
BIS (2-ETHYLHEXYL) PHTHALATE
Explanation
Bis(2-ethylhexyl)phthalate (DEHP) has not previously been
evaluated for an ADI for man by the Joint FAO/WHO Expert Committee on
Food Additives.
DEHP is used as a plasticizer in plastic film used in food
packaging. Dietary exposure to DEHP occurs through migration of the
plasticizer from packaging to food, and from the environment, where it
exists as a contaminant in drinking water and fish.
It has the following chemical formula:
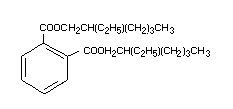 BIOLOGICAL DATA
BIOCHEMICAL ASPECTS
Metabolism
DEHP was hydrolyzed to the mono-ester by preparations derived
from the intestine and liver from several animal species (rat, baboon,
ferret) and from human intestine (Lake et al., 1977; Rowland et al.,
1977; Kaneshima et al., 1978). Homogenates of rat kidney, lung and
liver, as well as liver microsomes and mitochrondria, hydrolyse DEHP
to MEHP and 2-ethylhexanol. Hydrolysis was more rapid with liver
preparations than lung or kidney preparations. Liver microsomal
preparations were more active than mitochondrial preparations, whereas
the cytosol fraction was inactive (Carter et al., 1974). DEHP was
rapidly hydrolysed by pancreatic lipase, and at a slower rate by rat
liver homogenate, plasma, and preparations from kidney and lung. No
o-phthalic acid could be detected in the reaction products (Daniel &
Bratt, 1974; Albro & Thomas, 1973).
The in vitro absorption of mono- and DEHP was studied using
isolated perfused rat intestins. All DEHP was converted to mono
(ethylhexyl) phthalate (MEHP) before it reached the serosal perfusing
solution. No metabolic transformation of MEHP was reported (White et
al., 1980).
Adult male CD rats were given two oral doses of 14C-DEHP at
24 hour intervals and 24 hour urine samples collected. Five major
metabolites were present after the initial dose, the initial metabolic
product, mono (2-ethylhexyl)phthalate being metabolized to 5-keto-2-
(ethylhexyl)phthalate and 2-carboxyl-2-(ethylpentyl)phthalate,
5-hydroxy-2-(ethylhexyl)-phthalate and 2-carboxymethyl-butylphthalate.
Less than 5% of the administered compound was hydrolyzed completely to
o-phthalic acid. Various tests for conjugation were negative (Albro
et al., 1973).
Male, 4-month old, CD rats, CD-1 mice, Syrian golden hamsters and
Hartley albino guinea pigs were administered by gavage 2 doses of DEHP
at 24-hour intervals, at levels ranging from 20-360 mg/kg b.w. Urine
was collected for 48 hours after the initial dose. No metabolites were
detected in urine from hamsters or guinea pigs that were not also
present in rat or mouse urine. In the mouse, guinea pig and hamster, a
significant proportion of DEHP metabolites were present as glucuronide
conjugates. Conjugates of taurine and sulfate were not detected.
Metabolites of DEHP in rats were not conjugated. Omega and omega-1
oxidation of the ethylhexyl moiety was less efficient in guinea pig
than in the other species, but guinea pigs appeared to produce
significantly more MEHP (Albro et al., 1982a,b). The enzymatic
processes normally associated with omega, omega-l, alpha- and
beta-oxidation of fatty acids could account for the major urinary
metabolites of DEHP in the rat. However, the structures of recently
identified minor metabolites of DEHP suggest that oxidation of
the aliphatic side chain can occur at positions more distant than
omega-1 from the methyl terminus. This type of oxidation at multiple
sites in an aliphatic side chain occurs commonly when alphatic
hydrocarbons, rather than fatty acids, are metabolized by microsomal
oxidases (Albro et al., 1983).
In separate experiments, single gavage doses of 14C-DEHP
were given to Fisher 344 rats, CD mice, Syrian golden hamsters and
Hartley albino guinea pigs and the urine analyzed for 11 different
metabolites. Species differences were noted in the distribution of
urinary metabolites. In the rat, 3 metabolites, not including MEHP
(trace), comprised 81% of all urinary metabolites of DEHP, while in
mice, 75% of all urinary metabolites were accounted for by 5
compounds, including MEHP (18.6%). In the guinea pig, MEHP accounted
for 71.2% of all urinary metabolites, while in the hamster, 92.4%
of administered radioactivity was distributed among 5 urinary
metabolites, not including MEHP (4.5%). Unreacted DEHP occurred at
levels of 0.5% or less in all species. 100% of urinary metabolites
were not-conjugated in rats, while 85% were non-conjugated in
hamsters. Mice and guinea pigs excreted 35-36% of urinary metabolites
in the non-conjugated form (Albro et al., 1982b). In other studies,
dogs excreted 3 major urinary metabolites, and pigs excreted 5 (Ikeda
et al., 1980).
In a study with African Green monkeys infused with palsma
containing about 5 mg of 14C-DEHP, more than 50% of the 14C was
excreted in the urine by 4 hr, and more than 70% by 24 hr. The major
metabolites were the 5-ethyl, isohexanol monoester of phthalic acid
and MEHP. More than 80% of the urinary metabolites were conjugated to
gluconide (Albro et al., 1981).
Two cancer patients received, by platelet concentration infusion,
94.7 mg DEPH in 4 hr and 174.3 mg DEHP in 1.5 hr. More than 50% of the
infused dose appeared as DEHP metabolites in the urine within 6 hr.
The predominant metabolite was the 5-ethyl, isohexanol monoester of
phthalic acid. Approximately 80% of urinary metabolites were
glucuronide conjugates. Eight other metabolites, including MEHP, were
identified. The half-life of DEHP in plasma was 30 ± 12 min.,
apparent distribution volume was 2819 ± 83 ml/m2 and clearance was 78
± 20 ml/min-m2 (Peck et al., 1978; Albro et al., 1981).
Absorption, distribution and excretion
Rats were given by gavage single doses of 14C-DEHP
ranging from 2.6 to 1900 mg/kg b.w. 35-55% of the labeled dose was
excreted in the feces and 42-57% in the urine. At low dose levels
(2.6-2.9 mg/kg b.w.), 9-14% of the dose appeared in the bile. No
significant amounts of 14C-C02 could be found in the expired air. At
high dose levels (1000 mg/kg2 b.w.) during an 8 day period, fecal
excretion accounted for 6.5-10.5% of the administered dose of 14C as
metabolites and 24.0-38.8% as unchanged DEHP. Most fecal metabolites
had been excreted by 48 hours after dosing. 53.5-70.2% of the dose was
excreted as MEHP or metabolites in the urine. Some 14C remained in
fatty tissue 8 days after dosing, but none was found in 10 other
tissues. In liver, kidney and skeletal muscle, radioactivity has
essentially disappeared between 48 and 96 hours after dosing (Daniel &
Bratt, 1974; Schulz & Rubin, 1973; Williams & Blanchfield, 1974a).
Beagle dogs, miniature Hormel swine and male Sprague-Dawley rats
were fed DEHP (650, 750 and 750 ppm, respectively) for 21-28 days and
then given an oral dose of 14C-DEHP. In rats, 85% of the administered
radioactivity was excreted in the urine and feces 24 hours after
dosing excretion of metabolites predominated in dogs and urinary
excretion predominated in pigs, while in rats, metabolites were
distributed equally between urine and feces. Elimination of
administered 14C-DEHP was 84% complete in rats by 24 hours after
dosing; elimination of metabolites in dogs and pigs was slower
(50% after 24 hr), but was complete in all species by 4 days after
dosing. Serum half-life for 14C-DEHP and metabolites was 1.2 hr in
dogs and 5.4 hr in pigs. Analysis of tissue and excreta 4 days after
dosing with 14C-DEHP indicates that DEHP was metabolized 96% in rats,
63% in dogs and 71% in pigs (Ikeda et al., 1980).
Male mice were given single oral doses of Di-(2-ethylhexyl)
(14C)phthalate (6.72 mg/animal) and examined by whole-body
autoradiography at intervals from 1 hr to 7 days after treatment. Most
of the radioactivity was excreted in the urine and feces in the first
24 hr. Excretion was complete within 3-5 days. Following absorption,
14C was widely distributed in the carcass, but none was found in the
central nervous system, skeleton, or thymus, and only rarely in the
testes. There was no evidence of tissue storage (Gaunt & Butterworth,
1982).
Groups of 3 male Fischer 344 rats were given daily gavage doses
of 1.8, 18 or 180 mg/kg b.w. 14C-DEHP, and sacrificed 1, 3, 10 and
12 days after initiation of treatment. Residual 14C in testes and
liver, as percent of administered dose, decreased with duration of
treatment and magnitude of dose, except for testes at day 12 (all dose
levels) in which 14C levels were higher than at day 10. In
experiments with a single dose (1.8-1000 mg/kg b.w.) of DEHP, no
accumulation occurred in the liver at doses below 400g/kg b.w. (Albro
et al., 1982b).
Wistar rats, 200 g, were administered a single dose of DEHP or
MEHP (10,000 mg/kg b.w.) by gastric intubation and samples of blood
and tissue collected 1, 3, 6, 24, 48 and 96 hr post-intubation. Levels
of DEHP in liver declined with a half life of 1 day, while levels in
epididymal fat declined more slowly (t1/2 = 6.5 days), t1/2 values for
DEHP in other tissues were less than that observed in liver. MEHP
levels declined much more slowly (t1/2 = 50 hr) than DEHP (8.3 hr) in
testes, but more rapidly (t1/2 = 67.6 hr for MEHP vs. 156 hr for DEHP)
in epididymal fat. In other tissues, levels of the two phthalates
showed similar rates of decline. In heart and lungs, the highest
levels of DEHP and MEHP occurred within 1 hr of dosing, while, in
other tissues, the highest levels occurred 6-24 hours after dosing
(Oishi & Hiraga, 1982).
Groups of 24 female rats were fed diets containing either 0.1% or
0.5% ppm 14C-DEHP for 35 and 49 days, respectively. In rats fed 0.1%,
DEHP 14C residues reached steady state levels of 40-50 ppm in liver
in 3 days, while steady state levels in fat (7-9 ppm), were attained
by 2 weeks. 14C levels in brain never exceeded 1 ppm, but in the
heart, levels ranged from 1 to 6 ppm. At the high dose level,
equilibrium levels of 14C were attained in the rats after 9-14 days
of feeding. Steady state levels of 14C in the rat tissues were 120
ppm in liver, 80 ppm in fat, 2-3 ppm in brain and 15-20 ppm in heart.
After removal of rats from the test diet, 14C declined with a half
life of 1-2 days in liver, and 3-5 days in fat (Daniel & Bratt, 1974).
4 male C57BL mice, 20 g, were given oral doses of 14C-DEHP
(2-ethylhexyl -1-14C, 9.6 mg/kg b.w.; and carbonyl-14C, 3.6 mg/kg
b.w.) and sacrificed 5, 14 or 30 days later. Whole body
autoradiography showed that in the males, either labeled form of DEHP
was retained at low levels in bone. The (carbonyl -14C) DEHP but not
the other labeled form, displayed marked retention in the skin,
cartilage and tendons. In a study with pregnant mice, administration
of the 14C labeled compounds at day 8 or 16 of gestation resulted in
14C-DEHP being concentrated mainly in the yolk sac. DEHP (carbonyl-
14C was present in the neuroepithelium of the embryo. Dosing at day
18 of gestation resulted in a more general distribution of the 14C
labeled compounds in the fetuses, 4 hr post-dosing, but 24 hr post-
dosing the 14C was primarily located in the renal pelvis, urinary
bladder, intestines and skeleton of the fetus (Lindgren et al., 1982).
Two adult men ingested single doses of 10 g and 5 g, respectively
of DEHP. Phthalic acid equivalent to approximately 4.5% of the
administered dose was recovered from the urine in the 24 hr period
after dosing, the major portion being excreted 5 to 7 hours after
dosing (Schaffer et al., 1945).
Effects on enzymes and other biochemical parameters
When rats were fed DEHP (0, 1, 2 or 4%) for 4 weeks, liver
glycogen decreased markedly, while hepatic fatty acid synthesis
increased 2-fold. Treatment-related alterations in levels of
intermediates in carbohydrate metabolism indicate that gluconeogenesis
was inhibited at a point between 3-phosphoglycerate and fructose
1,6-diphosphate (Sakurai et al., 1978).
When male Wistar rats (80-95 gm) were fed 0.5% DEHP for 10 days,
livers of treated animals were enlarged, and showed reduced levels of
glucogen and triglycerides and increased levels of phospholipids.
These treatment-related changes were suggestive of metabolic
transformation from glycolysis to lipolysis as an energy source
(Yanagita et al., 1978).
Male Fisher-344 rats were fed diets containing either 100 ppm or
1.0% DEHP for 11 days. On days 1, 6 or 10, rats were given a tracer
dose of 14C DEHP and sacrificed 10 or 1 day later, respectively.
Purified protein, RNA and DNA fractions were obtained from the liver
and assayed for bound radioactivity. Binding was detectable only when
the ethylhexyl moiety of DEHP was labeled (Albro et al., 1982b).
Male F-344 rats, 150-180 g, fed 2% 2-ethylhexanol for 3 weeks
showed increased liver size, increased activity of hepatic peroxisome-
associated enzymes (catalase and carnitine acetyl transferase) and
hypolipidemia. These DEHP-induced effects are similar to those
produced by hypolipidemic drugs such as clofibrate (Moody & Reddy,
1978).
Special studies on peroxisomal proliferation
Adult male, Sprague-Dawley rats were fed for 2 weeks with diets
containing 2% DEHP or DEHP metabolites (MEHP, phthalic acid,
2-ethylhexanol and 2-ethylhexyl benzoate). At sacrifice, livers were
removed and samples taken for electron microscopy and subcellular
fractionation (mitochondria and microsomes). DEHP and MEHP stimulated
peroxisome enzymes (palmitoyl CoA oxidation, 350-600%), mitochondria
(protein, 238-272%; carnitine-acetyl transferase, 1000-2900%) and
microsomes (cytochrome P-450, 63-68%; NADHP-cytochrome C reductase,
59-73%). Cytochrome C oxidase was stimulated by MEHP and EH (75-91%)
but not by DEHP. 2-elkyl hexanol stimulated mitochondrial protein
synthesis by 75%. Phthalic acid and ethylhexyl benzoate had little or
no effect on marker enzymes. Ultrastructural examination revealed
induction of mitochondrial and peroxisomal proliferation by DEHP and
MEHP, without induction of the endoplasmic reticulum. Other
metabolites had no ultrastructurally visible effect (Ganning et al.,
1982, 1983; Ganning & Dallner, 1981).
Rats (120 g) fed diets containing 2% (w/w) di-(ethylhexyl)-
phthalate (DEHP) for up to 4 weeks, showed a marked increase in
activities of enzymes of peroxisomal beta-oxidation and of catalase
(liver). The time required to reach 50% maximal induction for enzymes
of peroxisomal beta-oxidation was 5-7 days, whereas that for
catalase was 3 days. After withdrawal of DEHP, activities of enzymes
of the beta-oxidation system and of catalase decrease to the control
level with a half-life of 2-3 days. Mitochondrial marker enzymes
(3-hydroxyacyl-CoA dehydrogenase, 3-ketoacyl-CoA thiolase
(non-specific) and acetoacetyl-CoA specific thiolase) also were
markedly increased in treated animals. These activities decreased
after withdrawal of treatment (Miyazawa et al., 1980; Ohno et al.,
1981).
Special studies on mutagenicity
In a number of laboratories, DEHP did not include reverse
mutations of the base pair substitution type in Salmonella
typhimurium with or without metabolic activation (Kirby et al.,
1983; Kozumbo et al., 1982; Ruddick et al., 1981; Simmon et al., 1977;
Warren et al., 1982; Yoshikawa et al., 1983; Zeiger et al., 1982).
Neither was 8-azaguanine resistance in S. typhimurium affected by
DEHP (Seed, 1982). Tomita et al., (1982) reported a weak positive
reaction for DEHP in the S. typhimurium system.
No mutations (tryptophan reversion) occurred in Eschericia coli
exposed to DEHP in vitro (Tomita et al., 1982; Yoshikawa et al.,
1983) or in DNA repair capability of Bacillus subtilis (Tomita et
al., 1982) exposed to DEHP.
DEHP did not show mutagenic effects in a number of mammalian cell
systems: L5178Y mouse lymphoma (forward mutation), Kirby et al.
(1983); rat hepatocyte (unscheduled DNA synthesis), Hodgson, (1982);
human lymphocyte (chromosomal aberrations), Turner et al. (1974);
human fetal lung cells (aneuploidy), Stenchever et al. (1976); Chinese
hamster fibroblast (chromosomal aberrations), Abe & Sazaki (1977),
Ishidate & Odashima (1977); Chinese hamster ovary (sister chromatid
exchange, sister chromatid exchange HGPRT reversion), Phillips et al.
(1982).
DEHP, MEHP and 2-ethylhexanol were tested for mutagenicity in the
following systems: Ames Salmonella/microsome plate test, mouse
lymphoms forward mutation assay, in vitro transformation of Balb/3T3
cells (with and without activation by primary rat hepatocytes), mouse
micronubleus test, and unscheduled DNA synthesis in rat hepatocytes.
No mutagenic activity was observed (Anonymous, 1982a-m).
MEHP was shown to be mutagenic in S. thyphimurium (histidine
reversion), in E. coli (tryptophan reversion) and in B. subtilus
(DNA repair) (Tomita et al., 1982). The same workers also found MEHP
to be mutagenic in Chinese hamster embryo cells (chromosome
aberration, 8-azaguanine resistance, 6-thioguanine resistance, sister
chromatid exchange). Phillips et al. (1982) found MEHP to be
mutagenic to Chinese hamster ovary cells (chromosomal damage, sister
chromatid exchange), but non-mutagenic toward HGPRT reversion. Kirby
et al. (1983) found MEHP to be non-mutagenic in the L5178Y mouse
lymphoma system (forward mutation).
2-ethylhexanol was shown to be weakly mutagenic in
S. typhimurium (8-azaguanine resistance, Seed, 1982), but was
non-mutagenic in L5178Y mouse lymphoma (Kirby et al., 1983), rat
hepatocyte (Hodgson et al., 1982) and Chinese hamster ovary (Phillips
et al., 1982).
Pregnant Syrian golden hamsters were given DEHP orally at 3.75,
7.5 or 15 g/kg b.w. or MEHP at 0.375, 0.750 or 1.5 g/kg b.w. on day 11
of gestation. In embryonic cells removed on day 12 and examined after
15-20 days of culture in vitro, there were significant increases in
aberrant metaphase cells from all treatment groups except the one that
received the lowest dose of DEHP. Aberrations included single
chromatid gaps, isochromatid gaps, single chromatid or isochromatid
breaks and chromatid exchanges. There were also significant increases
in the rate of morphological transformation of the cells at the two
highest doses of DEHP and the two lowest doses of MEHP, and a marginal
increase in 8-azaguanine and 6-thioguanine resistance (Tomita et al.,
1982).
No significant increases in clastogenic changes were observed in
the bone marrow cells of male Fischer 344 rats that had received
0.5-5.0 g DEHP/kg b.w./day, 0.01-0.14 g MEHP/kg b.w./day or
0.02-2.21 g 2-ethylhexanol/kg b.w./day by gavage for 5 days (Putman
et al., 1983). Dominant lethal assays using male mice given DEHP
(2.5-9.9 g/kg), MEHP (50-200 mg/kg) or 2-ethylhexanol (250-1000 mg/kg)
by gavage in corn oil for 5 days produced negative results: fertility
indices and the average numbers of dead and total implants per
pregnancy were within normal ranges in all cases (Rushbrook, 1982).
Special studies on carcinogenicity
Groups of 50 male and female B6C3F1 mice were fed diets
containing 0, 0.3 or 0.6% Di(2-ethylhexyl)phthalate (DEHP) for 103 wk.
Treatment with DEHP did not affect survival or food consumption. Mean
body weight gains of treated female mice (0.3 and 0.6%) were less than
those of the corresponding controls. No other clinical signs of
toxicity were reported. At termination of the studies, chronic
inflammation of the kidney and seminiferous tubular degeneration were
observed in male mice (0.6% diet). No other nonneoplastic lesions were
detected in the treated groups at incidences greater than in the
corresponding controls.
Treatment with DEHP caused liver tumors in both sexes of mice.
Incidence data are as follows: for males, hepatocellular carcinoma
9/50, 14/48 and 19/50, hepatocellular adenomas 6/50, 11/48 and 10/50,
and for females hepatocellular carcinoma 0/50, 7/50 and 17/50 and
hepatocellular adenoma 1/50, 12/50 and 18/50, for the 0, 0.3% and 0.6%
groups, respectively. 20 of the 57 hepatocellular carcinomas
diagnosed in treated mice (sexes and doses combined) had metastisized
to the lung. No pulmonary tumors were observed in any control rats.
The incidences of liver tumors in control mice was comparable to that
in recent historical controls. The incidences of both male and female
mice with hepatocellular carcinomas were significantly greater
(p<0.05) at the 0.6% dose than in controls, and in female mice at the
lower dose (p<0.05) by pairwise comparison, and trend tests detected
significant (p<0.05) dose related effects in both sexes. The
incidence of hepatocellular adenomas in both male and female did not
differ from that of controls. However, the incidences of combined
adenomas and carcinomas was significantly increased for both sexes at
both doses (p<0.05), by pairwise comparisons and showed significant
dose related trends (Kluwe et al., 1982).
Groups of 50 male and female Fischer 344 rats were fed diets
containing 0, 0.6 or 1.2% di(2-ethylhexyl)phthalate (DEHP) for 103
consecutive weeks. Treatment with DEHP did not affect survival rates
or food consumption. Mean body weight gains of treated male rats
(0.6 and 1.2% diet) and female rats (0.2%), were less than those of
the corresponding controls. No clinical signs of toxicity were
reported. At autopsy, seminiferous tubular degeneration and
hypertrophy of cells in the anterior pituitary were observed in
high-dose male rats. Other nonneoplastic lesions were detected in the
treated groups at incidences similar to the corresponding controls.
Incidence data are as follows: for males, hepatocellular
carcinoma 1/50, 1/49 and 5/49, neoplastic nodules 2/50, 5/49 and 7/49,
and for females, hepatocellular carcinoma 0/50, 2/49 and 8/50 and
neoplastic nodules 0/50, 4/49 and 5/50 for the 0, 0.6 and 1.26 groups,
respectively. The incidence of female rats with hepatocellular
carcinomas the 1.2% group was greater than that in control by pairwise
comparison (p<0.01) and there was a significant (p<0.05) dose
related trend effect. The incidence of female rats with neoplastic
nodules was greater in the 1.2% than in controls (p<0.05) and showed
a significant (p<0.05) dose related trend effect. For male rats the
incidence of hepatocellular carcinomas showed a dose related trend
effect, but was not significantly increased by pairwise comparison.
The incidence of neoplastic nodules in the male rats did not show a
significant increase using either pairwise comparison or trend tests
(Kluwe et al., 1982).
Special studies on promotion of carcinogenicity
Weanling male B6C3F1 mice received a single i.p. injection
(80 mg/kg) of diethylnitrosamine (DEN) at 4 weeks of age, followed by
oral administration of phenobarbital (PB) or di(2-ethylhexyl)phthalate
(DEHP) 2 weeks after DEN injection and continued for up to 6 months.
PB was administered in drinking water at 500 ppm and DEHP in the feed
at 0.3%, 0.6% or 1.2%. Groups of mice were sacrificed at 2, 4 and 6
months after DEN exposure and the livers were examined. Few
preneoplastic foci were seen at 2, 4 or 6 months, in mice exposed to
DEN, PB or DEHP alone, while numerous foci and neoplasms were seen in
mice given DEHP or PB after DEN. In DEHP-exposed mice, the number of
foci did not increase between 4 and 6 months, but the foci increased
in mean diameter and volume throughout the experiment. Foci and tumors
appeared earlier in mice given higher dietary levels of DEHP than in
those given lower doses. By the end of the study the number of foci
per unit volume of liver was similar in mice given any dose of DEHP,
but their volume was dose-related; basophilic foci and neoplasms
predominated. The latter were more malignant in appearance than
neoplasms in PB-exposed mice. At 6 months, the neoplasms in high dose
DEHP-exposed mice were significantly larger than those in PB-exposed
mice. Histochemistry, however, revealed similarities between lesions
in mice exposed to PB or DEHP. PB given continuously for 6 months
revealed no initiating activity of DEHP given once by gavage and
followed by PB in drinking water (Ward et al., 1983).
Special studies on the metabolites
Male Sprague-Dawley rats, 250-350 gm, were given 14C-mono
(ethylhexyl) phthalate (MEHP), 69 mg/kg, by gavage. MEHP is a primary
metabolite of DEHP. The resulting urinary metabolites of MEHP were
similar to those of DEHP, and included an alcohol, a ketone, and an
acid from the side chain oxidation of MEHP, as well as a small amount
of o-phthalic acid (Chu et al., 1978).
Phthalic acid, a compound from which DEHP is formed by
esterification, was fed by intubation to male Wistar rats at dose
levels of 3.3 or 40 mg/kg b.w. 0.15% (as 14C02) of the administered
dose was detectable in expired air. Only unchanged phthalic acid could
be found in urine, feces or tissue. Elimination was complete by 24
hours after dosing (Williams & Blanchfield, 1974b).
After oral administration to rats, 2-ethylhexanol was rapidly
absorbed and excreted. Radioactivity from (1-14C)-2-ethylhexanol was
detected in the urine (80-82%), feces (8-9%) and expired C02 (6-7%).
The route of metabolism of 2-ethylhexanol appears to involve oxidation
of 2-ethylhexanoic acid, followed by w- and (w-1)-oxidation.
Urinary metabolites included 2-heptanone, 4-heptanone, 2-ethyl-5-
hydroxyhexanoic acid, 2-ethyl-1,6- hexanedioic acid and 2-ethyl-5-
hexanone. The rate of elimination of 2-ethylhexanol was the same for
a 9 ug as for an 83 mg dose, with essentially all of the 14C being
recovered within 28 hours after administration (Albro, 1975).
Groups each of ten Weanling Sprague-Dawley rats were fed diets
containing 0, 25, 100, 400, 1,600 or 6,400 ppm of mono-2-ethylhexyl
phthalate for 28 days. Decreased growth rate was observed in the
highest dose group. Increased heart and liver weights were observed in
animals of the 1,600 and 6,400 ppm groups. Minor alterations in serum
biochemical values included decreased SDH and calcium levels, and
elevated alkaline phosphatase activity in some treated groups (Chu et
al., 1981).
In 3-month and 6-month feeding studies, groups of ten male and
female Weanling Sprague-Dawley rats were fed diets containing 1, 5,
25, 125 or 625 ppm of mono-2-ethylhexyl phthalate. Growth rate and
food consumption were not affected at any dose level or time interval.
Relative organ weights of rats of both sexes were not altered in the
3-month period, but the liver weights of female rats were increased in
the 6-month study. Changes in clinical chemistry and hematological
values were minimal. These included lower LDH, SGOT, hemoglobin, and
hematocrit values in male rats at the 3-month period and reduced
potassium content at the 6-month period. Histological changes occurred
in treated male and female rats at both time intervals in the liver,
heart, and adrenals. Alteration in the liver consisted of midzonal and
perioportal eosinophilic cytoplasmic inclusions and vacuolations with
isolated binucleated and necrotoc hepatocytes. There was a mild
enlargement of myocardial nuclei and segmental deregistration of
myocardial striations in test animals. The adrenal glands exhibited
vacuolation of the zone fasciculata which was less severe in the
6-month study than the 3-month counterpart (Chu et al., 1981).
Special studies on reproduction and teratology
Mouse
Groups of 10-22 pregnant mice (ddY-Slc (SPF) strain, 8-9 weeks
old) were administered single oral doses (0, 50, 100, 500 mg/kg b.w.)
of DEHP on day 7 of gestation, and sacrificed on day 18. Dose-related
decreases were noted in fetal body weights, number of live fetuses and
implantations per dam. Gross and skeletal anomalies exhibited
dose-related increases. The only treatment effect noted in fetuses
from dams fed 50 mg/kg DEHP was decreased body weight (Nakamura
et al., 1979).
Groups of 3-8 pregnant mice (ddY-Slc (SPF) strain, 8-9 weeks
old) were administered single oral doses of DEHP ranging from 100 to
30,000 mg/kg b.w. on days 6, 7, 8, 9 or 10 of gestation. MEHP was
administered to a separate group of pregnant mice at levels from
100-1000 mg/kg b.w. on days 7, 8 or 9 of gestation. Dams were
sacrificed on day 18 of gestation. With DEHP-treated dams,
treatment-related decreases were noted in fetal body weight
(days 6-10) and number of live fetuses (days 7-9). Because of the
variance in dose levels, no clear-cut treatment effect was evident on
implantations per dam. Treatment-related increases were seen in gross
anomalies (days 7-10) and skeletal anomalies (days 6-8 and day 9, high
dose only).
With MEHP-treated dams, treatment-related decreases were noted in
the number of live fetuses (days 7-8) and increases in gross anomalies
(days 8-9) and skeletal anomalies (day 8). Decreases in fetal body
weight were only marginally related to treatment except for the high
dose dams on day 8 and day 9. No clear-cut treatment effect could be
seen on the number of implantations per dam (Yagi et al., 1980).
Teratogenic effects were observed in CBA mice following a single
oral administration of DEHP in doses representing 1/3-1/12 of the
acute LD50 dose (26.9 g/kg b.w.) on days 6 to 10 of gestation. Excess
fetal deaths were observed when higher doses were given on day 7 but
not when given on day 9 or 10. A significant number of external and
skeletal malformations were found in the group given 7.5 g/kg b.w. on
day 8 (Yagi et al., 1976).
Pregnant female mice (ICR-JCL strain, 8-16 weeks of age) were
distributed in groups varying from 8 to 15 and fed diets containing
0, 0.05, 0.1, 0.2, 0.4 and 1.0% DEHP throughout gestation. Dams were
sacrificed on day 18 of gestation. At dose levels above 0.1%, maternal
weight gain was significantly depressed, although mean food intake was
unaffected. Fetal mortality was also significantly increased in this
dose range. At dose levels above 0.05%, the percentage of resorptions
and dead fetuses exhibited a dose-related increase, and was 100% at
dietary DEHP levels of 0.4% and 1%. Several cases of neural tube
defects (spine bifida and exencephaly) occurred in fetuses from dams
fed 0.2% DEHP. Deficient lumbar rib and ossification of sternabrae was
noted in fetuses from dams fed 0.1% and 0.2% DEHP. Fetal survival was
too low at higher doses to permit meaningful data on incidence of
anomalies. No other skeletal anomalies or visceral anomalies could be
related to treament (Shiota et al., 1980).
Rat
Pregnant Wistar rats, 175-200 g, were randomly distributed into
groups of 15 and dosed by gavage with 0, 225, 450, or 900 mg MEHP/kg
b.w. on days 6-15 of gestation. Another similar experiment was
performed at lower doses (0, 50, 100 and 200 mg/kg b.w.) of MEHP.
Dose-related maternal mortality occurred at the higher doses (225,
450, 900 mg/kg b.w.). In addition, the number of litters per treatment
group decreased significantly at higher doses. Litter size and pup
weight were not significantly affected by treatment. Disturbances in
the placement of the sternebrae plates were noted in fetuses from dams
treated with 450 mg/kg b.w. MEHP. No other skeletal or visceral
anomalies could be related to treatment. In the low-dose experiment,
the only treatment-related effect was a reduction in maternal weight
gain at MEHP doses of 100 and 200 mg/kg b.w. The no-effect level for
MEHP in this study was 50 mg/kg b.w. (Ruddick et al., 1981).
Groups of 20 female Wistar rats (80-120 g) were given daily doses
of 0, 0.34 and 1.70 g/kg DEHP by gavage. After approximately 3 months
of treatment, 10 rats (150-180 g) per dose level were mated. During
gestation, test compound was discontinued. In a separate experiment,
groups of 10 female rats (150-180 g) received comparable doses of
DEHP, but only during gestation. All fetuses from treated dams were
live; no skeletal abnormalities were found. No examination of fetuses
for soft tissue abnormalities was carried out. In dams treated during
gestation, litter size was unaffected but resorptions increased
significantly and fetal weights decreased significantly at both
treatment levels. No significant effects on resorptions, litter size
or fetal weights occurred in dams treated prior to gestation
(Nikoronow et al., 1973).
Rabbit
Virgin female New Zealand, white rabbits (3.9 + 0.5 kg) were
artificially inseminated and injected with sodium monoethylhexyl
phthalate (MEHP, 1.14, 4.69 or 11.38 mg/kg) on days 6-18 of gestation.
When fetuses were removed on day 30, no treatment effect was evident
on sex ratio, fetal weight or litter size. The number of corpora lutea
or resorptions were not affected by treatment, nor were weights of
maternal adrenals, liver, kidneys, heart or lung. No skeletal or
visceral abnormalities occurred that could be ascribed to treatment.
However, the 2 high dose groups of dams experienced increased
mortality (22% and 33%) relative to controls (Thomas et al., 1979).
When pregnant rabbits were injected with MEHP (11.4 mg/kg) on
days 6-18 of gestation, phthalate levels were less than 1 ug/ml in
fetuses and in homogenates of placenta and uteri. Maternal serum
levels failed to reveal any bioaccumulation, and there was no
significant evidence of histological changes in fetal organs,
including liver, lungs and kidneys (Thomas et al., 1980).
Special studies on testicular pathology and effect on zinc
and testosterone levels
Groups of 10 male JCL:ICR mice and rats were fed a diet
containing 2% DEHP or 2% MEHP for 1 week. Both test compounds
depressed growth, testicular weight, and liver weight. Levels of
testosterone and zinc were depressed in the testes, and kidney zinc
levels were elevated in both treatment groups. Liver zinc content was
depressed in the DEHP treatment group, but unchanged in the MEHP group
(Oishi & Hiraga, 1980a,b). Similar effects were observed with
Sprague-Dawley rats given oral doses of DEHP or MEHP (Gray et al.,
1982; Oishi & Hiraga, 1980c,d).
Male Fischer 344 rats and B63CF mice were distributed into groups
of 50 and fed diets containing 0, 0.6% or 1.2% DEHP (rat); or 0, 0.3
or 0.6% (mice) for 103 weeks. High dose rats exhibited a 90% incidence
of severe seminiferous tubular degeneration and testicular atrophy in
comparison to an incidence of 5% or less in low dose rats or controls.
The tubules in the affected animals were devoid of spermatocytes and
germinal epithelium. Only Sertoli cells were observed on the basement
membrane. High dose mice experienced a 14% incidence of seminiferous
tubular degeneration, compared to an incidence of 4% or less in low
dose mice or controls (Kluwe et al., 1982). In another study,
0.2% DEHP fed to male CDE rats for 17 weeks, caused marked
histological changes in the testes (Gray et al., 1977).
Groups of 10 male Wistar rats of varying age (4, 10 or 15 weeks)
were given 2.8 g/kg b.w. DEHP by gavage for 10 consecutive days.
Treated 4 week-old rats showed uniform seminiferous tubular atrophy
comprising a loss of spermatids and spermatocytes. 10 week old rats
experienced a 5-10% incidence of seminiferous tubular atrophy, while
in 15-week old rats, no adverse effects of this type were reported. If
treatment was stopped prior to puberty, normal testicular weight and
histology were regained within 12 weeks. Recovery was slower and less
complete when treatment was discontinued after puberty (Gray and
Butterworth, 1980).
Groups of 7-8 Syrian hamsters (DSN strain) were given oral doses
of 4200 mg/kg b.w./day DEHP or 1000 mg/kg b.w./day MEHP for 9 days.
MEHP reduced mean testicular weight to 73% of control values and
induced atrophy in less than 50% of the seminiferous tubules. DEHP had
no significant effects on these parameters or on urinary zinc
excretion (Gray et al., 1982).
Acute toxicity
Animal Route LD50 References
(mg/kg b.w.)
mouse p.o. 33,500 Krauskopf, 1973
rat p.o. 26,000 Patty, 1967
rabbit p.o. 34,000 ibid
33,900 Shaffer et al., 1945
Guinea pig p.o. 26,300 Krauskopf, 1973
Short-term studies
A study was conducted on the effect of oral administration of
di-(2-ethylhexyl)phthalate (DEHP) at a dose level of 25,000 ppm for
7 and 21 days in young male and female Wistar albino rats. DEHP
increased liver size in both sexes and reduced the relative weight of
testes in male rats. Liver enlargement was accompanied by increases in
several marker enzyme activities (7-ethoxy coumarin-0-deethylase,
cytochrome P-450, alcohol dehydrogenase, aniline-4-hydroxylase, and
succinate dehydrogenase). However, glucose-6-phosphatase activity was
decreased in all treatment groups. DEHP produced no hepatic
histological changes in either sex but ultrastructural studies
indicated proliferation of the smooth endoplasmic reticulum, an
increase in the number of microbodies (peroxisomes), and mitochondrial
changes (Mangham et al., 1981).
When female rats were fed 5000 ppm 14C-DEHP for 49 days,
relative liver weights increased progressively during the first week
of treatment to a value approximately 50% above normal. Only a slight
increase in smooth endoplasmic reticulum was visible by electron
microscopy. When treatment was discontinued, liver weights returned to
normal within one week. Rats fed for 35 days on a 1000 ppm diet did
not experience alterations in liver weight relative to controls
(Daniel & Bratt, 1974).
Groups of 10 male and 10 female Wistar rats, 90-120 g, were
administered daily gavage doses of DEHP at levels of 0, 3400 ppm and
34,000 ppm for 90 days. At autopsy, gross pathological observations
were made: liver, kidney and spleen were weighed and examined
microscopically. High-dose rats displayed uncertainty of movements,
drowsiness, diarrhea and rapid weight loss, with a 75% mortality.
Gross autopsy revealed congestion of the small intestine and loss of
mucosa in the stomach and of some parts of the intestine. A
treatment-related increase in mean liver weight of low-dose rats was
observed, but no gross or microscopic alterations were seen in the
liver, kidneys or spleen (Nikonorow et al., 1973).
Groups of 15 male and 15 female rats were given diets containing
0 (control), 0.2, 1.0 or 2.0% di(2-ethylhexyl)-phthalate (DEHP) for 17
weeks. At the two higher treatment levels there was a reduced rate of
body-weight gain and food intake. A paired-feeding study showed that
the reduced food intake did not account fully for the reduced growth
rate. Hematologic studies showed decreased hematocrits in both sexes
in the 1 and 2% DEHP group, as well as lower hemoglobin levels in
males at these dose levels. Renal concentrating and diluting ability
was reduced in the females receiving 2% DEHP. The relative testes
weight of rats on the (2%) diet was markedly decreased and
histopathological examination revealed severe seminiferous tubular
atrophy and cessation of spermatogenesis. Although testes weight of
the rats fed 0.2% DEHP was not reduced, histological studies showed
evidence of decreased spermatogenesis. There were no other
histopathological changes attributable to DEHP treatment (Gray et al.,
1977).
Groups of 22-24 male and female "hybrid" guinea pigs, 49 days
old, were fed diets containing 0, 0.04 or 0.13% DEHP for 1 year.
Histopathological examination was performed on kidney, liver, lung,
spleen and testes; only liver and kidney were weighed. No treatment
effects were reported on mortality, growth, food consumption,
microscopic pathology or incidence of neoplasms. Treated females had
significantly increased relative liver weights (Carpenter et al.,
1953).
Groups of 6-7 male albino ferrets, 18 months old, were fed diets
containing 0 or 1% DEHP for 14 months. Treated animals experienced
significant growth depression and liver enlargement, with altered
liver morphology. Microscopic examination of sections of brain, heart,
adrenals, thyroid, trachea, esophagus, lung, kidney, and bladder did
not reveal any treatment effect. Mean testicular weights of treated
animals were reduced and 3/7 test animals exhibited a nearly complete
absence of germinal epithelium in testes. Significant treatment-
related effects were seen on activities of marked enzymes for
microsomes, and in drug metabolizing enzymes. Significant changes were
seen in mitochondrial marker enzymes of treated animals (Lake et al.,
1976).
8 dogs (4 Cocker Spaniels, 4 Wire-haired Terriers), 14-17 months
old, were randomly distributed by sex and breed into 2 groups (one
control, one administered DEHP by capsule). The treatment group
received 30 mg DEHP/kg b.w./day, 5 days a week, for 19 doses; then
60 mg/kg b.w./day for 240 additional doses. There was no effect on
growth. At autopsy liver and kidney weight were within normal range.
Gross and histopathological examination of lung, heart, liver,
stomach, small intestine, colon, cecum, spleen, adrenal, gonad,
bladder and thyroid gland of treated animals showed no treatment
related effects (Carpenter et al., 1953).
Long-term studies
Groups of 32 and female Sherman rats, 60 days old, were fed diets
containing 0, 0.04, 0.13 and 0.4% DEHP for up to 2 years (P1). At week
12, animals from the control and high dose levels were bred and their
offspring used to establish an F1 generation composed of 32 males and
32 females. After one year of treatment, groups of 9 to 17 rats from
each sode level (P1 generation) were sacrificed for "tissue and organ
weight" data. Remaining rats were culled to 8 males and 8 females per
group and survivors sacrificed after 2 years of treatment. The F1
generation was placed on test at 15 days of age and sacrificed after
1 year. Histopathological examination was performed on adrenal, heart,
kidney, intestine, liver, lung, spleen, testes, ovary and gross
lesions; only kidney and liver were weighed. At the 0.04% level of
DEHP, both P1 and F1 males exhibited growth depression and an
increased mean relative weight of liver and kidney. F1 females also
showed increased liver and kidney weights at this dose level. No
treatment related effects were reported on survival, food consumption,
microscopic pathology, incidence of neoplasms, hematology or fertility
(Carpenter et al., 1953).
OBSERVATIONS IN MAN
No information available.
Comments
Orally ingested DEHP is probably absorbed as MEHP, since DEHP is
rapidly hydrolyzed to MEHP and 2-ethylhexanol by extracts from the
intestine from several animal species and man. In addition, studies
with isolated perfused rat intestine showed that all DEHP was
converted to MEHP before it reached the serosal perfusing solution.
DEHP metabolites are rapidly distributed throughout the body and reach
a steady state level in the tissues after a period of exposure of up
to 2 weeks, with some accumulation in liver and fat. Tissue residues
are rapidly depleted when DEHP is removed from the diet. There is no
definitive information on the chemical nature of the tissue residues,
since the available distribution studies reflect only a distribution
of 14C, derived from 14C labeled DEHP. In experimental animals the
absorbed material is excreted almost equally in the urine and feces,
with very little being expired as CO2. Biliary excretion ranges from
9-11%. The urine contains up to 5 major metabolites, MEHP, and
4 oxidation products (beta-oxidation) of the MEHP. Only small amounts
appear as o-phthalic acid. In species other than rat (including monkey
and man), the metabolites are excreted mainly as glucuronides. Species
differences were also noted in the distribution of other urinary
metabolites. MEHP appeared in only trace amounts in rat urine, as
did unchanged DEHP in the urine of all species. In monkey and man,
MEHP is one of the major urinary metabolites. In rats, 6.5-10.5% of
administered DEHP appeared as fecal metabolites; most had been
excreted by 48 hr after dosing. Rats fed low amounts (10 ppm) of DEHP
had no detectable residues in feces. The secondary metabolites in
feces have not been identified.
DEHP and MEHP cause an increased liver weight with marked
proliferation of mitochondria and peroxisomes in the liver. The
peroxisomal enzymes that are increased the most are those associated
with beta-oxidation systems. There is less induction of microsomal
enzymes. These effects are reversible when DEHP is removed from the
diet.
DEHP was teratogenic in mice, but not in rats and rabbits. DEHP
at high dietary levels caused severe seminiferous tubular degeneration
testicular atrophy and cessation of spermatogenesis in rats and mice.
Similar effects were observed with MEHP. Reduced zinc levels in serum,
liver and testes were also reported. Other short-term effects of
dietary DEHP include liver enlargement and effects on the heart and
adrenals. DEHP and 2-ethylhexanol were not mutagenic in the Ames test
and a number of mammalian cell systems. In contrast, MEHP was
mutagenic in a number of systems. In lifetime feeding studies in rats
and mice, DEHP caused a significant increase in liver tumors in both
species.
EVALUATION
DEHP is a hepatocarcinogen in both rats and mice.
Provisional acceptance
The level of DEHP in the food contact material and the extent of
its migration into food should be kept at the lowest levels which are
technologically possible.
REFERENCES
ABE, S. & SASAKI, M. (1977) Chromosome aberrations and sister
chromatid exchanges in Chinese hamster cells exposed to various
chemicals. J. National Cancer Inst., 58: 1635-1641.
ALBRO, P.W. (1975) The metabolism of 2-ethylhexanol in rats.
Xenobiotica, 5: 625-636.
ALBRO, P.W. & THOMAS, R. (1973) Enzymatic hydrolysis of
di(2-ethylhexyl)phthalate by lipase. Biochem. Biophys. Acta, 306:
380-390.
ALBRO, P.W., THOMAS, R., & FISHBEIN, L. (1973) Metabolism of
diethylhexyl phthalate by rats. Isolation and characterization of the
urinary metabolites. J. Chromatogr., 76: 321-330.
ALBRO, P.W., HASS, J.R., PECK, C.C., ODOM, D.G., CORBETT, J.T.,
BAILEY, F.J., BLATT, H.F., & BARRETT, B.B. (1981) Identification
of the metabolites of di-2-ethylhexyl phthalate in urine from the
African Green monkey. Drug Metab. Disp., 9: 233-235.
ALBRO, P.W., JORDAN, S.T., SCHROEDER, J.L., & CORBETT, J.T. (1982a)
Chromatographic separation and quantitative determination of the
metabolites of di-(2-ethylhexyl)phthalate from urine of laboratory
animals. J. Chromatogr., 244(1): 65-79.
ALBRO, P.W., CORBETT, J.T., SCHROEDER, J.L., JORDAN, S., & MATTHEWS,
H.B. (1982b) Pharmacokinetics, interactions with macromolecules and
species differences in metabolism of DEHP. Environ. Health
Perspect., 45: 19-25.
ALBRO, P.W., TONDEUR, I., MARBURY, D., JORDAN, S., SCHROEDER, J. &
CORBETT, J.T. (1983) Polar metabolites of di-(2-ethylhexyl)phthalate
in the rat. Biochim. Biophys. Acta, 760: 273-92.
ANONYMOUS (1982a) Mutagenicity evaluation of di-2-ethylhexyl phthalate
in the Ames Salmonella/microsome plate test. Unpublished report from
Chemical Manufacturers of America, submitted to the World Health
Organization by the United States Food and Drug Administration.
ANONYMOUS (1982b) Mutagenicity evaluation of di-2-ethylhexyl phthalate
in the mouse lymphoma forward mutation assay. Ibid.
ANONYMOUS (1982c) Evaluation of di-2-ethylhexytl phthalate in the
in vitro transformation of Balb/3T3 cells assay. Ibid.
ANONYMOUS (1982d) Evaluation of di2-ethylhexyl phthalate in the
in vitro transformation of Balb/3T3 cells with metabolic activation
by primary rat hepatocytes. Ibid.
ANONYMOUS (1982e) Mutagenicity evaluation of di-2-ethylhexyl phthalate
in the mouse micronucleus test. Ibid.
ANONYMOUS (1982f) Evaluation of di-2-ethylhexyl phthalate in the
primary rat hepatocyte unscheduled DNA synthesis assay. Ibid.
ANONYMOUS (1982g) Mutagenicity evaluation of mono-2-ethylhexyl
phthalate (MEHP) in the Ames Salmonella/microsome plate test. Ibid.
ANONYMOUS (1982h) Mutagenicity evaluation of mono-2-ethylhexyl
phthalate (MEHP) in the mouse micronucleus test. Ibid.
ANONYMOUS (1982i) Evaluation of mono-2-ethylhexyl phthalate (MEHP) in
the in vitro transformation of Balb/3T3 cells assay. Ibid.
ANONYMOUS (1982j) Evaluation of mono-2-ethylhexyl phthalate (MEHP) in
the in vitro transformation of Balb/3T3 cells with metabolic
activation by primary rat hepatocytes. Ibid.
ANONYMOUS (1982k) Mutagenicity evaluation of 2-ethylhexanol (2-EH) in
the Ames Salmonella/microsome plate test. Ibid.
ANONYMOUS (1982l) Mutagenicity evaluation of 2-ethylhexanol (2-EH) in
the mouse micronucleus test. Ibid.
ANONYMOUS (1982m) Evaluation of 2-ethylhexanol in the in vitro
transformation of Balb/3T3 cells assay. Ibid.
CARPENTER, C.P., WELL, C.S., & SMYTH, H.F. Jr. (1953) Chronic oral
toxicity of di-&2-ethylhexyl)phthalate for rats, guinea pigs, and
dogs. Arch. Inc. Hyg. Occup. Med., 8: 219-226.
CARTER, J.E., ROLL, D.B., & PETERSON, R.V. (1974) The in vitro
hydrolysis of di-(2-ethylhexyl)phthalate by rat tissues. Drug Metab.
Disposition, 2: 341-344.
CHU, I., VILLENEUVE, D.C., SECOURS, V., FRANKLIN, C., ROCK, G., &
VIAU, A. (1978) Metabolism and tissue distribution of
mono-2-ethylhexyl phthalate in the rat. Drug Metab. Dispos., 6:
146-9.
CHU, I., SECOURS, V.E., MARINO, I.A., VILLENEUVE, D.C., & VALLI, V.E.
(1981) Sub-acute and sub-chronic toxicity of mono-2-ethylhexyl
phthalate in the rat. Arch. Environ. Contam. Toxicology, 10(3):
271-80.
DANIEL, J.W. & BRATT, H. (1974) The absorption, metabolism and tissue
distribution of di-2-ethylhexyl)phthalate in rats. Toxicology, 2:
51-65.
GANNING, A.E. & DALLNER, G. (1981) Induction of peroxisomes and
mitochondria by di-(2-ethylhexyl) phthalate. FEBS Lett., 130:
77-9.
GANNING, A.E., KLASSON, E., BERGMAN, A., BRUNK, U., & DALLNER, G.
(1982) Effect of phthalate ester metabolites on rat liver. Acta
Chem. Scnad., B36: 563-5.
GANNING, A.E., BRUNK, U., & DALLNER, G. (1983) Effects of dietary
di-(2-ethylhexyl) phthalate on the structure and function of rat
hepatocytes. Biochim. Biophys. Acta, 763(1): 72-82.
GAUNT, I.F. & BUTTERWORTH, K.R. (1982) Autoradiographic study of
orally administered di(2-ethylhexyl)phthalate in the mouse. Food
Chem. Toxicol., 20(2): 215-7.
GRAY, T.J. & BUTTERWORTH,. K.R. (1980) Testicular atrophy produced by
phthlate esters. Arch. Toxicol. (Suppl.), 4: 452-5.
GRAY, T.J., BUTTERWORTH, K.R., GAUNT, I.F., GRASSO, P., & GANGOLLI,
S.D. (1977) Short-term toxicity study of di-(2-ethylhexyl) phthalate
in rats. Food Cosmet. Toxicol., 15: 389-399.
GRAY, T.J., BEAMAND, J.A., LAKE, B.G., FOSTER, J.R., & GANGOLLI, S.D.
(1982) Peroxisome proliferation in cultured rat hepatocytes produced
by clofibrate and phthalate ester metabolites. Toxicol. Lett.,
10(2): 273-9.
HODGSON, J.R. (1982) Evaluation of di-(2-ethylhexyl) phthalate and
its major metabolites in the primary rat hepatocyte unscheduled DNA
synthesis assay. Environ. Mutagen., 4:388 (Abstract).
IKEDA, G.J., SAPIENZA, P.P., COUVILLION, J.L. FARBER, R.M., & VAN
LOON, E.J. (1980) Comparative distribution, excretion and metabolism
of di-(2-ethylhexyl) phthalate in rats, dogs and miniature pigs.
Food Cosmet. Toxicol., 18(6): 637-642.
ISHIDATE, M.Jr. & ODASHIMA, S. (1977) Chromosome tests with 134
compounds on Chinese hamster cells in vitro - A screening for
chemical carcinogens. Mutat. Res., 48: 337-354.
KANESHIMA, H., YAMAGUCHI, T., OKUI, T., & NAITOH, M. (1978) Studies on
the effects of phthalate esters on the biological system. Part. 2.
In vitro metabolism and biliary excretion of phthalate esters in
rats. Bull. Environ. Contam. Toxicol., 19: 502-9.
KIRBY, P.E., PIZZARELLO, R.F., LAWLOR, R.E., HAWORTH, S.R., HODGSON,
J.R. (1983) Evaluation of di-(2-ethylhexyl) phthalate and its major
metabolites in the Ames test and L5178Y mouse lymphoma mutagenicity
assay. Environ. Mutagen., 5(5): 657-63.
KOZUMBO, W.J., KROLL, R., & RUBIN, R.J. (1982) Assessment of the
mutagenicity of phthalate esters. Environ. Health Perspect, 45:
103-109.
KLUWE, W.M., HASEMAN, J.K., DOUGLAS, J.F., & HUFF, J.E. (1982) The
carcinogenicty of dietary di-(2-ethylhexyl) phthalate (DEHP) in Fisher
344 rats and B6C3F1 mice. J. Toxicol. Environ. Health, 10(4):
797-815.
KRAUSKOPF, L.F. (1973) Studies on the toxicity of phthalates via
ingestion. Environ. Health Perspect., 3: 61-72.
LAKE, B.G., BRANTOM, P.G., GANGOLLI, S.D., BUTTERWORTH, K.R., &
GRASSO, P. (1976) Studies on the effects of orally administered
di-(2-ethylhexyl) phthalate in the ferret. Toxicology, 6 341-356.
LINDGREN, A., LINDQUIST, N.G., LYD'EN, A., OLSSON, T., & ULLBERG, S.
(1982) A whole body autoradiographic study on the distribution of
14C-labelled di-(2-ethylhexyl)phthalate in mice. Toxicology, 23:
149-58.
MANGHAM, B.A., FOSTER, S.R., & LAKE, B.G. (1981) Comparison of
the hepatic and testicular effects of orally administered
di-(2-ethylhexyl)phthalate and dialkyl 79-phthalate in the rat.
Toxicol. Appl. Pharmacol., 61: 205-214.
MIYAZAWA, S., FURUTA, S., OSUMI, T., & HASHIMOTO, T. (1980) Turnover
of enzymes of peroxisomal beta-oxidation in rat liver. Biochim.
Biophys. Acta, 630(3): 367-74.
MOODY, D.E. & REDDY, J.K. (1978) Hepatic peroxisomal (microbody)
proliferation in rats fed plasticizers and related compounds.
Toxicol. Appl. Pharmacol., 45(2): 497-504.
NAKAMURA, Y., YAGI, Y., TOMITA, I., & TSUCHIKAWA, K. (1979)
Teratogenicity of di-(2-ethylhexyl)phthalate in mice. Toxicol.
Lett., 4: 113-117.
NIKORONOW, M., MAZUR, H., & PIEKACZ, H. (1973) Effect of orally
administered plasticizers and polyvinyl chloride stabilizers in the
rat. Toxicol. Appl. Pharmacol., 25: 253-259.
OHNO, S., OHTAKE, N., FUJII, Y., YAMABAYASHI, S., USUDA, N., & NAGATA,
T. (1981) Histochemical studies on peroxisomes of rat livers during
DEHP administration and after withdrawal on thick sections by means of
light microscopy and high voltage electron microscopy. Acta
Histochem. Cytochem., 14: 126-42.
OISHI, S. & HIRAGA, K. (1980a) Effect of phthalic acid esters on mouse
testes. Toxicol. Lett., 5: 413-416.
OISHI, S. & HIRAGA, K. (1980b) Effects of phthalate acid monoesters on
mouse testes. Toxicol. Lett., 6: 239-242.
OISHI, S. & HIRAGA, K. (1980c) Testicular atrophy induced by phthalic
acid esters: Effect on testosterone and zinc concentrations.
Toxicol. Appl. Pharmacol., 53: 35-41.
OISHI, S. & HIRAGA, K. (1980d) Testicular atrophy induced by phthalic
acid monoesters: Effects of zinc and testosterone concentrations.
Toxicology, 15: 197-202.
OISHI, S. & HIRAGA, K. (1982) Distribution and elimination of di-2-
ethylhexyl phthalate (DEHP) and mono-2-ethylhexyl phthalate (MEHP)
after a single oral administration of DEHP in rats. Arch. Toxicol.,
51: 149-55.
PATTY, F.A. (1967) Industrial Hygiene and Toxicology, 2nd Rev. Vol. 2.
pp. 1906-1910. Interscience, New York.
PECK, C.C., ALBRO, P.W., HASS, J.R., ODOM, D.G., BARRETT, B.B., &
BAILEY, F.J. (1978) Metabolism and excretion of the plasticizer
di-(2-ethylhexyl)phthalate in man. Clin. Res., 26: 101A
(Abstract).
PHILLIPS, B,J., JAMES, T.E., & GANGOLLI, S.D. (1982) Genotoxicity
studies of di-(2-ethylhexyl) phthalate and its metabolites in CHO
cells. Mutat. Res., 102(3): 297-304.
PUTMAN, D.L., MOORE, W.A., SCHECHTMAN, L.M., & HODGSON, J.R. (1983)
Cytogenetic evaluation of di-(2-ethylhexyl) phthalate and its major
metabolites in Fischer 344 rats. Environ. Mutagen., 5(2): 227-31.
ROWLAND, I.R., COTTRELL, R.C., & PHILLIPS, J.C. (1977) Hydrolysis of
phthalate esters by the gastrointestinal contents of the rat. Food
Cosmet. Toxicol., 15: 17-21.
RUDDICK, J.A., VILLENEUVE, D.C., CHU, I., NESTMANN, E., & MILLES, D.
(1981) An assessment of the teratogenicity in the rat and mutagenicity
in Salmonella of mono-2-ethylhexyl phthalate. Bull. Environ. Contam.
Toxicol., 27: 181-6.
RUSHBROOK, C.J. (1982) Dominant lethal study of di-(2-ethylhexyl)
phthalate and its major metabolites in ICR/SIM mice. Environ.
Mutagen., 4: 387 (Abstract).
SAKURAI, T., MIYAZAWA, S., & HASHIMOTO, T. (1978) Effects of
di-(2-ethylhexyl) phthalate administration of carbohydrate and fatty
acid metabolism in rat liver. J. Biochem., 83: 313-320.
SCHAFFER, C.B., CARPENTER, C.B., & SMITH, H.F. (1945) Acute and
subacute toxicity of DEHP with note upon its metabolites. J. Ind.
Hyg. Toxicol., 27: 130-135.
SCHULTZ, C.O. & RUBIN, R.J. (1973) Distribution, metabolism and
excretion of di-(2-ethylhexyl) phthalate in the rat. Environ. Health
Perspect., 3: 123-129.
SEED, J.L. (1982) Mutagenic activity of phthalate esters in bacterial
liquid suspension assays. Environ. Health Perspect., 45: 111-114.
SHIOTA, K., CHOU, M.J., & NISHIMURA, H. (1980) Embryotoxic effects of
di-(2-ethylhexyl) phthalate (DEHP) and di-n-butyl phthalate (DBP) in
mice. Environ. Res., 22: 245-253.
SIMMON, V.F., JAUHANEN, K., & TARDIFF, R.G. (1977) Mutagenic activity
of chemicals identified in drinking water. Dev. Toxicol. Environ.
Sci., 2: 249-258.
STENCHEVER, M.A., ALLEN, M.A., JEROMINSKI, L., & PETERSEN, R.V. (1976)
Effects of bi-(2-ethylhexyl) phthalate on chromosomes of human
leukocytes and human fetal lung cells. J. Pharm. Sci., 65:
1648-1651.
THOMAS, J.A., SCHEIN, L.G., GUPTA, P.K., McCAFFERTY, R.E., FELICE,
P.R., & DONOVAN, M.P. (1979) Failure of monoethylhexyl phthalate to
cause teratogenic effects in offspring of rabbits. Toxicol. Appl.
Pharmacol. 51: 523-528.
THOMAS, J.A., MARTIS, L., GIOVANETTO, S., McCAFFERTY, R.E., & DONOVAN,
M.P. (1980) Biodistribution of monoethylhexyl phthalate (MEHP) in
pregnant rabbits and neonatal rats. Fed. Proc., 39: 525
(Abstract).
TOMITA, I., NAKAMURA, Y., AOKI, N., & INUI, N. (1982)
Mutagenic/carcinogenic potential of DEHP and MEHP. Environ. Health
Perspect., 45: 119-25.
TURNER, J.H., PETRICCIANI, J.C., CROUCH, M.L., & WENGER, S. (1974) An
evaluation of the effects of diethylhexyl phthalate (DEHP) on
mitotically capable cells in blood packs. Transfusion, 14: 560-6.
WARD, J.M., RICE, J.M., CREASIA, D., LYNCH, P., & RIGGS, C. (1983)
Dissimilar patterns of promotion by di-(2-ethylhexyl) phthalate
and phenobarbital of hepatocellular neoplasia initiated by
diethylnitrosamine in B6C3F1 mice. Carcinogenesis, 4(8): 1021-9.
WARREN, J.R., LALWANI, N.D., & REDDY, J.K. (1982) Phthalate esters as
peroxisome proliferator carcinogens. Environ. Health Perspect.,
45: 35-40.
WHITE, R.D., CARTER, D.E., EARNEST, D., & MUELLER, J. (1980)
Absorption and metabolism of three phthalate diesters by the rat
small intestine. Food Cosmet. Toxicol., 18: 383-6.
WILLIAMS, D.T. & BLANCHFIELD, B.J. (1974a) Retention, excretion and
metabolism of di-(2-ethylhexyl) phthalate administered orally to the
rat. Bull. Environ. Contam. Toxicol., 11: 371-378.
WILLIAMS, D.T. & BLANCHFIELD, B.J. (1974b) Retention, excretion and
metabolism of phthalic acid administered orally to the rat. Ibid.,
12: 109-112.
YAGI, Y., NAKAMURA, Y., TOMITA, I., TSUCHIKAWA, K., & SHIMOI, N.
(1980) Teratogenic potential of di- and mono-(2-ethylhexyl)
phthalate in mice. J. Environ. Pathol. Toxicol., 4: 533-544.
YANAGITA, T., KOBAYASHI, K., & ENOMOTO, N. (1978) Accumulation of
hepatic phospholipids in rats fed di-(2-ethylhexyl) phthalate.
Biochem. Pharmacol., 27: 2283-2288.
YOSHIKAWA, K., TANAKA, A., YAMAHA, T., & KURATA, H. (1983)
Mutagenicity study of nine monoalkyl phthalates and a dialkyl
phthalate using Salmonella typhimurium and Escherichia coli. Food
Chem. Toxicol., Apr; 21(2): 221-3.
ZEIGER, E., HAWORTH, S., SPECK, W., & MORTALMANS, K. (1982) Phthalate
esters testing in the National Toxicology Programs Environmental
Mutagenesis Test Development Program. Environ. Health Perspect.
BIOLOGICAL DATA
BIOCHEMICAL ASPECTS
Metabolism
DEHP was hydrolyzed to the mono-ester by preparations derived
from the intestine and liver from several animal species (rat, baboon,
ferret) and from human intestine (Lake et al., 1977; Rowland et al.,
1977; Kaneshima et al., 1978). Homogenates of rat kidney, lung and
liver, as well as liver microsomes and mitochrondria, hydrolyse DEHP
to MEHP and 2-ethylhexanol. Hydrolysis was more rapid with liver
preparations than lung or kidney preparations. Liver microsomal
preparations were more active than mitochondrial preparations, whereas
the cytosol fraction was inactive (Carter et al., 1974). DEHP was
rapidly hydrolysed by pancreatic lipase, and at a slower rate by rat
liver homogenate, plasma, and preparations from kidney and lung. No
o-phthalic acid could be detected in the reaction products (Daniel &
Bratt, 1974; Albro & Thomas, 1973).
The in vitro absorption of mono- and DEHP was studied using
isolated perfused rat intestins. All DEHP was converted to mono
(ethylhexyl) phthalate (MEHP) before it reached the serosal perfusing
solution. No metabolic transformation of MEHP was reported (White et
al., 1980).
Adult male CD rats were given two oral doses of 14C-DEHP at
24 hour intervals and 24 hour urine samples collected. Five major
metabolites were present after the initial dose, the initial metabolic
product, mono (2-ethylhexyl)phthalate being metabolized to 5-keto-2-
(ethylhexyl)phthalate and 2-carboxyl-2-(ethylpentyl)phthalate,
5-hydroxy-2-(ethylhexyl)-phthalate and 2-carboxymethyl-butylphthalate.
Less than 5% of the administered compound was hydrolyzed completely to
o-phthalic acid. Various tests for conjugation were negative (Albro
et al., 1973).
Male, 4-month old, CD rats, CD-1 mice, Syrian golden hamsters and
Hartley albino guinea pigs were administered by gavage 2 doses of DEHP
at 24-hour intervals, at levels ranging from 20-360 mg/kg b.w. Urine
was collected for 48 hours after the initial dose. No metabolites were
detected in urine from hamsters or guinea pigs that were not also
present in rat or mouse urine. In the mouse, guinea pig and hamster, a
significant proportion of DEHP metabolites were present as glucuronide
conjugates. Conjugates of taurine and sulfate were not detected.
Metabolites of DEHP in rats were not conjugated. Omega and omega-1
oxidation of the ethylhexyl moiety was less efficient in guinea pig
than in the other species, but guinea pigs appeared to produce
significantly more MEHP (Albro et al., 1982a,b). The enzymatic
processes normally associated with omega, omega-l, alpha- and
beta-oxidation of fatty acids could account for the major urinary
metabolites of DEHP in the rat. However, the structures of recently
identified minor metabolites of DEHP suggest that oxidation of
the aliphatic side chain can occur at positions more distant than
omega-1 from the methyl terminus. This type of oxidation at multiple
sites in an aliphatic side chain occurs commonly when alphatic
hydrocarbons, rather than fatty acids, are metabolized by microsomal
oxidases (Albro et al., 1983).
In separate experiments, single gavage doses of 14C-DEHP
were given to Fisher 344 rats, CD mice, Syrian golden hamsters and
Hartley albino guinea pigs and the urine analyzed for 11 different
metabolites. Species differences were noted in the distribution of
urinary metabolites. In the rat, 3 metabolites, not including MEHP
(trace), comprised 81% of all urinary metabolites of DEHP, while in
mice, 75% of all urinary metabolites were accounted for by 5
compounds, including MEHP (18.6%). In the guinea pig, MEHP accounted
for 71.2% of all urinary metabolites, while in the hamster, 92.4%
of administered radioactivity was distributed among 5 urinary
metabolites, not including MEHP (4.5%). Unreacted DEHP occurred at
levels of 0.5% or less in all species. 100% of urinary metabolites
were not-conjugated in rats, while 85% were non-conjugated in
hamsters. Mice and guinea pigs excreted 35-36% of urinary metabolites
in the non-conjugated form (Albro et al., 1982b). In other studies,
dogs excreted 3 major urinary metabolites, and pigs excreted 5 (Ikeda
et al., 1980).
In a study with African Green monkeys infused with palsma
containing about 5 mg of 14C-DEHP, more than 50% of the 14C was
excreted in the urine by 4 hr, and more than 70% by 24 hr. The major
metabolites were the 5-ethyl, isohexanol monoester of phthalic acid
and MEHP. More than 80% of the urinary metabolites were conjugated to
gluconide (Albro et al., 1981).
Two cancer patients received, by platelet concentration infusion,
94.7 mg DEPH in 4 hr and 174.3 mg DEHP in 1.5 hr. More than 50% of the
infused dose appeared as DEHP metabolites in the urine within 6 hr.
The predominant metabolite was the 5-ethyl, isohexanol monoester of
phthalic acid. Approximately 80% of urinary metabolites were
glucuronide conjugates. Eight other metabolites, including MEHP, were
identified. The half-life of DEHP in plasma was 30 ± 12 min.,
apparent distribution volume was 2819 ± 83 ml/m2 and clearance was 78
± 20 ml/min-m2 (Peck et al., 1978; Albro et al., 1981).
Absorption, distribution and excretion
Rats were given by gavage single doses of 14C-DEHP
ranging from 2.6 to 1900 mg/kg b.w. 35-55% of the labeled dose was
excreted in the feces and 42-57% in the urine. At low dose levels
(2.6-2.9 mg/kg b.w.), 9-14% of the dose appeared in the bile. No
significant amounts of 14C-C02 could be found in the expired air. At
high dose levels (1000 mg/kg2 b.w.) during an 8 day period, fecal
excretion accounted for 6.5-10.5% of the administered dose of 14C as
metabolites and 24.0-38.8% as unchanged DEHP. Most fecal metabolites
had been excreted by 48 hours after dosing. 53.5-70.2% of the dose was
excreted as MEHP or metabolites in the urine. Some 14C remained in
fatty tissue 8 days after dosing, but none was found in 10 other
tissues. In liver, kidney and skeletal muscle, radioactivity has
essentially disappeared between 48 and 96 hours after dosing (Daniel &
Bratt, 1974; Schulz & Rubin, 1973; Williams & Blanchfield, 1974a).
Beagle dogs, miniature Hormel swine and male Sprague-Dawley rats
were fed DEHP (650, 750 and 750 ppm, respectively) for 21-28 days and
then given an oral dose of 14C-DEHP. In rats, 85% of the administered
radioactivity was excreted in the urine and feces 24 hours after
dosing excretion of metabolites predominated in dogs and urinary
excretion predominated in pigs, while in rats, metabolites were
distributed equally between urine and feces. Elimination of
administered 14C-DEHP was 84% complete in rats by 24 hours after
dosing; elimination of metabolites in dogs and pigs was slower
(50% after 24 hr), but was complete in all species by 4 days after
dosing. Serum half-life for 14C-DEHP and metabolites was 1.2 hr in
dogs and 5.4 hr in pigs. Analysis of tissue and excreta 4 days after
dosing with 14C-DEHP indicates that DEHP was metabolized 96% in rats,
63% in dogs and 71% in pigs (Ikeda et al., 1980).
Male mice were given single oral doses of Di-(2-ethylhexyl)
(14C)phthalate (6.72 mg/animal) and examined by whole-body
autoradiography at intervals from 1 hr to 7 days after treatment. Most
of the radioactivity was excreted in the urine and feces in the first
24 hr. Excretion was complete within 3-5 days. Following absorption,
14C was widely distributed in the carcass, but none was found in the
central nervous system, skeleton, or thymus, and only rarely in the
testes. There was no evidence of tissue storage (Gaunt & Butterworth,
1982).
Groups of 3 male Fischer 344 rats were given daily gavage doses
of 1.8, 18 or 180 mg/kg b.w. 14C-DEHP, and sacrificed 1, 3, 10 and
12 days after initiation of treatment. Residual 14C in testes and
liver, as percent of administered dose, decreased with duration of
treatment and magnitude of dose, except for testes at day 12 (all dose
levels) in which 14C levels were higher than at day 10. In
experiments with a single dose (1.8-1000 mg/kg b.w.) of DEHP, no
accumulation occurred in the liver at doses below 400g/kg b.w. (Albro
et al., 1982b).
Wistar rats, 200 g, were administered a single dose of DEHP or
MEHP (10,000 mg/kg b.w.) by gastric intubation and samples of blood
and tissue collected 1, 3, 6, 24, 48 and 96 hr post-intubation. Levels
of DEHP in liver declined with a half life of 1 day, while levels in
epididymal fat declined more slowly (t1/2 = 6.5 days), t1/2 values for
DEHP in other tissues were less than that observed in liver. MEHP
levels declined much more slowly (t1/2 = 50 hr) than DEHP (8.3 hr) in
testes, but more rapidly (t1/2 = 67.6 hr for MEHP vs. 156 hr for DEHP)
in epididymal fat. In other tissues, levels of the two phthalates
showed similar rates of decline. In heart and lungs, the highest
levels of DEHP and MEHP occurred within 1 hr of dosing, while, in
other tissues, the highest levels occurred 6-24 hours after dosing
(Oishi & Hiraga, 1982).
Groups of 24 female rats were fed diets containing either 0.1% or
0.5% ppm 14C-DEHP for 35 and 49 days, respectively. In rats fed 0.1%,
DEHP 14C residues reached steady state levels of 40-50 ppm in liver
in 3 days, while steady state levels in fat (7-9 ppm), were attained
by 2 weeks. 14C levels in brain never exceeded 1 ppm, but in the
heart, levels ranged from 1 to 6 ppm. At the high dose level,
equilibrium levels of 14C were attained in the rats after 9-14 days
of feeding. Steady state levels of 14C in the rat tissues were 120
ppm in liver, 80 ppm in fat, 2-3 ppm in brain and 15-20 ppm in heart.
After removal of rats from the test diet, 14C declined with a half
life of 1-2 days in liver, and 3-5 days in fat (Daniel & Bratt, 1974).
4 male C57BL mice, 20 g, were given oral doses of 14C-DEHP
(2-ethylhexyl -1-14C, 9.6 mg/kg b.w.; and carbonyl-14C, 3.6 mg/kg
b.w.) and sacrificed 5, 14 or 30 days later. Whole body
autoradiography showed that in the males, either labeled form of DEHP
was retained at low levels in bone. The (carbonyl -14C) DEHP but not
the other labeled form, displayed marked retention in the skin,
cartilage and tendons. In a study with pregnant mice, administration
of the 14C labeled compounds at day 8 or 16 of gestation resulted in
14C-DEHP being concentrated mainly in the yolk sac. DEHP (carbonyl-
14C was present in the neuroepithelium of the embryo. Dosing at day
18 of gestation resulted in a more general distribution of the 14C
labeled compounds in the fetuses, 4 hr post-dosing, but 24 hr post-
dosing the 14C was primarily located in the renal pelvis, urinary
bladder, intestines and skeleton of the fetus (Lindgren et al., 1982).
Two adult men ingested single doses of 10 g and 5 g, respectively
of DEHP. Phthalic acid equivalent to approximately 4.5% of the
administered dose was recovered from the urine in the 24 hr period
after dosing, the major portion being excreted 5 to 7 hours after
dosing (Schaffer et al., 1945).
Effects on enzymes and other biochemical parameters
When rats were fed DEHP (0, 1, 2 or 4%) for 4 weeks, liver
glycogen decreased markedly, while hepatic fatty acid synthesis
increased 2-fold. Treatment-related alterations in levels of
intermediates in carbohydrate metabolism indicate that gluconeogenesis
was inhibited at a point between 3-phosphoglycerate and fructose
1,6-diphosphate (Sakurai et al., 1978).
When male Wistar rats (80-95 gm) were fed 0.5% DEHP for 10 days,
livers of treated animals were enlarged, and showed reduced levels of
glucogen and triglycerides and increased levels of phospholipids.
These treatment-related changes were suggestive of metabolic
transformation from glycolysis to lipolysis as an energy source
(Yanagita et al., 1978).
Male Fisher-344 rats were fed diets containing either 100 ppm or
1.0% DEHP for 11 days. On days 1, 6 or 10, rats were given a tracer
dose of 14C DEHP and sacrificed 10 or 1 day later, respectively.
Purified protein, RNA and DNA fractions were obtained from the liver
and assayed for bound radioactivity. Binding was detectable only when
the ethylhexyl moiety of DEHP was labeled (Albro et al., 1982b).
Male F-344 rats, 150-180 g, fed 2% 2-ethylhexanol for 3 weeks
showed increased liver size, increased activity of hepatic peroxisome-
associated enzymes (catalase and carnitine acetyl transferase) and
hypolipidemia. These DEHP-induced effects are similar to those
produced by hypolipidemic drugs such as clofibrate (Moody & Reddy,
1978).
Special studies on peroxisomal proliferation
Adult male, Sprague-Dawley rats were fed for 2 weeks with diets
containing 2% DEHP or DEHP metabolites (MEHP, phthalic acid,
2-ethylhexanol and 2-ethylhexyl benzoate). At sacrifice, livers were
removed and samples taken for electron microscopy and subcellular
fractionation (mitochondria and microsomes). DEHP and MEHP stimulated
peroxisome enzymes (palmitoyl CoA oxidation, 350-600%), mitochondria
(protein, 238-272%; carnitine-acetyl transferase, 1000-2900%) and
microsomes (cytochrome P-450, 63-68%; NADHP-cytochrome C reductase,
59-73%). Cytochrome C oxidase was stimulated by MEHP and EH (75-91%)
but not by DEHP. 2-elkyl hexanol stimulated mitochondrial protein
synthesis by 75%. Phthalic acid and ethylhexyl benzoate had little or
no effect on marker enzymes. Ultrastructural examination revealed
induction of mitochondrial and peroxisomal proliferation by DEHP and
MEHP, without induction of the endoplasmic reticulum. Other
metabolites had no ultrastructurally visible effect (Ganning et al.,
1982, 1983; Ganning & Dallner, 1981).
Rats (120 g) fed diets containing 2% (w/w) di-(ethylhexyl)-
phthalate (DEHP) for up to 4 weeks, showed a marked increase in
activities of enzymes of peroxisomal beta-oxidation and of catalase
(liver). The time required to reach 50% maximal induction for enzymes
of peroxisomal beta-oxidation was 5-7 days, whereas that for
catalase was 3 days. After withdrawal of DEHP, activities of enzymes
of the beta-oxidation system and of catalase decrease to the control
level with a half-life of 2-3 days. Mitochondrial marker enzymes
(3-hydroxyacyl-CoA dehydrogenase, 3-ketoacyl-CoA thiolase
(non-specific) and acetoacetyl-CoA specific thiolase) also were
markedly increased in treated animals. These activities decreased
after withdrawal of treatment (Miyazawa et al., 1980; Ohno et al.,
1981).
Special studies on mutagenicity
In a number of laboratories, DEHP did not include reverse
mutations of the base pair substitution type in Salmonella
typhimurium with or without metabolic activation (Kirby et al.,
1983; Kozumbo et al., 1982; Ruddick et al., 1981; Simmon et al., 1977;
Warren et al., 1982; Yoshikawa et al., 1983; Zeiger et al., 1982).
Neither was 8-azaguanine resistance in S. typhimurium affected by
DEHP (Seed, 1982). Tomita et al., (1982) reported a weak positive
reaction for DEHP in the S. typhimurium system.
No mutations (tryptophan reversion) occurred in Eschericia coli
exposed to DEHP in vitro (Tomita et al., 1982; Yoshikawa et al.,
1983) or in DNA repair capability of Bacillus subtilis (Tomita et
al., 1982) exposed to DEHP.
DEHP did not show mutagenic effects in a number of mammalian cell
systems: L5178Y mouse lymphoma (forward mutation), Kirby et al.
(1983); rat hepatocyte (unscheduled DNA synthesis), Hodgson, (1982);
human lymphocyte (chromosomal aberrations), Turner et al. (1974);
human fetal lung cells (aneuploidy), Stenchever et al. (1976); Chinese
hamster fibroblast (chromosomal aberrations), Abe & Sazaki (1977),
Ishidate & Odashima (1977); Chinese hamster ovary (sister chromatid
exchange, sister chromatid exchange HGPRT reversion), Phillips et al.
(1982).
DEHP, MEHP and 2-ethylhexanol were tested for mutagenicity in the
following systems: Ames Salmonella/microsome plate test, mouse
lymphoms forward mutation assay, in vitro transformation of Balb/3T3
cells (with and without activation by primary rat hepatocytes), mouse
micronubleus test, and unscheduled DNA synthesis in rat hepatocytes.
No mutagenic activity was observed (Anonymous, 1982a-m).
MEHP was shown to be mutagenic in S. thyphimurium (histidine
reversion), in E. coli (tryptophan reversion) and in B. subtilus
(DNA repair) (Tomita et al., 1982). The same workers also found MEHP
to be mutagenic in Chinese hamster embryo cells (chromosome
aberration, 8-azaguanine resistance, 6-thioguanine resistance, sister
chromatid exchange). Phillips et al. (1982) found MEHP to be
mutagenic to Chinese hamster ovary cells (chromosomal damage, sister
chromatid exchange), but non-mutagenic toward HGPRT reversion. Kirby
et al. (1983) found MEHP to be non-mutagenic in the L5178Y mouse
lymphoma system (forward mutation).
2-ethylhexanol was shown to be weakly mutagenic in
S. typhimurium (8-azaguanine resistance, Seed, 1982), but was
non-mutagenic in L5178Y mouse lymphoma (Kirby et al., 1983), rat
hepatocyte (Hodgson et al., 1982) and Chinese hamster ovary (Phillips
et al., 1982).
Pregnant Syrian golden hamsters were given DEHP orally at 3.75,
7.5 or 15 g/kg b.w. or MEHP at 0.375, 0.750 or 1.5 g/kg b.w. on day 11
of gestation. In embryonic cells removed on day 12 and examined after
15-20 days of culture in vitro, there were significant increases in
aberrant metaphase cells from all treatment groups except the one that
received the lowest dose of DEHP. Aberrations included single
chromatid gaps, isochromatid gaps, single chromatid or isochromatid
breaks and chromatid exchanges. There were also significant increases
in the rate of morphological transformation of the cells at the two
highest doses of DEHP and the two lowest doses of MEHP, and a marginal
increase in 8-azaguanine and 6-thioguanine resistance (Tomita et al.,
1982).
No significant increases in clastogenic changes were observed in
the bone marrow cells of male Fischer 344 rats that had received
0.5-5.0 g DEHP/kg b.w./day, 0.01-0.14 g MEHP/kg b.w./day or
0.02-2.21 g 2-ethylhexanol/kg b.w./day by gavage for 5 days (Putman
et al., 1983). Dominant lethal assays using male mice given DEHP
(2.5-9.9 g/kg), MEHP (50-200 mg/kg) or 2-ethylhexanol (250-1000 mg/kg)
by gavage in corn oil for 5 days produced negative results: fertility
indices and the average numbers of dead and total implants per
pregnancy were within normal ranges in all cases (Rushbrook, 1982).
Special studies on carcinogenicity
Groups of 50 male and female B6C3F1 mice were fed diets
containing 0, 0.3 or 0.6% Di(2-ethylhexyl)phthalate (DEHP) for 103 wk.
Treatment with DEHP did not affect survival or food consumption. Mean
body weight gains of treated female mice (0.3 and 0.6%) were less than
those of the corresponding controls. No other clinical signs of
toxicity were reported. At termination of the studies, chronic
inflammation of the kidney and seminiferous tubular degeneration were
observed in male mice (0.6% diet). No other nonneoplastic lesions were
detected in the treated groups at incidences greater than in the
corresponding controls.
Treatment with DEHP caused liver tumors in both sexes of mice.
Incidence data are as follows: for males, hepatocellular carcinoma
9/50, 14/48 and 19/50, hepatocellular adenomas 6/50, 11/48 and 10/50,
and for females hepatocellular carcinoma 0/50, 7/50 and 17/50 and
hepatocellular adenoma 1/50, 12/50 and 18/50, for the 0, 0.3% and 0.6%
groups, respectively. 20 of the 57 hepatocellular carcinomas
diagnosed in treated mice (sexes and doses combined) had metastisized
to the lung. No pulmonary tumors were observed in any control rats.
The incidences of liver tumors in control mice was comparable to that
in recent historical controls. The incidences of both male and female
mice with hepatocellular carcinomas were significantly greater
(p<0.05) at the 0.6% dose than in controls, and in female mice at the
lower dose (p<0.05) by pairwise comparison, and trend tests detected
significant (p<0.05) dose related effects in both sexes. The
incidence of hepatocellular adenomas in both male and female did not
differ from that of controls. However, the incidences of combined
adenomas and carcinomas was significantly increased for both sexes at
both doses (p<0.05), by pairwise comparisons and showed significant
dose related trends (Kluwe et al., 1982).
Groups of 50 male and female Fischer 344 rats were fed diets
containing 0, 0.6 or 1.2% di(2-ethylhexyl)phthalate (DEHP) for 103
consecutive weeks. Treatment with DEHP did not affect survival rates
or food consumption. Mean body weight gains of treated male rats
(0.6 and 1.2% diet) and female rats (0.2%), were less than those of
the corresponding controls. No clinical signs of toxicity were
reported. At autopsy, seminiferous tubular degeneration and
hypertrophy of cells in the anterior pituitary were observed in
high-dose male rats. Other nonneoplastic lesions were detected in the
treated groups at incidences similar to the corresponding controls.
Incidence data are as follows: for males, hepatocellular
carcinoma 1/50, 1/49 and 5/49, neoplastic nodules 2/50, 5/49 and 7/49,
and for females, hepatocellular carcinoma 0/50, 2/49 and 8/50 and
neoplastic nodules 0/50, 4/49 and 5/50 for the 0, 0.6 and 1.26 groups,
respectively. The incidence of female rats with hepatocellular
carcinomas the 1.2% group was greater than that in control by pairwise
comparison (p<0.01) and there was a significant (p<0.05) dose
related trend effect. The incidence of female rats with neoplastic
nodules was greater in the 1.2% than in controls (p<0.05) and showed
a significant (p<0.05) dose related trend effect. For male rats the
incidence of hepatocellular carcinomas showed a dose related trend
effect, but was not significantly increased by pairwise comparison.
The incidence of neoplastic nodules in the male rats did not show a
significant increase using either pairwise comparison or trend tests
(Kluwe et al., 1982).
Special studies on promotion of carcinogenicity
Weanling male B6C3F1 mice received a single i.p. injection
(80 mg/kg) of diethylnitrosamine (DEN) at 4 weeks of age, followed by
oral administration of phenobarbital (PB) or di(2-ethylhexyl)phthalate
(DEHP) 2 weeks after DEN injection and continued for up to 6 months.
PB was administered in drinking water at 500 ppm and DEHP in the feed
at 0.3%, 0.6% or 1.2%. Groups of mice were sacrificed at 2, 4 and 6
months after DEN exposure and the livers were examined. Few
preneoplastic foci were seen at 2, 4 or 6 months, in mice exposed to
DEN, PB or DEHP alone, while numerous foci and neoplasms were seen in
mice given DEHP or PB after DEN. In DEHP-exposed mice, the number of
foci did not increase between 4 and 6 months, but the foci increased
in mean diameter and volume throughout the experiment. Foci and tumors
appeared earlier in mice given higher dietary levels of DEHP than in
those given lower doses. By the end of the study the number of foci
per unit volume of liver was similar in mice given any dose of DEHP,
but their volume was dose-related; basophilic foci and neoplasms
predominated. The latter were more malignant in appearance than
neoplasms in PB-exposed mice. At 6 months, the neoplasms in high dose
DEHP-exposed mice were significantly larger than those in PB-exposed
mice. Histochemistry, however, revealed similarities between lesions
in mice exposed to PB or DEHP. PB given continuously for 6 months
revealed no initiating activity of DEHP given once by gavage and
followed by PB in drinking water (Ward et al., 1983).
Special studies on the metabolites
Male Sprague-Dawley rats, 250-350 gm, were given 14C-mono
(ethylhexyl) phthalate (MEHP), 69 mg/kg, by gavage. MEHP is a primary
metabolite of DEHP. The resulting urinary metabolites of MEHP were
similar to those of DEHP, and included an alcohol, a ketone, and an
acid from the side chain oxidation of MEHP, as well as a small amount
of o-phthalic acid (Chu et al., 1978).
Phthalic acid, a compound from which DEHP is formed by
esterification, was fed by intubation to male Wistar rats at dose
levels of 3.3 or 40 mg/kg b.w. 0.15% (as 14C02) of the administered
dose was detectable in expired air. Only unchanged phthalic acid could
be found in urine, feces or tissue. Elimination was complete by 24
hours after dosing (Williams & Blanchfield, 1974b).
After oral administration to rats, 2-ethylhexanol was rapidly
absorbed and excreted. Radioactivity from (1-14C)-2-ethylhexanol was
detected in the urine (80-82%), feces (8-9%) and expired C02 (6-7%).
The route of metabolism of 2-ethylhexanol appears to involve oxidation
of 2-ethylhexanoic acid, followed by w- and (w-1)-oxidation.
Urinary metabolites included 2-heptanone, 4-heptanone, 2-ethyl-5-
hydroxyhexanoic acid, 2-ethyl-1,6- hexanedioic acid and 2-ethyl-5-
hexanone. The rate of elimination of 2-ethylhexanol was the same for
a 9 ug as for an 83 mg dose, with essentially all of the 14C being
recovered within 28 hours after administration (Albro, 1975).
Groups each of ten Weanling Sprague-Dawley rats were fed diets
containing 0, 25, 100, 400, 1,600 or 6,400 ppm of mono-2-ethylhexyl
phthalate for 28 days. Decreased growth rate was observed in the
highest dose group. Increased heart and liver weights were observed in
animals of the 1,600 and 6,400 ppm groups. Minor alterations in serum
biochemical values included decreased SDH and calcium levels, and
elevated alkaline phosphatase activity in some treated groups (Chu et
al., 1981).
In 3-month and 6-month feeding studies, groups of ten male and
female Weanling Sprague-Dawley rats were fed diets containing 1, 5,
25, 125 or 625 ppm of mono-2-ethylhexyl phthalate. Growth rate and
food consumption were not affected at any dose level or time interval.
Relative organ weights of rats of both sexes were not altered in the
3-month period, but the liver weights of female rats were increased in
the 6-month study. Changes in clinical chemistry and hematological
values were minimal. These included lower LDH, SGOT, hemoglobin, and
hematocrit values in male rats at the 3-month period and reduced
potassium content at the 6-month period. Histological changes occurred
in treated male and female rats at both time intervals in the liver,
heart, and adrenals. Alteration in the liver consisted of midzonal and
perioportal eosinophilic cytoplasmic inclusions and vacuolations with
isolated binucleated and necrotoc hepatocytes. There was a mild
enlargement of myocardial nuclei and segmental deregistration of
myocardial striations in test animals. The adrenal glands exhibited
vacuolation of the zone fasciculata which was less severe in the
6-month study than the 3-month counterpart (Chu et al., 1981).
Special studies on reproduction and teratology
Mouse
Groups of 10-22 pregnant mice (ddY-Slc (SPF) strain, 8-9 weeks
old) were administered single oral doses (0, 50, 100, 500 mg/kg b.w.)
of DEHP on day 7 of gestation, and sacrificed on day 18. Dose-related
decreases were noted in fetal body weights, number of live fetuses and
implantations per dam. Gross and skeletal anomalies exhibited
dose-related increases. The only treatment effect noted in fetuses
from dams fed 50 mg/kg DEHP was decreased body weight (Nakamura
et al., 1979).
Groups of 3-8 pregnant mice (ddY-Slc (SPF) strain, 8-9 weeks
old) were administered single oral doses of DEHP ranging from 100 to
30,000 mg/kg b.w. on days 6, 7, 8, 9 or 10 of gestation. MEHP was
administered to a separate group of pregnant mice at levels from
100-1000 mg/kg b.w. on days 7, 8 or 9 of gestation. Dams were
sacrificed on day 18 of gestation. With DEHP-treated dams,
treatment-related decreases were noted in fetal body weight
(days 6-10) and number of live fetuses (days 7-9). Because of the
variance in dose levels, no clear-cut treatment effect was evident on
implantations per dam. Treatment-related increases were seen in gross
anomalies (days 7-10) and skeletal anomalies (days 6-8 and day 9, high
dose only).
With MEHP-treated dams, treatment-related decreases were noted in
the number of live fetuses (days 7-8) and increases in gross anomalies
(days 8-9) and skeletal anomalies (day 8). Decreases in fetal body
weight were only marginally related to treatment except for the high
dose dams on day 8 and day 9. No clear-cut treatment effect could be
seen on the number of implantations per dam (Yagi et al., 1980).
Teratogenic effects were observed in CBA mice following a single
oral administration of DEHP in doses representing 1/3-1/12 of the
acute LD50 dose (26.9 g/kg b.w.) on days 6 to 10 of gestation. Excess
fetal deaths were observed when higher doses were given on day 7 but
not when given on day 9 or 10. A significant number of external and
skeletal malformations were found in the group given 7.5 g/kg b.w. on
day 8 (Yagi et al., 1976).
Pregnant female mice (ICR-JCL strain, 8-16 weeks of age) were
distributed in groups varying from 8 to 15 and fed diets containing
0, 0.05, 0.1, 0.2, 0.4 and 1.0% DEHP throughout gestation. Dams were
sacrificed on day 18 of gestation. At dose levels above 0.1%, maternal
weight gain was significantly depressed, although mean food intake was
unaffected. Fetal mortality was also significantly increased in this
dose range. At dose levels above 0.05%, the percentage of resorptions
and dead fetuses exhibited a dose-related increase, and was 100% at
dietary DEHP levels of 0.4% and 1%. Several cases of neural tube
defects (spine bifida and exencephaly) occurred in fetuses from dams
fed 0.2% DEHP. Deficient lumbar rib and ossification of sternabrae was
noted in fetuses from dams fed 0.1% and 0.2% DEHP. Fetal survival was
too low at higher doses to permit meaningful data on incidence of
anomalies. No other skeletal anomalies or visceral anomalies could be
related to treament (Shiota et al., 1980).
Rat
Pregnant Wistar rats, 175-200 g, were randomly distributed into
groups of 15 and dosed by gavage with 0, 225, 450, or 900 mg MEHP/kg
b.w. on days 6-15 of gestation. Another similar experiment was
performed at lower doses (0, 50, 100 and 200 mg/kg b.w.) of MEHP.
Dose-related maternal mortality occurred at the higher doses (225,
450, 900 mg/kg b.w.). In addition, the number of litters per treatment
group decreased significantly at higher doses. Litter size and pup
weight were not significantly affected by treatment. Disturbances in
the placement of the sternebrae plates were noted in fetuses from dams
treated with 450 mg/kg b.w. MEHP. No other skeletal or visceral
anomalies could be related to treatment. In the low-dose experiment,
the only treatment-related effect was a reduction in maternal weight
gain at MEHP doses of 100 and 200 mg/kg b.w. The no-effect level for
MEHP in this study was 50 mg/kg b.w. (Ruddick et al., 1981).
Groups of 20 female Wistar rats (80-120 g) were given daily doses
of 0, 0.34 and 1.70 g/kg DEHP by gavage. After approximately 3 months
of treatment, 10 rats (150-180 g) per dose level were mated. During
gestation, test compound was discontinued. In a separate experiment,
groups of 10 female rats (150-180 g) received comparable doses of
DEHP, but only during gestation. All fetuses from treated dams were
live; no skeletal abnormalities were found. No examination of fetuses
for soft tissue abnormalities was carried out. In dams treated during
gestation, litter size was unaffected but resorptions increased
significantly and fetal weights decreased significantly at both
treatment levels. No significant effects on resorptions, litter size
or fetal weights occurred in dams treated prior to gestation
(Nikoronow et al., 1973).
Rabbit
Virgin female New Zealand, white rabbits (3.9 + 0.5 kg) were
artificially inseminated and injected with sodium monoethylhexyl
phthalate (MEHP, 1.14, 4.69 or 11.38 mg/kg) on days 6-18 of gestation.
When fetuses were removed on day 30, no treatment effect was evident
on sex ratio, fetal weight or litter size. The number of corpora lutea
or resorptions were not affected by treatment, nor were weights of
maternal adrenals, liver, kidneys, heart or lung. No skeletal or
visceral abnormalities occurred that could be ascribed to treatment.
However, the 2 high dose groups of dams experienced increased
mortality (22% and 33%) relative to controls (Thomas et al., 1979).
When pregnant rabbits were injected with MEHP (11.4 mg/kg) on
days 6-18 of gestation, phthalate levels were less than 1 ug/ml in
fetuses and in homogenates of placenta and uteri. Maternal serum
levels failed to reveal any bioaccumulation, and there was no
significant evidence of histological changes in fetal organs,
including liver, lungs and kidneys (Thomas et al., 1980).
Special studies on testicular pathology and effect on zinc
and testosterone levels
Groups of 10 male JCL:ICR mice and rats were fed a diet
containing 2% DEHP or 2% MEHP for 1 week. Both test compounds
depressed growth, testicular weight, and liver weight. Levels of
testosterone and zinc were depressed in the testes, and kidney zinc
levels were elevated in both treatment groups. Liver zinc content was
depressed in the DEHP treatment group, but unchanged in the MEHP group
(Oishi & Hiraga, 1980a,b). Similar effects were observed with
Sprague-Dawley rats given oral doses of DEHP or MEHP (Gray et al.,
1982; Oishi & Hiraga, 1980c,d).
Male Fischer 344 rats and B63CF mice were distributed into groups
of 50 and fed diets containing 0, 0.6% or 1.2% DEHP (rat); or 0, 0.3
or 0.6% (mice) for 103 weeks. High dose rats exhibited a 90% incidence
of severe seminiferous tubular degeneration and testicular atrophy in
comparison to an incidence of 5% or less in low dose rats or controls.
The tubules in the affected animals were devoid of spermatocytes and
germinal epithelium. Only Sertoli cells were observed on the basement
membrane. High dose mice experienced a 14% incidence of seminiferous
tubular degeneration, compared to an incidence of 4% or less in low
dose mice or controls (Kluwe et al., 1982). In another study,
0.2% DEHP fed to male CDE rats for 17 weeks, caused marked
histological changes in the testes (Gray et al., 1977).
Groups of 10 male Wistar rats of varying age (4, 10 or 15 weeks)
were given 2.8 g/kg b.w. DEHP by gavage for 10 consecutive days.
Treated 4 week-old rats showed uniform seminiferous tubular atrophy
comprising a loss of spermatids and spermatocytes. 10 week old rats
experienced a 5-10% incidence of seminiferous tubular atrophy, while
in 15-week old rats, no adverse effects of this type were reported. If
treatment was stopped prior to puberty, normal testicular weight and
histology were regained within 12 weeks. Recovery was slower and less
complete when treatment was discontinued after puberty (Gray and
Butterworth, 1980).
Groups of 7-8 Syrian hamsters (DSN strain) were given oral doses
of 4200 mg/kg b.w./day DEHP or 1000 mg/kg b.w./day MEHP for 9 days.
MEHP reduced mean testicular weight to 73% of control values and
induced atrophy in less than 50% of the seminiferous tubules. DEHP had
no significant effects on these parameters or on urinary zinc
excretion (Gray et al., 1982).
Acute toxicity
Animal Route LD50 References
(mg/kg b.w.)
mouse p.o. 33,500 Krauskopf, 1973
rat p.o. 26,000 Patty, 1967
rabbit p.o. 34,000 ibid
33,900 Shaffer et al., 1945
Guinea pig p.o. 26,300 Krauskopf, 1973
Short-term studies
A study was conducted on the effect of oral administration of
di-(2-ethylhexyl)phthalate (DEHP) at a dose level of 25,000 ppm for
7 and 21 days in young male and female Wistar albino rats. DEHP
increased liver size in both sexes and reduced the relative weight of
testes in male rats. Liver enlargement was accompanied by increases in
several marker enzyme activities (7-ethoxy coumarin-0-deethylase,
cytochrome P-450, alcohol dehydrogenase, aniline-4-hydroxylase, and
succinate dehydrogenase). However, glucose-6-phosphatase activity was
decreased in all treatment groups. DEHP produced no hepatic
histological changes in either sex but ultrastructural studies
indicated proliferation of the smooth endoplasmic reticulum, an
increase in the number of microbodies (peroxisomes), and mitochondrial
changes (Mangham et al., 1981).
When female rats were fed 5000 ppm 14C-DEHP for 49 days,
relative liver weights increased progressively during the first week
of treatment to a value approximately 50% above normal. Only a slight
increase in smooth endoplasmic reticulum was visible by electron
microscopy. When treatment was discontinued, liver weights returned to
normal within one week. Rats fed for 35 days on a 1000 ppm diet did
not experience alterations in liver weight relative to controls
(Daniel & Bratt, 1974).
Groups of 10 male and 10 female Wistar rats, 90-120 g, were
administered daily gavage doses of DEHP at levels of 0, 3400 ppm and
34,000 ppm for 90 days. At autopsy, gross pathological observations
were made: liver, kidney and spleen were weighed and examined
microscopically. High-dose rats displayed uncertainty of movements,
drowsiness, diarrhea and rapid weight loss, with a 75% mortality.
Gross autopsy revealed congestion of the small intestine and loss of
mucosa in the stomach and of some parts of the intestine. A
treatment-related increase in mean liver weight of low-dose rats was
observed, but no gross or microscopic alterations were seen in the
liver, kidneys or spleen (Nikonorow et al., 1973).
Groups of 15 male and 15 female rats were given diets containing
0 (control), 0.2, 1.0 or 2.0% di(2-ethylhexyl)-phthalate (DEHP) for 17
weeks. At the two higher treatment levels there was a reduced rate of
body-weight gain and food intake. A paired-feeding study showed that
the reduced food intake did not account fully for the reduced growth
rate. Hematologic studies showed decreased hematocrits in both sexes
in the 1 and 2% DEHP group, as well as lower hemoglobin levels in
males at these dose levels. Renal concentrating and diluting ability
was reduced in the females receiving 2% DEHP. The relative testes
weight of rats on the (2%) diet was markedly decreased and
histopathological examination revealed severe seminiferous tubular
atrophy and cessation of spermatogenesis. Although testes weight of
the rats fed 0.2% DEHP was not reduced, histological studies showed
evidence of decreased spermatogenesis. There were no other
histopathological changes attributable to DEHP treatment (Gray et al.,
1977).
Groups of 22-24 male and female "hybrid" guinea pigs, 49 days
old, were fed diets containing 0, 0.04 or 0.13% DEHP for 1 year.
Histopathological examination was performed on kidney, liver, lung,
spleen and testes; only liver and kidney were weighed. No treatment
effects were reported on mortality, growth, food consumption,
microscopic pathology or incidence of neoplasms. Treated females had
significantly increased relative liver weights (Carpenter et al.,
1953).
Groups of 6-7 male albino ferrets, 18 months old, were fed diets
containing 0 or 1% DEHP for 14 months. Treated animals experienced
significant growth depression and liver enlargement, with altered
liver morphology. Microscopic examination of sections of brain, heart,
adrenals, thyroid, trachea, esophagus, lung, kidney, and bladder did
not reveal any treatment effect. Mean testicular weights of treated
animals were reduced and 3/7 test animals exhibited a nearly complete
absence of germinal epithelium in testes. Significant treatment-
related effects were seen on activities of marked enzymes for
microsomes, and in drug metabolizing enzymes. Significant changes were
seen in mitochondrial marker enzymes of treated animals (Lake et al.,
1976).
8 dogs (4 Cocker Spaniels, 4 Wire-haired Terriers), 14-17 months
old, were randomly distributed by sex and breed into 2 groups (one
control, one administered DEHP by capsule). The treatment group
received 30 mg DEHP/kg b.w./day, 5 days a week, for 19 doses; then
60 mg/kg b.w./day for 240 additional doses. There was no effect on
growth. At autopsy liver and kidney weight were within normal range.
Gross and histopathological examination of lung, heart, liver,
stomach, small intestine, colon, cecum, spleen, adrenal, gonad,
bladder and thyroid gland of treated animals showed no treatment
related effects (Carpenter et al., 1953).
Long-term studies
Groups of 32 and female Sherman rats, 60 days old, were fed diets
containing 0, 0.04, 0.13 and 0.4% DEHP for up to 2 years (P1). At week
12, animals from the control and high dose levels were bred and their
offspring used to establish an F1 generation composed of 32 males and
32 females. After one year of treatment, groups of 9 to 17 rats from
each sode level (P1 generation) were sacrificed for "tissue and organ
weight" data. Remaining rats were culled to 8 males and 8 females per
group and survivors sacrificed after 2 years of treatment. The F1
generation was placed on test at 15 days of age and sacrificed after
1 year. Histopathological examination was performed on adrenal, heart,
kidney, intestine, liver, lung, spleen, testes, ovary and gross
lesions; only kidney and liver were weighed. At the 0.04% level of
DEHP, both P1 and F1 males exhibited growth depression and an
increased mean relative weight of liver and kidney. F1 females also
showed increased liver and kidney weights at this dose level. No
treatment related effects were reported on survival, food consumption,
microscopic pathology, incidence of neoplasms, hematology or fertility
(Carpenter et al., 1953).
OBSERVATIONS IN MAN
No information available.
Comments
Orally ingested DEHP is probably absorbed as MEHP, since DEHP is
rapidly hydrolyzed to MEHP and 2-ethylhexanol by extracts from the
intestine from several animal species and man. In addition, studies
with isolated perfused rat intestine showed that all DEHP was
converted to MEHP before it reached the serosal perfusing solution.
DEHP metabolites are rapidly distributed throughout the body and reach
a steady state level in the tissues after a period of exposure of up
to 2 weeks, with some accumulation in liver and fat. Tissue residues
are rapidly depleted when DEHP is removed from the diet. There is no
definitive information on the chemical nature of the tissue residues,
since the available distribution studies reflect only a distribution
of 14C, derived from 14C labeled DEHP. In experimental animals the
absorbed material is excreted almost equally in the urine and feces,
with very little being expired as CO2. Biliary excretion ranges from
9-11%. The urine contains up to 5 major metabolites, MEHP, and
4 oxidation products (beta-oxidation) of the MEHP. Only small amounts
appear as o-phthalic acid. In species other than rat (including monkey
and man), the metabolites are excreted mainly as glucuronides. Species
differences were also noted in the distribution of other urinary
metabolites. MEHP appeared in only trace amounts in rat urine, as
did unchanged DEHP in the urine of all species. In monkey and man,
MEHP is one of the major urinary metabolites. In rats, 6.5-10.5% of
administered DEHP appeared as fecal metabolites; most had been
excreted by 48 hr after dosing. Rats fed low amounts (10 ppm) of DEHP
had no detectable residues in feces. The secondary metabolites in
feces have not been identified.
DEHP and MEHP cause an increased liver weight with marked
proliferation of mitochondria and peroxisomes in the liver. The
peroxisomal enzymes that are increased the most are those associated
with beta-oxidation systems. There is less induction of microsomal
enzymes. These effects are reversible when DEHP is removed from the
diet.
DEHP was teratogenic in mice, but not in rats and rabbits. DEHP
at high dietary levels caused severe seminiferous tubular degeneration
testicular atrophy and cessation of spermatogenesis in rats and mice.
Similar effects were observed with MEHP. Reduced zinc levels in serum,
liver and testes were also reported. Other short-term effects of
dietary DEHP include liver enlargement and effects on the heart and
adrenals. DEHP and 2-ethylhexanol were not mutagenic in the Ames test
and a number of mammalian cell systems. In contrast, MEHP was
mutagenic in a number of systems. In lifetime feeding studies in rats
and mice, DEHP caused a significant increase in liver tumors in both
species.
EVALUATION
DEHP is a hepatocarcinogen in both rats and mice.
Provisional acceptance
The level of DEHP in the food contact material and the extent of
its migration into food should be kept at the lowest levels which are
technologically possible.
REFERENCES
ABE, S. & SASAKI, M. (1977) Chromosome aberrations and sister
chromatid exchanges in Chinese hamster cells exposed to various
chemicals. J. National Cancer Inst., 58: 1635-1641.
ALBRO, P.W. (1975) The metabolism of 2-ethylhexanol in rats.
Xenobiotica, 5: 625-636.
ALBRO, P.W. & THOMAS, R. (1973) Enzymatic hydrolysis of
di(2-ethylhexyl)phthalate by lipase. Biochem. Biophys. Acta, 306:
380-390.
ALBRO, P.W., THOMAS, R., & FISHBEIN, L. (1973) Metabolism of
diethylhexyl phthalate by rats. Isolation and characterization of the
urinary metabolites. J. Chromatogr., 76: 321-330.
ALBRO, P.W., HASS, J.R., PECK, C.C., ODOM, D.G., CORBETT, J.T.,
BAILEY, F.J., BLATT, H.F., & BARRETT, B.B. (1981) Identification
of the metabolites of di-2-ethylhexyl phthalate in urine from the
African Green monkey. Drug Metab. Disp., 9: 233-235.
ALBRO, P.W., JORDAN, S.T., SCHROEDER, J.L., & CORBETT, J.T. (1982a)
Chromatographic separation and quantitative determination of the
metabolites of di-(2-ethylhexyl)phthalate from urine of laboratory
animals. J. Chromatogr., 244(1): 65-79.
ALBRO, P.W., CORBETT, J.T., SCHROEDER, J.L., JORDAN, S., & MATTHEWS,
H.B. (1982b) Pharmacokinetics, interactions with macromolecules and
species differences in metabolism of DEHP. Environ. Health
Perspect., 45: 19-25.
ALBRO, P.W., TONDEUR, I., MARBURY, D., JORDAN, S., SCHROEDER, J. &
CORBETT, J.T. (1983) Polar metabolites of di-(2-ethylhexyl)phthalate
in the rat. Biochim. Biophys. Acta, 760: 273-92.
ANONYMOUS (1982a) Mutagenicity evaluation of di-2-ethylhexyl phthalate
in the Ames Salmonella/microsome plate test. Unpublished report from
Chemical Manufacturers of America, submitted to the World Health
Organization by the United States Food and Drug Administration.
ANONYMOUS (1982b) Mutagenicity evaluation of di-2-ethylhexyl phthalate
in the mouse lymphoma forward mutation assay. Ibid.
ANONYMOUS (1982c) Evaluation of di-2-ethylhexytl phthalate in the
in vitro transformation of Balb/3T3 cells assay. Ibid.
ANONYMOUS (1982d) Evaluation of di2-ethylhexyl phthalate in the
in vitro transformation of Balb/3T3 cells with metabolic activation
by primary rat hepatocytes. Ibid.
ANONYMOUS (1982e) Mutagenicity evaluation of di-2-ethylhexyl phthalate
in the mouse micronucleus test. Ibid.
ANONYMOUS (1982f) Evaluation of di-2-ethylhexyl phthalate in the
primary rat hepatocyte unscheduled DNA synthesis assay. Ibid.
ANONYMOUS (1982g) Mutagenicity evaluation of mono-2-ethylhexyl
phthalate (MEHP) in the Ames Salmonella/microsome plate test. Ibid.
ANONYMOUS (1982h) Mutagenicity evaluation of mono-2-ethylhexyl
phthalate (MEHP) in the mouse micronucleus test. Ibid.
ANONYMOUS (1982i) Evaluation of mono-2-ethylhexyl phthalate (MEHP) in
the in vitro transformation of Balb/3T3 cells assay. Ibid.
ANONYMOUS (1982j) Evaluation of mono-2-ethylhexyl phthalate (MEHP) in
the in vitro transformation of Balb/3T3 cells with metabolic
activation by primary rat hepatocytes. Ibid.
ANONYMOUS (1982k) Mutagenicity evaluation of 2-ethylhexanol (2-EH) in
the Ames Salmonella/microsome plate test. Ibid.
ANONYMOUS (1982l) Mutagenicity evaluation of 2-ethylhexanol (2-EH) in
the mouse micronucleus test. Ibid.
ANONYMOUS (1982m) Evaluation of 2-ethylhexanol in the in vitro
transformation of Balb/3T3 cells assay. Ibid.
CARPENTER, C.P., WELL, C.S., & SMYTH, H.F. Jr. (1953) Chronic oral
toxicity of di-&2-ethylhexyl)phthalate for rats, guinea pigs, and
dogs. Arch. Inc. Hyg. Occup. Med., 8: 219-226.
CARTER, J.E., ROLL, D.B., & PETERSON, R.V. (1974) The in vitro
hydrolysis of di-(2-ethylhexyl)phthalate by rat tissues. Drug Metab.
Disposition, 2: 341-344.
CHU, I., VILLENEUVE, D.C., SECOURS, V., FRANKLIN, C., ROCK, G., &
VIAU, A. (1978) Metabolism and tissue distribution of
mono-2-ethylhexyl phthalate in the rat. Drug Metab. Dispos., 6:
146-9.
CHU, I., SECOURS, V.E., MARINO, I.A., VILLENEUVE, D.C., & VALLI, V.E.
(1981) Sub-acute and sub-chronic toxicity of mono-2-ethylhexyl
phthalate in the rat. Arch. Environ. Contam. Toxicology, 10(3):
271-80.
DANIEL, J.W. & BRATT, H. (1974) The absorption, metabolism and tissue
distribution of di-2-ethylhexyl)phthalate in rats. Toxicology, 2:
51-65.
GANNING, A.E. & DALLNER, G. (1981) Induction of peroxisomes and
mitochondria by di-(2-ethylhexyl) phthalate. FEBS Lett., 130:
77-9.
GANNING, A.E., KLASSON, E., BERGMAN, A., BRUNK, U., & DALLNER, G.
(1982) Effect of phthalate ester metabolites on rat liver. Acta
Chem. Scnad., B36: 563-5.
GANNING, A.E., BRUNK, U., & DALLNER, G. (1983) Effects of dietary
di-(2-ethylhexyl) phthalate on the structure and function of rat
hepatocytes. Biochim. Biophys. Acta, 763(1): 72-82.
GAUNT, I.F. & BUTTERWORTH, K.R. (1982) Autoradiographic study of
orally administered di(2-ethylhexyl)phthalate in the mouse. Food
Chem. Toxicol., 20(2): 215-7.
GRAY, T.J. & BUTTERWORTH,. K.R. (1980) Testicular atrophy produced by
phthlate esters. Arch. Toxicol. (Suppl.), 4: 452-5.
GRAY, T.J., BUTTERWORTH, K.R., GAUNT, I.F., GRASSO, P., & GANGOLLI,
S.D. (1977) Short-term toxicity study of di-(2-ethylhexyl) phthalate
in rats. Food Cosmet. Toxicol., 15: 389-399.
GRAY, T.J., BEAMAND, J.A., LAKE, B.G., FOSTER, J.R., & GANGOLLI, S.D.
(1982) Peroxisome proliferation in cultured rat hepatocytes produced
by clofibrate and phthalate ester metabolites. Toxicol. Lett.,
10(2): 273-9.
HODGSON, J.R. (1982) Evaluation of di-(2-ethylhexyl) phthalate and
its major metabolites in the primary rat hepatocyte unscheduled DNA
synthesis assay. Environ. Mutagen., 4:388 (Abstract).
IKEDA, G.J., SAPIENZA, P.P., COUVILLION, J.L. FARBER, R.M., & VAN
LOON, E.J. (1980) Comparative distribution, excretion and metabolism
of di-(2-ethylhexyl) phthalate in rats, dogs and miniature pigs.
Food Cosmet. Toxicol., 18(6): 637-642.
ISHIDATE, M.Jr. & ODASHIMA, S. (1977) Chromosome tests with 134
compounds on Chinese hamster cells in vitro - A screening for
chemical carcinogens. Mutat. Res., 48: 337-354.
KANESHIMA, H., YAMAGUCHI, T., OKUI, T., & NAITOH, M. (1978) Studies on
the effects of phthalate esters on the biological system. Part. 2.
In vitro metabolism and biliary excretion of phthalate esters in
rats. Bull. Environ. Contam. Toxicol., 19: 502-9.
KIRBY, P.E., PIZZARELLO, R.F., LAWLOR, R.E., HAWORTH, S.R., HODGSON,
J.R. (1983) Evaluation of di-(2-ethylhexyl) phthalate and its major
metabolites in the Ames test and L5178Y mouse lymphoma mutagenicity
assay. Environ. Mutagen., 5(5): 657-63.
KOZUMBO, W.J., KROLL, R., & RUBIN, R.J. (1982) Assessment of the
mutagenicity of phthalate esters. Environ. Health Perspect, 45:
103-109.
KLUWE, W.M., HASEMAN, J.K., DOUGLAS, J.F., & HUFF, J.E. (1982) The
carcinogenicty of dietary di-(2-ethylhexyl) phthalate (DEHP) in Fisher
344 rats and B6C3F1 mice. J. Toxicol. Environ. Health, 10(4):
797-815.
KRAUSKOPF, L.F. (1973) Studies on the toxicity of phthalates via
ingestion. Environ. Health Perspect., 3: 61-72.
LAKE, B.G., BRANTOM, P.G., GANGOLLI, S.D., BUTTERWORTH, K.R., &
GRASSO, P. (1976) Studies on the effects of orally administered
di-(2-ethylhexyl) phthalate in the ferret. Toxicology, 6 341-356.
LINDGREN, A., LINDQUIST, N.G., LYD'EN, A., OLSSON, T., & ULLBERG, S.
(1982) A whole body autoradiographic study on the distribution of
14C-labelled di-(2-ethylhexyl)phthalate in mice. Toxicology, 23:
149-58.
MANGHAM, B.A., FOSTER, S.R., & LAKE, B.G. (1981) Comparison of
the hepatic and testicular effects of orally administered
di-(2-ethylhexyl)phthalate and dialkyl 79-phthalate in the rat.
Toxicol. Appl. Pharmacol., 61: 205-214.
MIYAZAWA, S., FURUTA, S., OSUMI, T., & HASHIMOTO, T. (1980) Turnover
of enzymes of peroxisomal beta-oxidation in rat liver. Biochim.
Biophys. Acta, 630(3): 367-74.
MOODY, D.E. & REDDY, J.K. (1978) Hepatic peroxisomal (microbody)
proliferation in rats fed plasticizers and related compounds.
Toxicol. Appl. Pharmacol., 45(2): 497-504.
NAKAMURA, Y., YAGI, Y., TOMITA, I., & TSUCHIKAWA, K. (1979)
Teratogenicity of di-(2-ethylhexyl)phthalate in mice. Toxicol.
Lett., 4: 113-117.
NIKORONOW, M., MAZUR, H., & PIEKACZ, H. (1973) Effect of orally
administered plasticizers and polyvinyl chloride stabilizers in the
rat. Toxicol. Appl. Pharmacol., 25: 253-259.
OHNO, S., OHTAKE, N., FUJII, Y., YAMABAYASHI, S., USUDA, N., & NAGATA,
T. (1981) Histochemical studies on peroxisomes of rat livers during
DEHP administration and after withdrawal on thick sections by means of
light microscopy and high voltage electron microscopy. Acta
Histochem. Cytochem., 14: 126-42.
OISHI, S. & HIRAGA, K. (1980a) Effect of phthalic acid esters on mouse
testes. Toxicol. Lett., 5: 413-416.
OISHI, S. & HIRAGA, K. (1980b) Effects of phthalate acid monoesters on
mouse testes. Toxicol. Lett., 6: 239-242.
OISHI, S. & HIRAGA, K. (1980c) Testicular atrophy induced by phthalic
acid esters: Effect on testosterone and zinc concentrations.
Toxicol. Appl. Pharmacol., 53: 35-41.
OISHI, S. & HIRAGA, K. (1980d) Testicular atrophy induced by phthalic
acid monoesters: Effects of zinc and testosterone concentrations.
Toxicology, 15: 197-202.
OISHI, S. & HIRAGA, K. (1982) Distribution and elimination of di-2-
ethylhexyl phthalate (DEHP) and mono-2-ethylhexyl phthalate (MEHP)
after a single oral administration of DEHP in rats. Arch. Toxicol.,
51: 149-55.
PATTY, F.A. (1967) Industrial Hygiene and Toxicology, 2nd Rev. Vol. 2.
pp. 1906-1910. Interscience, New York.
PECK, C.C., ALBRO, P.W., HASS, J.R., ODOM, D.G., BARRETT, B.B., &
BAILEY, F.J. (1978) Metabolism and excretion of the plasticizer
di-(2-ethylhexyl)phthalate in man. Clin. Res., 26: 101A
(Abstract).
PHILLIPS, B,J., JAMES, T.E., & GANGOLLI, S.D. (1982) Genotoxicity
studies of di-(2-ethylhexyl) phthalate and its metabolites in CHO
cells. Mutat. Res., 102(3): 297-304.
PUTMAN, D.L., MOORE, W.A., SCHECHTMAN, L.M., & HODGSON, J.R. (1983)
Cytogenetic evaluation of di-(2-ethylhexyl) phthalate and its major
metabolites in Fischer 344 rats. Environ. Mutagen., 5(2): 227-31.
ROWLAND, I.R., COTTRELL, R.C., & PHILLIPS, J.C. (1977) Hydrolysis of
phthalate esters by the gastrointestinal contents of the rat. Food
Cosmet. Toxicol., 15: 17-21.
RUDDICK, J.A., VILLENEUVE, D.C., CHU, I., NESTMANN, E., & MILLES, D.
(1981) An assessment of the teratogenicity in the rat and mutagenicity
in Salmonella of mono-2-ethylhexyl phthalate. Bull. Environ. Contam.
Toxicol., 27: 181-6.
RUSHBROOK, C.J. (1982) Dominant lethal study of di-(2-ethylhexyl)
phthalate and its major metabolites in ICR/SIM mice. Environ.
Mutagen., 4: 387 (Abstract).
SAKURAI, T., MIYAZAWA, S., & HASHIMOTO, T. (1978) Effects of
di-(2-ethylhexyl) phthalate administration of carbohydrate and fatty
acid metabolism in rat liver. J. Biochem., 83: 313-320.
SCHAFFER, C.B., CARPENTER, C.B., & SMITH, H.F. (1945) Acute and
subacute toxicity of DEHP with note upon its metabolites. J. Ind.
Hyg. Toxicol., 27: 130-135.
SCHULTZ, C.O. & RUBIN, R.J. (1973) Distribution, metabolism and
excretion of di-(2-ethylhexyl) phthalate in the rat. Environ. Health
Perspect., 3: 123-129.
SEED, J.L. (1982) Mutagenic activity of phthalate esters in bacterial
liquid suspension assays. Environ. Health Perspect., 45: 111-114.
SHIOTA, K., CHOU, M.J., & NISHIMURA, H. (1980) Embryotoxic effects of
di-(2-ethylhexyl) phthalate (DEHP) and di-n-butyl phthalate (DBP) in
mice. Environ. Res., 22: 245-253.
SIMMON, V.F., JAUHANEN, K., & TARDIFF, R.G. (1977) Mutagenic activity
of chemicals identified in drinking water. Dev. Toxicol. Environ.
Sci., 2: 249-258.
STENCHEVER, M.A., ALLEN, M.A., JEROMINSKI, L., & PETERSEN, R.V. (1976)
Effects of bi-(2-ethylhexyl) phthalate on chromosomes of human
leukocytes and human fetal lung cells. J. Pharm. Sci., 65:
1648-1651.
THOMAS, J.A., SCHEIN, L.G., GUPTA, P.K., McCAFFERTY, R.E., FELICE,
P.R., & DONOVAN, M.P. (1979) Failure of monoethylhexyl phthalate to
cause teratogenic effects in offspring of rabbits. Toxicol. Appl.
Pharmacol. 51: 523-528.
THOMAS, J.A., MARTIS, L., GIOVANETTO, S., McCAFFERTY, R.E., & DONOVAN,
M.P. (1980) Biodistribution of monoethylhexyl phthalate (MEHP) in
pregnant rabbits and neonatal rats. Fed. Proc., 39: 525
(Abstract).
TOMITA, I., NAKAMURA, Y., AOKI, N., & INUI, N. (1982)
Mutagenic/carcinogenic potential of DEHP and MEHP. Environ. Health
Perspect., 45: 119-25.
TURNER, J.H., PETRICCIANI, J.C., CROUCH, M.L., & WENGER, S. (1974) An
evaluation of the effects of diethylhexyl phthalate (DEHP) on
mitotically capable cells in blood packs. Transfusion, 14: 560-6.
WARD, J.M., RICE, J.M., CREASIA, D., LYNCH, P., & RIGGS, C. (1983)
Dissimilar patterns of promotion by di-(2-ethylhexyl) phthalate
and phenobarbital of hepatocellular neoplasia initiated by
diethylnitrosamine in B6C3F1 mice. Carcinogenesis, 4(8): 1021-9.
WARREN, J.R., LALWANI, N.D., & REDDY, J.K. (1982) Phthalate esters as
peroxisome proliferator carcinogens. Environ. Health Perspect.,
45: 35-40.
WHITE, R.D., CARTER, D.E., EARNEST, D., & MUELLER, J. (1980)
Absorption and metabolism of three phthalate diesters by the rat
small intestine. Food Cosmet. Toxicol., 18: 383-6.
WILLIAMS, D.T. & BLANCHFIELD, B.J. (1974a) Retention, excretion and
metabolism of di-(2-ethylhexyl) phthalate administered orally to the
rat. Bull. Environ. Contam. Toxicol., 11: 371-378.
WILLIAMS, D.T. & BLANCHFIELD, B.J. (1974b) Retention, excretion and
metabolism of phthalic acid administered orally to the rat. Ibid.,
12: 109-112.
YAGI, Y., NAKAMURA, Y., TOMITA, I., TSUCHIKAWA, K., & SHIMOI, N.
(1980) Teratogenic potential of di- and mono-(2-ethylhexyl)
phthalate in mice. J. Environ. Pathol. Toxicol., 4: 533-544.
YANAGITA, T., KOBAYASHI, K., & ENOMOTO, N. (1978) Accumulation of
hepatic phospholipids in rats fed di-(2-ethylhexyl) phthalate.
Biochem. Pharmacol., 27: 2283-2288.
YOSHIKAWA, K., TANAKA, A., YAMAHA, T., & KURATA, H. (1983)
Mutagenicity study of nine monoalkyl phthalates and a dialkyl
phthalate using Salmonella typhimurium and Escherichia coli. Food
Chem. Toxicol., Apr; 21(2): 221-3.
ZEIGER, E., HAWORTH, S., SPECK, W., & MORTALMANS, K. (1982) Phthalate
esters testing in the National Toxicology Programs Environmental
Mutagenesis Test Development Program. Environ. Health Perspect.