) and whose mothers' residence in early pregnancy was in
an area where the THM levels were greater than 40 µg/litre (OR = 2.1;
95% CI = 1.1-4.0). Replication of these results in another geographic
area is required before causality can be assessed. Previous
epidemiological studies in New Jersey reported increased relative
risks of neural tube defects associated with the mother's residence in
areas with high THMs; however, these studies suffer from
methodological limitations, and their results are inconclusive.
1 Gallagher MD, Nuckols JR, Stallones L, & Savitz DA (1997) Exposure
to trihalomethanes and adverse pregnancy outcomes in Colorado
(Unpublished manuscript).
The Waller et al. (1998) study is well designed and well
conducted; as it is the first study to suggest an adverse reproductive
effect associated with a brominated by-product, results should be used
to support further research on spontaneous abortion risks and
drinking-water contaminants, including THM species, other DBPs and
other water contaminants. The authors discussed the weaknesses and
strengths of their study. One weakness is that water consumption was
not assessed at work, but investigators did consider risks separately
for women who worked outside the home, reporting that "results were
stronger in women not employed outside the home, for whom our home
based exposure assessment should be more precise" (Waller et al.,
1998). A major limitation is that DBPs other than THMs and other water
contaminants were not studied. A concern is how to interpret the
results of the Waller et al. (1998) study in light of the findings of
Swan et al. (1998).
Swan et al. (1998) analysed tapwater and bottled water
consumption in the same cohort studied by Waller et al. (1998) and
reported a dose-related increase in spontaneous abortions among
tapwater drinkers in Region I, but not in Region II or III. "Our prior
studies suggested that the relation between spontaneous abortion and
tapwater was independent of chlorination by-products, since the
strongest associations were seen in the two studies conducted in areas
served only by unchlorinated groundwater. Additionally, in the two
rodent studies we conducted, a trend toward increased rates of fetal
resorption was seen in rats drinking unchlorinated groundwater,
compared with bottled water. In our current study, as discussed in the
study by Waller et al., spontaneous abortion risk was increased by
exposure to specific chlorination by-products in all regions.
Nevertheless, we believe that the associations with cold tapwater and
bottled water presented here, which are specific to Region I, cannot
be explained by exposure to chlorination by-products, because the
association is seen in the absence of high levels of these chemicals"
(Swan et al., 1998). In a letter to the editor, Swan & Waller (1998)
suggested that there may be "some constituent in addition to THMs"
that is specific to tapwater in Region I but provided a poor
explanation of why this may be so. "Bromodichloromethane (or some
compound highly correlated with it) was the trihalomethane most
strongly associated with SAB [spontaneous abortion]" (Waller et al.,
1998). "Swan et al. found a dose-related increase in SABs among
tapwater drinkers in Region I, but not in Regions II or III. Exposure
to TTHM [total THMs] or bromodichloromethane does not entirely explain
this association, since a tapwater effect is still evident among
Region I women with low levels of both TTHM and bromodichloromethane.
Furthermore, the initial studies in Region I found the strongest
effect in areas served only by unchlorinated groundwater. Thus, it is
likely that other factors contributed to the tapwater effect described
by Swan et al." (Waller et al., 1998). Because of these concerns,
judgement about the interpretation of the results of Waller et al.
(1998) should be deferred until additional water quality data are
analysed for the cohort or another well designed and well conducted
epidemiological study finds similar results.
Although the study by Waller et al. (1998) does not provide
sufficient evidence for a cause-effect relationship between exposure
to THMs and early-term miscarriages, it does provide important new
information that should be pursued with additional research. Assessing
the causality of an observed epidemiological association requires
evidence from more than a single study, and additional information is
needed on other water exposures.
Several exploratory epidemiological studies have suggested that
certain adverse reproductive effects and developmental outcomes may be
associated with chloroform or THMs in drinking-water, but additional
studies are required to determine whether these observed associations
are spurious or due to possible bias. The latest study from New Jersey
(USA) reported a moderate association between neural tube defects and
THM levels of more than 40 µg/litre but no associations with HAAs,
HANs or nitrates in drinking-water. The Waller et al. (1998) and Klotz
& Pyrch (1998) studies require replication in another area before
results can be properly interpreted.
5.4 Summary
Epidemiological studies have not identified an increased risk of
cardiovascular disease associated with chlorinated or chloraminated
drinking-water.
Based on the entire cancer-chlorinated drinking-water
epidemiology database, there is better evidence for an association
between exposure to chlorinated surface water and bladder cancer than
for other types of cancer. However, the latest published study (Cantor
et al., 1998) notes several inconsistencies in results among the
studies for smokers/non-smokers and males/females, and the evidence is
still considered insufficient to allow a judgement as to whether the
association is causal and which water contaminants may be important.
Evidence for a role of THMs is weak. Poole (1997) also notes that "The
basic conclusion of the present report is that the hypothesis of a
causal relationship between consumption of chlorination by-products
and the risk of any cancer, including bladder cancer and rectal
cancer, is still an open question."
The overall findings of Cantor et al. (1998) support the
hypothesis of an association between bladder cancer and duration of
use of chlorinated surface water or groundwater and estimated THM
exposures, but aspects of these results caution against a simple
interpretation and raise additional questions about the nature of the
association. An increase in bladder cancer risk was found with
duration of chlorinated groundwater use, as well as with total
duration of chlorinated drinking-water (surface water plus
groundwater) use, with relative risks similar to those observed with
chlorinated surface water. This finding is unexpected, because the
levels of by-products from most chlorinated groundwaters are much
lower than those in treated surface water. In addition, risk was found
to increase with duration of chlorinated surface water use among
ever-smokers, but not never-smokers, and among men, but not women.
This raises questions of internal consistency, as well as consistency
with other findings. In contrast, Cantor et al. (1998) found
associations for both sexes, primarily among never-smokers. Cantor et
al. (1985) noted: "In Ontario, King and Marrett noted somewhat higher
risk estimates for never-smokers associated with duration of
chlorinated surface water. In Colorado, McGeehin et al. reported
similar patterns of risk among smokers and never-smokers, and among
men and women. Finally, in a case-control study from Washington
County, Maryland, Freedman et al. reported results that parallel the
current findings, namely that the risk associated with chlorinated
surface water was primarily observed among men and among smokers.
Reasons for differences among these observations and differences with
results from our study are unclear. A possible explanation for the
apparent discrepancies in findings for smokers and never-smokers among
studies may reside in water quality and water treatment differences in
the respective study areas, with resulting variations in the chemical
composition of byproduct mixtures. Nevertheless, results should not
differ by sex."
The existing epidemiological data are insufficient to allow a
conclusion that the observed associations between bladder or any other
cancer and chlorinated drinking-water or THMs are causal or provide an
accurate estimate of the magnitude of risk.
Any association between exposure to chlorinated surface water,
THMs or the mutagenicity of drinking-water and cancer of the colon,
rectum, pancreas, brain and other sites cannot be evaluated at this
time because of inadequate epidemiological evidence. However, the
findings from well conducted studies associating bladder cancer with
chlorinated water and THMs cannot be completely dismissed, even though
inconsistencies have been noted for risks among men and women and
among smokers and non-smokers. Because of the large number of people
exposed to chlorinated drinking-water, it is important to resolve this
issue using studies designed with sound epidemiological principles.
Additional studies to resolve the questions about the associations
that have been reported for chlorinated surface water, THMs, fluid and
tapwater consumption and bladder cancer and reproductive and
developmental effects must focus on the resolution of various problems
noted in previous studies, especially consideration of exposures to
other DBPs.
The existing epidemiological data are insufficient to allow the
importance of the observed associations of chlorinated drinking-water
or THMs and adverse pregnancy outcomes to be assessed. Although
several studies have suggested that increased risks of neural tube
defects and miscarriage may be associated with THMs or selected THM
species, additional studies are needed to determine whether the
observed associations are spurious.
A recently convened scientific panel (US EPA, 1997) concluded
that the results of published epidemiological studies do not provide
convincing evidence that DBPs cause adverse pregnancy outcomes. The
panel recommended that additional studies be conducted, specifically
that the Waller et al. (1998) study be expanded to include additional
exposure information about by-products other than THMs and that a
similar study be conducted in another geographic area.
6. RISK CHARACTERIZATION
It should be noted that the use of chemical disinfectants in
water treatment usually results in the formation of chemical
by-products, some of which are potentially hazardous. However, the
risks to health from these by-products at the levels at which they
occur in drinking-water are extremely small in comparison with the
risks associated with inadequate disinfection. Thus, it is important
that disinfection not be compromised in attempting to control such
by-products.
6.1 Characterization of hazard and dose-response
6.1.1 Toxicological studies
6.1.1.1 Chlorine
A WHO Working Group for the Guidelines for drinking-water
quality (WHO, 1993) considered chlorine. This Working Group
determined a tolerable daily intake (TDI) of 150 µg/kg of body weight
for free chlorine. This TDI is derived from a NOAEL of approximately
15 mg/kg of body weight per day in 2-year studies in rats and mice
(NTP, 1992), incorporating an uncertainty factor of 100 (10 each for
intra- and interspecies variation). There are no new data that
indicate that this TDI should be changed.
6.1.1.2 Monochloramine
A WHO Working Group for the Guidelines for drinking-water
quality considered monochloramine (WHO, 1993). This Working Group
determined a TDI of 94 µg/kg of body weight based on a NOAEL of
approximately 9.4 mg/kg of body weight per day, the highest dose
tested, in a 2-year bioassay in rats (NTP, 1992), incorporating an
uncertainty factor of 100 (10 each for intra- and interspecies
variation). There are no new data that indicate that this TDI should
be changed.
6.1.1.3 Chlorine dioxide
Chlorine dioxide chemistry in drinking-water is complex, but the
major breakdown product in drinking-water is chlorite. In establishing
a specific TDI for chlorine dioxide, data on both chlorine dioxide and
chlorite can be considered, given the rapid hydrolysis to chlorite.
Therefore, an oral TDI for chlorine dioxide is 30 µg/kg of body
weight, based on the NOAEL of 2.9 mg/kg of body weight per day for
neurodevelopmental effects of chlorite in rats (CMA, 1997).
6.1.1.4 Trihalomethanes
Cancer following chronic exposure is the primary hazard of
concern for this class of DBPs. Owing to the weight of evidence
indicating that chloroform can induce cancer in animals only after
chronic exposure to cytotoxic doses, it is clear that exposures to low
concentrations of chloroform in drinking-water do not pose
carcinogenic risks. The NOAEL for cytolethality and regenerative
hyperplasia in mice was 10 mg/kg of body weight per day after
administration of chloroform in corn oil for 3 weeks (Larson et al.,
1994b). Based on the mode of action evidence for chloroform
carcinogenicity, a TDI of 10 µg/kg of body weight was derived using
the NOAEL for cytotoxicity in mice and applying an uncertainty factor
of 1000 (10 each for inter- and intraspecies variation and 10 for the
short duration of the study). This approach is supported by a number
of additional studies. This TDI is similar to the TDI derived in the
Guidelines for drinking-water quality (WHO, 1998), which was based
on a 7.5-year study in dogs. In this study, beagle dogs were given
chloroform in a toothpaste base in gelatin capsules, 6 days per week
for 7.5 years, at 0, 15 or 30 mg/kg of body weight per day. Slight
hepatotoxicity was observed at 15 mg/kg of body weight per day
(Heywood et al., 1979). Incorporating an uncertainty factor of 1000
(10 each for intra- and interspecies variation and 10 for use of a
LOAEL rather than a NOAEL and a subchronic study), a TDI of 13 µg/kg
of body weight (corrected for 6 days per week dosing) was derived.
Among the brominated THMs, BDCM is of particular interest because
it produces tumours in rats and mice and at several sites (liver,
kidney and large intestine) after corn oil gavage (NTP, 1987). The
induction of colon tumours in rats by BDCM (and by bromoform) is also
interesting because of the epidemiological associations with
colo-rectal cancer (see section 5.3.1). BDCM and the other brominated
THMs are also weak mutagens (IARC, 1991, 1999; Pegram et al., 1997).
It is generally assumed that mutagenic carcinogens will produce linear
dose-response relationships at low dose, as mutagenesis is generally
considered to be an irreversible and cumulative effect.
In a 2-year bioassay, BDCM given by corn oil gavage induced
tumours (in conjunction with cytotoxicity and increased proliferation)
in the kidneys of mice and rats at doses of 50 and 100 mg/kg of body
weight per day, respectively (NTP, 1987). The large intestine tumours
in rats occurred after exposure to both 50 and 100 mg/kg of body
weight per day. Using the incidence of kidney tumours in male mice
from this study, quantitative risk estimates have been calculated,
yielding a slope factor1 of 4.8 × 10-3 [mg/kg of body weight per
day]-1 and a calculated dose of 2.1 µg/kg of body weight per day for
a risk level of 10-5 (IRIS, 1993). A slope factor of 4.2 × 10-3
[mg/kg of body weight per day]-1 (2.4 µg/kg of body weight per day
for a 10-5 risk) was derived based on the incidence of large
intestine carcinomas in the male rat. IARC (1991, 1999) has classified
BDCM in Group 2B (possibly carcinogenic to humans).
1 Slope factors given here do not incorporate a surface area to body
weight correction.
DBCM and bromoform were studied in long-term bioassays. In a
2-year corn oil gavage study, DBCM induced hepatic tumours in female
mice, but not in rats, at a dose of 100 mg/kg of body weight per day
(NTP, 1985). In previous evaluations, it has been suggested that the
corn oil vehicle may play a role in the induction of tumours in female
mice (WHO, 1996). A small increase in tumours of the large intestine
in rats was observed in the bromoform study at a dose of 200 mg/kg of
body weight per day. No neoplastic effects were associated with
exposure of mice to chloroform (NTP, 1989a). The slope factors based
on these tumours are 6.5 × 10-3 [mg/kg of body weight per day]-1 for
DBCM or 1.5 µg/kg of body weight per day for 10-5 risk (IRIS, 1992)
and 1.3 × 10-3 [mg/kg of body weight per day]-1 or 7.7 µg/kg of body
weight per day for 10-5 risk for bromoform (IRIS, 1991).
These two brominated THMs are weakly mutagenic in a number of
assays, and they were by far the most mutagenic DBPs of the class in
the GST-mediated assay system (DeMarini et al., 1997; Pegram et al.,
1997). Because they are the most lipophilic THMs, additional concerns
about whether corn oil may have affected their bioavailability in the
long-term studies should be considered. A NOAEL for DBCM of 30 mg/kg
of body weight per day has been established in a 13-week corn oil
gavage study, based on the absence of histopathological effects in the
liver of rats (NTP, 1985). A TDI for DBCM of 30 µg/kg of body weight
was derived based on the NOAEL for liver toxicity of 30 mg/kg of body
weight per day and an uncertainty factor of 1000 (10 each for inter-
and intraspecies variation and 10 for the short duration of the study
and possible carcinogenicity). IARC (1991, 1999) has classified DBCM
in Group 3 (not classifiable as to its carcinogenicity to humans).
A NOAEL for bromoform of 25 mg/kg of body weight per day can be
derived on the basis of the absence of liver lesions in rats after 13
weeks of dosing by corn oil gavage (NTP, 1989a). A TDI for bromoform
of 25 µg/kg of body weight was derived based on this NOAEL for liver
toxicity and an uncertainty factor of 1000 (10 each for inter- and
intraspecies variation and 10 for the short duration of the study and
possible carcinogenicity). IARC (1991, 1999) has classified bromoform
in Group 3 (not classifiable as to its carcinogenicity to humans).
6.1.1.5 Haloacetic acids
The induction of mutations by DCA is very improbable at the low
doses that would be encountered in chlorinated drinking-water. The
available data indicate that DCA differentially affects the
replication rates of normal hepatocytes and hepatocytes that have been
initiated (Pereira & Phelps, 1996). The dose-response relationships
are complex, with DCA initially stimulating division of normal
hepatocytes. However, at the lower chronic doses used in animal
studies (but still very high relative to those that would be derived
from drinking-water), the replication rate of normal hepatocytes is
eventually sharply inhibited. This indicates that normal hepatocytes
eventually down-regulate those pathways that are sensitive to
stimulation by DCA. However, the effects in altered cells,
particularly those that express high amounts of a protein that is
immunoreactive to a c-Jun antibody, do not seem to be able to
down-regulate this response (Stauber & Bull, 1997). Thus, the rates of
replication in the pre-neoplastic lesions with this phenotype are very
high at the doses that cause DCA tumours to develop with a very low
latency. Preliminary data suggest that this continued alteration in
cell birth and death rates is also necessary for the tumours to
progress to malignancy (Bull et al., 1990). This interpretation is
supported by studies that employ initiation/promotion designs as well
(Pereira, 1996).
Based upon the above considerations, it is suggested that the
currently available cancer risk estimates for DCA be modified by
incorporation of newly developing information on its comparative
metabolism and modes of action to formulate a biologically based
dose-response model. These data are not available at this time, but
they should become available within the next 2-3 years.
The effects of DCA appear to be closely associated with doses
that induce hepatomegaly and glycogen accumulation in mice. The LOAEL
for these effects in an 8-week study in mice was 0.5 g/litre,
corresponding to approximately 100 mg/kg of body weight per day, and
the NOAEL was 0.2 g/litre, or approximately 40 mg/kg of body weight
per day (Kato-Weinstein et al., 1998). A TDI of 40 µg/kg of body
weight has been calculated by applying an uncertainty factor of 1000
to this NOAEL (10 each for inter- and intraspecies variation and 10
for the short duration of the study and possible carcinogenicity).
IARC (1995) has classified DCA in Group 3 (not classifiable as to its
carcinogenicity to humans).
TCA is one of the weakest activators of the PPAR known (Issemann
& Green, 1990). It appears to be only marginally active as a
peroxisome proliferator, even in rats (DeAngelo et al., 1989).
Furthermore, treatment of rats with high levels of TCA in
drinking-water does not induce liver tumours (DeAngelo et al., 1997).
These data strongly suggest that TCA presents little carcinogenic
hazard to humans at the low concentrations found in drinking-water.
From a broader toxicological perspective, the developmental
effects of TCA are the end-point of concern (Smith et al., 1989a;
Saillenfait et al., 1995). Animals appear to tolerate concentrations
of TCA in drinking-water of 0.5 g/litre (approximately 50 mg/kg of
body weight per day) with little or no signs of adverse effect. At 2
g/litre, the only sign of adverse effect appears to be hepatomegaly.
The hepatomegaly is not observed in mice at doses of 0.35 g of TCA per
litre in drinking-water, estimated to be equivalent to 40 mg/kg of
body weight per day (Pereira, 1996).
In a study by Smith et al. (1989a), soft tissue anomalies were
observed at approximately 3 times the rate in controls at the lowest
dose administered of 330 mg/kg of body weight per day. At this dose,
the anomalies were mild and would clearly be in the range where
hepatomegaly (and carcinogenic effects) would occur. Considering the
fact that the PPAR interacts with cell signalling mechanisms that can
affect normal developmental processes, a common mechanism underlying
hepatomegaly and the carcinogenic and developmental effects of this
compound should be considered.
The TDI for TCA is based on a NOAEL estimated to be 40 mg/kg of
body weight per day for hepatic toxicity in a long-term study in mice
(Pereira, 1996). Application of an uncertainty factor of 1000 to the
estimated NOAEL (10 each for inter- and intraspecies variation and 10
for possible carcinogenicity) gives a TDI of 40 µg/kg of body weight.
IARC (1995) has classified TCA in Group 3 (not classifiable as to its
carcinogenicity to humans).
Data on the carcinogenicity of brominated acetic acids are too
preliminary to be useful in risk characterization. Data available in
abstract form suggest, however, that the doses required to induce
hepatocarcinogenic responses in mice are not dissimilar to those of
the chlorinated acetic acids (Bull & DeAngelo, 1995). In addition to
the mechanisms involved in DCA- and TCA-induced cancer, it is possible
that increased oxidative stress secondary to their metabolism might
contribute to their effects (Austin et al., 1996; Parrish et al.,
1996).
There are a significant number of data on the effects of DBA on
male reproduction. No effects were observed in rats at doses of
2 mg/kg of body weight per day for 79 days, whereas an increased
retention of step 19 spermatids was observed at 10 mg/kg of body
weight per day. Higher doses led to progressively more severe effects,
including marked atrophy of the seminiferous tubules at 250 mg/kg of
body weight per day, which was not reversed 6 months after treatment
was suspended (Linder et al., 1997b). A TDI of 20 µg/kg of body weight
was determined by allocating an uncertainty factor of 100 (10 each for
inter- and intraspecies variation) to the NOAEL of 2 mg/kg of body
weight per day.
6.1.1.6 Chloral hydrate
In a 2-year study, chloral hydrate at 1 g/litre of drinking-water
(166 mg/kg of body weight per day) induced liver tumours in male mice
(Daniel et al., 1992a). Lower doses have not been evaluated. Chloral
hydrate has been shown to induce chromosomal anomalies in several
in vitro tests but has been largely negative when evaluated in
vivo (IARC, 1995). It is probable that the liver tumours induced by
chloral hydrate involve its metabolism to TCA and/or DCA. As discussed
previously, these compounds are considered to act as tumour promoters.
IARC (1995) has classified chloral hydrate in Group 3 (not
classifiable as to its carcinogenicity to humans).
Chloral hydrate administered to rats for 90 days in
drinking-water induced hepatocellular necrosis at concentrations of
1200 mg/litre and above, with no effect being observed at 600 mg/litre
(approximately 60 mg/kg of body weight per day) (Daniel et al.,
1992b). Hepatomegaly was observed in male mice at doses of 144 mg/kg
of body weight per day administered by gavage for 14 days. No effect
was observed at 14.4 mg/kg of body weight per day in the 14-day study,
but mild hepatomegaly was observed when chloral hydrate was
administered in drinking-water at 70 mg/litre (16 mg/kg of body weight
per day) in a 90-day follow-up study (Sanders et al., 1982). An
uncertainty factor of 1000 (10 each for inter- and intraspecies
variation and 10 for the use of a LOAEL instead of a NOAEL) applied to
this value gives a TDI of 16 µg/kg of body weight.
6.1.1.7 Haloacetonitriles
Without appropriate human data or an animal study that involves a
substantial portion of an experimental animal's lifetime, there is no
generally accepted basis for estimating carcinogenic risk from the
HANs.
Data developed in subchronic studies provided some indication of
NOAELs for the general toxicity of DCAN and DBAN. NOAELs of 8 and 23
mg/kg of body weight per day were identified in 90-day studies in rats
for DCAN and DBAN, respectively, based on decreased body weights at
the next higher doses of 33 and 45 mg/kg of body weight per day,
respectively (Hayes et al., 1986).
A Working Group for the WHO Guidelines for drinking-water
quality considered DCAN and DBAN (WHO, 1993). This Working Group
determined a TDI of 15 µg/kg of body weight for DCAN based on a NOAEL
of 15 mg/kg of body weight per day in a reproductive toxicity study in
rats (Smith et al., 1989b) and incorporating an uncertainty factor of
1000 (10 each for intra- and interspecies variation and 10 for the
severity of effects). Reproductive and developmental effects were
observed with DBAN only at doses that exceeded those established for
general toxicity (about 45 mg/kg of body weight per day) (Smith et
al., 1987). A TDI of 23 µg/kg of body weight was calculated for DBAN
based on the NOAEL of 23 mg/kg of body weight per day in the 90-day
study in rats (Hayes et al., 1986) and incorporating an uncertainty
factor of 1000 (10 each for intra- and interspecies variation and 10
for the short duration of the study). There are no new data that
indicate that these TDIs should be changed.
LOAELs for TCAN were identified at 7.5 mg/kg of body weight per
day for embryotoxicity and 15 mg/kg of body weight per day for
developmental effects in rats (Smith et al., 1988). However, later
studies suggest that these responses were dependent upon the vehicle
used (Christ et al., 1996). No TDI can be established for TCAN.
There are no data useful for risk characterization purposes for
other members of the HANs.
6.1.1.8 MX
The mutagen MX has recently been studied in a long-term study in
rats in which some carcinogenic responses were observed (Komulainen et
al., 1997). These data indicate that MX induces thyroid and bile duct
tumours. An increased incidence of thyroid tumours was seen at the
lowest dose of MX administered (0.4 mg/kg of body weight per day). The
induction of thyroid tumours with high-dose chemicals has long been
associated with halogenated compounds. The induction of thyroid
follicular tumours could involve modifications in thyroid function or
mutagenic mode of action. A dose-related increase in the incidence of
cholangiomas and cholangiocarcinomas was also observed, beginning at
the low dose in female rats, with a more modest response in male rats.
The increase in cholangiomas and cholangiocarcinomas in female rats
was utilized to derive a slope factor for cancer. The 95% upper
confidence limit for a 10-5 lifetime risk based on the linearized
multistage model was calculated to be 0.06 µg/kg of body weight per
day.
6.1.1.9 Chlorite
The primary and most consistent finding arising from exposure to
chlorite is oxidative stress resulting in changes in the red blood
cells (Heffernan et al., 1979a; Harrington et al., 1995a). This
end-point is seen in laboratory animals and, by analogy with chlorate,
in humans exposed to high doses in poisoning incidents. There are
sufficient data available to estimate a TDI for humans exposed to
chlorite, including chronic toxicity studies and a two-generation
reproductive toxicity study. Studies in human volunteers for up to 12
weeks did not identify any effect on blood parameters at the highest
dose tested, 36 µg/kg of body weight per day (Lubbers & Bianchine,
1984; Lubbers et al., 1984a). Because these studies do not identify an
effect level, they are not informative for establishing a margin of
safety.
In a two-generation study in rats, a NOAEL of 2.9 mg/kg of body
weight per day was identified based on lower auditory startle
amplitude, decreased absolute brain weight in the F1 and F2
generations and altered liver weights in two generations (CMA, 1997).
Application of an uncertainty factor of 100 to this NOAEL (10 each for
inter- and intraspecies variation) gives a TDI of 30 µg/kg of body
weight. This TDI is supported by the human volunteer studies.
6.1.1.10 Chlorate
Like chlorite, the primary concern with chlorate is oxidative
damage to red blood cells. Also like chlorite, 0.036 mg/kg of body
weight per day of chlorate for 12 weeks did not result in any adverse
effect in human volunteers (Lubbers et al., 1981). Although the
database for chlorate is less extensive than that for chlorite, a
recent well conducted 90-day study in rats is available, which
identified a NOAEL of 30 mg/kg of body weight per day based on thyroid
gland colloid depletion at the next higher dose of 100 mg/kg of body
weight per day (McCauley et al., 1995). A TDI is not derived because a
long-term study is in progress, which should provide more information
on chronic exposure to chlorate.
6.1.1.11 Bromate
Bromate is an active oxidant in biological systems and has been
shown to cause an increase in renal tumours, peritoneal mesotheliomas
and thyroid follicular cell tumours in rats and, to a lesser extent,
hamsters, and only a small increase in kidney tumours in mice. The
lowest dose at which an increased incidence of renal tumours was
observed in rats was 6 mg/kg of body weight per day (DeAngelo et al.,
1998).
Bromate has also been shown to give positive results for
chromosomal aberrations in mammalian cells in vitro and in vivo
but not in bacterial assays for point mutation. An increasing body of
evidence, supported by the genotoxicity data, suggests that bromate
acts by generating oxygen radicals in the cell.
In the WHO Guidelines for drinking-water quality, the
linearized multistage model was applied to the incidence of renal
tumours in a 2-year carcinogenicity study in rats (Kurokowa et al.,
1986a), although it was noted that if the mechanism of tumour
induction is oxidative damage in the kidney, application of the
low-dose cancer model may not be appropriate. The calculated upper 95%
confidence interval for a 10-5 risk was 0.1 µg/kg of body weight per
day (WHO, 1993).
The no-effect level for the formation of renal cell tumours in
rats is 1.3 mg/kg of body weight per day (Kurokowa et al., 1986a). If
this is used as a point of departure from linearity and if an
uncertainty factor of 1000 (10 each for inter- and intraspecies
variation and 10 for possible carcinogenicity) is applied, a TDI of 1
µg/kg of body weight can be calculated. This compares with the value
of 0.1 µg/kg of body weight per day associated with an excess lifetime
cancer risk of 10-5.
At present, there are insufficient data to allow a decision as to
whether bromate-induced tumours are a result of cytotoxicity and
reparative hyperplasia or a genotoxic effect. IARC (1986, 1987) has
assigned potassium bromate to Group 2B: the agent is possibly
carcinogenic to humans.
6.1.2 Epidemiological studies
Epidemiological studies must be carefully evaluated to ensure
that observed associations are not due to bias and that the design is
appropriate for an assessment of a possible causal relationship.
Causality can be evaluated when there is sufficient evidence from
several well designed and well conducted studies in different
geographic areas. Supporting toxicological and pharmacological data
are also important. It is especially difficult to interpret
epidemiological data from ecological studies of disinfected
drinking-water, and these results are used primarily to help develop
hypotheses for further study.
Results of analytical epidemiological studies are insufficient to
support a causal relationship for any of the observed associations. It
is especially difficult to interpret the results of currently
published analytical studies because of incomplete information about
exposures to specific water contaminants that might confound or modify
the risk. Because inadequate attention has been paid to assessing
water contaminant exposures in epidemiological studies, it is not
possible to properly evaluate increased relative risks that were
reported. Risks may be due to other water contaminants or to other
factors for which chlorinated drinking-water or THMs may serve as a
surrogate.
6.2 Characterization of exposure
6.2.1 Occurrence of disinfectants and disinfectant by-products
Disinfectant doses of several milligrams per litre are typically
employed, corresponding to doses necessary to inactivate
microorganisms (primary disinfection) or to maintain a distribution
system residual (secondary disinfection). A necessary ingredient for
an exposure assessment is DBP occurrence data. Unfortunately, there
are few published international studies that go beyond case-study or
regional data.
Occurrence data suggest, on average, an exposure in chlorinated
drinking-water to total THMs of about 35-50 µg/litre, with chloroform
and BDCM being the first and second most dominant species. Exposure to
total HAAs can be approximated by a total HAA concentration (sum of
five species) corresponding to about one-half of the total THM
concentration (although this ratio can vary significantly); DCA and
TCA are the first and second most dominant species. In waters with a
high bromide to TOC ratio and/or a high bromide to chlorine ratio,
greater formation of brominated THMs and HAAs can be expected. When a
hypochlorite solution (versus chlorine gas) is used, chlorate may also
occur in the hypochlorite solution and be found in chlorinated water.
DBP exposure in chloraminated water is a function of the mode of
chloramination, with the sequence of chlorine followed by ammonia
leading to the formation of (lower levels of) chlorine DBPs (i.e.,
THMs and HAAs) during the free-chlorine period; however, the
suppression of chloroform and TCA formation is not paralleled by a
proportional reduction in DCA formation.
All factors being equal, bromide concentration and ozone dose are
the best predictors of bromate formation during ozonation, with about
a 50% conversion of bromide to bromate. A study of different European
water utilities showed bromate levels in water leaving operating water
treatment plants of less than the detection limit (2 µg/litre) up to
16 µg/litre. The brominated organic DBPs formed upon ozonation
generally occur at low levels. The formation of chlorite can be
estimated by a simple percentage (50-70%) of the applied chlorine
dioxide dose.
6.2.2 Uncertainties of water quality data
A toxicological study attempts to extrapolate a laboratory
(controlled) animal response to a potential human response; one
possible outcome is the estimation of cancer risk factors. An
epidemiological study attempts to link human health effects (e.g.,
cancer) to a causative agent or agents (e.g., a DBP) and requires an
exposure assessment.
The chemical risks associated with disinfected drinking-water are
potentially based on several routes of exposure: (i) ingestion of DBPs
in drinking-water; (ii) ingestion of chemical disinfectants in
drinking-water and the concomitant formation of DBPs in the stomach;
and (iii) inhalation of volatile DBPs during showering. Although the
in vivo formation of DBPs and the inhalation of volatile DBPs may be
of potential health concern, the following discussion is based on the
premise that the ingestion of DBPs present in drinking-water poses the
most significant chemical health risk.
Human exposure is a function of both DBP concentration and
exposure time. More specifically, human health effects are a function
of exposure to complex mixtures of DBPs (e.g., THMs versus HAAs,
chlorinated versus brominated species) that can change
seasonally/temporally (e.g., as a function of temperature and nature
and concentration of NOM) and spatially (i.e., throughout a
distribution system). Each individual chemical disinfectant can form a
mixture of DBPs; combinations of chemical disinfectants can form even
more complex mixtures. Upon their formation, most DBPs are stable, but
some may undergo transformation by, for example, hydrolysis. In the
absence of DBP data, surrogates such as chlorine dose (or chlorine
demand), TOC (or UVA254) or bromide can be used to indirectly estimate
exposure. While TOC serves as a good surrogate for organic DBP
precursors, UVA254 provides additional insight into NOM
characteristics, which can vary geographically. Two key water quality
variables, pH and bromide, have been identified as significantly
affecting the type and concentrations of DBPs that are produced.
An exposure assessment should first attempt to define the
individual types of DBPs and resultant mixtures likely to form as well
as their time-dependent concentrations, as affected by their stability
and transport through a distribution system. For epidemiological
studies, some historical databases exist for disinfectant (e.g.,
chlorine) doses, possibly DBP precursor (e.g., TOC) concentrations,
and possibly total THM (and in some cases, THM species)
concentrations. In contrast to THMs, which have been monitored over
longer time frames because of regulatory scrutiny, monitoring data for
HAAs (and HAA species), bromate and chlorite are much more recent and
hence sparse. However, DBP models can be used to simulate missing or
past data (e.g., concentrations of HAAs can be predicted using data on
THM concentrations). Another important consideration is documentation
of past changes in water treatment practice.
6.2.3 Uncertainties of epidemiological data
Even in well designed and well conducted analytical studies,
relatively poor exposure assessments were conducted. In most studies,
duration of exposure to disinfected drinking-water and the water
source were considered. These exposures were estimated from
residential histories and water utility or government records. In only
a few studies was an attempt made to estimate a study participant's
water consumption and exposure to either total THMs or individual THM
species. In only one study was an attempt made to estimate exposures
to other DBPs. In evaluating some potential risks, i.e., adverse
outcomes of pregnancy, that may be associated with relatively short
term exposures to volatile by-products, it may be important to
consider the inhalation as well as the ingestion route of exposure
from drinking-water. In some studies, an effort was made to estimate
both by-product levels in drinking-water for etiologically relevant
time periods and cumulative exposures. Appropriate models and
sensitivity analysis such as Monte Carlo simulation can be used to
help estimate these exposures for relevant periods.
A major uncertainty surrounds the interpretation of the observed
associations, as exposures to a relatively few water contaminants have
been considered. With the current data, it is difficult to evaluate
how unmeasured DBPs or other water contaminants may have affected the
observed relative risk estimates.
More studies have considered bladder cancer than any other
cancer. The authors of the most recently reported results for bladder
cancer risks caution against a simple interpretation of the observed
associations. The epidemiological evidence for an increased relative
risk of bladder cancer is not consistent -- different risks are
reported for smokers and non-smokers, for men and women, and for high
and low water consumption. Risks may differ among various geographic
areas because the DBP mix may be different or other water contaminants
are also present. More comprehensive water quality data must be
collected or simulated to improve exposure assessments for
epidemiological studies.
7. RISK CONCLUSIONS AND COMPARISONS
Chlorination of drinking-water has been a cornerstone of efforts
to prevent the spread of waterborne disease for almost a century
(Craun et al., 1993). It is important to retain chlorination as an
inexpensive and efficacious process unless a clear public health
concern arises to eliminate it. It is uncertain that alternative
chemical disinfectants reduce these estimated risks significantly
(Bull & Kopfler, 1991).
Identifying the safest way of producing drinking-water requires
more conclusive toxicological or epidemiological evidence than is
available today. It is important to recognize that there is a sizeable
set of data already present on this issue and that resolution of this
problem will not simply come from an expansion of that database. The
focus must be elevated from questions of individual by-products and
routine toxicological testing to a much more systematic approach
towards the resolution of these larger issues.
7.1 Epidemiological studies
The epidemiological associations between chlorinated
drinking-water and human cancer have been subjected to several recent
reviews, and the conclusions remain controversial. The small to medium
relative risks for all the tumour sites studied (relative risks or
odds ratios almost always less than 2) and uncertainty related to the
magnitude and type of human exposures make it difficult to conclude
that real risks result from the ingestion of chlorinated
drinking-water.
7.2 Toxicological studies
Toxicological studies are best suited for developing information
on individual by-products or known combinations of by-products. The
deficiencies in the present toxicological database are outlined below.
7.2.1 Diversity of by-products
Significant qualitative and quantitative differences in the
toxicological properties of DBPs have been demonstrated, depending
upon whether they have some bromine substitution. Among the THMs, BDCM
is of particular interest because it produces tumours in both rats and
mice at several sites (NTP, 1987). Moreover, its potency calculated
under the assumptions of the linearized multistage model is an order
of magnitude greater than that of chloroform (Bull & Kopfler, 1991).
DBCM produced liver tumours only in mice (NTP, 1985), but bromoform
produced colon tumours in rats (NTP, 1989a). The fact that both BDCM
and bromoform given in corn oil vehicle induce colon cancer in animals
is of interest because of the epidemiological associations seen with
colo-rectal tumours and consumption of chlorinated water.
As with the THMs, however, a full complement of brominated and
mixed bromochlorinated acetates are produced with the chlorination of
drinking-water. These compounds have received little attention. While
the chlorinated HAAs appear to be without significant genotoxic
activity, the brominated HAAs appear to induce oxidative damage to
DNA. Increases in the 8-OH-dG levels in hepatic DNA were observed with
both acutely administered oral doses (Austin et al., 1996) and more
prolonged exposures in drinking-water (Parrish et al., 1996). This
activity increased with the degree of bromine substitution. Therefore,
it cannot be concluded that the brominated HAAs are the mechanistic
equivalents of the chlorinated HAAs.
Association of mutagenic activity with the chlorination of
drinking-water was first observed by Cheh et al. (1980). While some of
the major DBPs are mutagenic, they are much too weak as mutagens to
account for this activity. By far the largest individual contributor
to this activity is the compound referred to as MX. This compound has
been variously reported to account for up to 57% of the mutagenic
activity produced in the chlorination of drinking-water (Meier et al.,
1985a,b; Hemming et al., 1986; Kronberg & Vartiainen, 1988). MX has
recently been shown to be a carcinogen in rats (Komulainen et al.,
1997). As with other classes of DBPs, brominated analogues and
structurally related compounds that could be of importance are
produced in the chlorination of drinking-water (Daniel et al., 1991b;
Suzuki & Nakanishi, 1995).
7.2.2 Diversity of modes of action
It is important to recognize that the ways in which DBPs induce
cancer are quite different. All of the modern work that has come
forward on chloroform (Larson et al., 1994a,b, 1996) would strongly
undermine the hypothesis that chloroform is contributing to the
cancers observed in epidemiological studies. These toxicological
results make a convincing case that tumorigenic responses in both the
mouse liver and rat kidney are dependent upon necrosis and reparative
hyperplasia. There is no basis for associating this type of target
organ damage with the consumption of chlorinated drinking-water. On
the other hand, brominated THMs are mutagenic (Zeiger, 1990; Pegram et
al., 1997). It is generally assumed that mutagenic carcinogens will
produce linear dose-response relationships at low doses, as
mutagenesis is generally considered to be an irreversible and
cumulative effect.
In the HAA class, significant differences in mode of action have
been demonstrated for DCA and TCA. Despite the close structural
resemblance of DCA and TCA and their common target organ (liver cancer
induction), it is becoming clear that the mechanisms by which they act
are different. TCA is a peroxisome proliferator, and the tumour
phenotype and genotype seen in mice are consistent with this being the
mode of action by which it acts. However, DCA clearly produced tumours
at doses below those that are required for peroxisome proliferation
(DeAngelo et al., 1989, 1996; Daniel et al., 1992a; Richmond et al.,
1995). The tumour phenotypes that DCA and TCA produce in mice are very
different (Pereira, 1996; Pereira & Phelps, 1996; Stauber & Bull,
1997). From a risk assessment standpoint, however, one would question
whether either DCA or TCA, alone, is likely to present significant
cancer risk to humans at the low levels found in drinking-water.
Neither compound appears to influence the carcinogenic process by a
mutagenic mechanism (Stauber & Bull, 1997; Harrington-Brock et al.,
1998; Stauber et al., 1998). Although different mechanisms appear to
be involved, the mode of action for both compounds appears to be
tumour promotion. In experiments of short duration, they tend to
increase replication rates of normal hepatocytes; with more extended
exposures or very high doses, however, they tend to depress
replication rates (Carter et al., 1995; Stauber & Bull, 1997). There
is evidence to suggest that the depression of cell replication is
paralleled by depressed rates of apoptosis (Snyder et al., 1995).
It is noteworthy that there is little support in the animal data
for certain target organs that are prominently associated with
chlorinated drinking-water in epidemiological studies (e.g., bladder
cancer). Therefore, the possibility has to be left open that the
carcinogenic effect of DBPs may be dependent on genetically determined
characteristics of a target organ (or tissue) that make it more
susceptible than the same organ in test animals. This problem can be
resolved only by conducting toxicological studies in the appropriate
human tissues and by developing much stronger epidemiological
associations to guide these studies.
The epidemiological studies can contribute to the resolution of
the problem by (i) better identifying the drinking-water conditions
that are associated with bladder or colo-rectal cancer, (ii) focusing
on those characteristics of susceptibility that may increase the
sensitivity at these target sites, and (iii) determining if
interactions between biomarkers of susceptibility at these sites
contribute to the epidemiological associations with disinfection of
drinking-water. These "tasks" need to be accomplished sequentially. If
epidemiological studies can provide insights into the first two tasks,
then the experimental scientists can work much more profitably with
epidemiologists to address the third task.
7.2.3 Reproductive, developmental and neurotoxic effects
Much of this review has focused on questions related to chemical
carcinogenesis, in part because that is where the bulk of the
experimental data are found. There are other toxicological effects
associated with some DBPs that could be of importance. Recently
published epidemiological data (Waller et al., 1998) suggest the
possibility that increased spontaneous abortion rates may be related
to DBPs in drinking-water.
Reproductive effects in females have been principally
embryolethality and fetal resorptions associated with the HANs (Smith
et al., 1988, 1989b). The dihaloacetates, DCA and DBA, have both been
associated with effects on male reproduction, marked primarily by
degeneration of the testicular epithelium (Toth et al., 1992; Linder
et al., 1994a,b). Some effects on reproductive performance are noted
at doses of DBA as low as 10 mg/kg of body weight per day. Dogs
display testicular degeneration when administered doses of DCA of this
same magnitude (Cicmanec et al., 1991).
7.3 Risks associated with mixtures of disinfectant by-products
Disinfected drinking-water is a very complex mixture of
chemicals, most of which have not been identified. Studies on
individual DBPs may not represent the risk posed by the mixture.
Research on complex mixtures was recently reviewed by ILSI (1998).
Studies of simple combinations of chemicals provided positive results,
but only at concentrations so much greater than those that occur in
drinking-water as to be irrelevant. Studies utilizing complex mixtures
of chemicals as they could be isolated from water or produced by
chlorinating high concentrations of humic or fulvic acids produce
little convincing evidence of adverse effect. A variety of
methodological issues prevent our being too comfortable with that
conclusion (ILSI, 1998). Moreover, the effort never developed to the
point that the diverse qualities of water in various parts of the
country could be taken into account.
To be efficient, toxicological research needs to have a focus,
and the research performed to solve this problem must be
hypothesis-driven. This means that hypotheses of interactions would be
based on knowledge of the toxic properties of individual by-products
and would be subjected to experimental test. This can be much more
efficient than designing complex multifactorial studies of all
possible combinations of by-products produced by a disinfectant. An
additional nuance would be to develop hypotheses as explicit tests of
epidemiological findings. This would ensure that resources are
appropriately directed and would provide a research agenda that would
progress in a predictive way. At some stage, a hypothesis loses
credibility or becomes recognized as being as close to "truth" as can
be achieved experimentally.
Similarly, epidemiological studies must begin to focus on what is
known about the toxicology of individual DBPs. Testing of hypotheses
about certain adverse health effects should begin with some
understanding of which DBPs are known to produce an effect of interest
in experimental systems. Epidemiologists then need to focus on those
mechanisms of toxicity and interactions that are likely to be
important at low doses (i.e., those that can be logically extrapolated
to dose levels encountered from drinking disinfected drinking-water).
Finally, information of this type should be used to develop new
parameters that can be incorporated into the design of epidemiological
studies.
8. CONCLUSIONS AND RECOMMENDATIONS
Disinfection is unquestionably the most important step in the
treatment of water for drinking-water supplies. The microbial quality
of drinking-water should not be compromised because of concern over
the potential long-term effects of disinfectants and DBPs. The risk of
illness and death resulting from exposure to pathogens in
drinking-water is very much greater than the risks from disinfectants
and DBPs. Where local circumstances require that a choice be made
between microbiological limits or limits for disinfectants and DBPs,
the microbiological quality must always take precedence. Efficient
disinfection must never be compromised.
The microbiological quality of drinking-water is of paramount
importance and must receive priority over any other considerations in
relation to drinking-water treatment. However, the use of any chemical
disinfectant results in the formation of by-products that themselves
may be of health significance. A thorough understanding of how these
DBPs form and the factors that control their formation is valuable in
achieving a successful balance between satisfactory inactivation of
pathogens and the minimization of DBP formation. The microbiological
quality of drinking-water should always receive priority over the
minimization of DBPs.
Where it is possible, without compromising the microbiological
quality of drinking-water, steps should be taken to minimize the
concentrations of DBPs produced by the disinfectant(s) in use.
Strategies to minimize exposure to DBPs should focus on the
elimination of precursors through source water protection. Not only is
this often the most efficient method of reducing DBP concentrations,
but it will also assist in improving the microbiological quality of
the water. Where treatment is required, DBP control strategies should
emphasize DBP organic precursor (TOC) removal.
8.1 Chemistry
Chlorine and alternative chemical disinfectants (ozone, chlorine
dioxide and chloramine) all lead to the formation of DBPs. However,
between disinfectants or combinations thereof, there are differences
in DBP groups, species and mixtures that may affect human health. Key
water quality determinants of DBPs include TOC, bromide and pH. Based
on the current knowledge of both occurrence and health effects, the
DBPs of most concern include total THMs and THM species, total HAAs
and HAA species, bromate and chlorite.
8.2 Toxicology
None of the chlorination by-products studied to date is a potent
carcinogen at concentrations normally found in drinking-water.
The toxicology of the DBPs suggests that the likelihood of
adverse effects is not significantly different between the described
disinfectant options.
Toxicological information on mode and mechanism of action of
disinfectants and their by-products is the major limitation for
understanding the potential health risks at low doses.
8.3 Epidemiology
Epidemiological studies have not identified an increased risk of
cardiovascular disease associated with chlorinated or chloraminated
drinking-water.
The hypothesis of a causal relationship between consumption of
chlorination by-products and the increased relative risk of any cancer
remains an open question. There is insufficient epidemiological
evidence to support a causal relationship between bladder cancer and
exposures to chlorinated drinking-water, THMs, chloroform or other THM
species. The epidemiological evidence is inconclusive and equivocal
for an association between colon cancer and long duration of exposure
to chlorinated drinking-water, THMs or chloroform. There is
insufficient epidemiological information to properly interpret the
observed risks for rectal cancer and the risks for other cancers
observed in single analytical studies.
The results of currently published studies do not provide
convincing evidence that chlorinated water or THMs cause adverse
pregnancy outcomes.
9. RESEARCH NEEDS
9.1 Chemistry of disinfectants and disinfectant by-products
There is a need to consider the requirements of developing and
developed countries. While future research needs are articulated
below, technology transfer is important in implementing past research
into practice. The disinfection practice common to all countries is
chlorination; thus, chlorine DBPs should be the primary focus.
* There is a need for a study to develop more DBP occurrence data
from an international perspective; such an effort should also
compile information on DBP precursors (TOC and bromide).
* Because of the expertise required to measure certain DBPs (e.g.,
HAAs) and the deficiencies of historical databases, there is a need
to develop improved models for predicting DBP formation and
precursor removal, allowing the use of DBPs (e.g., chloroform) or
surrogates (e.g., TOC) that are more simple to measure. These
models can also be used to predict the formation of DBPs from the
use of a particular disinfectant and the factors that control the
appearance and formation of these DBPs, thus allowing appropriate
control strategies to be developed and better assessment of
exposure.
* Although HAAs have been monitored for several years, this
monitoring effort has been based on measurements of the sum of five
or six species. There is a need to develop more information on the
occurrence of the nine species of HAAs.
* For analytical reasons, non-polar organic by-products (measurable
by GC) and ionic by-products (measurable by IC) have received more
attention. There is a need to develop analytical methods for polar
by-products and to define their occurrence.
* A significant percentage of TOX remains unaccounted for by specific
halogenated DBPs; there is a need to identify these compounds.
* Given the health effects data on bromate and the anticipated higher
ozone doses that will be required to inactivate Cryptosporidium,
more data are needed on bromate formation in low-bromide waters
when high ozone doses are used.
* More information is needed on the composition of NOM to assist in
determining the type and extent of DBP formation that can be
expected in a given water and how well treatment processes will
achieve precursor removal.
* There is a need to develop a better understanding of how water
quality parameters affect the extent of bromine incorporation into
DBPs.
* To assist small water supply systems in minimizing DBP formation,
there is a need to develop simple, easily operated treatment
systems for the removal of NOM from source waters.
9.2 Toxicology
* Toxicological research needs to be focused to be effective. One
approach to accomplish this is to design experiments to determine
if a biological basis can be established for the epidemiological
findings.
* Further toxicological characterization of the effects of DBPs at
low doses is needed. These studies should be directed at
understanding the mechanisms that are operative at these doses in
humans.
* There are at least three areas that need the attention of both
toxicologists and epidemiologists: risks posed by brominated
by-products, how risks are modified by pH, and risks posed by the
use of a disinfectant as an oxidizing agent during drinking-water
treatment.
* A relatively small fraction of DBPs has received substantive
toxicological study. Future studies should be directed at those
by-products that occur with high frequency and at relatively high
concentrations.
* Little attention has been paid to those individuals in a population
who possess sensitivities to particular chemicals and/or modes of
action because of genetic and lifestyle factors. An example is
major polymorphic differences in enzymes that metabolize DBPs.
* Humans are exposed to complex mixtures of disinfectants and DBPs.
It is becoming apparent that chemicals with like mechanisms
interact in an additive way at low concentrations. Little
information exists on the potential non-additive interaction of
chemicals with different mechanisms.
9.3 Epidemiology
* Additional studies to evaluate cancer risks should be analytical
and should include a more comprehensive assessment of
drinking-water exposures, especially for DBPs, for etiologically
relevant time periods. The interpretation of the results from
currently conducted studies of both cancer and adverse pregnancy
outcomes suffers from lack of knowledge about exposures to other
DBPs and other water contaminants. It may be possible to assess
additional water exposures for study participants in several
recently conducted studies of cancer and reproductive risks. If
this can be done, a better estimate of exposure to water
contaminants and DBPs will be available for additional analyses of
risks in these study populations. Depending on the results of these
reanalyses, the need for additional studies of possible cancer
risks can be better evaluated.
* It is important to improve the quality of future epidemiological
studies with adequate and appropriate exposure information. This
underscores the need to include in the planning and conduct of
epidemiological studies individuals who are knowledgeable about DBP
chemistry.
* Additional studies are needed to better assess the importance of
the observed association between DBPs and early miscarriage or
neural tube defects. Research should continue on possible
reproductive and developmental effects associated with
drinking-water disinfection.
* The most important research need is to improve the assessment of
drinking-water exposures for epidemiological studies. There is a
need for improved models that can be used to estimate exposures to
various specific by-products and mixtures of by-products. There is
also a need to collect more complete information about individual
water consumption and activity patterns that may influence exposure
assessments. Better and more complete exposure information will
improve the sensitivity of epidemiological studies.
10. PREVIOUS EVALUATIONS BY INTERNATIONAL BODIES
A number of disinfectants and DBPs have been considered by IARC. On
the basis of the available published data, the most recent
classification of these chemicals is as follows:
Disinfectants
Hypochlorite salts: Group 3 (1991)
Disinfectant by-products
Trihalomethanes
Bromodichloromethane: Group 2B (1999)
Dibromochloromethane: Group 3 (1999)
Bromoform: Group 3 (1999)
Haloacetic acids
Dichloroacetic acid: Group 3 (1995)
Trichloroacetic acid: Group 3 (1995)
Haloacetaldehyde
Chloral and chloral hydrate: Group 3 (1995)
Haloacetonitriles
Bromochloroacetonitrile: Group 3 (1999)
Chloroacetonitrile: Group 3 (1999)
Dibromoacetonitrile: Group 3 (1999)
Dichloroacetonitrile: Group 3 (1999)
Trichloroacetonitrile: Group 3 (1999)
Other disinfectant by-products
Potassium bromate: Group 2B (1987)
Sodium chlorite: Group 3 (1991)
In addition, IARC has classified chlorinated drinking-water in
Group 3 (1991).
Disinfectants and DBPs were evaluated in the Guidelines for
drinking-water quality, and the following guideline values
recommended (WHO, 1993, 1996, 1998):
Disinfectants
Chlorine (hypochlorous acid and hypochlorite): 5 mg/litre (1993)
Monochloramine: 3 mg/litre (1993)
Disinfectant by-products
Trihalomethanes
Bromodichloromethane: 60 µg/litre for an excess lifetime
cancer risk of 10-5 (1993)
Dibromochloromethane: 100 µg/litre (1993)
Bromoform: 100 µg/litre (1993)
Chloroform: 200 µg/litre (1998)
Haloacetic acids
Dichloroacetic acid: 50 µg/litre (provisional) (1993)
Trichloroacetic acid: 100 µg/litre (provisional) (1993)
Haloacetaldehyde
Chloral hydrate: 10 µg/litre (provisional) (1993)
Haloacetonitriles
Dibromoacetonitrile: 100 µg/litre (provisional) (1993)
Dichloroacetonitrile: 90 µg/litre (provisional) (1993)
Trichloroacetonitrile: 1 µg/litre (provisional) (1993)
Other disinfectant by-products
Bromate: 25 µg/litre (provisional) for an excess lifetime
cancer risk of 7 × 10-5 (1993)
Chlorite: 200 µg/litre (provisional) (1993)
Cyanogen chloride (as cyanide): 70 µg/litre (1993)
Formaldehyde: 900 µg/litre (1993)
2,4,6-Trichlorophenol: 200 µg/litre for an excess lifetime
cancer risk of 10-5 (1993)
JECFA (FAO/WHO, 1993) evaluated potassium bromate and concluded
that it was genotoxic and carcinogenic.
The International Programme on Chemical Safety (IPCS) published
an evaluation of chloroform in its Environmental Health Criteria
Monograph series (WHO, 1994).
The IPCS Concise International Chemical Assessment Document
(CICAD) on chloral hydrate (in press) considers the dose level of 16
mg/kg in the study of Sanderson et al. (1982) to be a NOAEL rather
than a LOAEL (as in the present document), and therefore uses studies
in humans as the basis of a tolerable intake. On the basis of a LOAEL
of 11 mg/kg, and using an uncertainty factor of 10 for intraspecies
variation and 10 for conversion of LOAEL to NOAEL, the CICAD derived a
tolerable intake of 0.1 mg/kg.
REFERENCES
Abbas R & Fisher JW (1997) A physiologically-based pharmacokinetic
model for trichloroethylene and its metabolites, chloral hydrate,
trichloroacetate, dichloroacetate, trichloroethanol and
trichloroethanol glucuronide in B6C3F1 mice. Toxicol Appl Pharmacol,
147: 15-30.
Abbas RR, Seckel CS, Kidney JK, & Fisher JW (1996) Pharmacokinetic
analysis of chloral hydrate and its metabolism in B6C3F1 mice. Drug
Metab Dispos, 24: 1340-1346.
Abdel-Rahman MS, Couri D, & Bull RJ (1980) Kinetics of ClO2 and
effects of ClO2, ClO2-, and ClO3- in drinking water on blood
glutathione and hemolysis in rat and chicken. J Environ Pathol
Toxicol, 3: 431-449.
Abdel-Rahman MS, Berardi MR, & Bull RJ (1982a) Effect of chlorine and
monochloramine in drinking water on the developing rat fetus. J Appl
Toxicol, 2(3): 156.
Abdel-Rahman MS, Couri D, & Bull RJ (1982b) Metabolism and
pharmacokinetics of alternate drinking water disinfectants. Environ
Health Perspect, 46: 19-23.
Abdel-Rahman MS, Waldron DM, & Bull RJ (1983) A comparative kinetics
study of monochloramine and hypochlorous acid in rat. J Appl Toxicol,
3: 175-179.
Abdel-Rahman MS, Couri D, & Bull RJ (1984a) Toxicity of chlorine
dioxide in drinking water. J Am Coll Toxicol, 3: 277-284.
Abdel-Rahman MS, Couri D, & Bull RJ (1984b) The kinetics of chlorite
and chlorate in the rat. J Am Coll Toxicol, 3: 261-267.
Acharya S, Mehta K, Rodrigues S, Pereira J, Krishnan S, & Rao CV
(1995) Administration of subtoxic doses of t-butyl alcohol and
trichloroacetic acid to male Wistar rats to study the interactive
toxicity. Toxicol Lett, 80: 97-104.
Ade P, Guastadisegni C, Testai E, & Vittozzi L (1994) Multiple
activation of chloroform in kidney microsomes from male and female
DBA/2J mice. J Biochem Toxicol, 9: 289-295.
Adler ID (1993) Synopsis of the in vivo results obtained with the 10
known or suspected aneugens tested in the CEC collaborative study.
Mutat Res, 287(1): 131-137.
Agarwal AK & Mehendele HM (1983) Absence of potentiation of bromoform
hepatotoxicity and lethality by chlordecone. Toxicol Lett,
15: 251-257.
Ahmed AE, Soliman SA, Loh JP, & Hussein GI (1989) Studies on the
mechanism of haloacetonitriles toxicity: Inhibition of rat hepatic
glutathione- S-transferases in vitro. Toxicol Appl Pharmacol, 100:
271-279.
Ahmed AE, Jacob S, & Loh JP (1991) Studies on the mechanism of
haloacetonitriles toxicity: quantitative whole body autoradiographic
distribution of [2-14C]chloroacetonitrile in rats. Toxicology, 67:
279-302.
Aida Y, Takada K, Uchida O, Yasuhara K, Kurokawa Y, & Tobe M (1992a)
Toxicities of microencapsulated tribromomethane, dibromochloromethane
and bromodichloromethane administered in the diet to Wistar rats for
one month. J Toxicol Sci, 17: 119-133.
Aida Y, Yasuhara K, Takada K, Kurokawa Y, & Tobe M (1992b) Chronic
toxicity of microencapsulated bromodichloromethane administered in the
diet to Wistar rats. J Toxicol Sci, 17: 51-68, [Erratum] 17:167.
Aieta EM & Berg JD (1986) A review of chlorine dioxide in drinking
water treatment. J Am Water Works Assoc, 78(6): 62-72.
Alavanja M, Goldstein I, & Susser M (1978) Case-control study of
gastrointestinal and urinary tract cancer mortality and drinking water
chlorination. In: Jolley RL, Gorchev H, & Hamilton DH ed. Water
chlorination: Environmental impact and health effects. Ann Arbor,
Michigan, Ann Arbor Science Publishers, vol 2, pp 395-409.
Allen JW, Collins BW, & Evansky PA (1994) Spermatid micronucleus
analyses of trichloroethylene and chloral hydrate effects in mice.
Mutat Res, 323(1-2): 81-88.
Almeyda J & Levantine A (1972) Cutaneous reactions to barbiturates,
chloral hydrate and its derivatives. Br J Dermatol, 86: 313-316.
Ames RG & Stratton JW (1987) Effect of chlorine dioxide water
disinfection on hematologic and serum parameters of renal dialysis.
Arch Environ Health, 42(5): 280-285.
Ammann P, Laethem CL, & Kedderis GL (1998) Chloroform-induced
cytolethality in freshly isolated male B6C3F1 mouse and F344 rat
hepatocytes. Toxicol Appl Pharmacol, 149(2): 217-225.
Amy G, Chadik PA, & Chowdhury ZK (1987) Developing models for
predicting THM formation potential and kinetics. J Am Water Works
Assoc, 79(7): 89-96.
Amy G, Siddiqui M, Ozekin K, & Westerhoff P (1993) Threshold levels
for bromate ion formation in drinking water. In: Proceedings of the
International Water Supply Association Workshop, Paris. Boston,
Massachusetts, Blackwell Scientific Publications, pp 169-180.
Amy G, Siddiqui M, Zhai W, & Debroux J (1994) Survey on bromide in
drinking water and impacts on DBP formation. Denver, Colorado,
American Water Works Association (Report No. 90662).
Amy G, Siddiqui M, Ozekin K, Zhu HW, & Wang C (1998) Empirically-based
models for predicting chlorination and ozonation by-products:
Trihalomethanes, haloacetic acids, chloral hydrate, and bromate.
Cincinnati, Ohio, US Environmental Protection Agency
(EPA-815-R-98-005).
Anders MW, Stevens JL, Sprague RW, Shaath Z, & Ahmed AE (1978)
Metabolism of haloforms to carbon monoxide. II. In vivo studies.
Drug Metab Dispos, 6: 556-560.
Andrews SA & Ferguson MJ (1995) Minimizing DBP formation while
ensuring Giardia control. In: Minear RA & Amy GL ed. Disinfection
by-products in water treatment. Chelsea, Michigan, Lewis Publishers,
Inc.
Andrews SA, Huch PM, Chute AJ, Bolton JR, & Anderson WA (1996) UV
oxidation for drinking water-feasibility studies for addressing
specific water quality issues. In: Proceedings of the Water Quality
Technology Conference, New Orleans, LA. Denver, Colorado, American
Water Works Association.
Angel P & Karin M (1991) The role of Jun, Fos, and the AP-1 complex in
cell proliferation and transformation. Biochim Biophys Acta, 1072:
126-157.
Anna CH, Maronpot RR, Pereira MA, Foley JF, Malarkey DE, & Anderson MW
(1994) Ras proto-oncogene activation in dichloroacetic-,
trichloroethylene- and tetrachloroethylene-induced liver tumors in
B6C3F1 mice. Carcinogenesis, 15: 2255-2261.
APHA (American Public Health Association) (1995) Standard methods for
the examination of water and wastewater, 19th ed. Washington, DC,
American Public Health Association/American Water Works
Association/Water Pollution Control Federation.
Aschengrau A, Zierler S, & Cohen A (1989) Quality of community
drinking water and the occurrence of spontaneous abortions. Arch
Environ Health, 44(5): 283-290.
Aschengrau A, Zierler S, & Cohen A (1993) Quality of community
drinking water and the occurrence of late adverse pregnancy outcomes.
Arch Environ Health, 48(2): 105-114.
Ashby J, Mohammed R, & Callander RD (1987) N-Chloropiperidine and
calcium hypochlorite: Possible examples of toxicity-dependent
clastogenicity, in vitro. Mutat Res, 189: 59-68.
Austin EW & Bull RJ (1997) The effect of pretreatment with
dichloroacetate and trichloroacetate on the metabolism of
bromodichloroacetate. J Toxicol Environ Health, 52: 367-383.
Austin EW, Okita JR, Okita RT, Larson JL, & Bull RJ (1995)
Modification of lipoperoxidative effects of dichloroacetate and
trichloroacetate is associated with peroxisome proliferation.
Toxicology, 97: 59-69.
Austin EW, Parrish JM, Kinder DH, & Bull RJ (1996) Lipid peroxidation
and formation of 8-hydroxydeoxyguanosine from acute doses of
halogenated acetic acids. Fundam Appl Toxicol, 31: 77-82.
AWWARF (1991) Disinfection by-products database and model project.
Denver, Colorado, American Water Works Association Research
Foundation.
Bailar JC (1995) The practice of meta-analysis. J Clin Epidemiol,
48(1): 149-157.
Bailey PS (1978) Ozonation in organic chemistry. New York, London,
Academic Press.
Balster RL & Borzelleca JF (1982) Behavioral toxicity of
trihalomethane contaminants of drinking water in mice. Environ Health
Perspect, 46: 127-136.
Banerji AP & Fernandes AO (1996) Field bean protease inhibitor
mitigates the sister-chromatid exchanges induced by bromoform and
depresses the spontaneous sister-chromatid exchange frequency of human
lymphocytes in vitro. Mutat Res, 360: 29-35.
Bean JA, Isacson P, Hausler WJ, & Kohler J (1982) Drinking water and
cancer incidence in Iowa I. Trends and incidence by source of drinking
water and size of municipality. Am J Epidemiol, 116(6): 912-923.
Bempong MA & Scully FE Jr (1985) Mutagenicity and clastogenicity of
N-chloropiperidine. J Environ Pathol Toxicol, 6: 241-251.
Bempong MA, Montgomery C, & Scully FE Jr (1980) Mutagenic activity of
N-chloropiperidine. J Environ Pathol Toxicol, 4: 345-354.
Bercz JP & Bawa R (1986) Iodination of nutrients in the presence of
chlorine based disinfectants used in drinking water treatment. Toxicol
Lett, 34(2-3): 141-147.
Bercz JP, Jones L, Garner L, Murray D, Ludwig A, & Boston J (1982)
Subchronic toxicity of chlorine dioxide and related compounds in
drinking water in the nonhuman primate. Environ Health Perspect,
46: 47-55.
Bercz JP, Jones LL, Harrington RM, Bawa R, & Condie L (1986)
Mechanistic aspects of ingested chlorine dioxide on thyroid function:
Impact of oxidants on iodide metabolism. Environ Health Perspect,
69: 249-255.
Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K, Khorrami,
P, Henderson GN, De Marco T, & Chatterjee K (1994) Improved
hemodynamic function and mechanical efficiency in congestive heart
failure with sodium dichloroacetate. J Am Coll Cardiol, 23: 1617-1624.
Bhat HK, Kanz MF, Campbell GA, & Ansari GAS (1991) Ninety day toxicity
study of chloroacetic acids in rats. Fundam Appl Toxicol, 17: 240-253.
Bhunya SP & Behera BC (1987) Relative genotoxicity of trichloroacetic
acid (TCA) as revealed by different cytogenetic assays: Bone marrow
chromosome aberration, micronucleus and sperm-head abnormality in the
mouse. Mutat Res, 188: 215-221.
Bignami M, Conti G, Conti L, Crebelli R, Misuraca F, Puglia AM,
Randazzo R, Sciandrello G, & Carere A (1980) Mutagenicity of
halogenated aliphatic hydrocarbons in Salmonella typhimurium,
Streptomyces coelicolor and Aspergillus nidulans. Chem-Biol
Interact, 30: 9-23.
Blackshear PJ, Holloway PA, & Alberti KM (1974) The metabolic effects
of sodium dichloroacetate in the starved rat. Biochem J, 142: 279-286.
Blazak WF, Meier JR, Stewart BE, Blachman DC, & Deahl JT (1988)
Activity of 1,1,1- and 1,1,3-trichloroacetones in a chromosomal
aberration assay in CHO cells and the micronucleus and spermhead
abnormality assays in mice. Mutat Res, 206: 431-438.
Bloxham CA, Wright N, & Hoult JG (1979) Self-poisoning by sodium
chlorate: Some unusual features. Clin Toxicol, 15: 185-188.
Bolyard M & Fair PS (1992) Occurrence of chlorate in hypochlorite
solutions used for drinking water treatment. Environ Sci Technol,
26(8): 1663-1665.
Bolyard M, Fair PS, & Hautman DP (1993) Sources of chlorate ion in
U.S. drinking water. J Am Water Works Assoc, 85: 81-88.
Borzelleca JF & Carchman RA (1982) Effects of selected organic
drinking water contaminants on male reproduction. Research Triangle
Park, North Carolina, US Environmental Protection Agency (EPA
600/1-82-009; NTIS PB82-259847).
Bove FJ, Fulcomer MC, Koltz JB, Esmart J, Dufficy EM, & Zagraniski RT
(1992a) Report on phase IV-A: public drinking water contamination and
birthweight fetal deaths, and birth defects, a cross-sectional study.
Trenton, New Jersey, New Jersey Department of Health.
Bove FJ, Fulcomer MC, Koltz JB, Esmart J, Dufficy EM, Zagraniski RT, &
Savrin JE (1992b) Report on phase IV-B: public drinking water
contamination and birthweight and selected birth defects, a
case-control study. Trenton, New Jersey, New Jersey Department of
Health.
Bove FJ, Fulcomer MC, Klotz JB, Esmart J, Dufficy EM, & Savrin JE
(1995) Public drinking water contamination and birth outcomes. Am J
Epidemiol, 141(9): 850-862.
Bowman FJ, Borzelleca JF, & Munson AE (1978) The toxicity of some
halomethanes in mice. Toxicol Appl Pharmacol, 44: 213-215.
Brennan RJ & Schiestl RH (1998) Chloroform and carbon tetrachloride
induce intra-chromosomal recombination and oxidative free radicals in
Saccharomyces cerevisiae. Mutat Res, 397(2): 271-278.
Brenniman GR, Vasilomanolakis-Lagos J, Amsel J, Namekata T, & Wolff AH
(1980) Case-control study of cancer deaths in Illinois communities
served by chlorinated or nonchlorinated water. In: Jolley RL, Brungs
WA, Cumming RB, & Jacobs VA ed. Water chlorination: Environmental
impact and health effects. Ann Arbor, Michigan, Ann Arbor Science
Publishers, Inc., vol 3, pp 1/043-1/057.
Bronzetti G, Galli A, Corsi C, Cundari E, Del Carratore R, Nieri R, &
Paolini M (1984) Genetic and biochemical investigation on chloral
hydrate in vitro and in vivo. Mutat Res, 141: 19-22.
Brown DM, Langley PF, Smith D, & Taylor DC (1974) Metabolism of
chloroform - 1. The metabolism of [14C]chloroform by different
species. Xenobiotica, 4: 151-163.
Brown-Woodman PD, Hayes LC, Huq F, Herlihy C, Picker K, & Webster WS
(1998) In vitro assessment of the effect of halogenated
hydrocarbons: Chloroform, dichloromethane, and dibromoethane on
embryonic development of the rat. Teratology, 57(6): 321-333.
Bruchet A, Costentin E, Legrand MF, & Malleviale J (1992) Influence of
the chlorination of natural nitrogenous organic compounds on tastes
and odours in finished drinking waters. Water Sci Technol,
25(2): 323-333.
Brunborg G, Holme JA, Soderlund EJ, & Dybing E (1990) Organ-specific
genotoxic effects of chemicals: The use of alkaline elution to detect
DNA damage in various organs of in vivo exposed animals. Prog Clin
Biol RES, 340D: 43-52.
Brunborg G, Holme JA, Soderlund EJ, Hongslo JK, Vartiainen T, Lotjonen
S, & Becher G (1991) Genotoxic effects of the drinking water mutagen
3-chloro-4-(dichloromethyl)-5-hydroxy-2[5H]-furanone (MX) in mammalian
cells in vitro and in rats in vivo. Mutat Res, 260: 55-64.
Bruschi SA & Bull RJ (1993) In vitro cytotoxicity of mono-, di- and
trichloroacetate and its modulation by peroxisome proliferation.
Fundam Appl Toxicol, 21: 366-375.
Brusick D (1986) Genotoxic effects in cultured mammalian cells
produced by low pH treatment conditions and increased ion
concentrations. Environ Mutagen, 8: 879-886.
Bull RJ (1980) Health effects of alternate disinfectants and their
reaction products. J Am Water Works Assoc, 72: 299-303.
Bull RJ (1982a) Health effects of drinking water disinfectants and
disinfectant by-products. Environ Sci Technol, 16: 554A-559A.
Bull RJ (1982b) Toxicological problems associated with alternative
methods of disinfection. J Am Water Works Assoc, 74: 642-648.
Bull RJ (1992) Toxicology of drinking water disinfection. In: Lippman
M ed. Environmental toxicants: Human exposures and their health
effects. New York, Van Nostrand Reinhold, pp 184-230.
Bull RJ (1993) Toxicology of disinfectants and disinfectant
by-products. In: Craun GF ed. Safety of water disinfection: Balancing
chemical and microbial risks. Washington, DC, ILSI Press, pp 239-256.
Bull RJ & DeAngelo AB (1995) Carcinogenic properties of brominated
haloacetates - Disinfection by-products in drinking water: Critical
issues in health effects research. Washington, DC, International Life
Sciences Institute, p 29.
Bull RJ & Robinson M (1985) Carcinogenic activity of haloacetonitrile
and haloacetone derivatives in the mouse skin and lung. In: Jolley RL,
Bull RJ, & Davis WP ed. Water chlorination: Chemistry, environmental
impact and health effects. Chelsea, Michigan, Lewis Publishers, Inc.,
vol 5, pp 221-236.
Bull RJ & Kopfler FC (1991) Health effects of disinfectants and
disinfection by-products. Denver, Colorado, American Water Works
Association Research Foundation and American Water Works Association.
Bull RJ, Meier JR, Robinson M, Ringhand HP, Laurie RD, & Stober JA
(1985) Evaluation of mutagenic and carcinogenic properties of
brominated and chlorinated haloacetonitriles: By-products of
chlorination. Fundam Appl Toxicol, 5: 1065-1074.
Bull RJ, Sanchez IM, Nelson MA, Larson JL, & Lansing AL (1990) Liver
tumor induction in B6C3F1 mice by dichloroacetate and
trichloroacetate. Toxicology, 63: 341-359.
Bull RJ, Templin M, Larson JL, & Stevens DK (1993) The role of
dichloroacetate in the hepatocarcinogenicity of trichloroethylene.
Toxicol Lett, 68: 203-211.
Bull RJ, Birnbaum LS, Cantor KP, Rose JB, Butterworth BE, Pegram R, &
Tuomisto J (1995) Water chlorination: Essential process or cancer
hazard? Fundam Appl Toxicol, 28: 155-166.
Burton-Fanning FW (1901) Poisoning by bromoform. Br Med J, 18 May:
1202-1203.
Butler TC (1948) The metabolic fate of chloral hydrate. J Pharmacol
Exp Ther, 92: 49-58.
Butler TC (1949) Reduction and oxidation of chloral hydrate by
isolated tissues in vitro. J Pharmacol Exp Ther, 95: 360-362.
Butterworth BE, Templin MV, Constan AA, Sprankle CS, Wong BA, Pluta
LJ, Everitt JI, & Recio L (1998) Long-term mutagenicity studies with
chloroform and dimethylnitrosamine in female lacI transgenic B6C3F1
mice. Environ Mol Mutagen, 31(3): 248-256.
Cabana BE & Gessner PK (1970) The kinetics of chloral hydrate
metabolism in mice and the effect thereon of ethanol. J Pharmaocol Exp
Ther, 174: 260-275.
Canada (1993) Guidelines for Canadian drinking water quality. Ottawa,
Ontario, Health Canada, Health Protection Branch.
Cantor KP (1997) Drinking water and cancer. Cancer Causes Control,
8(3): 292-308.
Cantor KP, Hoover R, & Hartge P (1985) Drinking water source and
bladder cancer: A case-control study. In: Jolley RL, Bull RJ, & Davis
WP ed. Water chlorination: Chemistry, environmental impact, and health
effects. Chelsea, Michigan, Lewis Publishers, Inc., vol 5, pp 145-152.
Cantor KP, Hoover R, & Hartge P (1987) Bladder cancer, drinking water
source, and tap water consumption: A case-control study. J Natl Cancer
Inst, 79(1): 1269-1279.
Cantor KP, Hoover R, & Hartge P (1990) Bladder cancer, tap water
consumption, and drinking water source. In: Jolley RL, Condie LW, &
Johnson JD ed. Water chlorination: Chemistry, environmental impact,
and health effects. Chelsea, Michigan, Lewis Publishers, Inc., vol 6,
pp 411-419.
Cantor KP, Lynch CF, & Hildesheim M (1995) Chlorinated drinking water
and risk of bladder, colon, and rectal cancers: A case-control study
in Iowa. In: Disinfection by-products in drinking water: Critical
issues in health effects research. Washington, DC, International Life
Sciences Institute, p 133.
Cantor KP, Lynch CF, & Hildesheim M (1996) Chlorinated drinking water
and risk of glioma: a case-control study in Iowa, USA. Epidemiology,
7(suppl 4): S83.
Cantor KP, Lynch CF, Hildesheim ME, Dosemeci M, Lubin J, Alavanja M, &
Craun G (1998) Drinking water source and chlorination byproducts: Risk
of bladder cancer. Epidemiology, 9(1): 21-28.
Carlo GL & Mettlin CJ (1980) Cancer incidence and trihalomethane
concentrations in a public drinking water system. Am J Public Health,
70: 523-525.
Carlson M & Hardy D (1998) Controlling DBPs with monochloramine.
Effect of water quality conditions on controlling disinfection
by-products with chloramines. J Am Water Works Assoc, 90(2): 95-106.
Carlton BD, Bartlett A, Basaran AH, Colling K, Osis I, & Smith MK
(1986) Reproductive effects of alternative disinfectants. Environ
Health Perspect, 69: 237-241.
Carlton BD, Habash DL, Basaran AH, George EL, & Smith MK (1987) Sodium
chlorite administration in Long-Evans rats: Reproductive and endocrine
effects. Environ Res, 42: 238-245.
Carlton BD, Basaran AH, Mezza LE, George EL, & Smith MK (1991)
Reproductive effects in Long-Evans rats exposed to chlorine dioxide.
Environ Res, 56: 170-177.
Carraro F, Klein S, Rosenblatt JI, & Wolfe RR (1989) The effect of
dichloroacetate on lactate concentration in exercising humans. J Appl
Physiol, 66: 591-597.
Carter JH, Carter HW, & DeAngelo AB (1995) Biochemical, pathologic and
morphometric alterations induced in male B6C3F1 mouse liver by
short-term exposure to dichloroacetic acid. Toxicol Lett, 81: 55-71.
Cattley RC, DeLuca J, Elcombe C, Fenner-Crisp P, Lake BG, Marsman DS,
Pastoor TA, Popp JA, Robinson DE, Schwetz B, Tugwood J, & Wahli W
(1998) Do peroxisome proliferating compounds pose a hepatocarcinogenic
hazard to humans? Regul Toxicol Pharmacol, 27: 47-60.
Cavanagh JE, Weinberg HS, Gold A, Sangalah R, Marbury D, & Glaze WH
(1992) Ozonation by-products: identification of bromohydrins from the
ozonation of natural waters with enhanced bromide levels. Environ Sci
Technol, 26(8): 1658-1662.
Chang J-HS, Vogt CR, Sun GY, & Sun AY (1981) Effects of acute
administration of chlorinated water on liver lipids. Lipids, 16:
336-340.
Chang LW, Daniel FB, & DeAngelo AB (1992) Analysis of DNA strand
breaks induced in rodent liver in vivo, hepatocytes in primary
culture, and a human cell line by chlorinated acetic acids and
chlorinated acetaldehydes. Environ Mol Mutagen, 20: 277-288.
Cheh AM, Skochdopole J, Koski P, & Cole L (1980) Nonvolatile mutagens
in drinking water: Production by chlorination and destruction by
sulfite. Science, 207: 90-92.
Chipman JK, Davies JE, Parson JL, Mair J, O'Neil G, & Fawell JK (1998)
DNA oxidation by potassium bromate: a direct mechanism or linked to
lipid peroxidation? Toxicology, 126(2): 93-102.
Cho DH, Hong JT, Chin K, Cho TS, & Lee BM (1993) Organotropic
formation and disappearance of 8-hydroxydeoxyguanosine in the kidney
of Sprague-Dawley rats exposed to Adriamycin and KBrO3. Cancer Lett,
74: 141-145.
Chow BM & Roberts PV (1981) Halogenated by-product formation by
chlorine dioxide and chlorine. J Environ Eng Div, 107: 609-618.
Christ SA, Read EJ, Stober JA, & Smith MK (1996) Developmental effects
of trichloroacetonitrile administered in corn oil to pregnant
Long-Evans rats. J Toxicol Environ Health, 47: 233-247.
Christman RF, Norwood DS, Millington DS, & Johnson JD (1983) Identity
and yields of major halogenated products of aquatic fulvic acid
chlorination. Environ Sci Technol, 17: 625-628.
Chu I, Secours VE, Marino I, & Villeneuve DC (1980) The acute toxicity
of four trihalomethanes in male and female rats. Toxicol Appl
Pharmacol, 5: 351-353.
Chu I, Villeneuve DC, Secours VE, Becking GC, & Valli VE (1982)
Toxicity of trihalomethanes: I. The acute and subacute toxicity of
chloroform, bromodichloromethane, chlorodibromomethane, and bromoform
in rats. J Environ Sci Health, B17: 205-224.
Cicmanec JL, Condie LW, Olson GR, & Wang SR (1991) 90-day toxicity
study of dichloracetate in dogs. Fundam Appl Toxicol, 17: 376-389.
Clark RM, Adams JQ, & Kyjins BW (1994) DBP control in drinking water:
cost and performance. J Environ Eng, 120(4): 759-771.
Claude J, Kunze E, & Frentzel-Beyne R (1986) Life-style and
occupational risk factors in cancer of the lower urinary tract. Am J
Epidemiol, 124: 578.
Claus TH & Pilkis SJ (1977) Effect of dichloroacetate and glucagon on
the incorporation of labeled substrates into glucose and on pyruvate
dehydrogenase in hepatocytes from fed and starved rats. Arch Biochem
Biophys, 182: 52-63.
CMA (1997) Sodium chlorite: drinking water rat two-generation
reproductive toxicity study. Washington, DC, Chemical Manufacturers
Association (Quintiles Report CMA/17/96).
Cole WJ, Mitchell RG, & Salamonsen RF (1975) Isolation,
characterization and quantitation of chloral hydrate as a transient
metabolite of trichloroethylene in man using electron capture gas
chromatography and mass fragmentography. J Pharm Pharmacol, 27:
167-171.
Coleman WE, Munch JW, Kaylor WH, Streicher RP, Ringhand HP, & Meier JR
(1984) Gas chromatography/mass spectroscopy analysis of mutagenic
extracts of aqueous chlorinated humic acid. A comparison of the
byproducts to drinking water contaminants. Environ Sci Technol, 18:
674-681.
Coleman WE, Munch JW, & Kopfler FC (1992) Ozonation/post-chlorination
of humic acids: a model for predicting drinking water DBPs. J Ozone
Sci Eng, 14: 349-355.
Condie LW, Smallwood CL, & Laurie RD (1983) Comparative renal and
hepatotoxicity of halomethanes: bromodichloromethane, bromoform,
chloroform, dibromochloromethane and methylene chloride. Drug Chem
Toxicol, 6: 563-578.
Connor MJ & Gillings D (1974) An empiric study of ecological
inference. Am J Public Health, 74: 555-559.
Connor PM, Moore GS, Calabrese EJ, & Howe GR (1985) The renal effects
of sodium chlorite in the drinking water of C57L/J male mice.
J Environ Pathol Toxicol Oncol, 6: 253-260.
Conolly RB & Butterworth BE (1995) Biologically based dose response
model for hepatic toxicity: a mechanistically based replacement for
traditional estimates of noncancer risk. Toxicol Lett, 82: 901-906.
Cooper WJ, Meyer LM, Bofill CC, & Cordal E (1983) Quantitative effects
of bromine on the formation and distribution of trihalomethanes in
groundwater with a high organic content. In: Jolley RL, Brungs WA,
Cotruvo JA, Cumming RB, Mattice JS, & Jacobs VA ed. Water
chlorination: Environmental impacts and health effects - Book 1:
Chemistry and water treatment. Ann Arbor, Michigan, Ann Arbor Science
Publishers, vol 4, pp 285-296.
Cooper WJ, Zika RG, & Steinhauer MS (1985) Bromide-oxidant
interactions and THM formation: a literature review. J Am Water Works
Assoc, 77(4): 116-121.
Corley RA, Mendrala AL, Smith FA, Staats DA, Gargas ML, Conolly RB,
Andersen ME, & Reitz RH (1990) Development of a physiologically based
pharmacokinetic model for chloroform. Toxicol Appl Pharmacol, 103:
512-527.
Cosby NC & Dukelow WR (1992) Toxicology of maternally ingested
trichloroethylene (TCE) on embryonal and fetal development in mice and
of TCE metabolites on in vitro fertilization. Fundam Appl Toxicol,
19: 268-274.
Cotter JL, Fader RC, Lilley C, & Herndon DN (1985) Chemical
parameters, antimicrobial activities, and tissue toxicity of 0.1 and
0.5% sodium hypochlorite solutions. Antimicrob Agents Chemother, 28:
118-122.
Coude FX, Saudubray JM, DeMaugre F, Marsac C, Leroux JP, & Charpentier
C (1978) Dichloroacetate as treatment for congenital lactic acidosis.
N Engl J Med, 299: 1365-1366.
Couri D & Abdel-Rahman MS (1980) Effect of chlorine dioxide and
metabolites on glutathione dependent system in rat, mouse and chicken
blood. J Environ Pathol Toxicol, 3: 451-460.
Couri D, Miller CH, Bull RJ, Delphia JM, & Ammar EM (1982a) Assessment
of maternal toxicity, embryotoxicity and teratogenic potential of
sodium chlorite in Sprague-Dawley rats. Environ Health Perspect,
46: 25-29.
Couri D, Abdel-Rahman MS, & Bull RJ (1982b) Toxicological effects of
chlorine dioxide, chlorite and chlorate. Environ Health Perspect,
46: 13-17.
Cove DJ (1976) Chlorate toxicity in Aspergillus nidulans: Studies of
mutants altered in nitrate assimilation. Mol Gen Genet, 146: 147-159.
Crabb DW & Harris RA (1979) Mechanism responsible for the hypoglycemic
actions of dichloroacetate and 2-chloropropionate. Arch Biochem
Biophys, 198: 145-152.
Crabb DW, Yount EA, & Harris RA (1981) The metabolic effects of
dichloroacetate. Metabolism, 30: 1024-1039.
Cragle DL, Shy CM, Struba RJ, & Stiff EJ (1985) A case-control study
of colon cancer and water chlorination in North Carolina. In: Jolley
RL, Bull RJ, & Davis WP ed. Water chlorination: Chemistry,
environmental impact, and health effects. Chelsea, Michigan, Lewis
Publishers, Inc., vol 5, pp 153-159.
Craun GF (1985) Epidemiologic studies of organic micro-pollutants in
drinking water. Sci Total Environ, 47: 461.
Craun GF (1991) Epidemiologic studies of organic micropollutants in
drinking water. In: Hutzinger O ed. The handbook of environmental
chemistry - Volume 5A: Water pollution. Berlin, Heidelberg, New York,
Springer-Verlag, p 1-44.
Craun GF, Clark RM, Doull J, Grabow W, Marsh GM, Okun DA, Sobsey MD,
& Symons JM (1993) In: Craun GF ed. Safety of water disinfection:
Balancing chemical and microbial risks - Conference conclusions.
Washington, DC, ILSI Press, pp 657-667.
Crebelli R, Conti G, Conti L, & Carere A (1985) Mutagenicity of
trichloroethylene, trichloroethanol and chloral hydrate in
Aspergillus nidulans. Mutat Res, 155: 105-111.
Crozes G, White P, & Marshall M (1995) Enhanced coagulation: its
effects on NOM removal and chemical costs. J Am Water Works Assoc,
87(1): 78-89.
Crump KS, Hoel D, Langley H, & Peto R (1976) Fundamental carcinogenic
processes and their implications to low dose risk assessment. Cancer
Res, 36: 2973, 2979-2357.
Crump KS & Guess HA (1982) Drinking water and cancer: Review of recent
epidemiological findings and assessment of risks. Annu Rev Public
Health, 3: 339.
Cucinell SA, Odessky L, Weiss M, & Dayton PG (1966) The effect of
chloral hydrate on bishydroxycoumarin metabolism. J Am Med Assoc,
197: 366-368.
Curry SH, Chu P-I, Baumgartner TG, & Stacpoole PW (1985) Plasma
concentrations and metabolic effects of intravenous sodium
dichloroacetate. Clin Pharmacol Ther, 37: 89-93.
Curry SH, Lorenz A, Chu P-I, Limacher M, & Stacpoole PW (1991)
Disposition and pharmacodynamics of dichloroacetate (DCA) and oxalate
following oral DCA doses. Biopharm Drugs Dispos, 12(5): 375-390.
Cunliffe DA (1991) Bactericidal nitrification in chloraminated water
supplies. App Environ Microbiol, 57(11): 3399-3402.
Dalhamn T (1957) Chlorine dioxide: Toxicity in animal experiments and
industrial risks. Arch. Ind Health, 15: 101-107.
Daniel FB, Schenck KM, Mattox JK, Lin EL, Haas DL, & Pereira MA (1986)
Genotoxic properties of haloacetonitriles: Drinking water by-products
of chlorine disinfection. Fundam Appl Toxicol, 6: 447-453.
Daniel FB, Robinson M, Condie LW, & York RG (1990a) Ninety-day oral
toxicity study of dibromochloromethane in Sprague-Dawley rats. Drug
Chem Toxicol, 13: 135-154.
Daniel FB, Condie LW, Robinson M, Stober JA, York RG, Olson GR, & Wang
S-R (1990b) Comparative 90-day subchronic toxicity studies on three
drinking water disinfectants, chlorine, monochloramine and chlorine
dioxide, in the Sprague-Dawley rats. J Am Water Works Assoc,
82: 61-69.
Daniel FB, Ringhand HP, Robinson M, Stober JA, Olson GR, & Page NP
(1991a) Comparative subchronic toxicity of chlorine and monochloramine
in the B6C3F1 mouse. J Am Water Works Assoc, 83: 68-75.
Daniel FB, Olson GR, & Stober JA (1991b) Induction of gastrointestinal
tract nuclear anomalies in B6C3F1 mice by
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone and
3,4-(dichloro)-5-hydroxy-2(5H)-furanone. Environ Mol Mutagen,
17: 32-39.
Daniel FB, DeAngelo AB, Stober JA, Olson GR, & Page NP (1992a)
Hepatocarcinogenicity of chloral hydrate, 2-chloroacetaldehyde, and
dichloroacetic acid in male B6C3F1 mouse. Fundam Appl Toxicol,
19: 159-168.
Daniel FB, Robinson M, Stober JA, Page NP, & Olson GR (1992b)
Ninety-day toxicity study of chloral hydrate in the Sprague-Dawley
rat. Drug Chem Toxicol, 15: 217-232.
Daniel FB, Robinson M, Stober JA, Olson GR, & Page NR (1993a) Toxicity
of 1,1,1-trichloro-2-propanone in Sprague-Dawley rats. J Toxicol
Environ Health, 39: 383-393.
Daniel FB, Meier JR, & DeAngelo AB (1993b) Advances in research on
carcinogenic and genotoxic by-products of chlorine disinfection:
Chlorinated hydroxyfuranones and chlorinated acetic acids. Ann Ist
Super Sanita, 29: 279-291.
Das R & Blanc PD (1993) Chlorine gas exposure and the lung: A review.
Toxicol Ind Health, 9: 439-455.
Davis ME (1990) Subacute toxicity of trichloroacetic acid in male and
female rats. Toxicology, 63: 63-72.
DeAngelo AB, Daniel FB, McMillan L, Wernsing P, & Savage RE (1989)
Species and strain sensitivity to the induction of peroxisome
proliferation by chloroacetic acids. Toxicol Appl Pharmacol,
101: 285-298.
DeAngelo AB, Daniel FB, Stober JA, & Olson GR (1991) The
carcinogenicity of dichloroacetic acid in the male B6C3F1 mouse.
Fundam Appl Toxicol, 16: 337-347.
DeAngelo AB, Daniel FB, Most BM, & Olson GR (1996) The carcinogenicity
of dichloroacetic acid in the male Fischer 344 rat. Toxicology,
114: 207-221.
DeAngelo AB, Daniel FB, Most BM, & Olson GR (1997) Failure of
monochloroacetic acid and trichloroactic acid administered in the
drinking water to produce liver cancer in male F344/N rats. J Toxicol
Environ Health, 52: 425-445.
DeAngelo AB, George MH, Kilburn SR, Moore TM, & Wolf DC (1998)
Carcinogenicity of potassium bromate administered in the drinking
water to male B6C3F1 mice and F344/N rats. Toxicol Pathol,
26(5): 587-594.
De Biasi A, Sbraccia M, Keizer J, Testai E, & Vittozzi L (1992) The
regioselective binding of CHCl3 reactive intermediates to microsomal
phospholipids. Chem-Biol Interact, 85: 229-242.
de Groot H & Noll T (1989) Halomethane hepatotoxicity: Induction of
lipid peroxidation and inactivation of cytochrome P-450 in rat liver
microsomes under low oxygen partial pressures. Toxicol Appl Pharmacol,
97: 530-537.
De Leer EW, Damste JS, Erkelens C, & de Galan L (1985) Identification
of intermediates leading to chloroform and C-4 diacids in the
chlorination of humic acid. Environ Sci Technol, 19: 512-522.
De Leer EW, Bagerman T, van Schaik P, Zuydeweg CWS, & de Galan L
(1986) Chlorination of omega-cyanoalkanoic acids in aqueous medium.
Environ Sci Technol, 20(12): 1218-1223.
Dechert S & Dekant W (1996) The possible role of
S-chloromethylglutathione in the genotoxicity of dichloromethane.
Toxicologist, 30: 93 (abstract).
Delehanty JM, Imai N, & Liang CS (1992) Effects of dichloroacetate on
hemodynamic responses to dynamic exercise in dogs. J Appl Physiol,
72: 515-520.
DeMarini DM, Perry E, & Shelton ML (1994) Dichloroacetic acid and
related compounds: Induction of prophage in E. coli and mutagenicity
and mutation spectra in Salmonella TA100. Mutagenesis, 9: 429-437.
DeMarini DM, Abu-Sharkra A, Felton CF, Patterson KS, & Shelton ML
(1995) Mutation spectra in Salmonella of chlorinated, chloraminated,
or ozonated drinking water extracts: Comparison to MX. Environ Mol
Mutagen, 26: 270-285.
DeMarini DM, Shelton ML, Warren SH, Ross TM, Shim JY, Richard AM, &
Pegram RA (1997) Glutathione S-transferase-mediated induction of GC
to AT transitions by halomethanes in Salmonella. Environ Mol
Mutagen, 30: 440-447.
Dick D, Ng KM, Sauder DN, & Chu I (1995) In vitro and in vivo
percutaneous absorption of 14C-chloroform in humans. Hum Exp Toxicol,
14: 260-265.
Dix KJ, Kedderis GL, & Borghoff SJ (1997) Vehicle-dependent oral
absorption and target tissue dosimetry of chloroform in male rats and
female mice. Toxicol Lett, 91: 197-209.
Dlyamandoglu V & Selleck RE (1992) Reactions and products of
chloramination. Environ Sci Technol, 26(4): 808.
Douglas GR, Nestmann ER, McKague AB, San RHC, Lee EG, Liu-Lee VW, &
Kobel DJ (1985) Determination of potential hazads from pulp and paper
mills: Mutagenicity and chemical analysis. In: Stich HF ed.
Carcinogens and mutagens in the environment - Volume 5: The workplace.
Boca Raton, Florida, CRC Press, Inc., pp 151-166.
Doyle TJ, Zheng W, Cerhan JR, Hong CP, Sellars TA, Kushi LH, & Folsom
AR (1997) The association of drinking water source and chlorination
by-products with cancer incidence among postmenopausal women in Iowa:
a prospective cohort study. Am J Public Health, 87(7): 1168-1176.
Druckrey H (1968) [Chlorinated drinking water, toxicity tests
involving seven generations of rats.] Food Cosmet Toxicol, 6: 147-154.
Drugan JK, Khosravi-Far R, White MA, Der CJ, Sung YJ, Hwang YW, &
Campbell SL (1996) Ras interaction with two distinct binding domains
of Raf-1 may be required for Ras transformation. J Biol Chem,
271: 233-237.
Dwelle EH (1903) Fatal bromoform poisoning. J Am Med Assoc, 41: 1540.
Eaton JW, Kolpin CF, & Swofford HS (1973) Chlorinated urban water: a
cause of dialysis induced hemolytic anemia. Science, 181: 463-464.
Engerholm BA & Amy GL (1983) A predictive model for chloroform
formation with chlorination of humic substances. Water Res, 17: 1797.
Enzer M & Whittington FM (1983) The actions of dichloroacetic acid on
blood glucose, liver glycogen and fatty acid synthesis in
obese-hyperglycemic (ob/ob) and lean mice. Horm Metab Res,
15: 225-229.
Epstein DL, Nolen GA, Randall JL, Christ SA, Read EJ, Stober JA, &
Smith MK (1992) Cardiopathic effects of dichloroacetate in the fetal
Long-Evans rat. Teratology, 46: 225-235.
Exon JH, Koller LD, O'Reilly CA, & Bercz JP (1987) Immunotoxicologic
evaluation of chlorine-based drinking water disinfectants, sodium
hypochlorite and monochloramine. Toxicology, 44: 257-269.
Eyre RJ, Stevens DK, Parker JC, & Bull RJ (1995) Acid-labile adducts
to protein can be used as indicators of the cysteine S-conjugate
pathway of trichloroethene metabolism. J Toxicol Environ Health,
46: 443-464.
FAO/WHO (1989) Toxicological evaluations of certain food additives and
contaminants prepared by the thirty-third meeting of the Joint FAO/WHO
Expert Committee on Food Additives. Geneva, World Health Organization,
pp 25-36 (WHO Food Additives Series 24).
FAO/WHO (1993) Toxicological evaluations of certain food additives and
contaminants prepared by the thirty-ninth meeting of the Joint FAO/WHO
Expert Committee on Food Additives. Geneva, World Health Organization
(WHO Food Additives Series 30).
Farber B & Abramow A (1985) Acute laryngeal edema due to chloral
hydrate. Isr J Med Sci, 21: 858-859.
Fawell JK (1990) The fate and significance of mutagenic by-products of
chlorination in vivo. Marlow, Buckinghamshire, Foundation for Water
Research.
Fayad NM (1993) Seasonal variations of THMs in Saudi Arabian drinking
water. J Am Water Works Assoc, 85: 46-50.
Fekadu K, Parzefall W, Kronberg L, Franzen R, Schulte-Hermann R, &
Knasmuller S (1994) Induction of genotoxic effects by
chlorohydroxyfuranones, byproducts of water disinfection, in
E. coli K-12 cells recovered from various organs of mice. Environ
Mol Mutagen, 24: 317-324.
Feng TH (1966) Behaviour of organic chloramines in disinfection.
J Water Pollut Control Fed, 38(4): 614-628.
Ferguson LR, Morcombe P, &Triggs CN (1993) The size of
cytokinesis-blocked micronuclei in human peripheral blood lymphocytes
as a measure of aneuploidy induction by Set A compounds in the EEC
trial. Mutat Res, 287(1): 101-112.
Ferreira-Gonzalez A, DeAngelo AB, Nasim S, & Garrett CT (1995) Ras
oncogene activation during hepatocarcinogenesis in B6C3F1 male mice
by dichloroacetic and trichloroacetic acids. Carcinogenesis,
16: 495-500.
Fidler IJ (1977) Depression of macrophages in mice drinking
hyperchlorinated water. Nature (Lond), 270: 735-736.
Fidler IJ, Barnes Z, Fogler WE, Kirsh R, Bugelski P, & Poste G (1982)
Involvement of macrophages in the eradication of established
metastasis following intravenous injection of liposomes containing
macrophage activators. Cancer Res, 42: 496-501.
Fisher N, Hutchinson JB, Berry R, Hardy J, Ginocchio AV, & Waite V
(1979) Long-term toxicity and carcinogenicity studies of the bread
improver potassium bromate: 1. Studies in rats. Food Cosmet Toxicol,
17(1): 33-39.
Fisher JW, Whittaker TA, Taylor DH, Clewell HJ, & Andersen ME (1989)
Physiologically-based pharmacokinetic modeling of the pregnant rat: A
multiroute exposure model for trichloroethylene and its metabolite,
trichloroacetic acid. Toxicol Appl Pharmacol, 99: 395-414.
Fisher JW, Gargas ML, Allen BC, & Andersen ME (1991) Physiologically
based pharmacokinetic modelling with trichloroethylene and its
metabolite, trichloroacetic acid, in the rat and mouse. Toxicol Appl
Pharmacol, 109: 183-195.
Flaten TP (1992) Chlorination of drinking water and cancer incidence
in Norway. Int J Epidemiol, 21: 6-15.
Fleischacker SJ & Randtke SJ (1983) Formation of organic chlorine in
public water supplies. J Am Water Works Assoc, 75(3): 132-138.
Fox AW, Sullivan BW, Buffini JD, Neichin ML, Nicora R, Hoehler FK,
O'Rourke R, & Stoltz RR (1996) Reduction of serum lactate by
dichloroacetate, and human pharmacokinetic-pharmacodynamic
relationships. J Pharmacol Exp Ther, 279: 686-693.
Fox TR, Schumann AM, Watanabe PG, Yano BL, Maher VM, & McCormick JJ
(1990) Mutational analysis of the H-ras oncogene in spontaneous
C57BL/6 × C3H/He mouse liver tumors and tumors induced with genotoxic
and nongenotoxic hepatocarcinogens. Cancer Res, 50: 4014-4019.
Fox AW, Yang X, Murli H, Lawlor TE, Cifone MA, & Reno FE (1996)
Absence of mutagenic effects of sodium dichloroacetate. Fundam Appl
Toxicol, 32: 87-95.
Freedman MD, Cantor KP, Lee NL, Chen L, Lei H, Ruhl CE, & Wang SS
(1997) Bladder cancer and drinking water: a population-based
case-control study in Washington County, Maryland (United States).
Cancer Causes Control, 8: 738-744.
French AS, Copeland CB, Andrews DL, Williams WC, Riddle MM, & Luebke
RW (1998) Evaluation of the potential immunotoxicity of chlorinated
drinking water in mice. Toxicology, 125: 53-58.
French AS, Copeland DB, Andrews DL, Williams WC, Riddle MM, & Luebke
RW (1999) Evaluation of the potential immunotoxicity of
bromodichloromethane in rats and mice. J Toxicol Environ Health,
56(5): 297-310.
Fu L-J, Johnson EM, & Newman LM (1990) Prediction of the development
toxicity hazard potential of halogenated drinking water disinfection
by-products tested by the in vitro hydra assay. Regul Toxicol
Pharmacol, 11: 213-219.
Fujie K, Aoki T, & Wada M (1990) Acute and subacute cytogenetic
effects of the trihalomethanes on rat bone marrow cells in vivo.
Mutat Res, 242: 111-119.
Fujie K, Aoki T, Ito Y, & Maeda S (1993) Sister-chromatid exchanges
induced by trihalomethanes in rat erythroblastic cells and their
suppression by crude catechin extracted from green tea. Mutat Res,
300: 241-246.
Furnus CC, Ulrich MA, Terreros MC, & Dulout FN (1990) The induction of
aneuploidy in cultured Chinese hamster cells by propionaldehyde and
chloral hydrate. Mutagenesis, 5: 323-326.
Fuscoe JC, Afshari AJ, George MH, DeAngelo AB, Tice RR, Salman R, &
Allen JW (1996) In vivo genotoxicity of dichloroacetic acid:
Evaluation with the mouse peripheral blood micronucleus assay and the
single cell gel assay. Environ Mol Mutagen, 27: 1-9.
Galal-Gorchev H & Morris JC (1965) Formation and stability of
bromamide, bromimide and nitrogen tribromide in aqueous solution.
Inorg Chem, 4(6): 899-905.
Gao P & Pegram RA (1992) In vitro hepatic microsomal lipid and
protein binding by metabolically activated bromodichloromethane.
Toxicologist, 12: 421.
Gao P, Thorton-Manning JR, & Pegram RA (1996) Protective effects of
glutathione on bromodichloromethane in vivo toxicity and in vitro
macromolecular binding in Fischer 344 rats. J Toxicol Environ Health,
49: 149-159.
Garrett ER & Lambert HJ (1973) Pharmacokinetics of trichloroethanol
and metabolites and interconversions among variously referenced
pharmacokinetic parameters. J Pharm Sci, 62: 550-572.
Gemma S, Faccioli S, Chieco P, Sbraccia M, Testai E, & Vittozzi L
(1996) In vivo CHCl3 bioactivation, toxicokinetics, toxicity, and
induced compensatory cell proliferation in B6C3F1 male mice. Toxicol
Appl Pharmacol, 141: 394-402.
Gessner PK & Cabana BE (1970) A study of the interaction of the
hypnotic effects and of the toxic effects of chloral hydrate and
ethanol. J Pharmacol Exp Ther, 174: 247-259.
Giller S, Le Curieux F, Gauthier L, Erb F, & Marzin D (1995)
Genotoxicity assay of chloral hydrate and chloropicrine. Mutat Res,
348: 147-152.
Giller S, Le Curieux F, Erb F, & Marzin D (1997) Comparative
genotoxicity of halogenated acetic acids found in drinking water.
Mutagenesis, 12: 321-328.
Gilli G (1990) Water disinfection: Relationship between ozone and
aldehyde production. J Ozone Sci Eng, 12: 116-125.
Gilman AG, Rall TW, Neis AS, & Taylor P (1991) Goodman and Gilman's
pharmacological basis of therapeutics, 8th ed. New York, Pergamon
Press, pp 364-365.
Ginwalla AS & Mikita MA (1992) Reaction products of Suwannee River
fulvic acid with chloramine: characterization of products via 15-N
NMR. Environ Sci Technol, 26(6): 1148-1150.
Glaze WH, Weinberg HS, & Cavanagh JE (1993) Evaluating the formation
of brominated DBPs during ozonation. J Am Water Works Assoc,
85: 96-103.
Goldberg SJ, Lebowitz MD, & Graver EJ (1990) An association of human
congenital cardiac malformations and drinking water contaminants. J Am
Coll Cardiol, 16: 155-164.
Golden RJ, Holm SE, Robinson DE, Julkunen PH, & Reese EA (1997)
Chloroform mode of action: Implications for cancer risk assessment.
Regul Toxicol Pharmacol, 26(2): 142-145.
Goldsworthy TL & Popp JA (1987) Chlorinated hydrocarbon-induced
peroxisomal enzyme activity in relation to species and organ
carcinogenicity. Toxicol Appl Pharmacol, 88: 225-233.
Gonzalez-Leon A, Schultz IR, & Bull RJ (1997) Pharmacokinetics and
metabolism of dichloroacetate in the F344 rat after prolonged
administration in drinking water. Toxicol Appl Pharmacol,
146(2): 189-195.
Gordon G (1993) The chemical aspects of bromate control in ozonated
drinking water containing bromide ion. In: Proceedings of the
International Water Supply Association Workshop, Paris. Boston,
Massachusetts, Blackwell Scientific Publications, pp 41-49.
Gordon G & Emmert GL (1996) Bromate ion formation in water when
chlorine dioxide is photolyzed in the presence of bromide ion. In:
Proceedings of the Water Quality Technology Conference, New Orleans,
LA. Denver, Colorado, American Water Works Association.
Gordon G & Rosenblatt A (1996) Gaseous, chlorine-free chlorine dioxide
for drinking water. In: Proceedings of the Water Quality Technology
Conference, New Orleans, LA. Denver, Colorado, American Water Works
Association.
Gordon WP, Soderlund EJ, Holme JA, Nelson SD, Iyer L, Rivedal E, &
Dybing E (1985) The genotoxicity of 2-bromoacrolein and
2,3-dibromopropanal. Carcinogenesis, 6: 705-709.
Gordon G, Slootmaekers B, Tachiyashiki S, & Wood DW (1990) Minimizing
chlorite ion and chlorate ion in water treated with chlorine dioxide.
J Am Water Works Assoc, 82(4): 160-165.
Gordon G, Adam L, & Bubnis B (1995) Minimizing chlorate ion formation.
J Am Water Works Assoc, 87(6): 97-106.
Gordon G, Adams LC, Bubnis BP, Kuo C, Cushing RS, & Sakaji RH (1997)
Predicting liquid bleach decomposition. J Am Water Works Assoc,
89(4): 142-149.
Gottlieb MS, Carr JK, & Morris DT (1981) Cancer and drinking water in
Louisiana: Colon and rectal. Int J Epidemiol, 10: 117-125.
Gottlieb MS, Carr JK, & Clarkson JR (1982) Drinking water and cancer
in Louisiana: A retrospective mortality study. Am J Epidemiol,
116: 652-667.
Gray ET, Margerum DW, & Huffman RP (1979) Chloramine equilibria and
the kinetics of disproportionation in aqueous solution. In: Brinkman
FE & Bellama JM ed. Organometals and organometalloids, occurrence and
fate in the environment. Washington, DC, American Chemical Society, pp
264-277 (ACS Symposium Series No. 82).
Green T & Hathway DE (1978) Interactions of vinyl chloride with
rat-liver in vivo. Chem-Biol Interact, 22: 211-224.
Greenland S (1987) Quantitative methods in the review of epidemiologic
literature. Epidemiol Rev, 9: 1-30.
Greenland S (1994) Invited commentary - A critical look at some
popular meta-analytic methods. Am J Epidemiol, 140: 290-296.
Greenland S & Robins J (1994a) Invited commentary - Ecologic studies:
Biases, misconceptions, and counterexamples. Am J Epidemiol,
139(8): 747-760.
Greenland S & Robins J (1994b) Accepting the limits of ecologic
studies. Am J Epidemiol, 139(8): 769-771
Guastadisegni C, Balduzzi M, & Vittozzi L (1996) Preliminary
characterization of phospholipid adducts formed by [14C]-CHCl3
reactive intermediates in hepatocyte suspensions. J Biochem Toxicol,
11: 21-25.
Guastadisegni C, Guidoni L, Balduzzi M, Viti V, Di Consiglio E, &
Vitozzi L (1998) Characterization of a phospholipid adduct formed in
Sprague Dawley rats by chloroform metabolism: NMR studies. J Biochem
Mol Toxicol, 12(2): 93-102.
Haberer K (1994) [Survey of disinfectants use and occurrence of
disinfectant by-products in German waterworks.] Wasser Abwasser,
135: 409-417 (in German).
Haessler R, Davis RF, Wolff RA, Kuzume K, Shangraw R, & Van Winkle DM
(1996) Dichloroacetate reduces plasma lactate levels but does not
reduce infarct size in rabbit myocardium. Shock, 5: 66-71.
Halestrap AP (1975) The mitochondrial pyruvate carrier: Kinetics and
specificity for substrates and inhibitors. Biochem J, 148: 85-96.
Halestrap AP (1978) Pyruvate and ketone-body transport across the
mitochondrial membrane: Exchange properties, pH-dependence, and
mechanism of the carrier. Biochem J, 172: 377-387.
Haller JF & Northgraves WW (1955) Chlorine dioxide and safety. TAPPI,
38: 199-202.
Hara A, Yamamoto H, Deyashiki Y, Nakayama T, Oritani H, & Sawada H
(1991) Aldehyde dismutation catalyzed by pulmonary carbonyl reductase:
kinetic studies of chloral hydrate metabolism to trichloroacetic acid
and trichloroethanol. Biochim Biophys Acta, 1075: 61-67.
Harada K, Ichiyama T, Ikeda H, Ishihara T, & Yoshida K (1997) An
autopsy case of acute chloroform intoxication after intermittent
inhalation for years. Nippon Hoigaku Zasshi, 51(4): 319-323.
Harrington RM, Shertzer HG, & Bercz JP (1985) Effects of chlorine
dioxide on the absorption and distribution of dietary iodide in the
rat. Fundam Appl Toxicol, 5: 672-678.
Harrington RM, Shertzer HG, & Bercz JP (1986) Effects of chlorine
dioxide on thyroid function in the African green monkey and the rat.
J Toxicol Environ Health, 19: 235-242.
Harrington RM, Romano RR, Gates D, & Ridgway P (1995a) Subchronic
toxicity of sodium chlorite in the rat. J Am Coll Toxicol, 14: 21-33.
Harrington RM, Romano RR, & Irvine L (1995b) Developmental toxicity of
sodium chlorite in the rabbit. J Am Coll Toxicol, 14: 108-118.
Harrington-Brock K, Doerr CL, & Moore M (1995) Mutagenicity and
clastogenicity of 3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone
(MX) in L5178y/TK+/--3.7.2C mouse lymphoma cells. Mutat Res, 348:
105-110.
Harrington-Brock K, Doerr CL, & Moore MM (1998) Mutagenicity of three
disinfection by-products: di- and trichloroacetic acid and chloral
hydrate in L5178Y/TK+/--3.7.2C mouse lymphoma cells. Mutat Res,
413: 265-276.
Harris RH (1974) Implications of cancer-causing substances in
Mississippi River water. Washington, DC, Environmental Defense Fund.
Hasegawa R, Takahashi M, & Kobulo T (1986) Carcinogenicity study of
sodium hypochlorite in F344 rats. Food Chem Toxicol, 24: 1295-1302.
Hautman DP & Bolyard M (1993) Using ion chromatography to analyze
inorganic disinfection by-products. J Am Water Works Assoc,
85(10): 88-93.
Hayashi M, Kishi M, Sofuni T, & Ishidate M (1988) Micronucleus tests
in mice on 39 food additives and eight miscellaneous chemicals. Food
Chem Toxicol, 26: 487-500.
Hayashi M, Sutou S, Shimada H, Sato S, Sasaki YF, & Wakata A (1989)
Difference between intraperitoneal and oral gavage application in the
micronucleus test. Mutat Res, 223: 329-344.
Hayatsu H, Hoshimo H, & Kawazoe Y (1971) Potential carcinogenicity of
sodium hypochlorite. Nature (Lond), 233: 495.
Hayes JR, Condie LW, & Borzelleca JF (1986) Toxicology of
haloacetonitriles. Environ Health Perspect, 69: 183-202.
Heffernan WP, Guion C, & Bull RJ (1979a) Oxidative damage to the
erythrocyte induced by sodium chlorite, in vivo. J Environ Pathol
Toxicol, 2: 1487-1499.
Heffernan WP, Guion C, & Bull RJ (1979b) Oxidative damage to the
erythrocyte induced by sodium chlorite, in vitro. J Environ Pathol
Toxicol, 2: 1501-1510.
Hegi ME, Fox TR, Belinsky SA, Devereux TR, & Anderson MW (1993)
Analysis of activated protooncogenes in B6C3F1 mouse liver tumors
induced by ciprofibrate, a potent peroxisome proliferator.
Carcinogenesis, 14: 145-149.
Heiskanen K, Lindstrom-Seppa P, Haataja L, Vaittinen SL, Vartiainen T,
& Komulainen H (1995) Altered enzyme activities of xenobiotic
biotransformation in kidneys after subchronic administration of
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H) furanone (MX) to rats.
Toxicology, 100: 121-128.
Helliwell M & Nunn J (1979) Mortality in sodium chlorate poisoning. Br
Med J, April 28(6171): 1119.
Helma C, Kronberg L, Ma TH, & Knasmuller S (1995) Genotoxic effects of
the chlorinated hydroxyfuranones
3-chloro-4-(dichloromethyl)-5-hydroxy-2[5H]-furanone and
3,4-dichloro-5-hydroxy-2[5H]-furanone in Tradescantia micronucleus
assays. Mutat Res, 346: 181-186.
Hemming J, Holmbom B, Reunanen M, & Tikkanen L (1986) Determination of
the strong mutagen
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone in chlorinated
drinking and humic waters. Chemosphere, 5: 549-556.
Herbert V, Gardner A, & Colman N (1980) Mutagenicity of
dichloroacetate, an ingredient of some formulations of pangamic acid
(trade-named "vitamin B15"). Am J Clin Nutr, 33: 1179-1182.
Herren-Freund SL, Pereira MA, Khoury MD, & Olson G (1987) The
carcinogenicity of trichloroethylene and its metabolites,
trichloroacetic acid and dichloroacetic acid, in mouse liver. Toxicol
Appl Pharmacol, 90: 183-189.
Hewitt WR, Brown EM, & Plaa GL (1983) Acetone-induced potentiation of
trihalomethane toxicity in male rats. Toxicol Lett, 16: 285-296.
Heywood R, Sortwell RJ, Kelly PJ, & Street AE (1972) Toxicity of
sodium chlorate to the dog. Vet Rec, 90: 416-418.
Heywood R, Sortwell RJ, Noel PR, Street AE, Prentice DE, Roe FJ,
Wadsworth PF, Worden AN, & Van Abbe NJ (1979) Safety evaluation of
toothpaste containing chloroform. III. Long-term study in beagle dogs.
J Environ Pathol Toxicol, 2: 835-851.
Hildesheim ME, Cantor KP, Lynch CF, Dosemeci M, Lubin J, Alavanja M, &
Craun G (1998) Drinking water source and chlorination byproducts: risk
of colon and rectal cancers. Epidemiology, 9(1): 28-36.
Hill AB (1965) Environment and disease: Association or causation? Proc
R Soc Med, 58: 295-300.
Hirsch IA & Zauder HL (1986) Chloral hydrate: A potential cause of
arrhythmias. Anesth Analg, 65: 691-692.
Hobara T, Kobayashi H, Kawamoto T, Iwamoto S, & Sakai T (1987)
Extrahepatic metabolism of chloral hydrate, trichloroethanol and
trichloroacetic acid in dogs. Pharmacol Toxicol, 61: 58-62.
Hoehn RC, Dietrich AM, Farmer WS, Orr MP, Lee RG, Aieta EM, Wood DW, &
Gordon G (1990) Household odours associated with the use of chlorine
dioxide. J Am Water Works Assoc, 82(4): 166-172.
Hogan MD, Chi PY, Hoel DG, & Mitchell TJ (1979) Association between
chloroform levels in finished drinking water supplies and various
site-specific cancer mortality rates. J Environ Pathol Toxicol,
2: 873-887.
Hoigne J & Bader H (1994) Kinetics of reactions of chlorine dioxide in
water - I. Rate constants for inorganic and organic compounds. Water
Res, 28(1): 45-56.
Hoigne J, Bader H, Haag WR, & Staehelin J (1985) Rate constants of
reactions of ozone with organic and inorganic compounds in water: III.
Inorganic compounds and radicals. Water Res, 19(8): 993-1004.
Holmbom BR, Voss RH, Mortimer RD, & Wong A (1984) Fractionation,
isolation and characterization of Ames mutagenic compounds in kraft
chlorination effluents. Environ Sci Technol, 18: 333-337.
Horth H, Fawell JK, James CP, & Young WF (1991) The fate of the
chlorination-derived mutagen MX in vivo. Marlow, Buckinghamshire,
Foundation for Water Research.
Howe GR, Burch JD, & Miller AB (1977) Artificial sweeteners and human
bladder cancer. Lancet, 2: 578-581.
Hoz S, Basch H, Wolk JL, Rappoport Z, & Goldberg M (1991) Calculated
double bond stabilization by bromine and chlorine: Relevance to the
kBr/kCl element effect. J Org Chem, 56: 5424-5426.
Huebner W & Jung F (1941) [About the theory of chlorate intoxication.]
Schweiz Med Wochenschr, 71: 247-250 (in German).
Hussein GI & Ahmed AE (1987) Studies on the mechanism of
haloacetonitrile acute toxicity: Establishment of toxic doses and
target organs of toxicity in rats. Toxicologist, 7: 148.
Hutton PH & Chung FI (1992) Simulating THM formation potential in
Sacramento Delta. J Water Res Planning Manage, 118(5): 513-542 (Parts
I & II).
Hyttinen JM & Jansson K (1995) PM2 DNA damage induced by
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX). Mutat Res,
348: 183-186.
Hyttinen JM, Myohanen S, & Jansson K (1996) Kinds of mutations induced
by 3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX) in the
hprt gene of Chinese hamster ovary cells. Carcinogenesis,
17: 1179-1181.
IARC (1986) Potassium bromate. In: Some naturally occurring and
synthetic food components, furocoumarins and ultraviolet radiation.
Lyon, International Agency for Research on Cancer, pp 207-220 (IARC
Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to
Humans, Volume 40).
IARC (1987) Overall evaluations of carcinogenicity: An updating of
IARC Monographs 1 to 42. Lyon, International Agency for Research on
Cancer (IARC Monographs on the Evaluation of the Carcinogenic Risk of
Chemicals to Humans, Supplement 7).
IARC (1991) Chlorinated drinking-water; chlorination by-products; some
other halogenated compounds; cobalt and cobalt compounds. Lyon,
International Agency for Research on Cancer (IARC Monographs on the
Evaluation of the Carcinogenic Risk of Chemicals to Humans, Volume
52).
IARC (1995) Dry cleaning, some chlorinated solvents and other
industrial chemicals. Lyon, International Agency for Research on
Cancer (IARC Monographs on the Evaluation of Carcinogenic Risks to
Humans, Volume 63).
IARC (1999) Re-evaluation of some organic chemicals, hydrazine and
hydrogen peroxide. Lyon, International Agency for Research on Cancer
(IARC Monographs on the Evaluation of Carcinogenic Risks to Humans,
Volume 71).
Ijsselmuiden CB, Gaydos C, Feighner B, Novakoski WL, Serwadda D, Caris
V, & Comstock GW (1992) Cancer of the pancreas and drinking water: A
population-based case-control study in Washington County, Maryland. Am
J Epidemiol, 136: 836-842.
ILSI (1995) Disinfection by-products in drinking water: Critical
issues in health effects research - Proceedings of a workshop, Chapel
Hill, NC, 23-25 October 1995. Washington, DC, International Life
Sciences Institute.
ILSI (1997) An evaluation of EPA's proposed guidelines for carcinogen
risk assessment using chloroform and dichloroacetate as case studies:
Report of an expert panel. Washington, DC, International Life Sciences
Institute.
ILSI (1998) The toxicity and risk assessment of complex mixtures in
drinking water. Washington, DC, International Life Sciences Institute.
INERIS (1996) Study of acute toxicity of chlorite dioxide administered
to rats by vapour inhalation: Determination of the 50% lethal
concentration (LC50/4 hrs). Paris, Elf Atochem SA.
IPCS (1994) Environmental health criteria 163: Chloroform. Geneva,
World Health Organization, International Programme on Chemical Safety.
IRIS (1991) Integrated risk information system - Bromoform.
Washington, DC, US Environmental Protection Agency.
IRIS (1992) Integrated risk information system - Dibromochloromethane.
Washington, DC, US Environmental Protection Agency.
IRIS (1993) Integrated risk information system - Bromodichloromethane.
Washington, DC, US Environmental Protection Agency.
Isaac RA & Morris JC (1983) Transfer of active chlorine from
chloramine to nitrogenous organic compounds. Environ Sci Technol,
17: 738-742.
Ishidate M (1987) Data book of chromosomal aberrations in vitro.
Tokyo, Life-Science Information Center, p 383.
Ishidate M, Sofuni T, Yoshikawa K, & Hayashi M (1982) Studies on the
mutagenicity of low boiling organohalogen compounds. Tokyo, Medical
and Dental University (Unpublished intraagency report to the National
Institute of Hygienic Sciences, Tokyo).
Ishidate M, Sofuni T, Yoshikawa K, Hayashi M, Nohmi T, Sawada M, &
Matsuoka A (1984) Primary mutagenicity screening of food additives
currently used in Japan. Food Chem Toxicol, 22: 623-636.
Ishiguro Y, Santodonato J, & Neal MW (1988) Mutagenic potency of
chlorofuranones and related compounds in Salmonella. Environ Mol
Mutagen, 11: 225-234.
Islam MS, Zhao L, Zhou J, Dong L, McDougal JN, & Flynn GL (1996)
Systemic uptake and clearance of chloroform by hairless rats following
dermal exposure: I. Brief exposure to aqueous solutions. Risk Anal,
16: 349-357.
Issemann I & Green S (1990) Activation of a member of the steroid
hormone receptor superfamily by peroxisome proliferators. Nature
(Lond), 347: 645-650.
Jacobsen JS, Perkins CP, Callahan JT, Sambamurti K, & Humayun HZ
(1989) Mechanisms of mutagenesis by chloroacetaldehyde. Genetics,
121: 213-222.
Jamison KC, Larson JL, Butterworth BE, Harden R, Skinner BL, & Wolf DC
(1996) A non-bile duct origin for intestinal crypt-like ducts with
periductular fibrosis induced in livers of F344 rats by chloroform
inhalation. Carcinogenesis, 17: 675-682.
Jansson K & Hyttinen JM (1994) Induction of gene mutation in mammalian
cells by 3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX), a
chlorine disinfection by-product in drinking water. Mutat Res,
322: 129-132.
Jansson K, Maki-Paakkanen J, Vaittinen SL, Vartiainen T, Komulainen H,
& Tuomisto J (1993) Cytogenetic effects of
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX) in rat
peripheral lymphocytes in vitro and in vivo. Mutat Res,
229: 25-28.
Jansson K, Hyttinen JM, Niittykoski M, & Maki-Paakkanen J (1995)
Mutagenicity in vitro of 3,4-dichloro-5-hydroxy-2(5H)-furanone
(mucochloric acid), a chlorine disinfection by-product in drinking
water. Environ Mol Mutagen, 25: 284-287.
Jensen OM, Wahrendorf J, Knudsen JB, & Sorensen BL (1986) The
Copenhagen case-control study of bladder cancer: II. Effect of coffee
and other beverages. Int J Cancer, 37: 651.
Johnson JD & Jensen JN (1986) THM and TOX formation: routes, rates and
precursors. J Am Water Works Assoc, 78(4): 156-162.
Jones PS, Savory R, Barratt P, Bell AR, Gray TJ, Jenkins NA, Gilbert
DJ, Copeland NG, & Bell DR (1995) Chromosomal localisation,
inducibility, tissue-specific expression and strain differences in
three murine peroxisome-proliferator-activated-receptor genes. Eur
J Biochem, 233: 219-226.
Jow L & Mukherjee R (1995) The human peroxisome proliferator-activated
receptor (PPAR) subtype NUC1 represses the activation of hPPAR and
thyroid hormone receptors. J Biol Chem, 270: 3836-3840.
Jung F (1947) [About the theory of chlorate intoxication III.] Arch
Exp Pathol Pharmakol, 204: 157-165 (in German).
Jung F (1965) [On the reaction of methemoglobin with potassium
chlorate.] Acta Biol Med Ger, 15: 554-568 (in German).
Kanitz S, Franco Y, Patrone V, & Caltabellotta (1996) Association
between drinking water disinfection and somatic parameters at birth.
Environ Health Perspect, 104(5): 516-520.
Kaplan HL, Forney RB, Hughes FW, Jain NC, & Crim D (1967) Chloral
hydrate and alcohol metabolism in human subjects. J Forensic Sci,
12: 295-304.
Kaplan HL, Jain NC, Forney RB, & Richards AB (1969) Chloral
hydrate-ethanol interactions in the mouse and dog. Toxicol Appl
Pharmacol, 14: 127-137.
Kasai H, Nishimura S, Kurokawa Y, & Hayashi Y (1987) Oral
administration of the renal carcinogen, potassium bromate,
specifically produces 8-hydroxydeoxyguanosine in rat target organ DNA.
Carcinogenesis, 8: 1959-1961.
Katayama Y & Welsh FA (1989) Effect of dichloroacetate on regional
energy metabolites and pyruvate dehydrogenase activity during ischemia
and reperfusion in gerbil brain. J Neurochem, 52: 1817-1822.
Kato-Weinstein J, Lingohr MK, Orner GA, Thrall BD, & Bull RJ (1998)
Effects of dichloroacetate treatment on carbohydrate metabolism in
B6C3F1 mice. Toxicology, 130(2-3): 141-154.
Katz R, Tai CN, Deiner RM, McConnell RF, & Semonick DE (1981)
Dichloroacetate, sodium: 3-month oral toxicity studies in rats and
dogs. Toxicol Appl Pharmacol, 57: 273-287.
Kavanaugh MC, Trussell AR, Cromer J, & Trussell RR (1980) An empirical
kinetic model of THM formation: application to meet the proposed THM
standards. J Am Water Works Assoc, 72(10): 578-582.
Kawamoto T, Hobara T, Kobayashi H, Iwamoto S, Sakai T, Takano T, &
Miyazaki Y (1987) The metabolic ratio as a function of chloral hydrate
dose and intracellular redox as a function of chloral hydrate dose and
intracellular redox state in the perfused rat liver. Pharmacol
Toxicol, 60: 325-329.
Kawachi T, Komatsu T, Kada T, Ishidate M, Sasaki M, Sugiyami T, Tazima
Y, & Williams GM ed. (1980) In: The predictive value of short-term
screening tests in carcinogenicity evaluation. Amsterdam, Oxford, New
York, Elsevier Science Publishers, pp 253-267.
Keegan TE, Simmons JE, & Pegram RA (1998) NOAEL and LOAEL
determinations of acute hepatoxicity for chloroform and
bromodichloromethane delivered in an aqueous vehicle to F344 rats.
J Toxicol Environ Health, 55(1): 65-75.
Keller DA & Heck HD (1988) Mechanistic studies on chloral toxicity:
relationship to trichloroethylene carcinogenesis. Toxicol Lett,
42: 183-191.
Ketcha MM, Stevens DK, Warren DA, Bishop CT, & Brashear WT (1996)
Conversion of trichloroacetic acid to dichloroacetic acid in
biological samples. J Anal Toxicol, 20: 236-241.
Kevekordes S, Porzig J, Gebel T, & Dunkelberg H (1998) [Mutagenicity
of mixtures of halogenated aliphatic hydrocrabons and polycyclic
aromatic hydrocarbons in the Ames test with TA98 and TA100.] Zent.bl
Hyg U.weltmed, 200(5-6): 531-531 (in German).
Kimura S, Osaka H, Saitou K, Ohtuki N, Kobayashi T, & Nezu A (1995)
Improvement of lesions shown on MRI and CT scan by administration of
dichloroacetate in patients with Leigh syndrome. J Neurol Sci,
134: 103-107.
King WD & Marrett LD (1996) Case-control study of bladder cancer and
chlorination by-products in treated water. Cancer Causes Control,
7(6): 596-604.
Kirmeyer GJ, Foust GW, Pierson GC, & Simmler JJ (1993) Optimizing
chloramine treatment. Denver, Colorado, American Water Works
Association Research Foundation.
Kirmeyer GJ, Foust GW, Pierson GC, & Simmler JJ (1995) Occurrence of
nitrification in chloraminated water systems. Denver, Colorado,
American Water Works Association Research Foundation.
Kitchin KT & Brown JL (1994) Dose-response relationship for rat liver
DNA damage caused by 49 rodent carcinogens. Toxicology, 88: 31-49.
Klinefelter GR, Suarez JD, Roberts NL, & DeAngelo AB (1995)
Preliminary screening for the potential of drinking water disinfection
byproducts to alter male reproduction. Reprod Toxicol, 9: 571-578.
Klotz J & Pyrch L (1998) Neural tube defects and drinking water
disinfection. Epidemiology, 10(4): 383-390.
Klotz J, Pyrch L, Haltmeier P, Trimbath L, & Weisel C (1996) Neural
tube defects and chlorination by-products in drinking water.
Epidemiology, 7(suppl 4): S63 (abstract).
Knasmuller S, Zohrer E, Kronberg L, Kundi M, Franzen R, &
Schulte-Hermann R (1996) Mutational spectra of Salmonella
typhimurium revertants induced by chlorohydroxyfuranones, byproducts
of chlorine disinfection of drinking water. Chem Res Toxicol,
9: 374-381.
Knecht KT & Mason RP (1991) The detection of halocarbon-derived
radical adducts in bile and liver of rats. Drug Metab Dispos,
19: 325-331.
Koch B, Crofts EW, Schimpff WK, & Davis MK (1988) Analysis of
halogenated DBPs by capillary chromatography. In: Proceedings of the
Water Quality Technology Conference, St Louis, MO. Denver, Colorado,
American Water Works Association.
Koch B, Krasner SW, Sclimenti MJ, & Schimpff WK (1991) Predicting the
formation of DBPs by the simulated distribution system. J Am Water
Works Assoc, 83(10): 62-70.
Koch-Weser J, Sellers EM, Udall JA, Griner PF, & Rickles FR (1971)
Chloral hydrate and warfarin therapy. Ann Intern Med, 75(1): 141-142.
Koepke UC, Coon JM, & Triolo AJ (1974) Effect of paraoxon on the
hypnotic action of chloral hydrate. Toxicol Appl Pharmacol, 30: 36-51.
Koivusalo M, Jaakkola JJ, Vartiainen T, Hakulinen T, Karjalainen S,
Pukkala E, & Tuomisto J (1994a) Drinking water mutagenicity in past
exposure assessment of the studies on drinking water and cancer:
Application and evaluation in Finland. Environ Res, 64: 90-101.
Koivusalo M, Jaakkola JJ, Vartiainen T, & Hakulinen S (1994b) Drinking
water mutagenicity and gastrointestinal and urinary tract cancers: An
ecological study in Finland. Am J Public Health, 84: 1223-1228.
Koivusalo M, Vartiainen T, Hakulinen S, Pukkala E, & Jaakkola JJ
(1995) Drinking water mutagenicity and leukemia, lymphomas, and
cancers of the liver, pancreas and soft tissue. Arch Environ Health,
50: 269-276.
Koivusalo M, Pukkala E, Vartiainen T, Jaakkola JJ, Hakulinen S, &
Tuomisto J (1996) Drinking water chlorination and cancer - a cohort
study in Finland. Epidemiology, 7(suppl 4): S82 (abstract).
Koivusalo M, Pukkala E, Vartiainen T, Jaakkola JJ, Hakulinen S, &
Tuomisto J (1997) Drinking water chlorination and cancer - a
historical cohort study in Finland. Cancer Causes Control, 8: 192-200.
Komulainen H, Vaittinen SL, Vartiainen T, Lotjonen S, Paronen P, &
Tuomisto J (1992) Pharmacokinetics in rat of
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX), a drinking
water mutagen, after a single dose. Pharmacol Toxicol, 70: 424-428.
Komulainen H, Huuskonen H, Kosma VM, Lotjonen S, & Vartiainen T (1994)
Toxic effects and excretion in urine of
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX) in the rat
after a single oral dose. Arch Toxicol, 68: 398-400.
Komulainen H, Kosma VM, & Vaittinen S (1997) Carcinogenicity of the
drinking water mutagen
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX). J Natl
Cancer Inst, 89: 848-856.
Koransky W (1952) [A contribution to the theory of chlorate
oxidation.] Arch Exp Pathol Pharmakol, 215S: 483-491 (in German).
Korz V & Gattermann R (1997) Behavioral alterations in male golden
hamsters exposed to chlorodibromomethane. Pharmacol Biochem Behav,
58(3): 643-647.
Kraft J & van Eldik R (1989) Kinetics and mechanisms of the iron
(III)-catalyzed autooxidation of sulfur(IV) oxides in aqueous
solution. Inorg Chem, 28(12): 2297-2315.
Kramer MD, Lynch CF, Isacson P, & Hanson JW (1992) The association of
waterborne chloroform with intrauterine growth retardation.
Epidemiology, 3: 407-413.
Krasner SW, McGuire MJ, Jacangelo JG, Patania NL, Reagan KM, & Aieta
ME (1989) The occurrence of disinfection by-products in US drinking
water. J Am Water Works Assoc, 81(8): 41-53.
Krasner SW, Glaze WH, Weinberg HS, Daniel PA, & Najm IN (1993)
Formation and control of bromate during ozonation of waters containing
bromide. J Am Water Works Assoc, 85: 73-81.
Krasner SW, Scliminti MJ, & Chin R (1995) The impact of TOC and
bromide on chlorination by-product formation: Disinfection by-products
in water treatment. Boca Raton, Florida, CRC Press, Inc.
Kringstad KP, Ljungquist PO, de Sousa F, & Stromberg LM (1981)
Identification and mutagenic properties of some chlorinated aliphatic
compounds in the spent liquor from kraft pulp chlorination. Environ
Sci Technol, 15: 562-566.
Krishna S, Agbenyega T, Angus BJ, Bedu-Addo G, Ofori-Amanfo G,
Henderson G, Szwandt IS, O'Brien R, & Stacpoole PW (1995)
Pharmacokinetics and pharmacodynamics of dichloroacetate in children
with lactic acidosis due to severe malaria. Q J Med, 88: 341-349.
Kristiansen NK, Froshaug M, Aune KT, & Becher G (1994) Identification
of halogenated compounds in chlorinated seawater and drinking water
produced offshore using n-pentane extraction and open-loop stripping
technique. Environ Sci Technol, 28: 1669-1673.
Kroll RB, Robinson GD, & Chung JH (1994a) Characterization of
trihalomethane (THM)-induced renal dysfunction in the rat: I. Effects
of THM on glomerular filtration and renal concentrating ability. Arch
Environ Contam Toxicol, 27: 1-4.
Kroll RB, Robinson GD, & Chung JH (1994b) Characterization of
trihalomethane (THM)-induced renal dysfunction in the rat: I. Relative
potency of THMs in promoting renal dysfunction. Arch Environ Contam
Toxicol, 27: 5-7.
Kronberg L & Vartiainen T (1988) Ames mutagenicity and concentration
of the strong mutagen
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone and its geometric
isomer (E)-2-chloro-3-(dichloromethyl)-4-oxo-butenoic acid in
chlorine-treated tap water. Mutat Res, 206: 177-182.
Kronberg L, Holmbon B, Reunanen M, & Tikkanen L (1988) Identification
and quantification of the Ames mutagenic compound
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone and of its
geometric isomer (E)-2-chloro-3-(dichloromethyl)-4-oxobutenoic acid in
chlorine treated humic water and drinking water extracts. Environ Sci
Technol, 22(9): 1097-1103.
Kronberg L, Karlsson S, & Sjoholm R (1993) Formation of
ethenocarbaldehyde derivatives of adenosine and cytidine in reactions
with mucochloric acid. Chem Res Toxicol, 6: 495-499.
Kruithof JC & Meijers RT (1993) Presence and formation of bromate ion
in Dutch drinking water treatment. In: Proceedings of the
International Water Supply Association Workshop, Paris. Boston,
Massachusetts, Blackwell Scientific Publications, pp 125-133.
Kurlemann G, Paetzke I, Moller H, Masur H, Schuierer G, Weglage J, &
Koch HG (1995) Therapy of complex I deficiency: Peripheral neuropathy
during dichloroacetate therapy. Eur J Pediatr, 154: 928-932.
Kurokawa Y, Hayashi Y, Maekawa A, Takahashi M, Kokubo T, & Odashima S
(1983) Carcinogenicity of potassium bromate administered orally to
F344 rats. J Natl Cancer Inst, 71: 965-972.
Kurokawa Y, Takamura N, Matsushima Y, Imazawa T, & Hayashi Y (1984)
Studies on the promoting and complete carcinogenic activities of some
oxidizing chemicals in skin carcinogenesis. Cancer Lett, 24: 299-304.
Kurokawa Y, Imazawa T, Matsushima M, Takamura N, & Hayashi Y (1985a)
Lack of promoting effect of sodium chlorate and potassium chlorate in
two-stage rat renal carcinogenesis. J Am Coll Toxicol, 4: 331-337.
Kurokawa Y, Aoki S, Imazawa T, Hayashi Y, Matsushima Y, & Takamura N
(1985b) Dose-related enhancing effect of potassium bromate on renal
tumorigenesis in rats initiated with
N-ethyl- N-hydroxyethyl-nitrosamine. Jpn J Cancer Res (Gann),
76: 583-589.
Kurokawa Y, Aoki S, Matsushima Y, Takamura N, Imazawa T, & Hayashi Y
(1986a) Dose-response studies on the carcinogenicity of potassium
bromate in F344 rats after long-term oral exposure. J Natl Cancer
Inst, 77: 977-982.
Kurokawa Y, Takayama S, Konishi Y, Hiasa Y, Asahina S, Takahashi M,
Maekawa A, & Hayashi Y (1986b) Long-term in vivo carcinogenicity
tests of potassium bromate, sodium hypochlorite, and sodium chlorite
conducted in Japan. Environ Health Perspect, 69: 221-235.
Kurokawa Y, Matsushima Y, Takamura N, Imazawa T, & Hayashi Y (1987a)
Relationship between the duration of treatment and the incidence of
renal cell tumors in male F344 rats administered potassium bromate.
Jpn J Cancer Res (Gann), 78: 358-364.
Kurokawa Y, Takamura N, Matsuoka C, Imazawa T, Matsushima Y, Onodera
H, & Hayashi Y (1987b) Comparative studies on lipid peroxidation in
the kidney of rats, mice, and hamsters and on the effect of cysteine,
glutathione, and diethyl maleate treatment on mortality and
nephrotoxicity after administration of potassium bromate. J Am Coll
Toxicol, 6: 489-501.
Kurokawa Y, Maekawa A, Takahashi M, & Hayashi Y (1990) Toxicity and
carcinogenicity of potassium bromate - A new renal carcinogen. Environ
Health Perspect, 87: 309-335.
Kuwahara T, Ikehara Y, Kanatsu K, Doi T, Nagai H, Nakayashiki H,
Tamura T, & Kawai C (1984) Two cases of potassium bromate poisoning
requiring long-term hemodialysis therapy for irreversible tubular
damage. Nephron, 37: 278-280.
Lacey JH & Randle PJ (1978) Inhibition of lactate gluconeogenesis in
rat kidney by dichloroacetate. Biochem J, 170: 551-560.
Lake BG (1995) Peroxisome proliferation: current mechanisms relating
to non-genotoxic carcinogenesis. Toxicol Lett, 82/83: 673-681.
LaLonde RT & Xie S (1992) A study of inactivation reactions of
N-acetylcysteine with mucochloric acid, a mutagenic product of the
chlorination of humic substances in water. Chem Res Toxicol,
5: 618-624.
LaLonde RT & Xie S (1993) Glutathione and N-acetylcysteine
inactivations of mutagenic 2(5H)-furanones from the chlorination of
humics in water. Chem Res Toxicol, 6: 445-451.
LaLonde RT, Cook GP, Perakyla H, & Dence CW (1991a) Effect on
mutagenicity of the stepwise removal of hydroxyl group and chlorine
atoms from 3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone: 13C
NMR chemical shifts as determinants of mutagenicity. Chem Res Toxicol,
4: 35-40.
LaLonde RT, Cook GP, Perakyla H, & Bu L (1991b) Structure-activity
relationships of bacterial mutagens related to
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone: An emphasis on
the effect of stepwise removal of chlorine from the dichloromethyl
group. Chem Res Toxicol, 4: 540-545.
LaLonde RT, Leo H, Perakyla H, Dence CW, & Farrell RP (1992)
Associations of the bacterial mutagenicity of halogenated
2(5H)-furanones with their MNDO-PM3 computed properties and mode of
reactivity with sodium borohydride. Chem Res Toxicol, 5: 392-400.
Lambert GH, Muraskas J, Anderson CL, & Myers TF (1990) Direct
hyperbilirubinemia associated with chloral hydrate administration in
the newborn. Pediatrics, 86: 277-281.
Lange AL & Kawczynski E (1978) Controlling organics: the Contra Costa
County Water District experience. J Am Water Works Assoc, 70(11): 63.
Laptook AR & Rosenfeld CR (1984) Chloral hydrate toxicity in a preterm
infant. Pediatr Pharmacol, 4: 161-165.
Larson JL & Bull RJ (1992) Metabolism and lipoperoxidative activity of
trichloroacetate and dichloroacetate in rats and mice. Toxicol Appl
Pharmacol, 115: 268-277.
Larson RA & Rockwell AL (1979) Chloroform and chlorophenol production
by decarboxylation of natural acids during aqueous chlorination.
Environ Sci Technol, 13(3): 325-329.
Larson JL, Wolf DC, & Butterworth BE (1994a) Induced cytolethality and
regenerative cell proliferation in the livers and kidneys of male
B6C3F1 mice given chloroform by gavage. Fundam Appl Toxicol,
23: 537-543.
Larson JL, Wolf DC, & Butterworth BE (1994b) Induced cytotoxicity and
cell proliferation in the hepatocarcinogenicity of chloroform in
female B6C3F1 mice: Comparison of administration by gavage in corn
oil vs. ad libitum in drinking water. Fundam Appl Toxicol,
22(1): 90-102.
Larson JL, Wolf DC, & Butterworth BE (1995a) Induced regenerative cell
proliferation in livers and kidneys of male F-344 rats given
chloroform in corn oil by gavage or ad libitum in drinking water.
Toxicology, 95: 73-86.
Larson JL, Wolf DC, Mery S, Morgan KT, & Butterworth BE (1995b)
Toxicity and cell proliferation in the liver, kidneys, and nasal
passages of female F-344 rats induced by chloroform administered by
gavage. Food Chem Toxicol, 33: 443-456.
Larson JL, Templin MV, Wolf DC, Jamison KC, Leininger JR, Mery S,
Morgan KT, Wong BA, Conolly RB, & Butterworth BE (1996) A 90-day
chloroform inhalation study in female and male B6C3F1 mice:
implications for cancer risk assessment. Fundam Appl Toxicol,
30: 118-137.
Laurie RD, Bercz JP, Wessendarp TK, & Condie LW (1986) Studies of the
toxic interactions of disinfection by-products. Environ Health
Perspect, 69: 203-207.
Lawrence WH, Dillingham EO, Turner JE, & Autian J (1972) Toxicity
profile of chloroacetaldehyde. J Pharm Sci, 61: 19-25.
Lawrence CE, Taylor PR, Trock BJ, & Reilly AA (1984) Trihalomethanes
in drinking water and human colorectal cancer. J Natl Cancer Inst,
72: 563-568.
Leavitt SA, DeAngelo AB, George MH, & Ross JA (1997) Assessment of the
mutagenicity of dichloroacetic acid in lacI transgenic B6C3F1 mice.
Carcinogenesis, 18: 2101-2106.
LeBel GL, Benoit FM, & Williams DT (1995) Variation of chlorinated
DBPs in water from treatment plants using three different disinfection
processes. In: Proceedings of the Conference on Water Quality
Technology, New Orleans, LA.
Le Curieux F, Gauthier L, Erb F, & Marzin D (1995) The use of the SOS
chromotest, the Ames-fluctuation test and the newt micronucleus test
to study the genotoxicity of four trihalomethanes. Mutagenesis,
10: 333-341.
Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL,
Fernandez-Salguero PM, Westphal H, & Gonzalez FJ (1995) Targeted
disruption of the isoform of the peroxisome proliferator-activated
receptor gene in mice results in abolishment of the pleiotropic
effects of peroxisome proliferators. Mol Cell Biol, 15: 3012-3022.
Legube B, Bourbigot M, Bruchet A, Deguin A, Montiel A, & Matia L
(1993) Bromide ion/bromate ion survey on different European water
utilities. In: Proceedings of the International Water Supply
Association Workshop, Paris. Boston, Massachusetts, Blackwell
Scientific Publications.
Leuschner J & Leuschner F (1991) Evaluation of the mutagenicity of
chloral hydrate in vitro and in vivo. Drug Res, 41: 1101-1103.
Lewandowski ED & White LT (1995) Pyruvate dehydrogenase influences
postischemic heart function. Circulation, 91: 2071-2079.
Lichtenberg R, Zeller WP, Gatson R, & Hurley RM (1989) Clinical and
laboratory observations: Bromate poisoning. J. Pediatr, 114: 891-894.
Lilly PD, Simmons JE, & Pegram RA (1994) Dose-dependent vehicle
differences in the acute toxicity of bromodichloromethane. Fundam Appl
Toxicol, 23: 132-140.
Lilly PD, Simmons JE, & Pegram RA (1996) Effect of subchronic corn oil
gavage on the acute toxicity of orally administered
bromodichloromethane. Toxicol Lett, 87: 93-102.
Lilly PD, Ross TM, & Pegram RA (1997a) Trihalomethane comparative
toxicity: Acute renal and hepatic toxicity of chloroform and
bromodichloromethane following aqueous gavage. Fundam Appl Toxicol,
40: 101-110.
Lilly PD, Andersen ME, Ross TM, & Pegram RA (1997b) Physiologically
based estimation of in vivo rates of bromodichloromethane
metabolism. J Toxicol, 124: 141-152.
Lilly PD, Andersen ME, Ross TM, & Pegram RA (1998) A physiologically
based pharmacokinetic description of the oral uptake, tissue dosimetry
and rates of metabolism of bromodichloromethane in the male rat.
Toxicol Appl Pharmacol, 150: 205-217.
Lin EL, Daniel FB, Herren-Freund SL, & Pereira MA (1986)
Haloacetonitriles: Metabolism, genotoxicity, and tumor-initiating
activity. Environ Health Perspect, 69: 67-71.
Lin EL, Reddy TV, & Daniel FB (1992) Macromolecular adduction by
trichloroacetonitrile in the Fischer 344 rat following oral gavage.
Cancer Lett, 62: 1-9.
Lin EL, Mattox JK, & Daniel FB (1993) Tissue distribution, excretion,
and urinary metabolites of dichloroacetic acid in the male Fischer 344
rat. J Toxicol Environ Health, 38: 19-32.
Linder RE, Strader LF, Slott VL, & Suarez JD (1992) Endpoints of
spermatoxicity in the rat after short duration exposures to fourteen
reproductive toxicants. Reprod Toxicol, 6: 491-505.
Linder RE, Klinefelter GR, Strader LF, Suarez JD, & Dyer CJ (1994a)
Acute spermatogenic effects of bromoacetic acids. Fundam Appl Toxicol,
22: 422-430.
Linder RE, Klinefelter GR, Strader LF, Suarez JD, Roberts NL, & Dyer
CJ (1994b) Spermatotoxicity of dibromoacetic acid in rats after 14
daily exposures. Reprod Toxicol, 8: 251-259.
Linder RE, Klinefelter GR, Strader LF, Narotosky MG, Suarez JD,
Roberts NL, & Perreault SD (1995) Dibromoacetic acid affects
reproductive competence and sperm quality in the male rat. Fundam Appl
Toxicol, 28: 9-17.
Linder RE, Klinefelter GR, Strader LF, Suarez JD, & Roberts NL (1997a)
Spermatoxicity of dichloroacetic acid. Reprod Toxicol, 11: 681-688.
Linder RE, Klinefelter GR, Strader LF, Veeramachaneni DR, Roberts NL,
& Suarez JD (1997b) Histopathological changes in the testes of rats
exposed to dibromoacetic acid. Reprod Toxicol, 11: 47-56.
Lindstrom AB, Pleil JD, & Berkoff DC (1997) Alveolar breath sampling
and analysis to assess trihalomethane exposures during competitive
swimming training. Environ Health Perspect, 105(6): 636-642.
Lipscomb JC, Mahle DA, Brashear WT, & Barton HA (1995) Dichloroacetic
acid: Metabolism in cytosol. Drug Metab Dispos, 23: 1202-1205.
Lock EA, Gyte A, Widdowson P, Simpson M, & Wyatt I (1995)
Chloropropionic acid-induced alterations in glucose metabolic status:
possible relevance to cerebellar cell necrosis. Arch Toxicol,
69: 640-643.
Lopaschuk GD & Saddik M (1992) The relative contribution of glucose
and fatty acids to ATP production in hearts reperfused following
ischemia. Mol Cell Biochem, 116: 111-116.
Loveday KS, Anderson BE, Resnick MA, & Zeiger E (1990) Chromosome
aberration and sister chromatid exchange tests in Chinese hamster
ovary cells in vitro: V. Results with 46 chemicals. Environ Mol
Mutagen, 16: 272-303.
Lubbers JR & Bianchine JR (1984) Effects of the acute rising dose
administration of chlorine dioxide, chlorate and chlorite to normal
healthy adult male volunteers. J Exp Pathol Toxicol Oncol, 5: 215-228.
Lubbers JR, Chauhan S, & Bianchine JR (1981) Controlled clinical
evaluations of chlorine dioxide, chlorite and chlorate in man. Fundam
Appl Toxicol, 1: 334-338.
Lubbers JR, Chauan S, & Bianchine R (1982) Controlled clinical
evaluations of chlorine dioxide, chlorite, and chlorite in man.
Environ Health Perspect, 46: 57-62.
Lubbers JR, Chauhan S, Miller JK, & Bianchine JR (1984a) The effects
of chronic administration of chlorine dioxide, chlorite and chlorate
to normal healthy adult male volunteers. J Exp Pathol Toxicol Oncol,
5: 229-238.
Lubbers JR, Chauhan S, Miller JK, & Bianchine JR (1984b) The effects
of chronic administration of chlorite to glucose-6-phosphate
dehydrogenase deficient healthy adult male volunteers. J Environ
Pathol Toxicol Oncol, 5: 239-242.
Lue JN, Johnson CE, & Edwards DL (1988) Drug experience: Bromate
poisoning from ingestion of professional hair-care neutralizer. Clin
Pharm, 7: 66-70.
Lukas G, Vyas KH, Brindle SD, Le Sher AR, & Wagner WE (1980)
Biological disposition of sodium dichloroacetate in animals and humans
after intravenous administration. J Pharm Sci, 69: 419-421.
Lukasewycz MT, Bieringer CM, Liukkonen RJ, Fitzsimmons ME, Corcoran
HF, Sechoing L, & Carlson RM (1989) Analysis of inorganic and organic
chloramines: derivatization with 2-mercaptobenzothiazole. Environ Sci
Technol, 23(2): 196-199.
Luknitskii FI (1975) The chemistry of chloral. Chem Rev, 75: 82-94.
Lykins BW & Clark RM (1988) GAC for removing THMs. Washington, DC, US
Environmental Protection Agency (EPA 600/9-88/004).
Lynch AM & Parry JM (1993) The cytochalasin-B micronucleus/kinetochore
assay in vitro: Studies with 10 suspected aneugens. Mutat Res,
287(1): 71-86.
Lynch CF, Woolson RF, O'Gorman T, & Cantor KP (1989) Chlorinated
drinking water and bladder cancer: effect of misclassification on risk
estimates. Arch Environ Health, 44: 252-259.
Lynch CF, VanLier S, & Cantor KP (1990) A case-control study of
multiple cancer sites and water chlorination in Iowa. In: Jolley RL,
Condie LW, & Johnson JD ed. Water chlorination: Chemistry,
environmental impact and health effects. Chelsea, Michigan, Lewis
Publishers, Inc., vol 6, p 387.
McCabe LJ (1975) Association between halogenated methanes in drinking
water and mortality (NORS data). Cincinnati, Ohio, US Environmental
Protection Agency, Water Quality Division, 4 pp.
McCauley PT, Robinson M, Daniel FB, & Olson GR (1995) The effects of
subchronic chlorate exposure in Sprague-Dawley rats. Drug Chem
Toxicol, 18: 185-199.
McGeehin M, Reif J, Becker J, & Mangione E (1993) A case-control study
of bladder cancer and water disinfection in Colorado. Am J Epidemiol,
138(7): 492-501 (Abstract 127).
McGregor DB, Brown AG, Howgate S, McBride D, Riach C, & Caspary WJ
(1991) Responses of the L5178Y mouse lymphoma cell forward mutation
assay: V. 27 coded chemicals. Environ Mol Mutagen, 17: 96-219.
McGuire MJ & Meadow RG (1988) AWWARF trihalomethane survey. J Am Water
Works Assoc, 80(1): 61-68.
Mackay JM, Fox V, Griffiths K, Fox DA, Howard CA, Coutts C, Wyatt I, &
Styles JA (1995) Trichloroacetic acid: investigation into the
mechanism of chromosomal damage in the in vitro human lymphocyte
cytogenetic assay and the mouse bone marrow micronucleus test.
Carcinogenesis, 16(5): 1127-1133.
McVeigh JJ & Lopaschuk GD (1990) Dichloroacetate stimulation of
glucose oxidation improves recovery of ischemic rat hearts. Am J
Physiol, 259: H1079-H1085.
Mailhes JB, Aardema MJ, & Marchetti F (1993) Investigation of
aneuploidy induction in mouse oocytes following exposure to
vinblastine-sulfate, pyrimethamine, diethylstilbestrol, or chloral
hydrate. Environ Mol Mutagen, 22(2): 107-114.
Maki-Paakkanen & Jansson K (1995) Cytogenetic effects in the
peripheral lymphocytes and kidney cells of rats exposed to
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX) orally on
three consecutive days. Mutat Res, 343: 151-156.
Man KC & Brosnan JT (1982) Inhibition of medium and short-chain fatty
acid oxidation in rat heart mitochondria by dichloroacetate.
Metabolism, 31: 744-748.
Marcus PM, Savitz DA, Millikan RC, & Morgenstern H (1998) Female
breast cancer and trihalomethanes in drinking water in North Carolina.
Epidemiology, 9(2): 156-160.
Maronpot RR, Fox T, Malarkey DE, & Goldsworthy TL (1995) Mutations in
the ras proto-oncogene: clues to etiology and molecular pathogenesis
of mouse liver tumors. Toxicology, 101: 125-156.
Marshall EK & Owens AH (1954) Absorption, excretion, and metabolic
fate of chloral hydrate and trichloroethanol. Bull Johns Hopkins Hosp,
95: 1-18.
Mather GG, Exon JH, & Koller LD (1990) Subchronic 90 day toxicity of
dichloroacetic and trichloroacetic acid in rats. Toxicology,
64: 71-80.
Mathews JM, Troxler PS, & Jeffcoat AR (1990) Metabolism and
distribution of bromodichloromethane in rats after single and multiple
oral doses. J Toxicol Environ Health, 30: 15-22.
Matsuoka A, Hayashi M, & Ishidate M (1979) Chromosomal aberration
tests on 29 chemicals combined with S9 mix in vitro. Mutat Res,
66: 277-290.
Mayers DJ, Hindmarsh KW, Sankaran K, Gorecki DK, & Kasian GF (1991)
Chloral hydrate disposition following single-dose administration to
critically ill neonates and children. Dev Pharmacol Ther, 16: 71-77.
Meier JR (1988) Genotoxic activity of organic chemicals in drinking
water. Mutat Res, 196: 211-245.
Meier JR, Lingg RD, & Bull RJ (1983) Formation of mutagens following
chlorination of humic acid: A model for mutagen formation during
drinking water. Mutat Res, 118: 25-41.
Meier JR, Ringhand HP, Coleman WE, Munch JW, Streicher RP, Kaylor WH,
& Schenk KM (1985a) Identification of mutagenic compounds formed
during chlorination of humic acid. Mutat Res, 157: 111-122.
Meier JR, Bull RJ, Stober JA, & Cimino MC (1985b) Evaluation of
chemicals used for drinking water disinfection for production of
chromosomal damage and sperm-head abnormalities in mice. Environ
Mutagen, 7: 201-211.
Meier JR, Ringhand HP, Coleman WE, & Schenck KM (1986) Mutagenic
by-products from chlorination of humic acid. Environ Health Perspect,
69: 101-107.
Meier JR, Knohl RB, Coleman WE, Ringhand HP, Munch JW, Kaylor WH,
Streicher RP, & Kopfler FC (1987a) Studies on the potent bacterial
mutagen, 3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone: aqueous
stability, XAD recovery and analytical determination in drinking water
and in chlorinated humic acid solutions. Mutat Res, 189: 363-373.
Meier JR, Blazek WF, & Knohl RB (1987b) Mutagenic and clastogenic
properties of 3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone: A
potent bacterial mutagen in drinking water. Environ Mol Mutagen,
10: 411-424.
Meier JR, Monarca S, Patterson KS, Villarini M, Daniel FB, Moretti M,
& Pasquini R (1996) Urine mutagenicity and biochemical effects of the
drinking water mutagen,
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX) following
repeated oral administration to mice and rats. Toxicology, 110: 59-70.
Mel HC, Jolly WL, & Latimer W (1953) The heat and free energy of
formation of bromate ion. J Am Chem Soc, 75: 3827.
Melnick RL, Kohn MC, Dunnick JK, & Leininger KR (1998) Regenerative
hyperplasia is not required for liver tumor induction in female
B6C3F1 mice exposed to trihalomethanes. Toxicol Appl Pharmacol,
148(1): 133-136.
Mengele K, Schwarzmeier J, Schmidt P, & Moser K (1969) [Clinical
features and investigations of the erythrocyte metabolism in case of
intoxication with sodium chlorate.] Int J Clin Pharmacol, 2: 120-125
(in German).
Merdink JL, Gonzalez-Leon A, Bull RJ, & Schultz IR (1998) The extent
of dichloroacetate formation from trichloroethylene, chloral hydrate,
trichloroacetate and trichloroethanol in B6C3F1. Toxicol Sci,
45(1): 33-41.
Merdink JL, Stenner RD, Stevens DK, Parker JC, & Bull RJ (1999) Effect
of enterohepatic circulation on the pharmacokinetics of chloral
hydrate in F344 rats. J Toxicol Environ Health, 57(5): 357-368.
Merlet N, Thibaud H, & Dore M (1985) Chloropicrin formation during
oxidative treatment in the preparation of drinking water. Sci Total
Environ, 47: 223-228.
Merrick BA, Smallwood CL, Meier JR, McKean DL, Kaylor WH, & Condie LW
(1987) Chemical reactivity, cytotoxicity, and mutagenicity of
chloropropanones. Toxicol Appl Pharmacol, 91: 46-54.
Merrick BA, Meier JR, Smallwood CL, McKean DL, & Condie LW (1990)
Biochemical mechanisms of in vitro chloropropanone toxicity. In:
Jolley RL, Condie LW, & Johnson JD ed. Water chlorination: Chemistry,
environmental impact and health effects. Ann Arbor, Michigan, Lewis
Publishers, Inc., vol 6, pp 329-339.
Michael GE, Miday RK, Bercz JP, Miller RG, Greathouse DG, Kraemer DF,
& Lucas JB (1981) Chlorine dioxide water disinfection: A prospective
epidemiological study. Arch Environ Health, 36: 20-27.
Miller WJ & Uden PC (1983) Characterization of nonvolatile aqueous
chlorination products of humic substances. Environ Sci Technol,
17(3): 150-152.
Miller LH, Brownstein MH, & Hyman AB (1966) Fixed eruption due to
chloral hydrate. Arch Dermatol, 94: 60-61.
Miltner RJ, Shukairy HM, & Summers RS (1992) Disinfection by-product
formation and control by ozonation and biotreatment. J Am Water Works
Assoc, 84(11): 53-62.
Mink FL, Coleman WE, Munch JW, Kaylor WH, & Ringhand HP (1983)
In vivo formation of halogenated reaction products following peroral
sodium hypochlorite. Bull Environ Contam Toxicol, 30: 394-399.
Mink FL, Brown TJ, & Rickabaugh J (1986) Absorption, distribution and
excretion of 14C-trihalomethanes in mice and rats. Bull Environ
Contam Toxicol, 37: 752-758.
MMWR (1991) Chlorine gas toxicity from mixture of bleach with other
cleaning products - California. Morb Mortal Wkly Rep, 40(36): 619-621.
Mobley SA, Taylor DH, Laurie RD, & Pfohl RJ (1990) Chlorine dioxide
depresses T3 uptake and delays development of locomotor activity in
young rats. In: Jolley RL, Condie LW, & Johnson JD ed. Water
chlorination: Chemistry, environmental impact and health effects. Ann
Arbor, Michigan, Lewis Publishers, Inc., vol 6, pp 347-360.
Monson RR (1980) Occupational epidemiology. Boca Raton, Florida, CRC
Press, Inc., p 88.
Montgomery Watson, Inc. (1993) Disinfection/DBP database for the
negotiated regulation. Denver, Colorado, American Water Works
Association.
Moore GS & Calabrese EJ (1980) The effects of chlorine dioxide and
sodium chlorite on erythrocytes of A/J and C57BL/J mice. J Environ
Pathol Toxicol, 4: 513-524.
Moore GS & Calabrese EJ (1982) Toxicological effects of chlorite in
the mouse. Environ Health Perspect, 46: 31-37.
Moore BB & Sherman M (1991) Chronic reactive airway disease following
acute chlorine gas exposure in an asymptomatic atopic patient. Chest,
100: 855-856.
Moore GS, Tuthill RW, & Polakoff DW (1979a) A statistical model for
predicting chloroform levels in chlorinated surface water supplies.
J Am Water Works Assoc, 71: 37-45.
Moore GW, Swift LL, Rabinowitz DR, Crofford OB, Oates JA, & Stacpoole
PW (1979b) Reduction of serum cholesterol in two patients with
homozygous familial hypercholesterolemia by dichloroacetate.
Atherosclerosis, 33: 285-293.
Moore GS, Calabrese EJ, & McGee M (1980a) Health effects of
monochloramine in drinking water. J Environ Sci Health, A15: 239-258.
Moore GS, Calabrese EJ, & Leonard DA (1980b) Effects of chlorite
exposure on conception rate and litters of A/J mice. Bull Environ
Contam Toxicol, 25: 689-696.
Moore GS, Calabrese EJ, & Forti A (1984) The lack of nephrotoxicity in
the rat by sodium chlorite, a possible byproduct of chlorine dioxide
disinfection in drinking water. J Environ Sci Health, A19: 643-661.
Moore TC, DeAngelo AB, & Pegram RA (1994) Renal toxicity of
bromodichloromethane and bromoform administered chronically to rats
and mice in drinking water. Toxicologist, 14: 281 (abstract).
Morimoto K & Koizumi A (1983) Trihalomethanes induce sister chromatid
exchanges in human lymphocytes in vitro and mouse bone marrow cells
in vivo. Environ Res, 32: 72-79.
Morris JC, Ram N, Baum B, & Wajon E (1980) Formation and significance
of N-chloro compounds in water supplies. Washington, DC, US
Environmental Protection Agency (EPA 600/2-80-031).
Morris RD, Audet AM, & Angelillo IF (1992) Chlorination, chlorination
by-products and cancer: a meta-analysis. Am J Public Health,
82: 955-963.
Morrow NM & Minear RA (1987) Use of regression models to link raw
water characteristics to THM concentration in drinking water. Water
Res, 21: 41.
Müller G, Spassovski M, & Henschler D (1974) Metabolism of
trichloroethylene in man: II. Pharmacokinetics of metabolites. Arch
Toxicol, 32: 283-295.
Muller SP, Wolna P, Wunscher U, & Pankow D (1997) Cardiotoxicity of
chlorodibromomethane and trichloromethane in rats and isolated rat
cardiac myocytes. Arch Toxicol, 71(12): 766-777.
Mullins PA & Proudlock RJ (1990) Assessment of nuclear anomalies in
mice after administration of MX. Medmenham, United Kingdom, Water
Research Centre (Report prepared by the Huntingdon Research Centre,
United Kingdom).
Munson AE, Sain LE, Sanders VM, Kauffman BM, White KL, Page DG, Barnes
DW, & Borzelleca JF (1982) Toxicology of organic water contaminants:
trichloromethane, bromodichloromethane, dibromochloromethane and
tribromomethane. Environ Health Perspect, 46: 117-126.
Murphy PA (1993) Quantifying chemical risk from epidemiology studies:
Application to the disinfectant by-product issue. In: Craun GF ed.
Safety of water disinfection: Balancing chemical and microbial risks.
Washington, DC, ILSI Press, pp 373-387.
Murphy PA & Craun GF (1990) A review of previous studies reporting
associations between drinking water disinfection and cancer. In:
Jolley RL, Condie LW, & Johnson JD ed. Water chlorination: Chemistry,
environmental impact and health effects. Chelsea, Michigan, Lewis
Publishers, Inc., vol 6, p 361.
Musil J, Knotek A, Chalupa J, & Schmidt P (1964) Toxicologic aspects
of chlorine dioxide application for the treatment of water containing
phenols. Scientific papers from Institute of Chemical Technology,
Prague. Technol Water, 8: 327-345.
Nakajima M, Kitazawa M, Oba K, Kitagawa Y, & Toyoda Y (1989) Effect of
route of administration in the micronucleus test with potassium
bromate. Mutat Res, 223: 399-402.
Nakajima T, Elovaara E, Okino T, Gelboin HV, Klockars M, Riihimaki V,
Aoyama T, & Vainio H (1995) Different contributions of cytochrome P450
2E1 and P450 2B1/2 to chloroform hepatotoxicity in rat. Toxicol Appl
Pharmacol, 133: 215-222.
Narotsky MG, Hamby BT, Mitchell DS, & Kavlock RJ (1993) Bromoform
requires a longer exposure period than carbon tetrachloride to induce
pregnancy loss in F-344 rats. Toxicologist, 13: 255.
Narotsky MG, Pegram RA, & Kavlock RJ (1997) Effect of dosing vehicle
on the developmental toxicity of bromodichloromethane and carbon
tetrachloride in rats. Fundam Appl Toxicol, 40: 30-36.
NAS (1980) Committee on Drinking Water - Drinking water and health.
Washington, DC, National Academy of Sciences, National Academy Press,
vol 3, 415 pp.
NAS (1986) Committee on Drinking Water - Drinking water and health.
Washington, DC, National Academy of Sciences, National Academy Press,
vol 6, 457 pp.
Natarajan AT, Duivenvoorden WC, Meijers M, & Zwanenburg TS (1993)
Induction of mitotic aneuploidy using Chinese hamster primary
embryonic cells: Test results of 10 chemicals. Mutat Res,
287(1): 47-56.
National Center for Environmental Assessment (NCEA) (1998) NCEA
position paper regarding risk assessment use of the results from the
published study. Am J Public Health, 82: 955-963.
Nelson MA & Bull RJ (1988) Induction of strand breaks in DNA by
trichloroethylene and metabolites in rat and mouse liver in vivo.
Toxicol Appl Pharmacol, 94: 45-54.
Nelson MA, Lansing AJ, Sanchez IM, Bull RJ, & Springer DL (1989)
Dichloroacetic acid and trichloroacetic acid-induced DNA strand breaks
are independent of peroxisome proliferation. Toxicology, 58: 239-248.
Nelson MA, Sanchez IM, Bull RJ, & Sylvester SR (1990) Increased
expression of c-myc and c-H-ras in dichloroacetate and
trichloroacetate-induced liver tumors in B6C3F1 mice. Toxicology,
64: 47-57.
Nestmann ER, Chu I, Kowbel DJ, & Matula TI (1980) Short-lived mutagen
in Salmonella produced by reaction of trichloroacetic acid and
dimethyl sulphoxide. Can J Genet Cytol, 22(1): 35-40.
Neutra RR & Ostro B (1992) An evaluation of the role of epidemiology
in assessing current and future disinfection technologies for drinking
water. Sci Total Environ, 127: 91-138.
Ni YC, Wong TY, Lloyd RV, Heinze TM, Shelton S, Casciano D, Kadlubar
FF, & Fu PP (1996) Mouse liver microsomal metabolism of chloral
hydrate, trichloroacetic acid, and trichloroethanol leading to
induction of lipid peroxidation via a free radical mechanism. Drug
Metab Dispos, 24: 81-90.
Nicholl TA, Lopaschuk GD, & McNeill JH (1991) Effects of free fatty
acids and dichloroacetate on isolated working diabetic heart. Am J
Physiol, 261: H1053-H1059.
Nieminski EC, Chaudhuri S, & Lamoreaux T (1993) The occurrence of DBPs
in Utah drinking waters. J Am Water Works Assoc, 85(9): 98-105.
Nishikawa A, Kinae N, Furukawa F, Mitsui M, Enami T, Hasegawa T, &
Takahashi M (1994) Enhancing effects of
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX) on cell
proliferation and lipid peroxidation in the rat gastric mucosa. Cancer
Lett, 85: 151-157.
Noack MG & Doerr RL (1978) Reactions of chlorine, chlorine dioxide and
mixtures thereof with humic acid: an interim report on water
chlorination. In: Jolley RL, Gorchev H, & Hamilton DH ed. Water
chlorination: Environmental impact and health effects. Ann Arbor,
Michigan, Ann Arbor Science Publishers, vol 2, pp 49-65.
Nordenberg A, Delisle G, & Izukawa T (1971) Cardiac arrhythmia in a
child due to chloral hydrate ingestion. Pediatrics, 47: 134-135.
NTP (1985) Toxicology and carcinogenesis studies of
chlorodibromomethane in F344/N rats and B6C3F1 mice (Gavage studies).
Research Triangle Park, North Carolina, US Department of Health and
Human Services, National Toxicology Program (NTP Technical Report
Series No. 282).
NTP (1987) Toxicology and carcinogenesis studies of
bromodichloromethane in F344/N rats and B6C3F1 mice. Research
Triangle Park, North Carolina, US Department of Health and Human
Services, National Toxicology Program (NTP Technical Report Series No.
321; NIH Publication No. 88-2537).
NTP (1989a) Toxicology and carcinogenesis studies of tribromomethane
(bromoform) in F344/N rats and B6C3F1 mice (Gavage studies). Research
Triangle Park, North Carolina, US Department of Health and Human
Services, National Toxicology Program (NTP Technical Report Series No.
350).
NTP (1989b) Bromoform: Reproduction and fertility assessment in Swiss
CD-1 mice when administered by gavage. Research Triangle Park, North
Carolina, US Department of Health and Human Services, National
Toxicology Program (NTP-89-068).
NTP (1992) Technical report on the toxicology and carcinogenesis
studies of chlorinated and chloraminated water in F344/N rats and
B6C3F1 mice. Research Triangle Park, North Carolina, US Department of
Health and Human Services, National Toxicology Program (NTP Technical
Report Series No. 392; NIH Publication No. 91-2847).
Nuckols JR, Stallones L, & Reif J (1995) Evaluation of the use of a
geographic information system in drinking water epidemiology. In:
Reivhard E & Zapponi G ed. Assessing and managing health risks from
drinking water contamination: Approaches and applications.
Wallingford, United Kingdom, International Association of Hydrological
Sciences, pp 111-122 (IAHS Publication No. 233).
Nudel DB, Peterson BJ, Buckley BJ, Kaplan NA, Weinhouse E, & Gootman N
(1990) Comparative effects of bicarbonate and dichloroacetate in
newborn swine with hypoxic lactic acidosis. Dev Pharmacol Ther,
15: 86-93.
Nunn JW, Davies JE, & Chipman JK (1997) Production of unscheduled DNA
synthesis in rodent hepatocytes in vitro, but not in vivo, by
3-chloro-(dichloromethyl)-5-hydroxy-2[5H]-furanone (MX). Mutat Res,
373: 67-73.
Nutley EV, Tcheong AC, Allen JW, Collins BW, Ma M, Lowe XR, Bishop JB,
Moore DH, & Wyrobek AJ (1996) Micronuclei induced in round spermatids
of mice after stem-cell treatment with chloral hydrate: Evaluations
with centromeric DNA probes and kinetochore antibodies. Environ Mol
Mutagen, 28(2): 80-89.
Oliver BG (1983) Dihaloacetonitriles in drinking water: algae and
fulvic acids as precursors. Environ Sci Technol, 15: 1075-1080.
Omichinski JG, Soderlund EJ, Dybing E, Pearson PG, & Nelson SD (1988)
Detection and mechanism of formation of the potent direct-acting
mutagen 2-bromoacrolein from 1,2-dibromo-3-chloropropionic acid.
Toxicol Appl Pharmacol, 92: 286-294.
Onodera H, Tanigawa H, Matsushima Y, Maekawa A, Kurokawa Y, & Hayashi
Y (1986) Eosinophilic bodies in the proximal renal tubules of rats
given potassium bromate. Bull Natl Inst Hyg Sci, 103: 15-20.
Orme J, Taylor DH, Laurie RD, & Bull RJ (1985) Effects of chlorine
dioxide on thyroid function in neonatal rats. J Toxicol Environ
Health, 15: 315-322.
Osgood C & Sterling D (1991) Dichloroacetonitrile, a by-product of
water chlorination induces aneuploidy in Drosophila. Mutat Res,
261(2): 85-91.
Page T, Harris RH, & Epstein SS (1976) Drinking water and cancer
mortality in Louisiana. Science, 193: 55-57.
Pankow D, Damme B, Wunscher U, & Bergmann K (1997)
Chlorodibromomethane metabolism to bromide and carbon monoxide in
rats. Arch Toxicol, 71(4): 203-210.
Paode RD, Amy GL, Krasner SW, Summers RS, & Rice EW (1997) Predicting
the formation of aldehydes. J Am Water Works Assoc, 89(6): 79-93.
Parnell MJ, Koller LD, Exon JH, & Arnzen JM (1986) Trichloroacetic
acid effects on rat liver peroxisomes and enzyme-altered foci. Environ
Health Perspect, 69: 73-79.
Parrish JM, Austin EW, Stevens DK, Kinder DH, & Bull RJ (1996)
Haloacetate-induced oxidative damage to DNA in the liver of male
B6C3F1 mice. Toxicology, 110: 103-111.
Paykoc ZV & Powell JF (1945) The effect of sodium trichloroacetate.
J Pharmacol Exp Ther, 85: 289-293.
Pegram RA, Lilly PD, Thornton-Manning JR, Simmons JE, McDonald A, &
Moore TC (1993) Diurnal cycle dependent renal and hepatic toxicity of
bromodichloromethane. Toxicologist, 13: 360.
Pegram RA, Andersen ME, Warren SH, Ross TM, & Claxton LD (1996)
Glutathione S-transferase-mediated mutagenicity of trihalomethanes.
Toxicologist, 30: 291.
Pegram RA, Andersen ME, Warren SH, Ross TM, & Claxton LD (1997)
Glutathione S-transferase-mediated mutagenicity of trihalomethanes
in Salmonella typhimurium: contrasting results with
bromodichloromethane and chloroform. Toxicol Appl Pharmacol,
144: 183-188.
Penn A, Lu MX, & Parkes JL (1990) Ingestion of chlorinated water has
no effect upon indicators of cardiovascular disease in pigeons.
Toxicology, 63(3): 301-313.
Pereira MA (1994) Route of administration determines whether
chloroform enhances or inhibits cell proliferation in the liver of
B6C3F1 mice. Fundam Appl Toxicol, 23: 87-92.
Pereira MA (1995) Effect of dichloroacetic acid and trichloroacetic
acid upon cell proliferation in B6C3F1 mice: Final project report.
Denver, Colorado, American Water Works Association Research
Foundation.
Pereira MA (1996) Carcinogenic activity of dichloroacetic acid and
trichloroacetic acid in the liver of female B6C3F1 mice. Fundam Appl
Toxicol, 31: 192-199.
Pereira MA & Grothaus M (1997) Chloroform in drinking water prevents
hepatic cell proliferation induced by chloroform administered by
gavage in corn oil to mice. Fundam Appl Toxicol, 37(1): 82-87.
Pereira MA & Phelps BJ (1996) Promotion by dichloroacetic acid and
trichloroacetic acid of N-methyl- N-nitrosourea-initiated cancer in
the liver of female B6C3F1 mice. Cancer Lett, 102: 133-141.
Pereira MA, Lin LH, Lippitt JM, & Herren SL (1982) Trihalomethanes as
initiators and promoters of carcinogenesis. Environ Health Perspect,
46: 151-156.
Pereira MA, Lin LH, & Mattox JK (1984) Haloacetonitrile excretion as
thiocyanate and inhibition of dimethylnitrosamine demethylase: A
proposed metabolic scheme. J Toxicol Environ Health, 13: 633-641.
Peters RJB (1991) The analysis of halogenated acetic acids in Dutch
drinking water. Water Res, 25(4): 473-477.
Peters CJ, Young RJ, & Perry R (1980) Factors influencing the
formation of haloforms in the chlorination of humic materials. Environ
Sci Technol, 14: 1391-1395.
Peters RJB, De Leer EWB, & De Galan L (1990) Dihaloacetonitriles in
Dutch drinking water. Water Res, 24(6): 797-800.
Pfeiffer EH (1978) Health aspects of water chlorination with special
consideration to the carcinogenicity of chlorine: II. Communication:
On the carcinogenicity. Zent.bl Bakteriol Hyg I. Abt Orig B,
166: 185-211.
Piantadosi S (1994) Invited commentary: Ecologic biases. Am J
Epidemiol, 139(8): 71-64.
Piantadosi S, Byar DP, & Green SB (1988) The ecological fallacy. Am J
Epidemiol, 18: 269-274.
Pilotto LS (1995) Disinfection of drinking water, disinfection
by-products and cancer: what about Australia? Aust J Public Health,
19(1): 89-93.
Pleil JD & Lindstrom AB (1997) Exhaled human breath measurement method
for assessing exposure to halogenated volatile organic compounds. Clin
Chem, 43(5): 723-730.
Pohl LR, Branchflower RV, Highet RJ, Martin JL, Nunn DS, Monks TJ,
George JW, & Hinson JA (1981) The formation of diglutathionyl
dithiocarbonate as a metabolite of chloroform, bromotrichloromethane,
and carbon tetrachloride. Drug Metab Dispos, 9: 334-339.
Polhuijs M, Te Koppele JM, Fockens E, & Mulder GJ (1989) Glutathione
conjugation of the alpha-bromoisovaleric acid enantiomers in the rat
in vivo and its stereoselectivity. Biochem Pharmacol, 38: 3957-3962.
Polhuijs M, Meijer DK, & Mulder GJ (1991) The fate of diastereomeric
glutathione conjugates of bromoisovalerylurea in blood in the rat
in vivo and in the perfused rat liver. Stereoselectivity in biliary
and urinary excretion. J Pharmacol Exp Ther, 256: 458-461.
Polhuijs M, Mulder GJ, Meyer DJ, & Ketterer B (1992) Stereoselective
conjugation of 2-bromocarboxylic acids and their urea derivatives by
rat liver glutathione transferase 12-12 and some other isoforms.
Biochem Pharmacol, 444: 1249-1253.
Poole C (1994) Editorial: Ecologic analysis as outlook and method. Am
J Public Health, 84(5): 715-716.
Poole C (1997) Analytical meta-analysis of epidemiologic studies of
chlorinated drinking water and cancer: quantitative review and
re-analysis of the work published by Morris et al., Am J Public
Health, 82: 955-963. Cincinnati, Ohio, US Environmental Protection
Agency, National Center for Environmental Assessment.
Poon R, Levalier P, & Tryphonas H (1997) Effects of subchronic
exposure of monochloramine in drinking water on male rats. Regul
Toxicol Pharmacol, 25(2): 166-175.
Potter CL, Chang LW, DeAngelo AB, & Daniel FB (1996) Effects of four
trihalomethanes on DNA strand breaks, renal hyaline droplet formation
and serum testosterone in male F344 rats. Cancer Lett,
106(2): 235-242.
Prieto R & Fernandez E (1993) Toxicity and mutagenesis by chlorate are
independent of nitrate reductase in Chlamydomonas reinhardtii. Mol
Gen Genet, 237: 429-438.
Proudlock RJ & Crouch MJ (1990) Nuclear anomaly test on MX (mouse
stomach). Medmenham, United Kingdom, Water Research Centre (Report
prepared by the Huntingdon Research Centre, United Kingdom).
Racey-Burns LA, Burns AH, Summer WR, & Shephard RE (1989) The effect
of dichloroacetate on the isolated no flow arrested rat heart. Life
Sci, 44: 2015-2023.
Rav-Acha C & Choshen E (1987) Aqueous reactions of chlorine dioxide
with hydrocarbons. Environ Sci Technol, 21(11): 1069-1074.
Raymond P & Plaa GL (1997) Effect of dosing vehicle on the
hepatotoxicity of carbon tetrachloride and nephrotoxicity of
chloroform in rats. J Toxicol Environ Health, 51(5): 463-476.
Reckhow DA & Singer PC (1985) Mechanisms of organic halide formation
during fulvic acid chlorination and implications with respect to
preozonation. In: Jolley RL, Bull RJ, & Davis WP ed. Water
chlorination: chemistry, environmental impact and health effects.
Chelsea, Michigan, Lewis Publishers, Inc., vol 5, pp 1229-1257.
Reckhow DA, Singer PC, & Malcolm RL (1990) Chlorination of humic
materials: by-product formation and chemical interpretation. Environ
Sci Technol, 24(11): 1655-1664.
Reddy TV, Chang LW, DeAngelo AB, Pereira MA, & Daniel FB (1992) Effect
of non-genotoxic environmental contaminants on cholesterol and DNA
synthesis in cultured primary rat hepatocytes. Environ Sci,
1: 179-189.
Reif JS, Hatch MC, Bracken M, Holmes LB, Schwetz BA, & Singer PC
(1996) Reproductive and developmental effects of disinfection
by-products in drinking water. Environ Health Perspect,
104(10): 1056-1061.
Revis NW, McCauley P, Bull R, & Holdsworth G (1986a) Relationship of
drinking water disinfectants to plasma cholesterol and thyroid hormone
levels in experimental studies. Proc Natl Acad Sci (USA),
83: 1485-1489.
Revis NW, McCauley P, & Holdsworth G (1986b) Relationship of dietary
iodide and drinking water disinfectants to thyroid function in
experimental animals. Environ Health Perspect, 69: 243-248.
Reynolds SH, Stowers SJ, Patterson RM, Maronpot RR, Aaronson SA, &
Anderson MW (1987) Activated oncogenes in B6C3F1 mouse liver tumors:
Implications for risk assessment. Science, 237: 1309-1316.
Richardson AP (1937) Toxic potentialities of continued administration
of chlorate for blood and tissues. J Pharmacol Exp Ther, 59: 101-113.
Richmond RE, DeAngelo AB, Potter CL, & Daniel FB (1991) The role of
hyperplastic nodules in dichloroacetic acid-induced
hepatocarcinogenesis in B6C3F1 mice. Carcinogenesis, 12: 1383-1387.
Richmond RE, Carter JH, Carter HW, Daniel FB, & DeAngelo AB (1995)
Immunohistochemical analysis of dichloroacetic acid (DCA)-induced
hepatocarcinogenesis in male Fischer (F344) rats. Cancer Lett,
92: 67-76.
Rijhsinghani KS, Abrahams C, Swerdlow MA, Rao KV, & Ghose T (1986)
Induction of neoplastic lesions in the livers of C57BL × C3HF1 mice
by chloral hydrate. Cancer Detect Prev, 9: 279-288.
Riley TJ, Cauley JA, & Murphy P (1995) Water chlorination and lipo-
and apolipoproteins: the relationship in elderly white women of
Pennsylvania. Am J Public Health, 85(4): 570-573.
Ringhand HP, Kaylor WH, Miller RG, & Kopfler FC (1989) Synthesis of
3-14C-3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone and its
use in a tissue distribution study in the rat. Chemosphere,
18: 2229-2236.
Risch HA, Burch JD, Miller AB, Hill GB, Steele R, & Howe GR (1988)
Dietary factors and the incidence of cancer of the urinary bladder. Am
J Epidemiol, 127(6): 1179-1191.
Risch HA, Burch JD, Miller AB, Hill GB, Steele R, & Howe GR (1992)
Editorial: Chlorinated water and cancer - Is a meta-analysis a better
analysis? Pediatr Alert, 17: 91.
Robinson M, Bull RJ, Schamer M, & Long RE (1986) Epidermal hyperplasia
in mouse skin following treatment with alternative drinking water
disinfectants. Environ Health Perspect, 69: 293-300.
Robinson M, Bull RJ, Olson GR, & Stober J (1989) Carcinogenic activity
associated with halogenated acetones and acroleins in the mouse skin
assay. Cancer Lett, 48: 197-203.
Roby MR, Carle S, Pereira MA, & Carter DE (1986) Excretion and tissue
disposition of dichloroacetonitrile in rats and mice. Environ Health
Perspect, 69: 215-220.
Rook JJ (1977) Chlorination reactions of fulvic acids in natural
waters. J Environ Sci Technol, 11: 478-482.
Rook JJ, Gras AA, van der Heijden BG, & de Wee J (1978) Bromide
oxidation and organic substitution in water treatment. J Environ Sci
Health, A13: 91-116.
Rosen JD, Segall Y, & Casida JE (1980) Mutagenic potency of
haloacroleins and related compounds. Mutat Res, 78: 113-119.
Rosenkranz HS (1973) Sodium hypochlorite and sodium perborate:
preferential inhibitors of DNA polymerase-deficient bacteria. Mutat
Res, 21: 171-174.
Rosenkranz HS, Gutter B, & Speck WT (1976) Mutagenicity and
DNA-modifying activity: A comparison of two microbial assays. Mutat
Res, 41: 61-70.
Rothman KJ (1986) Modern epidemiology. Boston, Massachusetts, Little
Brown Co.
Ruddick JA, Villeneuve DC, Chu I, & Valli VE (1983) A teratological
assessment of four trihalomethanes in the rat. J Environ Sci Health,
B18: 333-349.
Russo A & Levis AG (1992) Detection of aneuploidy in male germ cells
of mice by means of a meiotic micronucleus assay. Mutat Res,
281(3): 187-191.
Russo A, Pacchierotti F, & Metalli P (1984) Nondisjunction induced in
mouse spermatogenesis by chloral hydrate, a metabolite of
trichloroethylene. Environ Mutagen, 6: 695-703.
Russo A, Stocco A, & Majone F (1992) Identification of
kinetochore-containing (CREST+) micronuclei in mouse bone marrow
erythrocytes. Mutagenesis, 5(3): 195-197.
Sai K, Takagi A, Umemura T, Hasegawa R, & Kurokawa Y (1991) Relation
of 8-hydroxydeoxyguanosine formation in rat kidney to lipid
peroxidation, glutathione level and relative organ weight after a
single administration of potassium bromate. Jpn J Cancer Res (Gann),
82: 165-169.
Sai K, Hayashi M, Takagi A, Hasegawa R, Sofuni T, & Kurokawa Y (1992a)
Effects of antioxidants on induction of micronuclei in rat peripheral
blood reticulocytes by potassium bromate. Mutat Res, 269: 113-118.
Sai K, Uchiyama S, Ohno Y, Hasegawa R, & Kurokawa Y (1992b) Generation
of active oxygen species in vitro by the interaction of potassium
bromate with rat kidney cell. Carcinogenesis, 13: 333-339.
Sai K, Umemura T, Takagi A, Hasegawa R, & Kurokawa Y (1992c) The
protective role of glutathione, cysteine and vitamin C against
oxidative DNA damage induced in rat kidney by potassium bromate. Jpn
J Cancer Res (Gann), 83: 45-51.
Saijo T, Naito E, Ito M, Takeda E, Hashimoto T, & Kuroda Y (1991)
Therapeutic effect of sodium dichloroacetate on visual and auditory
hallucinations in a patient with MELAS. Neuropediatrics, 22: 166-167.
Saillenfait AM, Langonne I, & Sabate JP (1995) Developmental toxicity
of trichloroethylene, tetrachloroethylene and four of their
metabolites in rat whole embryo culture. Arch Toxicol, 70: 71-82.
Sakai M, Matsushimia-Hibiya Y, Nishizawa M, & Nishi S (1995)
Suppression of rat glutathione transferase P expression by peroxisome
proliferators: Interactions between Jun and peroxisome
proliferator-activated receptor alpha. Cancer Res, 55: 5370-5376.
Sanchez IM & Bull RJ (1990) Early induction of reparative hyperplasia
in the liver of B6C3F1 mice treated with dichloroacetate and
trichloroacetate. Toxicology, 64: 33-46.
Sanders VM, Kauffmann BM, White KL, Douglas KA, Barnes DW, Sain LE,
Bradshaw TJ, Borzelleca JF, & Munson AE (1982) Toxicology of chloral
hydrate in the mouse. Environ Health Perspect, 44: 137-146.
Sans RM, Jolly WW, & Harris RA (1980) Studies on the regulation of
leucine catabolism: Mechanism responsible for oxidizable substrate
inhibition and dichloroacetate stimulation of leucine catabolism by
the heart. Arch Biochem Biophys, 200: 336-345.
Savin JE & Cohn PD (1996) Comparison of bladder and rectal cancer
incidence with trihalomethanes in drinking water. Epidemiology,
7(suppl 4): S63.
Savitz DA, Andrews KW, & Pastore LM (1995) Drinking water and
pregnancy outcome in central North Carolina: Source, amount, and
trihalomethane levels. Environ Health Perspect, 103(6): 592-596.
Sbrana I, Di Sibio A, Lomi A, & Scarcelli V (1993) C-mitosis and
numerical chromosome aberration analyses in human lymphocytes: 10
known or suspected spindle poisons. Mutat Res. 287(1): 57-70.
Scatina J, Abdel-Rahman MS, Gerges SE, Khan MY, & Gona O (1984)
Pharmacodynamics of Alcide, a new antimicrobial compound, in rat and
rabbit. Fundam Appl Toxicol, 4: 479-484.
Schechter DS & Singer P (1995) Formation of aldehydes during
ozonation. J Ozone Sci Eng, 17(1): 53-69.
Schoonjans K, Watanabe M, Suzuki H, Mahfoudi A, Krey G, Wahli W,
Grimaldi P, Staels B, Yamamoto T, & Auwerx J (1995) Induction of the
acyl-coenzyme A synthetase gene by fibrates and fatty acids is
mediated by a peroxisome proliferator response element of the C
promoter. J Biol Chem, 270: 19 269-19 276.
Schultz IR, Merdink JL, Gonzalez-Leon A, & Bull RJ (1999) Comparative
toxicokinetics of chlorinated and brominated haloacetates in F344
rats. Toxicol Appl Pharmacol, 158(2): 103-114.
Schwartz S (1995) The fallacy of the ecological fallacy: the potential
misuse of a concept and the consequences. Am J Public Health,
84(5): 819-823.
Schwartz DA, Smith DD, & Lakshminarayan MD (1990) The pulmonary
sequelae associated with accidental inhalation of chlorine gas. Chest,
97: 820-825.
Sclimenti MJ, Krasner SW, Glaze WH, & Weinberg HS (1990) Ozone
disinfection by-products: optimization of the PFBHA derivatization
method for the analysis of aldehydes. In: Proceedings of the Water
Quality Conference, San Francisco, CA. Denver, Colorado, American
Water Works Association.
Scully FE, Mazina KE, Ringhand HP, Chess EK, Campbell JA, & Johnson JD
(1990) Identification of organic N-chloramines in vitro in stomach
fluid from the rat after chlorination. Chem Res Toxicol, 3: 301-306.
Segall Y, Kimmel EC, Dohn DR, & Casida JE (1985) 3-Substituted
2-halopropenals: mutagenicity, detoxification and formation from
3-substituted 2,3-dihalopropenal promutagens. Mutat Res, 158: 61-68.
Sellers EM & Koch-Weser J (1970) Potentiation of warfarin-induced
hypoprothrominemia by chloral hydrate. N Engl J Med, 283: 827-831.
Sellers EM, Lang M, Koch-Weser J, LeBlanc E, & Kalant H (1972)
Interaction of chloral hydrate and ethanol in man: I. Metabolism. Clin
Pharmacol Ther, 13: 37-49.
Shangraw RE, Winter R, Hromco J, Robinson T, & Gallaher EJ (1994)
Amelioration of lactic acidosis with dichloroacetate during liver
transplantation in humans. Anesthesiology, 81: 1127-1138.
Sheahan BJ, Pugh DM, & Winstanley EW (1971) Experimental sodium
chlorate poisoning in dogs. Res Vet Sci, 12: 387-389.
Sherer TT, Sylvester SR, & Bull RJ (1993) Differential expression of
c-erbA mRNAs in the developing cerebellum and cerevral cortex of the
rat. Biol Neonate, 63(1): 26-34.
Shih KL & Lederberg J (1976) Chloramine mutagenesis in Bacillus
subtilis. Science, 192: 1141-1143.
Shukairy HM & Summers RS (1992) The impact of preozonation and
biodegradation on disinfection by-product formation. Water Res,
26(9): 1217-1227.
Shy CM (1985) Chemical contamination of water supplies. Environ Health
Perspect, 62: 399-406.
Siddiqui MS (1992) Ozone-bromide interactions: Formation of DBPs.
Tucson, Arizona, University of Arizona (Ph.D. Dissertation).
Siddiqui MS (1996) Chlorine-ozone interactions: Formation of chlorate.
Water Res, 30(9): 2160-2170.
Siddiqui MS & Amy GL (1993) Factors affecting DBP formation during
ozone-bromide reactions. J Am Water Works Assoc, 85(1): 63-72.
Siddiqui MS, Amy GL, Ozekin K, Zhai W, & Westerhoff P (1994)
Alternative strategies for removing bromate. J Am Water Works Assoc,
86(10): 81-96.
Siddiqui MS, Amy GL, & Rice RG (1995) Bromate ion formation: a
critical review. J Am Water Works Assoc, 87(10): 58-70.
Siddiqui MS, Zhai W, Amy GL, & Mysore C (1996a) Bromate ion removal by
activated carbon. Water Res, 30(7): 1651-1660.
Siddiqui MS, Amy GL, Cooper W, Kurucz CN, Waite TD, & Nickelson GM
(1996b) Bromate ion removal by high energy electron beam irradiation.
J Am Water Works Assoc, 88(10): 90-101.
Siddiqui MS, Amy GL, & McCollum LJ (1996c) Bromate destruction by UV
irradiation and electric arc discharge. J Ozone Sci Eng,
18(3): 271-290.
Siddiqui MS, Amy GL, & Murphy BD (1997) Ozone enhanced removal of
natural organic matter. Water Res, 31(12): 3098-3106.
Silver W & Stier M (1971) Cardiac arrhythmias from chloralhydrate.
Pediatrics, 48(2): 332-333.
Silver EM, Kuttab SH, Hansan T, & Hassan M (1982) Structural
considerations in the metabolism of nitriles to cyanide in vivo.
Drug Metab Dispos, 10: 495-498.
Simmon VF & Tardiff RG (1978) The mutagenic activity of halogenated
compounds found in chlorinated drinking water. In: Water chlorination:
Environmental impacts and health effects. Ann Arbor, Michigan, Ann
Arbor Science Publishers, pp 417-431.
Singelmann E & Steffen C (1983) Increased erythrocyte rigidity in
chlorate poisoning. J Clin Pathol, 36: 719.
Singelmann E, Wetzel E, Adler G, & Steffen C (1984) Erythrocyte
membrane alterations as the basis of chlorate toxicity. Toxicology,
30: 135-147.
Singer PC (1993) Formation and characterization of DBPs. In: Craun GF
ed. Safety of water disinfection: balancing chemical and microbial
risks. Washington, DC, ILSI Press.
Singer PC (1994a) Control of disinfection by-products in drinking
water. J Environ Eng, 120(4): 727-744.
Singer PC (1994b) Impacts of ozonation on the formation of
chlorination and chloramination by-products. Denver, Colorado,
American Water Works Association Research Foundation.
Sketchell J, Peterson HG, & Christofi N (1995) Disinfection by-product
formation after biologically assisted GAC treatment of water supplies
with different bromide and DOC content. Water Res, 29(12): 2635-2642.
Slattery ML, West DW, & Robison LM (1988) Fluid intake and bladder
cancer in Utah. Int J Cancer, 42: 17-22.
Smith MK, George EL, Zenick H, Manson JM, & Stober JA (1987)
Developmental toxicity of halogenated acetonitriles: Drinking water
by-products of chlorine disinfection. Toxicology, 46: 83-93.
Smith MK, Randall JL, Tocco DR, York RG, Stober JA, & Read EJ (1988)
Teratogenic effects of trichloroacetonitrile in the Long-Evans rat.
Teratology, 38: 113-120.
Smith MK, Randall JL, Read EJ, & Stober JA (1989a) Teratogenic
activity of trichloroacetic acid in the rat. Teratology, 40: 445-451.
Smith MK, Randall JL, Stober JA, & Read EJ (1989b) Developmental
toxicity of dichloroacetonitrile: A by-product of drinking water
disinfection. Fundam Appl Toxicol, 12: 765-772.
Smith MK, Randall JL, Read EJ, & Stober JA (1992) Developmental
toxicity of dichloroacetate in the rat. Teratology, 46: 217-223.
Smith MK, Thrall BD, & Bull RJ (1997) Dichloroacetate (DCA) modulates
insulin signaling. Toxicologist, 36: 1133-1140.
Snyder MP & Margerum DW (1982) Kinetics of chlorine transfer from
chloramine to amines, amino acids, and peptides. Inorg Chem,
21: 2545-2550.
Snyder RD, Pullman J, Carter JH, Carter HW, & DeAngelo AB (1995)
In vivo administration of dichloroacetic acid suppresses spontaneous
apoptosis in murine hepatocytes. Cancer Res, 55: 3702-3705.
So BJ & Bull RJ (1995) Dibromoacetate (DBA) acts as a promoter of
abnormal crypt foci in the colon of F344 rats. Toxicologist, 15: 1242.
Sobti RC (1984) Sister chromatid exchange potential of the halogenated
hydrocarbons produced during water chlorination. Chromosome Inf Serv,
37: 17-19.
Sood C & O'Brien PJ (1993) Molecular mechanisms of
chloroacetaldehyde-induced cytotoxicity in isolated rat hepatocytes.
Biochem Pharmacol, 46: 1621-1626.
Sora S & Carbone MJ (1987) Chloral hydrate, methylmercury hydroxide
and ethidium bromide affect chromosomal segregation during meiosis of
Saccharomyces cerevisiae. Mutat Res, 190: 13-17.
Speitel GE, Symons JM, Diehl AC, Sorensen HW, & Cipparone LA (1993)
Effect of ozone dose and subsequent biodegration on removal of
disinfection by-products precursors. J Am Water Works Assoc,
85(5): 86-95.
Spengler SJ & Singer B (1988) Formation of interstrand cross-links in
chloroacetaldehyde treated DNA demonstrated by ethidium bromide
fluorescence. Cancer Res, 48: 4804-4806.
Spirtas R, Stewart PA, Lee JS, Marano DE, Forbes CD, Grauman DJ,
Pettigrew HM, Blair A, Hoover RN, & Cohen JL (1991) Retrospective
cohort study of workers at an aircraft maintenance facility: I.
Epidemiological results. Br J Ind Med, 48: 515-530.
Sprankle CS, Larson JL, Goldsworthy SM, & Butterworth BE (1996) Levels
of myc, fos, Ha-ras, met and hepatocyte growth factor mRNA during
regenerative cell proliferation in female mouse liver and male rat
kidney after a cytotoxic dose of chloroform. Cancer Lett, 101: 97-106.
Stacpoole PW (1989) The pharmacology of dichloroacetate. Metabolism,
38: 1124-1144.
Stacpoole PW & Felts JM (1970) Diisopropylammonium dichloroacetate
(DIPA) and sodium dichloroacetate (DCA): Effect on glucose and fat
metabolism in normal and diabetic tissue. Metabolism, 19: 71-78.
Stacpoole PW & Greene YJ (1992) Dichloroacetate. Diabetes Care,
15: 785-791.
Stacpoole PW, Moore GW, & Kornhauser DM (1978) Metabolic effects of
dichloroacetate in patients with diabetes mellitus and
hyperlipoproteinemia. N Engl J Med, 298: 526-530.
Stacpoole PW, Harwood HJ, & Varnado CE (1983) Regulation of rat liver
hydroxymethylglutaryl coenzyme A reductase by a new class of
noncompetitive inhibitors: Effects of dichloroacetate and related
carboxylic acids on enzyme activity. J Clin Invest, 72: 1575-1585.
Stacpoole PW, Harwood HJ, Cameron DF, Curry SH, Samuelson DA, Cornwell
PE, & Sauberlich HE (1990) Chronic toxicity of dichloroacetate:
Possible relation to thiamine deficiency in rats. Fundam Appl Toxicol,
14: 327-337.
Stacpoole PW, Wright EC, Baumgartner TG, Bersin RM, Buchalter S, Curry
SH, Duncan CA, Harman EM, Henderson GN, Jenkinson S, Lachin JM, Lorenz
A, Schneider SH, Siegel JH, Summer WR, Thompson D, Wolfe CL, &
Zorovich B (1992) A controlled clinical trial of dichloroacetate for
treatment of lactic acidosis in adults. N Engl J Med, 327: 1564-1569.
Stauber AJ & Bull RJ (1997) Differences in phenotype and cell
replicative behavior of hepatic tumors inducted by dichloroacetate
(DCA) and trichloroacetate (TCA). Toxicol Appl Pharmacol,
144(2): 235-246.
Stauber AJ, Bull RJ, & Thrall BD (1998) Dichloroacetate and
trichloroacetate promote clonal expansion of anchorage-independent
hepatocytes in vivo and in vitro. Toxicol Appl Pharmacol,
150: 287-294.
Steffen C & Seitz R (1981) Severe chlorate poisoning: Report of a
case. Arch Toxicol, 48: 281-288.
Steffen C & Wetzel E (1993) Chlorate poisoning: Mechanism of toxicity.
Toxicology, 84: 217-231.
Stenner RD, Merdink JL, Templin MV, Stevens DK, Springer DL, & Bull RJ
(1996) Enterohepatic recirculation of trichloroethanol glucuronide as
a significant source of trichloroacetic acid in the metabolism of
trichloroethylene. Drug Metab Dispos, 25: 529-535.
Stevens JL & Anders MW (1981) Metabolism of haloforms to cabon
monoxide: IV. Studies on the reaction mechanism in vivo. Chem-Biol
Interact, 37: 365-374.
Stevens AA, Slocum CJ, Seeger DR, & Robeck GG (1976) Chlorination of
organics in drinking water. J Am Water Works Assoc, 68(11): 615.
Stevens AA, Moore LA, & Slocum CJ (1989) By-products of chlorination
at ten operating utilities. In: Disinfection by-products: Current
perspectives. Denver, Colorado, American Water Works Association, pp
23-61.
Stevens DK, Eyre RJ, & Bull RJ (1992) Adduction of hemoglobin and
albumin in vivo by metabolites of trichloroethylene,
trichloroacetate and dichloroacetate in rats and mice. Fundam Appl
Toxicol, 19: 336-342.
Stocker KJ, Statham J, Howard WR, & Proudlock RJ (1997) Assessment of
the potential in vivo genotoxicity of three trihalomethanes:
chlorodibromomethane, bromodichloromethane and bromoform. Mutagenesis,
12(3): 169-173.
Strauss BS (1991) The "A-rule" of mutagen specificity: a consequence
of DNA polymerase bypass of noninstructional lesions. Bioessays,
13(2): 79-84.
Struba RJ (1979) Cancer and drinking water quality. Chapel Hill, North
Carolina, University of North Carolina (Thesis).
Styles JA, Wyatt I, & Coutts C (1991) Trichloroacetic acid: studies on
uptake and effects on hepatic DNA and liver growth in mice.
Carcinogenesis, 12: 1715-1719.
Suarez-Varela MM, Gonzalez AL, Perez ML, & Caraco EF (1994)
Chlorination of drinking water and cancer incidence. J Environ Pathol
Toxicol Oncol, 13(1): 39-41.
Suh DH & Abdel-Rahman MS (1983) Kinetics study of chloride in rat. J
Toxicol Environ Health, 12: 467-473.
Suh DH, Abdel-Rahman MS, & Bull RJ (1983) Effect of chlorine dioxide
and its metabolites in drinking water on fetal developmental in rats.
J Appl Toxicol, 3: 75-79.
Susser M (1994a) The logic in ecological: I. The logic of analysis. Am
J Public Health, 84(5): 825-829.
Susser M (1994b) The logic in ecological: II. The logic of design. Am
J Public Health, 84(5): 830-835.
Suzuki N & Nakanishi J (1995) Brominated analogues of MX
(3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone) in chlorinated
drinking water. Chemosphere, 30: 1557-1564.
Swan SH & Waller K (1998) Disinfection by-products and adverse
pregnancy outcomes: what is the agent and how should it be measured?
[Editorial; comment]. Epidemiology, 9(5): 479-481.
Swan SH, Waller K, Hopkins B, Windam G, Fenster L, Schaefer C, &
Neutra RR (1998) A prospective study of spontaneous abortion: relation
to amount and source of drinking water consumed in early pregnancy.
Epidemiology, 9(2): 126-139.
Symons JM & Worley KL (1995) An advanced oxidation process for DBP
control. J Am Water Works Assoc, 87(11): 66-75.
Symons JM, Stevens AA, Clark RM, Geldreich EE, Love OT, & De Marco J
(1981) Treatment techniques for controlling trihalomethanes in
drinking water. Washington, DC, US Environmental Protection Agency
(EPA 600/2-81-156).
Takamura N, Kurokawa Y, Matsushima Y, Imazawa T, Onodera H, & Hayashi
Y (1985) Long-term oral administration of potassium bromate in male
Syrian golden hamsters. Sci Rep Res Inst Tohoku Univ, 32(1-4): 43-46.
Tao L, Kramer PM, Latendresse JR, & Pereira MA (1996) Expression of
the protooncogenes, TGF-alpha and ß in the liver of B6C3F1 mice
treated with trichloroethylene, dichloroacetic and trichloroacetic
acid. Toxicologist, 15: 1495.
Taylor DH & Pfohl RJ (1985) Effects of chlorine dioxide on
neurobehavioral development of rats. In: Jolley RL, Bull RJ, & Davis
WP ed. Water chlorination: Chemistry, environmental impact and health
effects. Chelsea, Michigan, Lewis Publishers, Inc., vol 5, pp 355-364.
Temperman J & Maes R (1966) Suicidal poisoning by sodium chlorate: A
report of three cases. J Forensic Med, 13: 123-129.
Templin MV, Parker JC, & Bull RJ (1993) Relative formation of
dichloroacetate and trichloroacetate from trichloroethylene in male
B6C3F1 mice. Toxicol Appl Pharmacol, 123: 1-8.
Templin MV, Stevens DK, Stenner RD, Bonate PL, Tuman D, & Bull RJ
(1995) Factors affecting species differences in the kinetics of
metabolites of trichloroethylene. J Toxicol Environ Health,
44: 435-447.
Templin MV, Jamison KC, Wolf DC, Morgan KT, & Butterworth BE (1996a)
Comparison of chloroform-induced toxicity in the kidneys, liver, and
nasal passages of male Osborne-Mendel and F-344 rats. Cancer Lett,
104: 71-78.
Templin MV, Jamison KC, Sprankle CS, Wolf DC, Wong BA, & Butterworth
BE (1996b) Chloroform-induced cytotoxicity and regenerative cell
proliferation in the kidneys and liver of BDF1 mice. Cancer Lett,
108: 225-231.
Templin MV, Larson JL, Butterworth BE, Jamison KC, Leininger JR, Mery
S, Morgan KT, Wong BA, & Wolf DC (1996c) A 90-day chloroform
inhalation study in F-344 rats: profile of toxicity and relevance to
cancer studies. Fundam Appl Toxicol, 32: 109-125.
Templin MV, Constan AA, Wolf DC, Wong BA, & Butterworth BE (1998)
Patterns of chloroform-induced regenerative cell proliferation in
BDF1 mice correlate with organ specificity and dose-response of tumor
formation. Carcinogenesis, 19(1): 187-193.
Teng H & Veenstra J (1995) Disinfection by-products formed using four
alternative disinfectants as a function of precursor characteristics.
In: Minear R & Amy G ed. Disinfection by-products in water treatment.
Chelsea, Michigan, Lewis Publishers, Inc.
TERA (1998) Toxicology excellence for risk assessment - Health risk
assessment/ characterization of the drinking water disinfection
byproducts chlorine dioxide and chlorite (8W-0766-NTLX). Cincinnati,
Ohio.
Testai E, Di Marzio S, di Domenico A, Piccardi A, & Vittozzi L (1995)
An in vitro investigation of the reductive metabolism of chloroform.
Arch Toxicol, 70: 83-88.
Testai E, De Curtis V, Gemma S, Fabrizi L, Gervasi P, & Vittozzi L
(1996) The role of different cytochrome P450 isoforms in in vitro
chloroform metabolism. J Biochem Toxicol, 11: 305-312.
Thier R, Taylor JB, Pemble SE, Humphreys WG, Persmark M, Ketterer B, &
Guengerich FP (1993) Expression of mammalian glutathione
S-transferase 5-5 in Salmonella typhimurium TA1535 leads to
base-pair mutations upon exposure to dihalomethanes. Proc Natl Acad
Sci (USA), 90: 8576-8580.
Thier R, Pemble SE, Kramer H, Taylor JB, Guengerich FP, & Ketterer B
(1996) Human glutathione S-transferase T1-1 enhances mutagenicity of
1,2-dibromoethane, dibromomethane, and 1,2,3,4-diepoxybutane in
Salmonella typhimurium. Carcinogenesis, 17: 163-166.
Thomas EL, Jefferson MM, Bennett JJ, & Learn DB (1987) Mutagenic
activity of chloramines. Mutat Res, 188: 35-41.
Thornton-Manning JR, Gao P, Lilly PD, & Pegram RA (1993) Acute
bromodichloromethane toxicity in rats pretreated with cytochrome P450
inducers and inhibitors. Toxicologist, 13: 361 (abstract).
Thornton-Manning JR, Seely JR, & Pegram RA (1994) Toxicity of
bromodichloromethane in female rats and mice after repeated oral
dosing. Toxicology, 94: 3-18.
Thurman EM (1985) Developments in biochemistry: Organic geochemistry
of natural waters. Dordrecht, The Netherlands, Nijhoff M & Junk W
Publishers.
Tomasi A, Albano E, Biasi F, Slater TF, Vannini V, & Dianzani M (1985)
Activation of chloroform and related trihalomethanes to free radical
intermediates in isolated hepatocytes and in the rat in vivo as
detected by the ESR-spin trapping technique. Chem-Biol Interact,
55: 303-316.
Tong Z, Board PG, & Anders MW (1998) Glutathione transferase zeta
catalyzes the oxygenation of the carcinogen dichloroacetic acid to
glyoxylic acid. Biochem J, 331: 371-374.
Toth GP, Long RE, Mills TS, & Smith MK (1990) Effects of chlorine
dioxide on the developing rat brain. J Toxicol Environ Health,
31: 29-44.
Toth GP, Kelty KC, George EL, Read EJ, & Smith MK (1992) Adverse male
reproductive effects following subchronic exposure of rats to sodium
dichloroacetate. Fundam Appl Toxicol, 19: 57-63.
Toth PP, El-Shanti H, Elvins S, Rhead WJ, & Klein JM (1993) Transient
improvement of congenital lactic acidosis in a male infant with
pyruvate decarboxylase deficiency treated with dichloroacetate.
J Pediatr, 123: 427-430.
Tratnyek PG & Hoigne J (1994) Kinetics of reactions of chlorine
dioxide in water - II: quantitative structure-activity relationships
for phenolic compounds. Water Res, 28(1): 57-66.
Trehy ML & Bieber TI (1981) Detection, identification and quantitative
analysis of dihaloacetonitriles in chlorinated natural waters. In:
Keith LH ed. Advances in the identification and analysis of organic
pollutants in water. Ann Arbor, Michigan, Ann Arbor Science
Publishers, vol 2, pp 941-975.
Trehy ML, Yost RA, & Miles CJ (1986) Chlorination by-products of amino
acids in natural waters. Environ Sci Technol, 20: 1117-1122.
Trussell RR & Umphres MD (1978) The formation of trihalomethanes. J Am
Water Works Assoc, 70(11): 604.
Tuomisto J, Hythinen J, Jansson K, Komulainen H, Kosma VM,
Maki-Paakkanen J, Vaittinen SL, & Vartiainen T (1995) Genotoxicity and
carcinogenicity of MX and other chlorinated furanones. In:
Disinfection by-products in drinking water: Critical issues in health
effects research. Washington, DC, International Life Sciences
Institute, pp 30-31.
Tuppurainen K, Lotjonen S, Laatikainen R, Vartiainen T, Maran U,
Strandberg M, & Tamm T (1991) About the mutagenicity of chlorine
substituted furanones and halopropenals. A QSAR study using molecular
orbital indices. Mutat Res, 247: 97-102.
Tuthill RW & Moore GS (1980) Drinking water chlorination: A practice
related to cancer mortality. J Am Water Works Assoc, 72: 570-573.
Umemura T, Sai K, Takagi A, Hasegawa R, & Kurokawa Y (1993) A possible
role for cell proliferation in potassium bromate (KBrO3). J Cancer
Res Clin Oncol, 119: 463-469.
Urano K, Wada H, & Takemasa T (1983) Empirical rate equation for
trihalomethane formation with chlorination of humic substances in
water. Water Res, 17(12): 1797-1802.
US EPA (1994a) Workshop report and recommendations for conducting
epidemiologic research on cancer and exposure to chlorinated drinking
water. Cincinnati, Ohio, US Environmental Protection Agency, pp
2/13-2/19.
US EPA (1994b) Drinking water criteria for trihalomethanes.
Washington, DC, US Environmental Protection Agency, Office of Water.
US EPA (1997) Workshop report and recommendations for conducting
epidemiologic research on reproductive and developmental effects and
exposure to disinfected drinking water. Research Triangle Park, North
Carolina, US Environmental Protection Agency.
US EPA (1998) Dichloroacetic acid - Carcinogenicity identification
characterization summary. Washington, DC, US Environmental Protection
Agency, Office of Research and Development.
Vagnarelli P, De Sario A, & De Carli L (1990) Aneuploidy induced by
chloral hydrate detected in b human lymphocytes with Y97 probe.
Mutagenesis, 5: 591-592.
Vaittinen SL, Komulainen H, Kosma VM, Julkunen A, Maki-Paakkanen J,
Jansson K, Vartiainen T, & Tuomisto J (1995) Subchronic toxicity of
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX) in Wistar
rats. Food Chem Toxicol, 33(12): 1027-1037.
Valentine RL & Solomon WL (1987) Effect of inorganic composition on
the kinetics of chloramine decomposition In the presence of excess
ammonia. Water Res, 21: 1475-1483.
Van Duuren BL, Goldschmidt BM, Loewengart G, Smith AC, Melchionne S,
Seidman I, & Roth D (1979) Carcinogenicity of halogenated olefinic and
aliphatic hydrocarbons in mice. J Natl Cancer Inst, 63: 1433-1439.
van Heijst AN, Zimmerman AN, & Pikaar SA (1977) [Chloral hydrate - the
forgotten poison.] Ned Tijdschr Geneesk, 121(40): 1537-1539 (in
Dutch).
Van Hoof F, Janssens JG, & van Dijck H (1986) Formation of mutagenic
activity during surface water pre-ozonation and its removal in
drinking water treatment. Chemosphere, 14(5): 501-509.
Varanasi U, Chu RY, Huang Q, Castellon R, Yeldandi AV, & Reddy JK
(1996) Identification of a peroxisome proliferator-responsive element
upstream of the human peroxisomal fatty acyl coenzyme A oxidase gene.
J Biol Chem, 271: 2147-2155.
Varma MM, Ampy FR, Verma K, & Talbot WW (1988) In vitro mutagenicity
of water contaminants in complex mixtures. J Appl Toxicol, 8: 243-248.
Vartiainen T, Liimatainen A, Kauranen P, & Hiisvirta L (1988)
Relations between drinking water mutagenicity and water quality
parameters. Chemosphere, 17: 189-202.
Velazquez SF, McGinnis PM, Vater ST, Stiteler WS, Knauf LA, & Schoeny
RS (1994) Combination of cancer data in quantitative risk assessments:
case study using bromodichloromethane. Risk Anal, 14: 285-291.
Vena JE, Graham S, Freudenheim J, Marshall J, Zielezny M, Swanson M, &
Sufrin G (1993) Drinking water, fluid intake, and bladder cancer in
Western New York. Arch Environ Med, 48(3): 191-198.
Vesselinovitch SD, Rao KV, Mihailovich N, Rice JM, & Lombard LS (1974)
Development of broad spectrum of tumors by ethylnitrosourea in mice
and the modifying role of age, sex and strain. Cancer Res, 34:
2530-2538.
Vogt CR, Liao JC, Sun GY, & Sun AY (1979) In vivo and in vitro
formation of chloroform in rats with acute dosage of chlorinated water
and the effect of membrane function. In: Trace substances in
environmental health: XIII. Proceedings of the University of Missouri
13th Annual Conference on Trace Substances in Environmental Health.
Columbia, Missouri, University of Missouri, pp 453-459.
Von der Hude W, Scheutwinkel M, Gramlich U, Fisler B, & Basler A
(1987) Genotoxicity of three-carbon compounds evaluated in the SCE
test in vitro. Environ Mutagen, 9: 401-410.
von Gunten U & Hoigne J (1994) Bromate formation during ozonation of
bromide-containing waters: Interactions of ozone and hydroxyl radical
reactions. Environ Sci Technol, 28(7): 1234.
Voudrias EA, Dielmann LM, Snoeyink VL, Larson RA, McCreary JJ, & Chen
AS (1983) Reactions of chlorite ion with activated carbon and with
vanillic acid and indan adsorbed on activated carbon. Water Research,
17: 1107-1112.
Wahli W, Braissant O, & Desvergne B (1995) Peroxisome proliferator
activated receptors: transcriptional regulators of adipogenesis, lipid
metabolism and more Chem Biol, 2: 261-266.
Waller CL & McKinney JD (1993) Theoretical investigation into the
potential of halogenated methanes to undergo reproductive metabolism.
J Comput Chem, 14(12): 1575-1579.
Waller K, Swan SH, DeLorenze G, & Hopkins B (1998) Trihalomethanes in
drinking water and spontaneous abortion. Epidemiology, 9: 134-140.
Warr TJ, Parry EM, & Parry JM (1993) A comparison of two in vivo
mammalian cell cytogenetic assays for the detection of mitotic
aneuploidy using 10 known or suspected aneugens. Mutat Res,
287(1): 29-46.
Warshaw BL, Carter MC, Hymes LC, Bruner BS, & Rauber AP (1985) Bromate
poisoning from hair permanent preparations. Pediatrics, 76: 975-978.
Waskell L (1978) A study of the mutagenicity of anesthetics and their
metabolites. Mutat Res, 57: 141-153.
Watanabe M, Kobayashi H, & Ohta T (1994) Rapid inactivation of
3-chloro-4-(dichloromethyl)-5-hydroxy-2(5H)-furanone (MX), a potent
mutagen in chlorinated drinking water, by sulfhydryl compounds. Mutat
Res, 312: 131-138.
Water Research Centre (1980) Trihalomethanes in water: Seminar held at
the Lorch Foundation, Lane End, Buckinghamshire, UK, 16-17 January
1980. Medmenham, United Kingdom, Water Research Centre.
Weil I & Morris JC (1949) Kinetic studies of chloramine: The rates of
formation of monochloramine, N-chloromethylamine and
N-chlorodimethylamine. J Am Chem Soc, 71: 1664-1671.
Weinberg HS, Glaze WH, Krasner SW, & Sclimenti MJ (1993) Formation and
removal of aldehydes in plants that use ozone. J Am Water Works Assoc,
85(5): 72-85.
Weinberg HS, Yamada H, & Joyce RJ (1998) New, sensitive and selective
method for determining submicrogram/l levels of bromate in drinking
water. J Chromatogr, A804(1/2): 137-142.
Weiss NS (1996) Cancer in relation to occupational exposure to
trichloroethylene. Occup Environ Med, 53: 1-5.
Wells PG, Moore GW, Rabin D, Wilkinson GR, Oates JA, & Stacpoole PW
(1980) Metabolic effects and pharmacokinetics of intravenously
administered dichloroacetate in humans. Diabetologia, 19: 109-113.
Whitehouse S & Randle PJ (1973) Activation of pyruvate dehydrogenase
in perfused rat heart by dichloroacetate. Biochem J, 134: 651-653.
Whitehouse S, Cooper RH, & Randle PJ (1974) Mechanism of activation of
pyruvate dehydrogenase by dichloroacetate and other halogenated
carboxylic acids. Biochem J, 141: 761-774.
WHO (1993) Guidelines for drinking-water quality - Volume 1:
Recommendations. Geneva, World Health Organization.
WHO (1996) Guidelines for drinking-water quality - Volume 2: Health
criteria and other supporting information. Geneva, World Health
Organization.
WHO (1998) Guidelines for drinking-water quality - Addendum to Volume
2: Health criteria and other supporting information. Geneva, World
Health Organization.
Wigle DT, Mao Y, Semenci WR, Smith MH, & Toft P (1986) Contaminants in
drinking water and cancer risks in Canadian cities. Can J Public
Health, 77: 335-342.
Wilkins JR & Comstock GW (1981) Source of drinking water at home and
site-specific cancer incidence in Washington County, Maryland. Am J
Epidemiol, 114: 178-190.
Wilson JR, Mancini DM, Ferraro N, & Egler J (1988) Effect of
dichloroacetate on the exercise performance of patients with heart
failure. J Am Coll Cardiol, 12: 1464-1469.
Wlodkowski TJ & Rosenkranz HS (1975) Mutagenicity of sodium
hypochlorite for Salmonella typhimurium. Mutat Res, 31: 39-42.
Wolf CR, Mansuy D, Nastainczyk W, Deutschmann G, & Ullrich V (1977)
The reduction of polyhalogenated methanes by liver microsomal
cytochrome P-450. Mol Pharmacol, 13: 698-705.
Wones RG & Glueck CJ (1986) Effect of chlorinated drinking water on
human lipid metabolism. Environ Health Perspect, 69: 255-258.
Wones RG, Mieczkowski L, & Frohman LA (1990) Chlorinated drinking
water and human lipid and thyroid metabolism. In: Jolley RL, Condie
LW, & Johnson JD ed. Water chlorination: Chemistry, environmental
impact and health effects. Chelsea, Michigan, Lewis Publishers, Inc.,
vol 6, p 301.
Wones RG, Deck CC, & Stadler B (1993a) Lack of effect of drinking
water chlorine on lipid and thyroid metabolism in healthy humans.
Environ Health Perspect, 99: 375-381.
Wones RG, Deck CC, & Stadler B (1993b) Effects of drinking water
monochloramine on lipid and thyroid metabolism in healthy men. Environ
Health Perspect, 99: 369-374.
Wones RG, Deck CC, Stadler B, Roark S, Hogg E, & Frohman LA (1993c)
Lack of effect of drinking water chlorine on lipid and thyroid
metabolism in healthy humans. Environ Health Perspect, 99: 375-381.
Woodard G, Lange SW, Nelson KW, & Calvery HO (1941) The acute oral
toxicity of acetic, chloroacetic, dichloroacetic and trichloroacetic
acids. J Ind Hyg Toxicol, 23: 78-82.
Woodruff RC, Mason JM, Valencia R, & Zimmering S (1985) Chemical
mutagenesis testing in Drosophila: V. Results of 53 coded compounds
tested for the National Toxicology Program. Environ Mutagen,
7(5): 677-702.
Worley KL (1994) Oxidation of NOM using peroxide and visible-UV
irradiation. Houston, Texas, University of Houston, Civil and
Environmental Engineering Department (M.S. Thesis).
Wyatt I, Gyte A, Mainwaring G, Widdowson PS, & Lock EA (1996)
Glutathione depletion in the liver and brain produced by
2-chloropropionic acid: Relevance to cerebellar granule cell necrosis.
Arch Toxicol, 70: 380-389.
Xie Y & Reckhow DA (1993) Identification of trihaloacetaldehydes in
ozonated and chlorinated fulvic acid solutions. Analyst, 118: 71-72.
Xu W & Adler ID (1990) Clastogenic effects of known and suspect
spindle poisons studied by chromosome analysis in mouse bone marrow
cells. Mutagenesis, 5(4): 371-374.
Xu G, Stevens DK, & Bull RJ (1995) Metabolism of bromodichloroacetate
in B6C3F1 mice. Drug Metab Dispos, 23: 1412-1416.
Yamada H (1993) By-products of ozonation of low bromide waters and
reduction of the by-products by activated carbon. In: Ozone in water
and wastewater treatment - Proceedings of the Eleventh Ozone World
Congress, San Francisco, California, 29 August - 3 September 1993.
Lille, France, International Ozone Association, pp 9-58.
Yamamoto K, Fukushima M, & Oda K (1988) Disappearance rates of
chloramines in river water. Water Res, 22(1): 79-84.
Yang CY, Chiu HF, Cheng MF, & Tsai SS (1998) Chlorination of drinking
water and cancer mortality in Taiwan. Environ Res, 78(1): 1-6.
Yang TC & Neely WC (1986) Relative stoichiometry of the oxidation of
ferrous ion by ozone in aqueous solution. Anal Chem, 58: 1551-1555.
Yarington CT (1970) The experimental causticity of sodium hypochlorite
in the esophagus. Ann Otol Rhinol Laryngol, 79(5): 895-988.
Yllner S (1971) Metabolism of chloroacetate-14C in the mouse. Acta
Pharmacol Toxicol, 30: 69-80.
Yokose Y, Uchhida K, Nakae D, Shiraiwa K, Yamamoto K, & Konishi Y
(1987) Studies of carcinogenicity of sodium chlorite in B6C3F1 mice.
Environ Health Perspect, 76: 205-210.
Yoshida Y, Hirose Y, Konda S, Kitada H, & Shinoda A (1977) A
cytological study of Heinz body-hemolytic anemia. Report of a case of
sodium chlorate poisoning complicated by methemoglobinemia and acute
renal failure. Acta Haematol Jpn, 40: 147-151.
Young TB, Kanarek MS, & Tsiatis AA (1981) Epidemiologic study of
drinking water chlorination and Wisconsin female cancer mortality. J
Natl Cancer Inst, 67: 1191-1198.
Young TB, Wolf DA, & Kanarek MS (1987) Case-control study of colon
cancer and drinking water trihalomethanes in Wisconsin. Int J
Epidemiol, 16: 190-197.
Young TB, Kanarek MS, Wolf DA, & Wilson DA (1990) Case-control study
of colon cancer and volatile organics in Wisconsin municipal
groundwater supplies. In: Jolley RL, Condie LW, & Johnson JD ed. Water
chlorination: Chemistry, environmental impact and health effects.
Chelsea, Michigan, Lewis Publishers, Inc., vol 6, p 373.
Yount EA & Harris RA (1980) Studies on the inhibition of
gluconeogenesis by oxalate. Biochim Biophys Acta, 633: 122-133.
Yount EA, Felten SY, O'Connor BL, Peterson RG, Powell RS, Yum MN, &
Harris RA (1982) Comparison of the metabolic and toxic effects of
2-chloropropionate and dichloroacetate. J Pharmacol Exp Ther,
222: 501-508.
Zajdela F, Croisy A, Barbin A, Malaveille C, Tomatis L, & Bartsch H
(1980) Carcinogenicity of chloroethylene oxide, an ultimate reactive
metabolite of vinyl chloride, and bis(chloromethyl)ether after
subcutaneous administration and in initiation-promotion experiments in
mice. Cancer Res, 40: 352-356.
Zeiger E (1990) Mutagenicity of 42 chemicals in Salmonella. Environ
Mol Mutagen, 16(suppl 18): 32-54.
Zeighami EA, Watson AP, & Craun GF (1990a) Chlorination, water
hardness and serum cholesterol in forty-six Wisconsin communities. Int
J Epidemiol, 19: 49-58.
Zeighami EA, Watson AP & Craun GF (1990b) Serum lipid levels in
neighboring communities with chlorinated and nonchlorinated drinking
water. In: Jolley RL, Condie LW, & Johnson JD ed. Water chlorination:
Chemistry, environmental impact and health effects. Chelsea, Michigan,
Lewis Publishers, Inc., vol 6, p 421.
Zierler S, Danley RA, & Feingold L (1986) Type of disinfectant in
drinking water and patterns of mortality in Massachusetts. Environ
Health Perspect, 69: 275-279.
Zierler S, Feingold L, Danley RA, & Craun G (1988) Bladder cancer in
Massachusetts related to chlorinated and chloraminated drinking water:
A case-control study. Arch Environ Health, 43: 195-200.
Zierler S, Feingold L, Danley RA, & Craun GF (1990) A case-control
study of bladder cancer in Massachusetts among populations receiving
chlorinated and chloraminated drinking water. In: Jolley RL, Condie
LW, & Johnson JD ed. Water chlorination: Chemistry, environmental
impact and health effects. Chelsea, Michigan, Lewis Publishers, Inc.,
vol 6, p 399.
RESUME ET EVALUATION
Le chlore (Cl2) est largement utilisé partout dans le monde
comme désinfectant chimique et c'est lui qui constitue la principale
barrière à la contamination microbienne de l'eau de boisson. Les
qualités biocides remarquables du chlore sont toutefois quelque peu
atténuées par la formation, lors du processus de chloration, de
sous-produits de ce désinfectant (SPC) qui posent un problème de santé
publique. C'est d'ailleurs la raison pour laquelle on utilise de plus
en plus d'autres désinfectants chimiques comme l'ozone (O3), le
dioxyde de chlore (ClO2) et les chloramines (NH2Cl). Toutefois
chacun d'entre eux donne aussi naissance à des SPC. Il est certain
qu'aucun compromis n'est tolérable en ce qui concerne la qualité de
l'eau, mais il faut cependant mieux connaître la chimie, la
toxicologie et l'épidémiologie des désinfectants chimiques et de leurs
sous-produits afin d'établir avec plus de sûreté les risques (d'ordre
chimique ou microbien) que comporte la consommation d'eau et de
trouver un compromis entre le risque chimique et le risque
microbiologique. Il est possible de réduire le risque chimique dû aux
SPC sans nuire à la qualité microbiologique de l'eau.
1. Chimie des désinfectants et de leurs sous-produits
Les désinfectants les plus utilisés sont le chlore, l'ozone, le
dioxyde de chlore et la chloramine. Les propriétés physiques et
chimiques des désinfectants et de leurs SPC conditionnent leur
comportement dans l'eau ainsi que leur toxicologie et leur
épidémiologie. Tous les désinfectants envisagés ici sont des oxydants
solubles dans l'eau que l'on produit soit sur place (comme l'ozone)
soit ailleurs (comme le chlore). Ils sont introduits dans l'eau sous
forme de gaz (ozone) ou de liquide (hypochlorite par ex.) à des doses
de l'ordre de quelques mg par litre, soit seuls, soit en association.
Les SPC étudiés dans ce qui suit sont dosables par chromatographie en
phase gazeuse ou liquide et peuvent être classés en dérivés organiques
ou minéraux halogénés (chlorés ou bromés) ou non halogénés, volatils
ou non volatils. Une fois formés, les SPC peuvent être stables ou
instables (par exemple, être décomposés par hydrolyse).
Les SPC se forment par réaction des désinfectants sur un certain
nombre de précurseurs. Les matières organiques naturelles (NOM) que
l'on dose habituellement sous la forme de carbone organique total
(TOC) constituent le précurseur organique, tandis que les ions bromure
(Br-) sont les précurseurs minéraux. La formation des SPC dépend de
la nature de l'eau (teneur en carbone organique total, bromures, pH,
température, ammoniaque, alcalinité due à la présence de carbonates)
et des conditions de traitement (par exemple, dose de désinfectant,
durée de contact, élimination des matières organiques naturelles en
amont du point d'introduction du désinfectant, adjonction préalable de
désinfectant).
Sous la forme d'acide hypochloreux ou d'ion hypochlorite
(HOCl/OCl-), le chlore réagit sur l'ion bromure en l'oxydant en acide
hypobromeux ou ion hypobromite (HOBr/OBr-). L'acide hypochloreux (un
oxydant plus énergique) et l'acide hypobromeux (un agent
d'halogénation plus efficace) réagissent collectivement sur les NOM
pour former des SPC halogénés tels que trihalogénométhanes (THM),
acides halogénoacétiques (HAA), halogénoacétonitriles (HAN),
halogénocétones, hydrate de chloral et chloropicrine. En général, ces
SPC se classent par ordre d'importance selon la séquence THM > HAA
> HAN. La teneur relative en TOC, bromures et chlore influe sur la
distribution des différents THM (on en compte quatre espèces : le
chloroforme, le bromoforme, le bromodichlorométhane - BDCM - et le
dibromochlorométhane - DBCM), des HAA (jusqu'à neuf espèces chlorées
ou bromées) et des HAN (plusieurs espèces chlorées ou bromées).
Généralement les THM, HAA et HAN chlorés sont plus abondants que leurs
homologues bromés, mais c'est l'inverse dans les eaux à forte teneur
en bromures. On a identifié de nombreux SPC dérivant du chlore mais
une proportion importante des halogénures organiques totaux reste à
caractériser. Dans les solutions concentrées d'hypochlorite, le chlore
donne également naissance à des chlorates (ClO3-).
L'ozone peut réagir directement ou indirectement sur les bromures
pour donner des sous-produits bromés et notamment des bromates
(BrO3-). En présence de NOM, il se forme des sous-produits
organiques non halogénés au cours de l'ozonisation, par exemple des
aldéhydes, des cétoacides et des acides carboxyliques, les aldéhydes
(comme le formaldéhyde) étant prédominants. En présence de NOM et de
bromures, l'ozonisation conduit à la formation d'acide hypobromeux qui
conduit à son tour à la formation d'halogénures organiques comme le
bromoforme.
Les principaux SPC qui se forment en présence de dioxyde de
chlore sont les ions chlorite (ClO3-) et chlorate, sans formation
directe de SPC organohalogénés. A l'inverse des SPC des autres
désinfectants, les principaux sous-produits du dioxyde de chlore
proviennent de la décomposition du désinfectant lui-même et non pas
d'une réaction sur des précurseurs.
Lorsqu'on utilise de la chloramine comme désinfectant secondaire,
il se forme en général un dérivé azoté, le chlorure de cyanogène
(CNCl) et des SPC en quantité sensiblement moindre. Il se pose
également la question de la présence de nitrites (NO2-) dans les
réseaux de distribution traités par la chloramine.
Autant qu'on sache, ce sont les THM, les HAA, les bromates et les
chlorites qui sont les SPC les plus fréquents et les plus intéressants
du point de vue de leurs effets sur la santé.
Le groupe de SPC prédominant, lorsqu'on traite par le chlore, est
celui des THM, dont les représentants principaux sont, dans l'ordre,
le chloroforme et le BDCM. Viennent ensuite les HAA dont les espèces
les plus abondantes sont, dans l'ordre, l'acide dichloracétique (DCA)
et l'acide trichloracétique (TCA).
La transformation des bromures en bromates au cours de
l'ozonisation dépend, entre autres, de la teneur en NOM, du pH et de
la température. La concentration peut aller de teneurs indétectables
(2 µg/litre) à plusieurs dizaines de milligrammes par litre. Il est en
général tout à fait possible de prévoir la concentration en chlorites,
qui va d'environ 50 à 70 % de la dose de dioxyde de chlore utilisée.
Les SPC se présentent sous la forme de mélanges complexes qui
sont fonction du désinfectant utilisé, de la qualité de l'eau et des
traitements qu'elle subit. Les autres facteurs qui entrent en ligne de
compte sont l'utilisation simultanée ou successive de plusieurs
désinfectants ou oxydants. En outre, la composition de ces mélanges
peut changer selon la saison. Il est clair que les effets sur la santé
qui pourraient résulter de la présence de ces composés chimiques dans
l'eau dépendent du degré d'exposition aux mélanges de SPC.
Indépendamment de ces SPC (et des THM en particulier), on ne
possède que très peu de données sur la présence de SPC dans l'eau qui
sort des installations de traitement pour pénétrer dans le réseau
d'adduction. A partir des bases de données des laboratoires, on a mis
au point des modèles empiriques qui permettent de prévoir la
concentration en THM (THM totaux et différentes espèces de THM), HAA
(HAA totaux et différentes espèces de HAA) et en bromates. Ces modèles
peuvent être utilisés pour évaluer l'efficacité des traitements et
calculer à l'avance les conséquences d'une modification de ces
traitements. On peut aussi les utiliser lors d'une évaluation de
l'exposition, pour simuler des données manquantes ou anciennes (par
exemple, pour calculer la concentration en HAA d'après celle des THM).
On peut agir sur les SPC en agissant en amont, c'est-à-dire sur
le précurseur - notamment en l'éliminant - ou en choisissant une autre
méthode de désinfection. Par exemple, on peut éliminer les matières
organiques naturelles (NOM) par coagulation, passage sur granulés de
charbon actif, filtration sur membrane et biofiltration en présence
d'ozone. A part l'utilisation de membranes, il n'y a guère d'autres
possibilités pour se débarrasser efficacement des bromures. Si on veut
éviter d'avoir à traiter l'eau, il faut protéger et contrôler les
sources. L'élimination des SPC une fois formés n'est pas une solution
valable dans le cas des SPC organiques; par contre on peut éliminer
les bromates et les chlorites par passage sur charbon actif ou par
réduction. Il y a encore la possibilité de réduire la teneur en SPC en
associant les désinfectants de façon optimale, qu'on utilise comme
désinfectants primaires,ou comme désinfectants secondaires. La
tendance actuelle est à l'utilisation simultanée ou successive de
divers désinfectants; l'ozone est utilisée exclusivement comme
désinfectant primaire, les chloramines comme désinfectants
secondaires, tandis que le chlore et le dioxyde de chlore peuvent
l'être dans les deux cas.
2. Cinétique et métabolisme chez les animaux de laboratoire et
l'Homme
2.1 Désinfectants
Les désinfectants résiduels sont des substances chimiques
susceptibles de réagir sur les composés organiques présents dans la
salive et dans le contenu stomacal, entraînant la formation de
produits secondaires. Il existe d'importantes différences dans la
pharmacocinétique du 36Cl selon qu'il provient du chlore lui-même, de
la chloramine ou du dioxyde de chlore.
2.2 Trihalogénométhanes
Les THM sont absorbés, métabolisés et éliminés rapidement par les
mammifères après ingestion ou inhalation. Après absorption, c'est au
niveau des graisses, du foie et des reins que la concentration
tissulaire est la plus élevée. La demi-vie varie généralement de 0,5 à
3 h et la voie d'élimination principale est une métabolisation en
dioxyde de carbone. Pour que la toxicité se manifeste, il faut que les
THM subissent une activation métabolique en intermédiaires réactifs et
les trois espèces bromées sont métabolisées plus rapidement et en
proportion plus importante que le chloroforme. La principale voie
métabolique est dans tous les cas une oxydation par l'intermédiaire du
cytochrome P450 (CYP) 2E1, qui conduit à la formation de dérivés
carbonylés halogénés (par exemple le phosgène et ses homologues
bromés), après quoi ceux-ci peuvent être hydrolysés en dioxyde de
carbone ou se fixer à des macromolécules tissulaires. Il existe des
voies métaboliques secondaires, comme la déshalogénation réductrice
par l'intermédiaire de la CYP2B1/2/2E1 (qui conduit à la formation de
radicaux libres) ou la conjugaison avec le glutathion (GSH) par
l'intermédiaire de la glutathion- S-transférase (GST) T1-1, qui
entraîne la formation d'intermédiaires mutagènes. Les THM bromés ont
beaucoup plus de chances d'emprunter les voies métaboliques
secondaires que le chloroforme et la conjugaison de ce dernier composé
avec le GSH sous l'action de la GST peut s'effectuer à des
concentrations ou à des doses de chloroforme extrêmement élevées.
2.3 Acides halogénoacétiques
Le métabolisme et la cinétique des acides trihalogénoacétiques et
dihalogénoacétiques diffèrent sensiblement. Dans la mesure où ils sont
métabolisés, les principales réactions des acides trihalogénoacétiques
se produisent dans la fraction microsomique, alors que le métabolisme
des acides dihalogénoacétiques, qui s'effectue essentiellement sous
l'action des glutathion-transférases, se déroule à plus de 90 % dans
le cytosol. Le TCA a une demi-vie biologique de 50 h chez l'Homme. La
demi-vie des autres acides trihalogénoacétiques diminue sensiblement
avec la substitution par le brome et on peut déceler des quantités
mesurables d'acides dihalogénoacétiques comme produits à côté d'acides
trihalogénoacétiques bromés. La demi-vie des acides
dihalogénoacétiques est très courte à faible dose, mais elle augmente
très fortement avec la dose.
2.4 Halogénoaldéhydes et halogénocétones
On ne possède que des données limitées sur la cinétique de
l'hydrate de chloral. Ses deux principaux métabolites sont le
trichloréthanol et le TCA. Le trichloréthanol subit une
glucuronidation rapide puis il passe dans le circuit entérohépatique,
il est hydrolysé et enfin oxydé en TCA. La déchloration du
trichloréthanol ou de l'hydrate de chloral conduirait à la formation
de DCA. Le DCA pourrait être ensuite transformé en monochloracétate
(MCA), en glyoxalate, en glycolate et en oxalate, probablement par le
canal d'un intermédiaire réactif. On n'a trouvé aucune donnée
concernant les autres aldéhydes ou cétones halogénés.
2.5 Halogénoacétonitriles
Ni le métabolisme ni la cinétique des HAN n'ont été étudiés. Les
données quantitatives indiquent que parmi les produits du métabolisme
on trouve du cyanure, du formaldéhyde, ainsi que du cyanure et des
halogénures de formyle.
2.6 Dérivés halogénés de l'hydroxyfuranone
La 3-chloro-4-(dichlorométhyl)-5-hydroxy-2(5H)-furanone (MX) est,
parmi les hydroxyfuranones, celle qui a été le plus largement étudiée.
Il ressort des études sur l'animal que le radiomarqueur (14C) est
rapidement résorbé au niveau des voies digestives et qu'il passe dans
la circulation générale. On n'a pas procédé au dosage de la MX
elle-même dans le sang. Le radiomarqueur est en grande partie excrété
dans les urines et les matières fécales, mais c'est la voie urinaire
qui est la principale voie d'excrétion. Au bout de cinq jours, il ne
reste dans l'organisme qu'une proportion minime du radiomarqueur
initial.
2.7 Chlorites
Le 36Cl provenant de l'ion chlorite est rapidement absorbé. La
dose initiale de chlorite se retrouve à moins de 50 % sous forme de
chlorure dans les urines et en faible proportion sous sa forme
initiale. Il est probable qu'une proportion s'intègre à la réserve de
chlorures de l'organisme, mais comme on ne dispose pas de méthode
d'analyse permettant de caractériser les chlorites présents dans les
échantillons biologiques, on ne possède pas de données détaillées à ce
sujet.
2.8 Chlorates
Les chlorates ont un comportement analogue à celui des chlorites.
Les problèmes analytiques sont les mêmes.
2.9 Bromates
Les bromates sont rapidement absorbés et excrétés, principalement
dans les urines, sous forme de bromures. On peut les mettre en
évidence dans les urines à des doses supérieures ou égales à 5 mg/kg
de poids corporel. La concentration urinaire des bromates culmine au
bout d'environ 1 h et au bout de 2 h ils ne sont plus décelables dans
le plasma.
3. Toxicologie des désinfectants et de leurs sous-produits
3.1 Désinfectants
Le chlore, la chloramine et le dioxyde de chlore sont très
irritants pour les voies respiratoires. L'hypochlorite de sodium, qui
entre dans la composition de l'eau de Javel, est souvent la cause
d'intoxications chez l'Homme. Il s'agit toutefois de cas qui n'ont
rien à voir avec une exposition par consommation d'eau de boisson. On
a relativement peu étudié les effets toxiques que ces désinfectants
peuvent avoir sur l'Homme ou l'animal d'expérience lorsqu'ils sont
présents dans l'eau de boisson. Les quelques études dont on dispose
indiquent que le chlore, les solutions d'hypochlorite ainsi que la
chloramine et le dioxyde de chlore eux-mêmes ne sont probablement pas
à l'origine de cancers ou d'un quelconque effet toxique. On
s'intéresse surtout aux produits secondaires très divers qui résultent
de l'action du chlore et d'autres désinfectants sur les matières
organiques naturelles présentes dans presque toutes les eaux quelle
que soit leur origine.
3.2 Trihalogénométhanes
A la dose d'environ 0,5 mmol/kg de poids corporel, les THM se
révèlent hépatotoxiques et néphrotoxiques chez les rongeurs. Le
véhicule utilisée pour l'administration influe sensiblement sur la
toxicité. Les THM ont peu d'effets toxiques sur la reproduction ou le
développement, mais le BDCM s'est révélé capable de réduire la
mobilité des spermatozoïdes chez des rats à qui on en avait fait
consommer une dose quotidienne de 39 mg/kg p.c. dans leur eau de
boisson. Comme le chloroforme, le BDCM, lorsqu'il est administré dans
de l'huile de maïs, provoque des cancers du foie et du rein après une
exposition à forte dose pendant toute la vie. Au rebours du
chloroforme et du DBCM, le BDCM et le bromoforme provoquent des
tumeurs du côlon chez des rats a qui on les administre par gavage dans
de l'huile de maïs. Le BDCM provoque des cancers ayant ces trois
localisations et à des doses inférieures à celles des autres THM.
Depuis la publication en 1994 des Critères d'hygiène de
l'environnement relatifs au chloroforme, des études supplémentaires
sont venues compléter la somme de données selon lesquelles le
chloroforme n'est pas un cancérogène exerçant une action mutagène
directe par réaction sur l'ADN. En revanche, les THM bromés se
révèlent être faiblement mutagènes, probablement par suite de leur
conjugaison au GSH.
3.3 Acides halogénoacétiques
Les HAA exercent différents effets toxiques sur les animaux de
laboratoire. Dans les cas les plus inquiétants, il s'agit d'effets
cancérogènes ou encore d'effets sur la reproduction et le
développement. L'acide dichloracétique a des effets neurotoxiques
sensibles aux fortes doses utilisées dans un but thérapeutique. Les
effets cancérogènes sont limités au foie et aux fortes doses. Il
existe une somme de données qui tendent à prouver que les effets
tumorigènes de l'acide dichloracétique et de l'acide trichloracétique
tiennent au fait que ces composés agissent sur la division et la mort
cellulaire plutôt qu'à leur faible activité mutagène. Le stress
oxydatif est également une caractéristique de la toxicité des dérivés
bromés appartenant à ce groupe. A forte dose, les acides
dichloracétique et trichloracétique provoquent des malformations
cardiaques chez le rat.
3.4 Halogénoaldéhydes et halogénocétones
Chez le rat, l'hydrate de chloral provoque une nécrose hépatique
à des doses quotidiennes égales ou supérieures à 120 mg/kg de poids
corporel. Chez l'Homme, son effet dépresseur sur le système nerveux
central est probablement en rapport avec un métabolite, le
trichloréthanol. On ne possède que des données toxicologiques limitées
sur les autres cétones et aldéhydes halogénés. Une exposition au
chloracétaldéhyde entraîne des anomalies hématologiques chez le rat.
Chez la souris, on constate que l'exposition à la
1,1-dichloropropanone (1,1-DCPN) a des effets hépatotoxiques, effets
qui ne s'observent pas avec l'isomère 1,3 (1,3-DCPN).
La plupart des tests sur bactéries à la recherche de mutations
ponctuelles se sont révélés négatifs dans le cas de l'hydrate de
chloral et il en a été de même pour la recherche de lésions
chromosomiques in vivo. On a toutefois montré que l'hydrate de
chloral peut provoquer des aberrations chromosomiques structurales
in vivo et in vitro. L'hydrate de chloral provoquerait également
la formation de tumeurs hépatiques chez la souris. On ne sait pas très
bien si c'est l'hydrate de chloral lui-même ou ses métabolites qui
sont à l'origine des effets cancérogènes. Les deux métabolites de
l'hydrate de chloral, à savoir l'acide trichloracétique et l'acide
dichloracétique, se sont révélés capables de provoquer des tumeurs
hépatiques chez la souris.
Un certain nombre d'aldéhydes et de cétones se révèlent être
puissamment mutagènes pour les bactéries. On observés des effets
clastogènes dus aux chloropropanones. Dans une étude au cours de
laquelle les animaux ont été exposés leur vie durant à du
chloracétaldéhyde contenu dans leur eau de boisson, on a observé la
formation de tumeurs hépatiques. D'autres aldéhydes halogénés comme le
2-chloropropénal, se sont révélés avoir un effet tumoro-initiateur sur
la peau de souris. On n'a pas étudié les propriétés cancérogènes des
halogénocétones présentes dans l'eau de boisson, mais la 1,3-DCPN se
comporte comme un initiateur tumoral cutané chez la souris.
3.5 Halogénoacétonitriles
Jusqu'ici on n'a procédé qu'à des études toxicologiques limitées
sur ces composés. Certains d'entre eux sont mutagènes, mais ces effets
sont difficiles à mettre en rapport avec le fait que ces composés se
comportent comme des initiateurs tumoraux cutanés. On ne dispose que
d'études très limitées sur la cancérogénicité de ce groupe de
composés. Les premiers indices d'effets toxiques sur le développement
relevés dans le cas de certains de ces composés, sont semble-t-il en
grande partie attribuables au véhicule utilisé pour les administrer.
3.6 Dérivés halogénés de l'hydroxyfuranone
D'après les études expérimentales, les effets toxiques de la MX
sont essentiellement des effets cancérogènes et mutagènes. Plusieurs
études in vitro ont mis en évidence une activité mutagène de ce
composé sur cellules bactériennes et mammaliennes. Il provoque
également des aberrations chromosomiques et des lésions de l'ADN sur
des cellules hépatiques et testiculaires isolées et on observe
in vivo des échanges entre chromatides soeurs dans les lymphocytes
périphériques de rats. Une évaluation globale des données relatives à
la mutagénicité montre que la MX est mutagène in vitro et in
vivo. Une étude de cancérogénicité sur des rats a montré qu'il y
avait augmentation de la fréquence des tumeurs au niveau de plusieurs
organes.
3.7 Chlorites
L'action toxique des chlorites est due principalement aux lésions
qu'ils causent par oxydation aux érythrocytes (stress oxydatif) à des
doses ne dépassant pas 10 mg/kg de poids corporel. On a également
relevé des indices d'effets neurocomportementaux légers chez des
ratons à des doses quotidiennes de 5,6 mg/kg de poids corporel. En ce
qui concerne la génotoxicité, les données sont contradictoires. Chez
des animaux exposés de manière chronique à cet ion, on ne relève pas
d'augmentation dans la fréquence des tumeurs.
3.8 Chlorates
Sur le plan toxicologique, les chlorates sont analogues aux
chlorites, mais en tant qu'oxydants, leur pouvoir lésionnel est
moindre. Il ne semblent pas être tératogènes ni génotoxiques in
vivo. On ne possède pas de données provenant d'études de
cancérogénicité à long terme.
3.9 Bromates
A haute dose, les bromates provoquent des lésions au niveau des
tubules rénaux chez le rat. Administrés de manière chronique ils
provoquent des tumeurs rénales, péritonéales et thyroïdiennes chez le
rat aux doses supérieures ou égales à 6 mg/kg p.c. Le hamster est
moins sensible et les souris beaucoup moins. Les bromates sont
également génotoxiques in vivo à forte dose chez le rat. Le pouvoir
cancérogène se révèle être la conséquence du stress oxydatif subi par
la cellule.
4. Etudes épidémiologiques
4.1 Cardiopathies
Les études épidémiologiques ont mis en évidence une augmentation
du risque de cardiopathies qui serait liée à la consommation d'eau
traitée par le chlore ou la chloramine. On n'a pas effectué d'études
de ce genre sur les autres désinfectants.
4.2 Cancer
Les données épidémiologiques ne sont pas suffisantes pour qu'on
puisse parler d'une relation causale entre le cancer de la vessie et
la consommation prolongée d'eau traitée par le chlore ou une
exposition à des THM comme le chloroforme. Il en va de même pour le
cancer du côlon, les données épidémiologiques étant dans ce cas
équivoques et non concluantes. On ne dispose pas de renseignements
suffisants pour évaluer le risque de cancer du rectum ni le risque
d'autres cancers observés lors d'une seule et unique étude analytique.
Des études épidémiologiques diverses se sont efforcées d'évaluer
le risque de cancer résultant de la consommation d'eau chlorée. Deux
de ces études ont porté sur de l'eau traitée à la chloramine.
Plusieurs autres sont consacrées à l'évaluation de l'exposition aux
THM totaux ou à certains d'entre eux comme le chloroforme, mais elles
ne prennent pas en considération les autres SPC ou contaminants de
l'eau, lesquels ne sont pas forcément les mêmes selon qu'il s'agit de
nappes souterraines ou d'eaux superficielles. Dans un cas, on a évalué
le pouvoir mutagène de l'eau de boisson par un test sur Salmonella
typhimurium. On n'a pas essayé de déterminer le risque de cancer que
pourrait comporter la consommation d'eau désinfectée par l'ozone ou le
dioxyde de chlore.
Des études cas-témoins écologiques ou basées sur l'examen des
certificats de décès ont permis d'échafauder un certain nombre
d'hypothèses qui devront être approfondies par des études analytiques
prenant en considération la consommation individuelle d'eau de
boisson, compte tenu d'éventuels facteurs de confusion.
Selon les études analytiques auxquelles il a été procédé, il y
aurait une augmentation faible à modérée du risque de cancer de la
vessie, du côlon, du rectum, du pancréas, du sein, du cerveau ou du
poumon en cas de consommation prolongée d'eau traitée par le chlore.
Un certain nombre d'études ont relevé, individuellement, une
association entre l'exposition à l'eau chlorée et le cancer du
pancréas, du sein et du cerveau. Toutefois, pour déterminer si ces
associations épidémiologiques correspondent effectivement à un lien
causal, une seule et unique étude ne suffit pas. Par exemple, une
étude relevé une légère augmentation du risque relatif de cancer du
poumon lié à la consommation d'eau de surface, mais ce risque est trop
faible pour qu'on puisse exclure la présence d'un facteur de confusion
résiduel.
Selon une étude cas-témoins, il y a aurait une corrélation
modérément forte entre le cancer du rectum et la consommation
prolongée d'eau chlorée ou une exposition cumulée à des THM, mais les
études de cohortes portant sur ce problème n'ont relevé aucune
augmentation du risque ou tout du moins un risque trop faible pour
qu'on puisse exclure la présence de facteurs de confusion résiduels.
On a aussi trouvé que l'augmentation de la durée de consommation
d'eau chlorée à la chloramine était associée à une diminution du
risque de cancer de la vessie, mais rien sur le plan biologique ne
permet de considérer que l'eau traitée à la chloramine puisse avoir un
tel effet protecteur.
A en juger par plusieurs études, il y aurait une association
entre l'augmentation du risque de cancer de la vessie et la
consommation prolongée d'eau chlorée ou une exposition de longue durée
à des THM, mais pour le même type de cancer les comparaisons entre
fumeurs et non fumeurs ou entre hommes et femmes donnent des résultats
incohérents. Dans trois de ces études, on a étudié le risque découlant
d'une exposition aux THM. L'une d'entre elles n'a mis aucune
corrélation en évidence, une autre a trouvé que le risque relatif
était modérément accru pour les hommes, mais pas pour les femmes.
Selon la troisième étude, une légère augmentation du risque relatif
serait associée à une exposition cumulée à des THM évaluée à 1957-6425
µg de THM par année-litre. On a également fait état d'une association
modérée entre le risque de cancer et une exposition à des
concentrations de THM supérieures à 24, 49 ou 74 µg/litre, selon le
cas. Il n'a pas été constaté d'association entre l'augmentation du
risque de cancer de la vessie dans une cohorte de femmes et la
consommation d'eau de surface chlorée délivrée par les services
municipaux ou l'exposition à du chloroforme ou d'autres THM, mais la
durée de suivi a été très courte (8 ans), d'où la rareté des cas à
étudier.
Comme on s'est insuffisamment intéressé, d'un point de vue
épidémiologique, à la détermination de l'exposition aux contaminants
présents dans l'eau, il n'est pas possible d'évaluer correctement
l'accroissement du risque de cancer qui ressort de ces différentes
études. Il peut y avoir des risques particuliers dus à d'autres SPC, à
des mélanges de produits secondaires ou à d'autres contaminants, mais
il est également possible que cet accroissement du risque soit dû à
d'autres facteurs dont on attribuerait les effets à l'eau chlorée et
aux THM.
4.3 Grossesses à issue défavorable
Des études ont été consacrées au lien qu'il pourrait y avoir
entre des grossesses à issue défavorable et la consommation d'eau
chlorée ou l'exposition à des THM. Un groupe de scientifiques qui
s'est récemment réuni sous l'égide de l'Environmental Protection
Agency des Etats-Unis a passé en revue les études épidémiologiques
consacrées à cette question et conclu que les résultats publiés ne
prouvent pas de manière convaincante que l'eau chlorée ou les THM sont
responsables d'accidents au cours de la grossesse.
Les résultats des premières études sont difficiles à interpréter
à cause d'insuffisances d'ordre méthodologique et de biais probables.
Une étude cas-témoins récemment achevée mais non encore publiée
fait état d'une augmentation modérée du risque relatif de malformation
du tube neural chez les enfants dont la mère a résidé au début de sa
grossesse dans une zone où la concentration des THM était supérieure à
40 µg/litre. Il faudra confirmer ces résultats dans une autre zone
pour pouvoir procéder à une évaluation en bonne et due forme. Une
étude menée antérieurement dans la même région avait fait état d'une
association similaire, mais elle présentait des insuffisances sur le
plan méthodologique.
Un récente étude de cohorte a révélé l'existence d'un risque
accru d'avortement prématuré chez les femmes enceintes ayant consommé
de grandes quantités (au moins 5 verres par jour) d'eau du robinet
froide contenant >75 µg de THM par litre. En considérant le cas de
chaque espèce chimique en particulier, on a constaté que seule une
forte consommation de BDCM (>18 µg/litre) était associée à un
risque d'avortement. Comme il s'agit de la première étude qui fasse
état d'un effet génésique indésirable imputable à un produit
secondaire bromé, un groupe scientifique a recommandé de procéder à
une autre étude dans une région différente afin de voir s'il est
possible de reproduire ces résultats et il a également indiqué qu'il
serait souhaitable d'essayer d'évaluer l'exposition des membres de la
cohorte à d'autres contaminants.
5. Caractérisation du risque
Il est à noter que le traitement de l'eau par des désinfectants
conduit habituellement à la formation de sous-produits dont certains
peuvent être dangereux. Toutefois, aux concentrations auxquelles ils
sont présents dans l'eau de boisson, le risque que ces composés
représentent pour la santé est extrêmement faible par rapport à celui
d'une désinfection insuffisante. Il ne faut pas que les tentatives en
vue de réduire ces produits secondaires conduisent à une désinfection
insuffisante.
5.1 Caractérisation du risque et relation dose-réponse
5.1.1 Toxicologie
1) Chlore
Le Groupe de travail de l'OMS qui s'est réuni en 1993 pour
préparer les Directives pour l'eau de boisson s'est penché sur le
problème du chlore. Il a fixé une dose journalière tolérable (TDI)
pour cet élément égale à 150 µg/kg de poids corporel de chlore libre,
en se basant sur la dose sans effet observable (NOAEL) qui est
d'environ 15 mg/kg p.c. par jour selon des études de 2 ans sur le rat
et la souris, avec une marge d'incertitude correspondant à un facteur
100 (10 pour à chaque fois pour tenir compte des variation intra- et
interspécifiques). On ne possède pas de nouvelles données selon
lesquelles il y aurait lieu de modifier cette dose journalière
tolérable.
2) Monochloramine
Le Groupe de travail précité a également étudié le cas de la
monochloramine. Il a fixé à 94 µg/kg p.c. la dose journalière
tolérable de ce composé en s'appuyant sur la NOAEL d'environ 9,4 mg/kg
p.c. obtenue à l'issue d'une étude toxicologique de 2 ans sur le rat
(dose maximale utilisée), avec une marge d'incertitude correspondant à
un facteur 100 (10 à chaque fois pour tenir compte des variations
intra- et interspécifiques). On ne possède pas de nouvelles données
selon lesquelles il y aurait lieu de modifier cette dose journalière
tolérable.
3) Dioxyde de chlore
Dans l'eau, la chimie du dioxyde de chlore se révèle complexe,
mais le principal produit de décomposition est l'ion chlorite. Pour
fixer une dose journalière tolérable, on peut prendre en considération
les données relatives au dioxyde de chlore et aux chlorites. Dans ces
conditions, la dose journalière tolérable de dioxyde de chlore est
égale à 30 µg/kg p.c.; elle a été obtenue sur la base d'une NOAEL de
2,9 mg/kg p.c. par jour, le critère pris en compte étant les anomalies
du développement neural chez le rat provoquées par l'ion chlorite.
4) Trihalogénométhanes
C'est le cancer qui constitue le principal risque d'une
exposition prolongée à ce groupe de SPC. Vu que les données relatives
au chloroforme n'indiquent la possibilité d'apparition de cancers du
foie chez l'animal qu'après exposition de longue durée à des doses
cytotoxiques, il est clair qu'une exposition à de faibles
concentrations de ce composé dans l'eau de boisson ne fait guère
courir de risque de cancer. On a obtenu une NOAEL de 10 mg/kg p.c. par
jour avec comme critères la cytolétalité et l'hyperplasie
régénérative, après administration du composé à des souris dans de
l'huile de maïs sur une période de 3 semaines. Compte tenu des données
relatives au mode d'action du chloroforme en tant que cancérogène, on
a fixé à 10 µg/kg p.c. la dose journalière tolérable de ce composé en
se basant sur la NOAEL relative à la cytotoxicité obtenue sur la
souris, avec une marge d'incertitude correspondant à un facteur 1000
(un facteur 10 à chaque fois pour les variations intra- et
interspécifiques plus 10 pour tenir compte de la brève durée de
l'étude). Cette façon de faire est justifiée par un certain nombre
d'autres études. Cette dose journalière tolérable a une valeur du même
ordre que celle que figure dans la dernière édition des Directives
pour l'eau de boisson, valeur qui est tirée d'une étude de 1979 au
cours de laquelle des chiens ont été exposés pendant 7,5 années à du
chloroforme.
Parmi les THM bromés, le BDCM est particulièrement intéressant
car il provoque des tumeurs de diverses localisations chez le rat et
la souris (foie, rein, côlon) après administration par gavage dans de
l'huile de maïs. La formation de tumeurs coliques chez le rat sous
l'action du BDCM (et du bromoforme) est également intéressante en
raison de l'existence d'associations épidémiologiques avec le cancer
colo-rectal. On estime également que BDCM et les autres THM bromés
sont faiblement mutagènes. Il est généralement admis que les
cancérogènes dotés de propriétés mutagènes donnent lieu à une relation
dose-réponse linéaire à faible dose, la mutagenèse étant généralement
considérée comme un effet irréversible et cumulatif.
Lors d'une étude toxicologique de 2 ans, du BDCM a été administré
par gavage dans de l'huile de maïs à des souris et des rats. On a
observé la formation de tumeurs (ainsi qu'une cytotoxicité et une
augmentation de la prolifération cellulaire) au niveau du rein aux
doses quotidienne de 50 mg/kg p.c. chez les souris et de 100 mg/kg
p.c. chez le rat. Des tumeurs du côlon ont été observées chez le rat
après exposition à des doses quotidiennes de 50 et 100 mg/kg p.c. En
s'appuyant sur la valeur de l'incidence des tumeurs rénales obtenue
lors de cette étude, on a pu procéder à une estimation quantitative du
risque. On a ainsi obtenu un CSF (cancer slope factor) de 4,8 × 10-3
[mg/kg p.c. par jour]-1 et pour un niveau de risque de 10-5, la dose
est de 2,1 µg/kg p.c. par jour. En se fondant sur la valeur de
l'incidence des cancers du côlon chez le rat, on a obtenu un CSF égal
à 4,2 × 10-3 [mg/kg p.c. par jour]-1 (2,4 µg/kg p.c. par jour pour un
risque de 10-5). Le Centre international de recherche sur le cancer
(CIRC) a classé le BDCM dans le groupe 2B (peut-être cancérogène pour
l'Homme).
Le DBCM et le bromoforme ont fait l'objet d'études toxicologiques
à long terme. Lors d'une étude au cours de laquelle on a gavé des rats
et des souris pendant deux ans avec ces produits incorporés à de
l'huile de maïs, on a constaté que le DBCM provoquait des tumeurs
hépatiques chez les souris femelles - mais pas chez les rats - à la
dose quotidienne de 100 mg/kg p.c. Il avait été avancé, lors
d'évaluations antérieures, que l'huile de maïs aurait pu jouer un rôle
dans l'apparition de ces tumeurs chez les souris femelles. Dans le cas
du bromoforme, on a observé des tumeurs du côlon en légère
augmentation chez le rat à la dose quotidienne de 200 mg/kg p.c. La
valeur du CSF obtenue dans le cas de ces tumeurs est égale à
6,5 × 10-3 [mg/kg p.c. par jour]-1 dans le cas du DBCM pour un risque
de 10-5 et à 1,3 × 10-3 [mg/kg p.c. par jour]-1 ou 7,7 µg/kg p.c. par
jour dans le cas du bromoforme pour un risque de 10-5.
Ces deux THM bromés sont faiblement mutagènes dans un certain
nombre de tests et ce sont en tous cas de loin les SPC qui se révèlent
les plus mutagènes du groupe dans le système d'épreuve utilisant les
GST. Comme ce sont en outre les plus lipophiles, il faut se demander
si l'huile de maïs n'a pas réduit la biodisponibilité dans le cas des
études à long terme. En ce qui concerne le DBCM, on a obtenu une NOAEL
de 30 mg/kg p.c. par jour en se basant sur l'absence d'effets
histopathologiques sur le foie chez des rats qui avaient reçu par
gavage pendant 13 semaines ce composé incorporé à de l'huile de maïs.
Le CIRC a placé ce composé dans le groupe 3 (composés ne pouvant être
classés par rapport à leur cancérogénicité pour l'Homme). On a fixé
une dose journalière tolérable de 30 µg/kg p.c. en se basant sur la
NOAEL de 30 mg/kg p.c. j-1 (critère : hépatotoxicité) avec une marge
d'incertitude correspondant à un facteur 1000 (un facteur de 10 à
chaque fois pour tenir compte des variations intra- et
interspécifiques, plus un autre facteur de 10 pour prendre en compte
la faible durée de l'étude et un pouvoir cancérogène éventuel).
De même, il est possible d'obtenir pour le bromoforme une NOAEL
de 25 mg/kg p.c. j-1 en se basant sur l'absence de lésions hépatiques
chez le rat après 13 semaines d'administration par gavage dans de
l'huile de maïs. A partir de cette valeur, on a fixé à 25 µg/kg p.c.,
la dose journalière tolérable, avec une marge d'incertitude
correspondant à un facteur 1000 (un facteur de 10 à chaque fois pour
tenir compte des variations intra- et interspécifiques, plus un autre
facteur 10 pour prendre en compte de la courte durée de l'étude et de
la cancérogénicité éventuelle du produit). Le CIRC a placé le
bromoforme dans le groupe 3 (composés ne pouvant être classés par
rapport à leur cancérogénicité pour l'Homme).
5) Acides halogénoacétiques
Il est très improbable que le DCA provoque des mutations aux
faibles doses correspondant à sa concentration habituelle dans l'eau
de boisson chlorée. D'après les données disponibles, il affecte
différemment la vitesse de réplication des hépatocytes selon qu'il y a
eu ou non action d'un initiateur. Les relations dose-réponse sont
complexes, la DCA commençant par stimuler la division des hépatocytes
normaux. Toutefois, aux doses plus faibles utilisées dans les études à
long terme sur l'animal (ces doses sont tout de même beaucoup plus
élevées que les concentrations présentes habituellement dans l'eau de
boisson), la vitesse de réplication des hépatocytes normaux finit par
diminuer fortement. On peut en déduire que les hépatocytes normaux
finissent par provoquer une régulation négative des voies de
stimulation par la DCA. Cependant, les cellules modifiées, notamment
celles qui expriment de grandes quantités de protéines réagissant sur
les anticorps c-Jun, ne semblent pas capables de réguler négativement
cette réponse. C'est pourquoi les cellules porteuses de ce phénotype
se divisent très rapidement dans les lésions précancéreuses aux doses
de DCA provoquant l'apparition de tumeurs à très faible temps de
latence. D'après les données les premières données, il semblerait que
cette modification permanente de la naissance et de la mort
cellulaires soit également nécessaire pour qu'il y ait cancérisation
des lésions. Les études dans lesquelles on fait également intervenir
un initiateur ou un promoteur tumoral appuient cette interprétation.
En se fondant sur les considérations précédentes, on peut penser
que les estimations actuelles du risque de cancer imputable à l'acide
dichloracétique devront être modifiées pour tenir compte des nouveaux
résultats concernant le métabolisme et le mode d'action comparés de ce
composé, afin que l'on puisse élaborer un modèle dose-réponse qui
s'appuie sur les données biologiques. Ces résultats ne sont pas encore
disponibles, mais ils devraient l'être d'ici 2 à 3 ans.
Les effets de l'acide dichloracétique se révèlent être en rapport
très étroit avec les doses qui provoque une hépatomégalie et une
accumulation de glycogène chez la souris. La dose la plus faible
produisant un effet nocif observable (LOAEL) dans ce cas a été évaluée
à 0,5 g/litre lors d'une étude de 8 semaines sur la souris. Cette dose
correspond à environ 100 mg/kg p.c. par jour. La même étude a fourni
une NOAEL de 0,2 g/litre, soit environ 40 mg/kg j-1. On a fixé la
dose journalière tolérable à 40 µg par kg de poids corporel en
appliquant un coefficient d'incertitude de 1000 à cette NOAEL (un
facteur de 10 à chaque fois pour tenir compte des variations intra- et
interspécifiques plus un autre facteur de 10 pour prendre en compte la
courte durée d'étude et une cancérogénicité éventuelle). Le CIRC a
placé l'acide dichloracétique dans le groupe 3 (composés ne pouvant
être classés par rapport à leur cancérogénicité pour l'Homme).
L'acide trichloracétique est l'un des plus faibles activateurs
des récepteurs PPAR (peroxisome proliferator-activated receptors)
que l'on connaisse. En tant que proliférateur des peroxysomes, il n'a
qu'une activité marginale, même chez le rat. Par ailleurs, lorsqu'on
donne à des rats une eau de boisson à forte teneur en acide
trichloracétique, il ne se forme pas de tumeurs du foie. Tous ces
résultats incitent fortement à penser que l'acide trichloracétique ne
présente qu'un faible risque cancérogène pour l'Homme étant donné sa
faible concentration dans l'eau de boisson.
Si on considère le problème toxicologique dans sa généralité, ce
sont les effets de ce composé sur le développement qui constituent le
point d'aboutissement le plus préoccupant de son action toxique. Les
animaux de laboratoire présentent une bonne tolérance à ce composé à
la dose de 0,5 g/litre (soit une dose quotidienne d'environ 50 mg/kg
de poids corporel) et ne présentent guère d'effets indésirables à
cette concentration. A la dose de 2 g/litre, le seul effet indésirable
consiste en une hépatomégalie. Chez la souris, on n'observe pas
d'hépatomégalie à la dose de 0,35 g/litre, c'est à dire pour une dose
quotidienne de 40 mg/kg p.c.
Dans une autre étude, on a observé environ trois fois plus
d'anomalies au niveau des tissus mous que chez les témoins à la dose
la plus faible administrée, soit 330 mg/kg j-1. A cette dose, il
s'agissait d'anomalies légères et la dose est bien du même ordre que
celle qui provoque une hépatomégalie (et certains effets
cancérogènes). Etant donné qu'il y a interaction entre les récepteurs
PPAR et les mécanismes de signalisation cellulaire susceptibles
d'intervenir dans les processus normaux de développement, il faut
envisager l'existence d'un mécanisme commun à l'hépatomégalie, aux
effets cancérogènes et aux effets sur le développement.
La dose journalière tolérable d'acide trichloracétique est basée
sur une NOAEL de 40 mg par kg de poids corporel par jour, obtenue à la
suite d'une étude à long terme sur la souris, le critère retenu étant
l'hépatotoxicité. En se donnant une marge d'incertitude correspondant
à un facteur 1000 (un facteur de 10 à chaque fois pour tenir compte
des variations intra- et interspécifiques plus un autre facteur de 10
pour prendre en compte la cancérogénicité éventuelle du produit), on
arrive à une dose journalière tolérable de 40 µg/kg p.c. Le CIRC a
placé l'acide trichloracétique dans le groupe 3 (composés ne pouvant
être classés par rapport à leur cancérogénicité pour l'Homme).
Les données relatives à la cancérogénicité des acides
bromacétiques ont un caractère trop préliminaire pour permettre la
caractérisation du risque. Les données que l'on peut tirer des résumés
analytiques incitent cependant à penser que ces composés sont
susceptibles d'avoir des effets hépatocancérogènes chez la souris à
des doses qui ne sont pas tellement différentes ce celles des acides
chloracétiques. Outre des mécanismes analogues à ceux qui sont à la
base des processus malins dont sont responsables les acides
dichloracétique et trichloracétique, il est possible que le stress
oxydatif découlant de leur métabolisation joue un rôle dans leurs
effets.
On possède un nombre non négligeable de données concernant les
effets que l'acide dibromacétique exerce sur la fonction reproductrice
chez le mâle. On n'a pas observé d'effets chez des rats à la dose
quotidienne de 2 mg/kg de poids corporel sur une durée de 79 jours,
mais à la dose de 10 mg/kg, il y avait augmentation de la rétention
des spermatides au stade 19. A mesure que les doses ont été
augmentées, on a noté une aggravation des effets, notamment une
atrophie importante des tubes séminifères à 250 mg/kg j-1. Il n'y a
pas eu retour à la normale dans les six mois suivant la cessation de
l'exposition. Pour déterminer la dose journalière tolérable, on est
parti d'une NOAEL journalière de 2 mg/kg p.c. en se donnant une marge
d'incertitude correspondant à un facteur 100 (un facteur de 10 à
chaque fois pour tenir compte des variations intra- et
interspécifiques).
6) Hydrate de chloral
A la concentration de 1 g par litre d'eau de boisson (166 mg/kg
j-1), il y a eu formation de tumeurs hépatiques chez des souris
exposée à ce produit pendant 104 semaines. On n'a pas étudié les
effets des doses inférieures à cette valeur. Il a été montré que
l'hydrate de chloral provoquait l'apparition d'anomalies
chromosomiques dans un certain nombre de systèmes d'épreuve in
vitro, mais in vivo les résultats se sont révélés largement
négatifs. Il est probable que si l'hydrate de chloral provoque des
tumeurs du foie, c'est parce qu'il est métabolisé en acide
dichloracétique ou trichloracétique. Comme on l'a vu plus haut, ces
composés sont considérés comme des promoteurs tumoraux. Le CIRC a
placé l'hydrate de chloral dans le groupe 3 (composés qui ne peuvent
être classés par rapport à leur cancérogénicité pour l'Homme).
Administré à des rats pendant 90 jours dans leur eau de boisson,
l'hydrate de chloral a provoqué une nécrose des hépatocytes aux
concentrations supérieures ou égales à 1200 mg/litre. En revanche,
aucun effet n'a été observé à 600 mg/litre (soit environ 60 mg/kg
j-1) On a observé une hépatomégalie chez des souris à la dose
quotidienne de 144 mg/kg p.c., lorsque le produit était administré par
gavage sur une période de 14 jours. Lors de cette même étude, aucun
effet n'a été observé à la dose quotidienne de 14,4 mg/kg, mais une
étude de 90 jours effectuée dans le prolongement de la précédente a
montré qu'à la dose de 70 mg/litre dans l'eau de boisson (c'est-à-dire
16 mg/kg j-1), il y avait une légère hépatomégalie. En se donnant une
marge d'incertitude correspondant à un facteur 1000 (un facteur de 10
à chaque fois pour tenir compte des variation inter- et
intraspécifiques plus un autre facteur 10 pour prendre en
considération le fait qu'on est parti de la LOAEL au lieu de la
NOAEL), on aboutit à une dose journalière tolérable de 16 µg/kg de
poids corporel.
7) Halogénoacétonitriles
Faute de données humaines suffisantes ou d'une étude sur l'animal
qui porte sur une fraction substantielle de la vie des animaux de
laboratoire, on ne dispose d'aucun élément d'appréciation du risque
cancérogène qui fasse l'unanimité.
Les données tirées des études subchroniques permettent d'avoir un
certain nombre d'indications sur les NOAEL relatives à la toxicité
générale du dichloracétonitrile (DCAN) et du dibromacétonitrile
(DBAN). C'est ainsi que l'on a obtenu une NOAEL respectivement égale à
8 et 23 mg/kg p.c. j-1 pour le DCAN et le DBAN à l'occasion d'études
de 90 jours sur des rats et des souris, le critère pris en
considération étant une diminution du poids corporel notée aux doses
respectives de 33 et 45 mg/kg p.c. j-1.
Le groupe de travail de l'OMS qui s'est réuni en 1993 pour la
préparation des Directives de qualité pour l'eau de boisson a étudié
le cas du DCAN et du DBAN. Il a fixé à 15 µg/kg j-1 la dose
journalière tolérable de DCAN en se basant sur une NOAEL de 15 mg/kg
j-1 obtenue à la suite d'une étude sur la toxicité génésique du DCAN
chez le rat. Le groupe a considéré qu'il y avait une marge
d'incertitude correspondant à un facteur 1000 (un facteur de 10 à
chaque fois pour tenir compte des variation intra- et interspécifiques
plus un autre facteur 10 afin de prendre également en considération la
gravité des effets). En ce qui concerne le DBAN, on n'a observé
d'effets sur la reproduction et le développement qu'aux doses
dépassant celles entraînant une intoxication générale (soit environ 45
mg/kg p.c. j-1). En se basant sur la NOAEL de 23 mg/kg p.c. j-1 tirée
de l'étude de 90 jours sur le rat et en se donnant une marge
d'incertitude correspondant à un facteur 1000 (un facteur de 10 à
chaque fois pour tenir compte des variations intra- et
interspécifiques plus un autre facteur 10 en considération de la
brièveté de l'étude), on a abouti à une valeur de la dose journalière
tolérable égale à 23 µg/kg p.c. j-1). On ne dispose d'aucune donnée
nouvelle qui justifierait de modifier cette dose.
En ce qui concerne le trichloracétonitrile (TCA), on a obtenu une
LOAEL de 7,5 mg/kg j-1 en prenant l'embryotoxicité comme critère et
une LOAEL de 15 mg/kg j-1 en prenant en considération les effets sur
le développement. Des résultats ultérieurs incitent toutefois à penser
que les effets constatés étaient liés au véhicule utilisé.
On ne possède aucune donnée qui permettrait de caractériser le
risque imputable à d'autres halogénoacétonitriles.
8) MX
Le MX, qui possède des propriétés mutagènes, a récemment fait
l'objet d'une étude à long terme sur le rat au cours de laquelle on a
observé des effets cancérogènes. Selon cette étude, le MX provoque des
tumeurs de la thyroïde et des voies biliaires. A la dose la plus
faible administrée (0,4 mg/kg p.c. j-1), on a constaté que
l'incidence des tumeurs thyroïdiennes était en augmentation. On estime
depuis longtemps que la formation de tumeurs de la thyroïde après
exposition à de fortes doses de produits chimiques est une
caractéristique des composés halogénés. Il se pourrait que les tumeurs
folliculaires de la thyroïde soient la conséquence d'un dérèglement de
la fonction thyroïdienne ou d'une mutagenèse. On a également observé
un accroissement de l'incidence des cholangiomes et des
cholangiocarcinomes qui était lié à la dose; cet effet à commencé à se
manifester à faible dose chez les femelles; il était moins marqué chez
les mâles. C'est précisément cette augmentation de l'incidence des
cholangiomes et des cholangiocarcinomes chez les rattes que l'on a
utilisé pour déterminer la valeur du CSF (cancer slope factor). La
limite supérieure de confiance à 95 % pour un risque de 10-5 sur
toute la durée de l'existence a été trouvée égale à 0,06 µg/kg p.c.
j-1 sur la base d'un modèle linéaire multistade.
9) Chlorites
L'exposition aux chlorites conduit systématiquement à un stress
oxydatif qui en constitue le principal effet avec pour conséquence des
modifications au niveau des érythrocytes. Ce point d'aboutissement de
l'action toxique de l'ion chlorite s'observe chez les animaux de
laboratoire et, comme dans le cas des chlorates, chez les sujets
humains intoxiqués par une exposition à de très fortes doses. On
possède suffisamment de données pour déterminer la dose journalière
tolérable par l'Homme, données notamment fournies par des études de
toxicité chronique ainsi qu'une étude de toxicité génésique portant
sur deux générations. Les études menées sur des volontaires pendant
des périodes allant jusqu'à 12 semaines n'ont pas permis de mettre en
évidence le moindre effet hématologique, même à la dose la plus forte
utilisée, à savoir 36 µg/kg p.c. j-1. Ces études n'ayant pas permis
de déterminer la dose à partir duquel se manifestent des effets
toxiques, on ne peut rien en tirer quant à l'ordre de grandeur de la
marge de sécurité.
Une étude portant sur deux générations de rats a permis de fixer
la NOAEL à 2,9 mg/kg p.c. j-1, les critères retenus étant la
diminution de l'amplitude du tressaillement auditif, la réduction du
poids du cerveau dans les générations F1 et F2 et la modification du
poids du foie dans les deux générations. En estimant que la marge
d'incertitude correspond à un facteur 100 (un facteur de 10 à chaque
fois pour tenir compte des variations intra- et interspécifiques), on
en a déduit que la dose journalière tolérable était de 30 µg/kg p.c.
Les études effectuées sur des volontaires confirment cette valeur.
10) Chlorates
Comme dans le cas des chlorites, le principal problème d'ordre
toxicologique tient aux lésions oxydatives qui se produisent au niveau
des érythrocytes. De même que pour l'ion chlorite, on a constaté
qu'une dose quotidienne de chlorates de 0,036 mg/kg p.c. pendant
12 semaines ne provoquait aucun effet indésirable chez des
volontaires. La base de données relative aux chlorates est moins riche
que pour les chlorites mais on dispose cependant des résultats d'une
récente étude de 90 jours sur le rat, qui a permis de déterminer la
NOAEL en prenant comme critère une déplétion des colloïdes dans la
glande thyroïde. La valeur obtenue est de 30 mg/kg p.c. j-1. On est
dans l'attente des résultats d'une autre étude à long terme, aussi n'a
t-on pas cherché à fixer la valeur de la dose journalière tolérable
car cette étude devrait fournir des données plus complètes sur
l'exposition chronique aux chlorates.
11) Bromates
Les bromates se comportent comme des oxydants actifs vis-à-vis
des systèmes biologiques et on a montré qu'ils provoquaient une
augmentation du nombre de tumeurs rénales, de mésothéliomes
péritonéaux et de tumeurs folliculaires de la thyroïde chez le rat et,
dans une moindre mesure, chez le hamster. Chez la souris en revanche,
on ne constate qu'une légère augmentation des tumeurs du rein. La dose
la plus faible à laquelle on ait observé une augmentation de
l'incidence des tumeurs rénales chez le rat est égale à 6 mg/kg p.c.
j-1.
Il a également été montré que les bromates provoquaient des
aberrations chromosomiques dans des cellules mammaliennes in vitro
et in vivo, mais on n'a pas constaté de mutations ponctuelles lors
d'épreuves sur bactéries. Un ensemble de plus en plus important de
données, confirmées par des tests de génotoxicité, incitent à penser
que l'ion bromate agit en donnant naissance à des radicaux oxygénés à
l'intérieur de la cellule.
Dans les Directives de qualité pour l'eau de boisson
(avant-dernière édition), on a utilisé un modèle linéaire multistade
pour étudier l'incidence des tumeurs rénales lors d'une étude de 2 ans
sur des rats, mais il avait été cependant noté que si le mécanisme de
cancérogenèse repose un stress oxydatif des cellules rénales, il
n'était peut-être pas approprié d'utiliser un modèle de cancérisation
faisant intervenir de faibles doses. La limite supérieure de
l'intervalle de confiance à 95 % pour un risque de 10-5, a été
estimée à 0,1 µg/kg p.c. j-1.
Chez le rat, la dose quotidienne sans effet est de 1,3 mg/kg
p.c., si l'effet retenu est la formation de tumeurs rénales. Si l'on
considère qu'à partir de cette valeur la relation dose-réponse n'est
plus linéaire et en prenant une marge de sécurité correspondant à un
facteur 1000 (un facteur de 10 à chaque fois pour tenir compte des
variations intra- interspécifiques plus un facteur 10 pour prendre en
compte la cancérogénicité possible du composé), on peut fixer à 1
µg/kg p.c. la dose journalière tolérable. Cette valeur est à
rapprocher de la valeur de 0,1 µg/kg j-1 obtenue dans le cas d'un
risque de cancer de 10-5 sur toute la durée de l'existence.
Pour l'instant, on ne possède pas suffisamment de données pour
savoir si les tumeurs dues aux bromates s'expliquent par un phénomène
de cytotoxicité, par une hyperplasie réparatrice ou par un effet
génotoxique.
Le CIRC a placé le bromate de potassium dans le groupe 2B
(composés peut-être cancérogènes pour l'Homme).
5.1.2 Etudes épidémiologiques
Il faut procéder à une évaluation minutieuse des études
épidémiologiques afin de s'assurer qu'elles ne comportent aucun biais
et qu'elles sont effectivement conçues pour mettre en évidence une
relation causale éventuelle. On peut procéder à l'évaluation de cette
relation causale lorsqu'on dispose d'éléments d'appréciation
suffisants fournis par plusieurs études bien conçues menées dans
différentes régions. Il importe également de disposer de données
toxicologiques et pharmacologiques à l'appui de l'hypothèse que l'on
cherche à vérifier. Les données épidémiologiques fournies par les
études écologiques relatives à la désinfection de l'eau sont d'une
interprétation particulièrement difficile et on les utilise
essentiellement pour la formulation d'hypothèses à examiner plus
avant.
Les résultats des études épidémiologiques analytiques ne
permettent pas, à eux seuls, d'établir une relation de cause à effet
dans le cas de telle ou telle association observée. Les résultats des
études analytiques qui sont actuellement publiées sont
particulièrement difficiles à interpréter car ils ne donnent pas assez
de renseignements sur l'exposition aux à certains contaminants
présents dans l'eau, qui pourraient constituer des facteurs de
confusion ou tout au moins modifier le risque. Etant donné que l'on ne
s'est pas suffisamment préoccupé d'évaluer l'exposition à ces
contaminants, il n'est pas possible d'estimer correctement l'excès de
risque relatif observé. Il n'est en effet pas exclu que ce risque soit
dû à d'autres contaminants présents dans l'eau ou à d'autres facteurs
dont l'action serait attribuée à tort à l'eau chlorée et aux THM
qu'elle peut contenir.
5.2 Caractérisation de l'exposition
5.2.1 Fréquence des désinfectants et des sous-produits de chloration
On utilise le plus souvent des doses de désinfectants de
plusieurs milligrammes par litre, qui sont nécessaires pour inactiver
les germes présents dans l'eau (désinfection primaire) ou maintenir
une concentration résiduelle dans le réseau d'adduction (désinfection
secondaire).
Pour évaluer l'exposition, il faut savoir quels sont les SPC
présents et à quelle concentration. Il n'existe malheureusement guère
d'études publiées au niveau international qui aillent au-delà de la
simple étude cas-témoins ou dont la portée ne soit pas limitée à une
région.
L'étude des données disponibles indique, qu'en moyenne, il y a 35
à 50 µg/litre de THM totaux dans l'eau de boisson traitée par le
chlore et que les espèces chimiques prédominantes sont, dans l'ordre,
le chloroforme et le bromodichlorométhane. En ce qui concerne
l'exposition aux HAA, on peut dire qu'elle est approximativement égale
à la somme de cinq espèces chimiques appartenant à ce groupe, dont la
concentration totale serait à peu près la moitié de celle des THM
(cette proportion peut toutefois varier assez sensiblement); les
composés prédominants sont, dans l'ordre, l'acide dichloracétique et
l'acide trichloracétique. Dans les eaux où le rapport des bromates aux
composés organiques totaux est élevé ou encore où les bromures
prédominent par rapport aux chlorures, il y a vraisemblablement
formation plus importante de THM et de HAA bromés. Si on utilise une
solution d'hypochlorite de préférence à du chlore à l'état gazeux, le
processus de chloration peut entraîner la formation de chlorates.
L'exposition aux SPC qui se forment lorsqu'on traite l'eau par la
chloramine est fonction de la méthode employée pour générer la
chloramine; par exemple, si on commence par injecter du chlore puis de
l'ammoniac, il se forme moins de SPC (THM et HAA) pendant la période
où le chlore est à l'état libre; toutefois, à la diminution de la
formation de chloroforme et d'acide trichloracétique ne correspond pas
une diminution proportionnelle de la formation d'acide
dichloracétique.
Toutes choses égales par ailleurs, la concentration des bromures
et la dose d'ozone sont les paramètres qui permettent le mieux de
prévoir la formation de bromates au cours de l'ozonisation, le taux de
conversion des bromures en bromates étant d'environ 50%. Une étude
portant sur diverses compagnies européennes de distribution d'eau a
montré que la concentration des bromates dans l'eau quittant les
installations de traitement allait de valeurs inférieures à la limite
de détection (2 µg/litre) à 16 µg/litre. Les sous-produits bromés qui
se forment au cours du processus d'ozonisation sont généralement
présents à faible concentration. Si on utilise du dioxyde de chlore,
il est possible d'estimer la quantité de chlorites formés par une
simple proportion (50-70 %) de la dose utilisée.
5.2.2 Incertitudes des données relatives à la qualité de l'eau
Dans une étude toxicologique, on s'efforce de d'extrapoler à
l'Homme une réaction (contrôlée) observée sur un animal de
laboratoire. On peut par exemple en tirer une estimation du risque de
cancer. Dans une étude épidémiologique, on tente de relier des effets
sur la santé humaine (par exemple, la formation de cancers) à l'action
d'un ou de plusieurs agents (par exemple des SPC) et il faut pour cela
évaluer l'exposition à ces agents.
Il peut y avoir plusieurs modes d'exposition aux risques que
présente la consommation d'eau chlorée : i) l'ingestion de SPC avec
l'eau qui les contient; ii) l'ingestion de désinfectants chimiques
contenus dans l'eau de boisson et la formation intragastrique de SPC;
iii) l'inhalation de SPC volatils en prenant une douche. La formation
de SPC in vivo ou leur inhalation pendant une douche peut poser
problème sur le plan sanitaire, mais la discussion qui suit repose sur
l'hypothèse que l'ingestion des SPC présents dans l'eau de boisson
constitue le mode d'exposition le plus important.
L'exposition humaine aux SPC dépend à la fois de leur
concentration et de la durée de contact. Plus précisément, les effets
que ces composés peuvent exercer sur la santé humaine sont fonction de
l'exposition à des mélanges complexes de SPC (par exemple THM plutôt
que HAA, dérivés bromés plutôt que chlorés) dont la composition peut
varier au cours du temps, avec la saison (par ex. selon la
température, la nature et la concentration des matières organiques
naturelles) ou encore en différents points (à l'intérieur du réseau
d'adduction par exemple). Chaque désinfectant utilisé pour traiter
l'eau peut donner naissance à un mélange de SPC et un traitement
associant plusieurs désinfectants va conduire à la formation de
mélanges encore plus complexes. Lorsqu'ils se forment, la plupart de
ces SPC sont stables mais certains d'entre eux peuvent subir des
transformations, notamment par hydrolyse. Si l'on ne possède pas de
données sur les SPC, il est possible d'obtenir une évaluation
indirecte de l'exposition en utilisant à la place la dose de chlore
(ou la demande de chlore), la teneur en carbone organique total (ou
l'absorbance dans l'ultraviolet à 254 nm [UVA254]) ou encore la teneur
en bromures. Si on ne connaît pas la concentration en précurseurs
organiques des SPC, on peut lui substituer la teneur en carbone
organique total, mais l'absorbance à 254 nm donne une meilleure idée
des caractéristiques des matières organiques naturelles, qui peuvent
être soumises à des variations géographiques. Deux variables très
importantes qui affectent la qualité de l'eau, à savoir la teneur en
bromures et le pH, ont un effet sensible sur la nature et la
concentration des SPC formés.
Lorsqu'on cherche à évaluer l'exposition, il faut tout d'abord
s'efforcer de déterminer quels sont les SPC susceptibles de se former
et de coexister simultanément. Il faut ensuite connaître l'évolution
de leur concentration au cours du temps, qui va dépendre de leur
stabilité et de leur transport à travers le réseau de distribution. En
ce qui concerne les études épidémiologiques, il existe des bases de
données historiques sur la concentration des désinfectants (par
exemple le chlore) et éventuellement des précurseurs de SPC (par
exemple le carbone organique total) ou encore des THM totaux (parfois
sur certains THM bien déterminés). Contrairement aux THM qui sont
depuis longtemps sous surveillance pour des raisons réglementaires,
les données de surveillance concernant les HAA en général ou certains
membres de ce groupe, les bromates et les chlorites sont beaucoup plus
récentes et par conséquent peu abondantes. On peut toutefois faire
appel à la modélisation pour simuler des données manquantes ou
anciennes. La documentation relative à l'évolution des méthodes de
traitement de l'eau est également un élément important à prendre en
considération.
5.2.3 Incertitudes des données épidémiologiques
Même dans les études bien conçues et bien menées, on s'aperçoit
que l'évaluation de l'exposition laisse à désirer. Dans la plupart des
cas, on a pris en considération la durée de l'exposition et la source
d'eau. L'exposition a été déterminée en étudiant la situation générale
des habitations ainsi que les dossiers des compagnies de distribution
ou des services publics. Il n'existe que très peu d'études dans
lesquelles on ait tenté d'estimer la consommation d'eau des
participants et leur exposition à divers THM ou mélanges de THM. Il
n'y en a qu'une où l'on se soit efforcé de déterminer l'exposition aux
autres SPC. Lorsqu'on cherche à évaluer certains risques, par exemple
un risque d'issue défavorable pour une grossesse qui pourrait être lié
à une exposition relativement brève à des sous-produits volatils, il
n'est pas inutile de prendre en considération la voie respiratoire
aussi bien que la voie digestive. Dans un certain nombre d'études, on
a tenté d'estimer la teneur en SPC pendant la période étiologiquement
significative ainsi que l'exposition cumulée. Il existe des modèles
appropriés ou des analyses de simulation comme la méthode de Monte
Carlo qui permettent d'estimer l'exposition au cours des périodes
voulues.
L'incertitude est très importante en ce qui concerne
l'interprétation des associations observées, car on n'a pris en
considération que l'exposition à un nombre relativement restreint de
contaminants présents dans l'eau. Il est difficile, à la lumière des
données actuelles, de déterminer dans quelle mesure les SPC ou autres
contaminants qui n'ont pas été dosés ont pu influer sur l'estimation
du risque relatif.
Il y a eu davantage d'études consacrées au cancer de la vessie
qu'à tout autre cancer. Les auteurs des études les plus récemment
publiées au sujet du risque de cancer vésical mettent en garde contre
toute interprétation simpliste des associations observées. Les
éléments d'appréciation dont on dispose à l'appui d'un risque accru de
cancer de la vessie ne sont pas concordants - on trouve un risque
différent pour les fumeurs et les non fumeurs, pour les hommes et les
femmes et selon que la consommation d'eau est forte ou faible. Le
risque peut différer selon la région car les mélanges de SPC ne sont
pas forcément identiques ou parce que l'eau peut contenir d'autres
contaminants. Il faut rassembler des données beaucoup plus complètes
sur la qualité de l'eau - ou les obtenir le cas échéant par simulation
- pour permettre une meilleure évaluation de l'exposition en vue des
études épidémiologiques.
RESUMEN Y EVALUACION
El cloro (Cl2) se ha utilizado ampliamente en todo el mundo como
desinfectante químico, siendo la principal barrera contra los
contaminantes microbianos del agua de bebida. Las notables propiedades
biocidas del cloro se han visto neutralizadas en parte por la
formación, durante el proceso de cloración, de subproductos de los
desinfectantes (SPD) que son motivo de preocupación para la salud
pública. En consecuencia, se están utilizando cada vez más
desinfectantes químicos alternativos, como el ozono (O3), el dióxido
de cloro (ClO2) y las cloraminas (NH2Cl, monocloramina); sin
embargo, se ha demostrado que cada uno de ellos forma su propia serie
de SPD. Aunque no se puede comprometer la calidad microbiológica del
agua de bebida, es necesario conocer mejor la química, la toxicología
y la epidemiología de los desinfectantes químicos y de sus SPD
asociados, a fin de tener un mayor conocimiento de los riesgos para la
salud (microbianos y químicos) asociados con el agua de bebida y
buscar un equilibrio entre ambos riesgos. Es posible reducir el riesgo
químico que representan los SPD sin comprometer la calidad
microbiológica.
1. Química de los desinfectantes y de los subproductos de los
desinfectantes
Los desinfectantes químicos más utilizados son el cloro, el
ozono, el dióxido de cloro y la cloramina. Las propiedades físicas y
químicas de los desinfectantes y los SPD pueden influir en su
comportamiento en el agua de bebida, así como en su toxicología y su
epidemiología. Todos los desinfectantes químicos que se examinan en el
presente documento son oxidantes solubles en agua, que se producen
in situ (por ejemplo, el ozono) o ex situ (por ejemplo, el cloro).
Se administran en forma de gas (por ejemplo, el ozono) o de líquido
(por ejemplo, el hipoclorito) a dosis normales de varios miligramos
por litro, solos o bien mezclados. Los SPD que se examinan aquí se
pueden medir por cromatografía de gases o líquida y clasificarse como
orgánicos o inorgánicos, halogenados (clorados o bromados) o no
halogenados y volátiles o no volátiles. Al formarse, los SPD pueden
ser estables o inestables (por ejemplo, descomposición por
hidrólisis).
Los SPD se forman en la reacción de desinfectantes químicos con
precursores de los SPD. La materia orgánica natural, que normalmente
se mide por el carbono orgánico total, actúa como precursor orgánico,
mientras que el ión bromuro (Br-) es el precursor inorgánico. En la
formación de los SPD influyen la calidad del agua (por ejemplo, el
carbono orgánico total, el bromuro, el pH, la temperatura, el amoníaco
y la alcalinidad por carbonatos) y las condiciones del tratamiento
(por ejemplo, la dosis de desinfectante, el tiempo de contacto, la
eliminación de la materia orgánica natural antes de la aplicación del
desinfectante, la adición previa del desinfectante).
El cloro en forma de ácido hipocloroso/ión hipoclorito
(ClOH/ClO-) reacciona con el ión bromuro, oxidándolo a ácido
hipobromoso/ión hipobromito (BrOH/BrO-). El ácido hipocloroso
(oxidante más potente) y el ácido hipobromoso (agente halogenante más
eficaz) reaccionan conjuntamente con la materia orgánica natural para
formar SPD del cloro, entre ellos trihalometanos (THM), ácidos
haloacéticos (AHA), haloacetonitrilos (HAN), halocetonas, hidrato de
cloral y cloropicrina. El predominio de los grupos de SPD del cloro
suele disminuir en el orden siguiente: THM, AHA y HAN. Las cantidades
relativas de carbono orgánico total, bromuro y cloruro influyen en la
distribución por especies de THM (cuatro especies: cloroformo,
bromoformo, bromodiclorometano y dibromoclorometano), de los AHA
(hasta nueve especies cloradas/bromadas) y de los HAN (varias especies
cloradas/bromadas). En general predominan las especies de THM, AHA y
HAN cloradas sobre las bromadas, aunque se puede dar el caso contrario
en aguas con un alto contenido de bromuro. Si bien se han identificado
numerosos SPD específicos del cloro, hay un porcentaje considerable de
halógenos orgánicos totales que siguen sin conocerse. Otra de las
reacciones del cloro es la que da lugar a la formación de clorato
(ClO3-) en soluciones concentradas de hipocloritos.
El ozono puede reaccionar directa o indirectamente con el bromuro
para formar SPD de ozono bromados, incluido el ión bromato (BrO3-).
En presencia de materia orgánica natural, durante la ozonación se
producen SPD orgánicos no halogenados, como aldehídos, cetoácidos y
ácidos carboxílicos, predominando los aldehídos (por ejemplo, el
formaldehído). En presencia de materia orgánica natural y de bromuro,
la ozonación forma ácido hipobromoso, que a su vez da lugar a la
formación de compuestos organohalogenados bromados (por ejemplo, el
bromoformo).
Los principales SPD del dióxido de cloro son los iones clorito
(ClO2-) y clorato, sin formación directa de SPD organohalogenados. A
diferencia de otros desinfectantes, los SPD más importantes del
dióxido de cloro proceden de la descomposición del desinfectante, y no
de la reacción con los precursores.
El uso de la cloramina como desinfectante secundario suele dar
lugar a la formación de cloruro de cianógeno (ClCN), un compuesto de
nitrógeno, y concentraciones significativamente reducidas de SPD del
cloro. Un problema conexo es la presencia de nitrito (NO2-) en
sistemas de distribución cloraminados.
Teniendo en cuenta el conocimiento actual acerca de la presencia
y los efectos en la salud, los SPD de mayor interés son los THM, los
AHA, el bromato y el clorito.
Se ha comprobado que el grupo predominante de SPD del cloro son
los THM, siendo el cloroformo y el bromodiclorometano la primera y la
segunda especies. El segundo grupo más importante lo forman los AHA,
siendo el ácido dicloroacético y el ácido tricloroacético la primera y
la segunda especies.
La conversión de bromuro en bromato durante la ozonación depende
de la materia orgánica natural, el pH y la temperatura, entre otros
factores. Los niveles pueden oscilar entre valores inferiores al nivel
de detección (2 µg/litro) y varias decenas de miligramos por litro.
Las concentraciones de cloruro suelen ser muy previsibles, variando
entre el 50% y el 70% aproximadamente de la dosis de dióxido de cloro
administrada.
Los SPD están presentes en mezclas complejas que dependen del
desinfectante químico utilizado, de la calidad del agua y de las
condiciones del tratamiento; otros factores son la combinación/uso
secuencial de desinfectantes/oxidantes múltiples. Además, la
composición de estas mezclas puede cambiar con las estaciones. Es
evidente que los efectos químicos potenciales relacionados con la
salud serán función de la exposición a las mezclas de SPD.
Aparte de los SPD del cloro (en particular los THM), hay muy
pocos datos sobre la presencia de SPD en el agua tratada y los
sistemas de distribución. A partir de las bases de datos de
laboratorio, se han elaborado modelos empíricos para pronosticar las
concentraciones de THM (THM totales y sus especies), de AHA (AHA
totales y sus especies) y de bromato. Estos modelos se pueden utilizar
en la evaluación del rendimiento para predecir el efecto de los
cambios de tratamiento y en la evaluación de la exposición para
simular datos que falten o pasados (por ejemplo, para pronosticar las
concentraciones de AHA a partir de los datos relativos a los THM).
Los SPD se pueden controlar mediante la inspección y eliminación
de sus precursores o con prácticas de desinfección modificadas. La
materia orgánica natural se puede eliminar mediante coagulación,
carbono granular activado, filtración por membrana y biofiltración de
ozono. Si se exceptúa la utilización de membranas, hay pocas
posibilidades de eliminar el bromuro con eficacia. Si se desea evitar
el tratamiento del agua habrá que proteger y controlar las fuentes. No
es posible eliminar los SPD tras su formación en el caso de los SPD
orgánicos, mientras que el bromato y el clorito se pueden eliminar con
carbono activado o agentes reductores. Cabe prever que el uso óptimo
de combinaciones de desinfectantes, utilizados como desinfectantes
primarios y secundarios, permitirá controlar ulteriormente los SPD.
Hay una tendencia a la utilización simultánea o sucesiva de diversos
desinfectantes; el ozono se utiliza exclusivamente como desinfectante
primario, las cloraminas sólo como secundario, y tanto el cloro como
el dióxido de cloro en ambos casos.
2. Cinética y metabolismo en animales de laboratorio y en el ser
humano
2.1 Desinfectantes
Los desinfectantes residuales son productos químicos reactivos
que reaccionan con compuestos orgánicos presentes en la saliva y en el
contenido del estómago para formar subproductos. Hay diferencias
significativas en la farmacocinética del 36Cl según se obtenga a
partir del cloro, de la cloramina o del dióxido de cloro.
2.2 Trihalometanos
Tras la exposición oral o por inhalación, los mamíferos absorben,
metabolizan y eliminan con rapidez los THM. Después de la absorción,
las concentraciones tisulares más elevadas se producen en la grasa, el
hígado y los riñones. La semivida suele oscilar entre 0,5 y tres horas
y la vía primaria de eliminación es la metabolización en anhídrido
carbónico. La toxicidad de los THM sólo se manifiesta previa
activación metabólica a intermediarios reactivos, y las tres especies
bromadas se metabolizan con mayor rapidez y en mayor medida que el
cloroformo. La ruta metabólica más importante para todos los THM es la
oxidación a través del citocromo P450 (CYP)2E1, que da lugar a la
formación de dihalocarbonilos (es decir, fosgeno y compuestos análogos
bromados), que se pueden hidrolizar para formar anhídrido carbónico o
unirse a macromoléculas de los tejidos. Las vías metabólicas
secundarias son la deshalogenación por reducción a través del
citocromo CYP2B1/2/2E1 (que da lugar a la formación de radicales
libres) y la conjugación del glutatión mediante la
glutatión- S-transferasa T1-1, que forma intermediarios mutágenos.
Los THM bromados tienen muchas más probabilidades que el cloroformo de
seguir las vías secundarias, y la conjugación del cloroformo con el
glutatión mediante la glutatión- S-transferasa se puede producir
solamente a concentraciones o dosis extraordinariamente elevadas de
cloroformo.
2.3 Ûcidos haloacéticos
La cinética y el metabolismo de los ácidos dihaloacéticos y
trihaloacéticos difieren considerablemente. En la medida en que se
metabolizan, las principales reacciones de los ácidos trihaloacéticos
se producen en la fracción microsomal, mientras que más del 90% del
metabolismo de los ácidos dihaloacéticos, fundamentalmente por
intervención de las glutatión-transferasas, tiene lugar en el citosol.
El ácido tricloroacético tiene una semivida biológica de 50 horas en
el ser humano. La semivida de los otros ácidos trihaloacéticos
disminuye considerablemente con la sustitución del bromo, y con ácidos
trihaloacéticos bromados se pueden detectar cantidades mensurables de
ácidos dihaloacéticos como productos. La semivida de los ácidos
dihaloacéticos a dosis bajas es muy breve, pero se puede elevar
radicalmente con el aumento de las dosis.
2.4 Haloaldehídos y halocetonas
Se dispone de datos limitados sobre la cinética del hidrato de
cloral. Sus dos metabolitos principales son el tricloroetanol y el
ácido tricloroacético. El tricloroetanol sufre una glucuronidación
rápida, con circulación enterohepática, hidrólisis y oxidación para
dar ácido tricloroacético. La decloración del tricloroetanol o del
hidrato de cloral daría lugar a la formación de ácido dicloroacético.
Luego, éste se puede transformar de nuevo en monocloroacetato,
glioxalato, glicolato y oxalato, probablemente a través de un
intermediario reactivo. No se encontró información sobre los demás
haloaldehídos y halocetonas.
2.5 Haloacetonitrilos
No se han estudiado el metabolismo y la cinética de los
haloacetonitrilos. Los datos cualitativos indican que entre los
productos del metabolismo figuran el cianuro, el formaldehído, el
cianuro de formilo y los haluros de formilo.
2.6 Derivados halogenados de las hidroxifuranonas
El miembro del grupo de las hidroxifuranonas que se ha estudiado
más a fondo es la 3-cloro-4-(diclorometil)-5-hidroxi-2 (5H)-furanona
(MX). De los estudios en animales parece deducirse que la MX marcada
con 14C se absorbe con rapidez en el tracto gastrointestinal y pasa a
la circulación sistémica. No se ha determinado la concentración de la
propia MX en la sangre. La MX marcada se excreta fundamentalmente en
la orina y en las heces, siendo la orina la vía principal de
excreción. Es muy pequeña la concentración del producto inicial
radiomarcado que se retiene en el organismo después de cinco días.
2.7 Clorito
El 36Cl del clorito se absorbe con rapidez. Menos de la mitad de
la dosis pasa a la orina como cloruro y una pequeña proporción lo hace
en forma de clorito. Probablemente un porcentaje importante entra a
formar parte de la reserva de cloruros del organismo, pero no hay
información detallada debido a la falta de métodos analíticos para
valorar el clorito en muestras biológicas.
2.8 Clorato
El clorato se comporta de manera semejante al clorito. Existen
los mismos problemas analíticos.
2.9 Bromato
El bromato se absorbe y excreta con rapidez, fundamentalmente en
la orina, en forma de bromuro. Se detecta bromato en la orina a dosis
de 5 mg/kg de peso corporal o superiores. Las concentraciones de
bromato en la orina alcanzan un máximo cuando ha transcurrido
alrededor de una hora, y después de dos horas no es posible detectarlo
en el plasma.
3. Toxicología de los desinfectantes y de los subproductos de los
desinfectantes
3.1 Desinfectantes
El gas cloro, la cloramina y el dióxido de cloro son irritantes
respiratorios potentes. El hipoclorito de sodio (ClONa) se utiliza
también como blanqueante y con frecuencia produce intoxicación en el
ser humano. Sin embargo, estas exposiciones no guardan relación con la
exposición en el agua de bebida. Se han realizado relativamente pocas
evaluaciones de los efectos tóxicos de estos desinfectantes, cuando
están presentes en el agua, para los animales de experimentación o el
ser humano. Los estudios realizados al respecto parecen indicar que el
cloro, las soluciones de hipoclorito, la cloramina y el dióxido de
cloro probablemente no contribuyan como tales a la aparición de cáncer
o de cualquier efecto tóxico. La atención se ha concentrado en la
amplia variedad de subproductos que se originan en las reacciones del
cloro y de otros desinfectantes con la materia orgánica natural, que
se encuentra prácticamente en todas las fuentes de agua.
3.2 Trihalometanos
Los THM inducen citotoxicidad hepática y renal en roedores
expuestos a dosis de unos 0,5 mmoles/kg de peso corporal. El vehículo
de administración influye notablemente en la toxicidad de los THM.
Éstos tienen escasa toxicidad en la reproducción o el desarrollo, pero
se ha demostrado que el bromodiclorometano reduce la motilidad de los
espermatozoides en ratas que consumen 39 mg/kg de peso corporal al día
en el agua. Al igual que el cloroformo, cuando el bromodiclorometano
se administra en aceite de maíz induce la formación de cáncer de
hígado y de riñón tras la exposición a lo largo de toda la vida a
dosis altas. A diferencia del cloroformo y del dibromoclorometano, el
bromodiclorometano y el bromoformo inducen la formación de tumores en
el intestino grueso de ratas expuestas al aceite de maíz mediante
sonda. El bromodiclorometano induce la formación de tumores en los
tres lugares a los que llega y a dosis más bajas que los demás THM.
Desde la publicación en 1994 de los criterios de salud ambiental de la
OMS sobre el cloroformo, se han añadido a la suma de datos acumulados
nuevos estudios que indican que el cloroformo no es un carcinógeno
mutágeno capaz de reaccionar de forma directa con el ADN. En cambio,
los THM bromados parecen tener una débil acción mutágena,
probablemente debido a la conjugación con el glutatión.
3.3 Ûcidos haloacéticos
Los AHA tienen efectos toxicológicos diversos en los animales de
laboratorio. Los que suscitan mayor preocupación son los que tienen
efectos carcinógenos o en la reproducción y el desarrollo. Los efectos
neurotóxicos son importantes a las elevadas dosis de ácido
dicloroacético que se utilizan con fines terapéuticos. Los efectos
carcinógenos parecen limitarse al hígado y a las dosis altas. La mayor
parte de los datos indican que los efectos tumorígenos del ácido
dicloroacético y del ácido tricloroacético dependen de la modificación
de los procesos de división y muerte celular más que de su actividad
mutágena, que es muy débil. El choque oxidativo es también una
característica de la toxicidad de los análogos bromados comprendidos
en esta categoría. Tanto el ácido dicloroacético como el
tricloroacético provocan a dosis altas malformaciones cardíacas en
ratas.
3.4 Haloaldehídos y halocetonas
El hidrato de cloral induce necrosis hepática en ratas a dosis
iguales o superiores a 120 mg/kg de peso corporal al día. Su efecto
depresor del sistema nervioso central en el ser humano probablemente
esté relacionado con su metabolito tricloroetanol. Hay pocos datos de
toxicidad para los otros aldehídos y cetonas halogenados. La
exposición al cloroacetaldehído provoca en las ratas efectos
hematológicos. La exposición de los ratones a la 1,1-dicloropropanona,
pero no a la 1,3-dicloropropanona, produce toxicidad hepática.
El hidrato de cloral dio resultados negativos en la mayor parte
de las pruebas bacterianas de mutaciones puntuales, pero no en todas,
y en estudios in vivo sobre daños cromosómicos. Sin embargo, se ha
demostrado que puede inducir aberraciones cromosómicas estructurales
in vitro e in vivo. Se ha notificado que el hidrato de cloral
provoca tumores hepáticos en ratones. No está claro si es el compuesto
original o sus metabolitos los que inducen el efecto carcinógeno. Los
dos metabolitos del hidrato de cloral, el ácido tricloroacético y el
ácido dicloroacético, han provocado la inducción de tumores hepáticos
en ratones.
Algunos aldehídos y cetonas halogenados son potentes inductores
de mutaciones en bacterias. Se han notificado efectos clastógenos para
las propanonas cloradas. En un estudio con exposición de animales
durante toda su vida al cloroacetaldehído, agregado al agua de bebida,
se observó la formación de tumores hepáticos. Se han identificado
otros aldehídos halogenados, por ejemplo el 2-cloropropenal, como
inductores de la formación de tumores cutáneos en ratones. No se han
sometido a prueba las halocetonas para investigar la carcinogenicidad
en el agua de bebida. Sin embargo, en un estudio de carcinogenicidad
cutánea en ratones la 1,3-dicloropropanona actuó como inductora de
tumores.
3.5 Haloacetonitrilos
Se han realizado hasta ahora pocas pruebas con estos compuestos
para investigar los efectos toxicológicos. Algunos de los grupos son
mutágenos, pero no hay una relación clara de esos efectos con la
actividad de los productos químicos como inductores de tumores
cutáneos. Son muy pocos los estudios que se han realizado sobre la
carcinogenicidad de este tipo de sustancias. Parece que los indicios
precoces de toxicidad de los miembros de este grupo en el desarrollo
se deben fundamentalmente al vehículo utilizado para su
administración.
3.6 Derivados halogenados de la hidroxifuranona
Los estudios experimentales parecen indicar que los efectos de
mayor importancia de la MX son su mutagenicidad y su carcinogenicidad.
En varios estudios in vitro se ha comprobado que la MX es mutágena
en sistemas de prueba con bacterias y mamíferos. La MX provocó
aberraciones cromosómicas e indujo lesiones en el ADN de células
hepáticas y testiculares aisladas, así como el intercambio de
cromátidas hermanas en linfocitos periféricos de ratas expuestas
in vivo. Una evaluación general de los datos de mutagenicidad pone
de manifiesto que la MX es mutágena in vitro e in vivo. En un
estudio de carcinogenicidad en ratas se observó un aumento de la
frecuencia de tumores en varios órganos.
3.7 Clorito
La acción tóxica del clorito se produce fundamentalmente en forma
de daño oxidativo en los glóbulos rojos a dosis de apenas 10 mg/kg de
peso corporal. Hay indicios de efectos leves en el neurocomportamiento
de crías de rata con 5,6 mg/kg de peso corporal al día. Los datos
sobre la genotoxicidad del clorito son contradictorios. En estudios de
exposición crónicos, el clorito no produjo un aumento de tumores en
animales de laboratorio.
3.8 Clorato
La toxicidad del clorato es semejante a la del clorito, pero como
oxidante este compuesto causa menos lesiones. No parece ser teratógeno
o genotóxico in vivo. No hay datos de estudios de carcinogenicidad
prolongados.
3.9 Bromato
A dosis elevadas el bromato provoca lesiones en los túbulos
renales de las ratas expuestas. En estudios crónicos induce tumores en
el riñón, el peritoneo y el tiroides de las ratas sometidas a dosis de
6 mg/kg de peso corporal y superiores. Los hámsteres son menos
sensibles y los ratones considerablemente menos. El bromato a dosis
elevadas es también genotóxico in vivo en ratas . La
carcinogenicidad parece ser un efecto secundario del choque oxidativo
en la célula.
4. Estudios epidemiológicos
4.1 Enfermedades cardiovasculares
En los estudios epidemiológicos no se ha identificado un riesgo
mayor de enfermedad cardiovascular asociado con el agua de bebida que
contiene cloro o cloramina. No se han realizado estudios de otros
desinfectantes.
4.2 Cáncer
Las pruebas epidemiológicas no son suficientes para confirmar la
existencia de una relación causal entre el cáncer de vejiga y la
exposición prolongada al agua de bebida clorada, a los THM, al
cloroformo u a otras especies de THM. Los datos epidemiológicos son
poco concluyentes y equívocos con respecto a una asociación entre el
cáncer de colon y la exposición prolongada al agua clorada, a los THM,
al cloroformo u a otras especies de THM. La información no es
suficiente para permitir una evaluación de los riesgos observados de
cáncer de recto y los riesgos de otros tipos de cáncer detectados en
estudios analíticos aislados.
En varios tipos de estudios epidemiológicos se ha intentado
evaluar los riesgos de cáncer que pueden estar asociados con la
exposición al agua clorada. En dos estudios se examinó el agua con
cloramina. En varios estudios se ha intentado estimar la exposición a
la totalidad de los THM o al cloroformo y a las otras especies de THM,
pero en los estudios no se examinaron las exposiciones a otros SPD o a
otros contaminantes del agua, que pueden ser diferentes para las
fuentes de agua superficial y freática. En un estudio se examinó la
mutagenicidad del agua medida mediante la valoración de Salmonella
typhimurium. No se han realizado evaluaciones de los posibles
riesgos de cáncer que pueden estar asociados con el agua desinfectada
con ozono o dióxido de cloro.
Diversos estudios de casos y testigos basados en datos ecológicos
o en certificados de defunción han permitido formular hipótesis que
deberán evaluarse más detenidamente en estudios analíticos que tomen
en consideración el consumo individual de agua de bebida y los
posibles factores de confusión.
En los estudios analíticos se ha notificado un aumento de escaso
a moderado del riesgo relativo de cáncer de vejiga, colon, recto,
páncreas, mama, cerebro o pulmón asociado con la exposición prolongada
al agua clorada. En estudios individuales se notificaron asociaciones
entre la exposición al agua clorada y el cáncer de páncreas, de mama o
de cerebro; sin embargo, para determinar si esas asociaciones
epidemiológicas corresponden efectivamente a una relación causal se
necesitará más de un estudio. En un estudio se asoció un ligero
aumento del riesgo relativo de cáncer de pulmón con el consumo de
aguas superficiales, pero la magnitud del riesgo era demasiado pequeña
para descartar la presencia de un factor de confusión residual.
En un estudio de casos y testigos se notificó una asociación
moderadamente elevada entre el cáncer de recto y la exposición
prolongada al agua clorada o la exposición acumulativa a los THM, pero
en estudios de cohortes se ha comprobado que no aumenta el riesgo o es
demasiado pequeño para que se pueda descartar la presencia de un
factor de confusión residual.
La disminución del riesgo de cáncer de vejiga se asoció con la
mayor duración de la exposición al agua de bebida con cloramina, pero
no hay base biológica para suponer un efecto protector de este tipo de
agua.
Aunque en varios estudios se observó un aumento del riesgo de
cáncer de vejiga asociado con la exposición prolongada al agua clorada
y la exposición acumulativa a los THM, se notificaron resultados
contradictorios de los estudios sobre el riesgo de cáncer de vejiga
entre fumadores y no fumadores y entre hombres y mujeres. En tres de
estos estudios se examinó la exposición estimada a los THM. En uno de
ellos no se encontró ninguna relación con la exposición acumulativa
estimada a los THM; en otro estudio se asoció un aumento moderadamente
alto del riesgo relativo con una mayor exposición acumulativa a los
THM en los hombres, pero no en las mujeres. En el tercer estudio se
notificó un ligero aumento del riesgo relativo asociado con una
exposición acumulativa estimada de 1957-6425 µg de THM por litro al
año; se notificaron asimismo asociaciones de ligeras a moderadas para
la exposición a concentraciones de THM superiores a 24 µg/litro,
superiores a 49 µg/litro y superiores a 74 µg/litro. En una cohorte de
mujeres no se asoció un aumento del riesgo relativo de cáncer de
vejiga con la exposición al consumo de aguas superficiales municipales
cloradas, al cloroformo o a otras especies de THM, pero el período de
verificación de ocho años fue muy corto, por lo que se pudieron
estudiar pocos casos.
Habida cuenta de que en los estudios epidemiológicos no se ha
prestado la debida atención a la evaluación de la exposición a los
contaminantes del agua, no es posible valorar de manera adecuada el
aumento de los riesgos relativos que se notificaron. Los riesgos
específicos pueden deberse a otros SPD, a mezclas de subproductos u a
otros contaminantes del agua, o bien a otros factores cuyos efectos se
atribuirían al agua clorada o a los THM.
4.3 Resultados adversos en la gestación
En algunos estudios se ha examinado la posible correlación entre
el consumo de agua clorada o la exposición a los THM o especies de THM
y diversos resultados adversos del embarazo. Un grupo científico
especial convocado recientemente por la Agencia para la Protección del
Medio Ambiente de los Estados Unidos examinó los estudios
epidemiológicos y llegó a la conclusión de que los resultados de los
estudios publicados actualmente no prueban de manera convincente de
que el agua clorada o los THM provoquen resultados adversos en el
embarazo.
Los resultados de los primeros estudios son difíciles de
interpretar debido a las limitaciones metodológicas o a un probable
sesgo.
En un estudio de casos y testigos concluido recientemente pero
que todavía no se ha publicado se notifica un aumento moderado del
riesgo relativo de defectos del tubo neural en niños cuyas madres
residieron al comienzo del embarazo en una zona donde los niveles de
THM eran superiores a 40 µg/litro. Para evaluar de manera adecuada
esta asociación habría que confirmar los resultados en otra zona. En
un estudio realizado anteriormente en la misma zona geográfica se
notificó una asociación semejante, pero el estudio presentaba
limitaciones metodológicas.
En un reciente estudio de cohortes se encontró un riesgo mayor de
aborto asociado con un consumo elevado de agua (cinco o más vasos de
agua de grifo fría al día) con niveles altos (>75 µg/litro) de THM.
Cuando se examinaron THM concretos, sólo se asoció con el riesgo de
aborto un consumo elevado de agua con bromodiclorometano (>18
µg/litro). Teniendo en cuenta que éste es el primer estudio que parece
indicar un efecto reproductivo adverso asociado con un subproducto
bromado, un grupo científico especial recomendó que se realizase otro
estudio en una zona geográfica diferente para tratar de reproducir
esos resultados y que se hicieran nuevos esfuerzos para evaluar las
exposiciones de la cohorte a otros contaminantes del agua.
5. Caracterización del riesgo
Hay que señalar que el uso de desinfectantes químicos en el
tratamiento del agua suele dar lugar a la formación de subproductos
químicos, algunos de los cuales son potencialmente peligrosos. Sin
embargo, los riesgos para la salud de estos subproductos en las
concentraciones en las cuales se encuentran en el agua de bebida son
muy pequeños en comparación con los riesgos asociados con una
desinfección inadecuada. Así pues, es importante que al intentar
controlar dichos subproductos no se comprometa la desinfección.
5.1 Caracterización del peligro y de la relación dosis-respuesta
5.1.1 Estudios toxicológicos
1) Cloro
Un Grupo de Trabajo de la OMS reunido en 1993 para preparar las
Guías para la calidad del agua de bebida examinó el problema del
cloro. Fijó una ingesta diaria tolerable (IDT) de 150 µg/kg de peso
corporal para el cloro libre basada en una concentración sin efectos
adversos observados (NOAEL) de unos 15 mg/kg de peso corporal al día
en estudios de dos años en ratas y ratones y aplicando un factor de
incertidumbre de 100 (10 por la variación intraespecífica y 10 por la
interespecífica). No hay nuevos datos que indiquen la necesidad de
cambiar esta IDT.
2) Monocloramina
El mencionado Grupo de Trabajo de la OMS sobre las Guías para
la calidad del agua de bebida examinó el caso de la monocloramina.
Fijó una ingesta diaria tolerable (IDT) de 94 µg/kg de peso corporal
basada en una concentración sin efectos adversos observados (NOAEL) de
alrededor de 9,4 mg/kg de peso corporal al día, la dosis más alta
sometida a prueba, en una biovaloración de dos años en ratas y
aplicando un factor de incertidumbre de 100 (10 por la variación
intraespecífica y 10 por la interespecífica). No hay nuevos datos que
indiquen la necesidad de cambiar esta IDT.
3) Dióxido de cloro
La química del dióxido de cloro en el agua es compleja, pero el
principal producto de su degradación es el clorito. Al establecer una
IDT específica para el dióxido de cloro, se pueden considerar los
datos relativos al dióxido de cloro y al clorito, dada la rápida
hidrólisis a clorito. Por consiguiente, la IDT por vía oral para el
dióxido de cloro es de 30 µg/kg de peso corporal, basada en una NOAEL
de 2,9 mg/kg de peso corporal al día para los efectos del clorito en
el desarrollo neural de las ratas.
4) Trihalometanos
El riesgo de cáncer tras la exposición crónica es el principal
motivo de preocupación en relación con esta clase de subproductos.
Debido a que los datos relativos al cloroformo indican que puede
inducir cáncer en animales sólo tras la exposición crónica a dosis
citotóxicas, es evidente que la exposición a concentraciones bajas de
este compuesto en el agua de bebida no representa un riesgo
carcinógeno. La NOAEL para la citoletalidad y la hiperplasia
degenerativa en ratones fue de 10 mg/kg de peso corporal al día tras
la administración de cloroformo en aceite de maíz durante tres
semanas. Tomando como base los datos relativos al mecanismo de acción
para la carcinogenicidad del cloroformo, se obtuvo una IDT de 10 µg/kg
de peso corporal utilizando la NOAEL para la citotoxicidad en ratones
y aplicando un factor de incertidumbre de 1000 (10 por la variación
interespecífica, 10 por la intraespecífica y 10 por la brevedad del
estudio). Respaldan este enfoque varios estudios adicionales. Esta IDT
es semejante a la que se determinó en la edición de 1998 de las
Guías para la calidad del agua de bebida de la OMS, basada en un
estudio de 1979 en el cual se expusieron perros durante 7,5 años al
cloroformo.
Entre los THM bromados, reviste particular interés el
bromodiclorometano porque induce la formación de tumores en ratas y
ratones y en varios lugares (hígado, riñones, intestino grueso) tras
el suministro con sonda de aceite de maíz. La inducción de tumores de
colon en ratas por el bromodiclorometano (y por el bromoformo) es
también interesante debido a las asociaciones epidemiológicas con el
cáncer de colon y recto. El bromodiclorometano y los otros THM
bromados son también débilmente mutágenos. En general, se supone que
los carcinógenos mutágenos a dosis bajas producirán relaciones
dosis-respuesta lineales, puesto que se suele considerar que la
mutagénesis es un efecto irreversible y acumulativo.
En una biovaloración de dos años, el bromodiclorometano
administrado con aceite de maíz mediante sonda indujo la formación de
tumores (junto con citotoxicidad y un aumento de la proliferación) en
los riñones de ratones y ratas a dosis de 50 y 100 mg/kg de peso
corporal al día, respectivamente. Se observaron tumores en el
intestino grueso de ratas tras la exposición tanto a 50 como a 100
mg/kg de peso corporal al día. Utilizando la incidencia de tumores de
riñón en ratones macho observada en este estudio, se han calculado
estimaciones del riesgo cuantitativo, obteniéndose un factor CSF
(cancer slope factor) de 4,8 × 10-3 [mg/kg de peso corporal al
día]-1 y una dosis calculada de 2,1 µg/kg de peso corporal al día
para un nivel de riesgo de 10-5. Se estableció un CSF de 4,2 × 10-3
[mg/kg de peso corporal al día]-1 (2,4 µg/kg de peso corporal al día
para un riesgo de 10-5) sobre la base de la incidencia de carcinoma
en el intestino grueso de ratas macho. El Centro Internacional de
Investigaciones sobre el Cáncer (CIIC) ha clasificado el
bromodiclorometano en el grupo 2B (posiblemente carcinógeno para el
ser humano).
El dibromoclorometano y el bromoformo se estudiaron en
biovaloraciones prolongadas. En un estudio de dos años con aceite de
maíz administrado mediante sonda, el dibromoclorometano indujo la
formación de tumores hepáticos en ratones hembra, pero no en ratas, a
una dosis de 100 mg/kg de peso corporal al día. En evaluaciones
anteriores se había indicado que el aceite de maíz como vehículo podía
desempeñar una función en la inducción de tumores en ratones hembra.
En el estudio del bromoformo a dosis de 200 mg/kg de peso corporal al
día se observó un pequeño aumento del número de tumores del intestino
grueso de ratas. Los CSF basados en estos tumores son 6,5 × 10-3
[mg/kg de peso corporal al día]-1 para el dibromoclorometano, o de
1,5 µg/kg de peso corporal al día para un riesgo de 10-5, y de
1,3 × 10-3 [mg/kg de peso corporal al día]-1, ó 7,7 µg/kg de peso
corporal al día para un riesgo de 10-5 en el caso del bromoformo.
Estos dos trihalometanos bromados se comportaron como mutágenos
débiles en algunas valoraciones y fueron con diferencia los
subproductos más mutágenos del grupo en el sistema de evaluación
basado en la glutatión-S-transferasa. Puesto que éstos son los THM más
lipófilos, se deberían examinar aspectos adicionales para determinar
si en los estudios prolongados el aceite de maíz puede haber influido
en su biodisponibilidad. Se ha establecido una NOAEL para el
dibromoclorometano de 30 mg/kg de peso corporal al día basada en la
ausencia de efectos histopatológicos en el hígado de ratas después de
13 semanas de exposición con aceite de maíz administrado por sonda. El
CIIC ha clasificado el dibromoclorometano en el grupo 3 (no
clasificable en cuanto a su carcinogenicidad para el ser humano). Se
determinó una IDT para este producto de 30 µg/kg de peso corporal
basada en una NOAEL para la toxicidad hepática de 30 mg/kg de peso
corporal al día y un factor de incertidumbre de 1000 (10 por la
variación interespecífica, 10 por la intraespecífica y 10 por la
brevedad del estudio y la posible carcinogenicidad).
De igual forma, se puede determinar para el bromoformo una NOAEL
de 25 mg/kg de peso corporal al día sobre la base de la ausencia de
lesiones hepáticas en ratas después de 13 semanas de administración
mediante sonda con aceite de maíz. Se estableció para el bromoformo
una IDT de 25 µg/kg de peso corporal sobre la base de esta NOAEL para
la toxicidad hepática y un factor de incertidumbre de 1000 (10 por la
variación interespecífica, 10 por la intraespecífica y 10 por la
brevedad del estudio y la posible carcinogenicidad). El CIIC ha
clasificado el bromoformo en el grupo 3 (no clasificable en cuanto a
su carcinogenicidad para el ser humano).
5) Ûcidos haloacéticos
Es muy poco probable que el ácido dicloroacético induzca
mutaciones a dosis tan bajas como las que se encontrarían en el agua
clorada. Los datos disponibles indican que el ácido dicloroacético
influye de distinta manera en la velocidad de replicación de los
hepatocitos normales y de los hepatocitos en los que ha habido
iniciación. Las relaciones dosis-respuesta son complejas, estimulando
inicialmente el ácido dicloroacético la división de los hepatocitos
normales. Sin embargo, a las dosis crónicas más bajas utilizadas en
los estudios con animales (pero todavía muy altas en relación con las
que se encontrarían en el agua), la velocidad de replicación de los
hepatocitos normales se inhibe de forma radical con el tiempo. Esto
indica que a larga los hepatocitos normales regulan a la baja esas
vías que son sensibles al efecto estimulante del ácido dicloroacético.
Sin embargo, los efectos en células alteradas, particularmente en las
que sintetizan grandes cantidades de una proteína que es
inmunorreactiva frente a un anticuerpo c-Jun, no parecen capaces de
regular a la baja esta respuesta. Así pues, la velocidad de
replicación en las lesiones preneoplásicas con este fenotipo es muy
elevada a las dosis de ácido dicloroacético que provocan la aparición
de tumores, siendo el período de latencia muy bajo. Los datos
preliminares parecen indicar que esta alteración continuada en la
velocidad de formación y muerte celular es también necesaria en la
evolución de los tumores hacia la malignidad. Respaldan esta
interpretación estudios en los que se utilizan también modelos de
iniciación de la alteración/estímulo.
Teniendo en cuenta lo expuesto, se propone la modificación de las
estimaciones de riesgo de cáncer establecidas actualmente para el
ácido dicloroacético con la incorporación de la nueva información que
se está obteniendo sobre su metabolismo comparativo y los mecanismos
de acción para formular un modelo de dosis-respuesta con una base
biológica. En este momento no se dispone de esos datos, pero se podrán
obtener en el plazo de los próximos 2-3 años.
Los efectos del ácido dicloroacético parecen hallarse en estrecha
correlación con las dosis que inducen hepatomegalia y acumulación de
glucógeno en ratones. La concentración más baja con efectos adversos
observados (LOAEL) para estos efectos en un estudio de ocho semanas en
ratones fue de 0,5 g/litro, equivalentes a unos 100 mg/kg de peso
corporal al día, y la NOAEL fue de 0,2 g/litro, es decir, unos 40
mg/kg de peso corporal al día. Se ha calculado una IDT de 40 µg/kg de
peso corporal, aplicando a esta NOAEL un factor de incertidumbre de
1000 (10 por la variación interespecífica, 10 por la intraespecífica y
10 por la brevedad del estudio y la posible carcinogenicidad). El CIIC
ha clasificado el ácido dicloroacético en el grupo 3 (no clasificable
en cuanto a su carcinogenicidad para el ser humano).
El ácido tricloroacético es uno de los activadores más débiles
conocidos de los receptores PPAR (peroxisome proliferator-activated
receptors). Parece tener una actividad solamente marginal como
proliferador de los perixosomas, incluso en ratas. Además, el
tratamiento de ratas con concentraciones altas de ácido
tricloroacético en el agua de bebida no induce la formación de tumores
hepáticos. Estos datos indican claramente que el ácido tricloroacético
presenta poco peligro carcinógeno para el ser humano en las bajas
concentraciones que se encuentran en el agua.
Desde una perspectiva toxicológica más amplia, los efectos
finales más preocupantes del ácido tricloroacético son las
consecuencias en el desarrollo. Los animales parecen tolerar
concentraciones de ácido tricloroacético en el agua de bebida de 0 ,5
g/litro (unos 50 mg/kg de peso corporal al día) con poco o ningún
signo de efectos adversos. Con 2 g/litro, el único signo de efectos
adversos parece ser la hepatomegalia. Ésta no se observa en ratones a
una dosis de 0,35 g de ácido tricloroacético por litro de agua, que se
estima que es equivalente a 40 mg/kg de peso corporal al día.
En otro estudio se observaron anomalías en los tejidos blandos
con un índice aproximadamente tres veces superior a los testigos a la
dosis más baja administrada, 330 mg/kg de peso corporal al día. A esta
dosis las anomalías fueron leves, y la dosis es claramente de la misma
magnitud que la que provocaría hepatomegalia (y efectos carcinógenos).
Teniendo en cuenta el hecho de que el PPAR interacciona con los
mecanismos de señalización celular que pueden afectar a los procesos
de desarrollo normales, se debe considerar la posibilidad de que
exista un mecanismo común a la hepatomegalia, a los efectos
carcinógenos y a los efectos en el desarrollo provocados por este
compuesto.
La IDT para el ácido tricloroacético se basa en una NOAEL
estimada de 40 mg/kg de peso corporal al día para la toxicidad
hepática en un estudio prolongado en ratones. La aplicación de un
factor de incertidumbre de 1000 (10 por la variación interespecífica,
10 por la intraespecífica y 10 por la posible carcinogenicidad) a la
NOAEL estimada proporciona una IDT de 40 µg/kg de peso corporal. El
CIIC ha clasificado el ácido tricloroacético en el grupo 3 (no
clasificable en cuanto a su carcinogenicidad para el ser humano).
Los datos sobre la carcinogenicidad de los ácidos acéticos
bromados son demasiado preliminares para ser útiles en la
caracterización del riesgo. Los datos obtenibles de los resúmenes
analíticos parecen indicar, sin embargo, que las dosis necesarias para
inducir respuestas hepatocarcinógenas en ratones no son muy diferentes
de las de los ácidos acéticos clorados. Además de los mecanismos que
intervienen en la inducción de cáncer por los ácidos dicloroacético y
tricloroacético, es posible que contribuya a sus efectos un aumento
del choque oxidativo resultante de su metabolismo.
Hay datos abundantes sobre los efectos del ácido dibromoacético
en la función reproductiva masculina. No se observaron efectos en
ratas a una dosis de 2 mg/kg de peso corporal al día durante 79 días,
mientras que con 10 mg/kg de peso corporal al día se detectó una mayor
retención de espermátidas en la fase 19. A dosis más elevadas los
efectos fueron progresivamente más graves, en particular una atrofia
acentuada de los túbulos seminíferos con 250 mg/kg de peso corporal al
día, sin que se hubiera producido una reversión seis meses después de
la suspensión del tratamiento. Se estableció una IDT de 20 µg/kg de
peso corporal mediante la asignación de un factor de incertidumbre de
100 (10 por la variación intraespecífica y 10 por la interespecífica)
a la NOAEL de 2 mg/kg de peso corporal al día.
6) Hidrato de cloral
Con concentraciones de hidrato de cloral de 1 g/litro de agua
(166 mg/kg de peso corporal al día) se indujo la formación de tumores
hepáticos en ratones expuestos durante 104 semanas. No se han evaluado
dosis inferiores. Se ha demostrado que el hidrato de cloral ha
inducido anomalías cromosómicas en varias pruebas in vitro, pero dio
resultados en gran parte negativos cuando se evaluó in vivo. Es
probable que en la inducción de tumores hepáticos por el hidrato de
cloral intervenga la formación de ácido tricloroacético y/o ácido
dicloroacético en su metabolismo. Como se ha señalado más arriba, se
considera que estos compuestos actúan como estimulantes de la
formación de tumores. El CIIC ha clasificado el hidrato de cloral en
el grupo 3 (no clasificable en cuanto a su carcinogenicidad para el
ser humano).
La administración de hidrato de cloral en el agua a ratas durante
90 días indujo necrosis hepatocelular con concentraciones de
1200 mg/litro y superiores, sin que se observasen efectos con
600 mg/litro (unos 60 mg/kg de peso corporal al día). Se detectó
hepatomegalia en ratones que recibieron dosis de 144 mg/kg de peso
corporal al día mediante sonda durante 14 días. No se observaron
efectos con 14,4 mg/kg de peso corporal al día en un estudio de
14 días, pero se detectó una ligera hepatomegalia cuando se administró
hidrato de cloral en el agua en una concentración de 70 mg/litro
(16 mg/kg de peso corporal al día) en un estudio complementario de 90
días. La aplicación de un factor de incertidumbre de 1000 (10 por la
variación interespecífica, 10 por la intraespecífica y 10 por utilizar
una LOAEL en lugar de la NOAEL) a este valor permite establecer una
IDT de 16 µg/kg de peso corporal.
7) Haloacetonitrilos
Sin datos adecuados para el ser humano o un estudio en animales
de experimentación que comprenda una gran parte de su vida, no existe
una base unánimemente aceptada para la estimación del riesgo
carcinógeno de los haloacetonitrilos.
Los datos obtenidos en estudios subcrónicos proporcionan algunos
indicios de las NOAEL para la toxicidad general del
dicloroacetonitrilo y del dibromoacetonitrilo. Se identificaron unas
NOAEL de 8 y 23 mg/kg de peso corporal al día en estudios de 90 días
en ratas para el dicloroacetonitrilo y el dibromoacetonitrilo,
respectivamente, basadas en la disminución del peso corporal a las
siguientes dosis más altas de 33 y 45 mg/kg de peso corporal al día,
respectivamente.
Un Grupo de Trabajo de la OMS reunido en 1993 para preparar las
Guías para la calidad del agua de bebida examinó el
dicloroacetonitrilo y el dibromoacetonitrilo. Fijó una IDT de 15 µg/kg
de peso corporal para el primero sobre la base de una NOAEL de
15 mg/kg de peso corporal al día obtenida en un estudio de toxicidad
reproductiva en ratas y aplicando un factor de incertidumbre de 1000
(10 por la variación intraespecífica, 10 por la interespecífica y 10
por la intensidad de los efectos). Solamente se observaron efectos
reproductivos y en el desarrollo para el dibromoacetonitrilo a dosis
que superaban las establecidas para la toxicidad general (unos 45
mg/kg de peso corporal al día). Se calculó una IDT de 23 µg/kg de peso
corporal para el dibromoacetonitrilo basada en una NOAEL de 23 mg/kg
de peso corporal al día obtenida en un estudio de 90 días en ratas y
aplicando un factor de incertidumbre de 1000 (10 por la variación
intraespecífica, 10 por la interespecífica y 10 por la brevedad el
estudio). No hay datos nuevos que indiquen que deban cambiarse estas
IDT.
Se identificó una LOAEL para el tricloroacetonitrilo de 7,5 mg/kg
de peso corporal al día para la embriotoxicidad y de 15 mg/kg de peso
corporal al día para los efectos en el desarrollo. Sin embargo,
estudios posteriores parecen indicar que estas respuestas fueron
función del vehículo utilizado. No se puede establecer una IDT para el
tricloroacetonitrilo.
No hay datos útiles para la caracterización del riesgo imputable
a otros miembros del grupo de los haloacetonitrilos.
8) MX
Recientemente se ha estudiado el mutágeno MX en un estudio
prolongado en ratas en el cual se observaron algunas respuestas
carcinógenas. Estos datos indican que el MX induce la formación de
tumores de tiroides y de los conductos biliares. Se observó un aumento
de la incidencia de tumores de tiroides a la dosis más baja de MX
administrada (0,4 mg/kg de peso corporal al día). La inducción de
tumores de tiroides a dosis elevadas de productos químicos se ha
relacionado desde hace tiempo con los compuestos halogenados. En la
inducción de tumores foliculares tiroideos podrían intervenir
modificaciones en la función del tiroides o un mecanismo de acción
mutágeno. Se observó asimismo un aumento de la incidencia de
colangiomas y colangiocarcinomas relacionado con la dosis, comenzando
con la dosis baja en ratas hembra, con una respuesta menor en ratas
macho. Se utilizó el aumento de los colangiomas y colangiocarcinomas
en ratas hembra para obtener un factor CSF para el cáncer. Se calculó
un límite de confianza superior al 95% para un riesgo a lo largo de
toda la vida de 10-5 basado en un modelo línealizado en fases
múltiples de 0,06 µg/kg de peso corporal al día.
9) Clorito
El efecto principal y más constante derivado de la exposición al
clorito es el choque oxidativo que provoca cambios en los glóbulos
rojos. Este efecto final se observa en animales de laboratorio y, por
analogía con el clorato, en personas expuestas a dosis elevadas en
casos de intoxicación. Hay datos suficientes para estimar una IDT en
las personas expuestas al clorito, en particular estudios de toxicidad
crónica y un estudio de toxicidad reproductiva de dos generaciones. En
estudios de hasta 12 semanas con voluntarios no se identificó efecto
alguno en los parámetros sanguíneos a la dosis más alta utilizada, a
saber, 36 µg/kg de peso corporal al día. Debido a que en estos
estudios no se identifica una concentración con efectos, no se pueden
utilizar para el establecimiento de un margen de inocuidad.
En un estudio de dos generaciones en ratas se identificó una
NOAEL de 2,9 mg/kg de peso corporal al día basada en una disminución
de la reacción de sobresalto auditivo, la disminución del peso
absoluto del cerebro en las generaciones F1 y F2 y la alteración del
peso del hígado en dos generaciones. La aplicación a esta NOAEL de un
factor de incertidumbre de 100 (10 por la variación interespecífica y
10 por la intraespecífica) permite establecer una IDT de 30 µg/kg de
peso corporal. Los estudios con voluntarios confirman esta IDT.
10) Clorato
Al igual que en el caso del clorito, el principal motivo de
preocupación con el clorato es el daño oxidativo de los glóbulos
rojos. Al igual que con el clorito, 0,036 mg de clorato por kg de peso
corporal al día durante 12 semanas no provocaron efectos adversos en
voluntarios. Aunque la base de datos para el clorato es menos amplia
que para el clorito, en un estudio reciente de 90 días en ratas bien
realizado se identificó una NOAEL de 30 mg/kg de peso corporal al día
basada en la reducción de los coloides de la glándula tiroidea a la
dosis siguiente más alta de 100 mg/kg de peso corporal al día. No se
determina una IDT porque está en curso un estudio prolongado que debe
proporcionar más información sobre la exposición crónica al clorato.
11) Bromato
El bromato es un oxidante activo de los sistemas biológicos y se
ha demostrado que provoca un aumento de la aparición de tumores
renales, mesoteliomas peritoneales y tumores de las células
foliculares tiroideas en ratas, y en menor medida en hámsteres, y sólo
un pequeño aumento de los tumores de riñón en ratones. La dosis más
baja a la cual se observó un aumento de la incidencia de tumores
renales en ratas fue de 6 mg/kg de peso corporal al día.
Se ha demostrado asimismo que el bromato da resultados positivos
para las aberraciones cromosómicas en células de mamíferos in vitro
e in vivo, pero no en valoraciones bacterianas para las mutaciones
puntuales. Hay cada vez más pruebas, respaldadas por los datos de
genotoxicidad, que parecen indicar que la acción del bromato consiste
en generar radicales de oxígeno en la célula.
En las Guías para la calidad del agua de bebida de 1993 de la
OMS se aplicó el modelo linealizado en fases múltiples a la incidencia
de tumores renales en un estudio de carcinogenicidad de dos años en
ratas, aunque se observó que, si el mecanismo de inducción de tumores
es el daño oxidativo en el riñón, podría no ser adecuada la aplicación
del modelo de cáncer a dosis bajas. El intervalo de confianza superior
del 95% calculado para un riesgo de 10-5 fue de 0,1 µg/kg de peso
corporal al día.
La concentración sin efectos para la formación de tumores en las
células renales de ratas es de 1,3 mg/kg de peso corporal al día. Si
se considera que a partir de este valor la relación dosis-respuesta ya
no es lineal y se aplica un factor de incertidumbre de 1000 (10 por la
variación interespecífica, 10 por la intraespecífica y 10 por la
posible carcinogenicidad), se puede calcular una IDT de 1 µg/kg de
peso corporal, frente a un valor de 0,1 µg/kg de peso corporal al día
asociado con un exceso de riesgo de cáncer de 10-5 a lo largo de toda
la vida.
De momento no hay datos suficientes para saber si los tumores
inducidos por el bromato se deben a la citotoxicidad y la hiperplasia
reparativa o son un efecto genotóxico.
El CIIC ha incluido el bromato de potasio en el grupo 2B (posible
carcinógeno para el ser humano).
5.1.2 Estudios epidemiológicos
Los estudios epidemiológicos se deben evaluar con cuidado para
garantizar que las asociaciones observadas no se deban a un sesgo y
que el diseño sea adecuado para evaluar una posible relación causal.
La causalidad se puede evaluar cuando hay datos suficientes de
diversos estudios bien definidos y realizados en distintas zonas
geográficas. También es importante el respaldo de datos toxicológicos
y farmacológicos. Es especialmente difícil interpretar los datos
epidemiológicos de los estudios ecológicos del agua desinfectada, y
esos resultados se utilizan fundamentalmente como ayuda en la
formulación de hipótesis para un ulterior estudio.
Los resultados de los estudios epidemiológicos analíticos son
insuficientes para respaldar una relación causal en el caso de
cualquiera de las asociaciones observadas. Es especialmente difícil
interpretar los resultados de los estudios analíticos publicados
actualmente a causa de la información incompleta acerca de la
exposición a contaminantes específicos del agua que podrían constituir
un factor de confusión o modificar el riesgo. Debido a que en los
estudios epidemiológicos no se ha prestado suficiente atención a la
evaluación de la exposición a los contaminantes del agua, no es
posible evaluar de manera adecuada el aumento de los riesgos relativos
notificados. Los riesgos se pueden deber a otros contaminantes del
agua o a otros factores cuya acción se atribuiría sin razón al agua
clorada o a los THM.
5.2 Caracterización de la exposición
5.2.1 Presencia de desinfectantes y de subproductos de los
desinfectantes
Se suelen utilizar dosis de desinfectantes de varios miligramos
por litro, que son las dosis necesarias para inactivar los
microorganismos (desinfección primaria) o las dosis necesarias para
mantener una concentración residual en el sistema de distribución
(desinfección secundaria).
Para la evaluación de la exposición se necesita saber cuáles son
los SPD presentes en el agua y en qué concentración.
Desafortunadamente, hay pocos estudios internacionales publicados que
vayan más allá del estudio de casos o de los datos regionales.
Los datos disponibles parecen indicar la presencia, como
promedio, de alrededor de 35-50 µg de THM totales por litro en el agua
clorada, siendo el cloroformo y el bromodiclorometano la primera y
segunda especies predominantes. La exposición a los AHA se puede
calcular de manera aproximada mediante la concentración total de los
AHA (suma de cinco especies) correspondiente a alrededor de la mitad
de la concentración total de THM (aunque esta proporción puede variar
significativamente); los ácidos dicloroacético y tricloroacético son
la primera y la segunda especies más importantes. En el agua con una
proporción bromuro/carbono orgánico total elevada o con una proporción
bromuro/cloro elevada, cabe prever una mayor formación de THM y AHA
bromados. Cuando se utiliza una solución de hipoclorito (con
preferencia al cloro gaseoso), también se puede producir clorato
durante la cloración.
La exposición a los SPD en el agua cloraminada depende del
sistema de cloraminación, con la secuencia de cloro seguido de
amoníaco para dar lugar a la formación de (niveles más bajos de) SPD
del cloro (es decir, THM y AHA) durante el periodo de cloro libre; sin
embargo, la supresión de la formación de cloroformo y ácido
tricloroacético no se corresponde con una reducción proporcional de la
formación de ácido dicloroacético.
En igualdad de condiciones, la concentración de bromuro y la
dosis de ozono son los mejores indicadores de la formación de bromato
durante la ozonación, con una conversión de bromuro a bromato de
alrededor del 50%. En un estudio de diferentes servicios europeos de
abastecimiento de agua se puso de manifiesto que los niveles de
bromato en el agua al salir de las instalaciones de tratamiento
oscilaba entre una concentración inferior al límite de detección
(2 µg/litro) y 16 µg/litro. Los SPD orgánicos bromados que se forman
durante la ozonación se suelen encontrar en concentraciones bajas. La
formación de clorito se puede calcular como un simple porcentaje
(50%-70%) de la dosis de dióxido de cloro aplicada.
5.2.2 Incertidumbre de los datos sobre la calidad del agua
En un estudio toxicológico se intenta extrapolar una respuesta
(controlada) de animales de laboratorio a una respuesta humana
potencial; un posible resultado es la estimación de los factores de
riesgo de cáncer. En un estudio epidemiológico se intenta relacionar
los efectos en la salud humana (por ejemplo, cáncer) con uno o varios
agentes causales (por ejemplo, un SPD), para lo cual se necesita una
evaluación de la exposición.
Los riesgos químicos asociados con el agua de bebida desinfectada
se basan potencialmente en varias vías de exposición: i) ingestión de
SPD en el agua de bebida; ii) ingestión de desinfectantes químicos en
el agua de bebida y formación concomitante de SPD en el estómago; y
iii) inhalación de SPD volátiles durante la ducha. Aunque la formación
in vivo de SPD y la inhalación de estos SPD volátiles puede ser un
motivo potencial de preocupación para la salud, el examen que se
expone a continuación se basa en la premisa de que la ingestión de los
SPD presentes en el agua de bebida es la vía de exposición más
importante.
La exposición humana depende tanto de la concentración de SPD
como del tiempo de exposición. Más en concreto, los efectos en la
salud humana dependen de la exposición a mezclas complejas de SPD (por
ejemplo, THM más bien que AHA, especies cloradas más bien que
bromadas) que pueden sufrir cambios estacionales/temporales (por
ejemplo, en función de la temperatura, las características y la
concentración de la materia orgánica natural) y espaciales (es decir,
en la totalidad de un sistema de distribución). Cada desinfectante
químico concreto puede formar una mezcla de SPD; las combinaciones de
desinfectantes químicos pueden formar incluso mezclas más complejas.
Al formarse, la mayoría de los SPD son estables, pero algunos se
pueden transformar, por ejemplo, mediante hidrólisis. En ausencia de
datos sobre los SPD, se pueden utilizar en su lugar la dosis de cloro
(o la demanda de cloro), el carbono orgánico total (o la absorción
ultravioleta a 254 nm [UVA254]) o el bromuro para estimar
indirectamente la exposición. Aunque el carbono orgánico total es un
buen sustitutivo de los precursores orgánicos de los SPD, la UVA254
proporciona información adicional sobre las características de la
materia orgánica natural, que pueden variar en función de la zona
geográfica. Se han identificado dos variables fundamentales de la
calidad del agua, el pH y la concentración de bromuro, como factores
que influyen de manera considerable en el tipo y las concentraciones
de SPD que se forman.
En una evaluación de la exposición se debe intentar definir en
primer lugar los tipos específicos de SPD y las mezclas resultantes
que probablemente se formarán, así como sus concentraciones a lo largo
del tiempo, en las que influyen su estabilidad y el transporte a
través del sistema de distribución. Para los estudios epidemiológicos
existen algunas bases de datos históricos sobre la concentración de
desinfectantes (por ejemplo, de cloro) y posiblemente de los
precursores de los SPD (por ejemplo, el carbono orgánico total) o de
los THM totales (y, en algunos casos, especies de THM). En contraste
con los THM, que se han vigilado durante periodos de tiempo más largos
por razones reglamentarias, los datos de vigilancia de los AHA (y sus
especies), el bromato y el clorito, son mucho más recientes y, por
consiguiente, poco abundantes. Sin embargo, se pueden utilizar modelos
de SPD para simular los datos que faltan o datos pasados. Otro aspecto
importante es la documentación relativa a la evolución de las
prácticas de tratamiento de agua.
5.2.3 Incertidumbres de los datos epidemiológicos
Incluso en estudios analíticos bien diseñados y realizados, se
llevaron a cabo evaluaciones relativamente deficientes de la
exposición. En la mayor parte de los estudios se examinó la duración
de la exposición al agua de bebida desinfectada y a la fuente de agua.
Estas exposiciones se estimaron a partir de los historiales de los
residentes y de los registros de los servicios de agua de bebida o de
los gobiernos. Sólo en un pequeño número de estudios se intentó
estimar el consumo de agua de los participantes en un estudio y la
exposición a los THM totales o a especies aisladas de THM. Sólo en un
estudio se intentó estimar la exposición a otros SPD. Al evaluar
algunos riesgos potenciales, es decir, los resultados adversos en el
embarazo, que pueden estar asociados con exposiciones relativamente
breves a subproductos volátiles, puede ser importante examinar la
inhalación además de la vía de exposición por ingestión de agua de
bebida. En algunos estudios se intentó estimar tanto las
concentraciones de subproductos en el agua de bebida durante períodos
de tiempo importantes desde el punto de vista etiológico como la
exposición acumulativa. Se pueden utilizar modelos adecuados y
análisis de sensibilidad, como la simulación de Monte Carlo, para
estimar estas exposiciones durante los períodos que interese estudiar.
La incertidumbre es grande en lo que se refiere a la
interpretación de las asociaciones observadas, puesto que se han
examinado exposiciones a un número relativamente pequeño de
contaminantes del agua. Con los datos actuales, es difícil evaluar
cómo pueden haber influido las concentraciones de SPD o de otros
contaminantes sin dosificar en las estimaciones de los riesgos
relativos observados.
Se han dedicado más estudios al cáncer de vejiga que a cualquier
otro tipo de cáncer. Los autores de los resultados notificados más
recientemente para los riesgos de cáncer de vejiga advierten contra
una interpretación simplista de las asociaciones observadas. Los
indicios epidemiológicos de un aumento del riesgo relativo de cáncer
de vejiga no son concordantes: se notifican riesgos diferentes para
fumadores y no fumadores, para hombres y mujeres y para un consumo
alto y bajo de agua. Los riesgos pueden variar entre diversas zonas
geográficas debido a que las mezclas de SPD pueden ser distintas o
porque el agua puede contener también otros contaminantes. Hay que
recopilar, u obtener mediante simulación, datos más exhaustivos sobre
la calidad del agua a fin de mejorar la evaluación de la exposición
con miras a los estudios epidemiológicos.