
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
ENVIRONMENTAL HEALTH CRITERIA 46
GUIDELINES FOR THE STUDY OF GENETIC EFFECTS
IN HUMAN POPULATIONS
This report contains the collective views of an international group of
experts and does not necessarily represent the decisions or the stated
policy of the United Nations Environment Programme, the International
Labour Organisation, or the World Health Organization.
Published under the joint sponsorship of
the United Nations Environment Programme,
the International Labour Organisation,
and the World Health Organization
World Health Orgnization
Geneva, 1985
The International Programme on Chemical Safety (IPCS) is a
joint venture of the United Nations Environment Programme, the
International Labour Organisation, and the World Health
Organization. The main objective of the IPCS is to carry out and
disseminate evaluations of the effects of chemicals on human health
and the quality of the environment. Supporting activities include
the development of epidemiological, experimental laboratory, and
risk-assessment methods that could produce internationally
comparable results, and the development of manpower in the field of
toxicology. Other activities carried out by the IPCS include the
development of know-how for coping with chemical accidents,
coordination of laboratory testing and epidemiological studies, and
promotion of research on the mechanisms of the biological action of
chemicals.
ISBN 92 4 150186 5
The World Health Organization welcomes requests for permission
to reproduce or translate its publications, in part or in full.
Applications and enquiries should be addressed to the Office of
Publications, World Health Organization, Geneva, Switzerland, which
will be glad to provide the latest information on any changes made
to the text, plans for new editions, and reprints and translations
already available.
(c) World Health Organization 1985
Publications of the World Health Organization enjoy copyright
protection in accordance with the provisions of Protocol 2 of the
Universal Copyright Convention. All rights reserved.
The designations employed and the presentation of the material
in this publication do not imply the expression of any opinion
whatsoever on the part of the Secretariat of the World Health
Organization concerning the legal status of any country, territory,
city or area or of its authorities, or concerning the delimitation
of its frontiers or boundaries.
The mention of specific companies or of certain manufacturers'
products does not imply that they are endorsed or recommended by the
World Health Organization in preference to others of a similar
nature that are not mentioned. Errors and omissions excepted, the
names of proprietary products are distinguished by initial capital
letters.
CONTENTS
GUIDELINES FOR THE STUDY OF GENETIC EFFECTS IN HUMAN POPULATIONS
CONTRIBUTORS
PREFACE
1. INTRODUCTION
2. METHODOLOGICAL AND EPIDEMIOLOGICAL ISSUES
2.1. General considerations
2.2. Methodological issues
2.3. Epidemiological considerations - common components
2.3.1. Study samples
2.3.2. Comparison samples
2.3.3. Confounding variables
2.3.4. Interaction
2.3.5. Methods for estimating exposure
2.3.6. End-points
2.3.7. Dose-response relationships
2.3.8. Time-response relationships
2.3.9. Problems in sample size and interpretation of
findings
2.4. Long-term medical follow-up
2.4.1. Relevance and limitations
2.4.2. Personal identification
2.4.3. Starting-point records
2.4.4. End-point records
2.4.5. Methods required for integrating source files
3. MUTATIONS IN SOMATIC CELLS
3.1. General considerations
3.1.1. End-points
3.1.2. Study requirements
3.2. Chromosomal end-points
3.2.1. Structural aberrations
3.2.1.1 Methods of culture
3.2.1.2 Fixation and slide preparation
3.2.1.3 Analysis of cells
3.2.1.4 Data analysis
3.2.1.5 Radiation-induced aberrations and
estimates of exposure
3.2.1.6 Chemically-induced aberrations and
estimates of exposure
3.2.1.7 Conclusions
3.2.2. Sister-chromatid exchange (SCE)
3.2.2.1 Formation of sister-chromatid exchange
3.2.2.2 Relevance of sister-chromatid exchange
3.2.2.3 Factors potentially influencing SCE
frequency
3.2.2.4 Methods for sister-chromatid exchange
analysis
3.2.2.5 Data processing and presentation
3.2.2.6 Conclusions
3.3. Gene mutations
3.3.1. Principles and basis for the methods
3.3.1.1 Autoradiographic method
3.3.1.2 Cloning method
3.3.2. Relevance and limitations
3.3.2.1 Relevance
3.3.2.2 Limitations
3.3.3. Procedures for assay of TGr T-lys arising in vivo
in human beings
3.3.3.1 Autoradiographic method
3.3.3.2 Cloning method
3.3.4. Data presentation and analysis
3.3.4.1 Autoradiographic method
3.3.4.2 Cloning method
3.3.5. Conclusions
4. GERMINAL MUTATIONS
4.1. Introduction
4.1.1. Approaches for detecting germinal mutations
4.1.1.1 Detection of chromosomal mutations
4.1.1.2 The biochemical approach to detecting
point mutations
4.1.1.3 Indicator phenotypes
4.1.2. Methodological considerations and strategies
4.1.2.1 Sample acquisition and storage
4.1.2.2 Timing of studies
4.1.3. Summary
4.2. Germinal chromosomal abnormalities
4.2.1. Principles and basis of the method
4.2.2. Relevance and limitations
4.2.2.1 Studies of induced abortions
4.2.2.2 Studies of fetal deaths
4.2.2.3 Studies of prenatal diagnosis specimens
4.2.2.4 Studies of live births
4.2.2.5 Studies of indicator phenotypes of
chromosomal abnormalities
4.2.3. Procedures
4.2.3.1 Fetal specimens
4.2.3.2 Live births and other offspring
4.2.3.3 Detection of "indicator" phenotypes for
germinal chromosomal mutations - trisomies
4.2.3.4 Data presentation
4.3. Biochemical approaches to detecting gene mutations in
human populations
4.3.1. Biochemical methods for monitoring for gene
mutations
4.3.1.1 One-dimensional electrophoresi
4.3.1.2 Two-dimensional electrophoresi
4.3.1.3 Enzyme activity
4.3.1.4 Other biochemical approaches
4.3.2. Analytical strategy and methodological
considerations
4.3.2.1 One-dimensional electrophoresis
4.3.2.2 Two-dimensional electrophoresis
4.3.2.3 Enzyme activity
4.3.2.4 Sample acquisition and storage
4.3.3. Data management
4.3.4. Considerations in screening for germinal mutations
4.3.4.1 Sample size
4.3.4.2 Distinction between "true" and "apparent"
mutations
4.3.4.3 Implementation of gene mutation screening
programmes
4.3.5. Summary
4.4. Sentinel phenotypes
4.4.1. Introduction
4.4.2. Basis of method
4.4.3. Relevance and limitations
4.4.4. Procedures
4.4.4.1 Surveillance of sentinel anomalies
4.4.5. Data intrepretation
4.4.5.1 Surveillance of sentinel anomalies
4.4.6. Conclusions
4.5. Fetal death
4.5.1. Introduction
4.5.2. Procedures
4.5.3. Data processing and presentation
4.5.4. Conclusions
REFERENCES
CONTRIBUTORS
The following experts participated in the preparation of this
monograph:
Dr R.J. Albertinib, University of Vermont, College of Medicine,
Burlington, Vermont, USA
Dr K. Altlandb, Institute for Human Genetics, University of
Giessen, Giessen, Federal Republic of Germany
Dr A.V. Carranoa,b, Biomedical Sciences Division, L-452, Lawrence
Livermore National Laboratory, Livermore, California, USA
Dr A. Czeizelb, Department of Human Genetics, National Institute of
Hygiene, Budapest, Hungary
Dr G.R. Douglasa,b, WHO Collaborating Centre, Mutagenesis Section,
Department of National Health and Welfare, Ottawa, Ontario,
Canada
Dr H.J. Evansb, Medical Research Council, Clinical and Population
Cytogenetics Unit, Western General Hospital, Edinburgh,
Scotland, United Kingdom
Dr E.B. Hooka,b, Bureau of Maternal and Child Health, New York
State Department of Health, Albany, New York, and Department of
Pediatrics, Albany Medical College, Albany, New York, USA
Dr J.R. Millera,b,c, Central Research Division, Takeda Chemical
Industries, Ltd., Osaka, Japan
Dr H. Mohrenweisera,b, Department of Human Genetics, University of
Michigan, Ann Arbor, Michigan, USA
Dr H.B. Newcombeb, Deep River, Ontario, Canada
Dr R.J. Prestona,b, Biology Division, Oak Ridge National
Laboratory, Oak Ridge, Tennessee, USA
Dr. E.M.B. Smithb, International Programme on Chemical Safety,
World Health Organization, Geneva, Switzerland
Ms M. Smitha,b, Vital Statistics and Disease Registries Section,
Statistics Canada, Ottawa, Ontario, Canada
Dr D. Warburtonb, Department of Human Genetics and Development,
Columbia University, New York, New York
-------------------------------------------------------------------
a WHO/DNHW Consultation, July 27-28, 1981, Ann Arbor, Michigan,
USA
b WHO/DNHW Task Group Meeting, September 18-21, 1984, Ottawa,
Ontario, Canada
c Chairman, Ann Arbor and Ottawa meetings
NOTE TO READERS OF THE CRITERIA DOCUMENTS
Every effort has been made to present information in the
criteria documents as fully and as accurately as possible. In
the interest of all users of the environmental health criteria
documents, readers are kindly requested to communicate any errors
found to the Manager of the International Programme on Chemical
Safety, World Health Organization, Geneva, Switzerland, in order
that they may be included in corrigenda, which will appear in
subsequent volumes.
In addition, experts in any particular field dealt with in the
criteria documents are kindly requested to make available to the
WHO Secretariat any important published information that may have
inadvertently been omitted, so that it may be considered in the
event of updating and re-evaluation of the conclusions contained in
the criteria documents.
PREFACE
Monitoring and assessment of effects on human health from
exposure to environmental agents of all types, with monitoring to
confirm that control measures are working effectively, are key
aspects of the World Health Organization's Environmental Health
Programme. Without objective data on effects, attempts to assign
causes can only be speculative. Speculation does not provide a
reliable basis for remedial action and the control of health
hazards. Knowledge of the presence and identity of mutagenic
agents in the environment and the extent of exposure to them is
therefore extremely important in interpreting and using the results
obtained from monitoring populations for genetic effects.
Within the World Health Organization, there are a number of
programmes that are concerned with the monitoring and assessment of
human exposures and effects on health.
Control of environmental health hazards is one such programme.
This has the objective that, by 1989, the countries actively
involved will have formulated and developed national policies and
programmes for the protection of the health of their populations
against environmental hazards. A number of projects on the
monitoring of air and water quality, food contamination, and of
selected pollutants in human tissues are carried out by the World
Health Organization. These activities are complemented by a more
recent component, the Human Exposure Assessment Location (HEAL)
project, which is devoted directly to the monitoring of human
exposure to certain pollutants in all environmental media. All of
these monitoring projects are implemented in conjunction with the
United Nations Environment Programme's Global Environmental
Monitoring System (GEMS).
The International Programme on Chemical Safety is a cooperative
activity of the World Health Organization, the International Labour
Office, and the United Nations Environment Programme. It resulted
from concern expressed on the hazards of exposure to chemicals and
the pressing need to identify and assess them, so that control
measures could be applied and safe use achieved. One of the means
of achieving safe use is the preparation and dissemination of
documents of practical use to those involved in implementing
chemical safety. Publication in the International Programme on
Chemical Safety environmental health criteria series emphasizes the
importance of a systematic approach to monitoring human populations
for genetic effects as an integral part of environmental health
management.
These guidelines are intended as a source of practical
information on the design and conduct of genetic studies on human
populations exposed, or suspected of being exposed, to mutagenic
agents. As they are guidelines, they do no include comprehensive
protocols for studies. However, attention is directed to important
details that must be included, as well as pitfalls to be avoided.
It is envisaged that this document will be of use both to those
already involved in the assessment of human genetic hazards and to
those who wish to become better informed on this subject. Although
future requirements are not ignored, these guidelines principally
encompass methods that are considered practicable at present.
The original concept for these guidelines arose from the
recommendations of a WHO Consultation on Genetic Monitoring held in
Ottawa, Canada, on October 17, 1980, organized by the WHO
Collaborating Centre on Environmental Mutagenesis, Department of
National Health and Welfare (DNHW) Canada, and held in conjunction
with an International Conference on Chemical Mutagenesis,
Population Monitoring, and Genetic Risk Assessment. The drafting
of the monograph, implemented initially by the late DR K.C. BORA
(DNHW), and subsequently directed by DR G.R. DOUGLAS (DNHW), who
was also responsible for editing the final draft, began with a
WHO/DNHW Consultation held in Ann Arbor, Michigan, USA, on
July 27 - 28, 1981. This meeting produced a draft document
containing material that formed the basis for this monograph.
Subsequently, a joint WHO/DNHW Task Group Meeting, held in Ottawa,
Canada, on September 18 - 21, 1984, completed the project.
* * *
Financial and other support for the meeting was provided by the
Department of National Health and Welfare, Canada. The United
Kingdom Department of Health and Social Security covered the costs
of printing.
1. INTRODUCTION
The extent to which human somatic and germinal mutation
frequencies may be increased by exposure to ionizing radiation
and to the variety of chemicals that characterize modern societies
has been a matter of concern in recent years. Somatic mutations,
either genic or chromosomal, are not transmitted to the offspring
of an exposed individual. However, increases in the frequency of
these mutations may contribute to an increase in the frequency
of acquired disorders, for example, cancer. Increases in the
frequency of germinal mutations, genic or chromosomal, are likely
to contribute to inherited defects in the offspring of individuals
exposed to mutagenic agents. There is, therefore, a clear need to
develop and apply methods to study exposed populations at risk of
increased levels of somatic or germinal mutations.
Any effort to determine whether an increase in mutation rate
has occurred in a given population must contend with formidable
problems, one of the most significant of which is defining the
genetic end-points suitable for study. Germinal mutations, either
genic or chromosomal, can give rise to a plethora of phenotypes,
only a few of which are useful and meet the criteria required
for studies in mutation epidemiology. Likewise, using current
techniques, only a subset of somatic-cell mutations are amenable
to study. However, since it is much easier to obtain large samples
of somatic rather than germinal events, it would be ideal if some
measure of germinal mutations and the health effects of their
consequent phenotypes could be conveniently extrapolated from
studies of somatic events. Although such an extrapolation has
been attempted for the effects of ionizing radiation (Brewen et
al., 1975), it is not possible, at present, to attempt the same for
chemical effects. Despite these difficulties, it is essential that
efforts be made to initiate studies aimed at measuring somatic and
germinal genetic changes and assessing the relationships between
the two.
Although similar mutational changes will occur in somatic
and germ cells,the methods for detecting them are quite different.
An increase in the frequency of somatic mutations, genic or
chromosomal, can be established from relatively few individuals,
provided that a large number of cells is analysed from each sample
(Bloom, 1981). In contrast, determining an increase in the
frequency of mutation in germ cells by examining affected offspring
involves large study populations; the smaller the increase to be
detected (or excluded), the larger the sample needed (Neel, 1980;
Vogel & Altland, 1982). International cooperation may be necessary
to obtain adequate samples. However, because of the costs involved
in mounting large-scale epidemiological surveys, it is essential
that they should be designed to make the most efficient use of
resources and that the tests used should be designed to give
meaningful results. The general principles are the same for
all countries, and comparability can be achieved through
standardization of many of the procedures. Since the practical
details of the strategy and tactics in any one country will be
influenced by local circumstances, the emphasis in these guidelines
will be placed on general principles and examples of possible
procedures.
2. METHODOLOGICAL AND EPIDEMIOLOGICAL ISSUES
2.1. General Considerations
Certain general principles influence the design of
epidemiological studies for genetic effects and the nature of
the procedures for data gathering and analysis. The information
requirements are basically similar, whether changes in the genetic
make-up of somatic cells or germ cells in the exposed individuals,
or inherited changes affecting the offspring of such individuals
are being investigated. The key elements for any such study should
include:
(a) means of identifying the exposed (or affected) population
to be studied;
(b) availability of a reasonably similar comparison group, to
serve as the unexposed (or unaffected) controls;
(c) an idea of either the nature of the presumed mutagen, or,
at least, of the general source of the anticipated harm;
(d) prior knowledge of the likely end-points that could serve
as indicators of genetic damage;
(e) where possible, some separation into different levels of
exposure with which to investigate a likely dose-response
relationship; and
(f) a means of observing the time course of the response.
Not all of these will be equally available for any study.
However, none of the elements that are available should be
overlooked in the initial documentation or the subsequent
analysis. In general, the need to guard against oversights in
necessary information will tend to increase with the interval of
time between the causal events and the resulting expressions of
harm.
Wherever long-term follow-up is involved, particular attention
should be paid to the personal identification of the individuals so
that starting-point data and end-point data will be appropriately
and unambiguously matched.
2.2. Methodological Issues
Not all studies will have a broad information base at the
outset. However, useful work can still be done, even where there
is no prior indication of harmful exposures, no suggestion of which
persons might be exposed, and no clear idea of the end-point
effects to expect. In principle, watch can be kept over a wide
range of potential indicators of genetic damage, among large
populations, such as the new-born, and this should be continued
over a long period of time. In this broad kind of continuing
observation, it is the temporal changes in the frequency of
end-points, above familiar background rates, that alert the
investigator to a possible mutagenic effect. The term
"surveillance" is used to describe this type of activity.
Methods for determining the frequency of a number of end-points
are discussed in this monograph. Various techniques are used for
determining germinal mutations. Indicators include potential
germinal effects, adverse reproductive outcomes, or other health
problems that may have a genetic basis.
Because of size considerations, it will often be necessary to
combine data from many studies. This will be greatly facilitated
if all investigators adhere to standardized laboratory procedures
and record-keeping practices including identification of the
individuals, with standard nomenclature for causes of death,
congenital anomalies, and protein and chromosomal variants.
2.3. Epidemiological Considerations - Common Components
The following comments apply to all methods and end-points.
2.3.1. Study samples
The appropriate epidemiological study design for examining the
possible hazards of agents must be determined. A cohort design
will be used in most studies where there is known exposure to a
suspected mutagenic agent. Alternatively, a case-control study
design may be used when exposure is common (e.g., smoking,
caffeine), or rare phenotypes are the end-points of interest
(e.g., new mutant phenotypes). In a case-control study, groups
of individuals are selected on the basis of whether they have the
genetic end-point under study. Also, in a case-control study, a
number of exposures can be evaluated in relation to a selected end-
point, in contrast to a cohort study in which a number of diseases
are evaluated in relation to one or more exposures.
In determining the population to be studied, several different
approaches can be taken. For example, questionnaires can be used
to determine if individuals in a particular setting have been
exposed to a putative hazard, and to gather data on possible
controls, or existing data sources (e.g., vital records, census
data, employment and work history records, special registries,
populations of specific geographical locations) may be used.
Regardless of the approach used, sufficient personal identifying
information must be recorded regarding the individuals to allow for
follow-up and for searching any large files.
2.3.2. Comparison samples
The identification and use of appropriate controls is
essential. In both cohort and case-control studies, a basis for
comparison is required, in order to evaluate whether the outcome
observed differs from that expected, had there not been any
increased risk from the agent under study. In certain cases a
population may serve as its own control by sampling before, during,
and after exposure (internal controls in a longitudinal study).
Since the frequency of a given end-point may vary according to age,
sex, socioeconomic status, and other variables, it is important
that this is taken into consideration when making the comparisons.
2.3.3. Confounding variables
Confounding can be described as the mixing of effects caused by
variables that are associated with both the exposures and end-
points to be studied, or a distortion of the apparent effect of an
exposure on risk brought about by the association with other
factors that can influence the outcome. In this regard, the
following should be considered:
(a) variables known to cause mutagenic effects should be
identified as far as possible (e.g., cigarette smoking,
individuals known to have undergone radiotherapy);
(b) cases and controls should be appropriately matched (e.g.,
for age and sex) in such a way that they have the same
distribution with respect to known confounding variables;
and
(c) appropriate statistical analysis to partition confounding
variables should be used.
2.3.4. Interaction
If an exposure is associated with an increased risk of a
particular end-point, it is important to determine whether the
risk is additive to that of other known causes of disease, or
synergistic in its effect.
2.3.5. Methods for estimating exposure
The following methods can be used to estimate either the levels
of exposure, or values that are proportional to the levels of
exposure:
(a) estimates of length of exposure as well as ambient levels
of the agent may be the best or only estimate of dose in a
working environment; it is recognized that such estimates
are crude, but, in some situations, they may be all that
is available;
(b) direct measurement of an agent in the environment (e.g.,
air or water sampling) may be used;
(c) direct measurement of an agent or its metabolites in body
fluid and tissues to estimate body burden (e.g., blood,
urine, hair, teeth);
(d) observation of other pathological evidence of organ or
tissue damage (e.g., liver damage, chloracne) may be
helpful in estimating doses; and
(e) the use of questionnaires related to work histories and
lifestyles may assist in determining the exposure of
concern or other exposures.
In some cases, there may be ample time to obtain exposure
data, but, with emergency situations, the information may be only
transiently available. Thus, it must be collected early and the
people involved, recruited, or identified before they disperse.
2.3.6. End-points
Effects of somatic or germinal origin are considered in
sections 3 and 4 of this document, respectively.
2.3.7. Dose-response relationships
The existence of a dose-response relationship - that is, an
increase in disease incidence with increase in amount of exposure -
supports the view that an association is a causal one. Thus, it is
desirable to attempt to quantify exposure as far as possible.
However, under certain circumstances, in order to make the best use
of limited resources, initial studies could be restricted to those
with high exposure doses. Individuals so exposed would be compared
to an unexposed population. If no differences were found, the
study could be terminated, but it should be noted that, at high
doses, excess cell lethality might mask genetic effects. If
differences were observed, effects at intermediate dose levels
should be studied, paying special attention to possible confounding
synergistic effects.
Estimates of doses for populations that have been exposed
accidently will most likely be less accurate than for those exposed
occupationally and medically. In such cases, it may be more useful
to estimate the upper boundary of the dose (i.e., maximum possible
dose) rather than the mean, or to stratify the population into 2 or
3 groups whose bounds, though wide, probably do not overlap enough
to wipe out the suspected differences between them.
2.3.8. Time-response relationships
Consideration of the optimal sampling time is critical for
quantifying the somatic end-points in the exposed individual.
Appropriate sampling times should be selected to maximize an
effect, whenever this is possible. This is discussed further in
section 3.
2.3.9. Problems in sample size and interpretation of findings
The ability to detect an effect of a given incidence will
depend on the sample size that can be studied and the base-line
frequency of a given end-point in the unexposed population. The
more frequent the end-point, the smaller the sample size needed to
detect a given effect. The required sample size will also depend
on the chosen values of alpha, the probability of falsely rejecting
the null hypothesis, and beta, the probability of falsely accepting
the null hypothesis. These are sometimes known as Type 1 and Type
2 errors. It is customary, in most experimental situations, to
choose alpha = 0.05 and beta = 0.20 (2-tailed test). However, in
order to be more certain that a real effect is not missed, then an
alpha = 0.10 (or 0.05 for a one-tailed test) and beta = 0.05 could
be chosen. However, this choice will increase the sample size
required to rule out a given effect incidence.
It is possible, for given values of alpha and beta, to
calculate the sample size required to rule out a given effect
incidence, for an end-point of known frequency in the exposed
population. Conversely, the effect size detectable with a given
sample size can be calculated. Table 1 shows the relationship
between sample size and effect incidence for a given degree of
assurance that the effect is real.
In any study, a negative result should be expressed in terms of
the size of the effect that can be ruled out at a given power. For
example, "this study rules out a 2-fold increase in the frequency
of end-point X, with 80% confidence" or "this study has a 50% power
to detect a doubling of the frequency of end-point X".
Further considerations of sample size will be discussed under
the sections devoted to each specific test end-point.
Table 1. Sample sizes needed to detect a given increase in
frequency, with alpha = 0.05a
-----------------------------------------------------------
Frequency Sample size to detect Sample size to detect
of a doubling a tripling
end-points beta = 80 beta = 95 beta = 80 beta = 95
-----------------------------------------------------------
.000001 23 5551 082 39 001 131 7 850 356 13 000 369
.00001 2 355 076 3 900 058 785 021 1 300 011
.0001 235 475 389 951 78 487 129 976
.001 23 515 38 940 7834 12 972
.01 2319 3839 769 1272
.05 435 719 141 232
.10 200 329 62 102
.15 121 199 36 58
-----------------------------------------------------------
a Two-tailed probability without correction for continuity.
For discussion of continuity correction, and computation
formula, see Fleiss (1981).
2.4. Long-Term Medical Follow-up
People need to be identified uniformly on various files in
order to monitor human populations for delayed effects caused by
exposure to environmental agents. Epidemiological studies are
greatly facilitated by the existence in many countries of
centralized, computerized, accessible national registries relating
to health outcomes.
The kinds of data required are concerned with establishing
statistical associations, which may serve as pointers to possible
"troublespots", or with investigating suspected exposed groups. An
investigation involves defining the population cohort for study,
the end-points of interest, and the appropriate study methods for
carrying out individual long-term follow-up efficiently (WHO,
1983).
A number of existing population-based files, concerned with
health events, are available as end-point records that can be used
to measure various health effects among a cohort under study
(Bloom, 1981). Normally, follow-up will consist simply of using a
starting-point record, in its machine-readable form, to search by
computer for an end-point record relating to the same individual or
family. Procedures have been developed for doing this and for
analysing the results, without loss of personal privacy (Smith &
Newcombe, 1980).
2.4.1. Relevance and limitations
Lack of adequate recorded information and of organization and
use of historical data in a consistent fashion are two major
stumbling blocks in long-term epidemiological or genetic research.
The term "record linkage" has been used to describe the process
whereby two or more records relating to the same individual,
family, or event are brought together. The success of this
procedure depends on the quality, quantity, and discriminating
power of the items of personal identification on the machine-
readable records that are to be brought together.
Existing historical data should be more readily accessible
for statistical analysis, but with built-in safeguards so that
confidentiality of health records and personal privacy are not
compromised. Examples of relevant data are: a) mortality data;
b) birth defect monitoring programmes; c) health surveillance
programmes; d) vital records, such as marriages and births; e)
morbidity records; and f) cancer registries. In addition, where
new data are being collected (e.g., on suspected high-risk groups,
or on individuals who exhibit "early indicator" conditions), this
should be done in such a fashion that it is possible to make
international comparisons and to pool data.
2.4.2. Personal identification
Each record needs to contain enough information to indicate
unambiguously the particular individual and/or family to whom it
refers (Table 2). If a universal numbering system were available
for each individual in the population, and were in general use on
all medical and vital health records, the problem of matching would
almost disappear. In the absence of such a universal number, the
record should ideally contain full birth names, birth date,
birthplace, sex, mother's maiden surname, and place of residence.
Other useful items are full current surname and current address.
Since records being linked will seldom have been generated at
exactly the same time, and, since some of the items are subject to
change with time, it is desirable that the date of the event and
the type of event be known. For example, the date of event (e.g.,
hospital visit) may infer a "last-known alive date", which could
substantially reduce the amount of scanning where a mortality
search is being carried out.
To identify the family involved, the marital status of the
individual, and, if applicable, the spouse's birth surname,
forenames, and birthplace should be recorded. In the case of
birth, marriage, death, cancer registries, and especially genetic
registries, it is desirable to have parental birth names,
birthplaces, and birth dates (or ages).
Table 2. A list of items to be included on starting- and end-point
records to facilitate follow-up studiesa
-------------------------------------------------------------------
* 1. Surname
* 2. Previous surname (if any)
* 3. First given name
* 4. Second and other given names
* 5. Usual name (or nickname)
* 6. Sex
* 7. Birth date (year, month, day)
* 8. Birth province or country
* 9. Birth city or place
* 10. Father's surname
* 11. Father's first given name
* 12. Father's second given name
+ 13. Father's birth province or country
14. Father's birth city or place
+ 15. Father's birth date (or age)
* 16. Mother's maiden name
+ 17. Mother's first given name
+ 18. Mother's second given name
+ 19. Mother's birth province or country
20. Mother's birth city or place
+ 21. Mother's birth date (or age)
* 22. Marital status
* 23. Spouse's birth surname
* 24. Spouse's first given name
25. Spouse's second given name
26. Spouse's birth province or country
27. Spouse's birth city or place
28. Social Security Number or equivalent
29. Health Insurance Number
* 30. Place of residence - province or country
31. Place of residence - complete address including postal
code
* 32. Date of event
* 33a. Last known alive date (e.g., date of last contact)
33b. Date hired by company
33c. Date left company
34. Principal lifetime occupation - type of work, type of
business, length of time worked
-------------------------------------------------------------------
Table 2. (contd.)
-------------------------------------------------------------------
35. Other items where applicable (e.g., birth order of child,
status of birth, religion, race, etc.)
* 36. Control code to indicate the kind of event
* 37. A control code digit to indicate whether alternate entries
for the same event are being recorded (e.g., where an
individual may have alternate spellings for surname)
* 38. A unique number (where no other suitable number is
available)
39. Where applicable, an indicator to denote whether the
individual is known to be dead,the date of death, and the
province or country of death
-------------------------------------------------------------------
a From: Smith (1977).
*,+ Top priority would be given to collecting the items identified
with asterisks. For genetic studies, additional parental
variables will be required, particularly those indicated with
+. Information relating to diagnostic procedures, work
histories, exposure histories, etc., plus updates, are added to
the basic record. For marriages, the record should identify
the groom, bride, groom's parents and bride's parents.
Records in files can contain erroneous information and
omissions. Thus,in order to reduce the frequency of matching
errors, it is necessary to introduce a measure of "redundancy"
into the identifying information. Alternative entries can also be
created, where the surname of an individual may have an alternative
spelling (e.g., adoptions). It may also be useful to record
nicknames, race, principal lifetime occupation, and birth order.
2.4.3. Starting-point records
Where information is sought concerning the likely health
effects of possible harmful agents in the environment, and of
various social and economic circumstances, the chief limiting
factor is a shortage of starting-point records in a form suitable
for searching death records, cancer registries, etc., for evidence
of harm to the exposed individual or his/her offspring.
Collaboration and cooperation is required among a variety
of organizations to help resolve this problem (e.g., Regulatory
Agencies, Departments of Labour and Health, and research workers).
Various laboratory tests and studies are being carried out to
predict whether particular chemicals would be likely to cause
mutations or cancers in human beings exposed over long periods to
low or moderate doses. Human data relating to subpopulations can
serve as a potential starting point for a variety of studies, but
the records need to be uniformly stored and readily accessible.
Studies involving large cohorts can be carried out at a national
level. International cooperation is necessary in cases where the
numbers are small, and pooling of data is required. An example
would be to follow up people who show "early indicator" conditions.
For instance, individuals who show chromosomal breakage could be
identified, and followed up to see whether they subsequently
developed cancer or had reproductive problems (e.g., stillbirths)
or defective children. Similarly, children found by survey to have
protein variants could be followed up to see whether they developed
special health problems or died early.
Starting-point records can comprise a wide assortment of
administrative and other kinds of microdata files. These may
include ad hoc local files (e.g., nominal rolls created using
personnel, pay, and pension records), specialized centralized
registers (e.g., health surveillance registers), records of
exposure to certain agents, records of employment, special
survey records, vital registrations, or other sources.
2.4.4. End-point records
A number of health events are routinely documented in forms
that are centralized to a certain extent and are in a machine-
readable form that lends itself to studies in which a measure of
the health impact on a particular cohort is required. Included
among such sources are death registrations (Kinlen, 1980;
Patterson, 1980; Smith & Newcombe 1980), and special registers of
cancer, congenital anomalies, and other handicapping conditions.
Secondary sources that might be helpful include marriage records,
to give changes of name for follow-up and family composition, and
birth records to indicate offspring at potential risk. Both birth
and stillbirth records are of particular value, where fertility is
of interest, for indicating a potential health effect. Hospital
and medical insurance records, may be useful to help convert
locally-based registers into population-based ones, and to give
a measure of the social burden of disease.
2.4.5. Methods required for integrating source files
Computer methods have been developed to derive family
statistics relating to hereditary and congenital disease and terms
of other ill health (Newcombe, 1967, 1969, 1977; Smith, 1977,
1980). For example, combining records from the vital registration
system, certain specialized disease registers, and records from a
universal hospital insurance scheme made multi-generation and
cousin studies possible (Table 3 and 4). However, individual
follow-up is in much greater demand, primarily for detecting
delayed cancers.
For linking an individual with health and other relevant
records, a unique identity number assigned at birth would be
extremely useful. These are not available in many countries.
Where no unique number is available, the searching procedures
to be used in bringing records together may involve probabilistic
matching techniques. A generalized record linkage system and
probabilistic matching procedures have been developed and used for
such studies (Smith et al., 1980; Howe & Lindsay, 1981; Smith &
Newcombe, 1982).
Linkage projects have been developed or are being planned in a
number of international centres (Mi, 1967; Acheson, 1968; Skolnick,
1980; Beebee, 1981; Fox & Goldblatt, 1982; Smith, 1982), which
facilitate the establishment of: (a) mortality data bases; (b)
cancer incidence reporting systems; (c) death clearance of cancer
registers; (d) procedures for supplementing cancer registers with
hospital and medicare data; and (e) procedures for constructing
better handicap registers.
Table 3. Identifying information available in the magnetic tape files of vital and health records example from British Columbiaa
--------------------------------------------------------------------------------------------------------------------------------------
Event and its date and place Father Mother Child
Event Date City & Sur- Initials Age Province Maiden Initials Age Province Sur- Given Sex Birth Full City Province Otherb
(or Province name of birth surname of birth name names order birth of of birth
year*) date birth
--------------------------------------------------------------------------------------------------------------------------------------
Marriage X X X X X X X X X X (1)
Livebirth X X X X X X X X X X X X X X X X X (2)
Stillbirth X X X X X X X X X X X X X X X X X (2)
Death X X X X X X X X X X X X X X X (3)
Handicap X* X X X X X (4)
Congenital X* X X X X X (4)
malformation
surveillance
Hospital
Separation X X X X X X (5)
--------------------------------------------------------------------------------------------------------------------------------------
a From: Newcombe (1977).
b (1) Marital status of groom and bride.
(2) Legitimate or illegitimate status; singleton, twin, triplet, etc.
(3) If married women, own maiden name surname instead of mother's; marital status.
(4) Coded address representing city, school district and provincial health unit area; whether born in home province.
(5) Hospital code and admission number: maiden surname if different; marital status; address as city, street, and number; religion;
three initials of head of family, or spouse, or guardian; plus codes for relationship and whether address is the same.
Table 4. Example of the file organization for a sibship history of disease (fictitious)a,b
---------------------------------------------------------------------------------------------------------------------------------
Event (birth Year Father Mother Child
order in Surname Initials Province Age Maiden Initials Province Age Given Sex Birth Disease
parentheses) of birthc surname of birthc names year month day
---------------------------------------------------------------------------------------------------------------------------------
Marriage 1948 Cox JW 07 25 Bell MA 09 21
Live birth (1) 1950 Cox JW 07 27 Bell MA 09 23 Annie May F 50 01 09
Live birth (2) 1952 Cox JW 07 29 Bell MA 09 25 Brian John M 52 04 14
Reg. handicap 1952 Cox - - - - - - - Brian John M 52 04 14 Spina bifida
Hospitalization 1955 Cox - - - - - - - Brian John M 52 04 14 Spina bifida
Death 1956 Cox JW 07 - Bell MA 09 - Brian John M 52 04 14 Spina bifida
Stillbirth (3) 1954 Cox JW 07 31 Bell MA 09 27 Mary Jane F 54 07 26 Anencephaly
---------------------------------------------------------------------------------------------------------------------------------
a From: Newcombe (1977).
b Obtained by merging the individual histories of birth, ill health and death, with the sibship histories of parental marriages
and births.
c Coded from a 2-digit geographical code for all Canadian provinces and all major countries; 07 = Saskatchewan 09 = British
Columbia.
3. MUTATIONS IN SOMATIC CELLS
3.1. General Considerations
General reasons for studying somatic mutation in human
populations include:
(a) to determine if an unsuspected introduction of new (or
increase in already present) mutagens has occurred;
(b) to evaluate mutation frequencies in populations known (or
suspected) to be exposed to mutagens;
(c) to monitor for changes in frequency in populations as a
consequence of the removal of mutagens;
(d) to identify groups in the general population with high
frequencies of somatic mutations, so that studies can be
undertaken to attempt to detect the responsible factors;
and
(e) to define the heterogeneity of susceptibility to genotoxic
agents within the population.
3.1.1. End-points
Genetic damage can be assayed at the molecular, functional
gene, and chromosomal levels. At the molecular level, DNA damage
can be assessed in terms of adduct formation, strand breakage and
repair, or base sequence alterations. At present, the measurement
of adduct formation requires either prior knowledge of the nature
of the adduct or limited population sizes, and is, therefore, not
currently applicable as an assay for monitoring large populations.
DNA strand breakage and repair are usually transient phenomena
initiated at the time of insult and completed shortly afterwards,
making them less than ideal for population monitoring purposes.
Recent research developments have made it feasible to use
restriction enzyme mapping techniques to analyse alterations in
DNA base sequence (Botstein et al., 1980; Skolnick & Francke,
1982; Southern, 1982). This approach is being used to define gene
structure and identify polymorphisms for linkage analysis. The
same approach could equally well be used to screen for mutational
changes, but its application must await the availability of
appropriate batteries of DNA probes.
With current techniques, changes in gene function can be
detected by the acquisition of resistance to selective agents or
by the alteration or loss of cellular constituents detected by
immunological methods. Although these immunological approaches
hold considerable promise, the available data are insufficient
for application to routine monitoring. On the other hand,
methods for the detection of thioguanine-resistant peripheral
blood T-lymphocytes have been established and are being refined.
A detailed description of the principles and methods of this
approach is presented in section 3.3.
At the chromosomal level, genetic damage is observed as an
alteration either in chromosome number or in chromosome structure.
Although numerical changes represent a significant proportion of
human heritable genetic diseases, their consequences in somatic
cells are less well characterized. Moreover, estimates of
numerical changes in somatic cells are heavily influenced by
technical artifacts. Alterations in chromosome structure are more
accurately assessed in somatic cells and are observed cytologically
as chromosomal aberrations or sister chromatid exchanges (SCEs).
These are currently the most readily-available and widely-used end-
points for evaluating somatic mutations in human beings. There is
extensive literature concerning these methods (Office of Technology
Assessment, US Congress, 1983), which are discussed in section 3.2.
Micronuclei, which can result from structural or numerical
chromosome aberrations, have been used as an indicator of such
damage in human studies. Micronuclei have been enumerated in
cultured lymphocytes to detect the effects of ionizing radiation
(Krepinski & Heddle, 1983) and in the exfoliated epithelial cells
of persons exposed to chemical carcinogens (Stich et al., 1982).
However, this method, though showing potential for monitoring
genetic damage in human beings, has not yet been accepted or used
widely and so is not considered in detail.
3.1.2. Study requirements
Any study attempting to evaluate human populations for somatic
cell mutations should satisfy the following requirements:
(a) the study should include exposed groups and appropriate
concurrent controls;
(b) specimens from exposed and control individuals should be
gathered, handled, transported, and evaluated at the same
time and in the same way, using for example, the same
culture medium, serum, and reagents;
(c) samples should be coded and evaluated in such a way that
the the origin of the sample is not known, at the time of
evaluation;
(d) attempts should be made to quantify levels of exposure and
to determine dose-response relationships.
However, in some instances, concurrent controls may not be
necessary, for example, in longitudinal studies to determine
temporal changes in mutation frequencies. In these cases, any
possible confounding variables, such as changes in batch or source
of media, sera, or reagents used in the study should be minimized.
Reagents should be stored and, when there is a batch change,
comparative studies of selected individuals (or stored samples)
with new and old reagents should be undertaken to determine if
there are differences. It is recognized that it may be extremely
difficult to find appropriate controls in all instances, because of
various confounding factors that can influence somatic mutation
frequencies (e.g., age, sex, exposure to ionizing radiation,
medication, recent viral disease, smoking, etc). An appropriate
control group is one that is matched as closely as possible with
the exposed group for these confounding factors.
If it is impossible to find appropriate matched controls for
everyone in an exposed group, but it is necessary to study all
those exposed, the following alternatives are suggested:
(a) to obtain the best controls available and avoid the most
significant confounding factors;
(b) to stratify the cases, prior to analysis, into those with
well-matched controls and those with less well-matched
controls;
(c) to give the greatest weight in the analysis and
interpretation of results to the group with the well-
matched controls.
In emergency situations, when appropriate matched controls
are not available, it is important to obtain some control specimens
that will be sampled, transported, and cultured at the same time
as those from exposed individuals. Sources of such specimens
are members of study teams, assuming that they have not been
contaminated. Each batch of specimens, collected in emergency
circumstances and transported to the laboratory, should include a
specimen from at least one unexposed individual.
The questions arise as to when, and for how long after exposure
to a mutagen, individuals can usefully be studied. It may be
important to follow populations with elevated mutant frequencies
sequentially, to determine if such frequencies are changing.
Because of the long life span of the circulating peripheral
lymphocyte, some types of mutation can be detected many years after
the exposure. However, the persistence of these events cannot be
predicted with certainty and will vary with exposure duration,
agent, and other factors. Thus, for chromosomal aberration and
sister chromatid exchange measurements, samples should be taken as
soon as possible after exposure. For example, persons receiving
chemotherapy can have high frequencies of chromosomal aberration,
sister chromatid exchange (SCE), and gene mutation in the
lymphocytes. However, in the months following cessation of
treatment, the chromosomal aberration frequency decreases, often to
a level that may not be significantly different from that in the
controls. The frequency of SCE also diminishes in time. For gene
mutations, there may be a time interval between the mutational
event and the expression of resulting mutants. This interval will
vary from days to weeks and may vary with the markers. Thus,
following exposure, somatic mutant frequencies may remain the same,
decrease, or increase as a result of cell division, selection, or
migration in vivo. Similarly, persons therapeutically exposed to
ionizing radiation show elevated frequencies of chromosomal
aberration and gene mutation, but not SCE. In these patients, the
frequency of aberrations may remain elevated for many years after
exposure. At present, there are few data relevant to the
persistence of specific locus somatic mutants. It is recommended
that more than one end-point for somatic mutational change be
considered in any population study, because each end-point has
limitations with regard to the conclusions that may be drawn.
For example, negative findings for chromosomal aberrations do not
preclude the possibility of increases in somatic mutations of other
types. In a population presumably exposed to ionizing radiation,
chromosomal aberration studies are most appropriate, but SCE
could also be analysed as an indicator of exposure to confounding
chemical agents. Conversely, in a population exposed to chemicals,
SCE studies are more appropriate in terms of assay sensitivity, but
chromosomal aberration studies provide additional important
information.
Considerable effort can be expended in subject identification
and sample collection; therefore, sufficient blood should be
collected at one time to assay several somatic cell mutational end-
points. Even though it may be necessary to evaluate a number of
end-points in a particular study, it is worthwhile setting
priorities for their analysis. For example, slides could be
prepared for the determination of both chromosomal aberrations and
SCE, but only one end-point scored initially. This choice should
be based on the type of end-point most likely to be elevated. The
remaining slides can be scored later, according to the needs of the
study.
In addition, blood collected from mutagen-exposed and control
individuals should be sufficient to allow cryopreservation of
cells and other components for later study. This will make
more extensive evaluations possible as well as contribute to the
creation of a repository of material from mutagen-exposed human
beings for subsequent research.
3.2. Chromosomal End-Points
3.2.1. Structural aberrations
Chromosomal aberrations can be studied in any cycling cell
population, or in any non-cycling cell population that can be
stimulated by a mitogenic agent to enter the cell cycle. In
animals, there are several cell types that fit these criteria,
but for human studies, to all intents and purposes, there are only
two cell types that are practically suitable. These are the bone
marrow cells, which are a cycling population, and the peripheral
blood lymphocytes, which are normally non-cycling, but can be
stimulated to enter the cell cycle by in vitro culturing with a
mitogen such as phytohaemagglutinin (PHA). However, because of
the ease of obtaining blood samples, in contrast to bone marrow
samples, lymphocyte assays have been used in the majority of
studies on the induction of chromosomal aberrations in human
beings.
Since the observation by Moorehead et al. (1960) that
peripheral lymphocytes could be stimulated by PHA to enter the cell
cycle and be observed at metaphase, an enormous amount of data has
been obtained on the induction of chromosomal alterations by
radiation and chemical agents using this lymphocyte assay system
(Preston et al., 1981). It is often assumed that this is a simple
and informative assay for human population monitoring, providing
information on potential clastogenic (or mutagenic) exposures.
However, it should be emphasized that such a view should be
regarded with caution in the case of supposed chemical exposures,
and the obvious distinction from its use for radiation exposures
should be appreciated.
3.2.1.1. Methods of culture
There are numerous methods for culturing human lymphocytes.
Many components of the methods are optional, and depend on
individual preference. Those that are less variable are described
here. More complete methods can be found in Evans & O'Riordan
(1975), Bloom (1981), and Preston et al. (1981).
It appears to be advantageous to set up cultures from fresh
blood samples. However, this is not always possible, usually
because samples are taken at some distance from the laboratory and
have to be transported. It is still possible to achieve good
growth from samples taken several days before culturing. Although
samples can be maintained at 4 °C during shipping and storage, it
appears to be better to maintain them at room temperature whenever
possible (Dzik & Neckers, 1983).
Cultures can be established from whole blood (approximately
0.5 ml per culture), buffy coats, or purified lymphocytes.
However, if a large number of samples are to be handled at one
time, it is clearly advantageous to use whole blood. For buffy-
coat cultures, it is preferable to use the white-cell layer from
about 3 ml of centrifuged whole blood, thus larger total blood
samples are needed. One of the advantages of buffy-coat cultures
is that the cell fixation procedure is not hampered by the large
volume of red cells, present in whole-blood cultures, which are
often difficult to disperse. A possible disadvantage of whole-
blood cultures is that a small quantity of the agent under study,
or another contaminating agent, could be carried over with the
donor's serum. However, there seems little probability that this
would influence the results, and thus, it would not negate the use
of whole-blood cultures. It is also important to establish
cultures in duplicate, as a precautionary measure against culture
failure or sparseness of analysable cells.
A large number of different tissue culture media have been
successfully used, and the choice is a matter of personal
preference. This is also true for the serum type used, with the
added proviso that it should be virus-free. Several different
T-cell mitogens are also suitable, PHA being the most frequently
used. One additional point concerns the use of tissue culture
media containing a low concentration of folic acid. It has been
noted that the growth of human lymphocytes in such media (notably
TC 199) can result in the appearance of specific chromosome breaks
at so-called "fragile sites" (Jacky et al., 1983), or in an
increase in aberrations at possible fragile sites (Reidy et al.,
1983). It is recommended that, for population-monitoring studies,
low folate media should be avoided, though it is suggested that
parallel cultures with low folate media could still be used to
provide additional or different types of information.
While it is appropriate for each laboratory to use a medium and
serum that provides good lymphocyte growth conditions, it is most
important that each laboratory should determine the rate of
progression of mitogenically-stimulated cells through their first
and subsequent cell cycles with their specific culture conditions,
using blood samples from several individuals. The rate of cell
progression can be variable, depending on the mitogen, culture
medium, serum, and temperature (usually 37 °C in a 5% C02/95% air
environment).
The progression of cells can be measured by culturing in the
presence of bromodeoxyuridine (BrdU), sampling cells over a range
of times (for example, every 2 h, from 42 h to 54 h after mitogenic
stimulation), and staining the fixed preparations using a technique
for obtaining differentially-stained chromosomes (Goto et al.,
1978). In this way, the cells that have replicated their DNA once,
in the presence of BrdU, will contain evenly- and darkly-stained
chromatids. Those that have replicated twice, in the presence of
BrdU, will contain differentially-stained chromatids, one light
blue and one dark blue, when Giemsa stain is used. Cells that
progress through 3 or more cell cycles, in the presence of BrdU,
will contain some differentially-stained and some evenly but
lightly-stained chromosomes. In studies on chromosome aberrations,
it is important to analyse cells in their first metaphase after
mitogenic stimulation; thus, a fixation time should be chosen when
a high proportion of analysable cells are at the first division
stage. Generally, it is not feasible to use a fixation time when
all the cells are at this stage, because this requires very early
fixation (approximately 42 h after stimulation), when the number of
mitotic cells is too low. A compromise time (for example, 48 h) is
selected, when, for the average individual, about 90% of the cells
are at the first division stage. There is considerable variation
from individual to individual in the percentage of cells at the
first or subsequent division, even 48 h after culture initiation,
and it is good practice to check the proportion of first mitoses
in a series of cultures containing BrdU, separate from the series
established for aberration analysis. In this way, the possible
effects on aberration frequencies of analysing different
proportions of first division cells from different samples can be
ascertained. It is also possible to analyse chromosome aberrations
from cultures grown with BrdU, where preparations have been
differentially stained: cells showing no differentiation, and
cells that are clearly first metaphase (M1) can then be analysed.
The decision on selecting or rejecting samples with a high
proportion of 2nd divisions is optional, but should, at least, be
consistent. If resampling is possible, then this should be done
using an earlier fixation time than that for the first sample.
There is also a possibility that, at the standard fixation time for
any particular laboratory, there may be insufficient analysable
mitotic cells, as a result of an unusually long cell cycle, either
inherent or induced. The only solution to this problem is to
resample using a later fixation time.
3.2.1.2. Fixation and slide preparation
Many different methods are available for obtaining
conventional, banded or harlequin-stained metaphase preparations,
and essentially any one that produces well-spread complete
metaphases is acceptable (MacGregor & Varley, 1983). There is
little likelihood of this step of the assay influencing the
results.
3.2.1.3. Analysis of cells
There are many schemes available for the classification of
chromosome aberration types, and comprehensive descriptions can be
found in Savage (1975) and Bloom (1981). The following section
includes descriptions of the abberations most commonly observed in
control samples or samples from individuals exposed to radiation or
chemical agents.
(A) Chromosome-type aberrations
(1) Terminal and interstitial deletions
 It is not possible to distinguish between chromosome-type
terminal deletions and non-sister union isochromatid deletions
(Fig. 2D), and so, in cases of radiation exposure, when induced
aberrations are of the chromosome-type, it is appropriate to
classify all paired acentric fragments as terminal deletions
(Fig. 1A).
The small interstitial deletions appearing as paired dots are
classified as "minutes" (Fig. 1A). The larger interstitial
deletions in which there is a clear space in the centre of the ring
are usually classified as acentric rings. The distinction is not
particularly clear-cut, and, in general, is merely an indication of
the different sizes of interstitial deletions. Acentric fragments
associated with inter- or intrachanges are not classified as
terminal deletions.
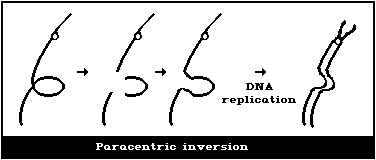
(2) Asymmetrical interchanges (usually dicentrics)
It is not possible to distinguish between chromosome-type
terminal deletions and non-sister union isochromatid deletions
(Fig. 2D), and so, in cases of radiation exposure, when induced
aberrations are of the chromosome-type, it is appropriate to
classify all paired acentric fragments as terminal deletions
(Fig. 1A).
The small interstitial deletions appearing as paired dots are
classified as "minutes" (Fig. 1A). The larger interstitial
deletions in which there is a clear space in the centre of the ring
are usually classified as acentric rings. The distinction is not
particularly clear-cut, and, in general, is merely an indication of
the different sizes of interstitial deletions. Acentric fragments
associated with inter- or intrachanges are not classified as
terminal deletions.
(2) Asymmetrical interchanges (usually dicentrics)
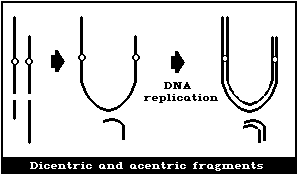
 It is assumed that a dicentric, analysed at the first in
vitro metaphase will be accompanied by an acentric fragment
(Fig. 1B) and a tricentric by 2 acentric fragments (Fig. 1C). A
cell with a dicentric and two acentric fragments is, by convention,
classified as a dicentric with its accompanying fragment and a
terminal deletion. The two acentric fragments could be the
result of incomplete rejoining during the formation of the
dicentric. However, although this cannot be ascertained, it has
been shown experimentally that its probability of occurrence is
rather low (< 10%) (Schmid & Bauchinger, 1980).
Asymmetrical interchanges, i.e., dicentrics, can be analysed
with greater efficiency than any other aberration type (> 95%),
and it is their frequency that is generally used for estimations of
radiation dose. For such determinations, a tricentric, for
example, is assumed to be equivalent to 2 dicentrics.
(3) Asymmetrical intrachange (centric ring)
It is assumed that a dicentric, analysed at the first in
vitro metaphase will be accompanied by an acentric fragment
(Fig. 1B) and a tricentric by 2 acentric fragments (Fig. 1C). A
cell with a dicentric and two acentric fragments is, by convention,
classified as a dicentric with its accompanying fragment and a
terminal deletion. The two acentric fragments could be the
result of incomplete rejoining during the formation of the
dicentric. However, although this cannot be ascertained, it has
been shown experimentally that its probability of occurrence is
rather low (< 10%) (Schmid & Bauchinger, 1980).
Asymmetrical interchanges, i.e., dicentrics, can be analysed
with greater efficiency than any other aberration type (> 95%),
and it is their frequency that is generally used for estimations of
radiation dose. For such determinations, a tricentric, for
example, is assumed to be equivalent to 2 dicentrics.
(3) Asymmetrical intrachange (centric ring)
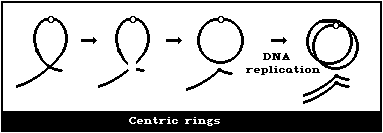 In asymmetrical intrachanges as in interchanges, a centric ring
is accompanied by an acentric fragment, and the same classification
scheme applies to these as for dicentrics ((2), above) (Fig. 1D).
(4) Symmetrical interchanges (reciprocal translocations)
In asymmetrical intrachanges as in interchanges, a centric ring
is accompanied by an acentric fragment, and the same classification
scheme applies to these as for dicentrics ((2), above) (Fig. 1D).
(4) Symmetrical interchanges (reciprocal translocations)
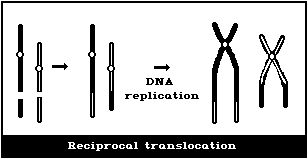 Symmetrical interchanges (Fig. 1E) are particularly difficult
to observe in conventionally-stained preparations, unless the
exchanged pieces produce 2 chromosomes, very different from the
normal karyotype. However, it is suggested that obvious
symmetrical interchanges should be recorded, but giving less weight
to their frequency than to that of most other aberration types.
It is not usually possible to observe these symmetrical
intrachanges (Figs. 1F, 1G) in conventionally-stained preparations,
unless a pericentric inversion produces a chromosome that is
distinctly different from the normal karyotype. Any obvious
symmetrical intrachanges should be recorded, but less weight given
to their frequency than to that of most other aberration types.
(5) Symmetrical intrachanges (peri- and paracentric
inversions)
Symmetrical interchanges (Fig. 1E) are particularly difficult
to observe in conventionally-stained preparations, unless the
exchanged pieces produce 2 chromosomes, very different from the
normal karyotype. However, it is suggested that obvious
symmetrical interchanges should be recorded, but giving less weight
to their frequency than to that of most other aberration types.
It is not usually possible to observe these symmetrical
intrachanges (Figs. 1F, 1G) in conventionally-stained preparations,
unless a pericentric inversion produces a chromosome that is
distinctly different from the normal karyotype. Any obvious
symmetrical intrachanges should be recorded, but less weight given
to their frequency than to that of most other aberration types.
(5) Symmetrical intrachanges (peri- and paracentric
inversions)
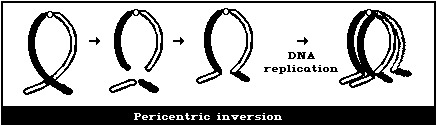
 (B) Chromatid-type aberrations
Chromatid-type aberrations are generally classified in the
same way as chromosome-type aberrations; the apparent unit of
involvement in a chromatid-type aberration is, in most cases, the
single chromatid, and not the whole chromosome, as seen for
chromosome-type aberrations.
(1) Terminal deletions
A terminal deletion is a distinct displacement of the chromatid
fragment distal to the lesion, or, if there is no displacement, the
width of the non-staining region between the centric and acentric
regions is greater than the width of a chromatid (Fig. 2A). The
latter definition is used to distinguish between terminal deletions
and achromatic lesions or "gaps" (section (2)).
(B) Chromatid-type aberrations
Chromatid-type aberrations are generally classified in the
same way as chromosome-type aberrations; the apparent unit of
involvement in a chromatid-type aberration is, in most cases, the
single chromatid, and not the whole chromosome, as seen for
chromosome-type aberrations.
(1) Terminal deletions
A terminal deletion is a distinct displacement of the chromatid
fragment distal to the lesion, or, if there is no displacement, the
width of the non-staining region between the centric and acentric
regions is greater than the width of a chromatid (Fig. 2A). The
latter definition is used to distinguish between terminal deletions
and achromatic lesions or "gaps" (section (2)).
 (2) Interstitial deletions
Chromatid-type interstitial deletions (Fig. 2B) are not as
readily observable as their chromosome-type counterpart, partly
because the small deleted fragment is often separated from the
deleted chromosome, and is not observed.
(2) Interstitial deletions
Chromatid-type interstitial deletions (Fig. 2B) are not as
readily observable as their chromosome-type counterpart, partly
because the small deleted fragment is often separated from the
deleted chromosome, and is not observed.
 (3) Achromatic lesions ("gaps")
Achromatic lesions or gaps are non-staining or very lightly
stained regions of chromosomes, present in one chromatid (single)
or, in both sister chromatids, at apparently identical loci
(double). If the non-staining region is of a width less than that
of a chromatid, the event is recorded as an achromatic lesion (Fig.
2C). This is clearly only a working definition. It is generally
suggested that achromatic lesions should be recorded, but always
separately from chromatid deletions. Their frequency should not
be included in the totals for aberrations per cell, since their
significance and relationship to other "true" aberration types is
not clear at present.
(3) Achromatic lesions ("gaps")
Achromatic lesions or gaps are non-staining or very lightly
stained regions of chromosomes, present in one chromatid (single)
or, in both sister chromatids, at apparently identical loci
(double). If the non-staining region is of a width less than that
of a chromatid, the event is recorded as an achromatic lesion (Fig.
2C). This is clearly only a working definition. It is generally
suggested that achromatic lesions should be recorded, but always
separately from chromatid deletions. Their frequency should not
be included in the totals for aberrations per cell, since their
significance and relationship to other "true" aberration types is
not clear at present.
 (4) Isochromatid deletions
Isochromatid deletions appear as exceptions to the class of
chromatid-type aberrations, since they involve both chromatids,
apparently with "breaks" at the same position on both. However, in
suitable material they can be shown to be induced by radiation in
the S and G2 phases of the cell cycle, as is the case for other
chromatid-type aberrations.
There are several possible types (Fig. 2D), depending on
the nature of the sister unions. If sister union occurs, it is
possible to distinguish isochromatid aberrations from chromosome-
type terminal deletions.
(4) Isochromatid deletions
Isochromatid deletions appear as exceptions to the class of
chromatid-type aberrations, since they involve both chromatids,
apparently with "breaks" at the same position on both. However, in
suitable material they can be shown to be induced by radiation in
the S and G2 phases of the cell cycle, as is the case for other
chromatid-type aberrations.
There are several possible types (Fig. 2D), depending on
the nature of the sister unions. If sister union occurs, it is
possible to distinguish isochromatid aberrations from chromosome-
type terminal deletions.
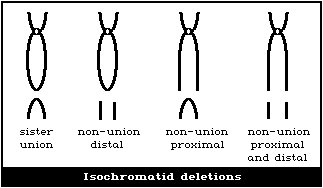 In mammalian cells, however, sister union is a rare event, and most
of the isochromatid deletions are of the non-union proximal and
distal type. The acentric fragment is most often not associated
with the deleted centric part of the chromosome. The convention
for analysis (discussed for chromosome-type terminal deletions,
above) is that, for radiation exposures, paired acentric fragments
are recorded as chromosome-type terminal deletions, but, for
chemical exposures or control samples, they are most appropriately
recorded as isochromatid deletions.
(5) Asymmetrical interchanges (interarm interchanges,
asymmetrical chromatid exchanges)
Asymmetrical interchanges are the chromatid-type equivalent to
chromosome-type dicentrics (Fig. 2E).
In mammalian cells, however, sister union is a rare event, and most
of the isochromatid deletions are of the non-union proximal and
distal type. The acentric fragment is most often not associated
with the deleted centric part of the chromosome. The convention
for analysis (discussed for chromosome-type terminal deletions,
above) is that, for radiation exposures, paired acentric fragments
are recorded as chromosome-type terminal deletions, but, for
chemical exposures or control samples, they are most appropriately
recorded as isochromatid deletions.
(5) Asymmetrical interchanges (interarm interchanges,
asymmetrical chromatid exchanges)
Asymmetrical interchanges are the chromatid-type equivalent to
chromosome-type dicentrics (Fig. 2E).
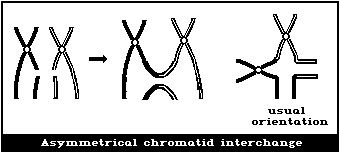 (6) Symmetrical interchanges (symmetrical chromatid
exchanges)
Symmetrical interchanges are the chromatid-type equivalent of
chromosome-type reciprocal translocations. In the case of
chromatid-type symmetrical exchanges, somatic pairing maintains an
association between the chromosomes involved in the exchange, and
thus they can be readily observed in the absence of any chromosome-
banding procedures (Fig. 2F).
(6) Symmetrical interchanges (symmetrical chromatid
exchanges)
Symmetrical interchanges are the chromatid-type equivalent of
chromosome-type reciprocal translocations. In the case of
chromatid-type symmetrical exchanges, somatic pairing maintains an
association between the chromosomes involved in the exchange, and
thus they can be readily observed in the absence of any chromosome-
banding procedures (Fig. 2F).
 (7) Asymmetrical and symmetrical intrachanges (interarm
intrachanges)
There are 2 forms of symmetrical and asymmetrical interarm
intfachanges but, when analysing metaphase cells, only one of each
is distinguishable (Fig. 2G). The symmetrical intrachange can be
observed in somatic pairing.
(7) Asymmetrical and symmetrical intrachanges (interarm
intrachanges)
There are 2 forms of symmetrical and asymmetrical interarm
intfachanges but, when analysing metaphase cells, only one of each
is distinguishable (Fig. 2G). The symmetrical intrachange can be
observed in somatic pairing.
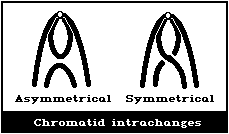 (8) Triradials
A triradial (3-armed configuration) can be described as the
interaction between one chromosome having an isochromatid deletion
and a second having a chromatid deletion. Many types of triradials
can be formed but, in mammalian cells, where the frequency of
sister union is low, the 2 illustrated are the most common (Fig.
2H).
(8) Triradials
A triradial (3-armed configuration) can be described as the
interaction between one chromosome having an isochromatid deletion
and a second having a chromatid deletion. Many types of triradials
can be formed but, in mammalian cells, where the frequency of
sister union is low, the 2 illustrated are the most common (Fig.
2H).
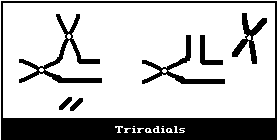 It is important to note that the different types of chromosome
aberration should be recorded separately. The results for any
sample should be presented as the aberration frequency per cell
for each aberration type, or frequency of aberrant cells for each
aberration type. It is not appropriate to express the results as
total aberrant cells or total breaks per cell, as this method of
presentation can hide important facets of the data. For the
statistical analysis of data, it is legitimate to combine
aberration classes into the general categories of chromatid-type
deletions, chromosome-type deletions, chromatid exchanges, and
chromosome exchanges. Occasionally, cells are seen with multiple
aberrations, and an attempt should be made to analyse such cells in
detail, rather than to record them simply in a category "multiple
aberration", since this classification will mean a loss of
information. If it proves impossible to analyse a cell, it should
be recorded in a category of "too many aberrations to analyse", and
not included in the total of cells scored.
The selection of cells to be analysed is particularly
important, to avoid observer bias. Such bias can result in either
a higher or lower measure of the unbiased aberration frequency,
depending on the nature of the selection. The generally accepted
method of cell selection is to scan slides at low magnification;
cells that appear to be suitable are then analysed at high
magnification. Once a cell is observed at high magnification,
it should only be rejected if: (a) the cell cannot be analysed
because of the number or complexity of aberrations; in this case,
it should be appropriately recorded, but not included in total
cells analysed; (b) the cell is not sufficiently well spread
when seen at high magnification, and the overlap of chromosomes
prohibits an accurate analysis; (c) non-chromosomal material, such
as dirt or stain crystals, not discernible at low magnification,
prevent complete analysis; (d) the cell contains fewer centromeres
than the observer's acceptable cut-off point; this may be the
analysis of cells with 46 ± 2 centromeres or only cells with 46
centromeres, either cut-off point is acceptable, provided that the
criterion is consistently applied.
It is particularly important in population-monitoring studies
for the samples to be analysed under code, and for the slides from
exposed and control individuals to be randomized in such a way that
the observer is unaware of the identity of the sample. There are
strong arguments in favour of more than one laboratory, with at
least 2 individuals in each laboratory, being responsible for the
analysis, in order to remove possible laboratory or individual
scoring bias, which could influence the results. Microscope
co-ordinates of every suspected aberrant cell should be recorded.
In order to provide further consistency, the classification of any
cell as aberrant by one individual should be confirmed by a second,
and it is advantageous to keep a photographic record of all
confirmed aberrant or otherwise suspect cells.
The number of cells to be analysed from any sample cannot be
fixed with any certainty, because it can be argued that this number
depends on the aberration frequency, and thus is retrospective. A
general suggestion would be to analyse 200 cells per sample.
The pros and cons of analysing banded preparations have been
discussed frequently, and do not warrant repetition. The amount of
time expended on the preparation and analysis of banded samples
does not seem to be justified by the increase in sensitivity of
analysis for aberration types detectable by banding. However, the
choice of scoring conventially-stained or banded preparations
should be left to the discretion of each laboratory.
3.2.1.4. Data analysis
Many statistical methods are available for the analysis of
data obtained from human population-monitoring studies. The
appropriateness of any particular method will depend, to some
extent, on the design of the study and the nature of the data
collected. Two examples of general approaches are given in Brewen
& Gengozian (1971) and Evans & O'Riordan (1975).
3.2.1.5. Radiation-induced aberrations and estimates of exposure
For a variety of reasons, it is sensible to discuss the use of
the lymphocyte assay for estimating exposure to ionizing radiation
separately from cases of chemical exposure. In some ways, it was
fortunate, but, in retrospect, perhaps also unfortunate, that all
the original studies on the analysis of chromosomal aberrations in
the lymphocytes of exposed or possibly-exposed persons applied to
radiation exposure. The fact that, in these cases, estimates of
exposure could be made with some accuracy has often led to the
assumption that similar exposure-estimating procedures could be
applied to persons, groups, or populations exposed environmentally
or occupationally to chemical agents. A clear note of caution in
accepting this assumption is made here, and will be discussed
further.
Ionizing radiation and a small number of chemical agents (e.g.,
streptonigrin, bleomycin, neocarzinosatin, cytosine arabinoside,
and 8-methoxycaffeine) are able to produce aberrations in all
stages of the cell cycle including chromosome-type aberrations in
G1 and chromatid-type aberrations in S and G2.
The peripheral lymphocytes, a subpopulation of which is
mitogenically stimulated when blood samples are placed in culture,
are essentially non-cycling, and are in the G0 stage of the cell
cycle, the term usually applied to non-cycling G1 cells. Thus,
following radiation exposure, chromosome-type aberrations will be
induced in these non-cycling G1 lymphocytes, and can be observed at
the first metaphase following mitogenic stimulation in culture.
The fact that aberrations can be produced in G1 cells means that
their frequency can be directly related to the dose received (i.e.,
aberration frequency is proportional to dose). In addition, it has
been shown for many species (Brewen & Gengozian, 1971; Preston et
al., 1972; Clemenger & Scott, 1973), including man (Brewen et al.,
1972), that the frequency of aberrations induced in vitro is the
same as the frequency induced by the same dose delivered in vivo.
This means that a standard dose-response curve, for any particular
radiation type, can be obtained for in vitro exposures, and can
then be used for estimating doses received by individuals as a
result of radiation accidents during medical or other environmental
exposures. The frequency of aberrations (usually dicentrics) is
measured in cultured blood samples from exposed individuals, and
then converted into a dose estimate from the standard dose-response
curve. There is extensive literature on the use of the lymphocyte
assay as a biological measurement of dose for many different types
of radiation (Bender & Gooch, 1966; Lloyd et al., 1983). However,
there are still some limiting factors in the use of the assay.
These include the length of time of blood sampling after exposure,
partial-body exposure, and non-homogeneous exposures.
It is also apparent that the lymphocyte assay can be used for
estimating doses following long-term radiation exposure (Evans et
al., 1979). Again, this is possible because the aberrations are
induced in G0 cells, and their frequency will be directly related
to exposure. Cells containing radiation-induced aberrations as
well as normal cells, will gradually be lost from the peripheral
lymphocyte pool, as a result of lymphocyte turnover. However, the
cells repopulating the peripheral pool will be derived mainly from
normal precursor cells, and not from those containing aberrations.
Many chromosome-type aberrations are lethal to the cell as the
result of loss of acentric fragments at division, or because of the
mechanical interference of the aberration with division. Thus,
during long-term exposures (months or years), the aberration
frequency will not be additive with time (or dose), but will reach
an equilibrium where new aberrations are formed and existing ones
are lost from the sampled population.
A similar argument holds for determining the length of time
after short-term exposure when samples can provide a measure of
the maximum aberration frequency or true induced frequency.
The lymphocyte turnover will result in loss of cells from the
peripheral pool, both normal and abnormal, and repopulation largely
by normal precursor cells. The aberration frequency appears to
stay constant for about 6 weeks after exposure (Brewen et al.,
1972).
Samples have been taken many years after exposure, for
example, with radiation-treated ankylosing spondylitis patients
(Buckton et al., 1978) and human beings exposed to the atomic bombs
in Hiroshima and Nagasaki (Awa et al., 1978). The fact that some
lymphocytes are very long-lived, in excess of 20 years (Awa et
al., 1978; Buckton et al., 1978), means that radiation-induced
aberrations can still be observed in cells that were present as
peripheral lymphocytes at the time of exposure. If it is assumed
that the aberration frequency declines exponentially with time, an
approximate estimate of the dose received can be made; the word
approximate is emphasized.
3.2.1.6. Chemically-induced aberrations and estimates of exposure
On the basis of the success of the lymphocyte assay for
estimating radiation exposures, it seemed appropriate to analyse
chromosome aberrations in blood samples from individuals,
occupationally exposed to single chemicals or complex mixtures
(Office of Technology Assessment, US Congress, 1983), to determine
whether there had been exposure to a clastogen, and to relate
aberration frequencies to dose level. However, the ability to
estimate dose levels from aberrations in populations exposed to
chemical mutagens is not reliable at present. More research is
necessary to make the assay more reliable and applicable, and to
make the results more readily interpretable.
The present low sensitivity of the assay for measuring the
frequency of chromosome aberrations following chemical exposure is
due to the mechanisms of induction of aberrations by chemical
agents. These mechanisms may also result in induced aberration
frequencies being only indirectly related to exposure dose, in
contrast to radiation-induced aberrations.
Chromosome aberrations induced by chemical treatments are
almost exclusively produced during the S-phase, irrespective of the
cell-cycle stage treated. Thus, the majority of aberrations will
be of the chromatid-type, although there are exceptions to this
general hypothesis (Evans & Vijayalaxmi, 1980; Preston & Gooch,
1981). Therefore, it is more appropriate to consider that the
probability of producing aberrations in G1 or G2 cells following
chemical treatments is low, but is considerably increased when
treated cells are in, or pass through, the S-phase.
Chemically-induced DNA damage in non-cycling lymphocytes will
not generally be converted into aberrations until the cells are
stimulated to re-enter the cell cycle in vitro, and undergo DNA
replication. Since repair of DNA damage can take place in G0 cells
as well as during the long first in vitro G1 stage, the aberration
frequency will not necessarily be proportional to the amount of
induced DNA damage but rather to the amount of DNA damage remaining
at the time of DNA replication. It is clear that the amount of DNA
damage, present at the time of replication, which has the potential
to be converted into aberrations, will depend on several factors
including: (a) the dose received; (b) the induced amount of the
particular types of DNA damage that can result in aberrations (a
value that can vary with the agent); (c) the amount of DNA repair
in G0 cells before sampling (i.e., time between exposure and
sampling); and (d) the amount of repair in G1 cells from mitogenic
stimulation to the first in vitro S-phase. Many of these will also
be subject to individual variation. The outcome is that, for most
chemical agents, only a proportion of induced DNA damage can be
converted into aberrations, at the time of replication. These DNA
repair factors all work to reduce the sensitivity of the lymphocyte
assay for measuring chemical exposure, and will result in the
aberration frequency being, at best, only indirectly proportional
to exposure.
If an increase in chromosome aberrations is observed in a
potentially exposed group when compared with a matched control
group, it is reasonable to conclude that there has been exposure to
a clastogenic agent, but no estimate of exposure level or of
subsequent adverse health effects (genetic or somatic) can be made.
On the other hand, if there is no difference in aberration
frequency between the possibly-exposed group and a matched control
group, it is not possible to rule out an exposure. However, in
this case, it may be possible, on the basis of previous experience,
to deduce a maximum exposure level that is not associated with a
detectable difference in aberration frequency.
3.2.1.7. Conclusions
Future research should enhance the sensitivity of the assay
system. In the meantime, it is suggested that care be taken in
the choice of groups to be considered for monitoring studies, with
particular regard to the usefulness of the information that might
be obtained, and to the likelihood of being able to draw definitive
conclusions. Cytogenetic monitoring can be an appropriate method
for the detection of chemical exposure, but, clearly, it cannot, at
present, be considered universally decisive. It should be used in
conjunction with other end-points.
3.2.2. Sister-chromatid exchange
3.2.2.1. Formation of sister-chromatid exchange
SCE results from the breakage and rejoining of DNA at
apparently homologous sites on the 2 chromatids of a single
chromosome. They were first visualized by Taylor et al. (1957)
who used tritiated thymidine to differentially label the DNA of
replicating cells and autoradiography to distinguish the silver
grain pattern on the 2 sister chromatids. Advances in
cytochemistry over the subsequent 15 years led to greatly
simplified procedures for visualizing these exchanges. Latt
(1973) demonstrated that bromodeoxy-uridine (BrdU), an analogue
of thymidine, when incorporated into DNA, could quench the
fluorescence of the fluorochrome Hoechst 33258. Perry & Wolff
(1974) found that incorporated BrdU also diminished the uptake
of Giemsa stain into the chromatin. In principle, SCEs can be
observed in any cell that has completed two, or the first of two,
replication cycles in the presence of BrdU. The most common
procedure for human lymphocytes is to grow the cells in BrdU for
two replication cycles.
SCEs are most efficiently induced by substances that form
covalent adducts with the DNA, or interfere with DNA precursor
metabolism or repair (Perry & Evans, 1975; Wolff, 1977; Perry,
1980; Carrano & Thompson, 1981; Latt, 1981). It is well documented
that SCEs are produced during DNA replication (Wolff et al., 1974)
and that the polarity of DNA is maintained in the process of
exchange (Taylor, 1958; Wolff & Perry, 1975). The molecular
mechanism by which the exchange is formed is not known but, since
there is an absolute requirement for DNA synthesis, hypotheses have
implicated the mechanics of this process in SCE formation (Bender
et al., 1974; Painter, 1980; Cleaver, 1981, Shafer, 1982).
Baseline SCE frequencies are increased in some human diseases
such as Bloom's syndrome (Chaganti et al., 1974) and multiple
sclerosis (Sutherland et al., 1980; Vijayalaxmi et al., 1983a).
Cells from patients with the inherited disease Xeroderma
pigmentosum are hypersensitive to the induction of SCEs by
ultraviolet radiation and alkylating agents (Wolff et al., 1975,
Wolff, 1977). There is no clear relation, however, between the
repair capacity of these cells and their sensitivity to SCE
induction (de Weerd-Kastelein et al., 1977).
3.2.2.2. Relevance of sister-chromatid exchange
An SCE represents the breakage of 4 strands of DNA (2 double
helices), a switch of the strands from one to the other arm of the
same chromosome, and the rejoining of the strands in their new
locations. The question is whether the breakage and rejoining
events occur without producing any modification in the genetic
code. There is evidence suggesting that this exchange process
might not always be error-free. In particular, a considerable
amount of information has been gathered concerning the relationship
between induced SCEs and other genetic effects in vitro and in
vivo.
Because the technique for enumerating SCEs is relatively
simple, this assay has been used quite extensively in genetic
toxicology studies. It has been amply demonstrated that the
frequency of SCE is dramatically increased when cells, or
animals, including human beings, are exposed to known mutagens
and carcinogens (Perry & Evans, 1975; Latt et al., 1981; Lambert
et al., 1982). Moreover, in early experiments using Chinese
hamster cells in culture, it was found that the induction of SCEs
was linearly related to the increase in single-gene mutations (HPRT
locus), when the cells were exposed to several substances each of
which differed in the type of lesion produced in DNA (Carrano et
al., 1978; Carrano & Thompson, 1982). The ratio of induced SCE to
induced mutation differed for each of the agents, suggesting that
each type of lesion formed is processed differently by the cell
favouring either the formation of SCEs or mutations. It is
possible that the same lesion is capable of producing both an SCE
and a mutation and/or that the lesions that form mutations are a
subset of those that form SCEs. The induction of mutations has
been shown recently to be correlated with the induction of SCEs in
mice for one chemical, ethyl nitrosourea (Jones et al., in press).
In this study, the induction of SCEs in mouse splenocytes,
following intraperitoneal administration of the chemical, was
linearly proportional to the increase in mutations at the HPRT
locus in the same splenocyte population, suggesting that the
mutation-SCE relation is not unique to in vitro studies.
Other investigations compared the induction of SCEs to either
the induction of transformation in vitro or induction of tumours
in vivo. In a series of experiments, the induction of SCEs was
shown to be linearly and positively correlated with the induction
of transformation of Syrian hamster embryo cells in vitro (Popescu
et al., 1981). This relationship held for exposure to 5 chemicals,
but not for X-irradiation. The ratio of induced SCEs to induced
transformation frequency varied for each chemical. This is
analogous with the results found for the SCE-to-mutation ratio
described above. Cheng et al. (1981) examined the induction of
SCEs in mouse bone marrow, regenerating liver, and alveolar
macrophages following intraperitoneal injection of ethyl carbamate
and related chemicals. The relative potencies in inducing SCE
parallelled the reported activities for the induction of lung
adenomas in mice. For ethyl carbamate, the doses for the
significant induction of SCEs in alveolar macrophages and lung
adenomas were similar. Further, the slopes of the dose responses
showed that the two end-points were of comparable sensitivity.
The results of early studies on rabbits demonstrated that it
was possible to measure an increase in SCEs in peripheral blood
lymphocytes for many months following repeated low-level exposure
to mitomycin C in vivo (Stetka et al., 1978). Increased SCEs in
human beings have been observed in cigarette smokers (Lambert
et al., 1978; Carrano, 1982) and in individuals undergoing
chemotherapy (Lambert et al., 1978; Musilova et al., 1979; Gillian
Clare et al., 1982). The results of these studies suggest that a
subpopulation of lymphocytes with high SCE frequencies may be
present for long periods after termination of exposure. These
cells may represent persistent damage in long-lived lymphocytes
or a sensitive subpopulation (Carrano & Moore, 1982). If this
is true, they may offer a more appropriate approach to the
quantification of persistent damage in vivo.
3.2.2.3. Factors potentially influencing SCE frequency
The baseline SCE frequency in human peripheral lymphocytes
averages about 7 - 10 per cell, but has been reported to range from
about 2 to 45 per cell in unexposed individuals (Alhadeff & Cohen,
1976; Lambert et al., 1976; Crossen et al., 1977; Galloway, 1977;
Carrano et al., 1980). Even within the same laboratory, the
baseline frequency may vary by a factor of two or more. Several
potential sources of variation have been identified and they
generally fall into 2 categories: a) culture factors associated
with the in vitro growth of the lymphocytes; and b) biological
factors associated with the genotype, lifestyle, or general health
of the individual. The biological factors usually cannot be
mitigated and must be accounted for in the selection of appropriate
control cohorts or ultimately in the statistical analysis.
(a) Culture-associated factors
A major source of variation can be attributed to the
concentration of BrdU relative to the number of lymphocytes in the
culture. The frequency of SCE has been shown to increase by as
much as 50%, when the BrdU concentration is raised 10-fold (Carrano
et al., 1980). The ability of BrdU to induce SCE has been well
documented in human and other cell systems (Wolff & Perry, 1974;
Lambert et al., 1976; Latt & Juergens, 1977; Mazrimas & Stetka,
1978) and it has been further shown that the SCE frequency can
increase if the BrdU concentration is held constant, but the
cell number decreased (Stetka & Carrano, 1977). The number of
lymphocytes that respond to mitogen and eventually incorporate
the analogue is generally not controllable, and, therefore, the
effect of BrdU can best be minimized by standardizing both the
concentration of BrdU and white blood cells at culture initiation.
Since the SCE increase above baseline following most long-term
exposures may be small, careful consideration of the influence of
BrdU is warranted.
Other culture-associated factors have been examined to
determine whether they influence the SCE frequency. One concern
has been the optimal time to harvest cells. In at least three
studies, it has been concluded that the baseline SCE frequency
does not depend on the time of harvest. Carrano et al. (1980)
found that, for any individual, the SCE frequency could vary by
as much as 30% as a function of culture time, but that the pattern
was not consistent among donors. Similarly, Beek & Obe (1979),
Becher et al. (1979), and Morimoto & Wolff (1980) did not observe
any significant alteration in the baseline SCE frequency as a
function of culture time. In a study by Snope & Rary (1979), for
4 individuals, the baseline SCE frequency observed in 58-h cultures
was about 30% less than in 70-h cultures. The authors interpreted
this to indicate that there were at least 2 subpopulations of
lymphocytes that differed in SCE frequency. Studies by Lindblad &
Lambert (1981) and Das & Sharma (1984) also revealed that the more
slowly cycling lymphocytes had a higher SCE frequency. The
possibility that this effect might be due to T- and B-lymphocyte
subpopulations has not been completely resolved. Santesson et al.
(1979) found that B-lymphocytes had a significantly lower SCE
frequency than T-lymphocytes, but Lindblad & Lambert (1981) could
not confirm this. In order to optimize an experimental design to
account for such an effect, all samples should, where possible, be
harvested at the same time.
Two other factors have been shown to influence the baseline SCE
frequency, i.e., the serum used and the culture temperature. Using
sera from 4 different sources, McFee & Sherrill (1981) showed that
the SCE frequency could increase by 50% as the serum concentration
increased. Kato & Sandberg (1977) also demonstrated an effect of
sera on the production of SCEs. The frequency of SCE in amphibian
cells increased almost 5-fold with increasing culture temperature
(Speit, 1980). In Chinese hamster cells, both lower (33 °C) and
higher (40 °C) temperatures increased the frequency of SCE by
approximately 60%. Because the alteration in temperature was
only effective in altering SCE frequencies while BrdU was being
incorporated, the author concluded that SCEs might arise via a
temperature-dependent disturbance of DNA replication. In a study
by Das & Sharma (1984), the frequency of SCEs in human lymphocytes
increased as a function of culture temperature and reached a
maximum at 40 °C. The authors concluded that temperature-dependent
DNA replication enzymes might be responsible for this effect.
In many human population studies, the cohort to be sampled
may be far away from the laboratory performing the cytogenetic
analysis, and it will often be necessary to ship the samples.
If blood is shipped commercially, recording thermometers in the
shipping container will provide some quality control. The effect
of blood storage on SCE frequency has been examined (Carrano et
al., 1980). No consistent alteration in SCE frequency was observed
for blood held at either 4° or 22 °C for up to 48 h. This was
confirmed by Sharma & Das (1984) for blood stored at 5 °C for up to
7 days, at 22 °C for 3 days, and at 37 °C for 2 days. However, it
should be noted that such storage can lead to an overall decrease
in lymphocyte numbers and modification of lymphocyte subclasses
(Dzik & Neckers, 1983). Thus, the samples should be placed in
culture as quickly as possible.
Finally, Deknudt & Kamra (1983) evaluated 4 different
mitogens for their effects on SCE frequency. They found that
PHA, concanavalin A (Con A), Wistaria floribunda (WFA), and lentil
lectin (LcH-A) extracts did not result in different baseline SCE
frequencies in the same donors. However, significant differences
among mitogens were noted for lymphocytes that had been exposed
to cyclophosphamide or mitomycin C. The use of a single T-cell
mitogen, such as PHA, for all studies is therefore recommended.
The experience accumulated from the use of lymphocyte cultures
suggests that there are many culture-associated factors that could
influence the SCE frequency. In order to minimize variation and
maximize sensitivity, the use of standardized procedures is
essential.
(b) Biological factors
Factors, unique to the individual being sampled, include such
confounding variables as sex, age, diet, genotype, medication, and
smoking. Each of these potentially plays a role in the induction
or expression of SCEs. Although some information is available on
the influence of these factors, the consequences are not always
clear-cut. Moreover, it is likely that they represent only a small
number of possible confounding factors.
Several investigators have compared the baseline frequency of
SCEs in male versus female donors and have not observed significant
differences (Galloway & Evans, 1975; Alhadeff & Cohen, 1976;
Crossen et al., 1977; Latt & Juergens, 1977; Morgan & Crossen,
1977; Cheng et al., 1979; de Arce, 1981; Waksvik et al., 1981;
Carrano & Moore, 1982; Livingston et al., 1983). The absence
of clear documentation of a sex difference for baseline SCE
frequencies suggests that this factor may not have to be matched in
a population study. However, that there is evidence for increased
SCE frequencies in women taking oral contraceptives (see below).
No studies relating SCEs to cyclic hormonal variation in the female
have been reported.
Another variable of concern in population studies is the age of
the donor, since it is not always possible to obtain accurate age
matching. The independence of age and baseline SCE frequency in
lymphocytes in adults has been observed (Galloway & Evans, 1975;
Morgan & Crossen, 1977; Hollander et al., 1978; Cheng et al., 1979;
Lambert & Lindblad, 1980; Carrano & Moore, 1982; Livingston et al.,
1983). Lower SCE frequencies have been found in infants (Seshadri
et al., 1982), in cord blood (Ardito et al., 1980), and in children
with a mean age of 1.5 years (Husgafvel-Pursiainen et al., 1980).
De Arce (1981) reported that people in the age range of 30 - 40
years had a higher number of SCEs per cell than those between the
ages of 0 - 10 or 60 - 70 years. Schmidt & Sanger (1981) found a
significant increase in SCE with age when comparing age groups of
1 - 18 months, 10 - 13 years, 26 - 32 years, and 63 - 85 years.
However, this might be due to differential BrdU incorporation
between cultures. With the exception of lower SCE frequencies in
new-born infants, the weight of evidence suggests that baseline SCE
frequencies do not change with age (at least up to 60 years).
It has been suggested that genetic factors play a role in the
baseline SCE frequency. When Cohen et al. (1982) compared SCE
frequencies, both within and among 12 pedigrees (2 generations),
they found significant differences between, but not within, the
families, suggesting a genetic contribution to baseline SCEs.
Pedersen et al. (1979) examined the cultured lymphocytes from 11
monozygotic and 9 dizygotic twins of the same sex. They did not
find any significant differences in the SCE frequency in
monozygotic versus dizygotic twins and concluded that genetic
factors do not play a role in baseline SCE frequency. Waksvik et
al. (1981) also did not find any genetic contribution to SCE
frequencies in their studies on twins. In order to determine
whether baseline SCE frequencies were related to ethnic origin,
SCEs were measured in adults of both sexes with no interracial
family backgrounds from Caucasian, American black, oriental, and
native American races (Butler, 1981). There was no significant
difference in the average frequency of SCEs in the 4 races.
Further research is needed to resolve the issue of genetic factors.
The potential influence of other factors on the frequencies of
human SCEs are worth considering. Because a large number of women
use oral contraceptives, care must be taken that this does not
confound the consequences of exposure to other agents. A study of
oral contraceptive users demonstrated a significant increase in
lymphocyte SCE associated with the daily intake of d-norgestrel or
ethenyl estradiol over a period of 6 - 24 months (Balakrishna
Murthy & Prema, 1979). Additional studies are needed to confirm
and extend these findings.
Despite knowledge of natural mutagens in food and the role of
food processing in creating mutagens (Nagao & Sugimura, 1981; Ames,
1983), little attention has been given to the role of diet in
causing baseline SCE variation. It has been reported that severe
protein calorie malnutrition in children is associated with
increased lymphocyte SCE frequencies (Balakrishna Murthy et al.,
1980). Following nutritional rehabilitation, SCE frequencies were
seen to decrease. Increased SCEs have also been reported in
chronic alcoholics (Butler & Sanger, 1981). For most human
studies, however, the nutrition factors are more likely to involve
long-term, low-level intake of caffeine, saccharin, other
additives, or well-cooked foods. Each of these factors has been
shown to be genotoxic in microbial, mammalian in vitro, or animal
assays, but there are no data on how these might influence SCE
frequency in human beings. Dietary information should be collected
in the initial questionnaire. Such information may be useful in
interpreting a population difference or can be used in large
composite data bases to search for diet-related factors. The use
of prescription and non-prescription medication, or vitamins,
should also be identified and appropriate information should be
collected before blood sampling, so that matched controls can be
employed.
Cigarette smoking is, perhaps, the most important confounding
factor in the interpretation of human SCE frequencies. The
preponderance of evidence indicates that individuals who smoke
have elevated SCE frequencies (Lambert et al., 1978; Ardito et al.,
1980; Hopkin & Evans, 1980; Lambert & Lindblad, 1980; Carrano,
1982; Husum et al., 1982; Livingston et al., 1983). However, this
effect has not been observed in all studies (Hollander et al.,
1978; Hedner et al., 1983). Since the elevation can be so marked,
it is important to control for cigarette smoking in any population
study.
Taken together, the culture and biological factors that
potentially confound the baseline SCE frequency in human beings
are numerous and contribute to individual variation.
3.2.2.4. Methods for sister-chromatid exchange analysis
The procedures for SCE analysis in human populations will
depend, to some extent, on the specific circumstances of the study.
The following generalized outline of a procedure may have to be
modified to fit special situations.
In order to identify any of the factors that may confound the
analysis of SCEs, it is important to obtain relevant information on
the background of each individual in the study population. This
information is best obtained through a questionnaire. The
questionnaire should attempt to determine the obvious exposure and
potential confounding factors. In addition to the questions
related to personal and family cancer incidence, birth defects,
known genetic disorders, and current health, there are specific
questions related to the interpretation of the SCE results. These
include: a) smoking history; b) medication and drug use; c) known
or suspected exposures to physical or chemical agents occurring
at work, at home, or during the pursuit of various hobbies or
recreation; and d) nutrition, particularly the adherence to special
diets or the use of artificial sweeteners or caffeine. Because a
complete list of agents that may directly or indirectly affect SCE
frequencies has not been established, it is better to collect too
much information than too little.
When the blood is collected, it should be mixed with heparin,
acid-citrate-dextrose, or another appropriate anticoagulant. As
already stated, the cultures should be established as soon as
possible after the blood is withdrawn, preferably within 24 h.
Just prior to establishing the culture, a total white blood count
and a slide for a differential should be made from the blood
sample. These parameters can serve as indicators of abnormally
high or low lymphocyte counts, factors that can have a bearing on
the observed frequency of SCEs.
It is important that a standard procedure be employed and,
although the exact procedure may differ among laboratories, there
are some common requirements. Because BrdU can induce SCEs in
human lymphocytes, both the BrdU concentration and the number
of lymphocytes potentially incorporating the drug, should be
stabilized. In order to ensure this, it is important that each
laboratory uses a fixed BrdU concentration and a consistent number
of lymphocytes per culture. Selection of culture medium, serum
type and concentration, whole blood versus purified lymphocyte
cultures, and duration of cultures is somewhat arbitrary and should
be based on experience. The serum lot should be pre-tested for its
potential to support lymphocyte growth and the reproducibility of
SCE frequencies in duplicate samples from the same donor. It is
recommended that the same serum lot be used for each population
study. The duration of culture should be such that it yields a
high proportion of second division cells so that the SCE frequency
is representative of a major fraction of the lymphocytes. Exposure
of the cultures to other than yellow or red light should be
avoided. Replicate cultures should be established for each
individual, and, when a large number of individuals is processed,
the inclusion of blood cultures from historical controls is useful.
At the appropriate culture time, the cells can be arrested in
mitosis with Colcemid(R), colchicine, or other suitable agents.
Several slides should be prepared from each culture and the
remaining cell pellet can be stored in absolute methanol:glacial
acetic acid (3:1) at a temperature of 4 °C or lower, for future
recall. The slides can be stained for analysis by fluorescence
alone (Latt, 1973), fluorescence plus Giemsa (Perry & Wolff, 1974),
or a suitable alternative. Once a staining procedure has been
standardized and adopted, it should be used throughout the whole
course of the study.
Slides should be coded and scored blind. The coded slides can
be scanned under low magnification (100 - 200X) and selected for
scoring on the basis of good staining and chromosome number. Only
well-differentiated metaphases should be accepted for scoring. It
is not essential for all the chromosomes in a cell to be counted.
Overlapping chromosomes can be disregarded and the remaining
chromosomes scored. At least 43 chromosomes should be analysed per
cell. The SCEs are counted for each chromosome in the cell and
expressed as SCE per chromosome or converted to the number of SCEs
per diploid cell. Although the precise number of cells to be
scored per individual will ultimately depend on the desired
statistical sensitivity, a minimum of 50 cells should be scored,
with 25 cells from each replicate culture.
3.2.2.5. Data processing and presentation
The standard statistical descriptors such as mean SCE per cell
or per chromosome, standard deviation or standard error, and number
of cells scored should be determined for each individual. For
heteroploid cells, the SCE frequency should be normalized to a
diploid cell. Population means and their errors should be
presented similarly and information on the number of cells scored
per sample as well as the number of people per group should be
given.
Student's t-test is commonly used for the statistical analysis
of population data. When comparing population groups, however, the
sample size is the number of people in each group, not the number
of cells scored for each group. Caution is advised with the use of
t-tests. Individual and/or population means may not be normally
distributed and hence the t-test may be inappropriate. Analysis of
variance or appropriate non-parametric statistics may be more
rigorous for the particular population under study (Carrano &
Moore, 1982). Data transformation can be useful in the statistical
analysis, if the mean and the variance are independent (DuFrain &
Garrand, 1981). No single statistical procedure can be recommended
for universal application. The investigators must carefully
examine the data and apply the most appropriate test for the
questions being addressed.
3.2.2.6. Conclusions
There are many examples of populations exposed to mutagens in
which large increases in SCE have been demonstrated. These studies
show that the end-point can be a very sensitive indicator of
chemical exposure. In contrast, SCEs are not a useful indicator of
human radiation exposure. From the many reported studies, it is
evident that further quantitative information on variations in
SCE level between people, and the factors that influence these
frequencies, are required. For this reason, marginal increases
above a baseline SCE frequency must be interpreted with caution.
Significantly increased levels are an indication that the
population has been exposed to a genotoxic agent; on the other
hand, failure to observe increased levels does not necessarily
indicate the absence of exposure. Any increase in SCE frequency
for an individual or group cannot be interpreted as an indication
that the individual or group is likely to suffer adverse health
consequences.
3.3. Gene Mutations
New procedures are being developed to detect mutations that
lead to the production of abnormal haemoglobins (Bigbee et al.,
1981), the loss of cell surface proteins (Bigbee et al., 1983), or
the loss or modification of enzymes used in the synthesis of DNA in
human somatic cells. The last of these procedures has been used
for human population studies and is considered in detail.
Resistance to 6-thioguanine is a marker that has been used
widely to detect somatic mutations occurring in vitro in cultured
mammalian cells (Hsie et al., 1979). Since similiar somatic
mutations occur in vivo, quantification of 6-thioguanine resistant
(TGr) cells in peripheral blood provides a measure of specific-
locus somatic mutations occurring in vivo.
In human beings, a naturally-occurring germinal mutation
of the X-chromosomal gene for hypoxanthine-guanine
phosphoribosyltransferase (HPRT) (EC 2.4.2.8) provides a prototype
for 6-thioguanine resistance at the somatic level (DeMars, 1971).
HPRT normally converts hypoxanthine and guanine to inosine
monophosphate, and it phosphorylates some purine analogues (e.g.,
6-thioguanine) to produce cytotoxic products (Caskey & Kruh, 1979).
Mutant cells, deficient in this enzyme, are unable to use exogenous
hypoxanthine as a purine source and are resistant to killing by
these purine analogues.
It is important to note that the different types of chromosome
aberration should be recorded separately. The results for any
sample should be presented as the aberration frequency per cell
for each aberration type, or frequency of aberrant cells for each
aberration type. It is not appropriate to express the results as
total aberrant cells or total breaks per cell, as this method of
presentation can hide important facets of the data. For the
statistical analysis of data, it is legitimate to combine
aberration classes into the general categories of chromatid-type
deletions, chromosome-type deletions, chromatid exchanges, and
chromosome exchanges. Occasionally, cells are seen with multiple
aberrations, and an attempt should be made to analyse such cells in
detail, rather than to record them simply in a category "multiple
aberration", since this classification will mean a loss of
information. If it proves impossible to analyse a cell, it should
be recorded in a category of "too many aberrations to analyse", and
not included in the total of cells scored.
The selection of cells to be analysed is particularly
important, to avoid observer bias. Such bias can result in either
a higher or lower measure of the unbiased aberration frequency,
depending on the nature of the selection. The generally accepted
method of cell selection is to scan slides at low magnification;
cells that appear to be suitable are then analysed at high
magnification. Once a cell is observed at high magnification,
it should only be rejected if: (a) the cell cannot be analysed
because of the number or complexity of aberrations; in this case,
it should be appropriately recorded, but not included in total
cells analysed; (b) the cell is not sufficiently well spread
when seen at high magnification, and the overlap of chromosomes
prohibits an accurate analysis; (c) non-chromosomal material, such
as dirt or stain crystals, not discernible at low magnification,
prevent complete analysis; (d) the cell contains fewer centromeres
than the observer's acceptable cut-off point; this may be the
analysis of cells with 46 ± 2 centromeres or only cells with 46
centromeres, either cut-off point is acceptable, provided that the
criterion is consistently applied.
It is particularly important in population-monitoring studies
for the samples to be analysed under code, and for the slides from
exposed and control individuals to be randomized in such a way that
the observer is unaware of the identity of the sample. There are
strong arguments in favour of more than one laboratory, with at
least 2 individuals in each laboratory, being responsible for the
analysis, in order to remove possible laboratory or individual
scoring bias, which could influence the results. Microscope
co-ordinates of every suspected aberrant cell should be recorded.
In order to provide further consistency, the classification of any
cell as aberrant by one individual should be confirmed by a second,
and it is advantageous to keep a photographic record of all
confirmed aberrant or otherwise suspect cells.
The number of cells to be analysed from any sample cannot be
fixed with any certainty, because it can be argued that this number
depends on the aberration frequency, and thus is retrospective. A
general suggestion would be to analyse 200 cells per sample.
The pros and cons of analysing banded preparations have been
discussed frequently, and do not warrant repetition. The amount of
time expended on the preparation and analysis of banded samples
does not seem to be justified by the increase in sensitivity of
analysis for aberration types detectable by banding. However, the
choice of scoring conventially-stained or banded preparations
should be left to the discretion of each laboratory.
3.2.1.4. Data analysis
Many statistical methods are available for the analysis of
data obtained from human population-monitoring studies. The
appropriateness of any particular method will depend, to some
extent, on the design of the study and the nature of the data
collected. Two examples of general approaches are given in Brewen
& Gengozian (1971) and Evans & O'Riordan (1975).
3.2.1.5. Radiation-induced aberrations and estimates of exposure
For a variety of reasons, it is sensible to discuss the use of
the lymphocyte assay for estimating exposure to ionizing radiation
separately from cases of chemical exposure. In some ways, it was
fortunate, but, in retrospect, perhaps also unfortunate, that all
the original studies on the analysis of chromosomal aberrations in
the lymphocytes of exposed or possibly-exposed persons applied to
radiation exposure. The fact that, in these cases, estimates of
exposure could be made with some accuracy has often led to the
assumption that similar exposure-estimating procedures could be
applied to persons, groups, or populations exposed environmentally
or occupationally to chemical agents. A clear note of caution in
accepting this assumption is made here, and will be discussed
further.
Ionizing radiation and a small number of chemical agents (e.g.,
streptonigrin, bleomycin, neocarzinosatin, cytosine arabinoside,
and 8-methoxycaffeine) are able to produce aberrations in all
stages of the cell cycle including chromosome-type aberrations in
G1 and chromatid-type aberrations in S and G2.
The peripheral lymphocytes, a subpopulation of which is
mitogenically stimulated when blood samples are placed in culture,
are essentially non-cycling, and are in the G0 stage of the cell
cycle, the term usually applied to non-cycling G1 cells. Thus,
following radiation exposure, chromosome-type aberrations will be
induced in these non-cycling G1 lymphocytes, and can be observed at
the first metaphase following mitogenic stimulation in culture.
The fact that aberrations can be produced in G1 cells means that
their frequency can be directly related to the dose received (i.e.,
aberration frequency is proportional to dose). In addition, it has
been shown for many species (Brewen & Gengozian, 1971; Preston et
al., 1972; Clemenger & Scott, 1973), including man (Brewen et al.,
1972), that the frequency of aberrations induced in vitro is the
same as the frequency induced by the same dose delivered in vivo.
This means that a standard dose-response curve, for any particular
radiation type, can be obtained for in vitro exposures, and can
then be used for estimating doses received by individuals as a
result of radiation accidents during medical or other environmental
exposures. The frequency of aberrations (usually dicentrics) is
measured in cultured blood samples from exposed individuals, and
then converted into a dose estimate from the standard dose-response
curve. There is extensive literature on the use of the lymphocyte
assay as a biological measurement of dose for many different types
of radiation (Bender & Gooch, 1966; Lloyd et al., 1983). However,
there are still some limiting factors in the use of the assay.
These include the length of time of blood sampling after exposure,
partial-body exposure, and non-homogeneous exposures.
It is also apparent that the lymphocyte assay can be used for
estimating doses following long-term radiation exposure (Evans et
al., 1979). Again, this is possible because the aberrations are
induced in G0 cells, and their frequency will be directly related
to exposure. Cells containing radiation-induced aberrations as
well as normal cells, will gradually be lost from the peripheral
lymphocyte pool, as a result of lymphocyte turnover. However, the
cells repopulating the peripheral pool will be derived mainly from
normal precursor cells, and not from those containing aberrations.
Many chromosome-type aberrations are lethal to the cell as the
result of loss of acentric fragments at division, or because of the
mechanical interference of the aberration with division. Thus,
during long-term exposures (months or years), the aberration
frequency will not be additive with time (or dose), but will reach
an equilibrium where new aberrations are formed and existing ones
are lost from the sampled population.
A similar argument holds for determining the length of time
after short-term exposure when samples can provide a measure of
the maximum aberration frequency or true induced frequency.
The lymphocyte turnover will result in loss of cells from the
peripheral pool, both normal and abnormal, and repopulation largely
by normal precursor cells. The aberration frequency appears to
stay constant for about 6 weeks after exposure (Brewen et al.,
1972).
Samples have been taken many years after exposure, for
example, with radiation-treated ankylosing spondylitis patients
(Buckton et al., 1978) and human beings exposed to the atomic bombs
in Hiroshima and Nagasaki (Awa et al., 1978). The fact that some
lymphocytes are very long-lived, in excess of 20 years (Awa et
al., 1978; Buckton et al., 1978), means that radiation-induced
aberrations can still be observed in cells that were present as
peripheral lymphocytes at the time of exposure. If it is assumed
that the aberration frequency declines exponentially with time, an
approximate estimate of the dose received can be made; the word
approximate is emphasized.
3.2.1.6. Chemically-induced aberrations and estimates of exposure
On the basis of the success of the lymphocyte assay for
estimating radiation exposures, it seemed appropriate to analyse
chromosome aberrations in blood samples from individuals,
occupationally exposed to single chemicals or complex mixtures
(Office of Technology Assessment, US Congress, 1983), to determine
whether there had been exposure to a clastogen, and to relate
aberration frequencies to dose level. However, the ability to
estimate dose levels from aberrations in populations exposed to
chemical mutagens is not reliable at present. More research is
necessary to make the assay more reliable and applicable, and to
make the results more readily interpretable.
The present low sensitivity of the assay for measuring the
frequency of chromosome aberrations following chemical exposure is
due to the mechanisms of induction of aberrations by chemical
agents. These mechanisms may also result in induced aberration
frequencies being only indirectly related to exposure dose, in
contrast to radiation-induced aberrations.
Chromosome aberrations induced by chemical treatments are
almost exclusively produced during the S-phase, irrespective of the
cell-cycle stage treated. Thus, the majority of aberrations will
be of the chromatid-type, although there are exceptions to this
general hypothesis (Evans & Vijayalaxmi, 1980; Preston & Gooch,
1981). Therefore, it is more appropriate to consider that the
probability of producing aberrations in G1 or G2 cells following
chemical treatments is low, but is considerably increased when
treated cells are in, or pass through, the S-phase.
Chemically-induced DNA damage in non-cycling lymphocytes will
not generally be converted into aberrations until the cells are
stimulated to re-enter the cell cycle in vitro, and undergo DNA
replication. Since repair of DNA damage can take place in G0 cells
as well as during the long first in vitro G1 stage, the aberration
frequency will not necessarily be proportional to the amount of
induced DNA damage but rather to the amount of DNA damage remaining
at the time of DNA replication. It is clear that the amount of DNA
damage, present at the time of replication, which has the potential
to be converted into aberrations, will depend on several factors
including: (a) the dose received; (b) the induced amount of the
particular types of DNA damage that can result in aberrations (a
value that can vary with the agent); (c) the amount of DNA repair
in G0 cells before sampling (i.e., time between exposure and
sampling); and (d) the amount of repair in G1 cells from mitogenic
stimulation to the first in vitro S-phase. Many of these will also
be subject to individual variation. The outcome is that, for most
chemical agents, only a proportion of induced DNA damage can be
converted into aberrations, at the time of replication. These DNA
repair factors all work to reduce the sensitivity of the lymphocyte
assay for measuring chemical exposure, and will result in the
aberration frequency being, at best, only indirectly proportional
to exposure.
If an increase in chromosome aberrations is observed in a
potentially exposed group when compared with a matched control
group, it is reasonable to conclude that there has been exposure to
a clastogenic agent, but no estimate of exposure level or of
subsequent adverse health effects (genetic or somatic) can be made.
On the other hand, if there is no difference in aberration
frequency between the possibly-exposed group and a matched control
group, it is not possible to rule out an exposure. However, in
this case, it may be possible, on the basis of previous experience,
to deduce a maximum exposure level that is not associated with a
detectable difference in aberration frequency.
3.2.1.7. Conclusions
Future research should enhance the sensitivity of the assay
system. In the meantime, it is suggested that care be taken in
the choice of groups to be considered for monitoring studies, with
particular regard to the usefulness of the information that might
be obtained, and to the likelihood of being able to draw definitive
conclusions. Cytogenetic monitoring can be an appropriate method
for the detection of chemical exposure, but, clearly, it cannot, at
present, be considered universally decisive. It should be used in
conjunction with other end-points.
3.2.2. Sister-chromatid exchange
3.2.2.1. Formation of sister-chromatid exchange
SCE results from the breakage and rejoining of DNA at
apparently homologous sites on the 2 chromatids of a single
chromosome. They were first visualized by Taylor et al. (1957)
who used tritiated thymidine to differentially label the DNA of
replicating cells and autoradiography to distinguish the silver
grain pattern on the 2 sister chromatids. Advances in
cytochemistry over the subsequent 15 years led to greatly
simplified procedures for visualizing these exchanges. Latt
(1973) demonstrated that bromodeoxy-uridine (BrdU), an analogue
of thymidine, when incorporated into DNA, could quench the
fluorescence of the fluorochrome Hoechst 33258. Perry & Wolff
(1974) found that incorporated BrdU also diminished the uptake
of Giemsa stain into the chromatin. In principle, SCEs can be
observed in any cell that has completed two, or the first of two,
replication cycles in the presence of BrdU. The most common
procedure for human lymphocytes is to grow the cells in BrdU for
two replication cycles.
SCEs are most efficiently induced by substances that form
covalent adducts with the DNA, or interfere with DNA precursor
metabolism or repair (Perry & Evans, 1975; Wolff, 1977; Perry,
1980; Carrano & Thompson, 1981; Latt, 1981). It is well documented
that SCEs are produced during DNA replication (Wolff et al., 1974)
and that the polarity of DNA is maintained in the process of
exchange (Taylor, 1958; Wolff & Perry, 1975). The molecular
mechanism by which the exchange is formed is not known but, since
there is an absolute requirement for DNA synthesis, hypotheses have
implicated the mechanics of this process in SCE formation (Bender
et al., 1974; Painter, 1980; Cleaver, 1981, Shafer, 1982).
Baseline SCE frequencies are increased in some human diseases
such as Bloom's syndrome (Chaganti et al., 1974) and multiple
sclerosis (Sutherland et al., 1980; Vijayalaxmi et al., 1983a).
Cells from patients with the inherited disease Xeroderma
pigmentosum are hypersensitive to the induction of SCEs by
ultraviolet radiation and alkylating agents (Wolff et al., 1975,
Wolff, 1977). There is no clear relation, however, between the
repair capacity of these cells and their sensitivity to SCE
induction (de Weerd-Kastelein et al., 1977).
3.2.2.2. Relevance of sister-chromatid exchange
An SCE represents the breakage of 4 strands of DNA (2 double
helices), a switch of the strands from one to the other arm of the
same chromosome, and the rejoining of the strands in their new
locations. The question is whether the breakage and rejoining
events occur without producing any modification in the genetic
code. There is evidence suggesting that this exchange process
might not always be error-free. In particular, a considerable
amount of information has been gathered concerning the relationship
between induced SCEs and other genetic effects in vitro and in
vivo.
Because the technique for enumerating SCEs is relatively
simple, this assay has been used quite extensively in genetic
toxicology studies. It has been amply demonstrated that the
frequency of SCE is dramatically increased when cells, or
animals, including human beings, are exposed to known mutagens
and carcinogens (Perry & Evans, 1975; Latt et al., 1981; Lambert
et al., 1982). Moreover, in early experiments using Chinese
hamster cells in culture, it was found that the induction of SCEs
was linearly related to the increase in single-gene mutations (HPRT
locus), when the cells were exposed to several substances each of
which differed in the type of lesion produced in DNA (Carrano et
al., 1978; Carrano & Thompson, 1982). The ratio of induced SCE to
induced mutation differed for each of the agents, suggesting that
each type of lesion formed is processed differently by the cell
favouring either the formation of SCEs or mutations. It is
possible that the same lesion is capable of producing both an SCE
and a mutation and/or that the lesions that form mutations are a
subset of those that form SCEs. The induction of mutations has
been shown recently to be correlated with the induction of SCEs in
mice for one chemical, ethyl nitrosourea (Jones et al., in press).
In this study, the induction of SCEs in mouse splenocytes,
following intraperitoneal administration of the chemical, was
linearly proportional to the increase in mutations at the HPRT
locus in the same splenocyte population, suggesting that the
mutation-SCE relation is not unique to in vitro studies.
Other investigations compared the induction of SCEs to either
the induction of transformation in vitro or induction of tumours
in vivo. In a series of experiments, the induction of SCEs was
shown to be linearly and positively correlated with the induction
of transformation of Syrian hamster embryo cells in vitro (Popescu
et al., 1981). This relationship held for exposure to 5 chemicals,
but not for X-irradiation. The ratio of induced SCEs to induced
transformation frequency varied for each chemical. This is
analogous with the results found for the SCE-to-mutation ratio
described above. Cheng et al. (1981) examined the induction of
SCEs in mouse bone marrow, regenerating liver, and alveolar
macrophages following intraperitoneal injection of ethyl carbamate
and related chemicals. The relative potencies in inducing SCE
parallelled the reported activities for the induction of lung
adenomas in mice. For ethyl carbamate, the doses for the
significant induction of SCEs in alveolar macrophages and lung
adenomas were similar. Further, the slopes of the dose responses
showed that the two end-points were of comparable sensitivity.
The results of early studies on rabbits demonstrated that it
was possible to measure an increase in SCEs in peripheral blood
lymphocytes for many months following repeated low-level exposure
to mitomycin C in vivo (Stetka et al., 1978). Increased SCEs in
human beings have been observed in cigarette smokers (Lambert
et al., 1978; Carrano, 1982) and in individuals undergoing
chemotherapy (Lambert et al., 1978; Musilova et al., 1979; Gillian
Clare et al., 1982). The results of these studies suggest that a
subpopulation of lymphocytes with high SCE frequencies may be
present for long periods after termination of exposure. These
cells may represent persistent damage in long-lived lymphocytes
or a sensitive subpopulation (Carrano & Moore, 1982). If this
is true, they may offer a more appropriate approach to the
quantification of persistent damage in vivo.
3.2.2.3. Factors potentially influencing SCE frequency
The baseline SCE frequency in human peripheral lymphocytes
averages about 7 - 10 per cell, but has been reported to range from
about 2 to 45 per cell in unexposed individuals (Alhadeff & Cohen,
1976; Lambert et al., 1976; Crossen et al., 1977; Galloway, 1977;
Carrano et al., 1980). Even within the same laboratory, the
baseline frequency may vary by a factor of two or more. Several
potential sources of variation have been identified and they
generally fall into 2 categories: a) culture factors associated
with the in vitro growth of the lymphocytes; and b) biological
factors associated with the genotype, lifestyle, or general health
of the individual. The biological factors usually cannot be
mitigated and must be accounted for in the selection of appropriate
control cohorts or ultimately in the statistical analysis.
(a) Culture-associated factors
A major source of variation can be attributed to the
concentration of BrdU relative to the number of lymphocytes in the
culture. The frequency of SCE has been shown to increase by as
much as 50%, when the BrdU concentration is raised 10-fold (Carrano
et al., 1980). The ability of BrdU to induce SCE has been well
documented in human and other cell systems (Wolff & Perry, 1974;
Lambert et al., 1976; Latt & Juergens, 1977; Mazrimas & Stetka,
1978) and it has been further shown that the SCE frequency can
increase if the BrdU concentration is held constant, but the
cell number decreased (Stetka & Carrano, 1977). The number of
lymphocytes that respond to mitogen and eventually incorporate
the analogue is generally not controllable, and, therefore, the
effect of BrdU can best be minimized by standardizing both the
concentration of BrdU and white blood cells at culture initiation.
Since the SCE increase above baseline following most long-term
exposures may be small, careful consideration of the influence of
BrdU is warranted.
Other culture-associated factors have been examined to
determine whether they influence the SCE frequency. One concern
has been the optimal time to harvest cells. In at least three
studies, it has been concluded that the baseline SCE frequency
does not depend on the time of harvest. Carrano et al. (1980)
found that, for any individual, the SCE frequency could vary by
as much as 30% as a function of culture time, but that the pattern
was not consistent among donors. Similarly, Beek & Obe (1979),
Becher et al. (1979), and Morimoto & Wolff (1980) did not observe
any significant alteration in the baseline SCE frequency as a
function of culture time. In a study by Snope & Rary (1979), for
4 individuals, the baseline SCE frequency observed in 58-h cultures
was about 30% less than in 70-h cultures. The authors interpreted
this to indicate that there were at least 2 subpopulations of
lymphocytes that differed in SCE frequency. Studies by Lindblad &
Lambert (1981) and Das & Sharma (1984) also revealed that the more
slowly cycling lymphocytes had a higher SCE frequency. The
possibility that this effect might be due to T- and B-lymphocyte
subpopulations has not been completely resolved. Santesson et al.
(1979) found that B-lymphocytes had a significantly lower SCE
frequency than T-lymphocytes, but Lindblad & Lambert (1981) could
not confirm this. In order to optimize an experimental design to
account for such an effect, all samples should, where possible, be
harvested at the same time.
Two other factors have been shown to influence the baseline SCE
frequency, i.e., the serum used and the culture temperature. Using
sera from 4 different sources, McFee & Sherrill (1981) showed that
the SCE frequency could increase by 50% as the serum concentration
increased. Kato & Sandberg (1977) also demonstrated an effect of
sera on the production of SCEs. The frequency of SCE in amphibian
cells increased almost 5-fold with increasing culture temperature
(Speit, 1980). In Chinese hamster cells, both lower (33 °C) and
higher (40 °C) temperatures increased the frequency of SCE by
approximately 60%. Because the alteration in temperature was
only effective in altering SCE frequencies while BrdU was being
incorporated, the author concluded that SCEs might arise via a
temperature-dependent disturbance of DNA replication. In a study
by Das & Sharma (1984), the frequency of SCEs in human lymphocytes
increased as a function of culture temperature and reached a
maximum at 40 °C. The authors concluded that temperature-dependent
DNA replication enzymes might be responsible for this effect.
In many human population studies, the cohort to be sampled
may be far away from the laboratory performing the cytogenetic
analysis, and it will often be necessary to ship the samples.
If blood is shipped commercially, recording thermometers in the
shipping container will provide some quality control. The effect
of blood storage on SCE frequency has been examined (Carrano et
al., 1980). No consistent alteration in SCE frequency was observed
for blood held at either 4° or 22 °C for up to 48 h. This was
confirmed by Sharma & Das (1984) for blood stored at 5 °C for up to
7 days, at 22 °C for 3 days, and at 37 °C for 2 days. However, it
should be noted that such storage can lead to an overall decrease
in lymphocyte numbers and modification of lymphocyte subclasses
(Dzik & Neckers, 1983). Thus, the samples should be placed in
culture as quickly as possible.
Finally, Deknudt & Kamra (1983) evaluated 4 different
mitogens for their effects on SCE frequency. They found that
PHA, concanavalin A (Con A), Wistaria floribunda (WFA), and lentil
lectin (LcH-A) extracts did not result in different baseline SCE
frequencies in the same donors. However, significant differences
among mitogens were noted for lymphocytes that had been exposed
to cyclophosphamide or mitomycin C. The use of a single T-cell
mitogen, such as PHA, for all studies is therefore recommended.
The experience accumulated from the use of lymphocyte cultures
suggests that there are many culture-associated factors that could
influence the SCE frequency. In order to minimize variation and
maximize sensitivity, the use of standardized procedures is
essential.
(b) Biological factors
Factors, unique to the individual being sampled, include such
confounding variables as sex, age, diet, genotype, medication, and
smoking. Each of these potentially plays a role in the induction
or expression of SCEs. Although some information is available on
the influence of these factors, the consequences are not always
clear-cut. Moreover, it is likely that they represent only a small
number of possible confounding factors.
Several investigators have compared the baseline frequency of
SCEs in male versus female donors and have not observed significant
differences (Galloway & Evans, 1975; Alhadeff & Cohen, 1976;
Crossen et al., 1977; Latt & Juergens, 1977; Morgan & Crossen,
1977; Cheng et al., 1979; de Arce, 1981; Waksvik et al., 1981;
Carrano & Moore, 1982; Livingston et al., 1983). The absence
of clear documentation of a sex difference for baseline SCE
frequencies suggests that this factor may not have to be matched in
a population study. However, that there is evidence for increased
SCE frequencies in women taking oral contraceptives (see below).
No studies relating SCEs to cyclic hormonal variation in the female
have been reported.
Another variable of concern in population studies is the age of
the donor, since it is not always possible to obtain accurate age
matching. The independence of age and baseline SCE frequency in
lymphocytes in adults has been observed (Galloway & Evans, 1975;
Morgan & Crossen, 1977; Hollander et al., 1978; Cheng et al., 1979;
Lambert & Lindblad, 1980; Carrano & Moore, 1982; Livingston et al.,
1983). Lower SCE frequencies have been found in infants (Seshadri
et al., 1982), in cord blood (Ardito et al., 1980), and in children
with a mean age of 1.5 years (Husgafvel-Pursiainen et al., 1980).
De Arce (1981) reported that people in the age range of 30 - 40
years had a higher number of SCEs per cell than those between the
ages of 0 - 10 or 60 - 70 years. Schmidt & Sanger (1981) found a
significant increase in SCE with age when comparing age groups of
1 - 18 months, 10 - 13 years, 26 - 32 years, and 63 - 85 years.
However, this might be due to differential BrdU incorporation
between cultures. With the exception of lower SCE frequencies in
new-born infants, the weight of evidence suggests that baseline SCE
frequencies do not change with age (at least up to 60 years).
It has been suggested that genetic factors play a role in the
baseline SCE frequency. When Cohen et al. (1982) compared SCE
frequencies, both within and among 12 pedigrees (2 generations),
they found significant differences between, but not within, the
families, suggesting a genetic contribution to baseline SCEs.
Pedersen et al. (1979) examined the cultured lymphocytes from 11
monozygotic and 9 dizygotic twins of the same sex. They did not
find any significant differences in the SCE frequency in
monozygotic versus dizygotic twins and concluded that genetic
factors do not play a role in baseline SCE frequency. Waksvik et
al. (1981) also did not find any genetic contribution to SCE
frequencies in their studies on twins. In order to determine
whether baseline SCE frequencies were related to ethnic origin,
SCEs were measured in adults of both sexes with no interracial
family backgrounds from Caucasian, American black, oriental, and
native American races (Butler, 1981). There was no significant
difference in the average frequency of SCEs in the 4 races.
Further research is needed to resolve the issue of genetic factors.
The potential influence of other factors on the frequencies of
human SCEs are worth considering. Because a large number of women
use oral contraceptives, care must be taken that this does not
confound the consequences of exposure to other agents. A study of
oral contraceptive users demonstrated a significant increase in
lymphocyte SCE associated with the daily intake of d-norgestrel or
ethenyl estradiol over a period of 6 - 24 months (Balakrishna
Murthy & Prema, 1979). Additional studies are needed to confirm
and extend these findings.
Despite knowledge of natural mutagens in food and the role of
food processing in creating mutagens (Nagao & Sugimura, 1981; Ames,
1983), little attention has been given to the role of diet in
causing baseline SCE variation. It has been reported that severe
protein calorie malnutrition in children is associated with
increased lymphocyte SCE frequencies (Balakrishna Murthy et al.,
1980). Following nutritional rehabilitation, SCE frequencies were
seen to decrease. Increased SCEs have also been reported in
chronic alcoholics (Butler & Sanger, 1981). For most human
studies, however, the nutrition factors are more likely to involve
long-term, low-level intake of caffeine, saccharin, other
additives, or well-cooked foods. Each of these factors has been
shown to be genotoxic in microbial, mammalian in vitro, or animal
assays, but there are no data on how these might influence SCE
frequency in human beings. Dietary information should be collected
in the initial questionnaire. Such information may be useful in
interpreting a population difference or can be used in large
composite data bases to search for diet-related factors. The use
of prescription and non-prescription medication, or vitamins,
should also be identified and appropriate information should be
collected before blood sampling, so that matched controls can be
employed.
Cigarette smoking is, perhaps, the most important confounding
factor in the interpretation of human SCE frequencies. The
preponderance of evidence indicates that individuals who smoke
have elevated SCE frequencies (Lambert et al., 1978; Ardito et al.,
1980; Hopkin & Evans, 1980; Lambert & Lindblad, 1980; Carrano,
1982; Husum et al., 1982; Livingston et al., 1983). However, this
effect has not been observed in all studies (Hollander et al.,
1978; Hedner et al., 1983). Since the elevation can be so marked,
it is important to control for cigarette smoking in any population
study.
Taken together, the culture and biological factors that
potentially confound the baseline SCE frequency in human beings
are numerous and contribute to individual variation.
3.2.2.4. Methods for sister-chromatid exchange analysis
The procedures for SCE analysis in human populations will
depend, to some extent, on the specific circumstances of the study.
The following generalized outline of a procedure may have to be
modified to fit special situations.
In order to identify any of the factors that may confound the
analysis of SCEs, it is important to obtain relevant information on
the background of each individual in the study population. This
information is best obtained through a questionnaire. The
questionnaire should attempt to determine the obvious exposure and
potential confounding factors. In addition to the questions
related to personal and family cancer incidence, birth defects,
known genetic disorders, and current health, there are specific
questions related to the interpretation of the SCE results. These
include: a) smoking history; b) medication and drug use; c) known
or suspected exposures to physical or chemical agents occurring
at work, at home, or during the pursuit of various hobbies or
recreation; and d) nutrition, particularly the adherence to special
diets or the use of artificial sweeteners or caffeine. Because a
complete list of agents that may directly or indirectly affect SCE
frequencies has not been established, it is better to collect too
much information than too little.
When the blood is collected, it should be mixed with heparin,
acid-citrate-dextrose, or another appropriate anticoagulant. As
already stated, the cultures should be established as soon as
possible after the blood is withdrawn, preferably within 24 h.
Just prior to establishing the culture, a total white blood count
and a slide for a differential should be made from the blood
sample. These parameters can serve as indicators of abnormally
high or low lymphocyte counts, factors that can have a bearing on
the observed frequency of SCEs.
It is important that a standard procedure be employed and,
although the exact procedure may differ among laboratories, there
are some common requirements. Because BrdU can induce SCEs in
human lymphocytes, both the BrdU concentration and the number
of lymphocytes potentially incorporating the drug, should be
stabilized. In order to ensure this, it is important that each
laboratory uses a fixed BrdU concentration and a consistent number
of lymphocytes per culture. Selection of culture medium, serum
type and concentration, whole blood versus purified lymphocyte
cultures, and duration of cultures is somewhat arbitrary and should
be based on experience. The serum lot should be pre-tested for its
potential to support lymphocyte growth and the reproducibility of
SCE frequencies in duplicate samples from the same donor. It is
recommended that the same serum lot be used for each population
study. The duration of culture should be such that it yields a
high proportion of second division cells so that the SCE frequency
is representative of a major fraction of the lymphocytes. Exposure
of the cultures to other than yellow or red light should be
avoided. Replicate cultures should be established for each
individual, and, when a large number of individuals is processed,
the inclusion of blood cultures from historical controls is useful.
At the appropriate culture time, the cells can be arrested in
mitosis with Colcemid(R), colchicine, or other suitable agents.
Several slides should be prepared from each culture and the
remaining cell pellet can be stored in absolute methanol:glacial
acetic acid (3:1) at a temperature of 4 °C or lower, for future
recall. The slides can be stained for analysis by fluorescence
alone (Latt, 1973), fluorescence plus Giemsa (Perry & Wolff, 1974),
or a suitable alternative. Once a staining procedure has been
standardized and adopted, it should be used throughout the whole
course of the study.
Slides should be coded and scored blind. The coded slides can
be scanned under low magnification (100 - 200X) and selected for
scoring on the basis of good staining and chromosome number. Only
well-differentiated metaphases should be accepted for scoring. It
is not essential for all the chromosomes in a cell to be counted.
Overlapping chromosomes can be disregarded and the remaining
chromosomes scored. At least 43 chromosomes should be analysed per
cell. The SCEs are counted for each chromosome in the cell and
expressed as SCE per chromosome or converted to the number of SCEs
per diploid cell. Although the precise number of cells to be
scored per individual will ultimately depend on the desired
statistical sensitivity, a minimum of 50 cells should be scored,
with 25 cells from each replicate culture.
3.2.2.5. Data processing and presentation
The standard statistical descriptors such as mean SCE per cell
or per chromosome, standard deviation or standard error, and number
of cells scored should be determined for each individual. For
heteroploid cells, the SCE frequency should be normalized to a
diploid cell. Population means and their errors should be
presented similarly and information on the number of cells scored
per sample as well as the number of people per group should be
given.
Student's t-test is commonly used for the statistical analysis
of population data. When comparing population groups, however, the
sample size is the number of people in each group, not the number
of cells scored for each group. Caution is advised with the use of
t-tests. Individual and/or population means may not be normally
distributed and hence the t-test may be inappropriate. Analysis of
variance or appropriate non-parametric statistics may be more
rigorous for the particular population under study (Carrano &
Moore, 1982). Data transformation can be useful in the statistical
analysis, if the mean and the variance are independent (DuFrain &
Garrand, 1981). No single statistical procedure can be recommended
for universal application. The investigators must carefully
examine the data and apply the most appropriate test for the
questions being addressed.
3.2.2.6. Conclusions
There are many examples of populations exposed to mutagens in
which large increases in SCE have been demonstrated. These studies
show that the end-point can be a very sensitive indicator of
chemical exposure. In contrast, SCEs are not a useful indicator of
human radiation exposure. From the many reported studies, it is
evident that further quantitative information on variations in
SCE level between people, and the factors that influence these
frequencies, are required. For this reason, marginal increases
above a baseline SCE frequency must be interpreted with caution.
Significantly increased levels are an indication that the
population has been exposed to a genotoxic agent; on the other
hand, failure to observe increased levels does not necessarily
indicate the absence of exposure. Any increase in SCE frequency
for an individual or group cannot be interpreted as an indication
that the individual or group is likely to suffer adverse health
consequences.
3.3. Gene Mutations
New procedures are being developed to detect mutations that
lead to the production of abnormal haemoglobins (Bigbee et al.,
1981), the loss of cell surface proteins (Bigbee et al., 1983), or
the loss or modification of enzymes used in the synthesis of DNA in
human somatic cells. The last of these procedures has been used
for human population studies and is considered in detail.
Resistance to 6-thioguanine is a marker that has been used
widely to detect somatic mutations occurring in vitro in cultured
mammalian cells (Hsie et al., 1979). Since similiar somatic
mutations occur in vivo, quantification of 6-thioguanine resistant
(TGr) cells in peripheral blood provides a measure of specific-
locus somatic mutations occurring in vivo.
In human beings, a naturally-occurring germinal mutation
of the X-chromosomal gene for hypoxanthine-guanine
phosphoribosyltransferase (HPRT) (EC 2.4.2.8) provides a prototype
for 6-thioguanine resistance at the somatic level (DeMars, 1971).
HPRT normally converts hypoxanthine and guanine to inosine
monophosphate, and it phosphorylates some purine analogues (e.g.,
6-thioguanine) to produce cytotoxic products (Caskey & Kruh, 1979).
Mutant cells, deficient in this enzyme, are unable to use exogenous
hypoxanthine as a purine source and are resistant to killing by
these purine analogues.
 TGr somatic cells arising in vivo can be shown to be mutants
by the stability of their phenotype and deficiency of HPRT activity
(Albertini, 1982; Albertini et al., 1982a; Morley et al., 1983b).
Also, molecular studies have demonstrated changes in the HPRT locus
at the DNA level (Albertini et al., in press).
3.3.1. Principles and basis for the methods
TGr T-lymphocytes (T-lys), present in human blood, can be
measured either by autoradiography to determine TGr T-ly variant
frequencies (Vfs), or by cloning to determine TGr T-ly mutant
frequencies (Mfs). Autoradiography is the earlier method (Strauss
& Albertini, 1977, 1979) and has been used for some human
population studies (Strauss & Albertini, 1979; Strauss et al.,
1979; Cole et al., 1982; Lange & Pranter, 1982; Morley et al.,
1982; Vijayalaxmi et al., 1983b; Albertini, 1983; Albertini et al.,
1983a); it is simple, inexpensive, and can be automated (Anmeus et
al., 1982; Zetterberg et al., 1982). Cloning is a more recent
development by which it is possible to recover the TGr cells for
characterization of the nature of the mutations (Albertini, 1982;
Albertini et al., 1982; Strauss, 1982; Morley et al., 1983a;
Vijayalaxmi & Evans, 1984). This characterization may be
necessary in order to quantify the different types of somatic
mutations responsible for a collection of mutants.
3.3.1.1. Autoradiographic method
The autoradiographic detection of rare TGr T-lys is based on
the ability of these cells to incorporate tritiated thymidine
(HTdR) in vitro following short-term lectin stimulation in the
presence of cytotoxic concentrations of 6-thioguanine. The vast
majority of human peripheral blood T-lys are in the G0 stage of the
cell cycle in vivo. When stimulated in vitro with lectins, such
as phytohaemagglutinin (PHA), T-lys are activated to early G1,
which is characterized by the acquisition of T-cell growth factor
(TCGF) receptors on their surfaces (Maizel et al., 1981). These
receptors are also called interleukin-2 (IL-2) receptors or Tac
antigens (Deeper et al., 1983). A series of cellular events
results in the production of interleukin-1, (IL-1) and IL-2, and
other factors in short-term peripheral blood mononuclear cell
cultures, with IL-2 driving the activated T-lys through G1 to DNA
synthesis and subsequent events in the cell cycle.
The initial round of lectin-stimulated T-ly DNA synthesis
in vitro is inhibited by 6-thioguanine. Inhibition probably is
at the activation step in 6-thioguanine sensitive cells and
occurs without incorporation of the analogue into DNA. The
interval of lectin stimulation before the addition of HTdR in
the autoradiographic assay is short, so that cell division in
vitro does not occur (Strauss & Albertini, 1979). Under proper
conditions, the T-lys that incorporate this label in the presence
of 6-thioguanine are TGr variants. These cells can be enumerated
by autoradiography and the TGr variant frequency (Vf) calculated.
3.3.1.2. Cloning method
Human T-lys are easily propagated for extended periods in vitro
by supplying TCGF to cells that have been activated with lectin or
antigen (Paul et al., 1981). Thus, mass populations derived from
peripheral blood can be developed and maintained. Moreover,
freshly-obtained human T-lys may be cloned directly in vitro, in
the presence or absence of 6-thioguanine. In the first case, large
cell innocula must be used, to ensure the presence of rare TGr
cells (Albertini, 1982; Albertini et al., 1982a; Strauss, 1982;
Morley et al., 1983a,b; Vijayalaxmi & Evans, 1984). Activating
intervals are short so that cell division does not occur in vitro
prior to exposure to cytotoxic concentrations of 6-thioguanine,
thus ensuring that the TGr cells recovered in vitro actually arose
in vivo. The number of TGr T-lys determined by cloning is used to
derive in vivo TGr T-ly mutant frequencies (Mfs).
3.3.2. Relevance and limitations
3.3.2.1. Relevance
The ability to measure TGr T-lys arising in vivo is important,
because it provides a genetic end-point for assessing human
genotoxicity that may be qualitatively different from somatic cell
chromosomal damage. Thus, the ability to measure gene mutation
provides a broader basis for human monitoring. Furthermore, gene
mutations, as assessed by TGr T-lys, are end-points that are
qualitatively different from other measures of in vivo DNA damage.
Measuring human somatic cell gene mutations occurring in vivo has
the following advantages:
(a) The effects of metabolic and kinetic factors are included
in measurements of somatic cell mutation occurring in vivo.
(b) Mutagenicity assays for in vivo events assess the genetic
effects of given environments, rather than the genotoxic
potential of targeted chemicals or other agents. Thus,
these assays can be used to measure the effects of
environmental mixtures in which the components and/or
their synergistic actions may not be known.
(c) Mutagenicity assays, in vivo, may define human population
heterogeneity in susceptibility to various mutagens/
carcinogens (Vijayalaxmi et al., 1983b). Such individual
susceptibilities may be important in assessing human
genotoxic risks.
(d) Mutagenicity assays, in vivo, have the potential for being
developed into a means of making quantitative human
genetic health risk assessments, provided that appropriate
correlations can be made between individual assay results
and individual health outcomes (Fowle et al., 1984).
3.3.2.2. Limitations
Although progress has been made in demonstrating that TGr T-lys
are mutant somatic cells, at least 3 major problems remain for
human in vivo somatic cell mutation assays. Two are concerned
with quantification and apply primarily to gene mutation assays.
The third applies to all human somatic cell mutation assays and the
relevance of the results for making estimates of human health
risks.
(a) The in vivo sensitivities of the human TGr T-ly assays in
terms of dose-response characteristics are not known at
present. However, because mutant cells are also
detectable in animals (Garcia & Couch, 1982; Gocke et al.,
1983; Recio et al., 1983; Jones et al., in press), these
studies may provide useful information.
(b) Although somatic cell mutations are the events of
relevance for human population monitoring, TGr T-ly assays
measure the frequency of somatic cell mutants, not the
number of mutations. In order to derive mutation
frequencies from mutant frequencies, it is necessary to
have information including cell pool sizes and
distributions in vivo, representativeness of test samples,
in vivo cell kinetics, and in vivo positive or negative
selection of mutants. One method for circumventing this
problem is to determine whether the mutants scored in a
mutagenicity assay are qualitatively heterogeneous, which
will yield a minimum number of independent mutational
events giving rise to the observed collection of mutants.
The ability to recover TGr T-lys from clonal assays makes
these determinations possible.
(c) Assays of genetic damage occurring in vivo in human
somatic cells have not been validated in that it has not
been possible to use the results as predictors of human
genotoxic health risks. Before such results can be used
as surrogate markers for human health outcomes, high-risk
populations will have to be monitored and correlations
made between the results from mutagenicity assays and
epidemiological outcomes. The importance of other factors
(e.g., immunotoxicity) in influencing human genotoxic
disease risks remains to be assessed.
3.3.3. Procedures for the assay of TGr T-lys arising in vivo in
human beings
3.3.3.1. Autoradiographic method
TGr T-ly Vfs for normal, control adults, as determined
autoradiographically by the method described here, rarely exceed
10 x 10-6. By contrast, chemotherapy-treated cancer patients
frequently have values several times higher (Strauss & Albertini,
1977, 1979; Lange & Pranter, 1982; Albertini, 1983). Other
laboratories report somewhat higher normal values, as well as a
positive correlation with age (Morley et al., 1982), indicating
that local ranges of normal values must be developed. In one
laboratory, approximately 75% of placental cord blood values fell
within the normal range for adults, while the remainder were
elevated (Albertini et al., 1983). Whether this reflects maternal
exposure to genotoxic agents, or an altered ratio between mutants
and mutations in the fetus remains to be determined.
The basic autoradiographic method for assessing TGr T-lys has
been described in detail by Albertini et al. (1982b) and Albertini
& Sylwester (1984). Peripheral blood mononuclear cells (MNCs),
which include the T-lys, are separated from whole blood by the
Ficoll-Hypaque method (Boyum, 1968). After washing, the MNCs are
suspended in a dimethyl sulfoxide-containing medium and aliquoted
into ampules for controlled freezing and storage. Freezing is
important for holding and shipping samples and serves also as one
method for eliminating the phenocopy effect of cycling cells as
described below. For assaying, MNCs are thawed, washed, and
suspended in appropriately supplemented medium for short-term
tissue culture. A minimum of 5 x 106 MNCs are cultured without
6-thioguanine (control culture), while several times this number of
cells are cultured with 2 x 10-4 M 6-thioguanine (test cultures).
All cultures contain phytohaemagglutinin (PHA), for activation of
T-lys, and are incubated under standard conditions. After
approximately 24 h (Strauss & Albertini, 1979), 3HTdR is added
to all cultures, and incubation continued for an additional 18 h.
Cultures are terminated by adding cold 0.1 M citric acid, to
prepare suspensions of free nuclei. The nuclei are then washed
and suspended in methanol-acetic acid fixative, counted, diluted
if necessary, and added to coverslips affixed to microscope slides.
Slides are dried, stained with aceto-orcein and subsequently
autoradiographed by standard methods. Slides are scored and Vfs
are calculated as described in section 3.3.4.1.
A subclass of 6-thioguanine-sensitive T-lys in human peripheral
blood may become labelled and scored as TGr (i.e., as variants),
when using the autoradiographic method, unless certain precautions
are taken (Albertini et al., 1982a,b; Albertini, 1982). These
phenocopies occur because, at any given time, a small minority of
human T-lys are in an activated state, in vivo. These cells have
the Tac antigen, when initially put into culture, and do not
require the activation step before proceeding to in vitro DNA
synthesis. Although ultimately 6-thioguanine sensitive, these
cells probably are not effectively blocked from accomplishing at
least one round of DNA synthesis in vitro, even in the presence
of 6-thioguanine. Thus, activated T-lys in the peripheral blood
constitute a potential source of phenocopies, when TGr T-ly Vfs are
determined by autoradiography. Cryopreservation appears to remove
this phenocopy effect by forcing the in vivo activated T-lys to
proceed to DNA synthesis in vitro at a time when a label is not
present (Albertini et al., 1982a,b). These cells are not scored as
variants. Cryopreservation is a critical step, although other
methods, such as prolonged incubation in 6-thioguanine, or
immunological elimination of Tac positive T-lys at the initiation
of culture, may accomplish the same purpose.
3.3.3.2. Cloning method
From the limited data available, normal control adults have
shown peripheral blood TGr T-ly Mfs of < 20 x 10-6 when based on
clonal assays in which single-cell cloning efficiencies achieved
values > 0.10. Similar findings from a number of laboratories
have been reported (Strauss & Albertini, 1979; Morley et al.,
1983a; Vijayalaxmi & Evans, 1984). Results for placental blood and
for mutagen-exposed adults remain to be determined. TGr T-ly Mfs
have been slightly higher than TGr T-ly Vfs, concurrently
determined (Albertini et al., 1984).
The cloning method for assessing TGr T-lys has been described
by Albertini et al. (1982a). Peripheral blood MNCs are obtained
from whole blood as for the autoradiographic method, but freezing
is not required. The MNCs are incubated with PHA in order to
activate the T-lys. The duration of activation is sufficiently
short to ensure that cell division does not occur. Activated cells
are then innoculated in limiting dilutions into the wells of
microtitre plates, in the presence or absence of 6-thioguanine
(approximately 10-5 M). In addition to the appropriate medium,
wells contain an optimum concentration of crude TCGF and feeder
cells. X-irradiated B-lymphoblastoid cells are well suited for use
as feeders. A TGr lymphoblast line is used to avoid interference
with subsequent HPRT enzyme assays of the recovered TGr T-lys. An
average of 1 activated cell per well is innoculated into the non-
selection wells (containing no 6-thioguanine); 105 activated cells
per well are innoculated into selection wells (containing 10-5 M
6-thioguanine). Wells are scored after approximately 2 weeks of
culture with one change of medium, by microscopy, by scintillation
spectrometry of HTdR incorporation (added during the last day of
culture), or by cell transfer and clonal expansion. In vitro
cloning efficiencies (CEs) of T-lys are determined from wells
receiving, on average, 1 cell per well. By assuming a Poisson
distribution of cells in wells, the average number of clonable
cells per well is derived from the P0 class of that distribution,
i.e., the observed fraction of wells without growing cells.
Similarly, the incidence of TGr cells in wells receiving 105
activated T-lys in 6-thioguanine is determined from the P0 class of
wells in the plates containing 6-thioguanine. The TGr T-ly Mf is
the incidence, divided by 105, corrected for the CE.
TGr T-lys obtained by cell transfer and clonal expansion may be
characterized for T-ly subset markers, stability of the TGr
phenotype, HGPRT enzyme activity, and, by using suitable cDNA
probes, for the nature of the molecular lesion at the DNA level
(Albertini et al., 1982a, in press).
For reasons not known at present, human T-lys, activated in
vitro as described, may fail to clone as efficiently when present
as single cells than when present in large numbers (i.e., 105
cells/well), even when the latter are selected in 6-thioguanine
(Albertini et al., 1984). Clonal assays with a single cell CE
of < 0.10 have the potential for yielding falsely elevated Mf
values, because of an underestimate of clonable cells in selection
wells. Current research is directed at increasing and/or
standardizing single-cell CEs. At present, however, only Mfs
determined in clonal assays achieving single cell CEs of 0.10 or
more should be considered valid.
3.3.4. Data presentation and analysis
3.3.4.1. Autoradiographic method
Slide scoring is done by one of 2 methods that give almost
identical results. One method involves determining labelling
indices (LIs) for both control (no 6-thioguanine) and test
(6-thioguanine containing) cultures for each individual. The LI of
test cultures (LIt) is determined by counting all labelled nuclei
on all slides made from test cultures, and dividing this number by
the total number of nuclei (determined in suspension) added to all
slides.
Number of labelled nuclei on all test slides
LIt = --------------------------------------------
Number of nuclei on all test slides
The LI of control cultures (LIc) is determined from a
differential count of 2500 nuclei on slides from control cultures.
Number of labelled nuclei per 2500 nuclei
LIc = -----------------------------------------
2500
The TGr T-ly Vf for each individual is calculated from the LI
of test cultures (LIt) divided by the LI of the control culture
(LIc).
LIt
Vf = ---
LIc
An alternative method of scoring autoradiographic assays has
recently been described for mouse TGr T-lys (Gocke et al., 1983),
and is equally applicable to human assays. Statistical methods for
deriving confidence intervals for variant frequencies are described
by Sylwester & Albertini (1984).
3.3.4.2. Cloning method
Cloning efficiencies (CEs) are calculated from the P0 class of
the Poisson distribution:
P0 = e-x, or
x = -ln P0 (control)
where x is the average number of clonable cells per well. When CE
is determined by single cell innocula, x = CE.
The incidence of TGr T-lys in selection wells, receiving on
average 105 cells per well, is also determined from the P0 class of
the Poisson distribution:
P0 = e-y, or
y = -ln P0 (test)
where y is the average number of clonable TGr T-lys per well.
The TGr T-ly Mf is determined from the incidence of TGr
T-lys per well divided by the average cell innocula, corrected
by the CE. When 10 cells are innoculated into selection wells:
-ln P0 (test)
Mf = -------------
CE x 105
Appropriate statistical procedures for handling Mf data,
differences between Mfs, etc. are being developed by methods
analogous to those referred to in section 3.3.4.1.
3.3.5. Conclusions
TGr T-lys arise in vivo, are present in human peripheral
blood, and their frequencies are measurable. Known mutagen
exposure increases the frequencies of these cells and their
characterization in vitro has shown them to be somatic mutants.
Thus, methods are available for measuring specific locus somatic
mutants occurring in vivo in human beings, for purposes of human
genetic monitoring. The 2 assays described here continue to be
developed. There are potential technical sources of error. Among
the limitations in the interpretation of the results of these
assays is the fact that the numbers of somatic mutants, and not the
numbers of somatic mutations, are being measured. The latter may be
of most interest for human population monitoring. However,
characterization of the recovered mutant cells, which is possible
in the cloning assay, may make it possible to estimate the minimum
number of mutations responsible for a given number of mutants.
Methods described here can be applied to the detection of
mutations, occurring in vivo at other genetic loci in human T-lys,
in order to broaden the base for human monitoring.
4. GERMINAL MUTATIONS
Methods and technical aspects of approaches available for
detecting germinal mutations in human populations are reviewed in
this section. It should serve as a guide, when such studies are
initiated, and as a reference for defining specific methods for
estimating human mutation rates.
4.1. Introduction
Germinal mutations include a spectrum of alterations in
either the structure or quantity of DNA in germinal cells. The
study of germinal mutations is the quantification of transmitted
genetic damage. Within the context of monitoring for induced
genetic damage, this is an important consideration, because it is
the offspring of exposed individuals, rather than the exposed
individuals themselves, that are the focus of concern. It is
generally assumed that a significant proportion of all mutations
have deleterious effects on both the health and the genetic
constitution of future generations. Thus, the increased genetic
risk and associated health effects are of ultimate concern,
following exposure to a putative mutagen, when possible increases
in germinal mutation rates are being ascertained.
Germinal mutations are usually classified in 2 categories.
Chromosomal mutations are operationally defined as changes in
either chromosome number or structure observable with standard
karyotypic techniques. More minute changes in DNA structure are
classified as gene mutations, often referred to as "point"
mutations.
4.1.1. Approaches for detecting germinal mutations
The 3 approaches to monitoring for germ cell mutations are
generally divided as follows: (a) chromosomal, (b) biochemical,
and (c) the indicator phenotype; each detects a different set of
end-points.
4.1.1.1. Detection of chromosomal mutations
Germinal chromosomal mutations occur in about 5% of recognized
conceptions (Hook, 1981a,b), making them more amenable than the
rarer specific locus mutations for study in small exposed
populations. Unbalanced chromosomal complements are almost always
associated with deleterious phenotypic effects, leading to fetal
death, livebirths with anomalies and mental retardation, and/or
sterility. Balanced chromosome complements may occur as mutations
without phenotypic effect. However, their deleterious effects will
be seen primarily in the next generation in individuals with
unbalanced chromosomal complements.
Chromosome abnormalities can originate in four different ways:
a) they may be inherited from a parent; b) they may result from
errors during gametogenesis (e.g., meiotic non-disjunction); c)
they may result from events during conception (e.g., dispermy
resulting in triploidy); d) they may result from events after
conception (e.g., mitotic non-disjunction). Only the second
category listed above is regarded as a germ-cell mutation in the
strict sense, but to avoid unnecessary complexity in the discussion
that follows, triploidy and tetraploidy will be considered in the
review of germinal events.
Chromosomal mutations can be subdivided into numerical and
structural anomalies.
(a) Numerical aberrations
Numerical chromosomal abnormalities include trisomy, monosomy,
triploidy, and tetraploidy. Most instances result from events that
occur during gametogenesis in a parent, or at the time of
fertilization, although strict proof of the time of origin is often
lacking.
Apart from triploidy and tetraploidy, numerical chromosome
abnormalities involving sex chromosomes without a mosaic 45, X cell
line, or involving autosomes are usually presumed to have resulted
from a germinal mutation. The only exceptions are situations in
which numerical abnormalities occur in only a single tissue, as
for example in some malignancies. It is often difficult, however,
to exclude formally the possibilities that they: a) have been
inherited from a parent who is a cryptic mosaic for the abnomal
line; or b) have resulted from a somatic event, mitotic non-
disjunction, early in the development of the organism.
The strongest risk factor known for numerical abnormalities is
older maternal age, which is highly associated with the frequency
of trisomy (Hassold et al., 1980, 1984). Paternal age seems to
have little, if any, effect (Hook, in press). In studies using
chromosomal markers, it has been demonstrated that at least 60% of
trisomy 21 appears to be the result of maternal 1st division non-
disjunction (Juberg & Mowrey, 1983).
Numerical abnormalities almost always occur in offspring
of parents who have normal chromosomal complements, and thus,
such abnormalities are presumably the result of germinal
mutations. Although there is not a great deal of experimental or
epidemiological evidence to link numerical chromosome abnormalities
with environmental agents, increases in such anomalies must be
considered as possible outcomes of exposure to possible mutagens.
An unknown proportion of trisomy or monosomy may result from post-
zygotic non-disjunction, or, in the case of trisomy, may be
inherited from parents who are cryptic carriers, and thus may not
be the result of germinal mutations in a strict sense. For
trisomy, the results of studies of the parental origin of the extra
chromosome suggest that a maximum of 25% of trisomies are due to
post-zygotic non-disjunction, and it may well be considerably less
(Juberg & Mowrey, 1983).
Evidence suggests that mosaic trisomies (47/46) do not
originate from post-zygotic events any more often than non-mosaic
trisomies (Hassold, 1982).
Ninety percent of numerical anomalies in recognized conceptuses
terminate as fetal deaths; thus, a study restricted to live births
will miss a major proportion of detectable abnormalities.
Only a few kinds of numerical anomalies commonly survive to
birth. The most common is Down's syndrome, trisomy 21, occurring
in about 1 in 1000 live births. Other numerical anomalies such as
trisomy 18 and trisomy 13 sometimes survive to occur as live
births, but these are much less common.
(b) Structural aberrations
The proportion of individuals with detected structural
cytogenetic abnormalities is likely to vary with technical
factors. Recent advances in the resolution of chromosome
substructure (high resolution banding) have reduced significantly
the size of detectable lesions and such advances appear likely to
continue. Thus, it is difficult to specify precisely the
proportion of recognized conceptuses with a structural abnormality.
With currently available techniques, these are much less frequent
than those with numerical abnormalities. Among live births, the
ratio of detected structural to numerical abnormalities is about
1:4 to 1:5 (Hook & Hamerton, 1977). Among fetal deaths, the ratio
is much lower, about 1:30 (Warburton et al., 1980). Unlike
numerical abnormalities, a significant fraction of detected
structural abnormalities are known to be inherited, so that
inferences concerning mutation are not possible unless both parents
are studied and found not be be carriers of the aberration present
in the offspring. The possibility of false assignment of paternity
must also be considered. Unlike trisomies, structural chromosome
abnormalities, in general, show little if any association with
parental age, although markers and X-isochromosomes may represent
exceptions. In instances in which the parental origin of a
structural mutation has been investigated, many have been found to
be predominantly paternal in origin, unlike trisomies (Magenis &
Chamberlin, 1981).
In general, mosaicism, involving a normal cell line and a cell
line with a structurally abnormal chromosome, is not attributable
to germinal cell mutation, but may be safely inferred to be of
post-zygotic origin (supernumerary markers are, however,
exceptions).
In terms of sensitivity to environmental factors, mutant
germinal structural chromosome aberrations are likely to be more
similar to germinal specific locus mutations than numerical
abnormalities, as both involve alterations in the structure of the
genetic material. Germinal chromosomal rearrangements, unlike
specific locus mutations, show little association with advanced
paternal age in human beings (Hook, in press) indicating that there
are etiological differences between these effects.
4.1.1.2. The biochemical approach to detecting point mutations
Many biochemical approaches for the study of germinal mutations
have been proposed (Bloom, 1981), but most are not feasible at
present. Two general approaches are currently used to study the
proteins in offspring of selected individuals. Electrophoresis is
used to detect a significant proportion of amino acid substitutions
while enzyme activity measurements are used to detect major losses
in protein function or protein quantity.
Studies involving genetic typing of large numbers of
individuals, either to analyse population structure or to develop
mutation monitoring programmes, have demonstrated the feasibility
of large-scale screening using one-dimenional electrophoretic
techniques (Harris et al., 1974; Neel et al., 1980a; Altland et
al., 1982). Through the introduction of high-resolution two-
dimensional electrophoresis (Klose, 1975; O'Farrell, 1975), the
number of proteins that can be studied in a single sample has been
increased to several hundred. The search for mutational events
resulting in loss of a functional gene product, using quantitative
techniques, is complementary to the electrophoretic assay, in that
this assay detects genetic events not normally detectable by
standard electrophoretic assays. Both the electrophoretic and
enzyme activity approaches have been used in studies to determine
the induced mutation rate in exposed mice (Johnson & Lewis 1981;
Johnson et al., 1981; Bishop & Feuers, 1982) and Drosophila (Racine
et al., 1980). Thus, it should be possible to acquire data on the
mutation rate in human populations and also to examine directly
some of the problems associated with extrapolation from
experimental animals.
One other biochemical approach has been proposed for detecting
mutational events in human populations. It is possible to study
mutations at the DNA level by employing the restriction enzyme
mapping approach. One potential advantage of the restriction
enzyme mapping approach (or other techniques that directly monitor
specific alterations in DNA structure) is the ability to examine
larger portions of the genome and thereby acquire increasing
amounts of data from each individual. Since this approach is
currently in the developmental stage, appropriate samples should be
retained, as far as possible, to take advantage of this or other
techniques that may be developed in the near future.
The major constraints in relation to the biochemical approach
include: (a) the large number of determinations and, to date, the
large populations necessary to detect statistically-significant
increases in the mutation rate; (b) the distinction between
"apparent" mutations and nonparentage; and (c) the problem of
confirming that a suspected variant is the result of an alteration
in DNA structure, i.e., is a transmissible trait.
4.1.1.3. Indicator phenotypes
Three types of indicator phenotypes are considered in this
section: Down's syndrome, fetal death, and sentinel phenotypes.
Down's syndrome will be discussed in section 4.3, which deals with
chromosomal mutations. The potential use of fetal death as an
expression of genetic damage arising from genic mutations will be
discussed in section 4.5. Although the term "sentinel phenotype"
is relatively new (Sutton, 1971), the notion of studying mutations
in human beings by counting the frequency of dominant traits is
credited to Danforth (1921). Mulvihill & Czeizel (1983) have
reviewed the current status of the concept.
As indicators of germinal genic mutations, sentinel phenotypes
are a significant health problem and are recorded in a variety of
health facilities. The sentinel phenotype is a clinical disorder
that: (a) occurs sporadically as a consequence of a single, highly
penetrant mutant gene, (b) is a dominant or X-linked trait of
considerable frequency and low fitness, and (c) is uniformly
expressed and accurately diagnosable with minimal effort, at or
near birth. Individuals with such traits are important for the
surveillance and monitoring of germinal genic mutations because
affected persons of unaffected parents arise from a new mutation.
Their use has severe disadvantages connected with the difficulty
of accurate nosological diagnosis arising from their genetic
heterogeneity and a general lack of clinical expertise. The
sentinel phenotype approach, like other strategies in mutation
epidemiology, is burdened with problems created by the inability
to link separate data files, the necessity to maintain
confidentiality, and the difficulty in collecting sufficiently
large study populations or study samples. The best course of
action for the present is to obtain field experiences and to
sustain critical discussion of the approach.
4.1.2. Methodological considerations and strategies
4.1.2.1. Sample acquisition and storage
Future access to study subjects may be limited; therefore, the
maximum quantity of sample, usually blood, should be obtained on
the initial contact. When more than one test is to be performed,
attempts should be made to coordinate the appropriate collection
procedures and minimize the number of different acquisitions. In
situations where fetal tissue is being collected for chromosomal
studies, it is suggested that, where possible, a sample of kidney
tissue should be collected and stored for possible biochemical
analysis.
Adequate samples should be stored for confirmation of any
observation and also for future analysis using new and/or refined
techniques. Lymphocytes should be stored in a manner that allows
for the retrieval of viable cells and the subsequent expansion of
the number of cells, thus providing material for future studies.
4.1.2.2. Timing of studies
With specific regard to mutation studies, following suspected
exposure to mutagens, women of child-bearing age who have been
exposed to a mutagen and wives of exposed men should be identified
and surveyed as soon as possible. Questionnaires or interviews
should be undertaken and used to obtain data on basic biological
and demographic variables (and information on other possible
mutagenic factors). The factors include, but are not necessarily
limited to, age, race, previous pregnancy history, smoking, and
drug use. Women should be encouraged to participate in a
continuing evaluation of future pregnancy outcomes. If a woman
thinks she is pregnant but is uncertain, pregnancy testing should
be carried out as soon as possible. A control population of
similar ethnic and socioeconomic background should be identified.
This could be done at the time of identification of women at risk
of pregnancy, or could be done after pregnancy is confirmed, using
as a control group, women pregnant at the same gestational age.
Each of these approaches has difficulties. To wait until the
exposed woman is pregnant before selecting a matched (pregnant)
control may result in the pregnancy of the exposed woman being
terminated by the time the control is chosen. It may be extremely
difficult to avoid biasing the results towards a more favourable
outcome in the controls. If controls are selected prior to the
pregnancy of the exposed woman, there is no certainty that the
exposed and control women will become pregnant at the same time,
if in fact they conceive at all. One possible approach is to
select several possible controls for each exposed woman, before
pregnancy, and follow the reproductive history of all of them.
Ideally, these controls should be matched as far as possible in
age, race, socioeconomic status, and previous reproductive history.
Infants, children, and young unmarried males, who have been
exposed should also be identified early and followed for possible
inclusion should a subsequent long-term study be undertaken (see
section on record linkage).
Women recruited into the study should be interviewed by
investigators, at least once a month, to ascertain if they are
pregnant or think they may be pregnant. If there is any question,
pregnancy testing should be carried out.
4.1.3. Summary
Given the current state of knowledge in the area of human
mutations, all 3 approaches, namely indicator phenotype,
chromosomal, and biochemical, to estimating germinal mutational
damage in human populations should be considered complementary.
They detect different types of genetic "damage" or "end-points",
have different degrees of relevance for estimating potential health
effects and probably different sensitivities. Therefore, a
collaborative monitoring exercise should employ all 3 in order to
obtain the maximum amount of data possible.
4.2. Germinal Chromosomal Abnormalities
4.2.1. Principles and basis of the method
Chromosomal aberrations associated with germinal cell mutation
have already been discussed in section 4.1.1.1.
Depending on their origin and consequences, chromosomal
mutations may be detected in gametes, the embryo or fetus, in
live births, or at later stages in life.
If a population is exposed to a mutagen, the most direct method
of detection of germinal chromosomal mutants would be examination
of the gametes themselves. At present, there are no methods for
evaluating human ova. Preparations of human sperm chromosomes can
be made, but these methods are difficult and time-consuming, and
few laboratories have yet been successful with this technique
(Martin et al., 1983). It is possible that improved techniques for
studying sperm chromosome constitution will be available in the
future, but, at present, this method cannot be regarded as
practical for most situations involving mutagenic exposures.
Also, at the present time, alterations in sperm morphology or
function cannot be regarded as a reliable index of germinal cell
mutation, nor can changes in the proportion of sperm showing double
"Y" bodies. Thus, the following discussion focuses on detection of
chromosomal mutations in the offspring of those exposed to known or
suspected mutagens.
A clinically-recognized pregnancy is generally diagnosed after
the first missed menses, at about 4 weeks of gestation. The
frequency of chromosome abnormalities at this point in gestation
has been estimated to be about 5% (Hook, 1981a). About 15% of
recognized pregnancies terminate in fetal death, approximately one-
third having a chromosome aberration (Harlap et al., 1980; Hook,
1981a). The rate of loss between conception and recognized
pregnancy is not known, but it is generally agreed to be very high
(Kline et al., 1980). The proportion of these early losses that is
associated with chromosomal aberration is unknown. The bulk of the
evidence from the use of sensitive human chorionic gonadotropin
serum assays, capable of diagnosing pregnancy immediately after
implantation (usually 7 days after conception), suggests that loss
between implantation and the first missed menses may be at least as
common as loss afterwards (Miller et al., 1980; Edmonds et al.,
1982), but not all results are in agreement (Whittaker et al.,
1983). Preimplantation losses are still unmeasurable. Only
clinically-recognized pregnancy in the usual sense will be dealt
with here, since the other types of study are unlikely to be
feasible on the necessary scale, and chromosome studies cannot
be performed on these early losses.
The proportion of chromosome abnormalities in fetal deaths
varies with gestational age, being highest (about 50%) in the
8 - 12 weeks range, and then decreasing with gestational age (to
about 7%) at more than 22 weeks (Warburton et al., 1980). The
kinds of chromosome abnormalities found in fetal deaths include
many rarely, or never, seen in live births, e.g., triploidy,
tetraploidy, and trisomy for most whole chromosomes. Monosomy X,
though occurring in only about 1 in 20 000 live births, occurs in
about 7% of early fetal deaths; triploidy occurs with about equal
frequency, as does trisomy 16, an anomaly never seen at term and
compatible only with very rudimentary embryonic development
(Warburton et al., 1980).
Chromosome abnormalities are found in about 7% of late fetal
deaths (including still births) (Sutherland et al., 1978). At
birth, the proportion has been found to be 0.6% (Hook & Hamerton,
1977). About 90% of all recognized conceptuses with chromosome
abnormalities terminate as fetal deaths.
Most recognized abnormalities involve numerical aberrations
that are presumptive mutants. Structural chromosome abnormalities
make up only about 5% of the chromosome abnormalities seen in fetal
deaths (Warburton et al., 1980). About 40% of structural
abnormalities found in fetal deaths or at amniocentesis are de novo
mutational events: the rest are inherited from a carrier parent
(Jacobs, 1981; Hook et al., 1984).
4.2.2. Relevance and limitations
4.2.2.1. Studies of induced abortions
Chromosome studies of induced abortions are a useful source of
data. Such investigations are, of course, only possible where
induced abortion is a legal option. Advantages are that the
frequency of chromosomal abnormalities is still high at the point
in gestation when most induced abortions are performed, that the
specimens are likely to be viable in culture, and that the
procedure can be scheduled for collection purposes. A disadvantage
is that it is likely to be more difficult to obtain specimens from
the controls than from the exposed population. If, for example,
exposed women were more likely than controls to provide specimens
of early abortions, the unadjusted rate of chromosome abnormalities
would appear, incorrectly, to be higher.
4.2.2.2. Studies of fetal deaths
Studies in which the products of conception from fetal deaths
are examined cytogenetically are likely to be the most productive
in terms of the proportion of chromosome abnormalities that are
detected. However, such studies are difficult because of problems
in specimen retrieval, culture failure, and the high cost of the
tissue culture procedures required. Usable specimens will be
obtainable from only a portion of cases, even with the best
retrieval systems, since viable fetal tissues are sometimes not
present at the time of expulsion. Organization of retrieval
systems can be a challenge, even in such ideal situations as a
teaching hospital, and are extremely difficult outside a medical
setting. Through special education, attempts can be made to
collect specimens from women having early abortions, where the
tissue is passed at home, and medical attention may not be sought.
In New York City, an interview study suggested that 40% of women
having a first trimester fetal death did not seek medical attention
(Kline et al., 1981). Factors affecting the probability of
specimen retrieval must always be examined to rule out biases that
would affect the outcome of the study.
When cytogenetic analysis is performed, further case loss
occurs because not all will be successfully karyotyped. This might
produce differences in studies done in different laboratories or at
different times. Thus, cases and controls should be studied at the
same time, in the same laboratory. Ideally, the laboratory
carrying out the study should not know if a specimen is from a
control or a case.
One further problem in interpreting studies of fetal death is
the possibility that an environmental agent might influence not
only the rate of occurrence of a genetic abnormality, but also the
probability that a conceptus with an abnormality can survive to a
particular point in gestation. If an exposure reduces the
viability of a conceptus with a chromosome abnormality, such as
monosomy X, so that it is lost even earlier in gestation, before
recognizable pregnancy, a reduction in that anomaly will be seen in
all recognized conceptuses. An exposure that postpones the fetal
death of a conceptus with abnormality from before the usual
recognition of pregnancy to later in gestation would result in
an increased proportion of detected abnormalities.
4.2.2.3. Studies of prenatal diagnosis specimens
In some locations, amniocentesis for prenatal diagnosis is
widely available. This procedure is usually done at 16 - 20 weeks
of gestation. In such areas, many women exposed to putative
mutagens might seek such procedures, though a much smaller
proportion of controls might do so. While data available from
such an outcome should be used in any analysis, this cannot be
regarded as a plausible sole source of pertinent data on chromosome
abnormalities. It should be noted that, if all women in a
population undergo amniocentesis, the expected proportion of
chromosomally abnormal fetuses is 1%.
4.2.2.4. Studies of live births
Studies of live-born infants and older children are easiest to
undertake, and cheapest to perform. However, because only a small
proportion would be expected to be affected (0.6%), a relatively
large-scale study would have to be undertaken to detect a
statistically-significant increase in the proportion affected. A
sample of 25 000 births is needed to detect a doubling of the
trisomy rate at birth ( P < 0.05)a.
-------------------------------------------------------------------
a For the purposes of discussion, cases with an extra sex
chromosome have been included.
Some inferences are possible in relation to large exposed
populations, based, in part, on phenotypic evaluation. Cytogenetic
study for mutation investigation might, for example, be carried out
only on children with major malformations at birth, i.e., about 2%.
In any event, such studies would be indicated on this group of
children for clinical reasons. This approach would reveal
primarily only unbalanced autosomal abnormalities, about 1/3 - 1/2
of all chromosomal abnormalities in live births (both structural
and numerical abnormalities would be detected). Such a restriction
in a cytogenetic study would result in perhaps 10 - 20% of those
evaluated being found to have an abnormality. This reduces 50-fold
the total number of cytogenetic studies required in the population,
but does not avoid the need for a large original exposed population
from which the selected group with malformations would be drawn.
4.2.2.5. Studies of indicator phenotypes of chromosomal abnormalities
Inferences in large populations might be possible on the basis
of simple enumeration of Down's syndrome individuals, 98% of whom
are likely to be trisomic or carry a de novo chromosome
rearrangement. The expected frequency is about 1 in 1000 births.
Similarly, cases of Patau syndrome and Edwards syndrome (resulting
from 47, +13 and 47, +18, respectively) might also be enumerated.
The expected frequencies, however, are much smaller (each about 1
in 10 000 live births (Hook & Hamerton, 1977), and the phenotypes
are not as useful an indication of karyotypic abnormality.
Some conclusions on the probability of karyotypic abnormality
can be reached through morphological examination and classification
of specimens from fetal death. A useful classification scheme,
suggested by Byrne (1983), provides categories reflecting the
degree of organization of development, as well as developmental
age. These categories range from an "empty sac" with no visible
embryo, to an apparently normal fetus greater than 30 mm. Studies
associating specimen morphology with karyotype have shown that,
while about 1/2 of the most poorly-developed specimens have a
chromosome abnormality, less than 2% of fetuses greater than 30 mm
in length and with no visible malformations externally, will have
a chromosome abnormality (Byrne, 1983). Thus, if resources are
limited, such specimens might be left unstudied without much loss
of information.
True hydatidiform moles represent a category of conception
where the chromosome constitution is usually 46,XX, reflecting 2
identical male haploid complements in an egg with no maternal
chromosomes. Although other rare karyotypes occur, single
morphological examination is sufficient to indicate the chromosome
abnormality with over 98% accuracy.
4.2.3. Procedures
4.2.3.1. Fetal specimens
In a prospective study, women can be provided with sterile
containers containing a balanced salt solution for collection of
specimens from early fetal deaths. Later fetal deaths are likely
to reach medical attention, and collection systems must be
organized at likely treatment areas. Specimen containers should
also be available at such possible collection points as clinics,
emergency rooms, and wards.
(a) Morphological examination
Specimens should be examined externally and described in a
classification scheme that reflects the degree of development
as well as developmental age. Fetal autopsies can be performed
on specimens of 30 mm or more. A careful description of the
classification scheme must be provided, with photographs, whenever
possible, of abnormal specimens.
(b) Karyotyping
Successful cultures can be obtained, up to 5 days after
expulsion, though such a delay is not recommended. Thus, transport
of samples over some distance is possible. As the products of
conception are almost never obtained in a sterile state, the use of
culture medium for storage is not recommended, since it will
encourage the growth of contaminants. Specimens should be kept
refrigerated (but not frozen), during storage or transport over
long distances.
Specimens from induced abortions should be obtained in a
sterile container from the operating room or clinic. Care must be
taken to prevent the usual routine fixation of such specimens.
A most important aspect of the procedure is the careful
examination of the specimen to ensure that only fetal tissues are
taken for culture. Some specimens will contain only decidua, and
cannot be used. Any fetal tissue can be used for karyotyping; if
available, fresh tissues from the embryo or fetus are most
desirable; however, the actual embryo may be very small for the
length of gestation, very macerated, or absent altogether. In this
case, fetal membranes (amnion and/or chorion), or placental villi,
carefully dissected away from maternal tissues, may be used.
Maternal cell contamination is an inevitable possibility in such
studies, but evidence suggests that it is infrequent in experienced
hands (Warburton et al., 1980; Hassold, 1982).
Fetal tissues may take from a few days to several weeks to
reach the stage where they can be karyotyped. Cover slip
preparations with in situ chromosome preparations allow faster
karyotyping, but may not yield as large a number of analysable
cells as cultures in flasks, which must be trypsinized before
harvesting (Byrne, 1983).
Banded chromosome analysis should be carried out on all
specimens, and on at least 10 cells from each culture analysed.
Mosaicism is not uncommon among cultures from spontaneous
abortions. More cells must be counted if a non-modal cell, not
accounted for by random loss, is found among the first 10 cells,
e.g., if a normal cell is found in an otherwise trisomic culture,
or a trisomic cell or a 45,X cell is found in an otherwise normal
culture. In general, the proportion of mosaics among all
abnormalities detected, is expected to be small (Warburton, 1980).
For the purposes of initial analyses, it is suggested that they be
classified with non-mosaic aberrations with the same abnormal line.
Almost all instances of numerical chromosome abnormalities
are presumably the result of a mutation in the most recent
generation. Thus, if monosomy, trisomy, triploidy, or tetraploidy
is discovered, then study of the parents to confirm a mutation is
not necessary, as the likelihood of mutation is perhaps 0.99.
However, this is not the case for structural cytogenetic
abnormalities. At about 18 weeks of gestation, for instance,
only about 40% of structural abnormalities are the result of a
recent mutation, 60% being inherited from carrier parents. Thus,
if a structural rearrangement is observed, study of the parents
should be undertaken to determine if this is the result of a
mutation first manifest in the offspring.
4.2.3.2. Live births and other offspring
Chromosome studies will normally be performed on PHA-stimulated
lymphocyte cultures from specimens of peripheral blood. Specimens
should be collected as soon as possible after birth in order to
minimize losses of neonatal deaths. Cord blood can be used, and is
easy to collect routinely.
Preservation of whole blood samples by deep-freezing (-70 °C.)
in DMSO will make possible a good retrieval of cells for chromosome
analysis (or other kinds of studies) for up to a year (Nakagone et
al., 1982). Furthermore, specimens can be collected and stored
before laboratory facilities have been organized for the study,
and, if necessary, can be transported to a laboratory some distance
away.
If a structural rearrangement is found, parental studies are
necessary.
4.2.3.3. Detection of "indicator" phenotypes for germinal chromosomal
mutations - trisomies
Cases of Down's syndrome can be identified through medical
records, vital statistics, registries, or education settings.
Identification is likely to be incomplete through any of these
routes, and care must be taken that both exposed and unexposed
women are equally likely to be discovered.
4.2.3.4. Data presentation
Results in those exposed should initially be presented
according to broad categories of events (Table 5) and also
within subcategories, when sample size allows, because different
chromosomal abnormalities may reflect different etiological
factors. Trisomy 21 and trisomy 16 may well result from different
mechanisms, for example, while increased triploidy might indicate
increased dispermy.
Data should be presented on "conceptions" followed from
detection of pregnancy, and results compared by category between
exposed and control groups. Results should be expressed as the
proportion of either the exposed or control group with a specific
abnormality. Data should be stratified by maternal age, or other
adjustment made for this variable, because of the very strong
association of maternal age with trisomies (Hassold et al., 1980,
1984). In addition, exploratory analysis should be undertaken
of other possible confounding variables such as ethnicity,
socioeconomic status, cigarette smoking, and alcohol consumption.
Any differences between exposed and control groups in these
variables should be adjusted for in the analysis by sample
stratification, or multivariate analyses. Results should also
be analysed according to whether the father, the mother, or both
parents have been exposed. If sufficient data are available by
exposure category, a dose-effect relationship should be sought.
With regard to structural rearrangement, cases in which one or
both parents cannot be studied should be scored separately, and
regarded as possible mutants. The mutation rate for structural
rearrangements should be reported as a range. The minimum boundary
excludes such cases as being of unknown status. The maximum
boundary includes such cases.
It should also be recognized that lack of an observed
difference during the first few years after exposure does not
preclude the possibility of late manifestations of effects. In one
study, it has been claimed that a radiation effect on trisomy 21
frequency first manifested itself 10 years after exposure (Alberman
et al., 1972).
A problem that may arise in the evaluation of results is that
of missing data. Pregnant exposed and control individuals may die,
or withdraw before the outcome of the pregnancy is established.
Moreover, if a fetal death occurs, tissue may not be collected and,
if collected, the specimen may not include fetal tissue. Even if
fetal tissue is set up in culture, the karyotyping procedures may
not be successful. In experienced laboratories, the proportion of
successful cultures has varied from 65% to 90% (Warburton, 1980).
Moreover, culture failures are likely to be preferentially higher
for fetuses with chromosome abnormalities because the greatest
proportion of culture failures occurs in specimens with poorly
"organized" morphogenesis, which have been dead for some time in
utero. Among specimens cultured successfully, the proportion of
cytogenetic abnormalities in specimens with poorly organized
morphologies is at least 50%, at least twice as high as that of
specimens whose morphogenesis is more normal (Byrne et al., in
press). Assuming that this occurs also among unsuccessful
cultures, then those of unknown outcome will contain a higher
proportion of chromosomally abnormal specimens than successful
cultures.
Another problem arises if the effect of exposure to a suspect
substance is not to increase the proportion of conceptuses with
abnormalities but only to alter the proportion of conceptuses whose
karyotypes can be determined. If, for example, such a detection
effect occurs selectively more often on fetuses of abnormal
karyotype, then there will be a bias towards an apparent mutagenic
effect of the substance.
Table 5. Types of germinal chromosomal abnormalities
-------------------------------------------------------------------
Numerical Aberrations: all presumed mutants
Monosomy (almost entirely X monosomy)
Trisomy 16
21
others
Triploidya
Tetraploidya
Structural aberrationsb
Unbalanced: Robertsonian
deletions and rings
markers and fragments
other
Balanced:
Robertsonian
inversions
reciprocal
-------------------------------------------------------------------
a Most triploidy results from dispermy, and tetraploidy from
mitotic non-disjunction. Thus, they might not be regarded
strictly as germinal cell mutations.
b Both parents must be shown to have normal chromosomes, before a
case can be regarded as new mutant.
For these reasons, inferences about differences between exposed
and control populations (on the basis of comparison of the observed
proportion of the abnormal among those studies) require explicit
assumptions about the conceptuses of unknown karyotype. The
characteristics of this group, for example, the proportion of them
among the total number of conceptuses in the sub-populations, their
fetal pathology, and their maternal age association, should be
investigated closely. If there are striking differences in the
nature of the conceptuses of unknown karyotype between exposed
and unexposed populations, great caution is necessary in drawing
conclusions from comparisons involving conceptuses of known
karyotype.
4.3. Biochemical Approaches to Detecting Gene Mutations in Human
Populations
4.3.1. Biochemical methods for monitoring for gene mutations
Two approaches have been used to study the proteins in
offspring of selected individuals. One is based on the detection
of differences in charge, shape, or size of proteins by one- or
two-dimensional electrophoretic techniques. In the other,
quantitative enzyme activity measurements are used to detect enzyme
variants associated with either the loss of enzyme function or the
absence of the enzyme protein.
4.3.1.1. One-dimensional electrophoresis
The introduction of new electrophoretic technology has
increased the proportion of variants detectable at the protein
level and the number of gene products that can be studied. Using
these techniques, it has been shown that, in contrast to previous
assumptions that only one-third of the amino acid substitutions
could be detected, many neutral change substitutions can also be
detected. This reflects the alterations in higher-order protein
structure associated with these later substitutions, which change
the charge distribution on the molecule. Experimental data suggest
that 80 - 90% of all amino acid substitutions may be detectable
with refined techniques (Johnson, 1976, 1977; Ramshaw et al., 1979;
Fuerst & Ferrell, 1980). The use of narrow-range pH gradients for
isoelectric focusing has also increased the detection and
resolution of the variants at many loci (Altland et al., 1979;
Chramback et al., 1980).
The number of gene products that can be studied by
electrophoretic analysis depends on the availability of techniques
to demonstrate selectively the position of the allele products
following electrophoretic separation. In addition to previously
developed protein specific stains (Harris & Hopkinson, 1976),
immunological techniques (e.g., immunoblotting and immunofixation)
(Chapuis-Cellier et al., 1980; Tsang et al., 1983) have become more
widely used, especially with the recent modifications. One
additional approach has involved several sequential steps of
electrophoresis to separate the protein of interest from the
remaining proteins, so that standard protein-staining techniques
can be used for visualization (Altland & Hackler, 1984).
The results of many studies involving genetic typings of large
numbers of individuals, either for population structure studies
or in the context of the development of mutation monitoring
programmes, have demonstrated the feasibility of sizeable screening
efforts using blood samples as the source of material (Harris et
al., 1974; Neel et al., 1980a,b). The products of at least 40 - 50
loci can be routinely screened by electrophoresis with either
starch and/or polyacrylamide as the support medium. For many
proteins, either support can be used, with polyacrylamide having
the advantage of requiring smaller sample aliquots and potentially
increased resolution. Isoelectric-focusing is feasible for many of
these proteins, especially when the potential for increased
resolution of some proteins is considered. Electrophoretic
techniques have been used in several human mutation screening
programmes (Neel et al., 1980a,b; Altland et al., 1982a,b).
They have also been used for estimating the mutation rate in mice
(Pretsch & Narayanan, 1979; Johnson & Lewis, 1983; Neel, 1983) and
Drosophila (Mukai & Cockerham, 1977; Voelker et al., 1980).
Electrophoretic mobility variants have been identified and the
genetic transmission of the trait to subsequent generations has
been confirmed.
4.3.1.2. Two-dimensional electrophoresis
The high resolution two-dimensional (2-D) electrophoretic
techniques described by O'Farrell (1975) with many subsequent
modifications (Dunn & Burghes, 1983a,b) has been used to separate
the large array of proteins found in cells and tissues. The
positions of the several hundred proteins in many types of samples
can be visualized by protein staining, especially when the highly
sensitive silver stains (Merril et al., 1981; Sammons et al., 1981)
or isotopic labelling of proteins in nucleated cells followed by
autoradiography or fluorography (McConkey et al., 1979; Thomas et
al., 1984) are used.
The ability of the 2-D system to resolve electrophoretic
mobility variants has been shown by Warner et al. (1982). In
other studies, the level of heterozygosity for polypeptides has
been examined in several types of human cells and tissues (McConkey
et al., 1979; Walton et al., 1979; Comings, 1982; Hamaguchi et al.,
1982). More recently, extensive studies have been reported on the
level of genetic variation in plasma proteins and other blood
proteins (Rosenblum et al., 1983, 1984). One advantage of the 2-D
approach over the single-dimension electrophoresis techniques is
the ability to study the gene products of at least several hundred
loci in each sample (individual). The 2-D electrophoresis
technique has been used to estimate the induced mutation rate
in mice (Klose, 1979; Marshall et al., 1984).
4.3.1.3. Enzyme activity
Much of the background to the search for enzyme deficiency
variants is derived from the study of metabolic diseases. The
extensive lists of inborn errors of metabolism indicate not only
the prevalence of genetic events associated with loss of enzyme
function but also the usefulness of quantitative enzyme assays as a
tool for studying this class of genetic events (Beutler, 1979; Kahn
et al., 1979; Miwa, 1979).
The search for mutational events resulting in loss of function,
owing to either loss or nonfunctionality of the gene product, is
complementary to the electrophoretic assays, in that it detects
genetic events, not normally detectable by standard electrophoretic
assays. Current data suggest that inherited rare enzyme deficiency
variants occur more frequently in human populations than rare
electrophoretic variants (Mohrenweiser, 1981; Mohrenweiser & Neel,
1981; Satoh et al., 1983). In addition, mutations resulting in
loss of enzyme function occur at least as frequently as
electrophoretically identifiable mutations in mice and Drosophila,
following exposure to mutagenic agents, either radiation or
chemical (Racine et al., 1980; Charles & Pretsch, 1981; Johnson &
Lewis, 1981).
4.3.1.4. Other biochemical approaches
Alterations in base sequence can be analysed using the
restriction enzyme mapping techniques (Botstein et al., 1980;
Skolnick & Francke, 1982; Southern, 1982). Most of the current
effort, in addition to defining gene structure, has been directed
towards identification of polymorphisms at restriction enzyme sites
and subsequent linkage analysis, but similar techniques could be
employed for screening for mutational events (Beaudet, 1983; Cooper
& Schmidtke, 1984 for recent listings of cloned human DNAs).
Although it is clear that it is now technically possible to
detect base sequence changes, incorporating this approach into
a monitoring protocol must await further developments that will
facilitate the generation of the quantity of data necessary for a
mutation screening programme.
In addition to the sensitivity of the method, another potential
advantage of an approach that studies mutations by examining the
DNA directly, is the ability to obtain very large quantities of
data from each individual. Thus, this method can be employed for
the study of small populations.
4.3.2. Analytical strategy and methodological considerations
Biochemical analyses of the samples collected from the F
population (offspring of exposed individuals) will be carried out
usually in a number of laboratories while population and health
data will be derived from various sources. It is unlikely that all
laboratories will be able to complete the entire battery of assays
or that the techniques will be identical in each laboratory or for
each study, thus each laboratory should receive samples from both
exposed and control groups. The key for the successful completion
of the analytical aspects of any study will be for each laboratory
to establish and maintain high standards of technical competence
and performance. Provision should be made for the exchange of
samples, as this will increase the amount of data obtained from
each individual and also serve to confirm the existence of
interesting observations. The basic technical procedures for
electrophoresis and enzyme activity assays, are available. These
techniques form the basis for establishing a new laboratory effort.
The methods described in each section are current routine
laboratory procedures; thus, the biochemical techniques for
monitoring germinal mutations in a human population are available.
4.3.2.1. One-dimensional electrophoresis
Approximately 40 - 50 blood proteins have been examined for
electrophoretic variation in many laboratories including the
laboratories at the University of Michigan, USA (Neel et al.,
1980a) and in Japan at the Radiation Effects Research Foundation
(RERF) (Neel et al., 1980b). Approximately half of the proteins
studied at the University of Michigan use polyacrylamide as the
support medium while the laboratory at RERF relies more on starch
as the support medium. Many of these proteins are also being
studied by isoelectric focusing techniques as a component of
mutation screening programmes in other laboratories (Altland et
al., 1982a,b).
Techniques are available for the electrophoretic analysis
of approximately 20 proteins, when the sample is obtained from
dried blood (Altland et al., 1979, 1982b; Metropolitan Police
Forensic Science Laboratory, 1980). The general principals of
electrophoretic separation and isozyme (protein) identification
using samples collected as dried stains are as described for other
blood samples.
High-speed one-dimensional electrophoretic screening
procedures have been developed for vertical polyacrylamide gel
electrophoresis, flat bed isoelectric focusing, and the sequential
combinations of these procedures. With these techniques, using
multiple sample handling procedures, 96 samples are analysed
simultaneously (Altland et al., 1982a). This is particularly
useful when large numbers of samples are being analysed.
An electrophoretic mobility variant is identified by an
alteration in the standard profile that is consistent with the
appearance of a new allele product. This decision process must
include previous knowledge of the protein subunit structure,
chromosomal location (e.g., hemizygous males) and factors such as
age, sex, etc., which are important in interpreting the data. The
new protein should have characteristics that indicate that it
differs from the original protein by an amino acid change. It
is important to confirm, using as many additional techniques as
possible, that any new variant is not the result of an artifactual
or secondary alteration in protein structure. Ultimately, such
confirmation would require amino acid or DNA sequencing studies.
This is important because of the low probability of obtaining data
to confirm genetic transmission of a new mutation in human
populations. The proteins that are being studied in the above-
mentioned studies have been selected because of the low frequency
of artifactual findings. But, as with any technique, it is
important for each laboratory to undertake appropriate control
experiments.
4.3.2.2. Two-dimensional electrophoresis
The 2-D electrophoresis techniques, used for human mutation
monitoring studies, have been described by Neel et al. (1983, 1984)
and modifications of the technique of O'Farrell (1975), by Anderson
& Anderson (1977) and Anderson et al. (1980). The blood sample is
fractionated into various components, e.g., plasma, erythrocyte
membranes, haemolysate, platelet, etc., the proteins of which are
then analysed. The proteins in the cell fraction to be studied are
solubilized in 4 - 8 M urea, which dissociates the multimeric
proteins into component subunits. The component polypeptides are
separated on the basis of charge by isoelectric-focusing in the
first dimension. Electrophoresis in the second dimension is in
the presence of sodium dodecylsulfate, so that the proteins are
separated in this dimension on the basis of apparent molecular
size. The sensitive silver-based staining techniques are most
often used for identifying the positions of the separated
polypeptides (Merril et al., 1981; Sammons et al., 1981).
Analysis of the results of 2-D electrophoresis can be scored by
visual inspection (Hanash et al., 1982; Rosenblum et al., 1983,
1984), though recently, significant progress has been achieved
in automating and computerizing the analysis of these gels
(Skolnick, 1982; Skolnick et al., 1982; Skolnick & Neel, in press).
Computerized analysis can increase the amount of data obtained from
each gel (the number of locus tests completed for each sample) and
also reduce the workload at this step.
4.3.2.3. Enzyme activity
The methods for detecting enzyme deficiency variants, defined
as a level of enzyme activity that is less than 65% of the mean
for the population and more than 3 standard deviation units below
the mean, have been described by Fielek & Mohrenweiser (1979),
Mohrenweiser & Fielek (1982), and Mohrenweiser (1983a). Additional
or alternative methods have been described by Bulfield & Moore
(1974), Krietsch et al. (1977), Eber et al. (1979), and Satoh et
al. (1983). The general analytical strategy for analysing large
groups of samples has been outlined by Mohrenweiser (1983b).
Enzymes that are: (a) present with reasonable levels of activity;
(b) primarily the gene product of a single locus; (c) not
influenced by environmental factors (e.g., nutritional status);
(d) easy to assay; and (e) exhibit little total variation among
individuals, are good candidates for inclusion in this approach
(Mohrenweiser, 1982; 1983a,b). The gene products of at least
12 - 14 loci can be routinely monitored for the presence of null
variants in erythrocytes.
Each of these biochemical approaches either has been, or is
being, used to obtain data for the estimation of radiation-induced
mutation rates in human populations. Detailed techniques are
available for each component in this section. The most significant
problem is obtaining the commitments (funding, study and control
populations, etc.) necessary to initiate a major mutagenic
study. Obtaining agreements on general approaches, necessary
standardization of technical aspects, and scoring of significant
events is a lesser problem.
4.3.2.4. Sample acquisition and storage
There are two general strategies for sample acquisition. The
first involves obtaining a large volume (up to 20 ml, although 5 ml
is an adequate sample) of whole blood. All members of a nuclear
family are sampled to the maximum extent possible, so that the
material for family studies to determine heritability of the
characteristic is routinely available. Experience has shown that
when the gene products of 40 - 50 loci are studied, family studies
are necessary in at least 10% of the families, even when only a
single child per family is studied. With the number of loci
studied with the 2-D electrophoresis technique, obviously the
percentage of family studies increases to include almost every
family. In either case, when the number of locus tests per sample
or family unit increases to this level, the work involved in
recontacting families for additional samples becomes a considerable
task.
The blood sample is fractionated into at least plasma, buffy
coat, and erythrocyte fractions. Each part is subdivided into a
fraction for routine analysis as well as a fraction for long-term
storage and potential additional analysis. An aliquot of red
cells is stored in glycerol-sorbitol solution and retained in
liquid nitrogen for blood group analysis in the event of a
potential mutation being observed. The "white cell" component
could be fractionated into several components, e.g., platelets,
polymorphonuclear leukocytes, etc., which could be used as samples
for 2-D electrophoretic studies. Alternatively, either all or
fractions of the lymphocyte samples could be retained as a source
of DNA for future restriction enzyme mapping studies. Another
alternative to the above protocol would be to store the white cell
fraction under such conditions that intact, viable cells could be
recovered and, with appropriate cell culturing techniques, the
number of cells expanded for use in future studies. In certain
situations, where a specific population is of special interest and
the potential for follow-up studies is limited, it may be advisable
to establish and store transformed lymphoblast cell lines in order
to maintain the genetic information contained in this population
for future studies. The second acquisition strategy involves
obtaining a smaller volume of sample, often in the form of a dried
blood stain, which is usually obtained in conjunction with other
studies, usually with new-born metabolic screening programmes.
Procedures have been established for screening for gene mutations
at 20 loci using a blood sample obtained on a Guthrie Test card.
With these techniques, 300 samples or 6000 locus tests per day can
be completed (Altland et al., 1982a,b; Vogel & Altland, 1982).
As the new-born metabolic screening programme in many countries
encompasses the total new-born population, this strategy appears
useful for establishing a surveillance progamme for determining the
human germinal mutation rate. The number of locus tests from each
individual is somewhat less than with the larger samples, thus the
frequency of family studies is less, though obviously as the number
of tests per sample increases, it may be important, whenever
possible, to obtain concurrent parental samples.
It has been suggested that children with congenital birth
defects could be of special interest for monitoring studies because
of a high gene mutation rate in such children (Dubinin & Altukhov,
1979). However, study has not been able to confirm a similar
increased or high mutation rate among a group of children with
apparently similar congenital birth defects (Neel & Mohrenweiser,
1984). The usefulness of the approach of targeting specific F
individuals within a population for detailed study, rather than
all F individuals, needs further study.
4.3.3. Data management
Neel et al. (1979) have developed a computer-based data
management system for handling the types and volume of data
generated in a population monitoring programme. This includes
a sample identification system with a letter designation for the
geographical site of sample acquisition, a number code for family
identification, and a single digit indicating family position.
This identification, which is the first entry into a computerized
data management system, is used for identification of all data
generated, thus it is used for linkage to the electrophoretic
and enzyme activity files, which are additional components of
the computer-based data management system. It is linked to family
identification (e.g., name) only when follow-up is necessary, in
order to maintain confidentiality of records. A similar data
management system is currently in operation at RERF. It is
important that the data-management system should be complete
and compatible with all aspects of the study (section 2).
4.3.4. Considerations in screening for germinal mutations
4.3.4.1. Sample size
The logistic problems associated with a germinal mutation
screening programme are significant because of the scale of the
effort. The magnitude of the effort reflects both the rarity of
mutational events (background mutation rates of 105 - 106) and
the need for statistical methods to ascertain generally small
(10 - 100% increase) differences between background and induced
mutation rates. The sample sizes necessary for any of these
approaches to monitoring depend on the baseline rate of the effect
under consideration and the magnitude of the increase in mutation
rates that can be left undetected.
Vogel (1970), Neel (1980), and Vogel & Altland (1982) have
calculated the sample sizes necessary for a research design
involving the contrasting of 2 samples. The samples can be either
collected consecutively, as when studying the possibility of a
changing mutation rate in a defined population, or simultaneously,
when contrasting a control group of children with a group at high
risk of mutation because of the occupational or other exposure
of their parents. The minimum sizes of the 2 samples are
determined by the magnitude of the errors, type I (alpha) and type
II (phi) that are considered acceptable. The minimum increase in
mutation rate that is to be detected (or the maximum to be deemed
insignificant) also influences the sample size necessary to detect
an increased mutation rate. Most geneticists would agree that
any monitoring programme should detect an increase of 50% above
background. Using type I and type II error limits of 0.05 and 0.20
and a mutation rate of 1 x 10-5, it would require some 7.5 million
observations (Neel, 1980). There is considerable disagreement on
how much effort should be devoted to detecting smaller increases in
mutation rate. Given the exponential nature of the relationship,
detecting an increase of 20% involves 2 samples 5 times as large
as those required to detect a 50% increase. It is obvious that,
although the set of assumptions and limits may change, generating
a data base necessary for estimating changes in human mutation
rates (or finding that at some limit of power, the rate has not
changed) requires many observations and considerable analytical
effort.
With the biochemical approach, in which many proteins are
examined in any given child, and with 2 loci at risk of mutation
for each polypeptide included in the study, the number of children
will be only a small fraction of the number of determinations.
For example, in a pilot study (Neel et al., 1980a), 50 proteins
were examined for mutations influencing electrophoretic behaviour.
Thus, the demonstration of a difference of 50% between the 2 groups
could be accomplished with 2 samples of approximately 90 000
children (it is assumed that each protein of a child is an
independent test of mutation and that each child constitutes 2
tests of each protein, i.e., both parents exposed). With the
two-dimensional gel electrophoresis method, which could permit
simultaneous monitoring of 500 (or more) different proteins of
the blood serum, erythrocytes, and/or leukocytes, with the same
assumptions as above, the size of the 2 samples is reduced to
approximately 7500 individuals.
4.3.4.2. Distinction between "true" and "apparent" mutations
Whenever a genetic trait is encountered in a child, neither
of whose parents is affected, the possibility that it is due to a
discrepancy between legal and biological parentage rather than the
result of mutation must be considered. This is less likely when
the mutation involves a major chromosomal abnormality rather than
a biochemical marker, because individuals carrying the former are
often sterile or have a severly reduced life expectancy. With the
sentinel phenotypes or biochemical variants, when an apparent
mutant is encountered it is always necessary to carry out extensive
genetic typings on both parents, and the child. At present, with
the battery of test traits usually available, including enzymes,
blood groups, and HLA, a child whose legal father is not the
biological parent can be detected with an assurance approaching
98%. Thus, in a situation characteristic of many countries, such
as the USA, where 2 - 3% of the children born in wedlock have
fathers other than the legal one and where variants of the type
under consideration have a frequency of 2 - 4/1000 examinations,
"false" mutations (i.e., undetected parentage exclusions) will have
a frequency not very different from the present expectation for
mutation.
It should be pointed out that recent developments, which could
be used in a study such as this mutation protocol, are also useful
for paternity studies. Many gene products studied using two-
dimensional electrophoretic techniques are characterized by
genetic polymorphisms useful in parentage studies, resulting in
an improvement in the ability to detect discrepancies between
legal and biological parentage. The study of DNA restriction
site polymorphisms will also decrease the frequency of undetected
non-parentage, thus the probability of an apparent mutation being
due to non-parentage should become very small with the increased
number of loci that can now be studied.
In the final analysis, there may always be some uncertainty as
to whether a particular apparent mutant is due to an undetected
discrepancy between legal and biological parentage. If the
control population has been properly chosen, however, the amount
of non-parentage could be demonstrated to be the same in the two
populations, so that this factor would be a constant diluent in the
2 samples. Furthermore, with a statistical approach, recently
developed by Rothman et al. (1981), under suitable conditions,
probabilities can be developed for parentage, and apparent
mutations ranked according to these probabilities.
4.3.4.3. Implementation of gene mutation screening programmes
Final decisions regarding analytical strategy and technical
approaches, beyond the points described above, must await further
information regarding the population of interest. The magnitude
of the study helps determine the number of laboratory centres
necessary to complete the study within a reasonable time.
Similarly, the size of the study population (and estimated mutation
rate) will dictate the amount of data that must be obtained from
each individual studied for the generation of a statistically
significant data base. The size of the study population (as well
as the age of the individuals from whom blood is obtained) will
also need to be considered when blood fractionation and allocation
procedures are finalized. Furthermore, techniques are continually
being refined and new approaches being developed, and it is unwise
to dictate the specific technical details until such time as the
commitment to proceed with a study has been made. At this point,
laboratories should be able, with relative ease, to define the
technical details for the laboratory and analytical protocols.
4.3.5. Summary
In many respects, with the limitations of the current state of
knowledge regarding the detection and subsequent consequences of
gene mutations, all practical approaches to estimating the germinal
mutation rate in human populations, following exposure to a
putative mutagen, should be considered to be complementary.
This is important as each technique differs in sensitivity, detects
different types of genetic damage or end-points, and has different
degrees of relevance for estimating potential health effects.
Therefore, it is logical in a collaborative monitoring exercise to
use all of the approaches in order to obtain the maximum possible
amount of data.
The technical aspects of a collaborative effort to use
biochemical methods to monitor selected human populations for
germinal mutation frequencies are available. Such programmes have
been conducted in the Federal Republic of Germany (Altland et al.,
1982b), Japan (Neel et al., 1980b; Satoh et al., 1983), and the USA
(Neel et al., 1980a; Mohrenweiser, 1981). Thus, feasibility has
been tested at a significant level of effort. There is no
technical reason for not expanding such an effort to other
populations of interest.
4.4. Sentinel Phenotypes
4.4.1. Introduction
A sentinel phenotype is a clinical disorder that occurs
sporadically as a consequence of a single, highly penetrant
mutant gene, which is a dominant or X-linked trait of considerable
frequency and low fitness, and is uniformly expressed and
accurately diagnosable with a minimal clinical effort, but a
relatively high probability of ascertainment (Mulvihill & Czeizel,
1983).
A new dominant mutation can manifest itself at any time in the
human life-span. In practice, it is worth distinguishing three
separate groups of sentinel phenotypes: sentinel anomalies,
easily observed at birth and generally recorded in birth defects
registries (Table 6); sentinel childhood tumours, which occur in a
well defined age-group and are recorded in childhood tumour
registries; and genetic disorders with delayed onset, which are
readily diagnosed and recorded in various types of genetic disease
registries (Table 7). For practical reasons, sentinel anomalies
are the easiest to handle.
4.4.2. Basis of method
Certain essential requirements for the surveillance or
monitoring of sentinel anomalies include:
(a) Sensitivity and specificity
Theoretically, all dominant phenotypes might be suitable for
these purposes. McKusick (1983) catalogues 1828 autosomal dominant
traits in man. These probably occur at a total birth prevalence
of 1% (Matsunaga, 1982). Nearly half of autosomal dominant
disorders may be caused by new mutations (Holmes et al., 1981).
Nevertheless, at present, no single dominant phenotype suits all,
or nearly all, the criteria of the above definition. However, the
22 anomalies listed in Table 6 comply satisfactorily. The main
problems are the validity of a diagnosis and the relatively
uncommon occurrence of all these candidate anomalies. If the
baseline prevalences are too low, both the specificity and
sensitivity of population screening are decreased (Hook, 1981b).
(b) Reliable and accurate diagnosis
A sentinel phenotype must be clearly distinguishable from
related disorders. However, each of the anomalies in Table 6 has
features that overlap with so many other anomalies that highly-
trained clinical experts must be involved in the diagnosis. In
general, these well-known genetic anomalies have a considerable
genetic heterogeneity. In addition, genocopies and sometimes
phenocopies exist for all sentinel anomaly candidates. The task
is to make a precise nosological diagnosis, i.e., to identify the
effect of the specific locus or sometimes of the given allele of
the specific locus.
Table 6. Candidate sentinel anomalies and the figures of the
Hungarian Programme per 10 000 births
-------------------------------------------------------------------
Congenital anomaly McKusick Expected
(s = syndrome) number rate
(1983)
-------------------------------------------------------------------
Achondroplasia 10 080 1.08
Acrocephalosyndactyly type I (Apert s.) 10 120 0.07
Acrocephalosyndactyly type V (Pfeiffer s.) 10 160 0.02
Aniridia, isolated 10 620 0.11
Van der Woude s. 11 930 0.02
Cleidocranial dysplasia 11 960 0.04
Contractural arachnodactyly 12 105 0.04
Crouzon s. 12 350 0.11
EEC s. 12 990 0.04
Holt-Oram s. 14 290 0.07
Treacher-Collins s. 15 450 0.04
Moebius s. 15 790 0.04
Nail-patella s. 16 120 0.04
Oculo-dento-digital (ODD s.) 16 420 0.02
Osteogenesis imperfecta, type I 16 620 0.22
Polysyndactyly preaxial IV. 17 420 0.22
Split hand and/or foot, typical 18 360 0.15
Spondyloepiphyseal dysplasia cong. 18 390 0.07
Thanatophoric dwarfism 18 760 0.04
Whistling face (Freeman-Sheldon s.) 19 370 0.02
Incontinentia pigmenti (Bloch-Sulzberger s.) 30 830 0.11
Oral-facial-digital s. (Gorlin-Psaume s. or 31 120 0.02
OFD I.)
-------------------------------------------------------------------
Total 2.59
-------------------------------------------------------------------
Cataracta 11 620 0.71
Ptosisa 17 830 0.66
-------------------------------------------------------------------
Grand total 3.96
-------------------------------------------------------------------
a Excluded from the final list because ptosis was rarely
ascertained and the nosological diagnosis of cataract was
impossible.
Table 7. Candidate sentinel phenotypes without reliable nenonatal
manifestations
--------------------------------------------------------------------
Disorder McKuskick Inheritance
number
(1983)
--------------------------------------------------------------------
Amelogenesis 10 450 ADa
Exostoses, multiple 13 370 AD
Fibrodysplasia ossificans progressiva 13 510 AD
Marfan's syndrome 15 470 AD
Myotonic dystrophy (Steinert disease) 16 090 AD
Neurofibromatosis (Recklinghausen disease) 16 220 AD
Polycystic kidneys 17 390 AD
Polyposis coli, I (familial) 17 510 AD
Polyposis intestinal, II (Peutz-Jeghers 17 520 AD
syndrome)
Polyposis intestinal, III (Gardner syndrome) 17 530 AD
Retinoblastoma (hereditary) 18 020 AD
Tuberous sclerosis 19 110 AD
von Hippel-Lindau syndrome 19 330 AD
Waardenburg syndrome 19 350 AD
Wilms' tumour (hereditary) 19 407 AD
Haemophilia A (Classical) 30 670 XRb
Haemophilia B (Christmas disease) 30 690 XR
Muscular dystrophy 331 020 XR
(Duchenne type)
Martin-Bell 30 955 XR
--------------------------------------------------------------------
a AD = autosomal dominant.
b XR = X-linked.
(c) Complete ascertainment
In countries where most births occur in hospitals and there are
neonatal check-ups, the easily-recognized congenital anomalies of
autosomal dominant origin can be used as sentinel anomalies,
particularly if a population-based registry or surveillance of
congenital anomalies is functioning.
(d) The knowledge of pedigree data
Since only a new occurrence of sentinel phenotype in a family
is a mutation, obtaining data from and/or clinical examination
of both parents is necessary to verify that neither is affected.
In practice, it is necessary to obtain clinical data on the
grandparents in order to exclude the possibility of non-penetrance
in the parents.
(e) Possibility of further action
After a significant increase in the rate of occurrence of one
or more mutations, it is essential to initiate an analytical
epidemiological study to identify the responsible mutagens and to
achieve the ultimate goal, i.e., prevention of further genetic
damage.
4.4.3. Relevance and limitations
The theoretical advantage of using sentinel phenotypes is
considerable. When a sentinel anomaly or sentinel childhood tumour
is recognized in an infant or child, in the absence of an affected
parent, there is little question that a germinal mutation has
occurred.
From the practical point of view, there are three advantages:
(a) Since sentinel phenotypes are diseases rather than
innocuous physical or biochemical traits or variants, the
affected person in a developed country will enter the
health-care system. Thus, affected persons will almost
certainly be registered in a recording system for reasons
other than their potential use in surveillance. Thus, the
so-called opportunistic approach is feasible (this term is
used to describe studies that are built on data, already
collected for other reasons).
(b) In the case of sentinel anomalies and sentinel childhood
tumours, parents with an affected infant or young child
are likely to be willing to cooperate in additional
investigations, with regard, for example, to family or
environmental histories.
(c) In sentinel phenotypes in which there is a low fitness,
there is a low probability of false paternity.
The main theoretical disadvantage is that the phenotype is not
an immediate gene product, but a distant manifestation of altered
DNA. Hence, intervening developmental events could obscure the
relationship between a mutagenic event and its expression as a
sentinel phenotype. In addition, dominant traits tend to have
greater variation in clinical manifestations than most recessive
and chromosomal syndromes. The main practical difficulty is that
most clinicians lack experience in the accurate diagnosis of these
rare disorders.
Since the expected incidence of mutant candidate sentinel
anomalies is 2 - 3 per 10 000 live births, a very large study
population is required in order to be able to detect any effects
in exposed groups of individuals.
4.4.4. Procedures
4.4.4.1. Surveillance of sentinel anomalies
A programme may be based on existing congenital anomaly
registries and surveillance systems. The target population
comprises all the new-born infants and, when possible, those up
to the age of one year. Several sources of ascertainment are
desirable. Three criteria of congenital anomaly surveillance
should be continuously checked:
(a) Completeness of ascertainment
In Hungary, the total birth prevalence of congenital
anomalies included in Chapter XIV "Congenital Anomaly" of the
International Classification of Diseases was estimated to be about
60 out of every 1000 new-born infants at, or after, birth (Czeizel
& Sankaranarayanan, 1984). Individual types of congenital
anomalies show a wide range of completeness of ascertainment.
(b) Validity of diagnosis
The validity of diagnosis may be known as a result of ad hoc
epidemiological studies. However, there may be a wide range of
misdiagnosis for different types of congenital anomalies.
(c) Base-line frequency
The baselines of all pregnancy outcomes (e.g., spontaneous and
induced abortions, still births, low-birthweight infants, etc.) and
other potentially confounding demographic variables (e.g., paternal
and maternal age) are also taken into consideration in evaluating
sentinel anomalies.
The surveillance of sentinel anomalies should consist of three
steps:
(1) Indexed patients with sentinel anomalies, notified to the
congenital anomalies registries and surveillance systems,
depending on their type of sentinel anomalies, should be
referred, with the help of their parents, to selected
participating paediatric, orthopaedic, or ophthalmological
institutions.
(2) Specialists in these institutions should examine these
indexed patients to confirm (or exclude) the nosological
diagnosis, to clarify family history (whether a sporadic
or familial case), to obtain data on environmental history
(with the help of a specifically-designed questionnaire),
and to give genetic counselling (if parents plan to have
babies in the future).
(3) The data obtained by experts at medical institutions
should be sent back to the programme director to be
evaluated.
4.4.5. Data interpretation
4.4.5.1. Surveillance of sentinel anomalies
Preliminary Hungarian experiences, based on data obtained
during 1980 - 82, are inconclusive. The registered prevalence at
birth of 24 sentinel anomalies was 2.6/10 000 births. The rate of
participation was only 41% (the main causes of case loss were the
death or severe condition of certain indexed patients) and the
diagnosis was confirmed in only 60% of the cases examined.
Nevertheless, as an average of 7 possible new mutations per year
was found over the 3-year period, during which nearly 430 000
births occurred, it is hoped that improving the completeness of
notification, participation rate, and diagnostic skills will
increase the number of new mutations identified in the future.
The possible causes of new mutations can be studied by
obtaining the environmental histories of the parents. The
environmental histories of the parents of familial cases can
provide adequate control data.
The main problem is that the basic data concerning even common
candidate sentinel anomalies are not known completely. Knowledge
of their frequency in different populations, the rate of prenatal
loss, definite figures on their penetrance,and a timetable of the
expression of their manifestations would improve the efficient use
of these sentinel anomalies.
The total prevalence at birth of the 24 sentinel anomalies
studied is not low. Assuming a Poisson distribution of events,
a background rate of 4.0 sentinel anomaly cases per 10 000 births,
and a 50% proportion of sporadicity, a sample size of 38 000 would
be needed to detect a doubling of mutation rate with probabilities
of type I and II errors at the 0.05 level. The null hypothesis
would be rejected, if 22 or more new mutations were seen.
The surveillance of sentinel anomalies has some other practical
benefits. Experts at referral centres have the opportunity to
improve their diagnostic skills, and to suggest prognosis and
treatment. From this aspect, verifying a genocopy or phenocopy is
important. Furthermore, surveillance may promote the detection of
teratogens. One of the main practical advantages of surveillance
is economy. In the Hungarian programme, an opportunistic and cost-
effective approach is used (Mulvihill & Czeizel 1983).
4.4.6. Conclusions
At present, most sentinel anomalies are not suitable for
surveillance, because of the inability to validate the diagnosis
and the incomplete ascertainment of some types. However, the
reliability of the diagnosis of sentinel anomalies will improve as
the number and skill of clinical geneticists increases. Thus, the
material in national or regional surveillance systems with a large
and unselected data base will become usable for studying sentinel
anomalies. The main argument for surveillance is that data are
already available in the registries and surveillance systems used
for other important medical purposes. In addition, the existence
of such programmes will improve existing medical care.
The monitoring of sentinel phenotypes has a specific purpose.
Defined populations, such as the offspring of self-poisoned
persons, epileptics, cancer patients (e.g., patients with acute
lymphoid leukaemia), or workers exposed to chemical compounds,
would be worth studying. However, the sentinel phenotype approach
is not useful for the monitoring of a small circumscribed new-born
population (< 50 000), exposed to known or suspected mutagens,
because such a small sample cannot yield statistically significant
results. Nevertheless, the importance of a opportunistic approach
should be stressed, because some types of industrial and disease
registries may provide data for this purpose.
There are a number of possibilities for improving the
feasibility of the surveillance and monitoring of sentinel
phenotypes. Because the birth prevalence of individual sentinel
anomalies is relatively low, international collaboration should be
encouraged. For example, the International Clearinghouse for Birth
Defects Monitoring Systems (Flynt & Hay, 1979) represents a huge
surveillance programme involving more than 24 countries and the
records of more than 2 million births per year. The majority of
the national and regional systems are able to evaluate the
occurrence of sentinel anomalies. However, the fact that most
sentinel anomalies do not have separate codes in the International
Classification of Disease is a serious technical limitation.
Unique codes need to be given to them so that international
comparisons of rates of sentinel anomalies can be made.
An important approach involves the use of sentinel childhood
tumours, mainly hereditary retinoblastomas and Wilms' tumours.
Patients with these disorders are diagnosed and, in general,
hospitalized in central institutions; thus, their records can
also be used for monitoring purposes. Although retinoblastoma
and Wilms' tumour are not usually detectable at birth, they have a
specific feature beyond those mentioned above. The vast majority
of cases of either type occur sporadically, and all sporadic
bilateral cases have an autosomal dominant origin, while most
sporadic unilateral cases are non-hereditary, persumably arising
from somatic mutation. Hence, these two types of tumour can be
used for surveillance of both germinal and somatic mutations.
Registries of individual genetic disorders (e.g., Huntington,
neurofibromatosis, polyposis coli) (Table 7), in several countries,
also offer a possibility for the surveillance of sentinel
phenotypes.
Improvement in diagnostic laboratory procedures has resulted
in progress in the surveillance of sentinel phenotypes (Vogel &
Altland, 1982). Recently, screening for the lipoproteinaemias has
become widespread and affords an opportunity for detecting common
familial hypercholesterolaemias of autosomal dominant origin.
Finally, some X-linked recessive traits and disorders, such as
Duchenne muscular dystrophy, Martin-Bell syndrome, or haemophilia
A, would be useful sentinel phenotypes, particularly if: (a) a
considerable fraction of cases were new mutations, and (b)
heterozygosity in the mothers could be detected reliably and
inexpensively by laboratory techniques.
Although, the monitoring of sentinel phenotypes cannot compete
with the efficacy of studying protein variants and the methods of
the new molecular genetics, simultaneous approaches can provide
possibilities for comparison and, in addition, the sentinel
phenotype approach may be more feasible in certain countries.
4.5. Fetal Death
4.5.1. Introduction
The proportion of human fetal deaths normally attributable to
germinal genic mutations is not known. If rare recessive lethals
are a common cause, studies on inbreeding have failed to show the
increase in fetal deaths expected (MacCluer, 1980). Dominant
lethal mutations acting in fetal life can occur, but their
contribution to fetal death is unknown and difficult to study with
present techniques. If this contribution were large, an increase
in fetal death with increasing paternal age might be expected.
Such an effect was seen in one study (Selvin & Garfinkel, 1976),
but was not seen in another study of late fetal deaths (Hatch,
1984).
The occurrence of a fetal death, particularly an early fetal
death, without any investigation of the products of conception, can
be considered as an indicator of germinal genic mutation. However,
any increase in FD could be due also to exposure to a teratogen
during pregnancy, or to interference with normal maternal hormonal
or immunological functions during pregnancy.
Teratogenic effects could be separated from genetic effects, if
the exposure could be allocated according to the sex of the exposed
parent, and whether it was pre-conceptional or post-conceptional.
Although the possibility exists that maternal exposure can occur
indirectly through paternal contact, most paternal exposures are
likely to affect fetal health via genetic changes transmitted in
the sperm. Pre-conceptional exposures only, in females, are also
most likely to induce genetic damage. However, it is not possible
to distinguish between germinal chromosome aberrations and genic
mutation using exposure data.
If morphological examination of the products of conception can
be carried out (section 4.2), further information can be gained.
Genetic effects, either chromosomal or genic mutations, are likely
to result in developmentally abnormal specimens, whereas factors
acting via interference with the maternal physiological response in
pregnancy are likely to result in developmentally normal specimens.
A change in the usual distribution of morphological types among
specimens of FD can be used to document an effect of an exposure:
a doubling of the number of "intact empty sacs" compared with
controls would confirm the validity of an observed rise in overall
rate of FD.
If the products of conception can be examined cytogenetically
(section 4.2), it may be possible to distinguish a class of FD
most probably due to genic mutation. A rise in developmentally
abnormal, but chromosomally normal specimens, after maternal
preconception, or paternal, exposures, might possible be attributed
to an increase in germinal genic mutations, lethal for fetal life.
4.5.2. Procedures
If exposed and unexposed populations are to be compared for
overall frequency of fetal death, the population(s) to be studied
must be clearly defined, and data collected on the total number of
births over 28 weeks of gestation, i.e., late fetal death (still
birth) and the total number of FDs. For purposes of comparison
between an exposed and unexposed population, bias should not be
introduced, if the ascertainment of either of these variables is
not complete, as long as the exposure status does not influence the
ascertainment. However, the higher the observed rate of early and
intermediate fetal death (spontaneous abortion) the greater the
power of the study, and the less likely it is to miss effects
limited to only particular subsets of the total (as is likely for
genetic effects). As most genetic effects will probably be
observed among the early spontaneous abortions, it is particularly
important to ascertain these as completely as possible. This is
not usually possible with most existing vital statistics sources
of fetal deaths. In a retrospective study, it will probably be
necessary to use either medical records or interviews to ascertain
reproductive history.
In a case-control study, efforts would be made to ascertain
spontaneous abortions through a medical treatment centre, and to
compare exposure histories between women who have spontaneous
abortions and women having live births. If the sample were
relatively small, cases and controls would probably have to be
matched for age, in order to control for this source of bias.
Finding controls at a point in gestation that matches the cases of
spontaneous abortion will also help to control for differences in
time of ascertainment, and differences in recall of events
occurring at the beginning of the pregnancy.
Data must be collected as carefully as possible on factors
known to influence the frequency of fetal death (e.g., maternal
age, socio-economic status, previous reproductive history, and
gestational age at abortion). Gestational age is usually estimated
from menstrual history. Even if data on the developmental stage of
the fetus is available, it should not be used to estimate
gestational age, since development often ceases some time before
abortion. Smoking and alcohol consumption histories would also be
useful, since these common exposures are associated with increased
abortion risk and might differ in exposed and unexposed
populations.
4.5.3. Data processing and presentation
Frequency is usually expressed as the number of fetal deaths
divided by the number of births over 28 weeks plus the number of
fetal deaths. Maternal age is the most important variable to be
controlled in any analysis, since it will influence the overall
frequency of abortion, and also the distribution of the kinds of
chromosome anomalies expected. Trisomies in spontaneous abortions
increase with maternal age in the same way as trisomies in live
births. On the other hand, monosomy X shows a decrease in
frequency with maternal age in spontaneous abortions. The risk
of abortion increases with the number of previous abortions in a
woman's history, and this variable should also be examined for
possible confounding effects.
4.5.4. Conclusions
(a) Fetal death can be used as a possible indicator of
germinal genetic mutation. Because of the high background
frequency, and concentration of anomalies, an increase can
be detected in a small sample size, e.g., a sample from
only 200 women will detect a doubling with 95% power.
(b) Without cytogenetic studies, it is not possible to define
the kind of genetic damage that has occurred (i.e.,
chromosomal or genic mutation) or to separate mutagenic
from teratogenic effects. However, defining the exposure
of the parent (pre-conceptional versus conceptional) may
make more inferences possible about the cause of a rise in
the frequency of fetal deaths.
(c) Estimates of the rate of fetal death may be imprecise
because of difficulties in identifying all causes, and in
determining the relevant population denominator of term
births. Data on factors known to influence rates of fetal
death, such as maternal age, socio-economic status, and
previous reproductive history, need to be collected, so
that possible biases can be explored in the analysis.
REFERENCES
ACHESON, E.D., ed. (1968) Record linkage in medicine,
Edinburgh, London, E & S Livingstone, Ltd.
ALBERMAN, E., POLANI, P.E., FRASER ROBERTS, J.A., SPICER,
C.C., ELLIOT, M., & ARMSTRONG, E. (1972) Parental exposure
to X-irradiation and Down's syndrome. Ann. hum. Genet.
(Lond.), 36: 195-208.
ALBERTINI, R.J. (1982) Studies with T-lymphocytes: an
approach to human mutagenicity monitoring. In: Bridges, B.A.,
Butterworth, B.E., & Weinstein, I.B., ed. Indicators of
genotoxic exposure, New York, Cold Spring Harbor Laboratory
Press, pp. 393-412 (Banbury Report No. 13).
ALBERTINI, R.J. (1983) Studies with 6-thioguanine-resistant
lymphocytes. In: DeSerres, F.J. & Sheridan, W., ed.
Utilization of mammalian specific locus studies in hazard
evaluation and estimation of genetic risk, New York, Plenum
Press, Vol. 28, pp. 11-27 (Environmental Science Research).
ALBERTINI, R.J. & SYLWESTER, D.L. (1984) 6-thioguanine
resistant lymphocytes in human blood. In: Kilbey, B.J.,
Legator, M., Nichols, W., & Ramel, C., ed. Handbook of
mutagenicity test procedures, Amsterdam, Elsevier Scientific
Publishing, pp. 357-372.
ALBERTINI, R.J., CASTLE, K.L., & BORCHERDING, W.R. (1982a)
T-cell cloning to detect the mutant 6-thioguanine-resistant
lymphocytes present in human peripheral blood. Proc. Natl
Acad. Sci. USA, 79: 6617-6621.
ALBERTINI, R.J., SYLWESTER, D.L., & ALLEN, E.F. (1982b) The
6-thioguanine resistant peripheral blood lymphocyte assay for
direct mutagenicity testing in man. In: Heddle, J.A., ed.
Mutagencity: new horizons in genetic toxicology, New York,
Academic Press, pp. 305-336.
ALBERTINI, R.J., DANNENBERG, B.D., SYLWESTER, D.L.,
PETTINGILL, C.A., & KELLEHER, P.C. (1983) Thioguanine
resistent (TGr) lymphocythes in humans: Placental blood
variant frequencies (VFs) are associated with exposure to
potential mutagens. Environ. Mutagen., 5: 401 (abstract).
ALBERTINI, R.J., CHASTENAY, B.J., O'NEILL, J.P., & GREENE,
C.J. (1984) T-cell cloning for human mutagenicity
monitoring. Environ. Mutagen., 6: 479 (abstract).
ALBERTINI, R.J., O'NEILL, H.P., HICKLAS, J.A., HEINTZ, N.H., &
KELLEHER, P.C. (in press) Southern blot analysis of HPRT
mutations occurring in vivo in human peripheral blood
T-lymphocytes. Environ. Mutagen. (abstract).
ALHADEFF, B. & COHEN, M.M. (1976) Frequency and distribution
of sister-chromatid exhanges in human peripheral lymphocytes.
Isr. J. med. Sci., 12: 1440-1447.
ALTLAND, K. & HACKLER, R. (1984) Concepts and applications
of double one-dimensional slab electrophoresis. In: Neuhoff,
V., ed. Electrophoresis 84, Berlin, Verlag Chemie, pp. 367-378.
ALTLAND, K., KAEMPFER, M., & GRANDA, H. (1979) Improved
screening test for abnormal hemoglobins from dried blood
samples. Hum Genet., 53: 97-100.
ALTLAND, K., KAEMPFER, M., & FORSSBOHM, M. (1982a) Mass
screening technique for detecting globin variant from newborn
blood samples. In: Bora, K.C., Douglas, G.R., & Nestmann,
E.R., ed. Chemical mutagenesis, human population monitoring
and genetic risk assessment, Amsterdam, Elsevier Biomedical
Press (Prog. Mutat. Res., Vol. 3, pp. 153-157).
ALTLAND, K., KAEMPFER, M., FORSSBOHM, M., & WERNER, W.
(1982b) Monitoring for changing mutation rates using blood
samples submitted for PKU screening. In: Bonne-Tamir, B.,
Cohen, T., & Goodman, R., ed. Human genetics, Part A. The
unfolding genome, New York, Alan R. Liss Inc., Vol. 103a,
pp. 227-287 (Prog. clin. biol. Res.).
AMES, B.N. (1983) Dietary carcinogens and anticarcinogens.
Oxygen radicals and degenerative diseases. Science, 221:
1256-1266.
ANDERSON, N.L. & ANDERSON, N.G. (1977) High resolution
two-dimensional electrophoresis of human plasma proteins.
Proc. Natl Acad. Sci. USA, 74: 5421-5425.
ANDERSON, N.L., EDWARDS, J.J., & GIOMETTI, C.S. (1980)
High-resolution two-dimensional electrophoretic mapping of
human proteins. In: Radola, B.J., ed. Electrophoresis 79:
advanced methods biochemical and clinical applications,
Berlin, New York, DeGruyter, pp. 313-328.
ANMEUS, H., MATSSON, P., & ZETTERBERG, G. (1982) Human
lymphoctyes resistant to 6-thioguanine: restrictions in the
use of a test for somatic mutations arising in vivo studies by
flow-cytometric enrichment of resistant cell nuclei. Mutat.
Res., 106: 163-178.
ARDITO, G., LAMBERTI, L., ANSALDI, E., & PONZETTO, P. (1980)
Sister-chromatid exchanges in cigarette-smoking human females
and their newborns. Mutat. Res., 78: 209-212.
AWA, A.A., SOFUMI, T., HONDA, T., ITON, M., NERISHI, S., &
OTAKE, M. (1978) Relationship between the radiation dose and
chromosome aberrations in atomic bomb survivors in Hiroshima
and Nagasaki. J. radiat. Res. (Tokyo), 19: 126-140.
BALAKRISHNA MURTHY, P. & PREMA, K. (1979) Sister-chromatid
exchanges in oral contraceptive users. Mutat. Res., 68:
149-152.
BALAKRISHNA MURTHY, P., BHASKARAM, P., & SRIKANTIA, S.G.
(1980) Sister-chromatid exchanges in protein-energy
malnutrition. Hum. Genet., 55: 405-406.
BEAUDET, A.L. (1983) Bibliography of cloned human and other
selected DNAs. Am. J. hum. Genet., 36: 235-249.
BECHER, R., SCHMIDT, C.G., THEISS, G., & HOSSFELD, D.K.
(1979) The rate of sister-chromatid exchange in normal human
bone marrow cells. Hum. Genet., 50: 213-216.
BEEBEE, G. (1981) Record linkage and needed improvements in
existing data sources. In: Petos, R. & Schneiderman, M., ed.
Quantification of occupational cancer, New York, Cold Spring
Harbor Laboratory, pp. 661-673 (Banbury Report No. 9).
BEEK, B. & OBE, G. (1979) Sister-chromatid exhanges in human
leukocyte chromosomes: spontaneous and induced frequencies in
early- and late-proliferating cells in vitro. Hum. Genet., 49:
51-61.
BENDER, M.A. & GOOCH, P.C. (1966) Somatic chromosome
aberrations induced by human whole-body irradiation: the
"Recuplex" criticality accident. Radiat. Res., 29: 568-582.
BENDER, M.A., GRIGGS, H.G., & BEDFORD, J.S. (1974)
Recombinational DNA repair and sister-chromatid exhanges.
Mutat. Res., 24: 117-123.
BEUTLER, E. (1979) Red cell enzyme defects as non-diseases
and diseases. Blood, 54: 1-7.
BIGBEE, W.L., BRANSCOMB, E.W., WEINTRAUB, H.B.,
PAPAYANNOPOULOU, Th., & STAMATOYANNOPOULOS, G. (1981) Cell
sorter immunofluorescene detection of human erythrocytes
labeled in suspension with antibodies specific for hemoglobin
S and C. J. immunol. Methods, 45: 117-127.
BIGBEE, W.L., VANDERLAAN, M., FONG, S.S.N., & JENSEN, R.H.
(1983) Monoclonal antibodies specific for the M- and N- forms
of human glycophorin A. Mol. Immunol., 20: 1353-1362.
BISHOP, J.B. & FEUERS, R.J. (1982) Development of a new
biochemical mutations test in mice based upon measurement of
enzyme activities. II. Test results with ethylmethanesulfonate
(EMS). Mutat. Res., 95: 263-271.
BLOOM, A.D., ed. (1981) Guidelines for studies of human
populations exposed to mutagenic and reproductive hazards,
New York, March of Dimes Birth Defects Foundation.
BOTSTEIN, D., WHITE, R.L., SKOLNICK, M., & DAVIS, R.W.
(1980) Construction of a genetic linkage map in man using
restriction fragment length polymorphism. Am. J. hum. Genet.,
32: 314-331.
BOYUM, A. (1968) Separation of leukocytes from blood and
bone marrow. Scand. J. clin. Lab. Invest., 21(Suppl. 97):
51-76.
BREWEN, J.G. & GENGOZIAN, N. (1971) Radiation-induced human
chromosome aberrations. II. Human in vitro irradiation
compared to in vitro and in vivo irradiation of marmoset
leukocytes. Mutat. Res., 13: 383-391.
BREWEN, J.G., PRESTON, R.J., & LITTLEFIELD, L.G. (1972)
Radiation-induced human chromosome aberration yields following
an accidental whole-body exposure to Co -rays. Radiat. Res.,
49: 647-656.
BREWEN, J.G., PRESTON, R.J., & GENGOZIAN, N. (1975) Analysis
of X-ray induced chromosome translocations in human and
marmoset spermatogonial cells. Nature (Lond.), 253: 468-470.
BUCKTON, K.E., HAMILTON, G.E., PATON, L., & LANGLANDS, A.D.
(1978) Chromosome aberrations in irradiated ankylosing
spondylitis patients. In: Evans, H.J. & Lloyd, D.C., ed.
Mutagen-induced chromosome damage in man, Edinburgh, Edinburgh
University Press, pp. 142-150.
BULFIELD, G. & MOORE, E.A. (1974) Semi-automated assays for
enzymopathies of carbohydrate metabolism in liver and
erythrocytes, using a reaction rate analyser. Clin. chim.
Acta, 53: 265-271.
BUTLER, M.G. (1981) Sister-chromatid exhange in 4 human
races. Mutat. Res., 91: 377-379.
BUTLER, M.G. & SANGER, W.G. (1981) Increased frequency of
sister-chromatid exchanges in alcoholics. Mutat. Res., 85:
71-76.
BYRNE, J. (1983) Fetal pathology/Laboratory manual. Birth
Defects: Original series, 19(2).
BYRNE, J., WARBURTON, D., KLINE, J., BLANC, W., & STEIN, Z.
(in press) Morphology of early fetal death and their
chronological characteristics. Teratology.
CARRANO, A.V. (1982) Sister-chromatid exchange as an
indicator of human exposure. In: Bridges, B.A., Butterworth,
B.E., & Weinstein, I.B., ed. Indicators of genotoxic exposure,
New York, Cold Spring Harbor Laboratory, pp. 307-318 (Banbury
Report No. 13).
CARRANO, A.V. & MOORE II, D.H. (1982) The rationale and
methodology for quantifying sister-chromatid exchange in
humans. In: Heddle, J.A., ed. Mutagenicity: new horizons in
genetic toxicology, New York, Academic Press, pp. 267-304.
CARRANO, A.V. & THOMPSON, L.H. (1981) Sister-chromatid
exchange and single-gene mutation. In: Wolff, S., ed.
Sister-chromatid exchange, New York, John Wiley & Sons, Inc.,
pp. 59-86.
CARRANO, A.V. & THOMPSON, L.H. (1982) Sister-chromatid
exchange and gene mutation. Cytogenet. Cell Genet., 33: 57-61.
CARRANO, A.V., THOMPSON, L.H., LINDL, P.A., & MINKLER, J.L.
(1978) Sister-chromatid exchanges as an indicator of
mutagenesis. Nature (Lond.), 271: 551-553.
CARRANO, A.V., MINKLER, J.L., STETKA, D.G., & MOORE II, D.H.
(1980) Variation in the baseline sister-chromatid exchange
frequency in human lymphocytes. Environ. Mutagen., 2: 325-337.
CASKEY, C.T. & KRUH, G.D. (1979) The HPRT locus. Cell, 16:
1-9.
CHAGANTI, R.S.K., SCHONBERG, S., & GERMAN, J. (1974) A many
fold increase in sister-chromatid exchanges in Blooms syndrome
lymphocytes. Proc. Natl Acad. Sci. USA, 71: 4508-4512.
CHAPUIS-CELLIER, C., FRANCINA, A., & ARNAUD, P. (1980)
Immunofixation after electrofocusing: application to the study
of the microheterogeneity and polymorphism of serum proteins.
In: Radola, B.J., ed. Electrophoresis 79, Berlin, Walter de
Gruyter, pp. 711-725.
CHARLES, D.A. & PRETSCH, W. (1981) A mutation affecting the
lactate dehydrogenase locus LDH-1 in the mouse. I. Genetical
and electrophoretic characterization. Biochem. Genet., 19:
301-307.
CHENG, W., MULVIHILL, J.J., GREENE, M.H., PICKLE, L.W., TSAI,
S., & WHANG-PENG, J. (1979) Sister-chromatid exhanges and
chromosome in chronic myelogenous leukaemia and cancer
families. Int. J. Cancer, 23: 8-13.
CHENG, M., CONNER, M.K., & ALARIE, Y. (1981) Potency of some
carbamates as multiple tissue sister-chromatid exchange
inducers and comparison with known carcinogenic activities.
Cancer Res., 41: 4489-4492.
CHRAMBACK, A., HJELMELAND, L., & NGUYEN, N.Y. (1980) Gel
electrofocusing with increased degrees of freedom. In: Radola,
B.J., ed. Electrophoresis 79, Berlin, Walter de Gruyter,
pp. 3-22.
CLEAVER, J.E. (1981) Correlations between sister-chromatid
exchange frequencies and replicon sizes. A model for the
mechanism of SCE production. Exp. cell Res., 136: 27-30.
CLEMENGER, J.F.P. & SCOTT, D. (1973) A comparison of
chromosome aberration yields in rabbit blood lymphocytes
irradiated in vitro and in vivo. Int. J. Radiat., 24: 487-496.
COHEN, M.M., MARTIN, A.O., OBER, C., & SIMPSON, S.J. (1982)
A family study of spontaneous sister-chromatid exchange
frequency. Am. J. hum. Genet., 34: 294-306.
COLE, R.J., AGHAMOHAMMADI, Z., & HENDERSEN, L. (1982)
Frequencies of genetic lesions in lymphocytes from newborn
humans. Prog. clin. Biol. Res., 109: 375-385.
COMINGS, D.E. (1982) Two-dimensional gel electrophoresis of
human brain proteins. III. Genetic and non-genetic variation
in 145 brains. Clin. Chem., 28: 798-804.
COOPER, D.N. & SCHMIDTKE, J. (1984) DNA restriction fragment
length polymorphisms and heterozygosity in the human genome.
Hum. Genet., 66: 1-16.
CROSSEN, P.E., DRETS, M.E., ARRIGHI, F.E., & JOHNSTON, D.A.
(1977) Analysis of the frequency and distribution of
sister-chromatid exchanges in cultured human lymphocytes.
Hum. Genet., 35: 345-352.
CZEIZEL, A. & SANKARANARAYANAN, K. (1984) The load of
genetic and partially genetic disorders in man. I. Congenital
anomalies: estimates of detriment in terms of years of life
lost and years of impaired life. Mutat. Res., 128(1): 73-103.
CZEIZEL, A., SZENTESI, I., & MOLNAR, G. (1984) Lack of
effect of self-poisoning on subsequent reproductive outcome.
Mutat. Res., 127: 175-182.
DANFORTH, G.H. (1921) The frequency of mutation and the
incidence of hereditary traits in man. Eugenics, genetics, and
the family. In: Scientific Papers of the 2nd International
Congress of Eugenics, New York, Vol. 1, pp. 120-128.
DAS, B.C. & SHARMA, T. (1984) Effects of temperature on the
frequency of sister-chromatid exchanges (SCEs) in peripheral
blood lymphocytes of man and muntjac. Environ. Mutagen., 6:
25-31.
DE ARCE, M.A. (1981) The effect of donor sex and age on the
number of sister-chromatid exchanges in human lymphocytes
growing in vitro. Hum. Genet., 57: 83-85.
DEEPER, J.M., LEONARD, W.J., ROBB, R.J., WALDMANN, T.A., &
GREENE, W.C. (1983) Blockade of the interleukin-2 receptor
by anti-Tac antibody: inhibition of human lymphocyte
activation. J. Immunol., 131: 690-696.
DEKNUDT, G. & KAMRA, O. (1983) Influence of various mitogens
on the yield of sister-chromatid exchanges, induced by
chemicals, in human lymphocytes. Mutat. Res., 111: 161-170.
DEMARS, R. (1971) Genetic studies of HG-PRT deficiency and
the Lech-Nyhan syndrome. Fed. Proc., 30: 944-945.
DE WEERD-KASTELEIN, E.A., KEIJZER, W., RAINALDI, G., &
BOOTSMA, D. (1977) Induction of sister-chromatid exchanges
in exeroderma pigmentosum cells after exposure to ultraviolet
light. Mutat. Res., 45: 253-261.
DUBININ, N.P. & ALTUKHOV, Y.P. (1979) Gene mutations (de
novo) found in electrophoretic studies of blood proteins of
infants with anomalous development. Proc. Natl Acad. Sci. USA,
76: 5226-5229.
DUFRAIN, R. & GARRAND, T. (1981) The influence of
incorporated halogenated analogues of thymidine on the
sister-chromatid exchange frequency in human lymphocytes.
Mutat. Res., 91: 233-238.
DUNN, M.J. & BURGHES, A.H.M. (1983a) High resolution
two-dimensional polyacrylamide gel electrophoresis. II.
Analyses and application. Electrophoresis, 4: 173-189.
DUNN, M.J. & BURGHES, A.H.M. (1983b) High resolution
two-dimensional polyacrylamide gel electrophoresis. I.
Methodological procedures. Electrophoresis, 4: 97-116.
EBER, S.W., DUNNWALD, M., BELSHRADSHY, B.H., BIDDLINGMARER,
F., SCHIEVELBERN, H., WEINMANN, H.M., & KRIETSCH, W.K.G.
(1979) Hereditary deficiency of triosephosphate isomerase in
four unrelated families. Eur. J. clin. Investig., 9: 195-202.
EDMONDS, D.K., LINDSAY, K.S., MILLER, J.F., WILLIAMSON, E., &
WOOD, P.J. (1982) Early embryonic mortality in women.
Fertil. Steril., 38: 447-453.
EVANS, H.J. & O'RIORDAN, M.L. (1975) Human peripheral blood
lymphocytes for the analysis of chromosome aberrations in
mutagen tests. Mutat. Res., 31: 135-148.
EVANS, H.J. & VIJAYALAXMI (1980) Storage enhances chromosome
damage after exposure of human leukocytes to mitomycin C.
Nature (Lond.), 284: 370-372.
EVANS, H.J., BUCKTON, K.E., HAMILTON, G.E., & CAROTHERS, A.
(1979) Radiation induced chromosome aberrations in nuclear
dockyard workers. Nature (Lond.), 277: 531-534.
FIELEK, S. & MOHRENWEISER, H.W. (1979) Erythrocyte enzyme
deficiencies assessed with a miniature centrifugal analyser.
Clin. Chem., 25: 384-388.
FLEISS, J.L. (1981) Statistical methods for rates and
proportions, New York, John Wiley and Sons.
FLYNT, J.W. & HAY, S. (1979) International clearinghouse for
birth defects monitoring systems. Contrib. Epid. Biostatist.,
1: 44.
FOWLE, J.R., III (1984) Workshop report: approaches for
improving mutagenicity risk assessment. Part I. Human
biomonitoring. In: US EPA Conference, 13-15 December, 1982,
Washington DC, US Environmental Protection Agency (Report EPA
600/9-84-016).
FOX, A.J. & GOLDBLATT, P.O. (1982) Longitudinal study of
socio-demographic mortality differentials, London, Her
Majesty's Stationary Office (Series LS No. 1).
FUERST, P.A. & FERRELL, R.E. (1980) The stepwise mutation
model: an experimental evaluation utilizing hemoglobin
variants. Genetics, 94: 185-201.
GALLOWAY, S.M. (1977) Ataxia telangiectasia: the effects of
chemical mutagens and X rays on sister-chromatid exchanges in
blood lymphocytes. Mutat. Res., 45: 343-349.
GALLOWAY, S.M. & EVANS, H.J. (1975) Sister-chromatid
exchanges in human chromosomes from normal individuals and
patients with ataxia telangiectasia. Cytogenet. Cell Genet.,
15: 17-29.
GARCIA, H.D. & COUCH, D.B. (1982) Detection of
phenotypically variant (6-thioguanine-resistant) rat spleen
lymphocytes. In: Bridges, B.A., Butterworth, B.E., &
Weinstein, I.B., ed. Indicators of genotoxic exposure,
New York, Cold Spring Harbor Laboratory Press, pp. 443-451
(Banbury Report No. 13).
GILLIAN CLARE, M., JONES, W.G., & TAYLOR, J.H. (1982)
Sister-chromatid exchanges in human lymphocytes exposed to
single cytotoxic drugs in vivo or in vitro. Eur. J. Cancer
clin. Oncol., 18: 979-989.
GOCKE, E., ECKHARDT, K., KING, M.T., & WILD, D. (1983)
Autoradiographic detection of 6-thioguanine resistant
lymphocytes of mice: a novel system in somatic mutagenesis
testing. Mutat. Res., 113: 455-465.
GOTO, K., MADEA, S., KANO, Y., & SUGEYAMA, T. (1978) Factors
involved in differential Giemsa staining of sister-chromatids.
Chromosoma, 66: 351-359.
HAMAGUCHI, H., YAMADA, M., SHIBASAKI, M., MUKAI, R., YABE, T.,
& KONDO, I. (1982) Genetic analysis of human lymphocyte
proteins by two-dimensional gel electrophoresis. Hum. Genet.,
62: 142-147.
HANASH, S.M., TUBERGEN, D.G., HEYN, R.M., NEEL, J.V., SANDY,
L., STEVENS, G.S., ROSENBLUM, B.B., & KRZESICKI (1982)
Two-dimensional gel electrophoresis of cell proteins in
childhood leukemia, with silver staining: a preliminary
report. Clin. Chem., 28: 1026-1030.
HARLAP, S., SHIONO, P.H., & RAMCHARON, S. (1980) A life
table of spontaneous abortions and the effects of age, parity,
and other variables. In: Porter, I.H. & Hook, E.G., ed. Human
embryonic and fetal death, New York, Academic Press,
pp. 145-158.
HARRIS, H. & HOPKINSON, D.A. (1976) Handbook of enzyme
electrophoresis in human genetics, New York, American Elsevier.
HARRIS, H., HOPKINSON, D.A., & ROBSON, E.B. (1974) The
incidence of rare alleles determining electrophoretic
variants data on 43 enzyme loci in man. Ann. Hum. Genet.
(Lond.), 37: 237-253.
HASSOLD, T. (1982) Mosatic trisomies in human spontaneous
abortions. Hum. Genet., 31: 31-35.
HASSOLD, T., JACOBS, P., KLINE, J., STEIN, Z., & WARBURTON,
D. (1980) Effect of maternal age on autosomal trisomies.
Ann. hum. Genet. (Lond.), 44: 29-36.
HASSOLD, T., WARBURTON, D., KLINE, J., & STEIN, Z. (1984)
The relationship of maternal age and trisomy among trisomic
spontaneous abortions. Am. J. hum. Genet., 36(6): 1349-1356.
HATCH, M. (1984) Male risk factors for spontaneous abortion.
Am. J. Epidem., 120(3): 499-500.
HEDNER, K., HOGSTEDT, B., KOLNIG, A.-M., MARK-VENDEL, E.,
STROMBECK, B., & MITELMAN, F. (1983) Sister-chromatid
exchanges and structural chromosome aberrations in relation to
smoking in 91 individuals. Hereditas, 98: 77-81.
HOLLANDER, D.H., TOCKMAN, M.S., LIANG, Y.W., BORGAONKAR, D.S.,
& FROST, J.K. (1978) Sister-chromatid exchanges in the
peripheral blood of cigarette smokers and in lung cancer
patients and the effect of chemotherapy. Hum. Genet., 44:
165-171.
HOLMES, L.B., VINENT, S.E., COOK, C., & COTE, K.R. (1981)
Surveillance of newborn infants for malformations due to
spontaneous germinal mutations. In: Hook, E.B. & Porter, I.H.,
ed. Population and biological aspects of human mutation,
New York, Academic Press, pp. 351-359.
HOOK, E.B. (1981a) Prevalance of chromosome abnormalities
during human gestation and implications for studies of
environmental mutagens. Lancet, 2: 169-172.
HOOK, E.B. (1981b) Human teratogenic and mutagenic markers
in monitoring aboutpoint sources of pollution. Environ. Res.,
25: 178-203.
HOOK, E.B. (in press) Paternal age and genetic outcomes. In:
Porter, I.H., ed. Proceedings of the 15th Birth Defects
Institute Symposium, Albany, New York, New York, Academic
Press.
HOOK, E.B. & HAMERTON, J.L. (1977) The frequency of
chromosome abnormalities detected in consecutive newborn
studies - Differences between studies - Results by sex and by
severity of phenotypic involvement. In: Hook, E.G. & Porter,
I.H., ed. Population cytogenetics: studies in humans,
New York, Academic Press, pp. 63-79.
HOOK, E.B., SCHREINEMACHERS, D.M., WILLEY, A.M., & CROSS,
P.K. (1984) Inherited structural cytogenetic abnormalities
detected incidentally in fetuses diagnosed prentally:
frequency, parental-age association, sex-ratio trends, and
comparisons with rates of mutants. Am. J. hum. Genet., 36:
422-443.
HOPKIN, J.M. & EVANS, H.J. (1980) Cigarette smoke-induced
DNA damage an lung cancer risks. Nature (Lond.), 283: 388-390.
HOWE, G.R. & LINDSAY, J.P. (1981) A generalized iterative
record linkage system for use in medical follow-up studies.
Comput. biomed. Res., 14: 327-340.
HSIE, A.W., O'NEILL, J.P., & McELHENY, V.K., ed. (1979)
Mammalian cell mutagenesis: the maturation of test systems,
New York, Cold Spring Harbor Laboratory Press (Banbury Report
No. 2).
HUSGAFVEL-PURSIAINEN, K., MAKI-PAAKKANEN, J., NORPPA, H., &
SORSA, M. (1980) Smoking and sister-chromatid exchange.
Hereditas, 92: 247-250.
HUSUM, B., WULF, H.C., & NIEBUHR, E. (1982) Increased
sister-chromatid exchange frequency in lymphocytes in healthy
cigarette smokers. Hereditas, 96: 85-88.
JACKY, P.F., BEEK, B., & SUTHERLAND, G.R. (1983) Fragile
sites in chromosomes: possible model for the study of
spontaneous chromosome breakage. Science, 220: 69-70.
JACOBS, P.A. (1981) Mutation rates for structural chromosome
rearrangements in man. Am. J. hum. Genet., 33: 44-54.
JOHNSON, G.B. (1976) Hidden alleles at the -glycerophosphate
dehydrogenase locus in colias butterflies. Genetics, 83:
149-167.
JOHNSON, G.B. (1977) Evaluation of the stepwise mutation
model of electrophoretic mobility: comparison of the gel
sieving behavior of alleles at the esterase - 5 locus of
Drosophila pseudoobscura. Genetics, 87: 139-157.
JOHNSON, F.M. & LEWIS, S.E. (1981) Electrophoretically
detected germinal mutations induced in the mouse by
ethylnitrosurea. Proc. Natl Acad. Sci. USA, 78: 3138-3141.
JOHNSON, F.M. & LEWIS, S.E. (1983) The detection of
ENU-induced mutants in mice by electrophoresis and the problem
of evaluating the mutation rate increase. In: DeSerres, F.J. &
Sheridan, W., ed. Utilization of mammalian specific locus
studies in hazard evaluation and estimation of genetic tisk,
New York, Plenum Press, pp. 95-108.
JOHNSON, F.M., ROBERTS, G.T., SHARMA, R.K., CHASALOW, F.,
ZWEIDINGER, R., MORGAN, A., HENDREN, R.W., & LEWIS, S.E.
(1981) The detection of mutants in mice by electrophoresis:
results of a model induction experiment with procarbazine.
Genetics, 97: 113-124.
JONES, I.M., BURKHART-SCHULTZ, K., & CARRANO, A.V. (in
press) A method to quantify spontaneous and in vivo induced
thioguanine resistant mouse lymphocytes. Mutat. Res.
JUBERG, R.C. & MOWREY, P.N. (1983) Origin of non-disjunction
in trisomy 21. Am. J. med. Genet., 16: 111-116.
KAHN, A., KAPLAN, J.C., & DREYFUS, J.C. (1979) Advances in
hereditrary red cell anomalies. Hum. Genet., 51: 1-27.
KATO, H. & SANDBERG, A.A. (1977) The efffect of sera on
sister-chromatid exchanges in vitro. Exp. cell Res., 109:
445-448.
KINLEN, L.J. (1980) The death registration system and the
national health service central regiater in Britain: their
value to epidemiological research. In: Cairns, J., Lyon, J.L.,
& Skonnick, M., ed. Cancer incidence in defined populations,
New York, Cold Spring Harbor Laboratories, pp. 437-442
(Banbury Report No. 4).
KLINE, J. & STEIN, Z. (in press) Very early pregnancy. In:
Dixon, R.L., ed. Target organ toxicity, New York, Raven Press .
KLINE, J., STEIN, Z., SUSSER, M., & WARBURTON, D. (1980)
Environmental influences on early reproductive loss in a
current New York City study. In: Porter, I.H. & Hook, E.B.,
ed. Human embryonic and fetal death, New York, Academic Press,
pp. 225-240.
KLINE, J., STEIN, Z., HATCH, M., & STROBINO, B. (1981)
The role of spontaneous abortion studies in environmental
research, Cincinnati, Ohio, Health Effects Research
Laboratory, 45268 (US EPA Cr 8067355010).
KLOSE, J. (1975) Protein mapping by combined isoelectric
focusing and electrophoresis of mouse tissue. A novel approach
to testing for induced point mutations in mammals.
Humangenetik, 26: 231-243.
KLOSE, J. (1979) Isoelectricfocusing and electrophoresis
combined as a method for defining new point mutations in the
mouse. Genetics, 92: 135-245.
KREPINSKI, A.B. & HEDDLE, J.A. (1983) Micronuclei as a rapid
and inexpensive measure of radiation-induced chromosomal
aberrations. In: Ishihora, T. & Sasaki, M.S., ed.
Radiation-induced chromosome damage in man, New York,
Alan R. Liss, Inc., pp. 93-109.
KRIETSCH, W.K.G., KRIETSCH, H., KAISER, W., DUNNWALD, M.,
KUNTZ, G.W.K., DUHM, I., & BUCHER, T. (1977) Hereditary
deficiency of phosphoglycerate kinase: a rare variant in
erythrocytes and leukocytes not associated with haemolytic
anemia. Eur. J. clin. Invest., 7: 427-435.
LAMBERT, B. & LINDBLAD, A. (1980) Sister-chromatid exchange
and chromosome aberrations in lymphocytes of laboratory
personnel. J. Toxicol. environ. Health, 6: 1237-1243.
LAMBERT, B., HANSSON, K., LINDSTEN, J., STEN, M., & WERELIUS,
B. (1976) Bromodeoxyuridine-induced sister-chromatid
exchanges in human lymphocytes. Hereditas, 82: 163-174.
LAMBERT, B., RINGBORG, U., HARPER, E., LINDBLAD, A. (1978)
Sister-chromatid exchanges in lymphocyte cultures of patients
receiving chemotherapy for malignant disorders. Cancer Treat.
Rep., 62: 1413-1419.
LAMBERT, B., LINDBLAD, A., HOLMBERG, L., & FRANCESCONI, D.
(1982) The use of sister-chromatid exchange to monitor human
populations for exposure to toxicologically harmful agents.
In: Wolff, S., ed. Sister-chromatid exchange, New York,
John Wiley & Sons, Inc., pp. 149-182.
LANGE, B.J. & PRANTER, J.E. (1982) The emergence of
6-thioguanine resistant lymphocytes in pediatric cancer
patients. Mutat. Res., 94: 487-499.
LATT, S.A. (1973) Microfluorometric detection of
deoxyribonucleic acid replication in human metaphase
chromosomes. Proc. Natl Acad. Sci. USA, 70: 3395-3399.
LATT, S.A. (1981) Sister-chromatid exchange formation.
Ann. Rev. Genet., 15: 11-55.
LATT, S.A. & JUERGENS, L.A. (1977) Determinants of
sister-chromatid exchange frequencies in human chromosomes.
In: Hook, E.B. & Porter, I.H., ed. Population cytogenetics:
studies in humans, New York, Academic Press, pp. 217-236.
LATT, S.A., ALLEN, J., BLOOM, S.E., CARRANO, A., FALKE, E.,
KRAM, D., SCHNEIDER, E., SCHRECK, R., TICE, R., WHITFIELD, B.,
& WOLFF, S. (1981) Sister-chromatid exchanges: a report of
the Gene-Tox program. Mutat. Res., 87: 17-62.
LINDBLAD, A. & LAMBERT, B. (1981) Relation between
sister-chromatid exchange, cell proliferation, and proportion
of B and T cells in human lymphocyte cultures. Hum. Genet.,
57: 31-34.
LIVINGSTON, G.K., CANNON, L.A., BISHOP, D.T., JOHNSON, P., &
FINEMAN, R.M. (1983) Sister-chromatid exchanges: variation
by age, sex, smoking, and breast cancer status. Cancer Genet.
Cytogenet., 8: 289-299.
LLOYD, D.C., PROSSER, J.S., MOQUET, J.E., & MALOWANY, D.J.
(1983) Doses in radiation accidents investigated by
chromosome aberration analysis. XIII. A review of cases
investigated: 1982, Harwell, Oxfordshire, National
Radiological Protection Board, 14 pp (Report No. NRPB-R148).
MACCLUER, J.W. (1980) Inbreeding and human fetal death. In:
Porter, I.H. & Hook, E.B., ed. Human embryonic and fetal
death, New York, Academic Press, pp. 241-260.
MACGREGOR H.C. & VARLEY, J.M. (1983) Working with animal
chromosomes, New York, John Wiley & Sons.
MCCONKEY, E.H., TAYLOR, B.J., & PHAN, D. (1979) Human
heterozygosity: a new estimate. Proc. Natl Acad. Sci. USA, 76:
6500-6504.
MCFEE, A.F. & SHERRILL, M.N. (1981) Mitotic response and
sister-chromatid exchanges in lymphocytes cultured in sera
from differenct sources. Experientia (Basel), 37: 27-29.
MCKUSICK, V.A. (1983) Mendelian inheritance in man:
catalogue of autosomal dominant, autosomal recessive, and
X-linked phenotypes, 6th ed., Baltimore, Maryland,
Johns Hopkins University Press.
MAGENIS, R.E. & CHAMBERLIN, J. (1981) The parental origin of
non-disjunction. In: De la Cruz, F., ed. Trisomy 21 (Down
Syndrome) research perspectives, Baltimore, Maryland,
University Park Press, pp. 77-94.
MAIZEL, A.L., MEHTA, S.R., HAUFT, S., FRANZINI, D., LACHMAN,
L.B., & FORD, R.J. (1981) Human T-lymphocyte/monocyte
interaction in response to lectin: kinetics of entry into
S-phase. J. Immunol., 127: 1058-1064.
MARSHALL, R.R., RAJ, A.S., GRANT, F.J., & HEDDLE, J.A.
(1984) The use of two-dimensional electrophoresis to detect
mutations induced in mouse spermatogoniaby ethylnitrosourea.
Can. J. Genet. Cytol., 25: 457-466.
MARTIN, R.H., BALKAN, W., BURNS, K., RODEMAKER, A.W., LIN,
C.C., & RUDD, N.L. (1983) The chromosome consitution of 1000
human spermatozoa. Hum. Genet., 63: 305-309.
MATSUNAGA, E. (1982) Perspectives in mutation epidemiology.
I. Incidence and prevalence of genetic disease (excluding
chromosomal aberrations) in human populations. Mutat. Res.,
99: 95-128.
MAZRIMAS, J.A. & STETKA, D.G. (1978) Direct evidence for the
role of incorporated BUdR in the induction of sister-chromatid
exchanges. Exp. cell Res., 117: 23-30.
MERRIL, C.R., GOLDMAN, D., SEDMAN, S.A., & EBERT, M.H.
(1981) Ultrasensitive stain for proteins in polyacrylamide
gels show regional variation in cerebrospinal fluid proteins.
Science, 211: 1437-1438.
METROPOLITAN POLICE FORENSIC SCIENCE LABORATORY (1980)
Biology methods manual, London.
MI, M.P. (1967) Record linkage and other genetic studies in
Hawaii. In: Crow, J.A. & Neel, J.V., ed. Proceedings of the
3rd International Congress on Human Genetics, Baltimore,
Maryland, Johns Hopkins University Press, pp. 489-496.
MILLER, J.F., WILLIAMSON, E., GLUE, J., GORDON, Y.B.,
GRUDZINSKAS, J.C., & SYKES, A. (1980) Fetal loss after
implantation: a prospective study. Lancet, 2: 554-556.
MIWA, S. (1979) Significance of the determination of red
cell enzyme activities. Am. J. Hematol., 6: 163-172.
MOHRENWEISER, H.W. (1981) Frequency of enzyme deficiency
variants in erythrocythes of newborn infants. Proc. Natl Acad.
Sci. USA, 78: 5046-5050.
MOHRENWEISER, H.W. (1982) Frequency of rare enzyme
deficiency variants: search for mutational events with human
health implications. In: Bora, K.C., Douglas, G.R., &
Nestmann, E.R., ed. Chemical mutagenesis, human population
monitoring and genetic risk assessment, Amsterdam, Elsevier
Biomedical Press, Vol. 3, pp. 51-68 (Prog. Mutat. Res.).
MOHRENWEISER, H.W. (1983a) Enzyme deficiency variants:
frequency and potential significance in human populations.
Isozymes: Curr. Topics Biol. Med. Res., 10: 51-68.
MOHRENWEISER, H.W. (1983b) Biochemical approaches to
monitoring human populations for germinal mutation rates. II.
Enzyme deficiency variants as a component of the estimated
genetic risk. In: DeSerres, F.J. & Sheridan, W., ed.
Utilization of mammalian specific locus studies in hazard
evaluation and estimation of genetic risk, New York, Plenum
Publishing Corporation, pp. 55-69.
MOHRENWEISER, H.W. & FIELEK, S. (1982) Elevated frequency of
carriers for triosephosphate isomerase deficiency in newborn
infants. Pediatr. Res., 16: 960-963.
MOHRENWEISER, H.W. & NEEL, J.V. (1981) Frequency of
thermostability variants and estimation of the total "rare"
variant frequency in human populations. Proc. Natl Acad. Sci.
USA, 78: 5729-5733.
MOOREHEAD, P.S., NOWELL, P.C., MELLMAN, W.J., BATTIPS, D.M., &
HUNGERFORD, D.A. (1960) Chromosome preparations of
leukocytes from human peripheral blood. Exp. cell Res., 20:
613-616.
MORGAN, W.F. & CROSSEN, P.E. (1977) The incidence of
sister-chromatid exchanges in cultured human lymphocytes.
Mutat. Res., 42: 305-312.
MORIMOTO, K. & WOLFF, S. (1980) Increase of sister-chromatid
exchanges and perturbations of cell division kinetics in human
lymphocytes by benzene metabolites. Cancer Res., 40: 1189-1193.
MORLEY, A.A., COX, S., & HOLLIDAY, R. (1982) Human
lymphocytes resistant to 6-thioguanine increase with age.
Mech. Aging Dev., 19: 21-26.
MORLEY, A.A., TRAINOR, K.J., & SESHADRI, R.S. (1983a)
Cloning of human lymphocytes using limiting dilution.
Exp. Hematol., 11: 418-424.
MORLEY, A.A., TRAINOR, K.J., SESHADRI, R., & RYALL, R.B.
(1983b) Measurement of in vivo mutations in human
lymphocytes. Nature (Lond.), 302: 155-156.
MUKAI, T. & COCKERHAM, C.C. (1977) Spontaneous mutations
rates at enzyme loci in Drosophila melanogaster. Proc. Natl
Acad. Sci. USA, 74: 2514-2517.
MULVIHILL, I.I. & CZEIZEL, A. (1983) Perspectives in
mutation epidemiology. VI. A 1983 view of sentinel phenotypes.
Mutat. Res., 123: 345-361.
MUSILOVA, J., MICHALOVA, K., & URBAN, J. (1979)
Sister-chromatid exchanges and chromosomal breakage in
patients treated with cytostatics. Mutat. Res., 67: 289-294
NAGAO, M. & SUGIMURA, T. (1981) Mutacarcinogens in
ordinarily cooked foods. In: Schoental, R. & Conners, T.A.,
ed. Dietary influences on cancer: traditional and modern,
Florida, CRC Press, Inc., pp. 201-218.
NAKAGONE, Y., YOKOCHI, T., MATSUBARA, T., & FUKADA, F.
(1982) Preservation of whole blood for chromosome analysis.
Cytogenet. cell Genet., 33: 254-255.
NEEL, J.V. (1980) Some considerations pertinent to
monitoring human populations for changing mutation rates.
Proc. XIV Int. Cong. Genet., 1: 225-238.
NEEL, J.V. (1983) Frequency of spontaneous and induced
"point" mutations in higher eukaryotes. J. Hered., 74: 2-15.
NEEL, J.V. & MOHRENWEISER, H.W. (1984) Failure to
demonstrate mutations affecting protein structure or function
in children with congenital defects or born prematurely.
Proc. Natl Acad. Sci. USA, 81: 5499-5503.
NEEL, J.V., MOHRENWEISER, H.W., SATOH, C., & HAMILTON, H.B.
(1979) A consideration of two biochemical approaches to
monitoring human populations for a change in germ cell
mutation rates. In: Berg, K., ed. Genetic damage in man caused
by environmental agents, New York, Academic Press, pp. 29-47.
NEEL, J.V., MOHRENWEISER, H.W., & MEISLER, M.M. (1980a) Rate
of spontaneous mutation at human loci encoding protein
structure. Proc. Natl Acad. Sci. USA, 77: 6037-6041.
NEEL, J.V., SATOH, C., HAMILTON, H.B., OTAKE, M., GORIKI, K.,
KAGEOKA, T., FUJITA, M., NERIISHI, S., & ASAKAWA, J. (1980b)
Search for mutations affecting protein structure in children
of atomic bomb survivors: preliminary report. Proc. Natl Acad.
Sci. USA, 77: 4221-4225.
NEEL, J.V., MOHRENWEISER, H.W., HANASH, S., ROSENBLUM, B.B.,
STERNBERG, S., WURZINGER, K., ROTHMAN, E., SATOH, C., GORIKI,
K., KRASTEFF, T., LONG, M., SKOLNICK, M., & KRZESICKI, R.
(1983) Biochemical approaches to monitoring human populations
for germinal mutation rates. I. Electrophoresis. In: DeSerres,
F.J. & Sheridan, W., ed. Utilization of mammalian specific
locus studies in hazard evaluation and estimation of genetic
risk, New York, Plenum Press, pp. 71-93.
NEEL, J.V., ROSENBLUM, B.B., SING, C.F., SKOLNICK, M.M.,
HANASH, S.M., & STERNBERG, S. (1984) Adapting
two-dimensional gel electrophoresis to the study of
humangermline mutation rates. In: Celis, J.E. & Bravo, R., ed.
Methods and applications of two-dimensional gel
electrophoresis of proteins, New York, Academic Press,
pp. 259-306.
NEWCOMBE, H.B. (1967) Record linking: the design of
efficient systems for linking records into individual and
family histories. Am. J. hum. Genet., 19: 335-359.
NEWCOMBE, H.B. (1969) The use of medical record linkage for
population studies. Method. Inform. Med., 8: 7-11.
NEWCOMBE, H.B. (1977) Methods of obtaining epidemiological
data for mutation risk estimation. In: Kilbey, B.J., Legator,
M., Nichols, W., & Ramel, C., ed. Handbook of mutagenicity
test procedures, Amsterdam, Elsevier/North Holland,
pp. 461-476.
O'FARRELL, P.H. (1975) High resolution two-dimensional
electrophoresis of proteins. J. Biol. Chem., 250: 4007-4021.
OFFICE OF TECHNOLOGY ASSESSMENT, US CONGRESS (1983) The role
of genetic testing in the prevention of occupational disease,
Washington DC, US Office of Technology Assessment, 243 pp
(GTA-BA-194).
PAINTER, R.B. (1980) A replication model for
sister-chromatid exchange. Mutat. Res., 70: 337-341.
PATTERSON, J.E. (1980) The establishment of a national death
index in the United States. In: Cairns, J., Lyon, J.L., &
Skolnick, M., ed. Cancer incidence in defined populations,
New York, Cold Spring Harbor Laboratories, pp. 443-451 (Banbury
Report No. 4).
PAUL, W.E., SREDNI, B., & SCHWARTZ, R.H. (1981) Long-term
growth and cloning of non-transformed lymphocytes. Nature
(Lond.), 294: 679-699.
PEDERSEN, C., OLAH, E., & MERRILD, U. (1979)
Sister-chromatid exchanges in cultured peripheral lymphocytes
from twins. Hum. Genet., 52: 281-294.
PERRY, P.E. (1980) Chemical mutagens and sister-chromatid
exchange. In: DeSerres F.J. & Hollaender, A., ed. Chemical
mutagens: principles and methods for their detection,
New York, Plenum Press, Vol. 6, pp. 1-39.
PERRY, P. & EVANS, H.J. (1975) Cytological detection of
mutagen-carcinogen exposure by sister-chromatid exchange.
Nature (Lond.), 258: 121-125.
PERRY, P. & WOLFF, S. (1974) New Giemsa method for the
differential staining of sister-chromatids. Nature (Lond.),
251: 156-158.
POPESCU, N.C., AMSBAUGH, S.C., & DIPAOLO, J.A. (1981)
Relationship of carcinogen-induced sister-chromatid exchange
and neoplastic cell transformation. Int. J. Cancer, 28: 71-77.
PRESTON, R.J. & GOOCH, P.C. (1981) The induction of
chromosome-type aberrations in G by methyl methanesulfonate
and 4-nitroquinoline-N-oxide and the non-requirement of an
S-phase for their production. Mutat. Res., 83: 395-402.
PRESTON, R.J., BREWEN, J.G., & JONES, K.P. (1972)
Radiation-induced chromosome aberrations in Chinese hamster
leukocytes. A comparison of in vivo and in vitro exposures.
Int. J. Radiat. Biol., 21: 397-450.
PRESTON, R.J., AU, W., BENDER, M.A., BREWEN, J.G., CARRANNO,
A.V., HEDDLE, J.A., MCFEE, A.F., WOLFF, S., & WASSOM, J.S.
(1981) Mammalian in vivo and in vitro cytogenetic assays: a
report of the US EPA's Gene-tox Program. Mutat. Res., 87:
143-188.
PRETSCH, W. & NARAYANAN, K.R. (1979) [Determination of
gene-mutations in mice by isoelectric focusing.] A. Physiol.
Chem., 360: 345-350 (in German).
RACINE, R.R., LANGLEY, C.H., & VOELKER, R.A. (1980) Enzyme
mutations induced by low-dose rate -irradiation in Drosophila:
frequency and characterization. Environ. Mutagen., 2: 167-177.
RAMSHAW, J.A.M., COYNE, J.A., & LEWONTIN, R.C. (1979) The
sensitivity of gel electrophoresis as a detector of genetic
variation. Genetics, 93: 1019-1037.
RECIO, L.B., CHASTENAY, B., ALBERTINI, R.J., & HSIE, A.W.
(1983) Detection of 6-thioguanine resistant splenocytes from
Chinese hamsters. Environ. Mutagen., 5: 401-402 (abstract).
REIDY, J.A., ZHOU, X., & CHEN, A.T.L. (1983) Folic acid and
chromosome breakage. I. Implications for genotoxicity studies.
Mutat. Res., 122: 217-221.
ROSENBLUM, B.B., NEEL, J.V., & HANASH, S.M. (1983)
Two-dimensional electrophoresis of plasma polypeptides reveals
"high" heterozygosity indices. Proc. Natl Acad. Sci. USA, 80:
5002-5006.
ROSENBLUM, B.B., NEEL, J.V., HANASH, S.M., JOSEPH, J.L., &
YEW, N. (1984) Identification of genetic variants in
erythrocyte lysate by two-dimensional gel electrophoresis.
Am. J. hum. Genet., 36: 601-612.
ROTHMAN, E.D., NEEL, J.V., & HOPPE, F.M. (1981) Assigning a
probability for paternity in apparent cases of mutation.
Am. J. hum. Genet., 33: 601-628.
SAMMONS, D.W., ADAMS, L.D., & NISHIZAWA, E.E. (1981) A
silver-based color development system for staining of
polypeptides in polyacrylamide gels. Electrophoresis, 2:
135-141.
SANTESSON, B., LINDAHL-KIESSLING, K., & MATTSSON, A. (1979)
SCE in B and T lymphocytes: possible implications for Bloom's
syndrome. Clin. Genet., 16: 133-135.
SATOH, C., NEEL, J.V., YAMASHITA, A., GORIKI, K., FUJITA, M.,
& HAMILTON, H.H. (1983) The frequency among Japanese of
heterozygotes for deficiency variants of 11 enzymes. Am. J.
hum. Genet., 33: 656-674.
SAVAGE, J.R.K. (1975) Classification and relationship of
induced chromosome structural changes. J. med. Genet., 12:
103-122.
SCHMIDT, M.A. & SANGER, W.G. (1981) Sister-chromatid
exchange in aged human lymphocytes. A brief note. Mech. Aging
Dev., 16: 67-70.
SELVIN, S. & GARFINKEL, J. (1976) Paternal age, maternal
age, and birth order and the risk of fetal loss. Hum. Biol.,
48: 223-230.
SESHADRI, R., BAKER, E., & SUTHERLAND, G.R. (1982)
Sister-chromatid exchange (SCE) analysis in mothers exposed to
DNA-damaging agents and their newborn infants. Mutat. Res.,
97: 139-146.
SHAFER, D.A. (1982) Alternate replication bypass mechanisms
for sister-chromatid exchange formation. Prog. Topics
Cytogenet., 2: 67-98.
SHARMA, T. & DAS, B.C. (1984) The effect of storage of blood
on the yield of x-ray-induced chromosome aberrations and
spontaneous sister-chromatid exchanges. Int. J. radiat. Biol.,
45: 151-158.
SKOLNICK, M. (1980) The Utah genealogical data base: a
resource for genetic epidemiology. In: Cairns, J., Lyon, J.L.,
& Skolnick, M., ed. Cancer incidence in defined populations,
New York, Cold Spring Harbor Laboratory, pp. 285-296 (Banbury
Report No. 4).
SKOLNICK, M.M. (1982) An approach to completely automatic
comparison of two-dimensional electrophoresis gels.
Clin. Chem., 28: 979-986.
SKOLNICK, M.M. & FRANCKE, U. (1982) Report of the committee
on human gene mapping by recombinant DNA techniques.
Cytogenet. cell Genet., 32: 194-204.
SKOLNICK, M.M. & NEEL, J.V. (in press) An algorithm for
comparing 2-D electrophoretic gels, with particular reference
to the study of mutation. Adv. hum. Genet.
SKOLNICK, M.M, STERNBERG, S.R., & NEEL, J.V. (1982) Computer
programs for adapting two-dimensional gels to the study of
mutation. Clin. Chem., 28: 969-978.
SMITH, M.E. (1977) The use of computers for studying the
origins and social cost of ill health. In: Shires, D.B. &
Wolf, H., ed. MEDINFO77: Proceedings of the Second World
Conference on Medical Informatics, Amsterdam, North Holland
Publishing Company, pp. 37-41.
SMITH, M.E. (1980) The present state of automated follow-up
in Canada. Part 1. Methodology and files. J. clin. Computing,
9: 1-18.
SMITH, M.E. (1982) Development of a national record linkage
program in Canada. 1982 survey research methods section.
Proceedings of the ASA : 303-308.
SMITH, M.E. & NEWCOMBE, H.B. (1980) Automated follow-up
facilities in Canada for monitoring delayed health effects.
Am. J. public Health, 70: 1261-1268.
SMITH, M.E. & NEWCOMBE, H.B (1982) Use of the Canadian
mortality data base for epidemiological follow-up. Can. J.
public Health, 73: 39-46.
SMITH, M.E., SILINS, J., & LINDSAY, J.P. (1980) The present
state of automated follow-up in Canada. Part II. The products.
J. clin. Computing, 9: 19-30.
SNOPE, A.J. & RARY, J.M. (1979) Cell-cycle duration and
sister-chromatid exchange frequency in cultured human
lymphocytes. Mutat. Res., 63: 345-349.
SOUTHERN, E.M. (1982) Application of DNA analysis to mapping
the human genome. Cytogenet. cell Genet., 32: 52-57.
SPEIT, G. (1980) Effects of temperature on sister-chromatid
exchanges. Hum. Genet., 55: 333-336.
STETKA, D.G. & CARRANO, A.V. (1977) The interaction of
Hoechst 33258 and BrdU-substituted DNA in the formation of
sister-chromatid exchanges. Chromosoma, 36: 21-31.
STETKA, D.G., MINKLER, J.L., & CARRANO, A.V. (1978)
Induction of long-lived chromosome damage, as manifested by
sister-chromatid exchange in lymphocytes of animals exposed to
mitomycin-C. Mutat. Res., 51: 383-396.
STICH, H.F., CURTIS, J.R., & PARIDA, B.B. (1982) Application
of the micronucleus test to exfoliated cells of high cancer
risk groups: tobacco chewers. Int. J. Cancer, 30: 553-559.
STRAUSS, G.H. (1982) Direct mutagenicity testing: the
development of a clonal assay to detect and quantitate mutant
lymphocytes arising in vivo. In: Bridges, B.A., Butterworth,
B.E., & Weinstein, I.B., ed. Indicators of genotoxic exposure,
New York, Cold Spring Harbor Laboratory Press, pp. 423-441
(Banbury Report No. 13).
STRAUSS, G.H. & ALBERTINI, R.J. (1977) 6-thioguanine
lymphocytes in human peripheral blood. In: Scott, D., Bridges,
B.A., & Sobels, F.H., ed. Progress in genetic toxicology,
Amsterdam, Elsevier/North Holland, pp. 327-334.
STRAUSS, G.H. & ALBERTINI, R.J. (1979) Enumeration of
6-thioguanine resistant peripheral blood lymphocytes in man as
a potential test for somatic cell mutations arising in vivo.
Mutat. Res., 61: 353-379.
STRAUSS, G.H., ALBERTINI, R.J., KRUSINSKI, P., & BAUGHMAN,
R.D. (1979) 6-Thioguanine resistant peripheral blood
lymphocytes in humans following psoralen long-wave light
therapy. J. Invest. Dermatol., 73: 211-216.
SUTHERLAND, G.R., CARTER, R.F., BAULD, R., SMITH, I.I., &
BAIN, A.D. (1978) Chromosome studies at the pediatric
necropsy. Ann. hum. Genet. (Lond.), 42: 173-181.
SUTHERLAND, G.R., BAKER, E., SESHADRI, R.S., & BLACK, A.
(1980) Increased sister-chromatid exchange in multiple
sclerosis. New Eng. J. Med., 303: 1126.
SUTTON, H.E. (1971) Workshop on monitoring of human
mutagenesis. Teratology, 4: 103-107.
SYLWESTER, D.L. & ALBERTINI, R.J. (in press) Confidence
intervals and sample size calculations to compare variant
frequencies. Environ. Mutagen.
TAYLOR, J.H. (1958) Sister-chromatid exchanges in
tritium-labeled chromosomes. Genetics, 43: 515-529.
TAYLOR, J.H., WOODS, P.S., & HUGHES, W.L. (1957) The
organization and duplication of chromosomes as revealed by
autoradiographic studies using tritium-labeled thymidine.
Proc. Natl Acad. Sci. USA, 43: 122-138.
THOMAS, G., MARTIN-PEREZ, J., SIEGMANN, M., & OTTO, A.M.
(1984) The effect of serum, EGF, PGF, and insulin on S6
phosphorylation and the initiation of protein and DNA
synthesis. Cell, 30: 235-242.
TSANG, V.C.W., PERALTA, J.M., & SIMMONS, A.R. (1983)
Enzyme-linked immunoelectrotransfer blot techniques for
studying the specificities of antigens and antibodies
separated by gel electrophoresis. Meth. Enzymol., 92: 377-391.
VIJAYALAXMI & EVANS, J.H. (1984) Measurement of spontaneous
and x-irradiation induced 6-thioguanine resistant human blood
lymphocytes. Mutat. Res., 125: 87-94.
VIJAYALAXMI, NEWTON, M.S., STEEL, C.M., EVANS, H.J., &
PENTLAND, B. (1983a) Spontaneous and mutagen-induced
sister-chromatid exchange in multiple sclerosis. J. med.
Genet., 20: 372-376.
VIJAYALAXMI, EVANS, J.H., RAY, J.H., & GERMAN, J. (1983b)
Bloom's Syndrome: evidence for an increased mutation frequency
in vivo. Science, 221: 851-853.
VOELKER, R.A., SCHAFFER, H.E., & MUKAI, T. (1980)
Spontaneous allozyme mutations in Drosophila: rate of
occurrence and nature of the mutants. Genetics, 94: 961-968.
VOGEL, F. (1970) Spontaneous mutation in man. In: Vogel, F.
& Rohrborn, G., ed. Chemical mutagenesis in mammals and man,
New York, Springer-Verlag, pp. 16-68.
VOGEL, F. & ALTLAND, K. (1982) Utilization of material from
PKU-screening programs for mutation screeing. In: Bora, K.C.,
Douglas, G.R., & Nestmann, ed. Chemical mutagenesis, human
population monitoring and genetic risk assessment, Amsterdam,
Elsevier Biomedical Press, Vol. 3, pp. 148-152 (Prog. Mutat.
Res.).
WAKSVIK, H., MAGNUS, P., & BERG, K. (1981) Effects of age,
sex, and genes on sister-chromatid exchange. Clin. Genet., 20:
449-454.
WALTON, K.E., STYER, D., & GUENSTEIN, E.I. (1979) Genetic
polymorphism in normal human fibroblasts as analyzed by
two-dimensional polyacrylamide gel electrophoresis. J. Biol.
Chem., 254: 7951-7960.
WARBURTON, D., STEIN, Z., KLINE, J., & SUSSER, M. (1980)
Chromosome abnormalities in spontaneous abortion: data from
the New York City Study. In: Porter, I.H. & Hook, E.B., ed.
Human embryonic and fetal death, New York, Academic Press,
pp. 23-33.
WARNER, L.A., NEEL, J.V., & MEISLER, M.H. (1982) Separation
of allelic variants by two-dimensional electrophoresis. Am. J.
hum. Genet., 34: 209-215.
WHITTAKER, P.G., TAYLOR, A., & LIND, T. (1983) Unsuspected
pregnancy loss in healthy women. Lancet, 1: 1126-1127.
WHO (1983) EHC 27: Guidelines on studies in environmental
epidemiology, Geneva, World Health Organization, 351 pp.
WOLFF, S. (1977) Sister-chromatid exchange. Ann. Rev.
Genet., 11: 183-201.
WOLFF, S. & PERRY, P. (1974) Differential Giemsa staining of
sister-chromatids and the study of sister-chromatid exchanges
without autoradiography. Chromosoma, 48: 341-353.
WOLFF, S. & PERRY, P. (1975) Insights on chromosome
structure from sister-chromatid exchanges and the lack of both
isolabelling and heterolabelling as determined by the FPG
technique. Exp. cell Res., 93: 23-30.
WOLFF, S., BODYCOTE, J., & PAINTER, R.B. (1974)
Sister-chromatid exchanges induced in Chinese hamster cells by
UV irradiation of different stages of the cell cycle: the
necessity for cells to pass through S. Mutat. Res., 25: 73-81.
WOLFF, S., BODYCOTE, J., THOMAS, G.H., & CLEAVER, J.E.
(1975) Sister-chromatid exchanges in xeroderma pigmentosum
cells that are defective in DNA excision repair or
post-replication repair. Genetics, 81: 349-355.
ZETTERBERG, G., ANMEUS, H., & MATSSON, P. (1982) Use of flow
cytometry to concentrate 6-thioguanine resistant variants of
human peripheral blood lymphocythes. In: Bridges, B.A.,
Butterworth, B.E., & Weinstein, I.B., ed. Indicators of
genotoxic exposures, New York, Cold Spring Harbor Laboratory
Press, pp. 413-420 (Banbury Report No. 13).
ZIK, W.H. & NECKERS, L. (1983) Lymphocyte subpopulations
altered during blood storage. New England J. Med., 309:
435-436.
TGr somatic cells arising in vivo can be shown to be mutants
by the stability of their phenotype and deficiency of HPRT activity
(Albertini, 1982; Albertini et al., 1982a; Morley et al., 1983b).
Also, molecular studies have demonstrated changes in the HPRT locus
at the DNA level (Albertini et al., in press).
3.3.1. Principles and basis for the methods
TGr T-lymphocytes (T-lys), present in human blood, can be
measured either by autoradiography to determine TGr T-ly variant
frequencies (Vfs), or by cloning to determine TGr T-ly mutant
frequencies (Mfs). Autoradiography is the earlier method (Strauss
& Albertini, 1977, 1979) and has been used for some human
population studies (Strauss & Albertini, 1979; Strauss et al.,
1979; Cole et al., 1982; Lange & Pranter, 1982; Morley et al.,
1982; Vijayalaxmi et al., 1983b; Albertini, 1983; Albertini et al.,
1983a); it is simple, inexpensive, and can be automated (Anmeus et
al., 1982; Zetterberg et al., 1982). Cloning is a more recent
development by which it is possible to recover the TGr cells for
characterization of the nature of the mutations (Albertini, 1982;
Albertini et al., 1982; Strauss, 1982; Morley et al., 1983a;
Vijayalaxmi & Evans, 1984). This characterization may be
necessary in order to quantify the different types of somatic
mutations responsible for a collection of mutants.
3.3.1.1. Autoradiographic method
The autoradiographic detection of rare TGr T-lys is based on
the ability of these cells to incorporate tritiated thymidine
(HTdR) in vitro following short-term lectin stimulation in the
presence of cytotoxic concentrations of 6-thioguanine. The vast
majority of human peripheral blood T-lys are in the G0 stage of the
cell cycle in vivo. When stimulated in vitro with lectins, such
as phytohaemagglutinin (PHA), T-lys are activated to early G1,
which is characterized by the acquisition of T-cell growth factor
(TCGF) receptors on their surfaces (Maizel et al., 1981). These
receptors are also called interleukin-2 (IL-2) receptors or Tac
antigens (Deeper et al., 1983). A series of cellular events
results in the production of interleukin-1, (IL-1) and IL-2, and
other factors in short-term peripheral blood mononuclear cell
cultures, with IL-2 driving the activated T-lys through G1 to DNA
synthesis and subsequent events in the cell cycle.
The initial round of lectin-stimulated T-ly DNA synthesis
in vitro is inhibited by 6-thioguanine. Inhibition probably is
at the activation step in 6-thioguanine sensitive cells and
occurs without incorporation of the analogue into DNA. The
interval of lectin stimulation before the addition of HTdR in
the autoradiographic assay is short, so that cell division in
vitro does not occur (Strauss & Albertini, 1979). Under proper
conditions, the T-lys that incorporate this label in the presence
of 6-thioguanine are TGr variants. These cells can be enumerated
by autoradiography and the TGr variant frequency (Vf) calculated.
3.3.1.2. Cloning method
Human T-lys are easily propagated for extended periods in vitro
by supplying TCGF to cells that have been activated with lectin or
antigen (Paul et al., 1981). Thus, mass populations derived from
peripheral blood can be developed and maintained. Moreover,
freshly-obtained human T-lys may be cloned directly in vitro, in
the presence or absence of 6-thioguanine. In the first case, large
cell innocula must be used, to ensure the presence of rare TGr
cells (Albertini, 1982; Albertini et al., 1982a; Strauss, 1982;
Morley et al., 1983a,b; Vijayalaxmi & Evans, 1984). Activating
intervals are short so that cell division does not occur in vitro
prior to exposure to cytotoxic concentrations of 6-thioguanine,
thus ensuring that the TGr cells recovered in vitro actually arose
in vivo. The number of TGr T-lys determined by cloning is used to
derive in vivo TGr T-ly mutant frequencies (Mfs).
3.3.2. Relevance and limitations
3.3.2.1. Relevance
The ability to measure TGr T-lys arising in vivo is important,
because it provides a genetic end-point for assessing human
genotoxicity that may be qualitatively different from somatic cell
chromosomal damage. Thus, the ability to measure gene mutation
provides a broader basis for human monitoring. Furthermore, gene
mutations, as assessed by TGr T-lys, are end-points that are
qualitatively different from other measures of in vivo DNA damage.
Measuring human somatic cell gene mutations occurring in vivo has
the following advantages:
(a) The effects of metabolic and kinetic factors are included
in measurements of somatic cell mutation occurring in vivo.
(b) Mutagenicity assays for in vivo events assess the genetic
effects of given environments, rather than the genotoxic
potential of targeted chemicals or other agents. Thus,
these assays can be used to measure the effects of
environmental mixtures in which the components and/or
their synergistic actions may not be known.
(c) Mutagenicity assays, in vivo, may define human population
heterogeneity in susceptibility to various mutagens/
carcinogens (Vijayalaxmi et al., 1983b). Such individual
susceptibilities may be important in assessing human
genotoxic risks.
(d) Mutagenicity assays, in vivo, have the potential for being
developed into a means of making quantitative human
genetic health risk assessments, provided that appropriate
correlations can be made between individual assay results
and individual health outcomes (Fowle et al., 1984).
3.3.2.2. Limitations
Although progress has been made in demonstrating that TGr T-lys
are mutant somatic cells, at least 3 major problems remain for
human in vivo somatic cell mutation assays. Two are concerned
with quantification and apply primarily to gene mutation assays.
The third applies to all human somatic cell mutation assays and the
relevance of the results for making estimates of human health
risks.
(a) The in vivo sensitivities of the human TGr T-ly assays in
terms of dose-response characteristics are not known at
present. However, because mutant cells are also
detectable in animals (Garcia & Couch, 1982; Gocke et al.,
1983; Recio et al., 1983; Jones et al., in press), these
studies may provide useful information.
(b) Although somatic cell mutations are the events of
relevance for human population monitoring, TGr T-ly assays
measure the frequency of somatic cell mutants, not the
number of mutations. In order to derive mutation
frequencies from mutant frequencies, it is necessary to
have information including cell pool sizes and
distributions in vivo, representativeness of test samples,
in vivo cell kinetics, and in vivo positive or negative
selection of mutants. One method for circumventing this
problem is to determine whether the mutants scored in a
mutagenicity assay are qualitatively heterogeneous, which
will yield a minimum number of independent mutational
events giving rise to the observed collection of mutants.
The ability to recover TGr T-lys from clonal assays makes
these determinations possible.
(c) Assays of genetic damage occurring in vivo in human
somatic cells have not been validated in that it has not
been possible to use the results as predictors of human
genotoxic health risks. Before such results can be used
as surrogate markers for human health outcomes, high-risk
populations will have to be monitored and correlations
made between the results from mutagenicity assays and
epidemiological outcomes. The importance of other factors
(e.g., immunotoxicity) in influencing human genotoxic
disease risks remains to be assessed.
3.3.3. Procedures for the assay of TGr T-lys arising in vivo in
human beings
3.3.3.1. Autoradiographic method
TGr T-ly Vfs for normal, control adults, as determined
autoradiographically by the method described here, rarely exceed
10 x 10-6. By contrast, chemotherapy-treated cancer patients
frequently have values several times higher (Strauss & Albertini,
1977, 1979; Lange & Pranter, 1982; Albertini, 1983). Other
laboratories report somewhat higher normal values, as well as a
positive correlation with age (Morley et al., 1982), indicating
that local ranges of normal values must be developed. In one
laboratory, approximately 75% of placental cord blood values fell
within the normal range for adults, while the remainder were
elevated (Albertini et al., 1983). Whether this reflects maternal
exposure to genotoxic agents, or an altered ratio between mutants
and mutations in the fetus remains to be determined.
The basic autoradiographic method for assessing TGr T-lys has
been described in detail by Albertini et al. (1982b) and Albertini
& Sylwester (1984). Peripheral blood mononuclear cells (MNCs),
which include the T-lys, are separated from whole blood by the
Ficoll-Hypaque method (Boyum, 1968). After washing, the MNCs are
suspended in a dimethyl sulfoxide-containing medium and aliquoted
into ampules for controlled freezing and storage. Freezing is
important for holding and shipping samples and serves also as one
method for eliminating the phenocopy effect of cycling cells as
described below. For assaying, MNCs are thawed, washed, and
suspended in appropriately supplemented medium for short-term
tissue culture. A minimum of 5 x 106 MNCs are cultured without
6-thioguanine (control culture), while several times this number of
cells are cultured with 2 x 10-4 M 6-thioguanine (test cultures).
All cultures contain phytohaemagglutinin (PHA), for activation of
T-lys, and are incubated under standard conditions. After
approximately 24 h (Strauss & Albertini, 1979), 3HTdR is added
to all cultures, and incubation continued for an additional 18 h.
Cultures are terminated by adding cold 0.1 M citric acid, to
prepare suspensions of free nuclei. The nuclei are then washed
and suspended in methanol-acetic acid fixative, counted, diluted
if necessary, and added to coverslips affixed to microscope slides.
Slides are dried, stained with aceto-orcein and subsequently
autoradiographed by standard methods. Slides are scored and Vfs
are calculated as described in section 3.3.4.1.
A subclass of 6-thioguanine-sensitive T-lys in human peripheral
blood may become labelled and scored as TGr (i.e., as variants),
when using the autoradiographic method, unless certain precautions
are taken (Albertini et al., 1982a,b; Albertini, 1982). These
phenocopies occur because, at any given time, a small minority of
human T-lys are in an activated state, in vivo. These cells have
the Tac antigen, when initially put into culture, and do not
require the activation step before proceeding to in vitro DNA
synthesis. Although ultimately 6-thioguanine sensitive, these
cells probably are not effectively blocked from accomplishing at
least one round of DNA synthesis in vitro, even in the presence
of 6-thioguanine. Thus, activated T-lys in the peripheral blood
constitute a potential source of phenocopies, when TGr T-ly Vfs are
determined by autoradiography. Cryopreservation appears to remove
this phenocopy effect by forcing the in vivo activated T-lys to
proceed to DNA synthesis in vitro at a time when a label is not
present (Albertini et al., 1982a,b). These cells are not scored as
variants. Cryopreservation is a critical step, although other
methods, such as prolonged incubation in 6-thioguanine, or
immunological elimination of Tac positive T-lys at the initiation
of culture, may accomplish the same purpose.
3.3.3.2. Cloning method
From the limited data available, normal control adults have
shown peripheral blood TGr T-ly Mfs of < 20 x 10-6 when based on
clonal assays in which single-cell cloning efficiencies achieved
values > 0.10. Similar findings from a number of laboratories
have been reported (Strauss & Albertini, 1979; Morley et al.,
1983a; Vijayalaxmi & Evans, 1984). Results for placental blood and
for mutagen-exposed adults remain to be determined. TGr T-ly Mfs
have been slightly higher than TGr T-ly Vfs, concurrently
determined (Albertini et al., 1984).
The cloning method for assessing TGr T-lys has been described
by Albertini et al. (1982a). Peripheral blood MNCs are obtained
from whole blood as for the autoradiographic method, but freezing
is not required. The MNCs are incubated with PHA in order to
activate the T-lys. The duration of activation is sufficiently
short to ensure that cell division does not occur. Activated cells
are then innoculated in limiting dilutions into the wells of
microtitre plates, in the presence or absence of 6-thioguanine
(approximately 10-5 M). In addition to the appropriate medium,
wells contain an optimum concentration of crude TCGF and feeder
cells. X-irradiated B-lymphoblastoid cells are well suited for use
as feeders. A TGr lymphoblast line is used to avoid interference
with subsequent HPRT enzyme assays of the recovered TGr T-lys. An
average of 1 activated cell per well is innoculated into the non-
selection wells (containing no 6-thioguanine); 105 activated cells
per well are innoculated into selection wells (containing 10-5 M
6-thioguanine). Wells are scored after approximately 2 weeks of
culture with one change of medium, by microscopy, by scintillation
spectrometry of HTdR incorporation (added during the last day of
culture), or by cell transfer and clonal expansion. In vitro
cloning efficiencies (CEs) of T-lys are determined from wells
receiving, on average, 1 cell per well. By assuming a Poisson
distribution of cells in wells, the average number of clonable
cells per well is derived from the P0 class of that distribution,
i.e., the observed fraction of wells without growing cells.
Similarly, the incidence of TGr cells in wells receiving 105
activated T-lys in 6-thioguanine is determined from the P0 class of
wells in the plates containing 6-thioguanine. The TGr T-ly Mf is
the incidence, divided by 105, corrected for the CE.
TGr T-lys obtained by cell transfer and clonal expansion may be
characterized for T-ly subset markers, stability of the TGr
phenotype, HGPRT enzyme activity, and, by using suitable cDNA
probes, for the nature of the molecular lesion at the DNA level
(Albertini et al., 1982a, in press).
For reasons not known at present, human T-lys, activated in
vitro as described, may fail to clone as efficiently when present
as single cells than when present in large numbers (i.e., 105
cells/well), even when the latter are selected in 6-thioguanine
(Albertini et al., 1984). Clonal assays with a single cell CE
of < 0.10 have the potential for yielding falsely elevated Mf
values, because of an underestimate of clonable cells in selection
wells. Current research is directed at increasing and/or
standardizing single-cell CEs. At present, however, only Mfs
determined in clonal assays achieving single cell CEs of 0.10 or
more should be considered valid.
3.3.4. Data presentation and analysis
3.3.4.1. Autoradiographic method
Slide scoring is done by one of 2 methods that give almost
identical results. One method involves determining labelling
indices (LIs) for both control (no 6-thioguanine) and test
(6-thioguanine containing) cultures for each individual. The LI of
test cultures (LIt) is determined by counting all labelled nuclei
on all slides made from test cultures, and dividing this number by
the total number of nuclei (determined in suspension) added to all
slides.
Number of labelled nuclei on all test slides
LIt = --------------------------------------------
Number of nuclei on all test slides
The LI of control cultures (LIc) is determined from a
differential count of 2500 nuclei on slides from control cultures.
Number of labelled nuclei per 2500 nuclei
LIc = -----------------------------------------
2500
The TGr T-ly Vf for each individual is calculated from the LI
of test cultures (LIt) divided by the LI of the control culture
(LIc).
LIt
Vf = ---
LIc
An alternative method of scoring autoradiographic assays has
recently been described for mouse TGr T-lys (Gocke et al., 1983),
and is equally applicable to human assays. Statistical methods for
deriving confidence intervals for variant frequencies are described
by Sylwester & Albertini (1984).
3.3.4.2. Cloning method
Cloning efficiencies (CEs) are calculated from the P0 class of
the Poisson distribution:
P0 = e-x, or
x = -ln P0 (control)
where x is the average number of clonable cells per well. When CE
is determined by single cell innocula, x = CE.
The incidence of TGr T-lys in selection wells, receiving on
average 105 cells per well, is also determined from the P0 class of
the Poisson distribution:
P0 = e-y, or
y = -ln P0 (test)
where y is the average number of clonable TGr T-lys per well.
The TGr T-ly Mf is determined from the incidence of TGr
T-lys per well divided by the average cell innocula, corrected
by the CE. When 10 cells are innoculated into selection wells:
-ln P0 (test)
Mf = -------------
CE x 105
Appropriate statistical procedures for handling Mf data,
differences between Mfs, etc. are being developed by methods
analogous to those referred to in section 3.3.4.1.
3.3.5. Conclusions
TGr T-lys arise in vivo, are present in human peripheral
blood, and their frequencies are measurable. Known mutagen
exposure increases the frequencies of these cells and their
characterization in vitro has shown them to be somatic mutants.
Thus, methods are available for measuring specific locus somatic
mutants occurring in vivo in human beings, for purposes of human
genetic monitoring. The 2 assays described here continue to be
developed. There are potential technical sources of error. Among
the limitations in the interpretation of the results of these
assays is the fact that the numbers of somatic mutants, and not the
numbers of somatic mutations, are being measured. The latter may be
of most interest for human population monitoring. However,
characterization of the recovered mutant cells, which is possible
in the cloning assay, may make it possible to estimate the minimum
number of mutations responsible for a given number of mutants.
Methods described here can be applied to the detection of
mutations, occurring in vivo at other genetic loci in human T-lys,
in order to broaden the base for human monitoring.
4. GERMINAL MUTATIONS
Methods and technical aspects of approaches available for
detecting germinal mutations in human populations are reviewed in
this section. It should serve as a guide, when such studies are
initiated, and as a reference for defining specific methods for
estimating human mutation rates.
4.1. Introduction
Germinal mutations include a spectrum of alterations in
either the structure or quantity of DNA in germinal cells. The
study of germinal mutations is the quantification of transmitted
genetic damage. Within the context of monitoring for induced
genetic damage, this is an important consideration, because it is
the offspring of exposed individuals, rather than the exposed
individuals themselves, that are the focus of concern. It is
generally assumed that a significant proportion of all mutations
have deleterious effects on both the health and the genetic
constitution of future generations. Thus, the increased genetic
risk and associated health effects are of ultimate concern,
following exposure to a putative mutagen, when possible increases
in germinal mutation rates are being ascertained.
Germinal mutations are usually classified in 2 categories.
Chromosomal mutations are operationally defined as changes in
either chromosome number or structure observable with standard
karyotypic techniques. More minute changes in DNA structure are
classified as gene mutations, often referred to as "point"
mutations.
4.1.1. Approaches for detecting germinal mutations
The 3 approaches to monitoring for germ cell mutations are
generally divided as follows: (a) chromosomal, (b) biochemical,
and (c) the indicator phenotype; each detects a different set of
end-points.
4.1.1.1. Detection of chromosomal mutations
Germinal chromosomal mutations occur in about 5% of recognized
conceptions (Hook, 1981a,b), making them more amenable than the
rarer specific locus mutations for study in small exposed
populations. Unbalanced chromosomal complements are almost always
associated with deleterious phenotypic effects, leading to fetal
death, livebirths with anomalies and mental retardation, and/or
sterility. Balanced chromosome complements may occur as mutations
without phenotypic effect. However, their deleterious effects will
be seen primarily in the next generation in individuals with
unbalanced chromosomal complements.
Chromosome abnormalities can originate in four different ways:
a) they may be inherited from a parent; b) they may result from
errors during gametogenesis (e.g., meiotic non-disjunction); c)
they may result from events during conception (e.g., dispermy
resulting in triploidy); d) they may result from events after
conception (e.g., mitotic non-disjunction). Only the second
category listed above is regarded as a germ-cell mutation in the
strict sense, but to avoid unnecessary complexity in the discussion
that follows, triploidy and tetraploidy will be considered in the
review of germinal events.
Chromosomal mutations can be subdivided into numerical and
structural anomalies.
(a) Numerical aberrations
Numerical chromosomal abnormalities include trisomy, monosomy,
triploidy, and tetraploidy. Most instances result from events that
occur during gametogenesis in a parent, or at the time of
fertilization, although strict proof of the time of origin is often
lacking.
Apart from triploidy and tetraploidy, numerical chromosome
abnormalities involving sex chromosomes without a mosaic 45, X cell
line, or involving autosomes are usually presumed to have resulted
from a germinal mutation. The only exceptions are situations in
which numerical abnormalities occur in only a single tissue, as
for example in some malignancies. It is often difficult, however,
to exclude formally the possibilities that they: a) have been
inherited from a parent who is a cryptic mosaic for the abnomal
line; or b) have resulted from a somatic event, mitotic non-
disjunction, early in the development of the organism.
The strongest risk factor known for numerical abnormalities is
older maternal age, which is highly associated with the frequency
of trisomy (Hassold et al., 1980, 1984). Paternal age seems to
have little, if any, effect (Hook, in press). In studies using
chromosomal markers, it has been demonstrated that at least 60% of
trisomy 21 appears to be the result of maternal 1st division non-
disjunction (Juberg & Mowrey, 1983).
Numerical abnormalities almost always occur in offspring
of parents who have normal chromosomal complements, and thus,
such abnormalities are presumably the result of germinal
mutations. Although there is not a great deal of experimental or
epidemiological evidence to link numerical chromosome abnormalities
with environmental agents, increases in such anomalies must be
considered as possible outcomes of exposure to possible mutagens.
An unknown proportion of trisomy or monosomy may result from post-
zygotic non-disjunction, or, in the case of trisomy, may be
inherited from parents who are cryptic carriers, and thus may not
be the result of germinal mutations in a strict sense. For
trisomy, the results of studies of the parental origin of the extra
chromosome suggest that a maximum of 25% of trisomies are due to
post-zygotic non-disjunction, and it may well be considerably less
(Juberg & Mowrey, 1983).
Evidence suggests that mosaic trisomies (47/46) do not
originate from post-zygotic events any more often than non-mosaic
trisomies (Hassold, 1982).
Ninety percent of numerical anomalies in recognized conceptuses
terminate as fetal deaths; thus, a study restricted to live births
will miss a major proportion of detectable abnormalities.
Only a few kinds of numerical anomalies commonly survive to
birth. The most common is Down's syndrome, trisomy 21, occurring
in about 1 in 1000 live births. Other numerical anomalies such as
trisomy 18 and trisomy 13 sometimes survive to occur as live
births, but these are much less common.
(b) Structural aberrations
The proportion of individuals with detected structural
cytogenetic abnormalities is likely to vary with technical
factors. Recent advances in the resolution of chromosome
substructure (high resolution banding) have reduced significantly
the size of detectable lesions and such advances appear likely to
continue. Thus, it is difficult to specify precisely the
proportion of recognized conceptuses with a structural abnormality.
With currently available techniques, these are much less frequent
than those with numerical abnormalities. Among live births, the
ratio of detected structural to numerical abnormalities is about
1:4 to 1:5 (Hook & Hamerton, 1977). Among fetal deaths, the ratio
is much lower, about 1:30 (Warburton et al., 1980). Unlike
numerical abnormalities, a significant fraction of detected
structural abnormalities are known to be inherited, so that
inferences concerning mutation are not possible unless both parents
are studied and found not be be carriers of the aberration present
in the offspring. The possibility of false assignment of paternity
must also be considered. Unlike trisomies, structural chromosome
abnormalities, in general, show little if any association with
parental age, although markers and X-isochromosomes may represent
exceptions. In instances in which the parental origin of a
structural mutation has been investigated, many have been found to
be predominantly paternal in origin, unlike trisomies (Magenis &
Chamberlin, 1981).
In general, mosaicism, involving a normal cell line and a cell
line with a structurally abnormal chromosome, is not attributable
to germinal cell mutation, but may be safely inferred to be of
post-zygotic origin (supernumerary markers are, however,
exceptions).
In terms of sensitivity to environmental factors, mutant
germinal structural chromosome aberrations are likely to be more
similar to germinal specific locus mutations than numerical
abnormalities, as both involve alterations in the structure of the
genetic material. Germinal chromosomal rearrangements, unlike
specific locus mutations, show little association with advanced
paternal age in human beings (Hook, in press) indicating that there
are etiological differences between these effects.
4.1.1.2. The biochemical approach to detecting point mutations
Many biochemical approaches for the study of germinal mutations
have been proposed (Bloom, 1981), but most are not feasible at
present. Two general approaches are currently used to study the
proteins in offspring of selected individuals. Electrophoresis is
used to detect a significant proportion of amino acid substitutions
while enzyme activity measurements are used to detect major losses
in protein function or protein quantity.
Studies involving genetic typing of large numbers of
individuals, either to analyse population structure or to develop
mutation monitoring programmes, have demonstrated the feasibility
of large-scale screening using one-dimenional electrophoretic
techniques (Harris et al., 1974; Neel et al., 1980a; Altland et
al., 1982). Through the introduction of high-resolution two-
dimensional electrophoresis (Klose, 1975; O'Farrell, 1975), the
number of proteins that can be studied in a single sample has been
increased to several hundred. The search for mutational events
resulting in loss of a functional gene product, using quantitative
techniques, is complementary to the electrophoretic assay, in that
this assay detects genetic events not normally detectable by
standard electrophoretic assays. Both the electrophoretic and
enzyme activity approaches have been used in studies to determine
the induced mutation rate in exposed mice (Johnson & Lewis 1981;
Johnson et al., 1981; Bishop & Feuers, 1982) and Drosophila (Racine
et al., 1980). Thus, it should be possible to acquire data on the
mutation rate in human populations and also to examine directly
some of the problems associated with extrapolation from
experimental animals.
One other biochemical approach has been proposed for detecting
mutational events in human populations. It is possible to study
mutations at the DNA level by employing the restriction enzyme
mapping approach. One potential advantage of the restriction
enzyme mapping approach (or other techniques that directly monitor
specific alterations in DNA structure) is the ability to examine
larger portions of the genome and thereby acquire increasing
amounts of data from each individual. Since this approach is
currently in the developmental stage, appropriate samples should be
retained, as far as possible, to take advantage of this or other
techniques that may be developed in the near future.
The major constraints in relation to the biochemical approach
include: (a) the large number of determinations and, to date, the
large populations necessary to detect statistically-significant
increases in the mutation rate; (b) the distinction between
"apparent" mutations and nonparentage; and (c) the problem of
confirming that a suspected variant is the result of an alteration
in DNA structure, i.e., is a transmissible trait.
4.1.1.3. Indicator phenotypes
Three types of indicator phenotypes are considered in this
section: Down's syndrome, fetal death, and sentinel phenotypes.
Down's syndrome will be discussed in section 4.3, which deals with
chromosomal mutations. The potential use of fetal death as an
expression of genetic damage arising from genic mutations will be
discussed in section 4.5. Although the term "sentinel phenotype"
is relatively new (Sutton, 1971), the notion of studying mutations
in human beings by counting the frequency of dominant traits is
credited to Danforth (1921). Mulvihill & Czeizel (1983) have
reviewed the current status of the concept.
As indicators of germinal genic mutations, sentinel phenotypes
are a significant health problem and are recorded in a variety of
health facilities. The sentinel phenotype is a clinical disorder
that: (a) occurs sporadically as a consequence of a single, highly
penetrant mutant gene, (b) is a dominant or X-linked trait of
considerable frequency and low fitness, and (c) is uniformly
expressed and accurately diagnosable with minimal effort, at or
near birth. Individuals with such traits are important for the
surveillance and monitoring of germinal genic mutations because
affected persons of unaffected parents arise from a new mutation.
Their use has severe disadvantages connected with the difficulty
of accurate nosological diagnosis arising from their genetic
heterogeneity and a general lack of clinical expertise. The
sentinel phenotype approach, like other strategies in mutation
epidemiology, is burdened with problems created by the inability
to link separate data files, the necessity to maintain
confidentiality, and the difficulty in collecting sufficiently
large study populations or study samples. The best course of
action for the present is to obtain field experiences and to
sustain critical discussion of the approach.
4.1.2. Methodological considerations and strategies
4.1.2.1. Sample acquisition and storage
Future access to study subjects may be limited; therefore, the
maximum quantity of sample, usually blood, should be obtained on
the initial contact. When more than one test is to be performed,
attempts should be made to coordinate the appropriate collection
procedures and minimize the number of different acquisitions. In
situations where fetal tissue is being collected for chromosomal
studies, it is suggested that, where possible, a sample of kidney
tissue should be collected and stored for possible biochemical
analysis.
Adequate samples should be stored for confirmation of any
observation and also for future analysis using new and/or refined
techniques. Lymphocytes should be stored in a manner that allows
for the retrieval of viable cells and the subsequent expansion of
the number of cells, thus providing material for future studies.
4.1.2.2. Timing of studies
With specific regard to mutation studies, following suspected
exposure to mutagens, women of child-bearing age who have been
exposed to a mutagen and wives of exposed men should be identified
and surveyed as soon as possible. Questionnaires or interviews
should be undertaken and used to obtain data on basic biological
and demographic variables (and information on other possible
mutagenic factors). The factors include, but are not necessarily
limited to, age, race, previous pregnancy history, smoking, and
drug use. Women should be encouraged to participate in a
continuing evaluation of future pregnancy outcomes. If a woman
thinks she is pregnant but is uncertain, pregnancy testing should
be carried out as soon as possible. A control population of
similar ethnic and socioeconomic background should be identified.
This could be done at the time of identification of women at risk
of pregnancy, or could be done after pregnancy is confirmed, using
as a control group, women pregnant at the same gestational age.
Each of these approaches has difficulties. To wait until the
exposed woman is pregnant before selecting a matched (pregnant)
control may result in the pregnancy of the exposed woman being
terminated by the time the control is chosen. It may be extremely
difficult to avoid biasing the results towards a more favourable
outcome in the controls. If controls are selected prior to the
pregnancy of the exposed woman, there is no certainty that the
exposed and control women will become pregnant at the same time,
if in fact they conceive at all. One possible approach is to
select several possible controls for each exposed woman, before
pregnancy, and follow the reproductive history of all of them.
Ideally, these controls should be matched as far as possible in
age, race, socioeconomic status, and previous reproductive history.
Infants, children, and young unmarried males, who have been
exposed should also be identified early and followed for possible
inclusion should a subsequent long-term study be undertaken (see
section on record linkage).
Women recruited into the study should be interviewed by
investigators, at least once a month, to ascertain if they are
pregnant or think they may be pregnant. If there is any question,
pregnancy testing should be carried out.
4.1.3. Summary
Given the current state of knowledge in the area of human
mutations, all 3 approaches, namely indicator phenotype,
chromosomal, and biochemical, to estimating germinal mutational
damage in human populations should be considered complementary.
They detect different types of genetic "damage" or "end-points",
have different degrees of relevance for estimating potential health
effects and probably different sensitivities. Therefore, a
collaborative monitoring exercise should employ all 3 in order to
obtain the maximum amount of data possible.
4.2. Germinal Chromosomal Abnormalities
4.2.1. Principles and basis of the method
Chromosomal aberrations associated with germinal cell mutation
have already been discussed in section 4.1.1.1.
Depending on their origin and consequences, chromosomal
mutations may be detected in gametes, the embryo or fetus, in
live births, or at later stages in life.
If a population is exposed to a mutagen, the most direct method
of detection of germinal chromosomal mutants would be examination
of the gametes themselves. At present, there are no methods for
evaluating human ova. Preparations of human sperm chromosomes can
be made, but these methods are difficult and time-consuming, and
few laboratories have yet been successful with this technique
(Martin et al., 1983). It is possible that improved techniques for
studying sperm chromosome constitution will be available in the
future, but, at present, this method cannot be regarded as
practical for most situations involving mutagenic exposures.
Also, at the present time, alterations in sperm morphology or
function cannot be regarded as a reliable index of germinal cell
mutation, nor can changes in the proportion of sperm showing double
"Y" bodies. Thus, the following discussion focuses on detection of
chromosomal mutations in the offspring of those exposed to known or
suspected mutagens.
A clinically-recognized pregnancy is generally diagnosed after
the first missed menses, at about 4 weeks of gestation. The
frequency of chromosome abnormalities at this point in gestation
has been estimated to be about 5% (Hook, 1981a). About 15% of
recognized pregnancies terminate in fetal death, approximately one-
third having a chromosome aberration (Harlap et al., 1980; Hook,
1981a). The rate of loss between conception and recognized
pregnancy is not known, but it is generally agreed to be very high
(Kline et al., 1980). The proportion of these early losses that is
associated with chromosomal aberration is unknown. The bulk of the
evidence from the use of sensitive human chorionic gonadotropin
serum assays, capable of diagnosing pregnancy immediately after
implantation (usually 7 days after conception), suggests that loss
between implantation and the first missed menses may be at least as
common as loss afterwards (Miller et al., 1980; Edmonds et al.,
1982), but not all results are in agreement (Whittaker et al.,
1983). Preimplantation losses are still unmeasurable. Only
clinically-recognized pregnancy in the usual sense will be dealt
with here, since the other types of study are unlikely to be
feasible on the necessary scale, and chromosome studies cannot
be performed on these early losses.
The proportion of chromosome abnormalities in fetal deaths
varies with gestational age, being highest (about 50%) in the
8 - 12 weeks range, and then decreasing with gestational age (to
about 7%) at more than 22 weeks (Warburton et al., 1980). The
kinds of chromosome abnormalities found in fetal deaths include
many rarely, or never, seen in live births, e.g., triploidy,
tetraploidy, and trisomy for most whole chromosomes. Monosomy X,
though occurring in only about 1 in 20 000 live births, occurs in
about 7% of early fetal deaths; triploidy occurs with about equal
frequency, as does trisomy 16, an anomaly never seen at term and
compatible only with very rudimentary embryonic development
(Warburton et al., 1980).
Chromosome abnormalities are found in about 7% of late fetal
deaths (including still births) (Sutherland et al., 1978). At
birth, the proportion has been found to be 0.6% (Hook & Hamerton,
1977). About 90% of all recognized conceptuses with chromosome
abnormalities terminate as fetal deaths.
Most recognized abnormalities involve numerical aberrations
that are presumptive mutants. Structural chromosome abnormalities
make up only about 5% of the chromosome abnormalities seen in fetal
deaths (Warburton et al., 1980). About 40% of structural
abnormalities found in fetal deaths or at amniocentesis are de novo
mutational events: the rest are inherited from a carrier parent
(Jacobs, 1981; Hook et al., 1984).
4.2.2. Relevance and limitations
4.2.2.1. Studies of induced abortions
Chromosome studies of induced abortions are a useful source of
data. Such investigations are, of course, only possible where
induced abortion is a legal option. Advantages are that the
frequency of chromosomal abnormalities is still high at the point
in gestation when most induced abortions are performed, that the
specimens are likely to be viable in culture, and that the
procedure can be scheduled for collection purposes. A disadvantage
is that it is likely to be more difficult to obtain specimens from
the controls than from the exposed population. If, for example,
exposed women were more likely than controls to provide specimens
of early abortions, the unadjusted rate of chromosome abnormalities
would appear, incorrectly, to be higher.
4.2.2.2. Studies of fetal deaths
Studies in which the products of conception from fetal deaths
are examined cytogenetically are likely to be the most productive
in terms of the proportion of chromosome abnormalities that are
detected. However, such studies are difficult because of problems
in specimen retrieval, culture failure, and the high cost of the
tissue culture procedures required. Usable specimens will be
obtainable from only a portion of cases, even with the best
retrieval systems, since viable fetal tissues are sometimes not
present at the time of expulsion. Organization of retrieval
systems can be a challenge, even in such ideal situations as a
teaching hospital, and are extremely difficult outside a medical
setting. Through special education, attempts can be made to
collect specimens from women having early abortions, where the
tissue is passed at home, and medical attention may not be sought.
In New York City, an interview study suggested that 40% of women
having a first trimester fetal death did not seek medical attention
(Kline et al., 1981). Factors affecting the probability of
specimen retrieval must always be examined to rule out biases that
would affect the outcome of the study.
When cytogenetic analysis is performed, further case loss
occurs because not all will be successfully karyotyped. This might
produce differences in studies done in different laboratories or at
different times. Thus, cases and controls should be studied at the
same time, in the same laboratory. Ideally, the laboratory
carrying out the study should not know if a specimen is from a
control or a case.
One further problem in interpreting studies of fetal death is
the possibility that an environmental agent might influence not
only the rate of occurrence of a genetic abnormality, but also the
probability that a conceptus with an abnormality can survive to a
particular point in gestation. If an exposure reduces the
viability of a conceptus with a chromosome abnormality, such as
monosomy X, so that it is lost even earlier in gestation, before
recognizable pregnancy, a reduction in that anomaly will be seen in
all recognized conceptuses. An exposure that postpones the fetal
death of a conceptus with abnormality from before the usual
recognition of pregnancy to later in gestation would result in
an increased proportion of detected abnormalities.
4.2.2.3. Studies of prenatal diagnosis specimens
In some locations, amniocentesis for prenatal diagnosis is
widely available. This procedure is usually done at 16 - 20 weeks
of gestation. In such areas, many women exposed to putative
mutagens might seek such procedures, though a much smaller
proportion of controls might do so. While data available from
such an outcome should be used in any analysis, this cannot be
regarded as a plausible sole source of pertinent data on chromosome
abnormalities. It should be noted that, if all women in a
population undergo amniocentesis, the expected proportion of
chromosomally abnormal fetuses is 1%.
4.2.2.4. Studies of live births
Studies of live-born infants and older children are easiest to
undertake, and cheapest to perform. However, because only a small
proportion would be expected to be affected (0.6%), a relatively
large-scale study would have to be undertaken to detect a
statistically-significant increase in the proportion affected. A
sample of 25 000 births is needed to detect a doubling of the
trisomy rate at birth ( P < 0.05)a.
-------------------------------------------------------------------
a For the purposes of discussion, cases with an extra sex
chromosome have been included.
Some inferences are possible in relation to large exposed
populations, based, in part, on phenotypic evaluation. Cytogenetic
study for mutation investigation might, for example, be carried out
only on children with major malformations at birth, i.e., about 2%.
In any event, such studies would be indicated on this group of
children for clinical reasons. This approach would reveal
primarily only unbalanced autosomal abnormalities, about 1/3 - 1/2
of all chromosomal abnormalities in live births (both structural
and numerical abnormalities would be detected). Such a restriction
in a cytogenetic study would result in perhaps 10 - 20% of those
evaluated being found to have an abnormality. This reduces 50-fold
the total number of cytogenetic studies required in the population,
but does not avoid the need for a large original exposed population
from which the selected group with malformations would be drawn.
4.2.2.5. Studies of indicator phenotypes of chromosomal abnormalities
Inferences in large populations might be possible on the basis
of simple enumeration of Down's syndrome individuals, 98% of whom
are likely to be trisomic or carry a de novo chromosome
rearrangement. The expected frequency is about 1 in 1000 births.
Similarly, cases of Patau syndrome and Edwards syndrome (resulting
from 47, +13 and 47, +18, respectively) might also be enumerated.
The expected frequencies, however, are much smaller (each about 1
in 10 000 live births (Hook & Hamerton, 1977), and the phenotypes
are not as useful an indication of karyotypic abnormality.
Some conclusions on the probability of karyotypic abnormality
can be reached through morphological examination and classification
of specimens from fetal death. A useful classification scheme,
suggested by Byrne (1983), provides categories reflecting the
degree of organization of development, as well as developmental
age. These categories range from an "empty sac" with no visible
embryo, to an apparently normal fetus greater than 30 mm. Studies
associating specimen morphology with karyotype have shown that,
while about 1/2 of the most poorly-developed specimens have a
chromosome abnormality, less than 2% of fetuses greater than 30 mm
in length and with no visible malformations externally, will have
a chromosome abnormality (Byrne, 1983). Thus, if resources are
limited, such specimens might be left unstudied without much loss
of information.
True hydatidiform moles represent a category of conception
where the chromosome constitution is usually 46,XX, reflecting 2
identical male haploid complements in an egg with no maternal
chromosomes. Although other rare karyotypes occur, single
morphological examination is sufficient to indicate the chromosome
abnormality with over 98% accuracy.
4.2.3. Procedures
4.2.3.1. Fetal specimens
In a prospective study, women can be provided with sterile
containers containing a balanced salt solution for collection of
specimens from early fetal deaths. Later fetal deaths are likely
to reach medical attention, and collection systems must be
organized at likely treatment areas. Specimen containers should
also be available at such possible collection points as clinics,
emergency rooms, and wards.
(a) Morphological examination
Specimens should be examined externally and described in a
classification scheme that reflects the degree of development
as well as developmental age. Fetal autopsies can be performed
on specimens of 30 mm or more. A careful description of the
classification scheme must be provided, with photographs, whenever
possible, of abnormal specimens.
(b) Karyotyping
Successful cultures can be obtained, up to 5 days after
expulsion, though such a delay is not recommended. Thus, transport
of samples over some distance is possible. As the products of
conception are almost never obtained in a sterile state, the use of
culture medium for storage is not recommended, since it will
encourage the growth of contaminants. Specimens should be kept
refrigerated (but not frozen), during storage or transport over
long distances.
Specimens from induced abortions should be obtained in a
sterile container from the operating room or clinic. Care must be
taken to prevent the usual routine fixation of such specimens.
A most important aspect of the procedure is the careful
examination of the specimen to ensure that only fetal tissues are
taken for culture. Some specimens will contain only decidua, and
cannot be used. Any fetal tissue can be used for karyotyping; if
available, fresh tissues from the embryo or fetus are most
desirable; however, the actual embryo may be very small for the
length of gestation, very macerated, or absent altogether. In this
case, fetal membranes (amnion and/or chorion), or placental villi,
carefully dissected away from maternal tissues, may be used.
Maternal cell contamination is an inevitable possibility in such
studies, but evidence suggests that it is infrequent in experienced
hands (Warburton et al., 1980; Hassold, 1982).
Fetal tissues may take from a few days to several weeks to
reach the stage where they can be karyotyped. Cover slip
preparations with in situ chromosome preparations allow faster
karyotyping, but may not yield as large a number of analysable
cells as cultures in flasks, which must be trypsinized before
harvesting (Byrne, 1983).
Banded chromosome analysis should be carried out on all
specimens, and on at least 10 cells from each culture analysed.
Mosaicism is not uncommon among cultures from spontaneous
abortions. More cells must be counted if a non-modal cell, not
accounted for by random loss, is found among the first 10 cells,
e.g., if a normal cell is found in an otherwise trisomic culture,
or a trisomic cell or a 45,X cell is found in an otherwise normal
culture. In general, the proportion of mosaics among all
abnormalities detected, is expected to be small (Warburton, 1980).
For the purposes of initial analyses, it is suggested that they be
classified with non-mosaic aberrations with the same abnormal line.
Almost all instances of numerical chromosome abnormalities
are presumably the result of a mutation in the most recent
generation. Thus, if monosomy, trisomy, triploidy, or tetraploidy
is discovered, then study of the parents to confirm a mutation is
not necessary, as the likelihood of mutation is perhaps 0.99.
However, this is not the case for structural cytogenetic
abnormalities. At about 18 weeks of gestation, for instance,
only about 40% of structural abnormalities are the result of a
recent mutation, 60% being inherited from carrier parents. Thus,
if a structural rearrangement is observed, study of the parents
should be undertaken to determine if this is the result of a
mutation first manifest in the offspring.
4.2.3.2. Live births and other offspring
Chromosome studies will normally be performed on PHA-stimulated
lymphocyte cultures from specimens of peripheral blood. Specimens
should be collected as soon as possible after birth in order to
minimize losses of neonatal deaths. Cord blood can be used, and is
easy to collect routinely.
Preservation of whole blood samples by deep-freezing (-70 °C.)
in DMSO will make possible a good retrieval of cells for chromosome
analysis (or other kinds of studies) for up to a year (Nakagone et
al., 1982). Furthermore, specimens can be collected and stored
before laboratory facilities have been organized for the study,
and, if necessary, can be transported to a laboratory some distance
away.
If a structural rearrangement is found, parental studies are
necessary.
4.2.3.3. Detection of "indicator" phenotypes for germinal chromosomal
mutations - trisomies
Cases of Down's syndrome can be identified through medical
records, vital statistics, registries, or education settings.
Identification is likely to be incomplete through any of these
routes, and care must be taken that both exposed and unexposed
women are equally likely to be discovered.
4.2.3.4. Data presentation
Results in those exposed should initially be presented
according to broad categories of events (Table 5) and also
within subcategories, when sample size allows, because different
chromosomal abnormalities may reflect different etiological
factors. Trisomy 21 and trisomy 16 may well result from different
mechanisms, for example, while increased triploidy might indicate
increased dispermy.
Data should be presented on "conceptions" followed from
detection of pregnancy, and results compared by category between
exposed and control groups. Results should be expressed as the
proportion of either the exposed or control group with a specific
abnormality. Data should be stratified by maternal age, or other
adjustment made for this variable, because of the very strong
association of maternal age with trisomies (Hassold et al., 1980,
1984). In addition, exploratory analysis should be undertaken
of other possible confounding variables such as ethnicity,
socioeconomic status, cigarette smoking, and alcohol consumption.
Any differences between exposed and control groups in these
variables should be adjusted for in the analysis by sample
stratification, or multivariate analyses. Results should also
be analysed according to whether the father, the mother, or both
parents have been exposed. If sufficient data are available by
exposure category, a dose-effect relationship should be sought.
With regard to structural rearrangement, cases in which one or
both parents cannot be studied should be scored separately, and
regarded as possible mutants. The mutation rate for structural
rearrangements should be reported as a range. The minimum boundary
excludes such cases as being of unknown status. The maximum
boundary includes such cases.
It should also be recognized that lack of an observed
difference during the first few years after exposure does not
preclude the possibility of late manifestations of effects. In one
study, it has been claimed that a radiation effect on trisomy 21
frequency first manifested itself 10 years after exposure (Alberman
et al., 1972).
A problem that may arise in the evaluation of results is that
of missing data. Pregnant exposed and control individuals may die,
or withdraw before the outcome of the pregnancy is established.
Moreover, if a fetal death occurs, tissue may not be collected and,
if collected, the specimen may not include fetal tissue. Even if
fetal tissue is set up in culture, the karyotyping procedures may
not be successful. In experienced laboratories, the proportion of
successful cultures has varied from 65% to 90% (Warburton, 1980).
Moreover, culture failures are likely to be preferentially higher
for fetuses with chromosome abnormalities because the greatest
proportion of culture failures occurs in specimens with poorly
"organized" morphogenesis, which have been dead for some time in
utero. Among specimens cultured successfully, the proportion of
cytogenetic abnormalities in specimens with poorly organized
morphologies is at least 50%, at least twice as high as that of
specimens whose morphogenesis is more normal (Byrne et al., in
press). Assuming that this occurs also among unsuccessful
cultures, then those of unknown outcome will contain a higher
proportion of chromosomally abnormal specimens than successful
cultures.
Another problem arises if the effect of exposure to a suspect
substance is not to increase the proportion of conceptuses with
abnormalities but only to alter the proportion of conceptuses whose
karyotypes can be determined. If, for example, such a detection
effect occurs selectively more often on fetuses of abnormal
karyotype, then there will be a bias towards an apparent mutagenic
effect of the substance.
Table 5. Types of germinal chromosomal abnormalities
-------------------------------------------------------------------
Numerical Aberrations: all presumed mutants
Monosomy (almost entirely X monosomy)
Trisomy 16
21
others
Triploidya
Tetraploidya
Structural aberrationsb
Unbalanced: Robertsonian
deletions and rings
markers and fragments
other
Balanced:
Robertsonian
inversions
reciprocal
-------------------------------------------------------------------
a Most triploidy results from dispermy, and tetraploidy from
mitotic non-disjunction. Thus, they might not be regarded
strictly as germinal cell mutations.
b Both parents must be shown to have normal chromosomes, before a
case can be regarded as new mutant.
For these reasons, inferences about differences between exposed
and control populations (on the basis of comparison of the observed
proportion of the abnormal among those studies) require explicit
assumptions about the conceptuses of unknown karyotype. The
characteristics of this group, for example, the proportion of them
among the total number of conceptuses in the sub-populations, their
fetal pathology, and their maternal age association, should be
investigated closely. If there are striking differences in the
nature of the conceptuses of unknown karyotype between exposed
and unexposed populations, great caution is necessary in drawing
conclusions from comparisons involving conceptuses of known
karyotype.
4.3. Biochemical Approaches to Detecting Gene Mutations in Human
Populations
4.3.1. Biochemical methods for monitoring for gene mutations
Two approaches have been used to study the proteins in
offspring of selected individuals. One is based on the detection
of differences in charge, shape, or size of proteins by one- or
two-dimensional electrophoretic techniques. In the other,
quantitative enzyme activity measurements are used to detect enzyme
variants associated with either the loss of enzyme function or the
absence of the enzyme protein.
4.3.1.1. One-dimensional electrophoresis
The introduction of new electrophoretic technology has
increased the proportion of variants detectable at the protein
level and the number of gene products that can be studied. Using
these techniques, it has been shown that, in contrast to previous
assumptions that only one-third of the amino acid substitutions
could be detected, many neutral change substitutions can also be
detected. This reflects the alterations in higher-order protein
structure associated with these later substitutions, which change
the charge distribution on the molecule. Experimental data suggest
that 80 - 90% of all amino acid substitutions may be detectable
with refined techniques (Johnson, 1976, 1977; Ramshaw et al., 1979;
Fuerst & Ferrell, 1980). The use of narrow-range pH gradients for
isoelectric focusing has also increased the detection and
resolution of the variants at many loci (Altland et al., 1979;
Chramback et al., 1980).
The number of gene products that can be studied by
electrophoretic analysis depends on the availability of techniques
to demonstrate selectively the position of the allele products
following electrophoretic separation. In addition to previously
developed protein specific stains (Harris & Hopkinson, 1976),
immunological techniques (e.g., immunoblotting and immunofixation)
(Chapuis-Cellier et al., 1980; Tsang et al., 1983) have become more
widely used, especially with the recent modifications. One
additional approach has involved several sequential steps of
electrophoresis to separate the protein of interest from the
remaining proteins, so that standard protein-staining techniques
can be used for visualization (Altland & Hackler, 1984).
The results of many studies involving genetic typings of large
numbers of individuals, either for population structure studies
or in the context of the development of mutation monitoring
programmes, have demonstrated the feasibility of sizeable screening
efforts using blood samples as the source of material (Harris et
al., 1974; Neel et al., 1980a,b). The products of at least 40 - 50
loci can be routinely screened by electrophoresis with either
starch and/or polyacrylamide as the support medium. For many
proteins, either support can be used, with polyacrylamide having
the advantage of requiring smaller sample aliquots and potentially
increased resolution. Isoelectric-focusing is feasible for many of
these proteins, especially when the potential for increased
resolution of some proteins is considered. Electrophoretic
techniques have been used in several human mutation screening
programmes (Neel et al., 1980a,b; Altland et al., 1982a,b).
They have also been used for estimating the mutation rate in mice
(Pretsch & Narayanan, 1979; Johnson & Lewis, 1983; Neel, 1983) and
Drosophila (Mukai & Cockerham, 1977; Voelker et al., 1980).
Electrophoretic mobility variants have been identified and the
genetic transmission of the trait to subsequent generations has
been confirmed.
4.3.1.2. Two-dimensional electrophoresis
The high resolution two-dimensional (2-D) electrophoretic
techniques described by O'Farrell (1975) with many subsequent
modifications (Dunn & Burghes, 1983a,b) has been used to separate
the large array of proteins found in cells and tissues. The
positions of the several hundred proteins in many types of samples
can be visualized by protein staining, especially when the highly
sensitive silver stains (Merril et al., 1981; Sammons et al., 1981)
or isotopic labelling of proteins in nucleated cells followed by
autoradiography or fluorography (McConkey et al., 1979; Thomas et
al., 1984) are used.
The ability of the 2-D system to resolve electrophoretic
mobility variants has been shown by Warner et al. (1982). In
other studies, the level of heterozygosity for polypeptides has
been examined in several types of human cells and tissues (McConkey
et al., 1979; Walton et al., 1979; Comings, 1982; Hamaguchi et al.,
1982). More recently, extensive studies have been reported on the
level of genetic variation in plasma proteins and other blood
proteins (Rosenblum et al., 1983, 1984). One advantage of the 2-D
approach over the single-dimension electrophoresis techniques is
the ability to study the gene products of at least several hundred
loci in each sample (individual). The 2-D electrophoresis
technique has been used to estimate the induced mutation rate
in mice (Klose, 1979; Marshall et al., 1984).
4.3.1.3. Enzyme activity
Much of the background to the search for enzyme deficiency
variants is derived from the study of metabolic diseases. The
extensive lists of inborn errors of metabolism indicate not only
the prevalence of genetic events associated with loss of enzyme
function but also the usefulness of quantitative enzyme assays as a
tool for studying this class of genetic events (Beutler, 1979; Kahn
et al., 1979; Miwa, 1979).
The search for mutational events resulting in loss of function,
owing to either loss or nonfunctionality of the gene product, is
complementary to the electrophoretic assays, in that it detects
genetic events, not normally detectable by standard electrophoretic
assays. Current data suggest that inherited rare enzyme deficiency
variants occur more frequently in human populations than rare
electrophoretic variants (Mohrenweiser, 1981; Mohrenweiser & Neel,
1981; Satoh et al., 1983). In addition, mutations resulting in
loss of enzyme function occur at least as frequently as
electrophoretically identifiable mutations in mice and Drosophila,
following exposure to mutagenic agents, either radiation or
chemical (Racine et al., 1980; Charles & Pretsch, 1981; Johnson &
Lewis, 1981).
4.3.1.4. Other biochemical approaches
Alterations in base sequence can be analysed using the
restriction enzyme mapping techniques (Botstein et al., 1980;
Skolnick & Francke, 1982; Southern, 1982). Most of the current
effort, in addition to defining gene structure, has been directed
towards identification of polymorphisms at restriction enzyme sites
and subsequent linkage analysis, but similar techniques could be
employed for screening for mutational events (Beaudet, 1983; Cooper
& Schmidtke, 1984 for recent listings of cloned human DNAs).
Although it is clear that it is now technically possible to
detect base sequence changes, incorporating this approach into
a monitoring protocol must await further developments that will
facilitate the generation of the quantity of data necessary for a
mutation screening programme.
In addition to the sensitivity of the method, another potential
advantage of an approach that studies mutations by examining the
DNA directly, is the ability to obtain very large quantities of
data from each individual. Thus, this method can be employed for
the study of small populations.
4.3.2. Analytical strategy and methodological considerations
Biochemical analyses of the samples collected from the F
population (offspring of exposed individuals) will be carried out
usually in a number of laboratories while population and health
data will be derived from various sources. It is unlikely that all
laboratories will be able to complete the entire battery of assays
or that the techniques will be identical in each laboratory or for
each study, thus each laboratory should receive samples from both
exposed and control groups. The key for the successful completion
of the analytical aspects of any study will be for each laboratory
to establish and maintain high standards of technical competence
and performance. Provision should be made for the exchange of
samples, as this will increase the amount of data obtained from
each individual and also serve to confirm the existence of
interesting observations. The basic technical procedures for
electrophoresis and enzyme activity assays, are available. These
techniques form the basis for establishing a new laboratory effort.
The methods described in each section are current routine
laboratory procedures; thus, the biochemical techniques for
monitoring germinal mutations in a human population are available.
4.3.2.1. One-dimensional electrophoresis
Approximately 40 - 50 blood proteins have been examined for
electrophoretic variation in many laboratories including the
laboratories at the University of Michigan, USA (Neel et al.,
1980a) and in Japan at the Radiation Effects Research Foundation
(RERF) (Neel et al., 1980b). Approximately half of the proteins
studied at the University of Michigan use polyacrylamide as the
support medium while the laboratory at RERF relies more on starch
as the support medium. Many of these proteins are also being
studied by isoelectric focusing techniques as a component of
mutation screening programmes in other laboratories (Altland et
al., 1982a,b).
Techniques are available for the electrophoretic analysis
of approximately 20 proteins, when the sample is obtained from
dried blood (Altland et al., 1979, 1982b; Metropolitan Police
Forensic Science Laboratory, 1980). The general principals of
electrophoretic separation and isozyme (protein) identification
using samples collected as dried stains are as described for other
blood samples.
High-speed one-dimensional electrophoretic screening
procedures have been developed for vertical polyacrylamide gel
electrophoresis, flat bed isoelectric focusing, and the sequential
combinations of these procedures. With these techniques, using
multiple sample handling procedures, 96 samples are analysed
simultaneously (Altland et al., 1982a). This is particularly
useful when large numbers of samples are being analysed.
An electrophoretic mobility variant is identified by an
alteration in the standard profile that is consistent with the
appearance of a new allele product. This decision process must
include previous knowledge of the protein subunit structure,
chromosomal location (e.g., hemizygous males) and factors such as
age, sex, etc., which are important in interpreting the data. The
new protein should have characteristics that indicate that it
differs from the original protein by an amino acid change. It
is important to confirm, using as many additional techniques as
possible, that any new variant is not the result of an artifactual
or secondary alteration in protein structure. Ultimately, such
confirmation would require amino acid or DNA sequencing studies.
This is important because of the low probability of obtaining data
to confirm genetic transmission of a new mutation in human
populations. The proteins that are being studied in the above-
mentioned studies have been selected because of the low frequency
of artifactual findings. But, as with any technique, it is
important for each laboratory to undertake appropriate control
experiments.
4.3.2.2. Two-dimensional electrophoresis
The 2-D electrophoresis techniques, used for human mutation
monitoring studies, have been described by Neel et al. (1983, 1984)
and modifications of the technique of O'Farrell (1975), by Anderson
& Anderson (1977) and Anderson et al. (1980). The blood sample is
fractionated into various components, e.g., plasma, erythrocyte
membranes, haemolysate, platelet, etc., the proteins of which are
then analysed. The proteins in the cell fraction to be studied are
solubilized in 4 - 8 M urea, which dissociates the multimeric
proteins into component subunits. The component polypeptides are
separated on the basis of charge by isoelectric-focusing in the
first dimension. Electrophoresis in the second dimension is in
the presence of sodium dodecylsulfate, so that the proteins are
separated in this dimension on the basis of apparent molecular
size. The sensitive silver-based staining techniques are most
often used for identifying the positions of the separated
polypeptides (Merril et al., 1981; Sammons et al., 1981).
Analysis of the results of 2-D electrophoresis can be scored by
visual inspection (Hanash et al., 1982; Rosenblum et al., 1983,
1984), though recently, significant progress has been achieved
in automating and computerizing the analysis of these gels
(Skolnick, 1982; Skolnick et al., 1982; Skolnick & Neel, in press).
Computerized analysis can increase the amount of data obtained from
each gel (the number of locus tests completed for each sample) and
also reduce the workload at this step.
4.3.2.3. Enzyme activity
The methods for detecting enzyme deficiency variants, defined
as a level of enzyme activity that is less than 65% of the mean
for the population and more than 3 standard deviation units below
the mean, have been described by Fielek & Mohrenweiser (1979),
Mohrenweiser & Fielek (1982), and Mohrenweiser (1983a). Additional
or alternative methods have been described by Bulfield & Moore
(1974), Krietsch et al. (1977), Eber et al. (1979), and Satoh et
al. (1983). The general analytical strategy for analysing large
groups of samples has been outlined by Mohrenweiser (1983b).
Enzymes that are: (a) present with reasonable levels of activity;
(b) primarily the gene product of a single locus; (c) not
influenced by environmental factors (e.g., nutritional status);
(d) easy to assay; and (e) exhibit little total variation among
individuals, are good candidates for inclusion in this approach
(Mohrenweiser, 1982; 1983a,b). The gene products of at least
12 - 14 loci can be routinely monitored for the presence of null
variants in erythrocytes.
Each of these biochemical approaches either has been, or is
being, used to obtain data for the estimation of radiation-induced
mutation rates in human populations. Detailed techniques are
available for each component in this section. The most significant
problem is obtaining the commitments (funding, study and control
populations, etc.) necessary to initiate a major mutagenic
study. Obtaining agreements on general approaches, necessary
standardization of technical aspects, and scoring of significant
events is a lesser problem.
4.3.2.4. Sample acquisition and storage
There are two general strategies for sample acquisition. The
first involves obtaining a large volume (up to 20 ml, although 5 ml
is an adequate sample) of whole blood. All members of a nuclear
family are sampled to the maximum extent possible, so that the
material for family studies to determine heritability of the
characteristic is routinely available. Experience has shown that
when the gene products of 40 - 50 loci are studied, family studies
are necessary in at least 10% of the families, even when only a
single child per family is studied. With the number of loci
studied with the 2-D electrophoresis technique, obviously the
percentage of family studies increases to include almost every
family. In either case, when the number of locus tests per sample
or family unit increases to this level, the work involved in
recontacting families for additional samples becomes a considerable
task.
The blood sample is fractionated into at least plasma, buffy
coat, and erythrocyte fractions. Each part is subdivided into a
fraction for routine analysis as well as a fraction for long-term
storage and potential additional analysis. An aliquot of red
cells is stored in glycerol-sorbitol solution and retained in
liquid nitrogen for blood group analysis in the event of a
potential mutation being observed. The "white cell" component
could be fractionated into several components, e.g., platelets,
polymorphonuclear leukocytes, etc., which could be used as samples
for 2-D electrophoretic studies. Alternatively, either all or
fractions of the lymphocyte samples could be retained as a source
of DNA for future restriction enzyme mapping studies. Another
alternative to the above protocol would be to store the white cell
fraction under such conditions that intact, viable cells could be
recovered and, with appropriate cell culturing techniques, the
number of cells expanded for use in future studies. In certain
situations, where a specific population is of special interest and
the potential for follow-up studies is limited, it may be advisable
to establish and store transformed lymphoblast cell lines in order
to maintain the genetic information contained in this population
for future studies. The second acquisition strategy involves
obtaining a smaller volume of sample, often in the form of a dried
blood stain, which is usually obtained in conjunction with other
studies, usually with new-born metabolic screening programmes.
Procedures have been established for screening for gene mutations
at 20 loci using a blood sample obtained on a Guthrie Test card.
With these techniques, 300 samples or 6000 locus tests per day can
be completed (Altland et al., 1982a,b; Vogel & Altland, 1982).
As the new-born metabolic screening programme in many countries
encompasses the total new-born population, this strategy appears
useful for establishing a surveillance progamme for determining the
human germinal mutation rate. The number of locus tests from each
individual is somewhat less than with the larger samples, thus the
frequency of family studies is less, though obviously as the number
of tests per sample increases, it may be important, whenever
possible, to obtain concurrent parental samples.
It has been suggested that children with congenital birth
defects could be of special interest for monitoring studies because
of a high gene mutation rate in such children (Dubinin & Altukhov,
1979). However, study has not been able to confirm a similar
increased or high mutation rate among a group of children with
apparently similar congenital birth defects (Neel & Mohrenweiser,
1984). The usefulness of the approach of targeting specific F
individuals within a population for detailed study, rather than
all F individuals, needs further study.
4.3.3. Data management
Neel et al. (1979) have developed a computer-based data
management system for handling the types and volume of data
generated in a population monitoring programme. This includes
a sample identification system with a letter designation for the
geographical site of sample acquisition, a number code for family
identification, and a single digit indicating family position.
This identification, which is the first entry into a computerized
data management system, is used for identification of all data
generated, thus it is used for linkage to the electrophoretic
and enzyme activity files, which are additional components of
the computer-based data management system. It is linked to family
identification (e.g., name) only when follow-up is necessary, in
order to maintain confidentiality of records. A similar data
management system is currently in operation at RERF. It is
important that the data-management system should be complete
and compatible with all aspects of the study (section 2).
4.3.4. Considerations in screening for germinal mutations
4.3.4.1. Sample size
The logistic problems associated with a germinal mutation
screening programme are significant because of the scale of the
effort. The magnitude of the effort reflects both the rarity of
mutational events (background mutation rates of 105 - 106) and
the need for statistical methods to ascertain generally small
(10 - 100% increase) differences between background and induced
mutation rates. The sample sizes necessary for any of these
approaches to monitoring depend on the baseline rate of the effect
under consideration and the magnitude of the increase in mutation
rates that can be left undetected.
Vogel (1970), Neel (1980), and Vogel & Altland (1982) have
calculated the sample sizes necessary for a research design
involving the contrasting of 2 samples. The samples can be either
collected consecutively, as when studying the possibility of a
changing mutation rate in a defined population, or simultaneously,
when contrasting a control group of children with a group at high
risk of mutation because of the occupational or other exposure
of their parents. The minimum sizes of the 2 samples are
determined by the magnitude of the errors, type I (alpha) and type
II (phi) that are considered acceptable. The minimum increase in
mutation rate that is to be detected (or the maximum to be deemed
insignificant) also influences the sample size necessary to detect
an increased mutation rate. Most geneticists would agree that
any monitoring programme should detect an increase of 50% above
background. Using type I and type II error limits of 0.05 and 0.20
and a mutation rate of 1 x 10-5, it would require some 7.5 million
observations (Neel, 1980). There is considerable disagreement on
how much effort should be devoted to detecting smaller increases in
mutation rate. Given the exponential nature of the relationship,
detecting an increase of 20% involves 2 samples 5 times as large
as those required to detect a 50% increase. It is obvious that,
although the set of assumptions and limits may change, generating
a data base necessary for estimating changes in human mutation
rates (or finding that at some limit of power, the rate has not
changed) requires many observations and considerable analytical
effort.
With the biochemical approach, in which many proteins are
examined in any given child, and with 2 loci at risk of mutation
for each polypeptide included in the study, the number of children
will be only a small fraction of the number of determinations.
For example, in a pilot study (Neel et al., 1980a), 50 proteins
were examined for mutations influencing electrophoretic behaviour.
Thus, the demonstration of a difference of 50% between the 2 groups
could be accomplished with 2 samples of approximately 90 000
children (it is assumed that each protein of a child is an
independent test of mutation and that each child constitutes 2
tests of each protein, i.e., both parents exposed). With the
two-dimensional gel electrophoresis method, which could permit
simultaneous monitoring of 500 (or more) different proteins of
the blood serum, erythrocytes, and/or leukocytes, with the same
assumptions as above, the size of the 2 samples is reduced to
approximately 7500 individuals.
4.3.4.2. Distinction between "true" and "apparent" mutations
Whenever a genetic trait is encountered in a child, neither
of whose parents is affected, the possibility that it is due to a
discrepancy between legal and biological parentage rather than the
result of mutation must be considered. This is less likely when
the mutation involves a major chromosomal abnormality rather than
a biochemical marker, because individuals carrying the former are
often sterile or have a severly reduced life expectancy. With the
sentinel phenotypes or biochemical variants, when an apparent
mutant is encountered it is always necessary to carry out extensive
genetic typings on both parents, and the child. At present, with
the battery of test traits usually available, including enzymes,
blood groups, and HLA, a child whose legal father is not the
biological parent can be detected with an assurance approaching
98%. Thus, in a situation characteristic of many countries, such
as the USA, where 2 - 3% of the children born in wedlock have
fathers other than the legal one and where variants of the type
under consideration have a frequency of 2 - 4/1000 examinations,
"false" mutations (i.e., undetected parentage exclusions) will have
a frequency not very different from the present expectation for
mutation.
It should be pointed out that recent developments, which could
be used in a study such as this mutation protocol, are also useful
for paternity studies. Many gene products studied using two-
dimensional electrophoretic techniques are characterized by
genetic polymorphisms useful in parentage studies, resulting in
an improvement in the ability to detect discrepancies between
legal and biological parentage. The study of DNA restriction
site polymorphisms will also decrease the frequency of undetected
non-parentage, thus the probability of an apparent mutation being
due to non-parentage should become very small with the increased
number of loci that can now be studied.
In the final analysis, there may always be some uncertainty as
to whether a particular apparent mutant is due to an undetected
discrepancy between legal and biological parentage. If the
control population has been properly chosen, however, the amount
of non-parentage could be demonstrated to be the same in the two
populations, so that this factor would be a constant diluent in the
2 samples. Furthermore, with a statistical approach, recently
developed by Rothman et al. (1981), under suitable conditions,
probabilities can be developed for parentage, and apparent
mutations ranked according to these probabilities.
4.3.4.3. Implementation of gene mutation screening programmes
Final decisions regarding analytical strategy and technical
approaches, beyond the points described above, must await further
information regarding the population of interest. The magnitude
of the study helps determine the number of laboratory centres
necessary to complete the study within a reasonable time.
Similarly, the size of the study population (and estimated mutation
rate) will dictate the amount of data that must be obtained from
each individual studied for the generation of a statistically
significant data base. The size of the study population (as well
as the age of the individuals from whom blood is obtained) will
also need to be considered when blood fractionation and allocation
procedures are finalized. Furthermore, techniques are continually
being refined and new approaches being developed, and it is unwise
to dictate the specific technical details until such time as the
commitment to proceed with a study has been made. At this point,
laboratories should be able, with relative ease, to define the
technical details for the laboratory and analytical protocols.
4.3.5. Summary
In many respects, with the limitations of the current state of
knowledge regarding the detection and subsequent consequences of
gene mutations, all practical approaches to estimating the germinal
mutation rate in human populations, following exposure to a
putative mutagen, should be considered to be complementary.
This is important as each technique differs in sensitivity, detects
different types of genetic damage or end-points, and has different
degrees of relevance for estimating potential health effects.
Therefore, it is logical in a collaborative monitoring exercise to
use all of the approaches in order to obtain the maximum possible
amount of data.
The technical aspects of a collaborative effort to use
biochemical methods to monitor selected human populations for
germinal mutation frequencies are available. Such programmes have
been conducted in the Federal Republic of Germany (Altland et al.,
1982b), Japan (Neel et al., 1980b; Satoh et al., 1983), and the USA
(Neel et al., 1980a; Mohrenweiser, 1981). Thus, feasibility has
been tested at a significant level of effort. There is no
technical reason for not expanding such an effort to other
populations of interest.
4.4. Sentinel Phenotypes
4.4.1. Introduction
A sentinel phenotype is a clinical disorder that occurs
sporadically as a consequence of a single, highly penetrant
mutant gene, which is a dominant or X-linked trait of considerable
frequency and low fitness, and is uniformly expressed and
accurately diagnosable with a minimal clinical effort, but a
relatively high probability of ascertainment (Mulvihill & Czeizel,
1983).
A new dominant mutation can manifest itself at any time in the
human life-span. In practice, it is worth distinguishing three
separate groups of sentinel phenotypes: sentinel anomalies,
easily observed at birth and generally recorded in birth defects
registries (Table 6); sentinel childhood tumours, which occur in a
well defined age-group and are recorded in childhood tumour
registries; and genetic disorders with delayed onset, which are
readily diagnosed and recorded in various types of genetic disease
registries (Table 7). For practical reasons, sentinel anomalies
are the easiest to handle.
4.4.2. Basis of method
Certain essential requirements for the surveillance or
monitoring of sentinel anomalies include:
(a) Sensitivity and specificity
Theoretically, all dominant phenotypes might be suitable for
these purposes. McKusick (1983) catalogues 1828 autosomal dominant
traits in man. These probably occur at a total birth prevalence
of 1% (Matsunaga, 1982). Nearly half of autosomal dominant
disorders may be caused by new mutations (Holmes et al., 1981).
Nevertheless, at present, no single dominant phenotype suits all,
or nearly all, the criteria of the above definition. However, the
22 anomalies listed in Table 6 comply satisfactorily. The main
problems are the validity of a diagnosis and the relatively
uncommon occurrence of all these candidate anomalies. If the
baseline prevalences are too low, both the specificity and
sensitivity of population screening are decreased (Hook, 1981b).
(b) Reliable and accurate diagnosis
A sentinel phenotype must be clearly distinguishable from
related disorders. However, each of the anomalies in Table 6 has
features that overlap with so many other anomalies that highly-
trained clinical experts must be involved in the diagnosis. In
general, these well-known genetic anomalies have a considerable
genetic heterogeneity. In addition, genocopies and sometimes
phenocopies exist for all sentinel anomaly candidates. The task
is to make a precise nosological diagnosis, i.e., to identify the
effect of the specific locus or sometimes of the given allele of
the specific locus.
Table 6. Candidate sentinel anomalies and the figures of the
Hungarian Programme per 10 000 births
-------------------------------------------------------------------
Congenital anomaly McKusick Expected
(s = syndrome) number rate
(1983)
-------------------------------------------------------------------
Achondroplasia 10 080 1.08
Acrocephalosyndactyly type I (Apert s.) 10 120 0.07
Acrocephalosyndactyly type V (Pfeiffer s.) 10 160 0.02
Aniridia, isolated 10 620 0.11
Van der Woude s. 11 930 0.02
Cleidocranial dysplasia 11 960 0.04
Contractural arachnodactyly 12 105 0.04
Crouzon s. 12 350 0.11
EEC s. 12 990 0.04
Holt-Oram s. 14 290 0.07
Treacher-Collins s. 15 450 0.04
Moebius s. 15 790 0.04
Nail-patella s. 16 120 0.04
Oculo-dento-digital (ODD s.) 16 420 0.02
Osteogenesis imperfecta, type I 16 620 0.22
Polysyndactyly preaxial IV. 17 420 0.22
Split hand and/or foot, typical 18 360 0.15
Spondyloepiphyseal dysplasia cong. 18 390 0.07
Thanatophoric dwarfism 18 760 0.04
Whistling face (Freeman-Sheldon s.) 19 370 0.02
Incontinentia pigmenti (Bloch-Sulzberger s.) 30 830 0.11
Oral-facial-digital s. (Gorlin-Psaume s. or 31 120 0.02
OFD I.)
-------------------------------------------------------------------
Total 2.59
-------------------------------------------------------------------
Cataracta 11 620 0.71
Ptosisa 17 830 0.66
-------------------------------------------------------------------
Grand total 3.96
-------------------------------------------------------------------
a Excluded from the final list because ptosis was rarely
ascertained and the nosological diagnosis of cataract was
impossible.
Table 7. Candidate sentinel phenotypes without reliable nenonatal
manifestations
--------------------------------------------------------------------
Disorder McKuskick Inheritance
number
(1983)
--------------------------------------------------------------------
Amelogenesis 10 450 ADa
Exostoses, multiple 13 370 AD
Fibrodysplasia ossificans progressiva 13 510 AD
Marfan's syndrome 15 470 AD
Myotonic dystrophy (Steinert disease) 16 090 AD
Neurofibromatosis (Recklinghausen disease) 16 220 AD
Polycystic kidneys 17 390 AD
Polyposis coli, I (familial) 17 510 AD
Polyposis intestinal, II (Peutz-Jeghers 17 520 AD
syndrome)
Polyposis intestinal, III (Gardner syndrome) 17 530 AD
Retinoblastoma (hereditary) 18 020 AD
Tuberous sclerosis 19 110 AD
von Hippel-Lindau syndrome 19 330 AD
Waardenburg syndrome 19 350 AD
Wilms' tumour (hereditary) 19 407 AD
Haemophilia A (Classical) 30 670 XRb
Haemophilia B (Christmas disease) 30 690 XR
Muscular dystrophy 331 020 XR
(Duchenne type)
Martin-Bell 30 955 XR
--------------------------------------------------------------------
a AD = autosomal dominant.
b XR = X-linked.
(c) Complete ascertainment
In countries where most births occur in hospitals and there are
neonatal check-ups, the easily-recognized congenital anomalies of
autosomal dominant origin can be used as sentinel anomalies,
particularly if a population-based registry or surveillance of
congenital anomalies is functioning.
(d) The knowledge of pedigree data
Since only a new occurrence of sentinel phenotype in a family
is a mutation, obtaining data from and/or clinical examination
of both parents is necessary to verify that neither is affected.
In practice, it is necessary to obtain clinical data on the
grandparents in order to exclude the possibility of non-penetrance
in the parents.
(e) Possibility of further action
After a significant increase in the rate of occurrence of one
or more mutations, it is essential to initiate an analytical
epidemiological study to identify the responsible mutagens and to
achieve the ultimate goal, i.e., prevention of further genetic
damage.
4.4.3. Relevance and limitations
The theoretical advantage of using sentinel phenotypes is
considerable. When a sentinel anomaly or sentinel childhood tumour
is recognized in an infant or child, in the absence of an affected
parent, there is little question that a germinal mutation has
occurred.
From the practical point of view, there are three advantages:
(a) Since sentinel phenotypes are diseases rather than
innocuous physical or biochemical traits or variants, the
affected person in a developed country will enter the
health-care system. Thus, affected persons will almost
certainly be registered in a recording system for reasons
other than their potential use in surveillance. Thus, the
so-called opportunistic approach is feasible (this term is
used to describe studies that are built on data, already
collected for other reasons).
(b) In the case of sentinel anomalies and sentinel childhood
tumours, parents with an affected infant or young child
are likely to be willing to cooperate in additional
investigations, with regard, for example, to family or
environmental histories.
(c) In sentinel phenotypes in which there is a low fitness,
there is a low probability of false paternity.
The main theoretical disadvantage is that the phenotype is not
an immediate gene product, but a distant manifestation of altered
DNA. Hence, intervening developmental events could obscure the
relationship between a mutagenic event and its expression as a
sentinel phenotype. In addition, dominant traits tend to have
greater variation in clinical manifestations than most recessive
and chromosomal syndromes. The main practical difficulty is that
most clinicians lack experience in the accurate diagnosis of these
rare disorders.
Since the expected incidence of mutant candidate sentinel
anomalies is 2 - 3 per 10 000 live births, a very large study
population is required in order to be able to detect any effects
in exposed groups of individuals.
4.4.4. Procedures
4.4.4.1. Surveillance of sentinel anomalies
A programme may be based on existing congenital anomaly
registries and surveillance systems. The target population
comprises all the new-born infants and, when possible, those up
to the age of one year. Several sources of ascertainment are
desirable. Three criteria of congenital anomaly surveillance
should be continuously checked:
(a) Completeness of ascertainment
In Hungary, the total birth prevalence of congenital
anomalies included in Chapter XIV "Congenital Anomaly" of the
International Classification of Diseases was estimated to be about
60 out of every 1000 new-born infants at, or after, birth (Czeizel
& Sankaranarayanan, 1984). Individual types of congenital
anomalies show a wide range of completeness of ascertainment.
(b) Validity of diagnosis
The validity of diagnosis may be known as a result of ad hoc
epidemiological studies. However, there may be a wide range of
misdiagnosis for different types of congenital anomalies.
(c) Base-line frequency
The baselines of all pregnancy outcomes (e.g., spontaneous and
induced abortions, still births, low-birthweight infants, etc.) and
other potentially confounding demographic variables (e.g., paternal
and maternal age) are also taken into consideration in evaluating
sentinel anomalies.
The surveillance of sentinel anomalies should consist of three
steps:
(1) Indexed patients with sentinel anomalies, notified to the
congenital anomalies registries and surveillance systems,
depending on their type of sentinel anomalies, should be
referred, with the help of their parents, to selected
participating paediatric, orthopaedic, or ophthalmological
institutions.
(2) Specialists in these institutions should examine these
indexed patients to confirm (or exclude) the nosological
diagnosis, to clarify family history (whether a sporadic
or familial case), to obtain data on environmental history
(with the help of a specifically-designed questionnaire),
and to give genetic counselling (if parents plan to have
babies in the future).
(3) The data obtained by experts at medical institutions
should be sent back to the programme director to be
evaluated.
4.4.5. Data interpretation
4.4.5.1. Surveillance of sentinel anomalies
Preliminary Hungarian experiences, based on data obtained
during 1980 - 82, are inconclusive. The registered prevalence at
birth of 24 sentinel anomalies was 2.6/10 000 births. The rate of
participation was only 41% (the main causes of case loss were the
death or severe condition of certain indexed patients) and the
diagnosis was confirmed in only 60% of the cases examined.
Nevertheless, as an average of 7 possible new mutations per year
was found over the 3-year period, during which nearly 430 000
births occurred, it is hoped that improving the completeness of
notification, participation rate, and diagnostic skills will
increase the number of new mutations identified in the future.
The possible causes of new mutations can be studied by
obtaining the environmental histories of the parents. The
environmental histories of the parents of familial cases can
provide adequate control data.
The main problem is that the basic data concerning even common
candidate sentinel anomalies are not known completely. Knowledge
of their frequency in different populations, the rate of prenatal
loss, definite figures on their penetrance,and a timetable of the
expression of their manifestations would improve the efficient use
of these sentinel anomalies.
The total prevalence at birth of the 24 sentinel anomalies
studied is not low. Assuming a Poisson distribution of events,
a background rate of 4.0 sentinel anomaly cases per 10 000 births,
and a 50% proportion of sporadicity, a sample size of 38 000 would
be needed to detect a doubling of mutation rate with probabilities
of type I and II errors at the 0.05 level. The null hypothesis
would be rejected, if 22 or more new mutations were seen.
The surveillance of sentinel anomalies has some other practical
benefits. Experts at referral centres have the opportunity to
improve their diagnostic skills, and to suggest prognosis and
treatment. From this aspect, verifying a genocopy or phenocopy is
important. Furthermore, surveillance may promote the detection of
teratogens. One of the main practical advantages of surveillance
is economy. In the Hungarian programme, an opportunistic and cost-
effective approach is used (Mulvihill & Czeizel 1983).
4.4.6. Conclusions
At present, most sentinel anomalies are not suitable for
surveillance, because of the inability to validate the diagnosis
and the incomplete ascertainment of some types. However, the
reliability of the diagnosis of sentinel anomalies will improve as
the number and skill of clinical geneticists increases. Thus, the
material in national or regional surveillance systems with a large
and unselected data base will become usable for studying sentinel
anomalies. The main argument for surveillance is that data are
already available in the registries and surveillance systems used
for other important medical purposes. In addition, the existence
of such programmes will improve existing medical care.
The monitoring of sentinel phenotypes has a specific purpose.
Defined populations, such as the offspring of self-poisoned
persons, epileptics, cancer patients (e.g., patients with acute
lymphoid leukaemia), or workers exposed to chemical compounds,
would be worth studying. However, the sentinel phenotype approach
is not useful for the monitoring of a small circumscribed new-born
population (< 50 000), exposed to known or suspected mutagens,
because such a small sample cannot yield statistically significant
results. Nevertheless, the importance of a opportunistic approach
should be stressed, because some types of industrial and disease
registries may provide data for this purpose.
There are a number of possibilities for improving the
feasibility of the surveillance and monitoring of sentinel
phenotypes. Because the birth prevalence of individual sentinel
anomalies is relatively low, international collaboration should be
encouraged. For example, the International Clearinghouse for Birth
Defects Monitoring Systems (Flynt & Hay, 1979) represents a huge
surveillance programme involving more than 24 countries and the
records of more than 2 million births per year. The majority of
the national and regional systems are able to evaluate the
occurrence of sentinel anomalies. However, the fact that most
sentinel anomalies do not have separate codes in the International
Classification of Disease is a serious technical limitation.
Unique codes need to be given to them so that international
comparisons of rates of sentinel anomalies can be made.
An important approach involves the use of sentinel childhood
tumours, mainly hereditary retinoblastomas and Wilms' tumours.
Patients with these disorders are diagnosed and, in general,
hospitalized in central institutions; thus, their records can
also be used for monitoring purposes. Although retinoblastoma
and Wilms' tumour are not usually detectable at birth, they have a
specific feature beyond those mentioned above. The vast majority
of cases of either type occur sporadically, and all sporadic
bilateral cases have an autosomal dominant origin, while most
sporadic unilateral cases are non-hereditary, persumably arising
from somatic mutation. Hence, these two types of tumour can be
used for surveillance of both germinal and somatic mutations.
Registries of individual genetic disorders (e.g., Huntington,
neurofibromatosis, polyposis coli) (Table 7), in several countries,
also offer a possibility for the surveillance of sentinel
phenotypes.
Improvement in diagnostic laboratory procedures has resulted
in progress in the surveillance of sentinel phenotypes (Vogel &
Altland, 1982). Recently, screening for the lipoproteinaemias has
become widespread and affords an opportunity for detecting common
familial hypercholesterolaemias of autosomal dominant origin.
Finally, some X-linked recessive traits and disorders, such as
Duchenne muscular dystrophy, Martin-Bell syndrome, or haemophilia
A, would be useful sentinel phenotypes, particularly if: (a) a
considerable fraction of cases were new mutations, and (b)
heterozygosity in the mothers could be detected reliably and
inexpensively by laboratory techniques.
Although, the monitoring of sentinel phenotypes cannot compete
with the efficacy of studying protein variants and the methods of
the new molecular genetics, simultaneous approaches can provide
possibilities for comparison and, in addition, the sentinel
phenotype approach may be more feasible in certain countries.
4.5. Fetal Death
4.5.1. Introduction
The proportion of human fetal deaths normally attributable to
germinal genic mutations is not known. If rare recessive lethals
are a common cause, studies on inbreeding have failed to show the
increase in fetal deaths expected (MacCluer, 1980). Dominant
lethal mutations acting in fetal life can occur, but their
contribution to fetal death is unknown and difficult to study with
present techniques. If this contribution were large, an increase
in fetal death with increasing paternal age might be expected.
Such an effect was seen in one study (Selvin & Garfinkel, 1976),
but was not seen in another study of late fetal deaths (Hatch,
1984).
The occurrence of a fetal death, particularly an early fetal
death, without any investigation of the products of conception, can
be considered as an indicator of germinal genic mutation. However,
any increase in FD could be due also to exposure to a teratogen
during pregnancy, or to interference with normal maternal hormonal
or immunological functions during pregnancy.
Teratogenic effects could be separated from genetic effects, if
the exposure could be allocated according to the sex of the exposed
parent, and whether it was pre-conceptional or post-conceptional.
Although the possibility exists that maternal exposure can occur
indirectly through paternal contact, most paternal exposures are
likely to affect fetal health via genetic changes transmitted in
the sperm. Pre-conceptional exposures only, in females, are also
most likely to induce genetic damage. However, it is not possible
to distinguish between germinal chromosome aberrations and genic
mutation using exposure data.
If morphological examination of the products of conception can
be carried out (section 4.2), further information can be gained.
Genetic effects, either chromosomal or genic mutations, are likely
to result in developmentally abnormal specimens, whereas factors
acting via interference with the maternal physiological response in
pregnancy are likely to result in developmentally normal specimens.
A change in the usual distribution of morphological types among
specimens of FD can be used to document an effect of an exposure:
a doubling of the number of "intact empty sacs" compared with
controls would confirm the validity of an observed rise in overall
rate of FD.
If the products of conception can be examined cytogenetically
(section 4.2), it may be possible to distinguish a class of FD
most probably due to genic mutation. A rise in developmentally
abnormal, but chromosomally normal specimens, after maternal
preconception, or paternal, exposures, might possible be attributed
to an increase in germinal genic mutations, lethal for fetal life.
4.5.2. Procedures
If exposed and unexposed populations are to be compared for
overall frequency of fetal death, the population(s) to be studied
must be clearly defined, and data collected on the total number of
births over 28 weeks of gestation, i.e., late fetal death (still
birth) and the total number of FDs. For purposes of comparison
between an exposed and unexposed population, bias should not be
introduced, if the ascertainment of either of these variables is
not complete, as long as the exposure status does not influence the
ascertainment. However, the higher the observed rate of early and
intermediate fetal death (spontaneous abortion) the greater the
power of the study, and the less likely it is to miss effects
limited to only particular subsets of the total (as is likely for
genetic effects). As most genetic effects will probably be
observed among the early spontaneous abortions, it is particularly
important to ascertain these as completely as possible. This is
not usually possible with most existing vital statistics sources
of fetal deaths. In a retrospective study, it will probably be
necessary to use either medical records or interviews to ascertain
reproductive history.
In a case-control study, efforts would be made to ascertain
spontaneous abortions through a medical treatment centre, and to
compare exposure histories between women who have spontaneous
abortions and women having live births. If the sample were
relatively small, cases and controls would probably have to be
matched for age, in order to control for this source of bias.
Finding controls at a point in gestation that matches the cases of
spontaneous abortion will also help to control for differences in
time of ascertainment, and differences in recall of events
occurring at the beginning of the pregnancy.
Data must be collected as carefully as possible on factors
known to influence the frequency of fetal death (e.g., maternal
age, socio-economic status, previous reproductive history, and
gestational age at abortion). Gestational age is usually estimated
from menstrual history. Even if data on the developmental stage of
the fetus is available, it should not be used to estimate
gestational age, since development often ceases some time before
abortion. Smoking and alcohol consumption histories would also be
useful, since these common exposures are associated with increased
abortion risk and might differ in exposed and unexposed
populations.
4.5.3. Data processing and presentation
Frequency is usually expressed as the number of fetal deaths
divided by the number of births over 28 weeks plus the number of
fetal deaths. Maternal age is the most important variable to be
controlled in any analysis, since it will influence the overall
frequency of abortion, and also the distribution of the kinds of
chromosome anomalies expected. Trisomies in spontaneous abortions
increase with maternal age in the same way as trisomies in live
births. On the other hand, monosomy X shows a decrease in
frequency with maternal age in spontaneous abortions. The risk
of abortion increases with the number of previous abortions in a
woman's history, and this variable should also be examined for
possible confounding effects.
4.5.4. Conclusions
(a) Fetal death can be used as a possible indicator of
germinal genetic mutation. Because of the high background
frequency, and concentration of anomalies, an increase can
be detected in a small sample size, e.g., a sample from
only 200 women will detect a doubling with 95% power.
(b) Without cytogenetic studies, it is not possible to define
the kind of genetic damage that has occurred (i.e.,
chromosomal or genic mutation) or to separate mutagenic
from teratogenic effects. However, defining the exposure
of the parent (pre-conceptional versus conceptional) may
make more inferences possible about the cause of a rise in
the frequency of fetal deaths.
(c) Estimates of the rate of fetal death may be imprecise
because of difficulties in identifying all causes, and in
determining the relevant population denominator of term
births. Data on factors known to influence rates of fetal
death, such as maternal age, socio-economic status, and
previous reproductive history, need to be collected, so
that possible biases can be explored in the analysis.
REFERENCES
ACHESON, E.D., ed. (1968) Record linkage in medicine,
Edinburgh, London, E & S Livingstone, Ltd.
ALBERMAN, E., POLANI, P.E., FRASER ROBERTS, J.A., SPICER,
C.C., ELLIOT, M., & ARMSTRONG, E. (1972) Parental exposure
to X-irradiation and Down's syndrome. Ann. hum. Genet.
(Lond.), 36: 195-208.
ALBERTINI, R.J. (1982) Studies with T-lymphocytes: an
approach to human mutagenicity monitoring. In: Bridges, B.A.,
Butterworth, B.E., & Weinstein, I.B., ed. Indicators of
genotoxic exposure, New York, Cold Spring Harbor Laboratory
Press, pp. 393-412 (Banbury Report No. 13).
ALBERTINI, R.J. (1983) Studies with 6-thioguanine-resistant
lymphocytes. In: DeSerres, F.J. & Sheridan, W., ed.
Utilization of mammalian specific locus studies in hazard
evaluation and estimation of genetic risk, New York, Plenum
Press, Vol. 28, pp. 11-27 (Environmental Science Research).
ALBERTINI, R.J. & SYLWESTER, D.L. (1984) 6-thioguanine
resistant lymphocytes in human blood. In: Kilbey, B.J.,
Legator, M., Nichols, W., & Ramel, C., ed. Handbook of
mutagenicity test procedures, Amsterdam, Elsevier Scientific
Publishing, pp. 357-372.
ALBERTINI, R.J., CASTLE, K.L., & BORCHERDING, W.R. (1982a)
T-cell cloning to detect the mutant 6-thioguanine-resistant
lymphocytes present in human peripheral blood. Proc. Natl
Acad. Sci. USA, 79: 6617-6621.
ALBERTINI, R.J., SYLWESTER, D.L., & ALLEN, E.F. (1982b) The
6-thioguanine resistant peripheral blood lymphocyte assay for
direct mutagenicity testing in man. In: Heddle, J.A., ed.
Mutagencity: new horizons in genetic toxicology, New York,
Academic Press, pp. 305-336.
ALBERTINI, R.J., DANNENBERG, B.D., SYLWESTER, D.L.,
PETTINGILL, C.A., & KELLEHER, P.C. (1983) Thioguanine
resistent (TGr) lymphocythes in humans: Placental blood
variant frequencies (VFs) are associated with exposure to
potential mutagens. Environ. Mutagen., 5: 401 (abstract).
ALBERTINI, R.J., CHASTENAY, B.J., O'NEILL, J.P., & GREENE,
C.J. (1984) T-cell cloning for human mutagenicity
monitoring. Environ. Mutagen., 6: 479 (abstract).
ALBERTINI, R.J., O'NEILL, H.P., HICKLAS, J.A., HEINTZ, N.H., &
KELLEHER, P.C. (in press) Southern blot analysis of HPRT
mutations occurring in vivo in human peripheral blood
T-lymphocytes. Environ. Mutagen. (abstract).
ALHADEFF, B. & COHEN, M.M. (1976) Frequency and distribution
of sister-chromatid exhanges in human peripheral lymphocytes.
Isr. J. med. Sci., 12: 1440-1447.
ALTLAND, K. & HACKLER, R. (1984) Concepts and applications
of double one-dimensional slab electrophoresis. In: Neuhoff,
V., ed. Electrophoresis 84, Berlin, Verlag Chemie, pp. 367-378.
ALTLAND, K., KAEMPFER, M., & GRANDA, H. (1979) Improved
screening test for abnormal hemoglobins from dried blood
samples. Hum Genet., 53: 97-100.
ALTLAND, K., KAEMPFER, M., & FORSSBOHM, M. (1982a) Mass
screening technique for detecting globin variant from newborn
blood samples. In: Bora, K.C., Douglas, G.R., & Nestmann,
E.R., ed. Chemical mutagenesis, human population monitoring
and genetic risk assessment, Amsterdam, Elsevier Biomedical
Press (Prog. Mutat. Res., Vol. 3, pp. 153-157).
ALTLAND, K., KAEMPFER, M., FORSSBOHM, M., & WERNER, W.
(1982b) Monitoring for changing mutation rates using blood
samples submitted for PKU screening. In: Bonne-Tamir, B.,
Cohen, T., & Goodman, R., ed. Human genetics, Part A. The
unfolding genome, New York, Alan R. Liss Inc., Vol. 103a,
pp. 227-287 (Prog. clin. biol. Res.).
AMES, B.N. (1983) Dietary carcinogens and anticarcinogens.
Oxygen radicals and degenerative diseases. Science, 221:
1256-1266.
ANDERSON, N.L. & ANDERSON, N.G. (1977) High resolution
two-dimensional electrophoresis of human plasma proteins.
Proc. Natl Acad. Sci. USA, 74: 5421-5425.
ANDERSON, N.L., EDWARDS, J.J., & GIOMETTI, C.S. (1980)
High-resolution two-dimensional electrophoretic mapping of
human proteins. In: Radola, B.J., ed. Electrophoresis 79:
advanced methods biochemical and clinical applications,
Berlin, New York, DeGruyter, pp. 313-328.
ANMEUS, H., MATSSON, P., & ZETTERBERG, G. (1982) Human
lymphoctyes resistant to 6-thioguanine: restrictions in the
use of a test for somatic mutations arising in vivo studies by
flow-cytometric enrichment of resistant cell nuclei. Mutat.
Res., 106: 163-178.
ARDITO, G., LAMBERTI, L., ANSALDI, E., & PONZETTO, P. (1980)
Sister-chromatid exchanges in cigarette-smoking human females
and their newborns. Mutat. Res., 78: 209-212.
AWA, A.A., SOFUMI, T., HONDA, T., ITON, M., NERISHI, S., &
OTAKE, M. (1978) Relationship between the radiation dose and
chromosome aberrations in atomic bomb survivors in Hiroshima
and Nagasaki. J. radiat. Res. (Tokyo), 19: 126-140.
BALAKRISHNA MURTHY, P. & PREMA, K. (1979) Sister-chromatid
exchanges in oral contraceptive users. Mutat. Res., 68:
149-152.
BALAKRISHNA MURTHY, P., BHASKARAM, P., & SRIKANTIA, S.G.
(1980) Sister-chromatid exchanges in protein-energy
malnutrition. Hum. Genet., 55: 405-406.
BEAUDET, A.L. (1983) Bibliography of cloned human and other
selected DNAs. Am. J. hum. Genet., 36: 235-249.
BECHER, R., SCHMIDT, C.G., THEISS, G., & HOSSFELD, D.K.
(1979) The rate of sister-chromatid exchange in normal human
bone marrow cells. Hum. Genet., 50: 213-216.
BEEBEE, G. (1981) Record linkage and needed improvements in
existing data sources. In: Petos, R. & Schneiderman, M., ed.
Quantification of occupational cancer, New York, Cold Spring
Harbor Laboratory, pp. 661-673 (Banbury Report No. 9).
BEEK, B. & OBE, G. (1979) Sister-chromatid exhanges in human
leukocyte chromosomes: spontaneous and induced frequencies in
early- and late-proliferating cells in vitro. Hum. Genet., 49:
51-61.
BENDER, M.A. & GOOCH, P.C. (1966) Somatic chromosome
aberrations induced by human whole-body irradiation: the
"Recuplex" criticality accident. Radiat. Res., 29: 568-582.
BENDER, M.A., GRIGGS, H.G., & BEDFORD, J.S. (1974)
Recombinational DNA repair and sister-chromatid exhanges.
Mutat. Res., 24: 117-123.
BEUTLER, E. (1979) Red cell enzyme defects as non-diseases
and diseases. Blood, 54: 1-7.
BIGBEE, W.L., BRANSCOMB, E.W., WEINTRAUB, H.B.,
PAPAYANNOPOULOU, Th., & STAMATOYANNOPOULOS, G. (1981) Cell
sorter immunofluorescene detection of human erythrocytes
labeled in suspension with antibodies specific for hemoglobin
S and C. J. immunol. Methods, 45: 117-127.
BIGBEE, W.L., VANDERLAAN, M., FONG, S.S.N., & JENSEN, R.H.
(1983) Monoclonal antibodies specific for the M- and N- forms
of human glycophorin A. Mol. Immunol., 20: 1353-1362.
BISHOP, J.B. & FEUERS, R.J. (1982) Development of a new
biochemical mutations test in mice based upon measurement of
enzyme activities. II. Test results with ethylmethanesulfonate
(EMS). Mutat. Res., 95: 263-271.
BLOOM, A.D., ed. (1981) Guidelines for studies of human
populations exposed to mutagenic and reproductive hazards,
New York, March of Dimes Birth Defects Foundation.
BOTSTEIN, D., WHITE, R.L., SKOLNICK, M., & DAVIS, R.W.
(1980) Construction of a genetic linkage map in man using
restriction fragment length polymorphism. Am. J. hum. Genet.,
32: 314-331.
BOYUM, A. (1968) Separation of leukocytes from blood and
bone marrow. Scand. J. clin. Lab. Invest., 21(Suppl. 97):
51-76.
BREWEN, J.G. & GENGOZIAN, N. (1971) Radiation-induced human
chromosome aberrations. II. Human in vitro irradiation
compared to in vitro and in vivo irradiation of marmoset
leukocytes. Mutat. Res., 13: 383-391.
BREWEN, J.G., PRESTON, R.J., & LITTLEFIELD, L.G. (1972)
Radiation-induced human chromosome aberration yields following
an accidental whole-body exposure to Co -rays. Radiat. Res.,
49: 647-656.
BREWEN, J.G., PRESTON, R.J., & GENGOZIAN, N. (1975) Analysis
of X-ray induced chromosome translocations in human and
marmoset spermatogonial cells. Nature (Lond.), 253: 468-470.
BUCKTON, K.E., HAMILTON, G.E., PATON, L., & LANGLANDS, A.D.
(1978) Chromosome aberrations in irradiated ankylosing
spondylitis patients. In: Evans, H.J. & Lloyd, D.C., ed.
Mutagen-induced chromosome damage in man, Edinburgh, Edinburgh
University Press, pp. 142-150.
BULFIELD, G. & MOORE, E.A. (1974) Semi-automated assays for
enzymopathies of carbohydrate metabolism in liver and
erythrocytes, using a reaction rate analyser. Clin. chim.
Acta, 53: 265-271.
BUTLER, M.G. (1981) Sister-chromatid exhange in 4 human
races. Mutat. Res., 91: 377-379.
BUTLER, M.G. & SANGER, W.G. (1981) Increased frequency of
sister-chromatid exchanges in alcoholics. Mutat. Res., 85:
71-76.
BYRNE, J. (1983) Fetal pathology/Laboratory manual. Birth
Defects: Original series, 19(2).
BYRNE, J., WARBURTON, D., KLINE, J., BLANC, W., & STEIN, Z.
(in press) Morphology of early fetal death and their
chronological characteristics. Teratology.
CARRANO, A.V. (1982) Sister-chromatid exchange as an
indicator of human exposure. In: Bridges, B.A., Butterworth,
B.E., & Weinstein, I.B., ed. Indicators of genotoxic exposure,
New York, Cold Spring Harbor Laboratory, pp. 307-318 (Banbury
Report No. 13).
CARRANO, A.V. & MOORE II, D.H. (1982) The rationale and
methodology for quantifying sister-chromatid exchange in
humans. In: Heddle, J.A., ed. Mutagenicity: new horizons in
genetic toxicology, New York, Academic Press, pp. 267-304.
CARRANO, A.V. & THOMPSON, L.H. (1981) Sister-chromatid
exchange and single-gene mutation. In: Wolff, S., ed.
Sister-chromatid exchange, New York, John Wiley & Sons, Inc.,
pp. 59-86.
CARRANO, A.V. & THOMPSON, L.H. (1982) Sister-chromatid
exchange and gene mutation. Cytogenet. Cell Genet., 33: 57-61.
CARRANO, A.V., THOMPSON, L.H., LINDL, P.A., & MINKLER, J.L.
(1978) Sister-chromatid exchanges as an indicator of
mutagenesis. Nature (Lond.), 271: 551-553.
CARRANO, A.V., MINKLER, J.L., STETKA, D.G., & MOORE II, D.H.
(1980) Variation in the baseline sister-chromatid exchange
frequency in human lymphocytes. Environ. Mutagen., 2: 325-337.
CASKEY, C.T. & KRUH, G.D. (1979) The HPRT locus. Cell, 16:
1-9.
CHAGANTI, R.S.K., SCHONBERG, S., & GERMAN, J. (1974) A many
fold increase in sister-chromatid exchanges in Blooms syndrome
lymphocytes. Proc. Natl Acad. Sci. USA, 71: 4508-4512.
CHAPUIS-CELLIER, C., FRANCINA, A., & ARNAUD, P. (1980)
Immunofixation after electrofocusing: application to the study
of the microheterogeneity and polymorphism of serum proteins.
In: Radola, B.J., ed. Electrophoresis 79, Berlin, Walter de
Gruyter, pp. 711-725.
CHARLES, D.A. & PRETSCH, W. (1981) A mutation affecting the
lactate dehydrogenase locus LDH-1 in the mouse. I. Genetical
and electrophoretic characterization. Biochem. Genet., 19:
301-307.
CHENG, W., MULVIHILL, J.J., GREENE, M.H., PICKLE, L.W., TSAI,
S., & WHANG-PENG, J. (1979) Sister-chromatid exhanges and
chromosome in chronic myelogenous leukaemia and cancer
families. Int. J. Cancer, 23: 8-13.
CHENG, M., CONNER, M.K., & ALARIE, Y. (1981) Potency of some
carbamates as multiple tissue sister-chromatid exchange
inducers and comparison with known carcinogenic activities.
Cancer Res., 41: 4489-4492.
CHRAMBACK, A., HJELMELAND, L., & NGUYEN, N.Y. (1980) Gel
electrofocusing with increased degrees of freedom. In: Radola,
B.J., ed. Electrophoresis 79, Berlin, Walter de Gruyter,
pp. 3-22.
CLEAVER, J.E. (1981) Correlations between sister-chromatid
exchange frequencies and replicon sizes. A model for the
mechanism of SCE production. Exp. cell Res., 136: 27-30.
CLEMENGER, J.F.P. & SCOTT, D. (1973) A comparison of
chromosome aberration yields in rabbit blood lymphocytes
irradiated in vitro and in vivo. Int. J. Radiat., 24: 487-496.
COHEN, M.M., MARTIN, A.O., OBER, C., & SIMPSON, S.J. (1982)
A family study of spontaneous sister-chromatid exchange
frequency. Am. J. hum. Genet., 34: 294-306.
COLE, R.J., AGHAMOHAMMADI, Z., & HENDERSEN, L. (1982)
Frequencies of genetic lesions in lymphocytes from newborn
humans. Prog. clin. Biol. Res., 109: 375-385.
COMINGS, D.E. (1982) Two-dimensional gel electrophoresis of
human brain proteins. III. Genetic and non-genetic variation
in 145 brains. Clin. Chem., 28: 798-804.
COOPER, D.N. & SCHMIDTKE, J. (1984) DNA restriction fragment
length polymorphisms and heterozygosity in the human genome.
Hum. Genet., 66: 1-16.
CROSSEN, P.E., DRETS, M.E., ARRIGHI, F.E., & JOHNSTON, D.A.
(1977) Analysis of the frequency and distribution of
sister-chromatid exchanges in cultured human lymphocytes.
Hum. Genet., 35: 345-352.
CZEIZEL, A. & SANKARANARAYANAN, K. (1984) The load of
genetic and partially genetic disorders in man. I. Congenital
anomalies: estimates of detriment in terms of years of life
lost and years of impaired life. Mutat. Res., 128(1): 73-103.
CZEIZEL, A., SZENTESI, I., & MOLNAR, G. (1984) Lack of
effect of self-poisoning on subsequent reproductive outcome.
Mutat. Res., 127: 175-182.
DANFORTH, G.H. (1921) The frequency of mutation and the
incidence of hereditary traits in man. Eugenics, genetics, and
the family. In: Scientific Papers of the 2nd International
Congress of Eugenics, New York, Vol. 1, pp. 120-128.
DAS, B.C. & SHARMA, T. (1984) Effects of temperature on the
frequency of sister-chromatid exchanges (SCEs) in peripheral
blood lymphocytes of man and muntjac. Environ. Mutagen., 6:
25-31.
DE ARCE, M.A. (1981) The effect of donor sex and age on the
number of sister-chromatid exchanges in human lymphocytes
growing in vitro. Hum. Genet., 57: 83-85.
DEEPER, J.M., LEONARD, W.J., ROBB, R.J., WALDMANN, T.A., &
GREENE, W.C. (1983) Blockade of the interleukin-2 receptor
by anti-Tac antibody: inhibition of human lymphocyte
activation. J. Immunol., 131: 690-696.
DEKNUDT, G. & KAMRA, O. (1983) Influence of various mitogens
on the yield of sister-chromatid exchanges, induced by
chemicals, in human lymphocytes. Mutat. Res., 111: 161-170.
DEMARS, R. (1971) Genetic studies of HG-PRT deficiency and
the Lech-Nyhan syndrome. Fed. Proc., 30: 944-945.
DE WEERD-KASTELEIN, E.A., KEIJZER, W., RAINALDI, G., &
BOOTSMA, D. (1977) Induction of sister-chromatid exchanges
in exeroderma pigmentosum cells after exposure to ultraviolet
light. Mutat. Res., 45: 253-261.
DUBININ, N.P. & ALTUKHOV, Y.P. (1979) Gene mutations (de
novo) found in electrophoretic studies of blood proteins of
infants with anomalous development. Proc. Natl Acad. Sci. USA,
76: 5226-5229.
DUFRAIN, R. & GARRAND, T. (1981) The influence of
incorporated halogenated analogues of thymidine on the
sister-chromatid exchange frequency in human lymphocytes.
Mutat. Res., 91: 233-238.
DUNN, M.J. & BURGHES, A.H.M. (1983a) High resolution
two-dimensional polyacrylamide gel electrophoresis. II.
Analyses and application. Electrophoresis, 4: 173-189.
DUNN, M.J. & BURGHES, A.H.M. (1983b) High resolution
two-dimensional polyacrylamide gel electrophoresis. I.
Methodological procedures. Electrophoresis, 4: 97-116.
EBER, S.W., DUNNWALD, M., BELSHRADSHY, B.H., BIDDLINGMARER,
F., SCHIEVELBERN, H., WEINMANN, H.M., & KRIETSCH, W.K.G.
(1979) Hereditary deficiency of triosephosphate isomerase in
four unrelated families. Eur. J. clin. Investig., 9: 195-202.
EDMONDS, D.K., LINDSAY, K.S., MILLER, J.F., WILLIAMSON, E., &
WOOD, P.J. (1982) Early embryonic mortality in women.
Fertil. Steril., 38: 447-453.
EVANS, H.J. & O'RIORDAN, M.L. (1975) Human peripheral blood
lymphocytes for the analysis of chromosome aberrations in
mutagen tests. Mutat. Res., 31: 135-148.
EVANS, H.J. & VIJAYALAXMI (1980) Storage enhances chromosome
damage after exposure of human leukocytes to mitomycin C.
Nature (Lond.), 284: 370-372.
EVANS, H.J., BUCKTON, K.E., HAMILTON, G.E., & CAROTHERS, A.
(1979) Radiation induced chromosome aberrations in nuclear
dockyard workers. Nature (Lond.), 277: 531-534.
FIELEK, S. & MOHRENWEISER, H.W. (1979) Erythrocyte enzyme
deficiencies assessed with a miniature centrifugal analyser.
Clin. Chem., 25: 384-388.
FLEISS, J.L. (1981) Statistical methods for rates and
proportions, New York, John Wiley and Sons.
FLYNT, J.W. & HAY, S. (1979) International clearinghouse for
birth defects monitoring systems. Contrib. Epid. Biostatist.,
1: 44.
FOWLE, J.R., III (1984) Workshop report: approaches for
improving mutagenicity risk assessment. Part I. Human
biomonitoring. In: US EPA Conference, 13-15 December, 1982,
Washington DC, US Environmental Protection Agency (Report EPA
600/9-84-016).
FOX, A.J. & GOLDBLATT, P.O. (1982) Longitudinal study of
socio-demographic mortality differentials, London, Her
Majesty's Stationary Office (Series LS No. 1).
FUERST, P.A. & FERRELL, R.E. (1980) The stepwise mutation
model: an experimental evaluation utilizing hemoglobin
variants. Genetics, 94: 185-201.
GALLOWAY, S.M. (1977) Ataxia telangiectasia: the effects of
chemical mutagens and X rays on sister-chromatid exchanges in
blood lymphocytes. Mutat. Res., 45: 343-349.
GALLOWAY, S.M. & EVANS, H.J. (1975) Sister-chromatid
exchanges in human chromosomes from normal individuals and
patients with ataxia telangiectasia. Cytogenet. Cell Genet.,
15: 17-29.
GARCIA, H.D. & COUCH, D.B. (1982) Detection of
phenotypically variant (6-thioguanine-resistant) rat spleen
lymphocytes. In: Bridges, B.A., Butterworth, B.E., &
Weinstein, I.B., ed. Indicators of genotoxic exposure,
New York, Cold Spring Harbor Laboratory Press, pp. 443-451
(Banbury Report No. 13).
GILLIAN CLARE, M., JONES, W.G., & TAYLOR, J.H. (1982)
Sister-chromatid exchanges in human lymphocytes exposed to
single cytotoxic drugs in vivo or in vitro. Eur. J. Cancer
clin. Oncol., 18: 979-989.
GOCKE, E., ECKHARDT, K., KING, M.T., & WILD, D. (1983)
Autoradiographic detection of 6-thioguanine resistant
lymphocytes of mice: a novel system in somatic mutagenesis
testing. Mutat. Res., 113: 455-465.
GOTO, K., MADEA, S., KANO, Y., & SUGEYAMA, T. (1978) Factors
involved in differential Giemsa staining of sister-chromatids.
Chromosoma, 66: 351-359.
HAMAGUCHI, H., YAMADA, M., SHIBASAKI, M., MUKAI, R., YABE, T.,
& KONDO, I. (1982) Genetic analysis of human lymphocyte
proteins by two-dimensional gel electrophoresis. Hum. Genet.,
62: 142-147.
HANASH, S.M., TUBERGEN, D.G., HEYN, R.M., NEEL, J.V., SANDY,
L., STEVENS, G.S., ROSENBLUM, B.B., & KRZESICKI (1982)
Two-dimensional gel electrophoresis of cell proteins in
childhood leukemia, with silver staining: a preliminary
report. Clin. Chem., 28: 1026-1030.
HARLAP, S., SHIONO, P.H., & RAMCHARON, S. (1980) A life
table of spontaneous abortions and the effects of age, parity,
and other variables. In: Porter, I.H. & Hook, E.G., ed. Human
embryonic and fetal death, New York, Academic Press,
pp. 145-158.
HARRIS, H. & HOPKINSON, D.A. (1976) Handbook of enzyme
electrophoresis in human genetics, New York, American Elsevier.
HARRIS, H., HOPKINSON, D.A., & ROBSON, E.B. (1974) The
incidence of rare alleles determining electrophoretic
variants data on 43 enzyme loci in man. Ann. Hum. Genet.
(Lond.), 37: 237-253.
HASSOLD, T. (1982) Mosatic trisomies in human spontaneous
abortions. Hum. Genet., 31: 31-35.
HASSOLD, T., JACOBS, P., KLINE, J., STEIN, Z., & WARBURTON,
D. (1980) Effect of maternal age on autosomal trisomies.
Ann. hum. Genet. (Lond.), 44: 29-36.
HASSOLD, T., WARBURTON, D., KLINE, J., & STEIN, Z. (1984)
The relationship of maternal age and trisomy among trisomic
spontaneous abortions. Am. J. hum. Genet., 36(6): 1349-1356.
HATCH, M. (1984) Male risk factors for spontaneous abortion.
Am. J. Epidem., 120(3): 499-500.
HEDNER, K., HOGSTEDT, B., KOLNIG, A.-M., MARK-VENDEL, E.,
STROMBECK, B., & MITELMAN, F. (1983) Sister-chromatid
exchanges and structural chromosome aberrations in relation to
smoking in 91 individuals. Hereditas, 98: 77-81.
HOLLANDER, D.H., TOCKMAN, M.S., LIANG, Y.W., BORGAONKAR, D.S.,
& FROST, J.K. (1978) Sister-chromatid exchanges in the
peripheral blood of cigarette smokers and in lung cancer
patients and the effect of chemotherapy. Hum. Genet., 44:
165-171.
HOLMES, L.B., VINENT, S.E., COOK, C., & COTE, K.R. (1981)
Surveillance of newborn infants for malformations due to
spontaneous germinal mutations. In: Hook, E.B. & Porter, I.H.,
ed. Population and biological aspects of human mutation,
New York, Academic Press, pp. 351-359.
HOOK, E.B. (1981a) Prevalance of chromosome abnormalities
during human gestation and implications for studies of
environmental mutagens. Lancet, 2: 169-172.
HOOK, E.B. (1981b) Human teratogenic and mutagenic markers
in monitoring aboutpoint sources of pollution. Environ. Res.,
25: 178-203.
HOOK, E.B. (in press) Paternal age and genetic outcomes. In:
Porter, I.H., ed. Proceedings of the 15th Birth Defects
Institute Symposium, Albany, New York, New York, Academic
Press.
HOOK, E.B. & HAMERTON, J.L. (1977) The frequency of
chromosome abnormalities detected in consecutive newborn
studies - Differences between studies - Results by sex and by
severity of phenotypic involvement. In: Hook, E.G. & Porter,
I.H., ed. Population cytogenetics: studies in humans,
New York, Academic Press, pp. 63-79.
HOOK, E.B., SCHREINEMACHERS, D.M., WILLEY, A.M., & CROSS,
P.K. (1984) Inherited structural cytogenetic abnormalities
detected incidentally in fetuses diagnosed prentally:
frequency, parental-age association, sex-ratio trends, and
comparisons with rates of mutants. Am. J. hum. Genet., 36:
422-443.
HOPKIN, J.M. & EVANS, H.J. (1980) Cigarette smoke-induced
DNA damage an lung cancer risks. Nature (Lond.), 283: 388-390.
HOWE, G.R. & LINDSAY, J.P. (1981) A generalized iterative
record linkage system for use in medical follow-up studies.
Comput. biomed. Res., 14: 327-340.
HSIE, A.W., O'NEILL, J.P., & McELHENY, V.K., ed. (1979)
Mammalian cell mutagenesis: the maturation of test systems,
New York, Cold Spring Harbor Laboratory Press (Banbury Report
No. 2).
HUSGAFVEL-PURSIAINEN, K., MAKI-PAAKKANEN, J., NORPPA, H., &
SORSA, M. (1980) Smoking and sister-chromatid exchange.
Hereditas, 92: 247-250.
HUSUM, B., WULF, H.C., & NIEBUHR, E. (1982) Increased
sister-chromatid exchange frequency in lymphocytes in healthy
cigarette smokers. Hereditas, 96: 85-88.
JACKY, P.F., BEEK, B., & SUTHERLAND, G.R. (1983) Fragile
sites in chromosomes: possible model for the study of
spontaneous chromosome breakage. Science, 220: 69-70.
JACOBS, P.A. (1981) Mutation rates for structural chromosome
rearrangements in man. Am. J. hum. Genet., 33: 44-54.
JOHNSON, G.B. (1976) Hidden alleles at the -glycerophosphate
dehydrogenase locus in colias butterflies. Genetics, 83:
149-167.
JOHNSON, G.B. (1977) Evaluation of the stepwise mutation
model of electrophoretic mobility: comparison of the gel
sieving behavior of alleles at the esterase - 5 locus of
Drosophila pseudoobscura. Genetics, 87: 139-157.
JOHNSON, F.M. & LEWIS, S.E. (1981) Electrophoretically
detected germinal mutations induced in the mouse by
ethylnitrosurea. Proc. Natl Acad. Sci. USA, 78: 3138-3141.
JOHNSON, F.M. & LEWIS, S.E. (1983) The detection of
ENU-induced mutants in mice by electrophoresis and the problem
of evaluating the mutation rate increase. In: DeSerres, F.J. &
Sheridan, W., ed. Utilization of mammalian specific locus
studies in hazard evaluation and estimation of genetic tisk,
New York, Plenum Press, pp. 95-108.
JOHNSON, F.M., ROBERTS, G.T., SHARMA, R.K., CHASALOW, F.,
ZWEIDINGER, R., MORGAN, A., HENDREN, R.W., & LEWIS, S.E.
(1981) The detection of mutants in mice by electrophoresis:
results of a model induction experiment with procarbazine.
Genetics, 97: 113-124.
JONES, I.M., BURKHART-SCHULTZ, K., & CARRANO, A.V. (in
press) A method to quantify spontaneous and in vivo induced
thioguanine resistant mouse lymphocytes. Mutat. Res.
JUBERG, R.C. & MOWREY, P.N. (1983) Origin of non-disjunction
in trisomy 21. Am. J. med. Genet., 16: 111-116.
KAHN, A., KAPLAN, J.C., & DREYFUS, J.C. (1979) Advances in
hereditrary red cell anomalies. Hum. Genet., 51: 1-27.
KATO, H. & SANDBERG, A.A. (1977) The efffect of sera on
sister-chromatid exchanges in vitro. Exp. cell Res., 109:
445-448.
KINLEN, L.J. (1980) The death registration system and the
national health service central regiater in Britain: their
value to epidemiological research. In: Cairns, J., Lyon, J.L.,
& Skonnick, M., ed. Cancer incidence in defined populations,
New York, Cold Spring Harbor Laboratories, pp. 437-442
(Banbury Report No. 4).
KLINE, J. & STEIN, Z. (in press) Very early pregnancy. In:
Dixon, R.L., ed. Target organ toxicity, New York, Raven Press .
KLINE, J., STEIN, Z., SUSSER, M., & WARBURTON, D. (1980)
Environmental influences on early reproductive loss in a
current New York City study. In: Porter, I.H. & Hook, E.B.,
ed. Human embryonic and fetal death, New York, Academic Press,
pp. 225-240.
KLINE, J., STEIN, Z., HATCH, M., & STROBINO, B. (1981)
The role of spontaneous abortion studies in environmental
research, Cincinnati, Ohio, Health Effects Research
Laboratory, 45268 (US EPA Cr 8067355010).
KLOSE, J. (1975) Protein mapping by combined isoelectric
focusing and electrophoresis of mouse tissue. A novel approach
to testing for induced point mutations in mammals.
Humangenetik, 26: 231-243.
KLOSE, J. (1979) Isoelectricfocusing and electrophoresis
combined as a method for defining new point mutations in the
mouse. Genetics, 92: 135-245.
KREPINSKI, A.B. & HEDDLE, J.A. (1983) Micronuclei as a rapid
and inexpensive measure of radiation-induced chromosomal
aberrations. In: Ishihora, T. & Sasaki, M.S., ed.
Radiation-induced chromosome damage in man, New York,
Alan R. Liss, Inc., pp. 93-109.
KRIETSCH, W.K.G., KRIETSCH, H., KAISER, W., DUNNWALD, M.,
KUNTZ, G.W.K., DUHM, I., & BUCHER, T. (1977) Hereditary
deficiency of phosphoglycerate kinase: a rare variant in
erythrocytes and leukocytes not associated with haemolytic
anemia. Eur. J. clin. Invest., 7: 427-435.
LAMBERT, B. & LINDBLAD, A. (1980) Sister-chromatid exchange
and chromosome aberrations in lymphocytes of laboratory
personnel. J. Toxicol. environ. Health, 6: 1237-1243.
LAMBERT, B., HANSSON, K., LINDSTEN, J., STEN, M., & WERELIUS,
B. (1976) Bromodeoxyuridine-induced sister-chromatid
exchanges in human lymphocytes. Hereditas, 82: 163-174.
LAMBERT, B., RINGBORG, U., HARPER, E., LINDBLAD, A. (1978)
Sister-chromatid exchanges in lymphocyte cultures of patients
receiving chemotherapy for malignant disorders. Cancer Treat.
Rep., 62: 1413-1419.
LAMBERT, B., LINDBLAD, A., HOLMBERG, L., & FRANCESCONI, D.
(1982) The use of sister-chromatid exchange to monitor human
populations for exposure to toxicologically harmful agents.
In: Wolff, S., ed. Sister-chromatid exchange, New York,
John Wiley & Sons, Inc., pp. 149-182.
LANGE, B.J. & PRANTER, J.E. (1982) The emergence of
6-thioguanine resistant lymphocytes in pediatric cancer
patients. Mutat. Res., 94: 487-499.
LATT, S.A. (1973) Microfluorometric detection of
deoxyribonucleic acid replication in human metaphase
chromosomes. Proc. Natl Acad. Sci. USA, 70: 3395-3399.
LATT, S.A. (1981) Sister-chromatid exchange formation.
Ann. Rev. Genet., 15: 11-55.
LATT, S.A. & JUERGENS, L.A. (1977) Determinants of
sister-chromatid exchange frequencies in human chromosomes.
In: Hook, E.B. & Porter, I.H., ed. Population cytogenetics:
studies in humans, New York, Academic Press, pp. 217-236.
LATT, S.A., ALLEN, J., BLOOM, S.E., CARRANO, A., FALKE, E.,
KRAM, D., SCHNEIDER, E., SCHRECK, R., TICE, R., WHITFIELD, B.,
& WOLFF, S. (1981) Sister-chromatid exchanges: a report of
the Gene-Tox program. Mutat. Res., 87: 17-62.
LINDBLAD, A. & LAMBERT, B. (1981) Relation between
sister-chromatid exchange, cell proliferation, and proportion
of B and T cells in human lymphocyte cultures. Hum. Genet.,
57: 31-34.
LIVINGSTON, G.K., CANNON, L.A., BISHOP, D.T., JOHNSON, P., &
FINEMAN, R.M. (1983) Sister-chromatid exchanges: variation
by age, sex, smoking, and breast cancer status. Cancer Genet.
Cytogenet., 8: 289-299.
LLOYD, D.C., PROSSER, J.S., MOQUET, J.E., & MALOWANY, D.J.
(1983) Doses in radiation accidents investigated by
chromosome aberration analysis. XIII. A review of cases
investigated: 1982, Harwell, Oxfordshire, National
Radiological Protection Board, 14 pp (Report No. NRPB-R148).
MACCLUER, J.W. (1980) Inbreeding and human fetal death. In:
Porter, I.H. & Hook, E.B., ed. Human embryonic and fetal
death, New York, Academic Press, pp. 241-260.
MACGREGOR H.C. & VARLEY, J.M. (1983) Working with animal
chromosomes, New York, John Wiley & Sons.
MCCONKEY, E.H., TAYLOR, B.J., & PHAN, D. (1979) Human
heterozygosity: a new estimate. Proc. Natl Acad. Sci. USA, 76:
6500-6504.
MCFEE, A.F. & SHERRILL, M.N. (1981) Mitotic response and
sister-chromatid exchanges in lymphocytes cultured in sera
from differenct sources. Experientia (Basel), 37: 27-29.
MCKUSICK, V.A. (1983) Mendelian inheritance in man:
catalogue of autosomal dominant, autosomal recessive, and
X-linked phenotypes, 6th ed., Baltimore, Maryland,
Johns Hopkins University Press.
MAGENIS, R.E. & CHAMBERLIN, J. (1981) The parental origin of
non-disjunction. In: De la Cruz, F., ed. Trisomy 21 (Down
Syndrome) research perspectives, Baltimore, Maryland,
University Park Press, pp. 77-94.
MAIZEL, A.L., MEHTA, S.R., HAUFT, S., FRANZINI, D., LACHMAN,
L.B., & FORD, R.J. (1981) Human T-lymphocyte/monocyte
interaction in response to lectin: kinetics of entry into
S-phase. J. Immunol., 127: 1058-1064.
MARSHALL, R.R., RAJ, A.S., GRANT, F.J., & HEDDLE, J.A.
(1984) The use of two-dimensional electrophoresis to detect
mutations induced in mouse spermatogoniaby ethylnitrosourea.
Can. J. Genet. Cytol., 25: 457-466.
MARTIN, R.H., BALKAN, W., BURNS, K., RODEMAKER, A.W., LIN,
C.C., & RUDD, N.L. (1983) The chromosome consitution of 1000
human spermatozoa. Hum. Genet., 63: 305-309.
MATSUNAGA, E. (1982) Perspectives in mutation epidemiology.
I. Incidence and prevalence of genetic disease (excluding
chromosomal aberrations) in human populations. Mutat. Res.,
99: 95-128.
MAZRIMAS, J.A. & STETKA, D.G. (1978) Direct evidence for the
role of incorporated BUdR in the induction of sister-chromatid
exchanges. Exp. cell Res., 117: 23-30.
MERRIL, C.R., GOLDMAN, D., SEDMAN, S.A., & EBERT, M.H.
(1981) Ultrasensitive stain for proteins in polyacrylamide
gels show regional variation in cerebrospinal fluid proteins.
Science, 211: 1437-1438.
METROPOLITAN POLICE FORENSIC SCIENCE LABORATORY (1980)
Biology methods manual, London.
MI, M.P. (1967) Record linkage and other genetic studies in
Hawaii. In: Crow, J.A. & Neel, J.V., ed. Proceedings of the
3rd International Congress on Human Genetics, Baltimore,
Maryland, Johns Hopkins University Press, pp. 489-496.
MILLER, J.F., WILLIAMSON, E., GLUE, J., GORDON, Y.B.,
GRUDZINSKAS, J.C., & SYKES, A. (1980) Fetal loss after
implantation: a prospective study. Lancet, 2: 554-556.
MIWA, S. (1979) Significance of the determination of red
cell enzyme activities. Am. J. Hematol., 6: 163-172.
MOHRENWEISER, H.W. (1981) Frequency of enzyme deficiency
variants in erythrocythes of newborn infants. Proc. Natl Acad.
Sci. USA, 78: 5046-5050.
MOHRENWEISER, H.W. (1982) Frequency of rare enzyme
deficiency variants: search for mutational events with human
health implications. In: Bora, K.C., Douglas, G.R., &
Nestmann, E.R., ed. Chemical mutagenesis, human population
monitoring and genetic risk assessment, Amsterdam, Elsevier
Biomedical Press, Vol. 3, pp. 51-68 (Prog. Mutat. Res.).
MOHRENWEISER, H.W. (1983a) Enzyme deficiency variants:
frequency and potential significance in human populations.
Isozymes: Curr. Topics Biol. Med. Res., 10: 51-68.
MOHRENWEISER, H.W. (1983b) Biochemical approaches to
monitoring human populations for germinal mutation rates. II.
Enzyme deficiency variants as a component of the estimated
genetic risk. In: DeSerres, F.J. & Sheridan, W., ed.
Utilization of mammalian specific locus studies in hazard
evaluation and estimation of genetic risk, New York, Plenum
Publishing Corporation, pp. 55-69.
MOHRENWEISER, H.W. & FIELEK, S. (1982) Elevated frequency of
carriers for triosephosphate isomerase deficiency in newborn
infants. Pediatr. Res., 16: 960-963.
MOHRENWEISER, H.W. & NEEL, J.V. (1981) Frequency of
thermostability variants and estimation of the total "rare"
variant frequency in human populations. Proc. Natl Acad. Sci.
USA, 78: 5729-5733.
MOOREHEAD, P.S., NOWELL, P.C., MELLMAN, W.J., BATTIPS, D.M., &
HUNGERFORD, D.A. (1960) Chromosome preparations of
leukocytes from human peripheral blood. Exp. cell Res., 20:
613-616.
MORGAN, W.F. & CROSSEN, P.E. (1977) The incidence of
sister-chromatid exchanges in cultured human lymphocytes.
Mutat. Res., 42: 305-312.
MORIMOTO, K. & WOLFF, S. (1980) Increase of sister-chromatid
exchanges and perturbations of cell division kinetics in human
lymphocytes by benzene metabolites. Cancer Res., 40: 1189-1193.
MORLEY, A.A., COX, S., & HOLLIDAY, R. (1982) Human
lymphocytes resistant to 6-thioguanine increase with age.
Mech. Aging Dev., 19: 21-26.
MORLEY, A.A., TRAINOR, K.J., & SESHADRI, R.S. (1983a)
Cloning of human lymphocytes using limiting dilution.
Exp. Hematol., 11: 418-424.
MORLEY, A.A., TRAINOR, K.J., SESHADRI, R., & RYALL, R.B.
(1983b) Measurement of in vivo mutations in human
lymphocytes. Nature (Lond.), 302: 155-156.
MUKAI, T. & COCKERHAM, C.C. (1977) Spontaneous mutations
rates at enzyme loci in Drosophila melanogaster. Proc. Natl
Acad. Sci. USA, 74: 2514-2517.
MULVIHILL, I.I. & CZEIZEL, A. (1983) Perspectives in
mutation epidemiology. VI. A 1983 view of sentinel phenotypes.
Mutat. Res., 123: 345-361.
MUSILOVA, J., MICHALOVA, K., & URBAN, J. (1979)
Sister-chromatid exchanges and chromosomal breakage in
patients treated with cytostatics. Mutat. Res., 67: 289-294
NAGAO, M. & SUGIMURA, T. (1981) Mutacarcinogens in
ordinarily cooked foods. In: Schoental, R. & Conners, T.A.,
ed. Dietary influences on cancer: traditional and modern,
Florida, CRC Press, Inc., pp. 201-218.
NAKAGONE, Y., YOKOCHI, T., MATSUBARA, T., & FUKADA, F.
(1982) Preservation of whole blood for chromosome analysis.
Cytogenet. cell Genet., 33: 254-255.
NEEL, J.V. (1980) Some considerations pertinent to
monitoring human populations for changing mutation rates.
Proc. XIV Int. Cong. Genet., 1: 225-238.
NEEL, J.V. (1983) Frequency of spontaneous and induced
"point" mutations in higher eukaryotes. J. Hered., 74: 2-15.
NEEL, J.V. & MOHRENWEISER, H.W. (1984) Failure to
demonstrate mutations affecting protein structure or function
in children with congenital defects or born prematurely.
Proc. Natl Acad. Sci. USA, 81: 5499-5503.
NEEL, J.V., MOHRENWEISER, H.W., SATOH, C., & HAMILTON, H.B.
(1979) A consideration of two biochemical approaches to
monitoring human populations for a change in germ cell
mutation rates. In: Berg, K., ed. Genetic damage in man caused
by environmental agents, New York, Academic Press, pp. 29-47.
NEEL, J.V., MOHRENWEISER, H.W., & MEISLER, M.M. (1980a) Rate
of spontaneous mutation at human loci encoding protein
structure. Proc. Natl Acad. Sci. USA, 77: 6037-6041.
NEEL, J.V., SATOH, C., HAMILTON, H.B., OTAKE, M., GORIKI, K.,
KAGEOKA, T., FUJITA, M., NERIISHI, S., & ASAKAWA, J. (1980b)
Search for mutations affecting protein structure in children
of atomic bomb survivors: preliminary report. Proc. Natl Acad.
Sci. USA, 77: 4221-4225.
NEEL, J.V., MOHRENWEISER, H.W., HANASH, S., ROSENBLUM, B.B.,
STERNBERG, S., WURZINGER, K., ROTHMAN, E., SATOH, C., GORIKI,
K., KRASTEFF, T., LONG, M., SKOLNICK, M., & KRZESICKI, R.
(1983) Biochemical approaches to monitoring human populations
for germinal mutation rates. I. Electrophoresis. In: DeSerres,
F.J. & Sheridan, W., ed. Utilization of mammalian specific
locus studies in hazard evaluation and estimation of genetic
risk, New York, Plenum Press, pp. 71-93.
NEEL, J.V., ROSENBLUM, B.B., SING, C.F., SKOLNICK, M.M.,
HANASH, S.M., & STERNBERG, S. (1984) Adapting
two-dimensional gel electrophoresis to the study of
humangermline mutation rates. In: Celis, J.E. & Bravo, R., ed.
Methods and applications of two-dimensional gel
electrophoresis of proteins, New York, Academic Press,
pp. 259-306.
NEWCOMBE, H.B. (1967) Record linking: the design of
efficient systems for linking records into individual and
family histories. Am. J. hum. Genet., 19: 335-359.
NEWCOMBE, H.B. (1969) The use of medical record linkage for
population studies. Method. Inform. Med., 8: 7-11.
NEWCOMBE, H.B. (1977) Methods of obtaining epidemiological
data for mutation risk estimation. In: Kilbey, B.J., Legator,
M., Nichols, W., & Ramel, C., ed. Handbook of mutagenicity
test procedures, Amsterdam, Elsevier/North Holland,
pp. 461-476.
O'FARRELL, P.H. (1975) High resolution two-dimensional
electrophoresis of proteins. J. Biol. Chem., 250: 4007-4021.
OFFICE OF TECHNOLOGY ASSESSMENT, US CONGRESS (1983) The role
of genetic testing in the prevention of occupational disease,
Washington DC, US Office of Technology Assessment, 243 pp
(GTA-BA-194).
PAINTER, R.B. (1980) A replication model for
sister-chromatid exchange. Mutat. Res., 70: 337-341.
PATTERSON, J.E. (1980) The establishment of a national death
index in the United States. In: Cairns, J., Lyon, J.L., &
Skolnick, M., ed. Cancer incidence in defined populations,
New York, Cold Spring Harbor Laboratories, pp. 443-451 (Banbury
Report No. 4).
PAUL, W.E., SREDNI, B., & SCHWARTZ, R.H. (1981) Long-term
growth and cloning of non-transformed lymphocytes. Nature
(Lond.), 294: 679-699.
PEDERSEN, C., OLAH, E., & MERRILD, U. (1979)
Sister-chromatid exchanges in cultured peripheral lymphocytes
from twins. Hum. Genet., 52: 281-294.
PERRY, P.E. (1980) Chemical mutagens and sister-chromatid
exchange. In: DeSerres F.J. & Hollaender, A., ed. Chemical
mutagens: principles and methods for their detection,
New York, Plenum Press, Vol. 6, pp. 1-39.
PERRY, P. & EVANS, H.J. (1975) Cytological detection of
mutagen-carcinogen exposure by sister-chromatid exchange.
Nature (Lond.), 258: 121-125.
PERRY, P. & WOLFF, S. (1974) New Giemsa method for the
differential staining of sister-chromatids. Nature (Lond.),
251: 156-158.
POPESCU, N.C., AMSBAUGH, S.C., & DIPAOLO, J.A. (1981)
Relationship of carcinogen-induced sister-chromatid exchange
and neoplastic cell transformation. Int. J. Cancer, 28: 71-77.
PRESTON, R.J. & GOOCH, P.C. (1981) The induction of
chromosome-type aberrations in G by methyl methanesulfonate
and 4-nitroquinoline-N-oxide and the non-requirement of an
S-phase for their production. Mutat. Res., 83: 395-402.
PRESTON, R.J., BREWEN, J.G., & JONES, K.P. (1972)
Radiation-induced chromosome aberrations in Chinese hamster
leukocytes. A comparison of in vivo and in vitro exposures.
Int. J. Radiat. Biol., 21: 397-450.
PRESTON, R.J., AU, W., BENDER, M.A., BREWEN, J.G., CARRANNO,
A.V., HEDDLE, J.A., MCFEE, A.F., WOLFF, S., & WASSOM, J.S.
(1981) Mammalian in vivo and in vitro cytogenetic assays: a
report of the US EPA's Gene-tox Program. Mutat. Res., 87:
143-188.
PRETSCH, W. & NARAYANAN, K.R. (1979) [Determination of
gene-mutations in mice by isoelectric focusing.] A. Physiol.
Chem., 360: 345-350 (in German).
RACINE, R.R., LANGLEY, C.H., & VOELKER, R.A. (1980) Enzyme
mutations induced by low-dose rate -irradiation in Drosophila:
frequency and characterization. Environ. Mutagen., 2: 167-177.
RAMSHAW, J.A.M., COYNE, J.A., & LEWONTIN, R.C. (1979) The
sensitivity of gel electrophoresis as a detector of genetic
variation. Genetics, 93: 1019-1037.
RECIO, L.B., CHASTENAY, B., ALBERTINI, R.J., & HSIE, A.W.
(1983) Detection of 6-thioguanine resistant splenocytes from
Chinese hamsters. Environ. Mutagen., 5: 401-402 (abstract).
REIDY, J.A., ZHOU, X., & CHEN, A.T.L. (1983) Folic acid and
chromosome breakage. I. Implications for genotoxicity studies.
Mutat. Res., 122: 217-221.
ROSENBLUM, B.B., NEEL, J.V., & HANASH, S.M. (1983)
Two-dimensional electrophoresis of plasma polypeptides reveals
"high" heterozygosity indices. Proc. Natl Acad. Sci. USA, 80:
5002-5006.
ROSENBLUM, B.B., NEEL, J.V., HANASH, S.M., JOSEPH, J.L., &
YEW, N. (1984) Identification of genetic variants in
erythrocyte lysate by two-dimensional gel electrophoresis.
Am. J. hum. Genet., 36: 601-612.
ROTHMAN, E.D., NEEL, J.V., & HOPPE, F.M. (1981) Assigning a
probability for paternity in apparent cases of mutation.
Am. J. hum. Genet., 33: 601-628.
SAMMONS, D.W., ADAMS, L.D., & NISHIZAWA, E.E. (1981) A
silver-based color development system for staining of
polypeptides in polyacrylamide gels. Electrophoresis, 2:
135-141.
SANTESSON, B., LINDAHL-KIESSLING, K., & MATTSSON, A. (1979)
SCE in B and T lymphocytes: possible implications for Bloom's
syndrome. Clin. Genet., 16: 133-135.
SATOH, C., NEEL, J.V., YAMASHITA, A., GORIKI, K., FUJITA, M.,
& HAMILTON, H.H. (1983) The frequency among Japanese of
heterozygotes for deficiency variants of 11 enzymes. Am. J.
hum. Genet., 33: 656-674.
SAVAGE, J.R.K. (1975) Classification and relationship of
induced chromosome structural changes. J. med. Genet., 12:
103-122.
SCHMIDT, M.A. & SANGER, W.G. (1981) Sister-chromatid
exchange in aged human lymphocytes. A brief note. Mech. Aging
Dev., 16: 67-70.
SELVIN, S. & GARFINKEL, J. (1976) Paternal age, maternal
age, and birth order and the risk of fetal loss. Hum. Biol.,
48: 223-230.
SESHADRI, R., BAKER, E., & SUTHERLAND, G.R. (1982)
Sister-chromatid exchange (SCE) analysis in mothers exposed to
DNA-damaging agents and their newborn infants. Mutat. Res.,
97: 139-146.
SHAFER, D.A. (1982) Alternate replication bypass mechanisms
for sister-chromatid exchange formation. Prog. Topics
Cytogenet., 2: 67-98.
SHARMA, T. & DAS, B.C. (1984) The effect of storage of blood
on the yield of x-ray-induced chromosome aberrations and
spontaneous sister-chromatid exchanges. Int. J. radiat. Biol.,
45: 151-158.
SKOLNICK, M. (1980) The Utah genealogical data base: a
resource for genetic epidemiology. In: Cairns, J., Lyon, J.L.,
& Skolnick, M., ed. Cancer incidence in defined populations,
New York, Cold Spring Harbor Laboratory, pp. 285-296 (Banbury
Report No. 4).
SKOLNICK, M.M. (1982) An approach to completely automatic
comparison of two-dimensional electrophoresis gels.
Clin. Chem., 28: 979-986.
SKOLNICK, M.M. & FRANCKE, U. (1982) Report of the committee
on human gene mapping by recombinant DNA techniques.
Cytogenet. cell Genet., 32: 194-204.
SKOLNICK, M.M. & NEEL, J.V. (in press) An algorithm for
comparing 2-D electrophoretic gels, with particular reference
to the study of mutation. Adv. hum. Genet.
SKOLNICK, M.M, STERNBERG, S.R., & NEEL, J.V. (1982) Computer
programs for adapting two-dimensional gels to the study of
mutation. Clin. Chem., 28: 969-978.
SMITH, M.E. (1977) The use of computers for studying the
origins and social cost of ill health. In: Shires, D.B. &
Wolf, H., ed. MEDINFO77: Proceedings of the Second World
Conference on Medical Informatics, Amsterdam, North Holland
Publishing Company, pp. 37-41.
SMITH, M.E. (1980) The present state of automated follow-up
in Canada. Part 1. Methodology and files. J. clin. Computing,
9: 1-18.
SMITH, M.E. (1982) Development of a national record linkage
program in Canada. 1982 survey research methods section.
Proceedings of the ASA : 303-308.
SMITH, M.E. & NEWCOMBE, H.B. (1980) Automated follow-up
facilities in Canada for monitoring delayed health effects.
Am. J. public Health, 70: 1261-1268.
SMITH, M.E. & NEWCOMBE, H.B (1982) Use of the Canadian
mortality data base for epidemiological follow-up. Can. J.
public Health, 73: 39-46.
SMITH, M.E., SILINS, J., & LINDSAY, J.P. (1980) The present
state of automated follow-up in Canada. Part II. The products.
J. clin. Computing, 9: 19-30.
SNOPE, A.J. & RARY, J.M. (1979) Cell-cycle duration and
sister-chromatid exchange frequency in cultured human
lymphocytes. Mutat. Res., 63: 345-349.
SOUTHERN, E.M. (1982) Application of DNA analysis to mapping
the human genome. Cytogenet. cell Genet., 32: 52-57.
SPEIT, G. (1980) Effects of temperature on sister-chromatid
exchanges. Hum. Genet., 55: 333-336.
STETKA, D.G. & CARRANO, A.V. (1977) The interaction of
Hoechst 33258 and BrdU-substituted DNA in the formation of
sister-chromatid exchanges. Chromosoma, 36: 21-31.
STETKA, D.G., MINKLER, J.L., & CARRANO, A.V. (1978)
Induction of long-lived chromosome damage, as manifested by
sister-chromatid exchange in lymphocytes of animals exposed to
mitomycin-C. Mutat. Res., 51: 383-396.
STICH, H.F., CURTIS, J.R., & PARIDA, B.B. (1982) Application
of the micronucleus test to exfoliated cells of high cancer
risk groups: tobacco chewers. Int. J. Cancer, 30: 553-559.
STRAUSS, G.H. (1982) Direct mutagenicity testing: the
development of a clonal assay to detect and quantitate mutant
lymphocytes arising in vivo. In: Bridges, B.A., Butterworth,
B.E., & Weinstein, I.B., ed. Indicators of genotoxic exposure,
New York, Cold Spring Harbor Laboratory Press, pp. 423-441
(Banbury Report No. 13).
STRAUSS, G.H. & ALBERTINI, R.J. (1977) 6-thioguanine
lymphocytes in human peripheral blood. In: Scott, D., Bridges,
B.A., & Sobels, F.H., ed. Progress in genetic toxicology,
Amsterdam, Elsevier/North Holland, pp. 327-334.
STRAUSS, G.H. & ALBERTINI, R.J. (1979) Enumeration of
6-thioguanine resistant peripheral blood lymphocytes in man as
a potential test for somatic cell mutations arising in vivo.
Mutat. Res., 61: 353-379.
STRAUSS, G.H., ALBERTINI, R.J., KRUSINSKI, P., & BAUGHMAN,
R.D. (1979) 6-Thioguanine resistant peripheral blood
lymphocytes in humans following psoralen long-wave light
therapy. J. Invest. Dermatol., 73: 211-216.
SUTHERLAND, G.R., CARTER, R.F., BAULD, R., SMITH, I.I., &
BAIN, A.D. (1978) Chromosome studies at the pediatric
necropsy. Ann. hum. Genet. (Lond.), 42: 173-181.
SUTHERLAND, G.R., BAKER, E., SESHADRI, R.S., & BLACK, A.
(1980) Increased sister-chromatid exchange in multiple
sclerosis. New Eng. J. Med., 303: 1126.
SUTTON, H.E. (1971) Workshop on monitoring of human
mutagenesis. Teratology, 4: 103-107.
SYLWESTER, D.L. & ALBERTINI, R.J. (in press) Confidence
intervals and sample size calculations to compare variant
frequencies. Environ. Mutagen.
TAYLOR, J.H. (1958) Sister-chromatid exchanges in
tritium-labeled chromosomes. Genetics, 43: 515-529.
TAYLOR, J.H., WOODS, P.S., & HUGHES, W.L. (1957) The
organization and duplication of chromosomes as revealed by
autoradiographic studies using tritium-labeled thymidine.
Proc. Natl Acad. Sci. USA, 43: 122-138.
THOMAS, G., MARTIN-PEREZ, J., SIEGMANN, M., & OTTO, A.M.
(1984) The effect of serum, EGF, PGF, and insulin on S6
phosphorylation and the initiation of protein and DNA
synthesis. Cell, 30: 235-242.
TSANG, V.C.W., PERALTA, J.M., & SIMMONS, A.R. (1983)
Enzyme-linked immunoelectrotransfer blot techniques for
studying the specificities of antigens and antibodies
separated by gel electrophoresis. Meth. Enzymol., 92: 377-391.
VIJAYALAXMI & EVANS, J.H. (1984) Measurement of spontaneous
and x-irradiation induced 6-thioguanine resistant human blood
lymphocytes. Mutat. Res., 125: 87-94.
VIJAYALAXMI, NEWTON, M.S., STEEL, C.M., EVANS, H.J., &
PENTLAND, B. (1983a) Spontaneous and mutagen-induced
sister-chromatid exchange in multiple sclerosis. J. med.
Genet., 20: 372-376.
VIJAYALAXMI, EVANS, J.H., RAY, J.H., & GERMAN, J. (1983b)
Bloom's Syndrome: evidence for an increased mutation frequency
in vivo. Science, 221: 851-853.
VOELKER, R.A., SCHAFFER, H.E., & MUKAI, T. (1980)
Spontaneous allozyme mutations in Drosophila: rate of
occurrence and nature of the mutants. Genetics, 94: 961-968.
VOGEL, F. (1970) Spontaneous mutation in man. In: Vogel, F.
& Rohrborn, G., ed. Chemical mutagenesis in mammals and man,
New York, Springer-Verlag, pp. 16-68.
VOGEL, F. & ALTLAND, K. (1982) Utilization of material from
PKU-screening programs for mutation screeing. In: Bora, K.C.,
Douglas, G.R., & Nestmann, ed. Chemical mutagenesis, human
population monitoring and genetic risk assessment, Amsterdam,
Elsevier Biomedical Press, Vol. 3, pp. 148-152 (Prog. Mutat.
Res.).
WAKSVIK, H., MAGNUS, P., & BERG, K. (1981) Effects of age,
sex, and genes on sister-chromatid exchange. Clin. Genet., 20:
449-454.
WALTON, K.E., STYER, D., & GUENSTEIN, E.I. (1979) Genetic
polymorphism in normal human fibroblasts as analyzed by
two-dimensional polyacrylamide gel electrophoresis. J. Biol.
Chem., 254: 7951-7960.
WARBURTON, D., STEIN, Z., KLINE, J., & SUSSER, M. (1980)
Chromosome abnormalities in spontaneous abortion: data from
the New York City Study. In: Porter, I.H. & Hook, E.B., ed.
Human embryonic and fetal death, New York, Academic Press,
pp. 23-33.
WARNER, L.A., NEEL, J.V., & MEISLER, M.H. (1982) Separation
of allelic variants by two-dimensional electrophoresis. Am. J.
hum. Genet., 34: 209-215.
WHITTAKER, P.G., TAYLOR, A., & LIND, T. (1983) Unsuspected
pregnancy loss in healthy women. Lancet, 1: 1126-1127.
WHO (1983) EHC 27: Guidelines on studies in environmental
epidemiology, Geneva, World Health Organization, 351 pp.
WOLFF, S. (1977) Sister-chromatid exchange. Ann. Rev.
Genet., 11: 183-201.
WOLFF, S. & PERRY, P. (1974) Differential Giemsa staining of
sister-chromatids and the study of sister-chromatid exchanges
without autoradiography. Chromosoma, 48: 341-353.
WOLFF, S. & PERRY, P. (1975) Insights on chromosome
structure from sister-chromatid exchanges and the lack of both
isolabelling and heterolabelling as determined by the FPG
technique. Exp. cell Res., 93: 23-30.
WOLFF, S., BODYCOTE, J., & PAINTER, R.B. (1974)
Sister-chromatid exchanges induced in Chinese hamster cells by
UV irradiation of different stages of the cell cycle: the
necessity for cells to pass through S. Mutat. Res., 25: 73-81.
WOLFF, S., BODYCOTE, J., THOMAS, G.H., & CLEAVER, J.E.
(1975) Sister-chromatid exchanges in xeroderma pigmentosum
cells that are defective in DNA excision repair or
post-replication repair. Genetics, 81: 349-355.
ZETTERBERG, G., ANMEUS, H., & MATSSON, P. (1982) Use of flow
cytometry to concentrate 6-thioguanine resistant variants of
human peripheral blood lymphocythes. In: Bridges, B.A.,
Butterworth, B.E., & Weinstein, I.B., ed. Indicators of
genotoxic exposures, New York, Cold Spring Harbor Laboratory
Press, pp. 413-420 (Banbury Report No. 13).
ZIK, W.H. & NECKERS, L. (1983) Lymphocyte subpopulations
altered during blood storage. New England J. Med., 309:
435-436.