
INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY
WORLD HEALTH ORGANIZATION
TOXICOLOGICAL EVALUATION OF CERTAIN
VETERINARY DRUG RESIDUES IN FOOD
WHO FOOD ADDITIVES SERIES: 43
Prepared by the Fifty-second meeting of the Joint FAO/WHO
Expert Committee on Food Additives (JECFA)
World Health Organization, Geneva, 2000
IPCS - International Programme on Chemical Safety
PRODUCTION AIDS
ESTRADIOL-17ß, PROGESTERONE, AND TESTOSTERONE
First draft prepared by
J. Leighton, S. Franceschi, G. Boorman, D.W. Gaylor, and J.G. McLean
Estradiol-17ß
Explanation
Biological data
Absorption, distribution, and elimination
Biotransformation
Hydroxylation
Conjugation
Biochemical parameters
Synthesis
Mechanism of action
Toxicological studies
Acute toxicity
Short-term studies of toxicity
Long-term studies of toxicity and carcinogenicity
Genotoxicity
Reproductive toxicity
Special studies on mechanism of action
Observations in humans
Therapeutic use
Estradiol-related genetic markers of carcinogenicity
Progesterone
Explanation
Biological data
Absorption, distribution, and excretion
Biotransformation
Biochemical parameters
Synthesis
Mechamism of action
Toxicological studies
Acute toxicity
Short-term studies of toxicity
Long-term studies of toxicity and carcinogencity
Genotoxicity
Reproductive toxicity
Observations in humans
Testosterone
Explanation
Biological data
Absorption, distribution, and elimination
Biotransformation
Biochemical parameters
Synthesis
Mechamism of action
Toxicological studies
Acute toxicity
Short-term studies of toxicity
Long-term studies of toxicity and carcinogenicity
Genotoxicity
Reproductive toxicity
Observations in humans
Epidemiological studies of women exposed to postmenopausal estrogen
therapy and hormonal contraceptives
Methods
Postmenopausal oestrogen therapy
Exposure
Human carcinogenicity
Breast cancer
Endometrial cancer
Cervical cancer
Ovarian cancer
Cancers of the liver and biliary tract
Colorectal cancer
Cutaneous malignant melanoma
Thyroid cancer
Summary and conclusions
Cardiovascular disease
Osteoporosis
Overall mortality
Hormonal contraceptives
Exposure
Human carcinogenicity
Breast cancer
Endometrial cancer
Cervical cancer
Ovarian cancer
Cancers of the liver and biliary tract
Colorectal cancer
Cutaneous malignant melanoma
Thyroid cancer
Summary and conclusions
Cardiovascular disease
Acute myocardial infarct
Stroke
Venous thromboembolism
Overall mortality
Meat intake and cancer risk
Comments and evaluation
Estradiol-17ß
Progesterone
Testosterone
References
The purpose of this monograph is to provide a review and summary
of the scientific information relative to a toxicological assessment
of the safety of three endogenous hormones, estradiol-17ß,
progesterone, and testosterone, with emphasis on information published
since the review of the Committee at its thirty-second meeting (Annex
1, reference 80). The biology and toxicology of the compounds and
metabolites formed endogenously and ingested orally are summarized. As
the pharmacokinetics and pharmaco-dynamics of synthetic steroidal and
nonsteroidal substances (e.g. diethyl-stilbestrol) differ
substantially, only a limited discussion of the pharmacology of these
compounds is presented. This review is not intended to be exhaustive
but to highlight the scientific literature that may be relevant to use
of the hormones from the point of view of food safety. The Committee
at its thirty-second meeting did not prepare toxicological monographs
on the natural hormones estradiol-17ß, progesterone, and testosterone.
1. ESTRADIOL-17b
1.1 Explanation
Estradiol benzoate (10-28 mg) or estradiol-17ß (estradiol; 8-24
mg) are administered to cattle as an ear-implant formulation to
increase the rate of weight gain (i.e. growth promotion) and to
improve feed efficiency. Estradiol valerate is administered by
subcutaneous or intramuscular injection to synchronize estrus in
cattle. Esters of estradiol are rapidly cleaved to estradiol in vivo
and are thus also considered to be endogenous substances, as the
residues produced are structurally identical to the estradiol produced
in animals and humans after hydrolysis.
Estradiol was reviewed previously by the Committee, at its
thirty-second meeting (Annex 1, reference 80), when it concluded that
the establishment of an acceptable residue level and an ADI was
'unnecessary'. This conclusion was based on studies of the patterns of
use of estradiol for growth promotion in cattle, the residues in
animals, analytical methods, toxicological data from studies in
laboratory animals, and clinical findings in human subjects. The
Committee further concluded that estradiol residues resulting from its
use for growth promotion in accordance with good husbandry practices
were unlikely to be a hazard to humans.
1.2 Biological data
1.2.1 Absorption, distribution, and excretion
Estradiol is generally considered to be inactive when
administered orally due to gastrointestinal and/or hepatic
inactivation. In a study to monitor its oral availability and to
identify the sites of metabolism, 14C-estradiol was infused into
selected portions of the gastrointestinal tract of gilts, and blood
samples were collected from the jugular and portal veins. The
concentration of free estrogens in the jugular vein was low (< 1%) at
all times after instillation of labelled estradiol, and it was
detectable only briefly. The concentration of conjugated estrogens in
the jugular vein peaked rapidly after instillation, particularly when
instilled into the lower gut. Approximately 60-90% of the radiolabel
in blood was present as glucuronide conjugates; smaller amounts of
sulfated compounds were detected, and approximately 1% as
diconjugates. The principal steroid identified after cleavage by
b-glucuronidase and sulfatase was estrone. The authors concluded that
conjugation occurs as estradiol crosses the mucosa of the
gastrointestinal tract, and free estradiol in the portal plasma is
conjugated during the first pass through the liver (Moore et al.,
1982). In companion studies, the authors concluded that the limiting
factor in absorption of conjugates was hydrolysis to free estrogen
(Pohland et al., 1982) and that a possible dose-limiting rate of
absorption was observed at the highest dose (4 mmol 3H-estradiol
glucuronide) (Coppoc et al., 1982).
Crystalline estradiol (10 mg in cocoa butter) was placed in the
stomachs of prepubertal gilts that had been held without food for 26
h, and blood samples were taken from the jugular and hepatic portal
veins for hormone measurements. The concentrations of estradiol,
estrone, estradiol glucuronide, and estrone sulfate in the hepatic
portal vein rose within 5 min and remained elevated for several hours.
Estradiol represented only 6% of the total estrogen measured during
the sampling period, indicating extensive pre-hepatic metabolism of
estradiol. In the periphery, the concentrations of estradiol
glucuronide, estrone glucuronide, and estrone sulfate, but not those
of estradiol or estrone, rose in the jugular vein, indicating that
most of the estradiol and estrone had been removed by the liver.
Infusion of bile containing estrogens into the duodenum resulted in
peaks of estrogen glucuronide and estrone glucuronide in the hepatic
portal and jugular veins within a few minutes, followed by a second
rise 180 min later. The first peak did not occur in bile extracted
with ether to remove free estradiol and estrone, and the second peak
did not occur in gilts given oral antibiotics before bile infusion.
The authors concluded that estrogens administered orally are
conjugated by the gut wall and pass to the liver, where they enter
either the bile pool for enterohepatic circulation or the bloodstream
(Ruoff & Dziuk, 1994).
Oral administration of 0.5 mg fine-particle estradiol in the
early follicular phase of the menstrual cycle to six fasting, female
volunteers resulted in a peak mean estradiol concentration of 211
pg/ml 4 h after administration (mean basal estradiol concentration,
138 pg/ml). The serum estrone concentrations also peaked at this time,
when the peak:baseline ratio of estrone was greater than that of
estradiol. Peaks were observed 4 h after dosing for estrone sulfate
and 6 h after dosing for estradiol sulfate; the peak for estrone
sulfate was always higher than that for estradiol sulfate. The
predominance of estrone over estradiol in serum after oral
administration of estradiol and comparison with serum concentrations
reached after vaginal administration indicate extensive first-pass,
probably intestinal, metabolism (Nahoul et al., 1993).
The distribution of estradiol in female Wistar rats was measured
in heart, liver, kidney, brain, and plasma by radioimmunoassay for 24
h after intravenous administration of 0.1 mg/kg bw or after
intragastric administration of 10 mg/kg bw. The concentration of
estradiol in liver was 20 times higher after intragastric than after
intravenous administration when equivalent plasma concentrations of
hormone were evaluated. Negligible differences were seen in the
estradiol concentrations of other tissues. The tissue concentrations
of estradiol were higher than those in plasma at all times. The
absolute bioavailability, as measured by comparison of the
dose-corrected values for the area under the integrated
concentration-time curve (AUC), was 8.3% after an intragastric dose of
10 mg/kg bw. The total clearance was 154 ml/min per kg bw. The
half-life of estradiol in liver was 2.6 h (Schleicher et al., 1998).
The uptake of estradiol by adipose tissue, a reservoir for estrogens,
was not investigated in this study.
Fourteen young women received a single dose of 2, 4, or 8 mg
estradiol orally or 0.3 mg intravenously. The 8-mg dose of estradiol
resulted in a 70-78% reduction in the AUC relative to expected
values for estradiol and for free and total estrone, suggesting
incomplete absorption at this dose. The absolute bioavailability of
the 4-mg dose was calculated to be 5%. The mean ratio of free
estrone:estradiol was 1 after intravenous injection and and 20 after
oral administration. In a two-comparment model, the AUC for young
women given a 0.3-mg dose intravenously was 4000 pg-h/ml; total
clearance was 22 ml/min per kg bw. Pharmacokinetic parameters showed
high intraindividual and interindividual variation,which limits the
therapeutic usefulness of oral preparations (Kuhnz et al., 1993).
Circulating estradiol is bound to sex hormone-binding globulin
(SHBG) and, to a lesser extent, serum albumin. Only 1-2% of
circulating estradiol is unbound; 40% is bound to SHBG and the
remainder to albumin (Carr, 1998). Plasma SHBG is secreted from the
liver; a similar, non-secretory form is present in many tissues,
including reproductive tissues and the brain. Adult rodent livers do
not produce the secretory form of SHBG (Reventos et al., 1993). Some
estrogen metabolites (2-methoxyestrone and 2-methoxy-estradiol) have
higher binding affinities for SHBG than estradiol itself (Philip &
Murphy, 1986), and other estrogens (estrone and estriol) do not bind
to this serum protein in humans (Renoir et al., 1980). Estradiol binds
to human SHBG with lower affinity than testosterone.
The plasma concentrations of SHBG are regulated; they may be
increased 5-10-fold by estrogens and decreased twofold by testosterone
(Griffin & Wilson, 1998). Thus, a 20-fold higher concentration of
total testosterone in men than in women results in a 40-fold
difference in free testosterone (Grumbach & Styne, 1998). Unliganded
plasma SHBG binds to either steroid or to SHBG-receptor; SHBG must
first bind to the receptor and then the steroid in order to act: SHBG
that is liganded to steroid cannot bind to the receptor (Hyrb et al.,
1990). The SHBG-receptor complex present on the membranes of target
tissues may be responsible for the interaction between the steroid
hormone and cAMP pathways (Rosner, 1991). These observations provide a
mechanistic explanation for the finding that some estrogenic effects
are rapid (milliseconds) and are possibly mediated in a non-genomic
manner. The intracellular form of the SHBG protein may sequester or
direct hormone to the target tissue.
Estrogens are eliminated in faeces and urine. The principal
metabolites found in urine are polyhydroxylated forms conjugated at C3
to glucuronic acid or sulfate. Elimination in bile is subject to
enterohepatic circulation, and 20% of estrogens may be lost through
faecal elimination. A high-fibre diet has been implicated in increased
elimination of estrogens by this route, probably by decreasing gut
transit time (Lewis et al., 1997). A high-fibre diet nonsignificantly
lowered the serum estradiol AUC in human volunteers given an oral dose
of estradiol glucuronide (Lewis et al., 1998).
Urinary and faecal metabolites of estrogens in animals and humans
have been studied for use as possible indicators of risk for
hormone-dependent cancers or for infertility. Quantitative and
qualitative differences between low-and high-risk populations and
alterations in metabolite profiles due to diet have been reported
(Michnovich & Bradlow, 1990; Aldercreutz et al., 1994; Ursin et al.,
1997). There is at present no consensus about the importance of
specific metabolites or metabolite ratios as prognostic factors, with
the possible exception of estriol as a marker of the well-being of the
feto-placental unit.
The terminal plasma half-life of estradiol after intravenous
adminis-tration to humans was 27 min; the volume of distribution was
calculated to be 0.082 l/kg bw (White et al., 1998). Elsewhere, the
plasma half-life of estradiol has been reported to be approximately 30
min (Wingard et al., 1991).
1.2.2 Biotransformation
The major metabolites of estradiol, progesterone, and
testosterone are shown in Figure 1.
1.2.2.1 Hydroxylation
Concern about the carcinogenicity of estrogens and, more
recently, the possible genotoxicity of estrogen metabolites has
sparked interest in establishing the pathways of estradiol metabolism,
and extensive reviews have been published (IARC, 1979; Zhu & Conney,
1998a). The two main competing, irreversible pathways for estradiol
hydroxylation are 2-or 4-hydroxylation and 16 alpha-hydroxylation
(Michnovicz et al., 1989), which have been implicated in both the
pathophysiology and the protective characteristics of estrogens. Minor
pathways of hydroxylation at other sites in the steroid metabolic
pathway have also been identified (Zhu & Conney, 1998a).
Hepatic hydroxylation of estradiol in humans and most other
species leads primarily to the formation of 2-hydroxyestradiol or
2-hydroxyestrone, with subsequent methylation; 4-hydroxy estrogens are
also formed, although to a lesser extent. In the alternative pathway,
the principal products are 16 alpha-hydroxyestrone and estriol, both of
which are estrogen agonists. The pathway of estradiol metabolism in
vitro was shown to be concentration-dependent; in hamster liver
microsomes, 16alpha-hydroxylation predominates at low (< 25 µmol/L)
concentrations, whereas 16 alpha-and C2-hydroxylation contributed
equally to estradiol metabolism at higher concentrations (Butterworth
et al., 1996). Human forms of cytochrome P450s (CYP) which catalyse
the 2-or 4-hydroxylation of estradiol and estrone include CYP1A2 and,
to a lesser extent, CYP3A4 and CYP2C9 (Shou et al., 1997; Yamazaki et
al., 1998). Estradiol and estrone 16a-hydroxylation is catalysed by
CYP1A2 (estradiol) and CYP3A4 (estradiol and estrone). CYP1B1
catalyses the 4-hydroxylation of estrone and estradiol and may be the
dominant enzymatic pathway for estrogen metabolism in some
extrahepatic tissues, particularly steroidogenic tissues and their
respective targets (Larsen et al., 1998; Zhu & Conney, 1998a).
While most estrogen metabolism occurs in the liver as
2-hydroxylation, extrahepatic metabolism occurs as well. Conflicting
reports have been published on the predominance of 2-and
4-hydroxylation of estradiol in Syrian hamster kidney. The major route
appears to be 2-hydroxy formation after catalysis by CYP1A1/2 and
CYP3A expressed in this tissue (Hammond et al., 1997; Sarabia et al.,
1997). Alternatively, 4-hydroxylation has been shown to predominate
over 2-hydroxylation in the hamster kidney (Weisz et al., 1992). The
4-hydroxy estradiol formed in this tissue is thought to be due to the
lack of specificity of the responsible CYPs, as a specific estrogen
4-hydroxylase (presumably CYP1B1) was not found in this tissue. CYP1B1
protein was also not found in human renal adenocarcinoma cells (Spink
et al., 1997). Rat pituitary, mouse, and human uterus and human
mammary gland are other tissues that express high levels of estrogen
4-hydroxylase (Liehr et al., 1995; Liehr & Ricci, 1996; Yager & Liehr,
1996; Larsen et al., 1998).
Significant differences in steroid metabolism are seen between
rodents and humans (IARC, 1979). Sex-specific regulation of CYPs has
been observed in rodent but not human liver, although sex differences
in the metabolism of xenobiotics are found in humans (Kedderis &
Mugford, 1998). Human but not mouse CYP1B1, recently identified as an
estrogen 4-hydroxylase, metabolizes estradiol (Savas et al., 1997).
Several mechanisms of CYP-mediated aromatic hydroxylation of
estrogens (estradiol and estrone) have been proposed, including
epoxide formation, direct oxygen insertion, and hydrogen abstraction.
Hydroxylation by hydrogen abstraction, electron delocalization, and
subsequent hydroxy radical addition has been proposed on the basis of
electronic considerations of oxidation of estrone and substrates with
additional aromaticity (2-napthol and equilenin) (Sarabia et al.,
1997).
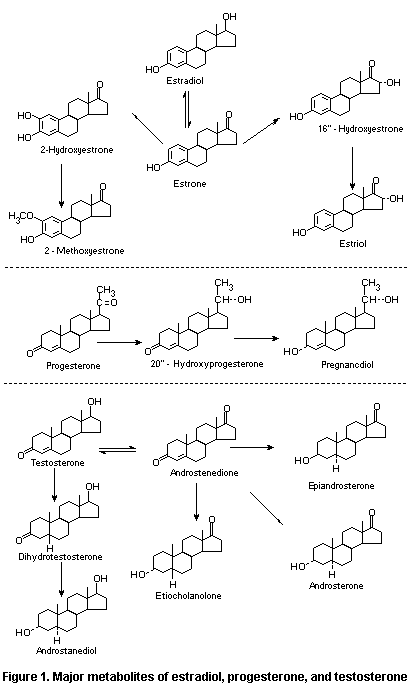 Hydroxyestrogens may be further modified by the action of
catechol-O-methyltransferase (COMT). High activity of COMT is found in
many tissues, including liver and kidneys, blood cells, endometrium,
and breast. A genetic polymorphism for this enzyme results in a
trimodal distribution of activity, but epidemological studies of the
polymorphism in relation to breast cancer risk have yielded
conflicting results (Lavigne et al., 1997; Millikan et al., 1998;
Thompson et al., 1998). The methylation of catecholestrogens
effectively prevents these compunds from entering the redox cycling
pathway, and 2-methoxyestradiol may be an antitumour agent (Zhu &
Conney, 1998a,b).
Methylation of 4-hydroxyestradiol by COMT is inhibited by
2-hydroxy-estradiol (Roy et al., 1990). Interestingly, tissues which
develop estradiol-induced tumours (rat pituitary, male Syrian hamster
kidney and mouse uterus) have very high concentrations of endogenous
catecholamines (up to 50-fold relative to other strains or species and
non-target tissues). Catecholamines in target tissues may inhibit or
compete for COMT-catalysed methylation, thus leading to increased
concentrations of hydroxylated metabolites of estradiol (Zhu and
Conney, 1998a).
1.2.2.2 Conjugation
Studies of the conjugation of estrogens with glucuronic acid or
sulfate have been reviewed in detail (IARC, 1979). Lysosomes from male
Syrian hamster livers and kidneys can catalyse the deconjugation of
estradiol and estrone glucuronides. The rates of deconjugation of
estrogen glucuronides were higher in kidney than in liver, by 56% for
estrone and 34% for estradiol. Treatment of hamsters for nine days
with subcutaneous implants containing 25 mg estradiol (releasing 61
µg/day) increased lysosomal estrone and estradiol 3ß-glucuronidase
activity in kidney by 15 and 25%, respectively, and by about 100% in
liver. Estradiol was deconjugated at negligible rates in both liver
and kidney (Zhu et al., 1996). Human liver microsomal sulfatases
convert estrone sulfate to estrone before 16 alpha-hydroxylation
(Huang et al., 1998). Estrone sulfate, the most abundant estrogen in
blood, and other estrogen conjugates may serve as a circulating
reservoir of estradiol, and regulation of deconjugation reactions may
affect intracellular estradiol concentrations.
Demethylation of catechol estrogens has also been reported. The
rates of demethylation of 2-and 4-methoxyestradiol were about equal in
kidney microsomes, but the rate of 2-methoxyestradiol demethylation in
liver was fivefold higher than that of 4-methoxyestradiol. Estradiol
treatment decreased hepatic 2-methoxyestradiol demethylation by about
20% relative to controls with little effect on 4-methoxyestradiol
demethylation, whereas the opposite was observed in kidney (Zhu
et al., 1996).
In the absence of conjugation, a pathway for further catalysis of
catechol estrogens has been suggested. Redox cycling of catechol
(hydroxyquinone) to quinone through semiquinone intermediates is
catalysed by oxidation of catechol estrogens by peroxidases or CYP1A1
lipid hydroperoxide cofactors. Reduction of the quinone to the
hydroquinone is catalysed by NADPH-dependent P450 reductase and other
enzymes. Oxygen radicals formed in this redox process may increase the
carbonyl content of proteins, formation of DNA 8-hydroxydeoxyguanosine
adducts, and lipid peroxidation. The authors concluded that redox
cycling is a critical step in estrogen-mediated carcinogenesis (Wang &
Liehr, 1994; Yager & Liehr, 1996).
1.2.3 Biochemical parameters
1.2.3.1 Synthesis
In mammals, estradiol, estrone, and estriol are synthesized from
steroid precursors in the gonads, adrenal cortex, and placenta or
through peripheral conversion of androgens in other tissues of
mammals. Cholesterol, obtained primarily from circulating low-density
lipoprotein, serves as the precursor for steroid biosynthesis,
although steroidogenic cells are capable of local cholesterol
synthesis de novo. In non-pregnant premenopausal women, the
principal estrogens found in the blood are estradiol and estrone.
Estradiol is synthesized and secreted primarily from ovarian granulosa
cells, whereas most estrone (approximately 40% of total estrogens) is
formed peripherally from estradiol and androstenedione. Estradiol and
estrone may be intra converted through the action of the enzyme
17ß-hydroxysteroid dehydrogenase. Estrone may be further metabolized
to estriol, primarily in the liver. Estriol is also formed in the
fetal liver and in the placenta from
16alpha-hydroxydehydroepiandrosterone sulfate, which is secreted from
the fetal adrenal and circulating dehydroepiandrosterone sulfate. At
least 90% of urinary estriol is derived from fetal sources (Carr,
1992). In men and postmenopausal women, the source of serum estradiol
is peripheral conversion of androgens by the enzyme aromatase. In men,
approximately 0.3% of plasma testosterone is aromatized to estradiol;
an additional contribution of approximately 25% of the total estradiol
may be due to testicular secretion (Griffin & Wilson, 1998). Estrone
is the predominant circulating estrogen in postmenopausal women,
formed by peripheral conversion of adrenal androgens in adipose
tissue.
Gonadal synthesis of estradiol is regulated by luteinizing and
follicle stimulating hormones secreted by the anterior pituitary
gland. The secretion of these two hormones is regulated by
gonadotropin-releasing hormone secreted by the hypothalamus, steroid
hormones, and other factors in a complex feedback loop which
effectively regulates the serum concentrations of hormone within a
physiological concentration range, which is particularly variable in
premenopausal women. The feedback loop is controlled by the dominant
circulating hormone (estradiol and progesterone in women, testosterone
in men). Feedback control for estradiol in men and for testosterone in
women is therefore not operative (Wilson et al., 1998). There is
evidence that this feedback loop exists in prepubertal children but is
quiescent.
In humans, plasma estradiol concentrations generally remain low
during the first 12 years of life. Around the time of menarche, rising
plasma concentrations of gonadotropins from the anterior pituitary
stimulate the ovary to produce estradiol. During a normal menstrual
cycle, plasma estradiol concentrations change very little throughout
the first half of the follicular phase but increase as the follicles
develop, reaching serum concentrations that are up to nine times
greater than the basal concentrations near mid-cycle. After the
mid-cycle surge, the estradiol concentrations fall precipitously.
During the luteal phase, the serum estradiol concentrations rise to a
plateau for 8-10 days, before declining. Should fertilization occur,
the corpus luteum formed from the dominant follicle after ovulation
remains active as the principal source of estradiol for the first 6-8
weeks of pregnancy. The corpus luteum is later supplanted by the
placenta as the site of estrogen synthesis. As the placenta lacks
17 alpha-hydroxylase, fetal and maternal circulating androgens are
necessary for placental estrogen synthesis. During pregnancy, the
feto-placental unit secretes a large quantity of estriol into the
maternal circulation, which is ultimately excreted in the urine.
The concentrations of circulating estrogens, their daily
production, and their metabolic clearance rates can be found in
previous reviews (IARC, 1979, specifically 'General remarks on sex
hormones') and in most textbooks of endocrinology (e.g. Braunstein,
1994; Goldfien & Monroe, 1994; Carr, 1998; Griffin & Wilson, 1998; see
also textbook appendices for summary tables). They are summarized in
Table 1. Somewhat different values can be calculated for the basal
production rate of estradiol in prepubertal boys and girls
(Angsusingha et al., 1974; IARC, 1979) when measurements of hormone in
urine or plasma are used as the basis for the calculation (4 or 12
mg/day).
1.2.3.2 Mechanism of action
The conventional view of the action of steroid sex hormones
involves interaction of the sex hormone with specific intracellular
receptors, which subsequently bind tightly to specific DNA sequences
in the genome (Malayer & Gorski, 1993). This tight nuclear binding
initiates transcription of specific genes, which ultimately leads to
physiological events. These include include development of
reproductive tissues, maturation of the ovarian follicle, development
of the uterus and vagina, and ductal development in the breast.
Estrogen withdrawal results in menstruation. In non-reproductive
tissues, estrogens may affect bone growth and prevent bone resorption,
and effects on the plasma lipid profile through action in the liver.
Estrogens typically promote cell growth or cell proliferation in
responsive cells in culture.
Table 1. Levels of circulating estrogens, metabolic clearance rates, and
daily production
Sex Age or phase Serum Metabolic Total daily
concentration clearance production
(pg/ml) (L/day) (mg/day)
Male 1400
Prepubertal < 10 < 0.014
12-16 years < 23 < 0.031
> 16 years 20-50 0.027-0.068
Female 1400
< 8 years < 7 < 0.01
2-12 8-18 0.01-0.024
12-14 16-34 0.02-0.09
14-16 20-68 0.03-0.09
Early follicular 20-100 0.03-0.14
Preovulatory 100-350 0.14-0.47
Luteal 100-350 0.14-0.47
Late pregnancy 18 000 24
Postmenopausal 10-30 0.01-0.04
It has been proposed that the carcinogenicity of estrogens is
distinct from its hormonal properties; however, since that hypothesis
originated, information on estrogen-receptor and ligand interactions
has been re-evualuated in the light of recent identification of
additonal estrogen receptor proteins. Three specific receptors have
now been identified for the endogenous ligand estradiol: the classical
receptor ER alpha, ERß, and ERß2. Analyses of ER alpha and ERß RNA
indicate that ER alpha is widely distributed and that ER ß is
prominent in prostate, ovary (localized to the granulosa cells of the
maturing ovary), epididymis, urinary bladder, uterus, lung, thymus,
colon, small intestine, vessel walls, pituitary glan, hypothalamus,
cerebellum, and brain cortex (Couse et al., 1997a; Kuiper et al.,
1998). These receptor isoforms differ in their agonist and antagonist
reactions to agents such as tamoxifen and to classes of agents
variously referred to as 'endocrine disruptors' or 'xenoestrogens' and
to endogenous estrogen metabolites such as 2-hydroxyestradiol.
Moreover, alpha-estrogen receptor knockout (alpha ERKO; Couse et al.,
1997a) and ERß-/- female mice (Krege et al., 1998) have very different
phenotypes. Alpha ERKO females lack the ability to complete
folliculogenesis, are infertile, and have multiple cystic,
haemorrhagic, and atretic follicles, whereas ERß-/- female mice
develop normally and are indistinguishable from their wild-type
littermates. These mice are fertile, but their fertility is
compromised, as demonstrated by reductions in litter size. Mammary
gland development is normal in ERß-/- mice, in contrast to the absence
of breast development beyond that of prepubertal females in alpha ERKO
mice. Male mice lacking ERß are also fertile (alpha ERKO males are
infertile) but develop prostate and bladder hyperplasia as they age.
These ERKO mice and the receptor-binding properties of various ligands
demonstrate that ER alpha and ERß have different functional
responsibilities.
Abundant evidence exists that hormonal carcinogenicity is linked
to the relative balance of various estrogens. Proliferation of mammary
glands and other reproductive (i.e. target) tissues is inextricable
linked to hormonal changes during the menstrual cycle, pregnancy, and
the initiation or cessation of cyclic menstruation. The cyclic
proliferation of tissues is correlated to the cellular content of the
estrogen receptor. In cultured cells, the growth of cells with
estrogen receptors (e.g. MCF-7 cells) but not those without estrogen
receptors (e.g. MDA-MB-231 cells) is dependent on the estradiol
concentration in serum. Molecular genetic studies of human cancer
indicate that the progression from a normal to malignant phenotype
requires activation of one or several oncogenes and/or inactivation of
tumour suppressor genes. These events require cell division (reviewed
by Bernstein & Ross, 1993; Russo & Russo, 1996; Tsai et al., 1998).
A functional role for catechol estrogens distinct from that for
estradiol has been suggested (reviewed by Zhu & Conney, 1998a,b).
Because of the extremely short half-lives of these compounds
(Schneider et al., 1984), they are unlikely candidates for circulating
hormones. Locally, 2-hydroxyestradiol reportedly stimulates
progesterone production in ovarian granulosa cells (Spicer et al.,
1990), and 2-hydroxyestrone has been shown to inhibit MCF-7 cell
proliferation in the presence of quinalizarin, a potent inhibitor of
COMT. This challenge can be overcome with physiological concentrations
of estradiol. No effects on cell growth were observed with
concentrations of 10-9 to 10-6 mol/L of 2-hydroxyestradiol in the
absence of methyltransferase inhibition (Schneider et al., 1984).
The endogenous metabolite 2-methoxyestradiol has been suggested
to be an antiangiogenic factor and tumour suppressor (Fotis et al.,
1994; Zhu & Conney, 1998a); however, the concentrations required to
induce apoptotis in cultured cells are about 10 times greater than
those observed in humans (Yue et al., 1997). Those authors suggested
that unless the concentration of 2-methoxyestradiol in the lipid phase
(i.e. membranes) exceeds that in the aqueous phase, as reported for
some lipophilic calcium blockers, the antiangiogenic properties of
2-methoxyestradiol are of little physiological relevance.
A role for catechol estrogens in implantation in the mouse uterus
has been suggested on the basis of the observation of increased
activity of 4-hydroxylase activity on day 4 of pregnancy (Paria et
al., 1990). 2-and 4-Hydroxyestradiol bind to the estrogen receptor
with reduced relative affinities of 23 and 26%, respectively (Schultze
et al., 1994). Additionally, a distinct signaling pathway separate
from the estrogen receptor may exist for 4-hydroxyestradiol. In
alpha ERKO mice, 4-hydroxyestradiol stimulated up-regulation of
lactoferrin mRNA and water imbibition (Das et al., 1997). In these
mice, the effects of 4-hydroxyestradiol could not be mimicked by
estradiol, nor could the effects be blocked by the anti-estrogen ICI
182,780; however, it has been postulated that the estrogenic effects
in the uterus in alpha ERKO mice are mediated through ERß (Krege et
al., 1998).
The physiological effects of estradiol that are reported not to
be receptor-mediated (i.e. not mediated via the classical receptor
mechanism) include those on myometrial, neuronal, pituitary, maturing
oocyte, and granulosa cells (Wehling, 1997). Estradiol may protect
against oxidative damage in neuronal cells (Behl et al., 1995), but
very high concentrations of estradiol were required for this effect:
10-5 but not 10-7 mol/L was effective in preventing cell death. At
physiological concentrations in blood, the antioxidant properties of
estradiol protect against oxidation of low-density lipoprotein (Rifici
& Kachadurian, 1992; Hoogerbrugge et al., 1998). Novel so-called
'scavestrogens', structurally related to 17 alpha-estradiol, have
antioxidant properties that protect against the radical-mediated death
of cultured cells (Blum-Degen et al., 1998). Other studies indicate
that the pro-oxidant and antioxidant properties of estrogens may be
dependent on structure and concentration. Estradiol, estriol, and
methoxyestrogen metabolites had only antioxidant properties. Catechol
estrogens showed pro-oxidant properties at low concentrations (5
pmol/L to 100 nmol/L), but antioxidant properties dominated at high
concentrations (0.5-50 µmol/L) (Markides et al., 1998). In addition to
oxidant and antioxidant effects, a rapid, non-genomic effect of
estradiol, possibly mediated by cAMP, Ca++, or ion channel gating,
has been postulated (reviewed by Moss et al., 1997).
1.3 Toxicological studies
1.3.1 Acute toxicity
The therapeutic dose of fine-particle estrogen given orally is
0.5-2 mg/day. No adverse effects were reported in children after
accidental ingestion of large doses of estrogen-containing oral
contraceptives (Physician's Desk Reference, 1999). An
electroencephalogram of a young woman who took 160 mg of estradiol
valerate (80 tablets of 2 mg), which is converted to estradiol-ß
in vivo, showed traces typical of subcortical disturbance on the
first day; however, the recording was normal one week later (Punnenon
& Salmi, 1983).
1.3.2 Short-term studies of toxicity
Estradiol was administered in the diet to female Crl:CD BR rats
at doses equal to 0. 0.003, 0.17, 0.69, or 4.1 mg/kg bw per day for 90
days. The end-points were chosen to evaluate both short-term and
reproductive toxicity and several mechanistic and biochemical
parameters. Administration of doses > 0.17 produced dose-dependent
increases in body weight, food consumption, and feed efficiency. At
0.69 and 4.1 mg/kg bw per day, minimal to mild non-regenerative
anaemia, lymphopenia, decreased serum cholesterol (at the high dose
only), and altered splenic lymphocyte subtypes were observed. Changes
in the weights of several organs were noted. Evidence of ovarian
malfunction (reduced corpora lutea and large antral folicles) was
found at doses > 0.17 mg/kg bw per day. Pathological changes in
males and females fed 0.69 or 4.1 mg/kg bw per day included
centrilobular hepatocellular hypertrophy, diffuse hyperplasia of the
pituitary gland, feminization of the male mammary gland, mammary gland
hyperplasia in females, cystic ovarian follicles, hypertrophy of the
endometrium and endometrial glands in the uterus, degeneration of the
seminiferous epithelium, and atrophy of the testes and acessory sex
glands (Biegel et al., 1998b).
1.3.3 Long-term studies of toxicity and carcinogenicity
The toxicity of estrogens, including estradiol and related
esters, has been reviewed extensively by working groups convened by
the IARC (1979, 1987). After reviewing studies of estradiol
administered orally to mice and by subcutaneous injection or
implantation in mice, rats, hamsters, guinea-pigs, and monkeys, the
group concluded that there is sufficient evidence for the
carcinogenicity of estradiol in experimental animals, noting that:
"Administration to mice increased the incidences of mammary,
pituitary, uterine, cervical, vaginal, testicular, lymphoid and bone
tumours. In rats, there was an increased incidence of mammary and/or
pituitary tumours. estradiol-17b produced a statistically
nonsignificant increase in the incidence of foci of altered
hepatocytes and hepatic nodules induced by partial hepatectomy and
administration of N-nitrosodiethylamine in rats. In hamsters, a high
incidence of malignant kidney tumours occurred in intact and castrated
males and in ovariectomized females, but not in intact females. In
guinea-pigs, diffuse fibromyomatous uterine and abdominal lesions were
observed.' The IARC working group concluded that the carcinogenic
effects of estrogens and progestogens were inextricably linked to the
hormonal milieu and to dose-effect relationships (IARC, 1987).
Hormonal effects on non-cancer end-points were not evaluated by the
groups.
The induction of renal tumours by various steroidal and
non-steroidal estrogens was examined in castrated male Syrian hamsters
treated subcutaneously for nine months with a capsule that released an
average of 110 µg of the hormone per day. The tumour incidences
associated with the hormonal activity of the substances tested are
presented in Table 2. These data demonstrate a good correlation among
the hormonal parameters progesterone receptor induction and serum
prolactin and relative estrogenic potency (estrogen receptor binding)
in hamster kidney. All animals trested with estrone, equilin plus
d-equilin, or Premarin(R) developed renal tumours, with combined
numbers of tumour foci in both kidneys of 15, 18, and 16, respectively
(Li et al., 1995).
Castrated adult male Syrian hamsters were treated for eight
months with subcutaneous pellets containing 20 mg estrogen or
anti-estrogen; the release rates in µg/day were as follows:
diethylstilbestrol, 156; ethinyl-estradiol, 215; estradiol, 96;
estradiol-17 alpha, 104; 17 alpha-ethinyl-11ß-methoxy-estradiol
(moxestrol), 104; and tamoxifen, 183. Treatment with ethinylestradiol
resulted in progressive dysplasia but no renal tumours, but dysplasia
was observed in the proximal tubules of the renal cortex, which was
uncommon in animals treated with diethylstilbestrol or estradiol.
Animals treated with estradiol, diethylstilbestrol, or moxestrol had a
tumour incidence of 100%, which was completely abolished by
concurrrent treatment with ethinylestradiol (Table 3). Simultaneous
administration of diethylstilbestrol and estradiol-17ß or estradiol-17
alpha (a noncarcinogenic estrogen) did not mitigate the
carcinogenicity of diethylstilbestrol (Li et al., 1998).
The carcinogenicity of estradiol and its metabolites was
investigated in groups of 20-35 B6C3F1 mice of each sex given a
single daily intraperitoneal injection of the compound on four
consecutive days starting at 12 days of age. They were then maintained
for approximately 18 months and killed. Of the catechol estrogens and
their quinones tested, only estrone-3,4-quinone was significantly
carcinogenic in the livers of male mice (Table 4). It was also highly
toxic, as most of the mice died from unknown causes shortly after
treatment. Estrone was protective against liver tumour formation in
this system, and few tumours were induced in female mice (Cavalieri et
al.,1997).
The tumour formation in the kidneys of male Syrian hamsters given
25-mg pellets containing estrogen or catechol estrogen:cholesterol
(90:10) by subcutaneous implantation and killed 175 days later was
studied histologically. Estradiol and 4-hydroxyestradiol each induced
renal tumours in four of five animals, whereas neither
2-hydroxyestradiol nor 2-methoxystradiol was carcinogenic. The lack of
carcinogenicity of 2-hydroxyestradiol was not due to failure of the
hormone to stimulate cell growth in vivo, as estradiol,
4-hydroxyestradiol, and 2-hydroxyestradiol supported the growth of
estrogen-dependent H-301 cells injected into male hamsters.
2-Methoxyestradiol was not effective in stimulating these cells. The
authors note that none of the three compounds was mutagenic in
Salmonella typhimurium TA100 strain (see below; Liehr et al., 1986).
The role of estrogen and its metabolites in tumour formation was
examined in castrated male Syrian hamsters implanted for 9-10 months
with pellets containing various estrogens (doses not given). The
results are shown in Table 5. The authors suggested that the
neoplastic changes seen in hamster kidney after continuous exposure to
estrogens are due to synergistic action between hormonal and
carcinogenic factors (Li & Li, 1989).
Castrated male Syrian hamsters received subcutaneously implanted
pellets containing diethylstilbestrol, alpha-dienestrol, hexestrol,
diethylstilbestrol 3,4-oxide, estradiol, estrone, ethinylestradiol,
equilin or (+)-equilenin, which released 100-210 µg/day, for a total
of nine months. The tumour incidence was 75-100%, except with
ethinylestradiol (20%) and (+)-equilenin (0%). The ability of
estrogens to cause renal tumours correlated well with their ability to
compete for estrogen receptor binding, with the notable exception of
ethinylestradiol (Li et al., 1983). The tumours induced by
ethinylestradiol are of a different origin than other hormone-induced
renal tumours (Oberley et al., 1991). The presence of estrogen
receptors, probably ER alpha, in interstitial cells of control and
estrogen-treated hamsters was confirmed by immuno-histochemical
staining and northern blotting. Receptors were also found in renal
corpuscles, arterial cells, and interstitial capillaries but not in
the tubular epithelia of the cortex, further indicating that the
tumours have an interstitial or mesenchymal origin (Bhat et al.,
1993). The presence of ERß, which was identified after the study, has
not been investigated.
Virgin female C3H/HeJ mice with a high titre of antibodies to the
mouse mammary tumour virus were fed diets that provided doses
equivalent to 0.015, 0.15, or 0.75 mg/kg bw per day estradiol from
week 6 to week 110 of age (Highman et al., 1978). The microscopic
findings in animals sacrificed after 52 weeks of feeding are shown in
Table 6. In a continuation of this study, the authors reported the
preneoplastic and neoplastic findings in mice sacrificed after up to
104 weeks on the estrogenic diets (Highman et al., 1980). The results
are shown in Tables 7 and 8. Uterine cervical adenosis may be a
precursor of cervical adenocarcinoma in C3H/HeJ mice and may therefore
serve as an early indicator of the uterine carcinogenicity of a
compound. In these experiments, high doses of estradiol increased the
incidence of adenosis but did not affect the incidence of ovarian
tubular adenomas. After 66-91 weeks of treatment, high doses of
estradiol also increased the incidence of mammary gland hyperplastic
alveolar nodules but not mammary adenocarcinomas. The authors reported
the occurrence of several other sporadic tumours at different sites in
both control and treated animals. They concluded that the incidence of
lesions in mice given estradiol was generally dose-dependent,
indicating that this compound either induces or facilitates the
development of these lesions (Highman et al., 1980)
Table 2. Estrogenicity and carcinogenicity of various steroidal and stilbene estrogens in Syrian hamster kidney
Estrogen % of competitive Induction of Serum % of animals No. of tumour
binding to estrogen progesterone prolactin with tumours foci in both
receptors due to receptors in kidney (ng/ml) kidneys
renal tumours (fmol/mg protein)
Steroidal
Estradiol-17ß 55 48 390 100 17
11ß-Methoxy ethinyloestradiol 52 60 330 100 22
16 alpha-Hydroxyestrone 48 45 390 38 3
11ß-Methoxyestradiol 30 35 390 25 2
11ß-Methylestradiol 14 18 150 0 0
Estradiol-17 alpha 34 6 130 0 0
Deoxestrone 14 8 94 0 0
Stilbene
Diethylstilbestrol 46 50 450 100 19
Indenestrol B 46 49 280 100 11
Indanestrol 10 29 100 0 0
From Li et al. (1995)
Table 3. Prevention of the carcinogenicity of estrogens by ethinylestradiol
Estrogen % tumour induction No. of tumour
nodules in both
kidneys
Diethylstilbestrol 100 15
Estradiol-17ß 100 13
Ethinylestradiol 10 2
Moxestrol 100 18
Diethylstilbestrol + ethinylestradiol 0 0
17ß-Estradiol + ethinylestradiol 0 0
Moxestrol + ethinylestradiol 0 0
Diethylstilbestrol + diethylstilbestrol 100 12
Diethylstilbestrol + 17ß-estradiol 100 12
Diethylstilbestrol + 17 alpha-estradiol 100 9
From Li et al. (1998)
Intact and ovariectomized or hysterectomized nulliparous female
C3H/HeJ mice, five months of age, received either estradiol at a dose
of 0.5 mg/L in drinking-water for one year or estradiol plus daily
injections of 0.1 mg 2-bromo-alpha-ergocryptine, an effective
suppressor of prolactin secretion. All mice were examined weekly for
mammary tumours. One year after the onset of treatment, all surviving
mice were sacrificed. The formation of mammary hyperplastic nodules
was highly significantly suppressed in mice that received both
estradiol and 2-bromo-alpha-ergocryptine, and the mammary tumour
incidence was slightly but significantly reduced in comparison with
that in animals receiving estradiol alone. The tumour incidence in
concurrent controls was not reported. In a separate study, nulliparous
30-day-old mice received estradiol in the drinking-water with or
without a daily injection of 0.1 mg 2-bromo-alpha-ergocryptine for
19-20 months. The effects on the mammary gland are shown in Table 9.
The authors concluded that at least a portion of the oncogenic
activity of estrogenic steroids on the mammary gland in rodents is
manifested through a stimulatory effect on prolactin secretion
(Welsch, 1976).
The results of short-and long-term studies of the carcinogenicity
of estrogens are summarized in Table 10.
Table 4. Carcinogenicity of estradiol and its metabolites in male
B6C3F1 mice
Compound Dose No. of mice with tumours/
(µmol/kg bw) total no. of animals (%)
Estradiol-17ß 30 6/20 (30)
2-Hydroxy-17ß-estradiol 30 10/28 (36)
4-Hydroxy-17ß-estradiol 30 10/24 (42)
17ß-Estradiol-2,3-quinone 7.5 8/26 (31)
17ß-Estradiol-3,4-quinone 7.5 4/21 (19)
Estrone 30 3/32 (9.4)
2-Hydroxyestrone 30 9/30 (30)
4-Hydroxyestrone 30 8/33 (24)
Estrone-2,3-quinone 7.5 12/25 (48)
Estrone-3,4-quinone 3.7 6/10 (60)
Benzo[a]pyrene 60 12/12 (100)
Solvent 7/19 (37)
Untreated 11/33 (33)
From Cavalieri et al. (1997)
1.3.4 Genotoxicity
A working group convened by IARC evaluated the results of tests
for genetic toxicity conducted with estradiol and concluded:
"Oestradiol-17ß did not induce chromosomal aberrations in bone-marrow
cells of mice treated in vivo. Unusual nucleotides were found in
kidney DNA of treated hamsters. It induced micronuclei but not
aneuploidy, chromosomal aberrations or sister chromatid exchanges in
human cells in vitro. In rodent cells in vitro, it induced
aneuploidy and unscheduled DNA synthesis but was not mutagenic and did
not induce DNA strand breaks or sister chromatid exchanges.
Oestradiol-17ß was not mutagenic to bacteria." The group also
concluded that the limited data available on estrone and estriol were
indicative of genotoxicity (IARC, 1987).
A mechanism has been proposed by which catechol estrogens
interact with DNA. Nonmethylated catechol estrogens can be oxidized to
a quinone which can bind to DNA; thus, 2-and 4-hydroxy estradiol
produce 2,3-and 3,4-quinones, respectively. Reaction of the
3,4-quinone of estrone or estradiol with deoxyguanosine (dG) at N7
resulted in loss of the deoxyribose moiety and thus induced
depurinating adducts. No adducts were observed after reaction of the
3,4-quinone with deoxyadenosine (dA). Reaction of estrone-2,3-quinone
with dG and dA produced a stable N2-dG or N6-dA adduct, the
deoxyribose group remaining intact. Formation of depurinating and
stable adducts in calf thymus DNA by activated catechol estrogens and
in mammary glands of female Sprague-Dawley rats injected with 200 nmol
4-hydroxy-estrone was confirmed by the 32P-postlabelling technique.
The authors suggested that the formation of depurinating adducts via
3,4-quinone followed by misreplication of unrepaired apurinic sites
are the critical steps in initiation of cancer by estrogens (Cavalieri
et al., 1997; Stack et al., 1998).
Studies of the genetic toxicology of estrogens are summarized in
Table 11.
The mutagenicity of estradiol and its metabolites was assessed in
Salmonella typhimurium strain TA100, but the authors concluded that
none of the compounds was mutagenic in this assay (Liehr et al.,
1986). Estradiol was evaluated in several short-term tests for
genotoxic potentiol and at 1, 10, or 100 µg/ml was found to cause
chromosomal aberrations and sister chromatid exchange in cultured
human lymphocytes with and without metabolic activation. In the
absence of metabolic activation, the lowest dose caused aberrations
after 72 h of treatment but not after 24 or 48 h; the intermediate
dose caused aberrations after 48 and 72 h but not after 24 h of
treatment. With a 6-h treatment, aberrations were observed at 10 and
100 µg/ml in the presence of metabolic activation but not in its
absence. Estradiol caused sister chromatid exchange at most doses with
or without metabolic activation (Dhillon & Dhillon, 1995).
Table 5. Carcinogenicity of estradiol and its metabolites in castrated
male Syrian hamsters
Compound No. of mice with tumours/
total no. of animals (%)
Estradiol-17ß 6/6 (100)
Estrone 8/10 (80)
Estriol 4/7 (57)
2-Hydroxy-17ß-estradiol 0/6 (0)
2-Hydroxyestrone 0/6 (0)
4-Hydroxy-17ß-estradiol 5/5 (100)
4-Hydroxyestrone 2/6 (33)
Ethinylestradiol 3/15 (20)
Equilin 6/8 (75)
(+)-Equilenin 0/9 (0)
From Li & Li (1989)
Table 6. Percent incidence of pathological changes in female mice given estradiol for 52 weeks
Dose No. of Effects in the Effects in the Effects in the
(mg/kg bw animals cervix uterine horn mammary gland
per day)
Adenosis in Adenosis in Glandular Hyperplastic Adeno- Osseus
upper third upper and hyperplasia alveolar carcinoma hyperplasia
lower thirds nodules
0 47 11 0 26 0 4 0
0.015 35 17 0 24 0 0 0
0.15 36 15 0 62 3 6 18
0.75 48 38 36 96 9 8 82
From Highman et al. (1977)
Table 7. Incidences (and%) of uterine cervical adenosis and ovarian tubular
adenomas in mice fed estradiol
Dose Week
(mg/kg bw
per day) 26 52 78 104
Adenosis Adenosis Adenosis Tubular Adenosis Tubular
adenoma adenoma
0 2/37 (5) 5/46 (11) 1/14 (7) 2/14 (14) 13/19 (16) 12/24 (50)
0.015 1/37 (3) 5/29 (17) 1/5 (20) 0/5 (0) 3/24 (12) 11/25 (44)
0.15 5/45 (110) 5/35 (14) 3/14 (21) 2/16 (12) 8/20 (40) 12/20 (60)
0.75 25/42 (60) 33/45 (73) 4/7 (57) 0/7 (0) 3/6 (50) 3/7 (43)
Table 8. Incidences (and%) of pathological changes in the mammary gland of female mice fed estradiol
Dose Hyperplastic alveolar nodules Adenocarcinomas
(mg/kg bw
per day) Weeks 0-39 Weeks 40-65 Weeks 66-91 Weeks 92-105 Weeks 0-39 Weeks 40-65 Weeks 66-91 Weeks 92-105
0 0/91 (0) 0/57 (0) 3/29 (10) 6/50 (12) 4/91 (4) 15/57 (26) 13/29 (45) 19/50 (38)
0.015 0/89 (0) 0/63 (0) 0/19 (0) 4/31 (13) 0/89 (0) 28/63 (44) 8/19 (42) 13/31 (42)
0.15 0/94 (0) 2/56 (4) 8/29 (28) 7/21 (33) 4/94 (4) 21/56 (37) 14/24 (58) 6/21 (29)
0.75 0/93 (0) 5/78 (6) 5/19 (26) 6/17 (35) 5/93 (5) 34/78 (44) 11/19 (58) 8/17 (47)
From Highman et al. (1980)
Table 9. Effects of treatment with estradiol with or without 2-bromo-alpha-ergocryptine on the number of mammary hyperplastic
nodules and mammary tumours in young nulliparous C3H/HeJ mice
Treatment No. of mice at No. of mice at No. of hyperplastic nodules No. of mice with
start of study end of study in inguinal mammary glands mammary tumours
Controls 100 16 3.1 11
Estradiol 100 12 4.8 27
Estradiol + 2-bromo-alpha-ergocryptine 100 28 2.8 9
Table 10. Summary of results of short-term and long-term studies
of the carcinogenicity of estrogens
Species Dose Findings Reference
Short-term study
Rats 0.003-4.12 mg/kg bw Histopathological changes, particularly at Biegel et al. (1998b)
per day for 90 days intermediate and high doses
Long-term studies
Mice Increased incidences of mammary, pituitary, IARC (1979)
uterine, vaginal, testicular, lymphoid, and
bone tumours
Rats Mammary and pituitary tumours; statistically IARC (1979)
nonsignificant increase in incidence of foci
of altered hepatocytes and hepatic nodules
after partial hepatotectomy and adminsitration
of N-nitrosodiethylamine
Hamsters Malignant renal tumours in intact and IARC (1979)
castrated males and in ovariectomized
but not intact females
Castrated male 111 µg/day 100% renal tumour incidence Li et al. (1995)
Syrian hamsters,
9 months
Castrated male 96 µg/day 100% incidence of renal tumours; modulated by Li et al. (1998)
Syrian hamsters, ethinylestradiol
8 months
B6C3F1 mice 4 daily Estrone was protective; estradiol-17ß did not Cavalieri et al.
intraperitoneal increase incidence over control (1997)
injections
(30 µmol/kg bw)
Table 10. (continued)
Species Dose Findings Reference
Male Syrian 25 mg subcutaneously, Estradiol-17ß and 4-hydroxy-17ß-estradiol Liehr et al. (1986)
hamsters 175 days produced tumours, but 2-hydroxy-17ß-estradiol
did not
Castrated male Not reported; Estradiol-17ß and 4-hydroxy-17ß-estradiol Li & Li (1989)
Syrian hamsters 9-10 months produced tumours, but 2-hydroxy-17ß-estradiol
did not
Castrated male 100-200 µg/day Ethinylestradiol produced a lower incidence of Li et al. (1983)
Syrian hamsters tumours than estradiol-17ß
Virgin C3H/HeJ 0.015-0.75 mg/kg bw Estradiol-17ß caused a dose-dependent increase Highman et al.
mice per day in feed in tumour incidence (1977, 1980)
C3H/HeJ mice 0.5 mg/l in Estradiol-17ß caused tumours Welsch (1976)
drinking-water
When Swiss albino mice were given a single intraperitoneal
injection of 100, 1000, or 10 000 µg/kg bw, the highest dose increased
the number of micronucleated polychromatic erythrocytes and the
frequency of sister chromatid exchange, although the
polychromatic:normochromatic erythrocyte ratio did not appear to be
affected. Estradiol did not cause reverse mutation in Salmonella
strains TA100, TA1535, TA97a or TA98, at concentrations of 1-10 000
µg/plate in the absence of metabolic activation and 1-1000 µg/plate in
its presence. In a host-mediated assay in which mice were given 100,
1000, or 10 000 µg/kg bw followed 2 h later by injection of
S. typhimurium, no change in the number of His+ revertants per
plate was observed (Dhillon & Dhillon, 1995).
Estradiol was evaluated in five tests for the induction of
micronuclei in bone marrow in vivo in female rats given three daily
subcutaneous doses of 20 µg/kg bw and in mice given a single
intraperitoneal injection of 10-150 mg/kg bw. The authors concluded
that estradiol was not genotoxic to the bone marrow of rodents (Ashby
et al., 1997).
In male B6C3F1 mice and male Fischer 344 rats that received
estradiol at 310, 620, or 1250 mg/kg bw in three injections, there was
no increase in the frequency of polychromatic erythrocytes. In male
and female B6C3F1 mice treated with various numbers of injections,
solvents, routes of administration, and killing schedules, no
significant increase in the frequency of micronuclei was observed
(Shelby et al., 1997).
The onset of genomic rearrangements was tested at 10-5 mol/L
estradiol in two X-ray-transformed cell lines (X-ray-9 and F-17a) and
two untransformed cell lines (10T1/2b and 10T1/2c). Genomic
rearrangements (deletions or additions in minisatellites) were
observed in the transformed but not the untransformed lines. No new
rearrangements were observed after withdrawal of estradiol (Paquette,
1996).
The ability of estradiol to induce morphological transformation,
gene mutations, chromosomal aberrations, sister chromotid exchange,
unscheduled DNA synthesis and other chromosomal changes was assessed
in Syrian hamster embryo cells. Cell growth was completely inhibited
at 10-30 µg/ml but was not affected at a concentration < 3 µg/ml.
Treatment of cells with 0.3-6 µg/ml did not affect their
colony-forming efficiency, but at 10 µg/ml colony formation was 53%
that of controls. Incubation with estradiol at 0.3-10 µg/ml for 48 h
induced a dose-dependent increase in the frequency of morphological
changes. Estradiol in this dose range also induced numerical changes
(chromosome gains and loses). The majority of these cells (94%) were
diploid. Estradiol had no other effects in this assay (Tsutsui et al.,
1987). No exogenous metabolic activation was used in these
experiments.
Table 11. Genetic toxicity of estrogens
End-point Test system Dose Result Reference
In vitro
Reverse mutation S. typhimurium TA100 50-1500 µg/plate Negative Liehr et al.
(1986)
Reverse mutation S. typhimurium TA100, 1-10 000 µg/plate -S9 Negative Dhillon & Dhillon
TA1535, TA97a, TA98 1-1000 µg/plate +S9 Negative (1995)
Chromosomal aberration, Cultured human 1-100 µg/ml Positive Dhillon & Dhillon
sister chromatid lymphocytes (1995)
exchange
Micronucleus formation Human cells Positive IARC (1987)
Aneuploidy, chromosomal Human cells Negative IARC (1987)
aberrations, sister
chromatid exchange
Aneuploidy, unscheduled Rodent cells Positive IARC (1987)
DNA synthesis
Mutagenicity, DNA damage, Rodent cells Negative IARC (1987)
sister chromatid
exchange
Cell transformation, Syrian hamster 0-10 µg/ml Positive Tsutsui et al.
numerical chromosomal embryo cells (-S9) (1987)
changes
Gene mutation, Syrian hamster 0-10 µg/ml Negative Tsutsui et al.
chromosomal aberration, embryo cells (-S9) (1987)
sister chromatid
exchange, unscheduled
DNA synthesis
Table 11. (continued)
End-point Test system Dose Result Reference
Numerical chromosomal Cultured human 0.05-75 µmol/l Positive Schuler et al.(1998)
changes lymphocytes
Chromosomal breakage Cultured human 0.05-75 µmol/l Negative Schuler et al.(1998)
lymphocytes
DNA damage pBR322 (-S9) 0.01-0.1 mmol/l Single-strand Yoshie & Ohshima
breaks with 2- (1998)
and 4-hydroxy
estradiol and
estrone; negative
with estradiol
and estrone
Microtubule disruption Chinese hamster 0-100 µmol/l EC50, 10 µmol/l Aizu-Yokota et al.
V79 cells (1995)
Adduct formation
Syrian hamster embryo cells 1 µg/ml (-S9) Increase with Hayashi et al.
estradiol and (1996)
2- and 4-hydroxy
estradiol
Syrian hamsters 2-150 mg/kg bw Increase in Han & Liehr
intraperitoneally kidney at 50 (1994a)
mg/kg bw; no
increase in liver
Male Syrian hamsters 50 mg/kg bw Increase in Han & Liehr
intraperitoneally kidney; no (1994a)
time dependence
Table 11. (continued)
End-point Test system Dose Result Reference
Male Syrian hamsters 100 mg/kg bw Increase in liver Han & Liehr
intraperitoneally 1-2 h after (1994a)
dosing but not
later
Male Syrian hamsters 25 mg Increase in Han & Liehr
subcutaneous implant kidney on day 3 (1994a)
but not day 6;
no hepatic
adducts;
substantial
differences in
adduct levels in
controls between
days 3 and 6
Male Syrian hamsters 100 mg/animal per Negative for Han & Liehr
day intraperitoneally kidney with (1994a)
for 3 days estradiol and
2- and 4-hydroxy
estradiol
NBL rats Dose not reported; Unidentified Han et al.
serum level 14 times adduct after 16 (1995)
that of control but not 8 weeks
of treatment
Mongrel dogs Dose not reported Decrease in Winter & Liehr
prostate adduct (1996)
level; increase
in carbonyl
content
Table 11. (continued)
End-point Test system Dose Result Reference
Human liver 2 mmol/l Negative Seraj et al.
(1996)
Rat liver 2 mg/kg bw per day Adducts in male Feser et al. (1996)
by gavage but not for female liver
14 days
Human breast tissue Positive Musarrat et al.
correlation (1996)
Human breast tissue No correlation Nagashima et al.
(1995)
In vivo
Micronucleus formation, Mouse bone marrow 100-10 000 µg/kg Positive at Dhillon & Dhillon
sister chromatid exchange bw as single highest dose (1995)
intraperitoneal
injection
Micronucleus formation Rat bone marrow 20 µg/kg bw as Negative Ashby et al.
three daily (1997)
subcutaneous
injections
Micronucleus formation Mouse bone marrow 10-10 mg/kg bw Negative Ashby et al.
as single (1997)
intraperitoneal
injection
Micronucleus formation Mouse and rat bone 0.1-10 mg/kg bw Negative Shelby et al.
marrow intraperitoneally (1997)
Table 11. (continued)
End-point Test system Dose Result Reference
Frequency of Male and female mice 310-1250 mg/kg bw Negative Shelby et al.
polychromatic as three (1997)
erythrocytes injections
Male Syrian hamster 5-150 mg/kg bw Negative Han & Liehr
intraperitoneally (1994a)
DNA damage Male Syrian hamster kidney 25 mg subcutaneously Single-strand Han & Liehr
and liver every two weeks breaks in kidney (1994a)
but not liver
DNA damage Male Syrian hamster 250 µg/animal per Single-strand Han & Liehr
day for 7 days by breaks with (1994a)
infusion estradiol and
4-hydroxy but
not 2-hydroxy
Chromosomal aberration Male Syrian hamster 20 mg via Positive in Banerjee et al.
subcutaneous capsule kidney (1994)
DNA damage NBL rat Subcutaneous capsules, Single-strand Ho & Roy (1994)
16 weeks; dose breaks in
not reported prostate with
estradiol +
testosterone
The effect of estradiol at 0.05-75 µmol/L was examined in
cultured human lymphocytes by multicolour fluorescence in-situ
hybridization. DNA probes for the centromere and adjacent
heterochromatin regions of chromosomes 1, 9, and 16 were used to
detect hyperdiploidy, polyploidy, and chromosomal breakage. Nonlinear
increases in hyperdiploidy but no chromosomal breakage was observed.
The authors concluded that induction of numerical changes in
chromosomes by estradiol followed a sublinear dose-response
relationship, probably with a threshold concentration. Binding of
estradiol to microtubules or saturation of detoxification mechanisms
are possible explanations for the observation (Schuler et al., 1998).
Male Syrian hamsters were treated with intraperitoneal injections
of 5, 15, or 150 mg/kg bw estradiol; subcutaneous implants containing
25 mg estradiol for two weeks; or continuous infusion of estradiol or
2-or 4-hydroxy-estradiol at 250 mg/animal per day for seven days. A
single intraperitoneal injection of estradiol had no effect on DNA
single-strand breaks in liver or kidney DNA, but the subcutaneous
implants increased the number of renal single-strand breaks by 10%; no
effect was seen in liver. Infusion of estradiol or 4-hydroxyestradiol,
but not 2-hydroxyestradiol, for one week resulted in a 9% increase in
the number of single-strand breaks relative to untreated controls. The
authors suggested that estrogen-induced carcinogenesis is mediated by
free-radical damage (Han & Liehr, 1994a).
Male Syrian hamsters received capsules containing 20 mg
diethyl-stilbestrol, estradiol, moxestrol, 17 alpha-estradiol, or
ß-dienestrol, and between 94 (dienestrol) and 140 (diethylstilbestrol)
µg/day were obsorbed daily from the pellet. Animals were sacrificed at
0.5, 1, 2, 3, 4, or 5 months (diethylstil-bestrol) or at 5 months (all
other treatments). Chromosomal aberrations but not exchanges in
hamster kidney DNA were cumulative with continued exposure to
diethylstilbestrol. The kidneys of estradiol-and moxestrol-treated
animals had chromosomal aberrations at frequencies similar to those
seen with diethylstilbestrol, whereas the frequency of chromosomal
aberrations in animals treated with the weaker estrogens were similar
to those of controls. The authors suggested that estrogen-induced
chromosomal aberrations are involved in tumorigenesis but that the
process does not involve metabolic activation, since moxestrol, which
is poorly metabolized, did induce chromosomal aberrations (Banerjee et
al., 1994).
2-Catechol estradiol and 4-catechol estrone at 0.01-0.1 mmol/L
induced strand breaks in the pBR322 plasmid, and the level was greatly
enhanced by a nitric oxide-releasing compound. The strand breaks could
be inhibited by antioxidants such as N-acetylcysteine and ascorbate
and by superoxide dismutase. Estradiol, estrone, O-methylcatechol
estrogens, and diethylstilbestrol did not induce strand breaks. The
authors suggest that NO mediates conversion of catechol estrogens to
quinones, and the oxygen radicals produced by the quinone/hydroquinone
redox system react with NO to form peroxynitrite, which causes strand
breaks (Yoshie & Ohshima, 1998).
Natural estrogens and their derivatives were tested for the
ability to induce microtubule disruption in Chinese hamster V79 cells
(which lack cytochrome P450 enzymes) at concentrations of 1-100
µmol/L. The EC50 values were 10 µmol/L for estradiol and 9 µmol/L for
17 alpha-estradiol. The most potent disrupting agent tested was
2-methoxyestradiol (EC50, 2 µmol/L). Preincubation of cells with 1
µmol/L taxol for 2 h protected them against microtubule disruption by
estradiol at doses up to 50 µmol/L. The authors concluded that some
natural estrogens cause microtubule disruption in a nongenomic manner
(Aizu-Yokota et al., 1995).
An increase in the frequency of DNA strand breakage and
accumulation of lipid peroxidation products in the dorsolateral but
not the ventral prostate were seen in four NBL rats treated
subcutaneously with testosterone plus estradiol, relative to control
rats. Treatment of castrated rats with testosterone resulted in a
slightly lower rate of strand breaks than in untreated controls. The
authors concluded that estradiol was responsible for the single-strand
breaks in these animals (Ho & Roy, 1994).
Studies of adducts
Male Syrian hamsters were injected intraperitoneally with 2, 10,
50, or 150 mg/kg bw estradiol and sacrificed 4 h later; with 50 mg/kg
bw and sacrificed 1-8 h later; or with 100 mg/kg bw and sacrificed 1-8
h later. Their livers and kidneys were examined for
8-hydroxy-2'-deoxyguanosine (8-OHdG) as a marker of hydroxy radical
interaction with DNA. Four hours after dosing with 50 mg/kg bw, the
renal 8-OHdG levels were double those of controls; adducts were not
determined in kidneys from animals treated at 150 mg/kg bw. No
dose-dependence was observed. The levels of hepatic DNA adducts in
treated animals were similar to those in controls. In hamsters treated
with 50 mg/kg estradiol and killed 1-8 h later, the level of renal DNA
adducts was greater than that in controls at 4 h but not at 1, 2, or 8
h after dosing; hepatic DNA adducts were not determined. In animals
injected with 100 mg/kg estradiol, the number of hepatic DNA adducts
was increased 1 and 2 h after dosing. Treatment of hamsters with
subcutaneous implants containing 25 mg estradiol for three or six days
increased the renal levels of 8-OHdG by 50% over that in controls by
day 3, but the levels were no different from those of controls in
animals implanted with estradiol for six days. No effect was observed
on the background level of liver DNA adducts at either time. Treatment
of hamsters for three days by intraperitoneal injection with 100
µg/animal per day estradiol or 2-or 4-hydroxyestradiol also had no
effect on renal DNA 8-OHdG levels. The authors concluded that the
mechanism of the carcinogenic action of estrogen occurs through
generation of free radicals via redox cycling of catechol estrogen
metabolites (Han & Liehr, 1994b). Substantial differences were
observed in the levels of adducts in untreated animals after three and
six days in the implant experiment. While no statistical analysis was
performed, the differences were sufficient to indicated a substantial
variation in the background level of adducts. The lack of dose-and
time-dependence of adduct formation in these experiments is
inconsistent with the hypothesis that estradiol is a genotoxic
carcinogen.
An unidentified adduct was observed in DNA isolated from the
dorsolateral prostate of NBL rats treated by subcutaneous
administration of estradiol and testosterone for 16 weeks but not 8
weeks. The circulating estradiol concentrations were increased
approximately 14 times over those of controls, while normal plasma
testosterone concentrations were maintained. The appearance of the
adduct correlated with the appearance of dysplastic lesions (Han
et al., 1995).
Incubation of Syrian hamster embryo cells with 1 µg/ml estradiol
or 2-or 4-hydroxy estradiol for 24 h induced DNA adduct formation in
parallel with cell transformation. The level of DNA adduct formation
was greatest with 4-hydroxy estradiol and then with 2-hydroxy
estradiol and estradiol. No exogenous metabolic activation was used in
these experiments. In later experiments, diethylstilbestrol increased
adduct formation in the absence but not in the presence of metabolic
activation (Hayashi et al., 1996).
Mongrel dogs were treated with capsules containing 5
alpha-dihydro-testosterone (DHT) and/or estradiol for 60 days. The
capsules were of uniform length (7 cm), but the quantity of hormone
used was not described. Blood sampoles were obtained for the
measurement of hormone. The 8-OHdG adduct levels in DNA from prostate
were reduced in all dogs that received DHT, whereas treatment with
estradiol or DHT plus estradiol had no effect. Free radical-induced
damage (carbonyl content) of proteins was observed in prostate tissue,
and the authors concluded that the damage was consistent with injury
by estrogen metabolites followed by DHT-stimulated growth of altered
prostatic cells (Winter & Liehr, 1996).
Sterol-initiated DNA adduct formation was examined in vitro by
32P-postlabelling. After exposure of human liver DNA to 2 mmol/L
steroid, several steroids but not estradiol, estrone, or estriol
formed DNA adducts. The presence of a carbonyl group at C17 (which
estradiol lacks) was strongly associated with DNA binding (Seraj et
al., 1996).
When three male and three female Han:Wistar rats given estradiol
at 2 mg/kg bw per day intragastrically as an aqueous microcrystalline
suspension for 14 days, an estrogen-specific DNA adduct was observed
by 32P-postlabelling in the livers of male but not female rats (Feser
et al., 1996).
To assess the hypothesis that estrogen-induced adduct formation
is related to estrogen-induced tumorigenesis in humans, DNA from
normal human breast tissue, benign tumours, and malignant tissue with
invasive ductal carcinoma was examined for the presence of 8-OHdG
adducts by a novel solid-phase immunoslot-blot assay with
adduct-specific antibodies. The amounts of 8-OHdG found in the three
tissues were 0.25, 0.98, and 2.4 pmol/µg DNA, respectively; 13 times
more endogenous formation of 8-OHdG was observed in MCF-7 cells which
undergo hormone-dependent cell growth and have estrogen receptor s
than in normal cultured human mammary epithelial cells, but no
difference in adduct levels was observed between normal cells and
MDA-MB 231 cells, which undergo receptor-independent growth and lack
estrogen receptors. 8-OHdG adduct levels also correlated to the
estrogen receptor status of the tissue, with higher adduct levels in
malignant tissue with estrogen receptors than in those without. Age
and smoking status did not correlate to the 8-OHdG content of DNA. The
authors concluded that accumulation of 8-OhdG adducts in DNA is
predictive of the risk for breast cancer and may be a major
contributor to the development of breast neoplasia (Musarrat
et al., 1996).
No difference in 8-OhdG adduct levels was found by
high-performance liquid chromatography-electrochemical detection in
breast cancer tissue and adjacent non-cancerous tissue, and no
correlation was found with expression of estrogen or progesterone
receptors or with clinical stage or histological grade (Nagashima et
al., 1995).
1.3.5 Reproductive toxicity
The embryotoxicity and teratogenicity of estradiol were reviewed
by a working group convened by IARC (1979), which concluded that
"Oestradiol-17ß has teratogenic actions on the genital tract and
possibly on other organs and impairs fertility."
Estradiol was administered in the feed to female Crl:CD BR rats
at doses equal to 0, 0.003, 0.17, 0.69, or 4.1 mg/kg bw per day in a
90-day, one-generation study. As no pups were born to dams at the two
highest doses, only three dose groups of the F1 generation were
assessed. The mean daily intakes of the F1 females were 0, 0.005 and
0.27 mg/kg bw per day, respectively. The F0 rats were 49 days of age
on test day 0, and serum hormones were evaluated after 7, 28, and 90
days of feeding; they were evaluated on postnatal day 98 for the F1
generation. In the F0 generation, estradiol at doses > 0.17 mg/kg bw
per day produced a dose-dependent increase in serum estradiol
concentration, and all doses produced a dose-dependent decrease in
serum progesterone concentration on test day 90, which correlated with
ovarian atrophy and lack of corpora lutea. The serum concentration of
luteinizing hormone was decreased at all times at > 0.69 mg/kg bw
per day and at 0.17 mg/kg bw per day on test day 90. Little change was
observed in the serum concentrations of follicle-stimulating hormone.
The serum concentration of prolactin was increased at 4.1 mg/kg bw per
day on test day 90. In the F1 generation on postnatal day 28, the
serum estradiol concentration was increased and that of progesterone
decreased at 0.27 mg/kg bw per day. No change in serum prolactin,
follicle-stimulating hormone, or luteinizing hormone concentration was
noted. Dietary estradiol caused marked effects on the estrus cycle at
0.17 mg/kg bw per day (F0) and 0.27 mg/kg bw per day (F1) and at
0.69 and 4.1 mg/kg bw per day (F0 generation) (Biegel et al., 1998b).
Information on the pups in this study was presented elsewhere.
The groups at the two highest doses produced no pups, and the weights
of the pups in the two remaining groups decreased relative to that of
controls; the weights of pups of the F0 generation at 0.003 mg/kg bw
per day (0.005 mg/kg bw per day for the F1 generation) recovered
after birth and remained similar to those of controls throughout the
study. The mean length of gestation in this dose group was
statistically nonsignificantly decreased, which the authors suggested
contributed to the decreased birth weight. The body weights of animals
at 0.27 mg/kg bw per day remained below control levels throughout the
study. Parenteral administration of estradiol did not affect the
anogenital distance in male or female pups. Onset of sexual maturity,
as measured by prepubertal separation in males and vaginal opening in
females, was delayed. Some of the histopathological findings were more
severe in the F1 generation than in the parent generation. The
authors concluded that additional studies were needed to define the
dose-response curve more accurately (Biegel et al., 1998a).
The average litter size of transgenic 'knockout' female mice
deficient in steroid 5 alpha-reductase type I (SRD5 alpha 1-/-,
wild-type C57Bl/6J/129Sv) is reduced in comparison with wild-type
controls (2.7 vs. 8 pups, respectively). In reductase-deficient
animals, the maternal serum estrogen concentrations were chronically
increased by two-to threefold relative to control animals. In control
animals, spikes in DHT and to a much smaller extent testosterone
concentrations occurred in maternal plasma on day 9 of gestation. In
the 5a-reductase-deficient animals, the androgen peaks at day 9 were
reversed. Oogenesis, fertilization, implantation, and placental
morphology appeared normal in reductase-deficient animals. Fetal loss
occurred between gestation days 10.75 and 11, commensurate with a
surge in placental androgen production. Minimal fetal loss was
observed on gestation day 10.5. To test the hypothesis that steroid
hormones contribute to fetal loss in reductase-deficient animals,
pregnant animals were treated with pellets containing various amounts
of steroid hormone. Table 12 shows the effect on mean embryo survival.
Bleeding was observed grossly in the uteri of control and experimental
animals. Administration of estrogen receptor antagonists or inhibitors
of aromatase prevented the excess fetal loss observed in the
reductase-deficient mice. Testosterone was mildly protective against
fetal loss in the knockout mice. The authors concluded that
5a-reductase guards against the toxic effects of estrogen during
pregnancy. They also noted that the human placenta, unlike the rodent
placenta, has high levels of aromatase, resulting in very high
concentrations of estrogens in the amniotic fluid. They speculated
that the human fetoplacental unit has developed a mechanism to protect
itself against estrogens (Mahendroo et al., 1997).
Pregnant female CF-1 mice were treated subcutaneously on
gestation day 13 with Silastic capsules containing 0, 25, 100, 200, or
300 µg estradiol. Male fetuses positioned in utero between a male and
female (MF males) were examined for treatment-related prostatic
effects. In some experiments, MF males were obtained from pregnant
dams killed on gestation day 19 and reared by foster dams. At the age
of seven months, MF males were castrated, implanted subcutaneously
with capsules containing 500 µg testosterone, and killed three weeks
later. The total concentration of estradiol in serum was increased in
a dose-dependent manner in treated MF fetuses collected on day 18,
with 94, 150, 230, 360, and 530 pg/ml in the animals at the five
doses, respectively. A 40% increase in the number of prostatic
glandular epithelial buds was found in MF males from dams treated with
25 µg estradiol, relative to controls. An increase in prostate size
was also noted, but the size of the individual buds was not changed;
prostate weight was increased in MF males at the low dose sacrificed
at eight months but was decreased at the high dose, resulting in an
inverted-U dose-response curve. The authors concluded that increased
fetal serum estrogen concentrations affect androgen regulation of
prostate differentiation, resulting in a permanent increase in the
number of prostatic androgen receptors and in prostate size (vom Saal
et al., 1997). The doses used in this study were below the NOEL.
Under conditions associated with reduced estrogen synthesis in
humans (aromatase deficiency, placental sulfatase deficiency, fetal
anencephaly), estrogen production and concentrations may be reduced by
80-90%. However, progesterone production and fetal development remain
normal, indicating that considerably more estrogen is produced during
normal pregnancy than is necessary (Fisher, 1998).
The embryotoxicity of steroidal and nonsteroidal estrogens was
examined in cultured whole embryos obtained from Sprague-Dawley rats.
Preliminary experiments resulted in steep dose-response curves for all
estrogens examined at doses ranging from 0.05 to 0.5 mmol/L. For
example, diethylstilbestrol had no effect at concentrations < 0.15
mmol/L but was lethal to 100% of cultured embryos at doses > 0.25
mmol/L. The concentrations tested resulted in low embryolethality
(2-20%). Estrogens had dysmorpho-genic effects at concentrations of
0.1-0.2 mmol/L. The commonest effect observed was hypoplasia of the
prosencephalon. Estradiol and estrone were markedly and statistically
significantly more toxic in the presence of metabolic activation (from
livers of pregnant and non-pregant females and Aroclor 1254-treated
adult male rats), but metabolic activation attenuated the embryotoxic
effects of ethinylestradiol, tamoxifen, and erythrohexestrol and had
no effect on other estrogens. In this system, estradiol was more
efficiently converted to catechol estrogens in male liver, but
Table 12. Effects of steroid hormones in 21-day release pellets on embryo survival
Pellet Dose Control females Srd5a1-/- females
treatment
No. Live Dead No. Live Dead
litters embryos embryos litters embryos embryos
None 4 8.0 0.4 8 3.2 5.1
Placebo 3 9.6 0.67 3 4.3 5.0
Androstenedione 0.5 4 3.3 4.5 5 1.0 7.8
Testosterone 0.5 2 8.8 1.5 5 6.2 3.2
Estradiol 0.5 2 0 7.0 5 0 8.4
0.08 5 0 7.4 6 0 7.8
0.02 2 0 5.0 6 0.3 6.3
0.01 2 0 11 6 0 6.8
0.005 2 0 8.5 5 0 5.0
0.0025 2 4.5 4.0 4 2.8 5.5
ethinylestradiol was converted to catechol estrogens approximately
three times more effectively than estradiol when metabolic activation
systems from pregnant and non-pregnant animals was used. The authors
concluded that the effects observed are independent of steroid
structure or estrogen activity and are strongly dependent on the
pathways and rates of biotransformation of some (but not all) of the
parent compounds (Beyer et al., 1989).
Ten mg of estradiol or testosterone were implanted subcutaneously
into groups of five and seven female Sprague-Dawley rats on day 10 of
pregnancy, whereas control pregnant rats were given dextran by the
same method. Implantation with estradiol or testosterone resulted in
complete resorption of embryos in all treated animals (Sarkar et al.,
1986).
A review of the birth certificates and hospital records of 7723
infants whose mothers had reported using oral contraceptives indicated
that these compounds present no major teratogenic hazard (Rothman &
Louik, 1978).
Studies of reproductive toxicity are summarized in Table 13.
1.3.6 Special studies of mechanisms of action
Estrogen-induced tumorigenesis has been the subject of two lines
of investigation: receptor-based effects and redox cycling and DNA
adduct formation leading to genetic damage. During the past decade,
considerable attention has been focused on understanding the molecular
basis of hormone receptor biology. Recently, transgenic animals that
overexpress or lack estrogen receptors (Couse et al., 1997b) or
aromatase (Fisher et al., 1998) have been developed. The role of
estrogens and other hormones in mammary neoplasia in rodents and their
relevance to human risk has been reviewed (Russo & Russo, 1996), and
it was noted that rodent models mimic some but not all of the complex
external and endogenous factors involved in initiation, promotion, and
progression of carcinogenesis. Tumour type and incidence are
influenced by the age, reproductive history, and the endocrine milieu
of the host at the time of exposure. The spontaneous incidence of
tumours differs in different strains of rats and mice. In rats, most
spontaneously developed neoplasias, with the exception of leukaemia,
are of endocrine organs or organs under endocrine control. Russo &
Russo (1996) concluded that mechanism-based toxicology is not yet
sufficient for human risk assessment, and the approach should be
coupled to and validated by traditional long-term bioassays.
The estrogen-responsive male Syrian hamster kidney model has been
widely used to study the carcinogenicity of estrogens in vivo.
Separation of carcinogenic from hormonal effects in male and
ovariectomized female Syrian hamster kidney has been reviewed (Yager &
Liehr, 1996). In hamsters treated chronically with relatively high
doses by subcutaneous implantation, certain potent synthetic estrogens
such as ethinylestradiol result in < 10% tumour incidence in kidney,
whereas treatment with other estrogens result in renal tumour
development in nearly all animals. Ethinylestradiol also acts at
different sites from other estrogens in the hamster kidney. Thus, the
estrogenicity of a compound is essential but not sufficient for renal
carcinogenesis in hamsters. Other factors that have been suggested to
contribute to carcinogenicity include cell-type-specific uptake and
differential estrogen metabolism (such as high 4-hydroxylation rates)
leading to estrogen-induced damage to cell macro-molecules (DNA
and protein). Similarly, the progesterone metabolite
5 alpha-pregnane-3,20-dione successfully competes with progesterone
for receptor binding and biological effectiveness in some tissues but
not others (Tsai et al., 1998). Since a second estrogen receptor
isoform (ERb) has recently been identified, the results described
below should be interpreted with caution.
The estrogen metabolite 4-hydroxyestradiol, but not
2-hydroxyestradiol, was tumorigenic in male hamster kidneys (Yager &
Liehr, 1996), the proposed mechanism of action being redox cycling
resulting in oxygen radical formation and subsequent damage to cell
macromolecules. It is not certain that this pathway is relevant
in vivo at physiological concentrations of estradiol. For example,
micromolar concentrations of estradiol are necessary to cause
microtubular disruption in Chinese hamster V79 cells, and these are
greatly in excess of the picomolar to nanomolar concentrations
normally found in serum. At higher concentrations, the lipophilicity
of estradiol and some metabolites (such as methoxy derivatives) and
their ability to intercalate into DNA and lipid membranes may be more
important from a toxicological perspective than the estrogenic
properties.
The hormonal (i.e. receptor-mediated) and carcinogenic (i.e.
genotoxic) properties of synthetic hormones were differentiated by
measuring the rates of catechol estrogen and methyl ester formation by
a weak carcinogen, 17a-ethinylestradiol, and by a strong hormonal
carcinogen, moxestrol. The rates of hydroxylation in comparison with
that for estradiol were 40-50% for moxestrol and 25-35% for
ethinylestradiol, the differences being apparent at longer reaction
times (i.e. 20 min but not 10 min). 2-Hydroxymoxestrol was a poor
substrate for COMT, proceeding at a rate of about 3% of the
methylation of 2-hydroxyestradiol. In hamster kidney cytosolic protein
extracts, 10 nmol/L progesterone decreased binding of 2 nmol/L
3H-progesterone by 78%, and 10 nmol/L ethinylestradiol inhibited it
by 35%. Interpretation of this result is complicated, as the assay was
performed with insufficient excess progesterone. Estradiol and
moxestrol had no effect. The authors suggested that the decreased
capacity of ethinylestradiol to form catechol metabolites and its
progestogen antagonist activity contribute to the low tumour incidence
seen with this compound (Zhu et al., 1993).
Table 13. Studies of the reproductive toxicity of estrogens
Species Dose Findings Reference
Mice, rats 0.1-35 mg/day Teratogenic IARC (1979)
subcutaneously
Female rats 0.003-4.1 mg/kg bw No NOEL identified Biegel et al.
per day orally for (1998b)
90 days
Transgenic mice Estrogen No NOEL; effects Mahendroo et
concentrations observed between days al. (1997)
reduced two- 10.75 and 11
to threefold
Mice 0-300 µg/animal No NOEL for effects on vom Saal et
fetal prostate al. (1997)
Cultured whole 0.5-0.5 mmol/l Dysmorphogenic effects Beyer et al.
embryos of at 0.1-0,2 mmol/L (1989)
Sprague-Dawley
rats
Sprague-Dawley 10-mg implant Embryo resorption Sarkar et al.
rats (1986)
Humans Acidental exposure No effect reported Rothman & Louk
(1978)
Several steroidal estrogens were tested at doses of 0.1-100
nmol/L for their ability to increase proliferation of primary renal
proximal tubular cells in culture. Most of the estrogens tested
(including 4-hydroxyestradiol and estrone and, to a lesser extent,
2-hydroxyestradio) increased cell proliferation at a concentration of
0.1-10 nmol/L but inhibited it at 100 nmol/L. The authors concluded
that the ability to induce cell proliferation is a more accurate
predictor of carcinogenicity in this system than estrogen-responsive
end-points or the amount of catechol metabolites generated (Li et al.,
1995).
To determine the role of estrogens in tubular renal damage and
the subsequent reparative cell proliferation, castrated adult male
Syrian hamsters were given subcutaneous pellets that released hormones
at the following rates (µg/day): diethylstilbestrol, 145; estradiol,
134, estrone, 104, ethinylestradiol, 154; tamoxifen, 141;
progesterone, 147; and DHT, 121. Diethylstilbestrol was administered
for one to nine months, while the other compounds were administered
for five months. The severity of tubular damage increased with
progressive estrogen treatment, with a prominent rise in the number of
secondary and tertiary lysosomes. The concentration of cathepsin D was
increased in estrogen-treated kidneys (by approximately 2.7-and
3.5-fold at four and five months, respectively) and paralleled the
rise in estrogen receptor content. Progesterone and DHT alone had no
effect, and concomitant treatment of animals with estrogen and either
tamoxifen or DHT mitigated the estrogenic effects. The primary form of
cathepsin D found in the kidneys of control and estrogen-treated
animals was the 52-kDa isoform, considered to be the inactive form of
the protein. The 31-and 27-kDa isoforms, believed to be the active
forms, were found in significant amounts only in the kidneys of
estrogen-treated animals, primary renal tumours, and their metastases.
The authors suggested that cathepsin D mediates renal tubular damage
as a first step in reparative cell proliferation (Li et al., 1997).
Estradiol- or diethylstilbestrol-induced growth of cultured
proximal renal tubular cells could be inhibited by ethinylestradiol.
Expression of estrogen-responsive protooncogene (c- myc, c- fos, and
c- jun) RNA and protein in kidneys was reduced in animals treated
concomitantly relative to that found in animals treated with estradiol
or diethylstilbestrol. The authors concluded that ethinylestradiol
interferes with estrogen receptor-mediated mitogenic pathways,
preventing gene dysregulation and tumour development. This effect does
not appear to be due to differential binding to estrogen receptors by
estrogenic substances (Li et al., 1998).
Other hormones, notably progesterone, testosterone, and
deoxycortico-sterone, and the antiestrogen tamoxifen prevent or
inhibit the growth of estrogen-induced renal tumours in Syrian
hamsters (reviewed by Yager & Liehr, 1996). Progesterone and tamoxifen
exert a protective effect on mammary carcinogenesis (Inoh et al.,
1985). A review of clinical data indicated that adjuvant progestogen
therapy for treatment of patients with metastatic renal-cell carcinoma
is not effective, indicating that carcinogenesis in the Syrian hamster
model is not representative of human renal carcinogenesis (Linehan et
al., 1997).
Rodent tissues that form estrogen-induced tumours have high
concentrations of the caetcholamine noradrenaline. In a study to test
the hypothesis that hydrogen peroxide formed by monoamine oxidase
deamination of catecholamines provides a source of free radicals, in
addition to that postulated to be provided by metabolic redox cycling
of catechol estrogen intermediates, Syrian hamsters and Sprague-Dawley
rats received 25 mg estradiol in a subcutaneous capsule for two weeks.
Treatment increased monoamine oxidase activity in hamster kidney but
not liver and had no effect on monoamine oxidase activity in rat liver
or kidney. The induction of hamster kidney monoamine oxidase activity
could be prevented by tamoxifen. The authors concluded that
receptor-mediated induction of monoamine oxidase, which deaminates
catecholamines, may increase production of hydrogen peroxide and
hydroxyl radicals, thus contributing to tumour initiation (Sarabia &
Liehr, 1998).
Male Syrian hamsters were treated with quercetin, an inhibitor of
COMT, in order to assess the potentiating effects of this compound on
renal tumorigenesis. All six animals treated with subcutaneous pellets
that released estradiol at 61 µg/day developed kidney tumours, but no
tumours were seen in hamsters treated with quercetin at 0.3 or 3% in
the diet for 5.7 or 6.5 months, respectively. Concomitant
administration of estradiol and quercetin increased the number of
large tumours and the incidence of metastases over that seen with
hormone treatment alone. Quercetin inhibited 2 and 4-catechol estrogen
methylation by 34 and 22%, respectively. The rates of redox cycling in
liver and kidney were not affected by treatment with quercetin or
estradiol (Zhu & Liehr, 1994).
Male Syrian hamsters received subcutaneously implanted pellets
containing 25 mg estradiol (which were replaced every three months)
for seven months. Dietary supplementation with 1% vitamin C decreased
estrogen-induced renal carcinogenesis by 50% in a small number of male
Syrian hamsters. In related experiments, the effect of estradiol
and/or vitamin C was examined on the renal activity of the detoxifying
enzymes quinone reductase, catalase, superoxide dismutase, glutathione
peroxidase, glutathione reductase, glucose-6-phosphate dehydrogenase,
and gamma-glutamyl transpeptidase. Glutathione peroxidase activity was
increased in the kidneys of hamsters treated with estradiol for 1
month (141% of control). Quinone reductase activity was reduced in the
kidneys of estradiol-treated animals (18% of control), but the
activity was partially restored by dietary supplementation with
vitamin C for one month (32% of control); in liver, concomitant
treatment with vitamin C and estradiol reduced the activity of this
enzyme (6% of control), and estradiol treatment alone caused a smaller
decrease in activity (68% of control). Differences in catalase
activities were observed after one month but not by seven months of
treatment. Vitamin C had no effect on the intensity or specificity of
estrogen-related kidney DNA adducts. The authors concluded that the
enzymatic changes observed in estradiol-and/or vitamin C-treated
animals were insufficient to account for the differences in renal
tumour incidence. The authors concluded that vitamin C inhibits
estrogen-induced carcinogenesis by reducing the concentration of
estrogen quinone metabolites (Liehr et al., 1989).
COMT is present in the epithelial cells of the proximal
convoluted tubules of the kidney, predominantly in the juxtamedullary
region, where estrogen-induced tumours arise. Treatment of male Syrian
hamsters with estradiol or ethinylestradiol for two or four weeks
altered the intensity, distribution, and subcellular location of
immunoreactivity to COMT. Staining for this enzyme in control animals
was largely of the soluble cytoplasm and nuclear membrane-bound forms,
whereas staining for the soluble form of nuclear COMT was observed in
estrogen-treated animals. No differences were observed between the two
estrogens or between animals treated with estrogen for two or four
weeks; no difference in nuclear location was observed between treated
and control animals. Estradiol-induced renal tumours did not stain for
COMT, and the nuclear signal present in human cells was lacking in
hamster kidney. The authors suggested that a change in the subcellular
distribution of COMT is a protective response to catechol estrogen
metabolic damage to the genome (Weiss et al., 1998).
Male Noble rats were treated with subcutaneous Silastic implants
containing testosterone and/or estradiol or diethylstilbestrol for 16
weeks. In estradiol-treated animals, the plasma testosterone
concentration, determined after three weeks of treatment, was
decreased more than 10-fold (from 4.8 ng/ml to < 0.3 ng/ml), whereas
the estradiol concentration was increased 4.5-fold (from 16 pg/ml to
75 pg/ml). Diethylstilbestrol but not estradiol caused a statistically
significant decrease in body weight, and both hormones decreased the
relative weight of the dorsolateral and ventral prostate and seminal
vesicles plus coagulating glands. The body weights of animals treated
for 16 weeks with testosterone and estradiol were significantly lower
than those of controls, the relative weights of the dorsolateral and
ventral prostate and seminal vesicles plus coagulating glands were
increased, and the testicular weight was decreased approximately
twofold. Multifocal epithelial dysplasia and marked inflammatory
changes were observed in the lateral prostate. No changes were seen in
the morphology of the ventral prostate seminal vesicle, coagulating
gland (anterior prostate), or ampullary gland. Implants of
testosterone plus diethylstilbestrol induced widespread dysplasia in
the ventral prostate and lesser or no ventral prostatic dysplasia. In
explant cultures, animals treated with testosterone plus
diethylstilbestrol or testosterone plus estradiol showed a reduced
ability to convert the 5 alpha,3ß-hydroxysteroid derivative of 3H-DHT
to the more polar 6 alpha-and 7 alpha-hydroxylated derivatives,
resulting in accumulation of 3ß-androstanediol. These metabolic
changes resulted in a threefold (testosterone plus estradiol) or
eightfold (testosterone plus diethylstilbestrol) increase in
accumulation of 3ß-androstanediol in the dysplastic ventral prostate;
no accumulation was observed in the explanted dorsolateral prostate.
In animals treated with testosterone plus diethylstilbestrol, the
ratio of estrone:estradiol was reversed in the ventral prostate,
whereas in animals treated with testosterone plus estradiol, estradiol
metabolism was decreased in the dysplastic dorsolateral prostate but
not in the ventral prostate. The authors concluded that differences
between target tissues in the bioavailability of the estrogen
component determines in which lobe prostate dysplasia develops (Ofner
et al., 1992).
Male Noble rats were treated with Silastic implants containing
testosterone and estradiol for 16 weeks since it had been reported
previously that such implants increase the plasma estradiol
concentration threefold while maintaining testosterone at
physiological concentrations. This treatment regimen produced
dysplasia in the dorsolateral prostate, without liver dysplasia.
Microsomes were prepared from the liver, ventral prostate, and
dorsolateral prostate of control and treated animals to determine
metabolic conversion of estradiol to catechol estrogens. Catechol
estrogen formation was observed at high levels in liver microsomal
incubates and low levels in prostate incubates. Treatment failed to
alter the extent or profile of hepatic estradiol metabolism, except
for a significant reduction in estriol production relative to
controls. A nonsignificant reduction in 2-hydroxyestradiol formation
was also observed. The authors concluded that catechol estrogen
formation is not a mediating step in estrogen-induced tumorigenesis
(Lane et al., 1997).
Mongrel dogs were treated for 60 days subcutaneously with DHT
and/or estradiol; however, the quantity of hormone in the implant and
the resulting plasma concentrations were not measured. Previous
studies had indicated that such implants maintain plasma hormone
concentrations at physiological levels. The activities of aryl
hydrocarbon hydroxylase, 7-ethoxycoumarin O-deethylase, and
estradiol 2-and 4-hydroxylase were elevated in the prostate glands of
animals treated with either hormone or their combination and were
either decreased or unchanged in liver and kidney. The increase
observed in estradiol-treated animals was substantially modified by
concomitant treatment with DHT. The activity of estrogen 2-hydroxylase
was increased tenfold and fourfold in animals given estradiol and
estradiol plus DHT, respectively. The activities of
7-ethoxycoumarin- O-deethylase, aryl hydrocarbon hydroxylase,
glutathione peroxidase I, and catalase were also increased in the
prostate. Hormones had variable effects on these enzymes in liver and
kidney. The free radical-generated carbonyl content of the prostate
increased 2.5-fold after treatment with estradiol and twofold with
treatment with estradiol plus DHT. No hormone-related effects on
carbonyl content were seen in kidney proteins, whereas DHT and the
combination with estradiol increased the carbonyl content by 60 and
150% over the control level. Treatment with estradiol alone resulted
in a substantial but nonsignificant decrease in the hepatic protein
carbonyl content relative to controls. In DNA hydrolysates, a 45%
decrease was observed in the 8-OHdG content in DNA from the prostate
of DHT-treated animals relative to control DNA; no changes were
observed in DNA from other tissues or in that from animals treated
with estradiol or estradiol plus DHT. The authors concluded that the
hormone-induced changes in the activities of catechol estrogen
synthetase and other enzymes were consistent with the postulated
generation of free radicals by redox cycling of catechol estrogen
specifically in the prostate. The protein damage observed is
consistent with induction of benign prostatic hyperplasia injury by
estrogen metabolites followed by growth stimulation of altered foci by
DHT (Winter & Liehr, 1996).
Adult male Noble rats received subpannicularimplantations of
pellets that released a daily mean average of 120 µg of testosterone
propionate and 110 µg of estradiol. Mammary gland ductal
adenocarcinomas were induced in 100% of the rats after eight to nine
months of treatment, whereas neither hormone alone was effective in
inducing adenocarcinomas. Testosterone propionate disrupted mammary
gland ducts and caused proliferation of stromal tissue, while
estradiol induced ductal epithelial growth and nodular atypical
hyperplasia. The authors concluded that androgens interact with
androgen receptors or progesterone receptors, or perhaps both, in
mammary glands induced by estradiol and testosterone propionate to
cause tumours, reduce the latency, enhance tumour size, and increase
the incidence to 100% (Liao et al., 1998).
1.4 Observations in humans
1.4.1 Therapeutic use
Estrogens, sometimes in conjunction with progestogens, are used
for contraception and for hormone replacement. They are among the most
commonly prescribed drugs and include the parent molecule, its esters,
and other synthetic steroidal and non-steroidal derivatives. Estrogens
can be delivered orally, by intramuscular injection, or
percutaneously. The main concern associated with use of estrogens for
these purposes is a possibly increased risk for cancer; other
side-effects may include hypertension, thromboembolic and other
vascular diseases, breakthrough bleeding, gall-bladder disorders,
nausea, migraine, and mood changes. The progestational component of
combined hormonal therapy may be responsible for some of these effects
(Williams & Stancel, 1996; Carr & Griffin, 1998).
The effects of various dosages of orally administered conjugated
estrogens, mestranol, and ethinylestradiol on the binding capacity of
serum corticosteroid-binding globulin (CBG)-binding capacity was
assessed in three healthy postmenopausal women, each of whom received
Premarin(R) orally at 0.3, 0.62, 1.2, or 2.5 mg/day for 14 days.
Each dose increased serum CBG-binding capacity, but the increase was
not significant at 0.3 mg/day. The serum concentration was
approximately doubled with a doubling of the dose. The authors
concluded that there may be a threshold concentration of exogenous
estrogens below which there is no associated increase in serum
concentrations of CBG and above which there is a dose-response
relationship between the exogenous estrogen and serum CBG-binding
capacity (Moore et al., 1978).
Twenty-three healthy postmenopausal women received one or more
two-week courses of estrogens orally, including fine-particle
estradiol (1, 2, or 10 mg/day) and conjugated estrogens (Premarin(R)
at 0.3, 0.62, 1.2, or 2.5 mg/day). The other estrogens that were
assessed included piperizine estrone sulfate, ethinylestradiol, and
diethylstilbestrol. Each dose was ingested by three women, and the
pre-treatment and post-treatment concentrations of
follicle-stimulating hormone, luteinizing hormone, CBG-binding
capacity, SHBG-binding capacity, angiotensinogen, estrone, and
estradiol were determined. The relative potency of the estrogen
preparations were assessed. Conjugated estrogens were 3.2 times more
potent than fine-particle estradiol in increasing SHBG-binding
capacity, and fine-particle estradiol was 1.9 times more potent than
piperizine estrone sulfate in inducing CBG-binding capacity, whereas
conjugated estrogens were 2.5 times more potent than piperizine
estrone sulfate. Doses < 1.7 mg equivalents of piperizine estrone
sulfate failed to increase CBG-binding capacity significaqntly.
Fine-particle estrogens and piperizine estrone sulfate were equipotent
in more potent than piperizine estrone sulfate. The angiotensinogen
response exceeded the baseline range only after administration of 2.2
mg equivalents piperizine estrone sulfate. The natural estrogen
preparations, piperizine estrone sulfate, fine-particle estradiol, and
conjugated estrogens were nearly equipotent on affecting serum
follicle-stimulating hormone concentrations. No dose-response
relationship for serum luteinizing hormone could be demonstrated with
any estrogen preparation (Mashchak et al., 1982).
See also section 4, 'Epidemiological studies of women exposed to
postmenopausal estrogen therapy and hormonal contraceptives'.
1.4.2 Estradiol-related genetic markers of carcinogenicity
The A2 allele of CYP17, the cytochrome P450 that catalyses the
rate-limiting step in androgen biosynthesis, has been suggested to be
a genetic marker for hormone-dependent disease such as polycystic
ovarian syndrome, anovulatory infertility, hirsutism, and breast
cancer. In a nested case-control study, no association was found
between higher basal transcription of the A2 allele and subsequent
development of breast cancer, stage of disease at diagnosis, or age at
menarche. No racial differences in genotype were observed among
Caucasian, African-American, Asian, and Hispanic women (Helzlsouer et
al., 1998).
The data on the effect of COMT polymorphism on human risk for
hormone-induced cancer are conflicting and are discussed elsewhere in
this report.
2. PROGESTERONE
2.1 Explanation
Progesterone (100 or 200 mg) in combination with estradiol
benzoate (10 mg or 20 mg) is administered to cattle as an ear implant
to increase the rate of weight gain (i.e. growth promotion) and
improve feed efficiency. Progesterone is also administered on an
intravaginal sponge to lactating and non-lactating dairy cows and
goats to synchronize estrus. Progesterone is considered to be an
endogenous substance, as exogenously administered progesterone is
structurally identical to the progesterone produced in animals and
humans.
2.2 Biological data
2.2.1 Absorption, distribution, and excretion
Progesterone is largely inactive when administered by the oral
route because of its low systemic bioavailability. High doses of
certain fine-particle forms have been studied for possible use in
contraception or in hormone replacement therapy, and the results are
discussed below.
The pharmacokinetics of orally administered fine-particle
progesterone have been reviewed (Sitruk-Ware et al., 1987). Two 100-mg
capsules of fine-particle progesterone given to women in the early
follicular phase resulted in a peak plasma concentration at the second
hour of 13 ng/ml. When compared with crystallized compound, the same
dose of fine-particle progesterone resulted in twice as the peak serum
concentration. The plasma profiles of the three major metabolites,
pregnanediol 3a-glucuronide, 17-hydroprogesterone, and
20a-dihydroprogesterone, mimicked that of progesterone.
Six fasting women received 100 mg of fine-particle progesterone
vaginally or orally in the early follicular phase of the menstrual
cycle. The maximal plasma concentration was reached 2 h after oral
administration and had returned to baseline by 6 h. Progesterone was
metabolized predominantly to 5a derivatives, regardless of the route
of administration; 5a-pregnanolone was found after oral but not
vaginal administration. The serum concentrations of
deoxycorticosterone and deoxycorticosterone sulfate were also
increased after oral administration of progesterone. The authors
concluded that ingested progesterone is metabolized mainly in the
intestine (Nahoul et al., 1993).
Seven subjects consumed several oral formulations containing 200
mg progesterone, and blood samples were collected after 0.5, 1, 2, 3,
4, and 6 h. A mean peak serum concentration of 30 ng/ml progesterone
was achieved with a fine-particle formulation in oil 2 h after
administration; the mean peak concentrations achieved with the other
formulations were 9.6 ng/ml at 4 h with plain-milled, 13 ng/ml at 3.2
h with fine-particle, 11 ng/ml at 4 h with plain-milled in oil, and 11
ng/ml at 4.1 h with fine-particle enteric coated capsules (Hargrove et
al., 1989).
Five postmenopausal women received 100 mg progesterone per day
orally for five days. The peak plasma progesterone concentrations,
7-11 ng/ml, were reached within 4 h after the last dose, and the
concentrations remained raised for 96 h. The concentration of the
metabolite 20a-dihydro-progesterone mimicked that of progesterone
closely, at 3-5.1 ng/ml. Other metabolites present in plasma included
pregnanediol-3alpha-glucuronide (550-1000 ng/ml) and
17-hydroxyprogesterone (1.4-3.2 ng/ml) (Whitehead et al., 1980).
The effect of food on progesterone absorption was tested in 15
normal postmenopausal women who received 200 mg fine-particle
progesterone for five days or placebo under fasting and non-fasting
conditions; 100, 200, or 300 mg fine-particle progesterone for five
days under fasting conditions; or 200 mg fine-particle progesterone or
50 mg progesterone in oil intramuscularly for two days. Ingestion of
food increased the integrated area under the curve of plasma
concentration-time and maximal plasma concentration but not the time
to the maximal concentration, and increased the bioavailability of
progesterone twofold. Absorption and elimination were independent of
dose over the range tested. The bioavailability of orally administered
progesterone was 8.6% that of intramuscularly administered compound
(Simon et al., 1993).
The bioavailability of 50-200 mg fine-particle progesterone was
studied in normally menstruating women treated by sublingual, oral
(capsule and tablet), vaginal, or rectal administration during the
follicular phase. Increased serum concentrations persisted for over 8
h in women given > 100 mg progesterone vaginally or rectally and over
24 h in those given 200-mg tablets orally. The authors concluded that
clinically relevant concentrations of progesterone can be reached with
fine-particle oral formulations (Maxson & Hargrove, 1985).
Progesterone is bound in serum to CBG and to albumin. Albumin has
a high capacity but low affinity for progesterone, while CBG has a
higher affinity but a lower capacity. Approximately 17% of serum
progesterone is bound to CBG and 80% to albumin, and 2.5% is in the
free state. CBG is synthesized and secreted by the liver and other
tissues, and fetal hepatocytes can synthesize it. A receptor for
unliganded CBG exists on the surface of target cell membranes, which,
when bound to steroid, can activate adenyl cyclase (Orth & Kovacs,
1998).
Progesterone is eliminated via the urine after conversion in the
liver to pregnane-3 alpha,20 alpha-diol (preganediol) and conjugation
to glucuronic acid at C3. Pregnanediol is the major urinary metabolite
of progesterone and serves as an index of progesterone secretion. The
metabolic clearance rate for progesterone is approximately 2200 l/day
(Goldfien & Monroe, 1994).
Elimination of progesterone after intravenous administration of
500 mg/kg bw to ovariectomized rats was described by a two-compartment
model, with distribution and elimination half-lives of 0.13 ± 0.024
(mean ± SD) and 1.2 ± 0.21 h, respectively, and a volume of
distribution of 2.4 l/kg bw (Gangrade et al., 1992). The plasma
half-life of progesterone has also been reported as 5 min (Williams &
Stancel, 1996).
In five ovariectomized gilts given 47.4 nmol progesterone,
bi-exponential clearance was observed, with mean half-lives of 2.5 and
34 min. The authors concluded that the half-life of the first
compartment indicated that the liver is the principal organ of
clearance. In two gilts given 44.7 or 64.6 nmol progesterone, a
secondary increase in the concentration of progesterone in portal and
mesenteric plasma was observed, indicating that progesterone may
undergo enterohepatic circulation (Symonds et al., 1994).
Six adult anestrous bitches were given progesterone
intramuscularly at 2 mg/kg bw, and the progesterone concentrations in
serum were monitored for 72 h. A peak serum concentration of 34 ng/ml
was reached 1.8 h after dosing, but by 72 h the serum concentrations
had reached the preinjection concentration of 0.9 ng/ml. The mean
half-life of absorption was 0.5 h and the mean half-life of
elimination 12 h. The authors used computer modelling to establish
that a dose of 3 mg/kg bw intramuscularly once a day would maintain
the serum concentration at > 10 ng/ml (Scott-Moncrieff et al., 1990).
2.2.2 Biotransformation
Progesterone is irreversibly converted for subsequent elimination
by hydroxylation to more polar and less active compounds, particularly
at C21, C6, and C16. Progesterone hydroxylation is catalysed by
members of various families of cytochromes P450 and other enzymes
found in hepatic and extrahepatic tissues. Human 6ß-hydroxylase
activity is catalysed most efficiently by CYP3A4 and to a lesser
extent by CYP2D6, whereas 16 alpha-hydroxylation is catalysed by
CYP3A4, 1A1, and 2D6 (Niwa et al., 1998).
Tissue and species differ in the metabolism of progesterone. For
example, human but not rat kidneys possess 6ß-hydroxylase activity.
Steroid 21-hydroxylase in liver is provided primarily by CYP2C5 in
rabbits (Kedderis & Mugford, 1998) and by CYP2C6 in rats (Endoh et
al., 1995), which are P450 isozymes that have not been found in human
liver. Rabbit liver CYP2C3 catalyses the 6ß-and 16 alpha-hydroxylation
of progesterone (Hsu et al., 1993), but this enzyme has also not been
found in human liver.
It is well recognized that progesterone can serve as a precursor
for other bioactive steroids, including hydroxylated progestogens,
corticosteroids, androgens, and estrogens. Progesterone in plasma may
be 21-hydroxylated to deoxycorticosterone enzymatically, independently
of the adrenal steroid 21-hydroxylase (Mellon & Miller, 1989).
Alternatively, progesterone may be reduced to 5
alpha-dihydroprogesterone (5 alpha-pregnane-3,20-dione) and further to
5 alpha-pregnan-3 alpha-ol-20-one, which may bind to the GABAA
receptor on cell surface membranes (reviewed by Baulieu, 1997; see
also below). Both of these compounds are secreted by the ovary and
both are also produced by progesterone metabolism in the hypothalamus
and pituitary (Mahesh et al., 1996).
Progesterone can be metabolized to 17 alpha-hydroxyprogesterone
by P450 17 alpha-hydroxylase and further to androstenedione. Cells
that do not express this enzyme cannot metabolize progesterone to
androgenic or estrogenic hormones. In human ovarian thecal cells,
17 alpha-hydroxypro-gesterone was not further metabolized to
androstenedione or to any other product, whereas pregnenolone and
17 alpha-hydroxypregnenolone were converted to dihydroepiandrosterone
and androsterone (McAllister et al., 1989). Thus, the conversion of
progesterone to androgen is a minor pathway in some tissues or during
particular stages of follicular development.
A substantial increase was noted in the ability of the rat uterus
to metabolize progesterone to 5 alpha-pregnane-3,20-dione and
3 alpha-hydroxy-5 alpha-pregnan-20-one (allopregnanolone) between days
11 and 21 of gestation when serum progesterone concentrations are
high. While the former compound actively binds progesterone receptors
in some tissues, this ability is apparently lacking in rat uterus
(Tsai et al., 1998). Some indirect evidence exists that uterine
contractility is mediated, at least in part, through GABAA receptors
(Mahesh et al., 1996).
2.2.3 Biochemical parameters
2.2.3.1 Synthesis
Progesterone is synthesized from steroid precursors in the
gonads, adrenal cortex, and placenta of mammals. Cholesterol obtained
primarily from circulating low-density lipoprotein serves as the
precursor for steroid biosynthesis, although steroidogenic cells are
capable of local de novo cholesterol synthesis. In non-pregnant women,
progesterone in the bloodstream is derived principally from secretion
by the granulosa and thecal cells of the ovary. Gonadal synthesis of
progesterone is regulated by luteinizing and follicle-stimulating
hormones secreted by the anterior pituitary. The secretion of these
hormones, in turn, is regulated by gonadotropin-releasing hormone,
steroid hormones, and other regulating factors in a complex feedback
loop which is not completely understood. During the follicular phase,
about half of the progesterone is of ovarian origin and the other half
of adrenal origin. During the luteal phase, production shifts
predominantly to progesterone of ovarian origin.
The concentrations of circulating progestogens, daily production,
and metabolic clearance rates have been reported in previous reviews
(IARC, 1979, specifically 'General remarks on sex hormones') and in
most textbooks of endocrinology (e.g. Braunstein, 1994; Goldfien &
Monroe, 1994; Carr, 1998; Griffin & Wilson, 1998).
2.2.3.2 Mechanism of action
In the conventional view of the action of steroid sex hormones,
they diffuse passively into the cell, interact with specific
intranuclear receptors, and bind to specific DNA sequence on the
genome (O'Malley et al., 1991). Ligand-receptor binding in conjunction
with interactions with other nuclear transcription factors initiates
transcription of a specific set of genes which are ultimately
responsible for the translation of hormonal action into physiological
events. Intracellular receptors for progesterone, a superfamily of
ligand-activated transcriptional factors, mediate most of its effects.
In humans, three specific progesterone-binding receptors have been
defined: the 116-kDa B-receptor, an N-terminal truncated 94-kDa
A-receptor, and the 60-kDa C-receptor; the last lacks the first zinc
finger but contains, as do isoforms A and B, the hormone-binding
domain (Leighton & Wei, 1998). Progesterone receptors, like other
steroid hormone receptors, form dimers when a hormone binds. The
various progesterone receptor isoforms have similar binding affinities
for progesterone but may modulate regulation of a defined set of
hormone-dependent genes. In addition, receptors for progesterone,
glucocorticoids, mineralocorticoids, and androgens bind to overlapping
DNA response elements and can thus regulate the same genes, adding
complexity to the biological function of the steroid (Tsai et al.,
1998). The specificity depends on the type or number of receptors
present in the cell and other DNA sequences outside the
steroid-binding domain.
Progesterone and estrogen interact to control the growth and
development of organ systems, primarily the female reproductive tract.
Progesterone inhibits estrogen-stimulated endometrial proliferation
and prepares the endometrium for implantation of the blastocytst. In
this process, the endometrium is converted from a proliferative to a
secretative phase, with accompanying morphological and biochemical
changes. Although progesterone stimulates formation of secretory
alveoli in the mammary gland, particularly during pregnancy,
progesterone itself has minimal effect in the absence of estrogen
priming, as in the uterus. Of the sex steroid hormones, progesterone
has the smallest identified distribution of receptor tissue (Williams
& Stancel, 1996) and perhaps the smallest cohort of transcriptionally
responsive genes. Receptor levels and hormone responsiveness vary
throughout the estrus cycle in all species: in the follicular phase,
the levels are low but increase in response to serum estrogen; the
levels decrease during the luteal phase in response to output of
progesterone by the corpus luteum. In rats, the progesterone receptor
levels increase throughout gestation despite high circulating
concentrations of progesterone, whereas in other species little change
or a decrease (guinea-pig) is noted (Tsai et al., 1998). An
estrogen-stimulated increase in progesterone receptors is essential
for progestogen activity in the uterus. Estrogen is required for the
maintenance of intracellular progesterone receptor levels, and
progesterone, in turn, decreases the estrogen receptor concentration
and the levels of progesterone receptor.
The metabolic effects of progesterone in humans include increased
plasma concentrations of cholesterol and low-density lipoprotein and
decreased high-density lipoprotein, and transient decreases in sodium
excretion due to its capacity to antagonize aldosterone. Progesterone
has well-known effects on basal body temperature. Hydroxylated
metabolites of progesterone, namely the 3 alpha-and 3 alpha,5 alpha
(allopregnanolone) derivatives, are suspected to act as
neuropharmacological agents through the GABAA receptor (Mahesh et
al., 1996; Wiebe et al., 1997; Concas et al., 1998; Smith et al.,
1998). Synthesis and accumulation of the progestogen-type
neuropharmacological agents are not related to plasma progesterone
concentrations (Concas et al., 1998).
2.3 Toxicological studies
The toxicological studies reviewed below were reported in the
scientific literature.
2.3.1 Acute toxicity
The therapeutic dose of fine-partcile progesterone is 400 mg/day
for 10 days 5Physicians' Desk Reference, 1999). A dose of 300 mg/day
taken by women at bedtime for 10 days per month was well tolerated,
the only specific side-effect being mild, transient drowsiness (de
Lignieres).
2.3.2 Short-term studies of toxicity
In order to clarify the nature of the vaginal response to
neonatal treatment with progesterone and to record the responses of
other target organs, including the mammary gland, to such treatment,
291 female BALB/cfC3H/Crgl mice bearing the mouse mammary tumour virus
received daily subcutaneous injections of 5 or 20 µg estradiol or 100
µg progesterone; 5 µg estradiol and 100 µg progesterone; or 20 µg
estradiol and 100 µg progesterone for five days beginning 36 h after
birth. One half of the mice were ovariectomized on day 40. Vaginal
smears were taken for 25 days when the mice were 40-50 days old. The
mice were killed and autopsied at the time of onset of mammary tumours
or at one year of age. The results are shown in Table 14. Mice
receiving progesterone alone showed ovary-dependent, persistent
vaginal cornification. When progesterone and estradiol were given
concurrently, the occurrence of persistent vaginal cornification was
significantly lower than in mice receiving estradiol alone.
Progesterone alone produced hyperplastic downgrowths and lesions of
both vaginal and cervical epithelia but to a lesser degree than in
mice treated neonatally with estrogen. When progesterone was given
concurrently with estradiol, the incidence of lesions was lower but
their severity was greater. The low doses of estradiol and
progesterone each resulted in an earlier age of onset and a higher
incidence of mammary tumours; this also occurred after both combined
estrogen plus progesterone treatments. In treated mice ovariectomized
on day 40, normal mammary development did not occur and mammary
tumours failed to appear, regardless of neonatal treatment. The
authors concluded that progesterone causes ovary-dependent, persistent
vaginal cornification and hyperplastic changes in the vaginal and
cervical epithelium, affects the incidence of these lesions in mice
treated simultaneously with estradiol, and significantly increases the
incidence of mammary tumours in mammary tumour-virus bearing mice
(Jones & Bern, 1977).
2.3.3 Long-term studies of toxicity and carcinogenicity
The toxicology of progesterone administered by subcutaneous or
intramuscular injection to mice, rabbits, and dogs or by subcutaneous
implantation in mice has been extensively reviewed (IARC, 1979).
Progesterone increased the incidences of ovarian, uterine, and mammary
tumours in mice and mammary gland tumours in dogs. Neonatal treatment
increased the occurrence of precancerous and cancerous lesions of the
genital tract and mammary tumorigenesis in female mice. The IARC
Working Group concluded that the carcinogenic effects of estrogens and
progestogens were inextricably linked to the hormonal milieu and to
dose-effect relationships (IARC, 1979, 1987)
The role of hormones, including progesterone, in mammary
neoplasia in rodents and their relevance to human risk assessment have
been reviewed (Russo & Russo, 1996), and it was noted that rodent
models mimic some but not all of the complex external and endogenous
factors involved in initiation, promotion, and progression of
carcinogenesis. Tumour type and incidence are influenced by the age,
reproductive history, and endocrine milieu of the host at the time of
exposure. The spontaneous incidence of tumours varies from strain to
strain of rats and mice. In rats, most spontaneously developed
neoplasias, with the exception of leukemia, are of endocrine organs or
organs under endocrine control. Russo & Russo (1996) concluded that
mechanism-based toxicology is not yet sufficient for human risk
assessment, and the approach should be coupled to and validated by
traditional long-term bioassays.
Table 14. Incidences of mammary tumours and nodules in mice treated neonatally with progesterone
Neonatal treatment No. bearing tumours/ Age at No. bearing nodules/
no. examined (%) appearance no. examined (%)
Estradiol Progesterone (months)
0 100 23/32 (72) 9.3 27/32 (84)
5 0 17/19 (89) 9.2 19/19 (100)
5 100 20/32 (63) 9.3 27/32 (84)
20 0 4/11 (30) 9.8 6/11 (55)
20 100 33/44 (75) 9.6 40/44 (91)
0 0 5/17 (29) 11.3 1/17 (6)
Female rabbits were treated with large doses of either
progesterone or testosterone for up to 763 days, and
hydroxyprogesterone caproate was given intramuscularly every other
week at an average dose of 13 mg/rabbit to 19 animals; testosterone
ethanate was given intramuscularly every other week at an average dose
of 15 mg to 21 animals. Rabbits treated with progesterone developed
numerous cysts of the endometrium, sometimes associated with atypical
hyperplasia. Active mammary secretion also occurred. After large doses
of testosterone, one animal developed two adenomatous polyps of the
endometrium, but there were no other noteworthy endometrial changes.
One control animal developed similar polyps. Neither hormone
significantly altered other tissues, including the ovary, adrenal,
thyroid, and pituitary glands. No precancerous endometrial changes or
cancers were found (Meissner & Sommers, 1966).
2.3.4 Genotoxicity
Genetic toxicity was not studied to support registration of
progesterone for use in cattle. A working group convened by IARC
examined the available information and concluded that: 'Progesterone
did not induce dominant lethal mutations in mice or chromosomal
aberrations in rats treated in vivo. It did not induce chromosomal
aberrations or sister chromatid exchanges in cultured human cells, nor
chromosomal aberrations or DNA strand breaks in rodent cells. Studies
on transformation of rodent cells in vitro were inconclusive: a
clearly positive result was obtained for rat embryo cells, a weakly
positive result for mouse cells and a negative result for Syrian
hamster embryo cells. Progesterone was not mutagenic to bacteria.'
(IARC, 1987).
Several studies on the genetic toxicity of progesterone have been
reported in the scientific literature since the IARC evaluation. After
exposure of DNA to 2 mmol/L progesterone, no DNA adducts were formed,
as determined by 32P-postlabelling. The presence of a carbonyl group
at C17, which progesterone lacks, was strongly associated with DNA
binding by other sterols (Seraj et al., 1996).
No significant increase in the frequency of structural or
numerical chromosomal aberrations was seen in Syrian hamster embryo
cells after treatment for 24 h with 3-30 µg/ml progesterone.
Progesterone was able to transform cells only at the highest dose
tested (30 µg/ml), but even at this dose, the frequency of
transformation was one half that found with 1 µg/ml benzo[ a]pyrene
(Tsutsui et al., 1995).
Progesterone was administered by gavage to female rats as a
single dose of 100 mg/kg bw three days before a two-thirds
hepatectomy, and the rats were killed for cell sampling two days after
the hepatectomy. The frequencies of micronucleated and binucleated
hepatocytes were determined in a suspension of liver cells isolated by
collagenase perfusion. The frequency of micronucleated hepatocytes was
3.5-fold higher in progesterone-treated animals than in controls, but
no increase in the frequency of binucleated hepatocytes was observed.
The increase in micronucleus frequency may be related to chromosomal
breakage or to loss of whole chromosomes. A dose of 100 mg/kg bw
progesterone administered by gavage weekly for six weeks, followed by
partial hepatocetomy, did not increase the number of
gamma-glutamyltranspeptidase-positive foci in liver. The authors noted
that similar findings were made under similar experimental conditions
with the non-genotoxic liver mitogen 4-acetylaminofluorene,
progesterone did not form DNA adducts in rat liver, and equivocal
responses were observed for DNA repair in primary rat heptocytes.
In vitro, progesterone was not mutagenic in bacteria, did not induce
chromosomal aberrations or sister chromosome exchange in cultured
human cells or chromosomal aberrations or DNA strand breaks in rodent
cells, inconclusive results were obtained with regard to
transformation of rodent cells and in the L5178Y TK+/- forward
mutation assay, and a slight but statistically significant increase in
the frqueny of sister chromatid exchange was observed in lymphocytes
from children. In vivo, progesterone did not induce dominant lethal
mutations in mice or chromosomal aberrations in rat bone marrow
(Martelli et al., 1998).
In three male and three female Han:Wistar rats given progesterone
at 100 mg/kg bw per day intragastrically as an aqueous
microcrystalline suspension for 14 days, no progesterone-specific
adducts were observed in the liver, as determined by 32P-postlabelling
(Feser et al., 1996).
2.3.5 Reproductive toxicity
Progesterone at doses of 5-200 mg was administered daily by
subcutaneous injection to 22 pregnant Long-Evans rats on days 15-20 of
gestation. The pups were surgically removed from the dams on day 22
and killed after 20 days. No difference in the anogenital distance was
observed between treated and control rats, and sex was easily
determined, whereas animals treated with concentrations of 1.5-2.5 mg
synthetic progestogens were all masculinized (Revesz et al., 1960).
Six pregnant rhesus monkeys were treated with progesterone in oil
intramuscularly at a dose of 50 mg daily for five days each week.
Treatment was begun on days 24-28 of gestation and was maintained
until delivery. The newborns were weighed, killed, and completely
autopsied, with appropriate histological sections. The offspring of
animals treated with progesterone (three males and thee females) were
delivered at about term, and no abnormalities were noted. In contrast,
most of the offspring of monkeys treated with synthetic progestogens
were born prematurely and were not viable; furthermore, all of the
females were masculinized. The authors concluded that progesterone in
comparatively large doses produces no abnormalities and does not
interfere with the normal conclusion of a pregnancy in rhesus monkeys
(Wharton & Scott, 1964).
2.4 Observations in humans
The commonest uses of progestogens are for contraception and for
hormone replacement therapy. For these purposes, synthetic
progestational agents are usually administered, which differ in
structure and function from natural progesterone (Williams & Stancel,
1996).
Changes in endometrial morphology was investigated in 43 patients
without ovaries who were given progesterone intravaginally,
intramuscularly, or orally. The response to intramuscular
administration in oil was heterogeneous, whereas the endometrial
morphology seen after intravaginal administration of fine-particle
progesterone closely matched that observed in the natural cycle, and
pregnancies were supported in two women. Orally administered
progesterone did not affect endometrial morphology (Bourgain et al.,
1990).
Sixty women with oligomenorrhea or amenorrhoea received a 10-day
course of 200 or 300 mg fine-particle progesterone orally. Withdrawal
bleeding occurred in 90% of the women receiving 300 mg progesterone,
58% receiving 200 mg, and 29% of those given placebo. Their lipid
concentrations were unchanged (Shangold et al., 1991).
Seven women with luteal phase defects, treatment with 200 mg
fine-particle progesterone orally three times a day was effective
(Frishman et al., 1995).
The side-effects occurring in women receiving fine-particle
progesterone orally as part of hormone replacement therapy have been
reviewed. They include minor changes in plasma lipoprotein profile in
some but not all studies but no effect on haemostatic parameters (such
as platelet aggregation and fibronolytic capacity) (Sitruk-Ware et
al., 1987).
Two groups of six healthy postmenopausal women were given 300 mg
of fine-particle progesterone once or twice a day on days 1-14 after
estrogen priming for 30 days. Endometrial biopsy samples were then
studied for effects on histological appearance, glycogen content,
ribosomal RNA, and nuclear estrogen receptors in glands, surface
epithelial, and stroma, and the samples from treated women were
compared with biopsy samples obtained before therapy. The group given
progesterone once a day showed incomplete secretory conversion,
whereas the other group showed full secretory conversion with
suppression of mitotic activity and the presence of a predecidual
reaction. Staining for glycogen was significantly increased in both
groups, by 124% at 300 mg/day and 291% at 600 mg/day. Ribosomal RNA
was decreased by 50 and 73% in the two groups, respectively. The
nuclear receptor content decreased in every region of the endometrium
in both groups; although the decrease in nuclear estrogen receptor
content in the stroma of the group given 300 mg/day did not reach
significance, this finding was attributed to insufficient samples (Kim
et al., 1996).
Postmenopausal women were treated for 21 of 28 days for five
years with percutaneous estradiol at a dose of 1.5 mg/day. Within the
first six months, some women received a dose of 3 mg/day estradiol for
25 of 28 days. The initial dose of progesterone was 200 mg/day orally
for the last 14 days of estradiol treatment. This dose was increased
to 300 mg/day for women in whom the desired clinical effect was not
achieved at the lower dose (i.e. withdrawal bleeding). The baseline
plasma concentration was 18 pg/ml, which increased to an average of
68-89 pg/ml at various times during the treatment period. During the
last 12 months of treatment, 2% of the women had episodes of irregular
bleeding, 21% had regular cyclic bleeding, and 77% had no bleeding.
The highest incidence of cyclic bleeding was observed with the
high-estradiol plus high-progesterone regimen (3 mg estradiol and 300
mg progesterone), and the highest incidence of amenorrhoea was seen in
women given the low-estradiol plus low-progesterone regimen. There was
no evidence of endometrial hyperplasia after five years of treatment
with estradiol and progesterone (Moyer et al., 1993).
See also section 4, 'Epidemiological studies of women exposed to
postmenopausal estrogen therapy and hormonal contraceptives'.
3. TESTOSTERONE
3.1 Explanation
Testosterone propionate (200 mg) in combination with estradiol
benzoate (20 mg) is administered to cattle as an ear implant to
increase the rate of weight gain (growth promotion) and to improve
feed efficiency. Testosterone propionate is rapidly cleaved to
testosterone in vivo and is thus considered an 'endogenous
substance', as the residues are structurally identical to testosterone
produced in animals and humans.
3.2 Biological data
3.2.1 Absorption, distribution, and excretion
Testosterone is generally considered to be inactive when given by
the oral route owing to gastrointestinal and/or hepatic inactivation.
Maintenance of physiological concentrations after injection of
testosterone is also difficult because of its rapid clearance (Griffin
& Wilson, 1998). There is little clinical interest in oral forms of
testosterone (i.e. fine-particle formulations), as the high doses
required to overcome first-pass metabolism in the liver result in a
high hepatic load (WHO, 1992).
Absorption of the ester testosterone undecanoate has been studied
for its possible use in testosterone replacement therapy. The plasma
concentrations of testosterone were measured by radioimmunoassay after
oral administration of 25 mg testosterone or 40 mg testosterone
undecanoate (approximately 25 mg testosterone) to young women, and
were compared with that in subjects receiving testosterone at 1.5
µg/kg bw by intravenous administration. Bioavailable testosterone
represented 3.6% of administered testosterone and 6.8% of administered
testosterone undecanoate. The low bioavailability of orally
aministered testosterone has been attributed to its high metabolic
clearance rate, 25 ml/min per kg. The estimated bioavailability
provides justification for the high dose (120-140 mg/day) considered
to be necessary to replace daily production (5-7 mg) (Tauber et al.,
1986). The long aliphatic side-chain of testosterone undecanoate is
thought to facilitate its lymphatic absorption and thus to avoid
first-pass metabolism (Butler et al., 1992; Griffin & Wilson, 1998).
In normal men, circulating androgens are bound to SHBG and to a
lesser extent to serum albumin. Only 1-2% of the testosterone in
circulation is unbound: 44% is bound to SHBG and 54% is bound to
albumin and other proteins (Griffin & Wilson, 1998). Testosterone
binds to albumin with lower affinity than to SHBG. Plasma SHBG is
secreted from the liver, but adult rodent livers do not produce the
secretory form of SHBG (Reventos et al., 1993). The plasma
concentration of SHBG is increased 5-10-fold by estrogens and
decreased twofold by testosterone (Griffin & Wilson, 1998). A
non-secretory form of SHBG is present in many tissues, including
reproductive tissues and the brain. A SHBG receptor on the surface of
some cell membranes binds unliganded SHBG; steroid hormone then binds
SHBG, which can then not bind receptor (Hryb et al., 1990). The
SHBG-receptor complex present on membranes of target tissues may be
responsible for the interaction between the steroid hormone and cAMP
pathways (Rosner, 1991). The intracellular form of SHBG may also
sequester or direct hormones to their target tissue.
When radiolabelled testosterone is administered intragastrically
to rats, about one fourth appears in bile within 12 h as glucuronide
and sulfate conjugates, indicating enterohepatic circulation (IARC,
1979). When testosterone is used as a substrate, hepatic
glycuronsyltransferase and sulfotransferase activities are higher in
men than in women and show extensive individual variation (Pacifici et
al., 1997). Individual but not sex differences were observed in the
activities of the human jejunal estrogen and dehydroepiandrosterone
sulfotransferases (Her et al., 1996).
About 90% of an administered dose of radiolabelled testosterone
is found in the urine, and about 6% undergoes enterohepatic
circulation and appears in the feces (Wilson, 1996). About 1% of
testosterone is secreted in urine as glucuronide, while the remainder
is secreted primarily as 17-ketosteroids (androsterone and
etiocholanolone). Smaller amounts of glucuronide and sulfate
conjugates of androstanediol and estrogens are found.
The plasma half-life of testosterone after intravenous
administration was 10 min (Tauber et al., 1986).
3.2.2 Biotransformation
The metabolism of testosterone has been reviewed (IARC, 1979;
Goldfien & Monroe, 1994; Wilson, 1996; Griffin & Wilson, 1998).
17ß-Hydroxysteroid dehydrogenase catalyses the reversible
oxido-reduction of androstenedione to testosterone, and of estrone to
estradiol and dehydroepi-androsterone to D5-androstenediol. Further
reductions in this pathway (5 alpha and 5 beta followed by 3 keto)
lead to the formation of androsterone and etiocholanolone, the
principal urinary products of testosterone.
Significant differences are found between the metabolic pathways
of testosterone in rodents and humans. Sex-specific regulation of
cytochrome P450s has not been found in human liver, although sex
differences in the metabolism of xenobiotics exist (Kedderis &
Mugford, 1998). Hepatic rat testosterone 7 alpha-hydroxylase (P450
2A1) and mouse testosterone 15 alpha-hydroxylase (P450 2A4) are not
found in human liver. Testosterone is converted to its 6ß form by
human liver CYP3A4 (Lacroix et al., 1997). The major pathways for
inactivation of testosterone in female mouse liver are hydroxylation
at the 16ß-, 6 alpha-, and 16 alpha-positions followed by conjugation
(Wilson & LeBlanc, 1998). Metabolism of testosterone in rat liver,
used as a diagnostic tool for P450 isozyme activity, produced
7 alpha-, 6ß-, 16 alpha-, and 2 alpha-hydroxytestosterones (Swales et
al., 1996). Testosterone 6ß-and 16 alpha-hydroxylation is carried out
in rat liver by CYP3A18 and by other members of the 3A family (Nagata
et al., 1996). No 16ß-hydroxylase (CYP2B1) activity was detected in
rat liver (Swales et al., 1996).
Extensive reductive metabolism of testosterone occurs in the
liver and extrahepatic tissues. The rat small intestine is capable of
oxidizing testosterone, and the enzyme is present in homogenates of
rat gastric and duodenal mucosa at significantly higher levels than in
jejunum; it was also found in the ileum and colon. The major portal
vein metabolite was androstenedione, indicaing that the gut rather
than the liver is primarily responsible for the metabolism of orally
administered testosterone (Farthing et al., 1982)
Testosterone is a precursor of other physiologically relevant
steroids. The active metabolite DHT, formed through the action of
5 alpha-reductase, is an important physiological substrate for the
androgen receptor. DHT binds with higher affinity to the androgen
receptor than testosterone and is considered to be the main cellular
mediator of androgen activity in some tissues. DHT is metabolized to
androsterone, androstanedione, and 3 alpha- and 3ß-androstanediol.
3ß-Hydroxysteroid dehydrogenase also catalyses the conversion of
dehydroepiandrosterone to androstenedione in the testosterone
biosynthetic pathway. Estradiol is formed by aromatization of the A
ring of testosterone. DHT cannot be converted to estrogen.
3.2.3 Biochemical parameters
3.2.3.1 Synthesis
Testosterone is synthesized in testicular Leydig cells, ovarian
thecal cells, and the adrenal cortex. The principal regulator of
gonadal steroidogenic synthesis is luteinizing hormone, secreted by
the anterior pituitary. DHT, derived from reduction of testosterone in
peripheral tissues, and D5-androstenediol are also physiologically
relevant androgens (Griffin & Wilson, 1998; Miyamoto et al., 1998).
Testosterone synthesis begins in the fetus during the first trimester
of pregnancy after stimulation by maternal chorionic gonadotropin. In
males, the plasma concentrations of testosterone are high during the
prenatal period, peak at around three months of age, and then remain
very low until about 11 years of age. The testosterone concentrations
reach a plateau at around 17 years of age, remain elevated for
decades, and then gradually decline (see Table 15). The blood
concentrations of testosterone oscillate in response to the pulsatile
secretion of luteinizing hormone throughout the day. Testosterone
decreases the pulsatile release of gonadotropin-releasing hormone from
the hypothalamus and luteinizing hormone from the pituitary by
negative feedback (Griffin & Wilson, 1998). A diurnal trend in the
blood concentrations of testosterone is found as well, the
concentrations in plasma being approximately 25% higher in the early
morning than in the afternoon (Orth & Kovacs, 1998).
Plasma testosterone concentrations also increase in females
during puberty, although to a lesser extent than in males. In adult
premenopausal women, 70-80% of circulating testosterone is derived
from conversion of dehydroxyepiandrosterone and androstenedione from
the adrenals and ovaries. In postmenopausal women, ovarian secretion
may account for up to 50% of testosterone production, as bilateral
oophorectomy results in a significant reduction in circulating
androgens. Women in their 40s have about 50% less circulating
androgens than women in their 20s, probably because of declining
adrenal dehydroxyepiandrosterone secretion (reviewed by Gelfand &
Wiita, 1997).
The concentrations of circulating androgens, their daily
production and metabolic clearance rates are given in reviews (e.g.
IARC, 1979, specifically 'General remarks on sex hormones') and in
most textbooks of endocrinology (e.g. Braunstein, 1994; Goldfien &
Monroe, 1994; Carr, 1998; Griffin & Wilson, 1998).
3.2.3.2 Mechanism of action
Testosterone and DHT readily diffuse into target cells and bind
with high affinity to the androgen receptor. Only one intracellular
receptor type has so far been identified, and molecular and genetic
analyses indicate that multiple androgen receptor subtypes are
unlikely to exist. The receptor-hormone complex binds to specific
target DNA sequences and initiates transcription of androgen-regulated
genes, translating exposure to hormones into physiological events
(Wilson, 1996).
Testosterone is necessary during pubertal development for the
initiation and maintenance of spermatogenesis and for the libido.
Androgens interact with other growth factors to regulate the growth
and differentiation of the prostate. Androgens also stimulate
erythropoietin production in the kidney, stimulate stem cells in the
haematopoietic system, increase nitrogen retention, and regulate male
hair growth, including balding in genetically susceptible individuals.
Androgens in conjunction with growth hormone may accelerate linear
growth during puberty. Androgens decrease serum SHBG concentrations
and shift the high-or low-density lipoprotein:cholesterol ratio
(Griffin & Wilson, 1998; WHO, 1992)
Testosterone and DHT produce different biological effects in
reproductive and non-reproductive organs and during development. In
tissues such as bone, muscle, and testis, testosterone is thought to
have a direct androgenic action, while conversion to DHT is essential
for androgenic action in other target tissues. In the fetus,
testosterone is responsible for development of the seminal vesicles
and epididymis and for virilization of the mesonephric ducts, whereas
DHT is required for external virilization.
3.3 Toxicological studies
The toxicological studies reviewed below were reported in the
scientific literature.
3.3.1 Acute toxicity
Fine-particle orally administered testosterone has not been
approved for clinical use. In studies with 400 mg of fine-particle
testosterone administered orally for 21 days to six healthy male
volunteers, testosterone was well tolerated, with few effects on
lmiver enzyme systems, but induction of the heaptic drug-metabolizing
system, including testosterone metabolism, was reported in all six
subjects (Johnsen et al., 1976).
In one report of an adverse event, a hypogonadal 21-year-old man
began treatment with 200-mg intramuscular injections of testosterone
enanthate every two weeks. He had a cerebral accident which was
attributed to treatment. His plasma testosterone concentration was 11
400 ng/dL, whereas the range in normal men is 200-1000 ng/dL. The
authors concluded that cerebral infarct is a potential complication of
over-zealous andrigen replacement (Nagelberg et al., 1986).
Table 15. Concentrations, metabolic clearance rates, and daily production of circulating androgens
Person Serum concentration Metabolic clearance Total daily production
(ng/ml) (l/day) (mg/day)
Male 690
Prepubertal 0.08-0.14 0.06-0.1
Pubertal 0.84-1.8 0.58-1.2
Adult 3-10 2.1-6.9
Female 690
Prepubertal 0.05-0.13 0.03-0.09
Pubertal 0.09-0.24 0.06-0.17
Adult 0.3-0.7 0.21-0.48
3.3.2 Short-term studies of toxicity
Six intact adult male baboons received weekly intramuscular
injections of 200 mg testosterone enanthate (equivalent to 8 mg/kg bw)
for up to 28 weeks, while two control animals received weekly
injections of the vehicle only. Quantitative increases in the weight
and volume of both prostatic lobes were seen after 15 weeks of
treatment, and by week 28 there was an increase in stromal tissue with
papillary ingrowth or invagination of glandular epithelium in the
caudal lobe of the prostate. The serum concentrations of testosterone
and DHT were significantly elevated, from 10 and 2-3 ng/ml to 30-40
and 5-6 ng/ml, respectively. The androstenedione concentrations were
increased by three to four times and that of estradiol from 20 to
80-90 pg/ml. The authors concluded that these steroids play a direct
role in inducing early benign prostate hypertrophy in baboons and that
their observations were similar to those in human benign prostate
hypertrophy (Karr et al., 1984).
3.3.3 Long-term studies of toxicity and carcinogenicity
The effects of subcutaneously injected or implanted testosterone
and its esters have been reviewed extensively (IARC, 1979). The
working group convened by IARC concluded that: "There is sufficient
evidence for the carcinogenicity of testosterone in experimental
animals. In the absence of adequate data in humans, it is reasonable,
for practical purposes, to regard testosterone as if it presented a
carcinogenic risk to humans."
The relevance of animal models to human prostate disorders has
been reviewed (WHO, 1992). Besides humans, dogs are the only animals
that develop prostatic cancer and benign prostatic hyperplasia at a
high frequency. In this model, long-term treatment with androgens and
estrogens is required to produce hyperplasia, although such synergism
is not observed in other species. ACI rats spontaneously develop
histologically evident prostatic cancer which does not progress to
clinically relevant disease when pharmaco-logically relevant amounts
of exogenous androgen are administered. Prostate cancer has been
induced only in the Noble and Lobund-Wistar strains of rat.
The role of hormones, including androgens, in the development of
mammary neoplasia in rodents and their relevance to human risk
assessment have been reviewed (Russo & Russo, 1996). Endogenous
androgens are necessary for mammary development in rodents, and it was
noted that rodent models mimic some but not all of the complex
external and endogenous factors involved in initiation, promotion, and
progression of carcinogenesis. Tumour type and incidence are
influenced by the age, reproductive history, and the endocrine milieu
of the host at the time of exposure. The spontaneous incidence of
tumours differs in different strains of rats and mice. In rats, most
spontaneous neoplasias, with the exception of leukaemia, are of
endocrine organs or organs under endocrine control. Russo & Russo
(1996) concluded that mechanism-based toxicology is not yet sufficient
for human risk assessment, and the approach should be coupled to and
validated by traditional long-term bioassays.
Fischer 344 rats were given 3,2'-dimethyl-4-aminobiphenyl (a
prostate carcinogen) at 50 mg/kg bw 10 times at two-week intervals,
and then, from week 20, testosterone propionate and/or
diethylstilbestrol by subcutaneous Silastic implant for 40 weeks, as
seven cycles of 30 days' treatment and 10 days' withdrawal.
Intermittent administration of testosterone resulted in suppression of
the development of ventral prostate adenocarcinomas and slight
(non-significant) increases in the incidences of invasive carcinomas
of the lateral prostate and seminal vesicles. Diethylstilbestrol
completely suppressed tumorigenesis, and the combination with
testosterone propionate inhibited prostate tumour development (Cui
et al., 1998).
Hydroxyprogesterone caproate was given intramuscularly every
other week at an average dose of 13 mg to 19 female rabbits, and
testosterone ethanate was given intramuscularly every other week at an
average dose of 15 mg to 21 animals; both treatments were given for up
to 763 days. Rabbits treated with progesterone developed numerous
endometrial cysts, sometimes associated with atypical hyperplasia;
active mammary secretion was also seen. Treatment with testosterone
induced two adenomatous polyps of the endometrium in one animal, but
no other noteworthy endometrial changes were found and one control
animal developed similar polps. Neither significantly altered other
tissues such as the ovary, adrenal, thyroid, or pituitary gland. No
precancerous endometrial changes or cancers were found (Meissner &
Sommers, 1966).
3.3.4 Genotoxicity
No studies of genetic toxicity were available to the IARC working
group (IARC, 1979). Summaries of more recent studies from the
scientific literature are presented below.
Testosterone at concentrations of 1-100 µg/ml had weak
transforming activity, but with no dose-response relationship, in
Syrian hamster embryo cells. It inhibited the transforming potential
of benzo[ a]pyrene when incubated concomitantly or sequentially. The
transforming effect of testosterone was amplified by phorbol acetate
but was completely inhibited by dexamethasone (Lasne et al., 1990).
The growth of Syrian hamster embryo cells was reduced by
incubation with testosterone or testosterone propionate at 10-30 µg/ml
in a dose-dependent manner, and the two compounds were equipotent in
producing morphological transformation. The propionate induced
dose-dependent cell transformation at concentrations of 1-30 µg/ml,
while testosterone was effective only at 30 µg/ml. At that
concentration, the transformation frequency induced by testosterone
and the propionate was one-half that induced by 1 µg/ml
benzo[a]pyrene. Neither steroid significantly increased the frequency
of chromosomal aberrations or aneuploidy. Testosterone did not induce
gene mutations at the hprt or Na+/K+ ATPase locus (Tsutsui et
al., 1995).
When testosterone and other steroids were added at a final
concentration of 2 mmol/L to DNA obtained from human surgical
resections, rat liver, HepG2 cells, and calf thymus, testosterone did
not form adducts with naked DNA. Furthermore, no adducts were observed
in DNA isolated from HepG2 cells incubated with 10-100 µmol/L
testosterone for 24 h. The presence of a carbonyl group at C17 (which
testosterone lacks) was predictive of DNA reactivity (Seraj et al.,
1996). DNA strand breaks were observed in the dorsolateral prostate of
Nobel rats treated with a combination of testosterone and estradiol
but not in those treated with testosterone alone (Ho & Roy, 1994).
NBL/Cr rats received two Silastic implants subcutaneously, one of
which contained testosterone and the other estradiol, at the age of
eight to nine weeks, and were killed 8, 16, or 24 weeks after the
start of treatment. The quantity of steroid in the implant was not
described, but the release rate was predicted to increase the
estradiol concentrations by approximately 14-fold while maintaining
normal plasma testosterone concentrations. Adduct levels were
determined by 32P-postlabelling. The prostate glands were found to
contain two major endogenous adducts and several minor spots,
irrespective of treatment, but in treated animals the relative levels
of the two adducts were decreased from 1 and 10 × 109 to 0.5 and 3 ×
109 in the ventral but not in the dorsolateral or anterior prostate.
A major adduct spot was observed in the dorsolateral prostate of
animals treated with both hormones for 16 and 24 weeks but not in
those treated for 8 weeks or in control animals. The presence of this
adduct coincided with the occurrence of dysplastic lesions in the
dorsolateral prostate. It appears to be different from the indirect
estrogen-DNA adducts found in the kidneys of male hamsters treated
with estrogen implants and from I-compounds. The authors suggested
that testosterone enhances prostatic carcinogenesis, perhaps by
generating estradiol through peripheral conversion (Han et al., 1995).
3.3.5 Reproductive toxicity
Complete resorption of embryos was seen in female Sprague-Dawley
rats that received 10 mg estradiol (five rats) or testosterone (seven
rats) as a subcutaneous implant on day 10 of gestation. No effect was
seen in control pregnant rats given dextran by the same route (Sarkar
et al., 1986).
3.4 Observations in humans
Androgens are used therapeutically in men with deficient
testicular function to replace normal testosterone. In women, an
estrogen-androgen combination is used in postmenopausal hormone
replacement therapy (Sands & Studd, 1995; Wilson, 1996). The effects
of excess androgens particularly in young boys and women include
virilizing effects (such as hair loss, deepened voice, acne, growth of
facial hair, and menstrual irregularities); estrogenic effects (such
as gynecomastia in men), and toxic effects (such as oedema and
hepatotoxic effects, especially with 17 alpha substitutions). The
serious side-effects of orally administered androgens are particularly
common in the liver in the treatment of aplastic anaemia but are also
observed during treatment for other conditions, including
hypogonadism. The complications include blood-filled cysts and
heaptomas. Increasing the plasma concentration of testosterone to
above physiological levels results in decreased secreation of
follicle-stimulating and luteinizing hormones and decreased testicular
volume and sperm production (Wilson, 1996; Griffin & Wilson, 1998).
In postmenopausal women treated with androgen replacement therapy
at doses up to 10 mg/day for more than six months, the virilizing
effects of orally administered alkylated andrigens such as
methyltestosterone were found to be deoendent on dose and duration.
The masculinizing effects abated when exposure to the andrigens was
decreased or eliminated (reviewed by Gelfand & Wiita, 1997).
Methylation of testosterone at the 17a position effectively reduces
its first-pass metabolism in the liver, thus increasing its
bioavailability. It can thus reach the systemic circulation when given
at therapeutic concentrations (Friffin & Wilson, 1998).
Testosterone undecanotate, the only oral testosterone ester
preparation available, has been used successfully for the management
of delayed puberty in boys by administration of 40 mg daily for 15-21
months. No side-effects were observed, but progression of
virilization, testicular growth, and acceleration of growth associated
with puberty were observed (Butler et al., 1992).
Subcutaneously implanted pellets containing 100 mg crystalline
testosterone release the compound at a rate of 1 mg/day, which falls
to 0.5 mg by three months and 0.35 mg at six months. In postmenopausal
women, such implants raised the serum testosterone concentration
threefold over that of controls. Development of downy facial hair,
hirsutism, acne, and, rarely, voice changes and clitoromegaly have
been reported as adverse effects. Estrogen and estrogen plus
testosterone implants lowered low-density lipoprotein cholesterol and
raised high-density lipoprotein cholesterol in the same proportion
(Sands & Studd, 1995).
The role of endogenous androgens in the development of benign
prostatic hyperplasia and prostate cancer and the effectiveness of
androgen ablation have been reviewed (WHO, 1992; Osterling et al.,
1997). These disorders arise from different parts of the prostate but
are highly associated statistically. The prevalence of histologically
discernible prostate cancer appears to be similar in various
geographic locations, but progression to clinical prostate cancer is
associated with environmental and genetic factors. These disorders
develop later in life only in men who had normal concentrations of
androgens throughout puberty and possibly up to 40 years. Differences
in serum testosterone concentrations have been studied as a possible
cause for the geographical differences in the occurrence of prostate
cancer. Dutch men were found to have a higher average plasma
testosterone concentration than Japanese men, which was statistically
associated with body weight. Nevertheless, the serum testosterone
concentrations of Japanese men with prostate cancer were not different
than those in men without this disease (WHO, 1992).
Epidemiological studies of the relationship between serum
concentrations of androgens and estrogens and prostate cancer have
yielded inconsistent but largely negative results (for references, see
Dorgan et al., 1998; Ross et al., 1998). Since a possible association
with markers of androgen metabolism (androstanediol glucuronide and
DHT) has been observed in some studies, a complex, androgen-dependent,
multi-gene model involving both androgen biosynthesis and metabolic
pathways has been suggested to be critical for susceptibility to
prostatic cancer (Ross et al., 1998).
The effectiveness of orally administered testosterone was
assessed in five eunuchs in a trial in which controls received a
placebo and neither the participants nor the investigators was aware
of which group received treatment. Initial studies indicated that a
dose of 25 mg testosterone had no effect on any clinical parameter,
including sexual desire, erection, ejaculation, and general
well-being. In a second study, a dose of 100 mg/day did not alter the
clinical parameters, whereas 400 mg/day (100 mg four times a day) was
fully effective (Johnsen et al., 1974). Oral administration of
testosterone at 30 mg/day did not restore sexual function to
castrates, while 100 mg/day had a slight effect; 20 mg of testosterone
propionate administered twice a week by injection had moderate
potency. Thus, testosterone is more than 16 times as active by
injection as it is orally (Foss & Camb, 1939).
4. EPIDEMIOLOGICAL STUDIES OF WOMEN EXPOSED TO POSTMENOPAUSAL
ESTROGEN THERAPY AND HORMONAL CONTRACEPTIVES
4.1 Methods
Although no large epidemiological studies on estradiol,
progesterone, or testosterone alone are available, millions of women
have been exposed, chiefly since the 1960s, to a range of exogenous
estrogens and/or progestogens in the course of postmenopausal estrogen
therapy or hormonal contraception. A few extensive reviews on these
compounds have been published recently (WHO, 1998; IARC, 1999) which
provide many more details of the studies summarized here. The findings
of a large number of case-control and cohort studies and clinical
trials in which the relationship between use of exogenous estrogens
and progestogens and the risk for disease was evaluated are reviewed.
Each study design has its strengths and weaknesses: cohort studies are
considered to be less susceptible to bias in recall or information,
but case-control studies are useful for investigating relatively rare
conditions. Whenever possible, the results of pooled analyses of
original data or meta-analyses of published results are given, in
order to provide the best estimate of the relationship between hormone
use and cancer risk. The strength of an association between exposure
to exogenous hormones and the development of disease is given, as is
customary in epidemiological studies, in terms of the relative risk
(RR), which is the ratio of the incidence rate of the disease among
subjects exposed to the factor to that among unexposed subjects. A
relative risk of 1.0 indicates no association, whereas a relative risk
significantly greater than 1.0 implies a positive association between
exposure to the factor and the development of disease; conversely, a
relative risk of less than 1.0 implies that exposure to the factor may
have a protective effect. In order to provide some indication of the
variation of relative risks, 95% confidence intervals (CIs) are
usually calculated. If the interval does not include 1.0, the finding
is considered to be statistically significant. In a few instances,
such as for breast cancer, the estimated excess number of breast
cancer among 10 000 women who had used hormones is also given.
4.2 Postmenopausal estrogen therapy
4.2.1 Exposure
The numbers of women who have used postmenopausal estrogen
therapy varies among countries and within regions of countries (IARC,
1999). The prevalence of use has been greater in the United States
than in most other countries, and use of estrogen therapy after the
menopause is rare in developing countries, although it is increasing.
Conjugated equine estrogens at a standard dose of 0.625 mg daily are
the most widely prescribed preparation for estrogen therapy in women
in the United States, but estradiol and its esters such as estradiol
valerate (1-2 mg daily) are more widely used in most of Europe (Table
16). Conjugated equine estrogens and estradiol compounds yield similar
plasma concentrations of estradiol and estrone. Progestogens such as
medroxyprogesterone acetate, levonorgestrel, and norethindrone acetate
(Table 17) are added to estrogens, typically for 10 days of each
21-day cycle, in combined regimens. Oral administration is the most
popular route, although percutaneous methods are becoming commoner;
use of injections, the first form of postmenopausal estrogen therapy,
has been declining.
4.2.2 Human carcinogenicity
4.2.2.1 Breast cancer
The relationship between estrogen use and breast cancer risk is
of paramount importance because of the frequency of this neoplasm, of
which there are approximately 800 000 new cases per year worldwide,
representing 21% of all cancers in women (Parkin et al., 1999).
Information on the relationship between postmenopausal estrogen
therapy and the risk for breast cancer is available from many
epidemiological studies (Colditz, 1998; IARC, 1999). A pooled analysis
of the original data for 52 705 women with breast cancer and 108 411
women without breast cancer from 51 studies in 21 countries showed a
small increase in risk in current and recent users with longer
duration of use (five years or more) (Collaborative Group on Hormonal
Factors in Breast Cancer, 1997).
The relative risk for having breast cancer was 1.02 (95% CI,
1.01-1.04) for each year of use among current users or those who had
ceased use one to four years previously and 1.4 (95% CI, 1.2-1.5) for
women who had used postmenopausal estrogen therapy for five years or
longer (average duration, 11 years). This increase is comparable with
the effect on risk for breast cancer of delaying menopause, since the
risk for women who have never used such preparations is 1.03 (95% CI,
1.02-1.03) for each additional year at menopause. Five or more years
after cessation of estrogen use there was no significant excess risk
for breast cancer overall or in relation to duration of use.
Of the many factors examined that might affect the relationship
between risk for breast cancer and estrogen use, only a woman's weight
and body-mass index had an effect: the increase in the risk for breast
cancer of current and recent users associated with long duration of
use was greater for women of lower weight than of higher weight or
higher body-mass index. There was no marked variation in the results
according to hormonal type or dose, but little information was
available about long duration of use of any specific preparation.
Cancers diagnosed in women who had ever used postmenopausal estrogen
therapy tended to be less advanced clinically than those diagnosed in
women who had never used them.
Table 16. Routes of administration and dosages of commonly used estrogens
Estrogen Route Dosage
Natural and equine estrogens
Conjugated equine estrogens Oral 0.3-2.5 mg/day
Intramuscular 25 mg/day
Intravenous 25 mg/day
Vaginal 2-4 g/day
Piperazine estrone sulfate Oral 0.625-2.5 mg/day
Vaginal 2-4 g/day
Estradiol Patch 0.05-0.1 mg every
3-4 days
Vaginal 1-4 g/day
Micronized, valerate Oral 1-2 mg/day
Valerate Intramuscular 10 mg/month
Cypionate Intramuscular 1-5 mg/month
Synthetic estrogens
Ethinylestradiol Oral 20-50 µg/day
Mestranol Oral 50-100 µg/day
Diethylstilbestrol Oral 1-5 mg/day
Quinestrol Oral 0.1 mg/day
Table 17. Routes of administration and dosages of commonly used progestogens
Progestogen Route Dosage
Progesterone
Suppositories Vaginal 25-200 mg/day
Rectal 25-200 mg/day
In oil Intramuscular 50-200 mg/day
Micronized Oral 100-300 mg/day
17 alpha-Hydroxy derivatives
Medroxyprogesterone acetate Oral 2.5-10 mg/day
Intramuscular 250-1000 mg/day or week
Megestrol acetate Oral 20-320 mg/day
17 alpha-Hydroxyprogesterone Intramuscular 125-250 mg/week
caproate
Table 17. (continued)
Progestogen Route Dosage
19-Nortestosterone derivatives
Ethynodiol diacetate Oral 1 mg/day
Norethindrone Oral 0.35-10 mg/day
Norethindrone acetate Oral 1-10 mg/day
Norethyndrol Oral 2.5-5 mg/day
Norgestrel Oral 0.3-0.5 mg/day
Levonorgestrel Oral 0.075-0.5 mg/day
Halogenated progesterone
Cyproterone acetate Oral 10-50 mg/day
Retroprogesterone
Dydrogesterone Oral 5-20 mg/day
In Europe and North America, the cumulative incidence of breast
cancer among women aged 50-70 who have never used postmenopausal
estrogen therapy is about 45 per 1000. The cumulative excess numbers
of breast cancers diagnosed per 1000 women of these ages who began use
of hormones at age 50 and used them for 5, 10, or 15 years were
estimated to be 2 (95% CI, 1-3), 6 (3-9), and 12 (5-20), respectively
(Collaborative Group on Hormonal Factors in Breast Cancer, 1997).
4.2.2.2 Endometrial cancer
The association with use of postmenopausal estrogen therapy is
most consistent for endometrial cancer (IARC, 1999). In the United
States, the incidence of endometrial cancer began to rise in the
1960s, reached a peak in the mid-1970s, and then declined until the
1990s. The increased incidence occurred primarily among postmenopausal
women and followed and then paralleled an increase in use of
postmenopausal estrogen therapy.
At least eight cohort and 30 case-control studies have been
conducted to investigate the impact of postmenopausal estrogen therapy
on the occurrence of endometrial cancer. Virtually all (6/8 cohort
studies and 29/30 case-control studies) reported elevated risks
associated with any use of postmenopausal estrogen therapy, and the
excess was statistically significant in the majority (IARC, 1999). The
published results from 30 such studies have been summarised by
meta-analytic methods (Table 18; Grady et al., 1995). The summary
relative risk for an increased incidence of this cancer was 2.3 (95%
CI, 2.1-2.5) for estrogen users when compared to non-users, and a much
higher relative risk was associated with prolonged duration of use
(RR = 9.5 for 10 or more years' use); the risk remained elevated five
or more years after discontinuation of use of unopposed (without the
addition of progestogen) estrogen therapy (RR = 2.3). Interruption of
estrogen use for five to seven days per month did not result in a
lower risk than daily use. Users of unopposed conjugated equine
estrogens had a greater increase in the relative risk of developing
endometrial cancer than users of synthetic estrogens. The risk for
dying from endometrial cancer was increased among users of unopposed
estrogens (RR = 2.7; 95% CI, 0.9-8). Cohort studies showed a decreased
risk for endometrial cancer among users of estrogen plus
progestogen(RR = 0.4), whereas case-control studies showed a small
increase (RR = 1.8). Information on the risk for endometrial cancer
among users of estrogen plus progestogen is, however, still limited
(Grady et al., 1995; IARC, 1999).
In conclusion, the risk for endometrial cancer increases
substantially with long duration of use of unopposed estrogens, and
the increased risk persists for several years after discontinuation of
use. Although not statistically significant, the risk of users of
unopposed estrogens of dying from endometrial cancer is also
increased, so that the association with use of estrogens cannot be
attributed exclusively to greater medical attention for users.
4.2.2.3 Cervical cancer
Only one cohort and two case-control studies are available on the
relationship between use of postmenopausal estrogen therapy and the
risk for invasive cervical cancer (IARC, 1999); in none of them were
the possible confounding effects of oncogenic human papillomaviruses
(the most important cause of cervical cancer, IARC, 1995) considered.
On balance, the limited evidence available suggests that
postmenopausal estrogen therapy is not associated with an increased
risk for invasive cervical carcinoma. The results provide some
suggestion that it is associated with a reduced risk for cervical
cancer, but that may be due to more active screening for preinvasive
disease among women who have received postmenopausal estrogen therapy.
4.2.2.4 Ovarian cancer
The four cohort and 12 case-control studies that addressed the
risk for ovarian cancer (largely epithelial) among women undergoing
postmenopausal estrogen therapy gave somewhat variable results (IARC,
1999). One cohort study and one large case-control study showed a
significant excess risk for ovarian cancer among women who used this
therapy. A pooled analysis of individual data from 12 studies in the
United States based on 2197 cases of invasive epithelial ovarian
cancer among white women and 8893 white controls (Whittemore et al.,
1992) gave pooled multivariate relative risks for invasive ovarian
cancer among women who had ever used postmenopausal estrogen therapy
for more than three months of 0.9 (95% CI, 0.7-1.3) in comparison with
hospital-based controls and 1.1 (95% CI, 0.9-1.4) in comparison with
population-based controls; no consistent duration-risk relationship
was seen. The relative risk for use for > 15 years was 0.5 (95% CI,
0.2-1.3) in the hospital-based and 1.5 (95% CI, 0.8-3.1) in the
population-based studies. The overall trend per year of use was 1.0
for both types of study, and both risk estimates were nonsignificant.
Allowance was made in the analysis for age, study, parity, and use of
combined oral contraceptives. The relative risk in a similar pooled
analysis of individual data for 327 cases of epithelial ovarian
tumours of borderline malignancy associated with any use of
postmenopausal estrogen therapy was 1.1 (95% CI, 0.7-2) (Harris et
al., 1992).
The original data from the four available European studies on
ovarian cancer (from Greece, Italy, and the United Kingdom) were also
reassessed. For a total of 1470 ovarian cancer cases and 327 controls,
a pooled relative risk of 1.7 (95% CI, 1.3-2.2) was found (Negri et
al., in press). Although this relative risk appears to be higher than
those reported from North America, it is not incompatible.
Table 18. Pooled relative risks (RRs) and 95% confidence intervals (CIs)
derived in a meta-analysis of studies of post-menopausal estrogen therapy
and endometrial cancer
Analysis RR 95% CI No. of studies
Any use of estrogens
All eligible studies 2.3a 2.1-2.5 29
Cohort studies 1.7a 1.3-2.1 4
Case-control studies 2.4a 2.2-2.6 25
Hospital controls 2.2 2.0-2.5 10
Gynaecologcial controls 3.3b 2,7-4.0 6
Community controlsc 2.4a 2.0-2.9 10
Dose of conjugated
estrogens (mg)
0.3 3,9 1.6-9.5 3
0.625 3.4 2.0-5.6 4
> 1.25 5.8 4.5-7.5 9
Duration of use (years)
< 1 1.4 1.0-1.8 9
1-5 2.8 2.3-3.5 12
5-10 5.9 4.7-7.5 10
> 10 9.5b 7.4-12 10
Table 18. (continued)
Analysis RR 95% CI No. of studies
Regimen
Intermittent and cyclic 3.0b 2.4-3.8 8
Continuous 2.9b 2.2-3.8 8
Type of estrogen
Conjugated 2.5d 2.1-2.9 9
Synthetice 1.3b 1.1-1.6 7
Time since last use
(years)
< 1 4.1b 2.9-5.7 3
1-4 3.7 2.5-5.5 3
> 5 2.3 1.8-3.1 5
Stage of tumour
0-1 4.2 3.1-5.7 3
2-4 1.4 0.8-2.4 3
Not invasive 6.2 4.5-8.4 4
Invasive 3.8b 2.9-5.1 6
Death from endometrial 2,7 0.9-8.0 3
cancer
a p for homogeneity, < 0.0001
b p for homogeneity, < 0.01
c Residential-, neighbourhood- and population-based controls
d p for homogeneity, < 0.005
e Primarily ethinylestradiol, estradiol valerate, estriol,
and other, unspecified estrogens; diethylstilbestrol and
estrogens combined with androgen were excluded, except
when such use was included with use of all synthetic estrogens
4.2.2.5 Cancers of the liver and biliary tract
Two cohort and two case-control studies that addressed the
association between use of postmenopausal estrogen therapy and the
risk for cancers of the liver or biliary tract showed no alteration in
risk.
4.2.2.6 Colorectal cancer
Seven cohort and 12 case-control studies provide information on
use of postmenopausal estrogen therapy and the risk for colorectal
cancer (Franceschi & La Vecchia, 1998a; IARC, 1999). The risk was not
increased and appeared to be reduced in half of the studies. In a
meta-analysis of the 20 independent estimates of the association
between any use of postmenopausal hormones and colorectal cancer
published up to December 1996 (Herbert-Croteau, 1998), the summary
relative risk was 0.85 (95% CI, 0.73-0.99), although there was
substantial heterogeneity among the studies. The protective effect of
hormones was found to be stronger in more recent publications. The
relative risks were lower for current or recent users (RR = 0.69; 95%
CI,0.52-0.91) and among users of > 5 years (RR = 0.73; 95% CI,
0.53-1) than for short-term users (RR = 0.88; 95% CI, 0.64-1.2). These
data suggest that use of postmenopausal estrogen therapy has a
protective effect against colorectal carcinogenesis. Inadequate
assessment of exposure, poor control of confounding factors such as
lifestyle, and changing patterns of use over time possibly contributed
to the results of studies conducted so far.
4.2.2.7 Cutaneous malignant melanoma
One cohort and nine case-control studies addressed the risk for
cutaneous malignant melanoma in relation to use of postmenopausal
estrogen therapy. Most suggested no alteration (IARC, 1999).
4.2.2.8 Thyroid cancer
Seven case-control studies on thyroid cancer and use of
postmeno-pausal estrogen therapy showed no clear effect on risk (IARC,
1999). La Vecchia et al. (1999) carried out a pooled analysis of the
original data from eight studies on estrogens and thyroid cancer,
comprising a total of 1305 cases and 2300 controls: 110 (8%) cases and
205 (9%) controls reported ever having used postmenopausal estrogen
therapy (RR = 0.78; 95% CI, 0.58-1.0).
4.2.2.9 Summary and conclusions
The working group convened by IARC (1999) concluded that there is
sufficient evidence in humans for the carcinogenicity of
postmenopausal estrogen therapy, largely based on the clear evidence
and substantial strength of the association with risk for endometrial
cancer. The association with breast cancer is weak but consistent with
biological mechanisms such as the adverse effects of delay in age at
menopause and obesity in postmenopausal women. Users of postmenopausal
estrogen therapy had no excess risk for cancers at other sites. The
evidence for the carcinogenicity of postmenopausal combined
estrogen-progestogen therapy was deemed to be limited.
4.2.3 Cardiovascular disease
Many cohort (Grodstein et al., 1996) and case-control (Rosenberg
et al., 1993) studies and three meta-analyses (Stampfer & Colditz,
1991; Grady et al., 1992, US Congress, 1995) have concluded that
postmenopausal estrogen therapy decreases the risk for coronary heart
disease by 35-50%. The predicted increase in the life expectancy of
hormone users, based on estimates from observational studies, is two
to three years (Grady et al., 1992). In a few studies of the outcomes
of estrogen therapy, with or without the addition of a progestogen, in
women with established coronary disease, lower risks were found among
users for a second infarct, death related to coronary heart disease,
and a second coronary artery stenosis, due to a reduction in lipids
and increased fibrinolysis (Petitti, 1998).
A pooled analysis (Hemminki & McPherson, 1997) of 22 randomized
trials involving 4124 women who received oestrogen therapy, placebo,
no therapy, or vitamins and minerals did not support the favourable
findings of the observational studies. The relative risk for women
taking hormones compared with those not taking them was 1.4 (95% CI,
0.48-4) for cardiovascular events without pulmonary embolus and deep
vein thrombosis and 1.6 (95% CI, 0.65-4.2) for cardiovascular events
with these aspects. The largest trial so far on the effect of
postmenopausal therapy with estrogen plus progestoge on the risk for
coronary heart disease involved 2763 women under the age of 80 (mean
age, 67) with established coronary disease (Hulley et al., 1998).
During an average follow-up period of 4.1 years, hormonal treatment
did not reduce the overall rate of coronary heart disease (RR = 0.99;
95% CI, 0.80-1.2) but did increase the frequency of thromboembolic
events and gall-bladder disease. The difference between the findings
of the observational studies and the trials may reflect some selection
bias in the former (Petitti, 1998). Trials, however, reflect
exclusively the short-term effects of postmenopausal estrogen therapy
on cardiovascular disease, while the long-term effects, reflected in
observational studies may be substantially different. An early
thrombogenic effect of estrogen may be counterbalanced by an
anti-atherogenic effect that occurs with longer duration of use
(Rosenberg et al., 1993).
4.2.4 Osteoporosis
Osteoporosis, a systemic skeletal disease characterized by low
bone mass and microarchitectural deterioration with a consequent
increase in bone fragility and susceptibility to fractures, affects an
estimated 75 million people in developed countries. Evidence that
postmenopausal estrogen therapy prevents fractures comes from cohort
and case-control studies, the typical relative risk for non-spinal
fracture being about 0.7 for women currently taking hormones. The
beneficial effect of postmenopausal estrogen therapy was more marked
in women who began therapy within five years after the menopause, and
it appears to be unaffected by age and concomitant progestogen
therapy. Evidence from randomized, controlled clinical trials of a
long-term favourable effect of postmenopausal estrogen therapy is
scanty (Eastell, 1998).
4.2.5 Overall mortality
The estimates of the excess numbers of cases of endometrial
cancer and, possibly, breast cancer diagnosed in women who used
postmenopausal estrogen therapy should be considered in the context of
other effects of such therapy on health (IARC, 1999). Use of
postmenopausal estrogens affects organs other than the breast and may
well decrease the incidences of coronary heart disease and
osteoporotic fractures, although it increases the incidence of venous
thromboembolism (Petitti, 1998).
A few studies of mortality in women showed an inverse association
with postmenopausal estrogen therapy use, most of the relative risks
ranging from 0.4 to 0.8 (Folsom et al., 1995; Sturgeon et al., 1995;
Grodstein et al., 1997; Schairer et al., 1997). In a prospective study
of nurses' health in the United States, current users of hormones had
a lower risk for death (RR = 0.63; 95% CI, 0.56-0.7) than subjects who
had never taken them, after adjustment for confounding variables;
however, the apparent benefit decreased with long-term use (RR = 0.8;
95% CI, 0.67-0.96 after > 10 years) because of an increase in the
rate of mortality from breast cancer. Current hormone users with
coronary risk factors (69% of the women) had the greatest reduction in
mortality (RR = 0.51; 95% CI, 0.45-0.57), and those at low risk had
substantially less benefit (RR = 0.89; 95% CI, 0.62-1.3). The rate of
mortality among women who use postmenopausal hormones is thus lower
than that of non-users, but the survival benefit diminishes with
longer duration of use and is lower for women at low risk of coronary
disease (Grodstein et al., 1997). Since women who use hormones may be
basically healthier than women who do not (Sturgeon et al., 1995), the
benefits attributed to use of estrogens in non-randomized studies of
postmenopausal estrogen users may be overestimated.
4.3 Hormonal contraceptives
4.3.1 Exposure
Oral contraceptives have been used since the early 1960s and are
now used by about 90 million women worldwide (IARC, 1979, 1999 and
related references). 'The pill' is given as a combination of an
estrogen and progestogen or, formerly, as sequential therapy.
Progestogen-only contraceptives have been available for over 20 years
(IARC, 1999) and are administered primarily by intramuscular depot
injections and subcutaneous implants in developing countries, where
there is widest use. Oral progestogen-only 'mini pills' are used
primarily in Europe and North America, but fewer women use these
preparations than parenterally administered progestogens and combined
oral contraceptives. The hormone content of combined preparations has
decreased over time, multiphase formulations have been marketed, and
various progestogens have been introduced (WHO, 1998). Formulations
containing 30-50 µg of estrogen, called 'low-dose' pills, are
available. Oral contraceptives may be used for emergency post-coital
contraception, and the components of oral contraceptives are used to
treat peri-and postmenopausal symptoms and number of other conditions.
Oral contraceptive preparations containing 50 µg of ethinylestradiol
contain 1, 2.5, 3, or 4 mg of norethisterone, while preparations
containing 30 µg of ethinylestradiol contain 150 or 250 µg of
levonorgestrel. The scientific validity of classifying different oral
contraceptive formulations by 'potency' is a controversial issue.
Combined oral contraceptives tend to have progestogenic, estrogenic,
and anti-oetrogenic effects which vary according to the target organ
and the hormonal content of the formulation (WHO, 1998). The estrogen
component of combined oral contraceptives is either ethinylestradiol
or mestranol and the progestogens used are chlormadinone acetate,
cyproterone acetate, desogestrel, ethynodiol diacetate, gestodene,
levonorgestrel, lynestrenol, megestrol, norethisterone, norethisterone
acetate, norethynodrel, and norgestrel. The estrogen used most
commonly is ethinylestradiol, and the most commonly used progestogens
are desogestrel, levonorgestrel, and norethisterone.
Use of oral contraceptives varies widely (IARC, 1999). They were
already being used extensively in the Netherlands, Sweden, the United
Kingdom, and the United States in the 1960s; extensive use of oral
contraceptives by adolescents was documented in Sweden and the United
Kingdom as early as 1964. In contrast, very little use of oral
contraceptives is reported in Japan, the countries of the former
Soviet Union, and most developing countries. The type of oral
contraceptives prescribed also differs among countries, and both the
type of oral contraceptive and the doses of estrogens and progestogens
have changed between and within countries over time.
It is important to stress that use of oral contraceptives is a
relatively recent human activity, and the health benefits and adverse
effects in women have not yet been followed over a complete
generation, even though these preparations are some of the most widely
used and best studied drugs in the world. Women who began using oral
contraceptives before the age of 20 in the 1960s are only now reaching
the ages (50-60 years) at which the incidences of most malignancies
begin to increase.
Estrogens and progestogens belonging to the same chemical groups
may have different estrogenic, androgenic, and progestogenic effects.
Little is known about the long-term health risks and potential
protective effects of the individual components. The effects become
increasingly complex as women grow older and may be exposed to
different types and doses of hormones, starting with oral
contraceptives and progressing to postmenopausal hormone therapy.
4.3.2 Human carcinogenicity
4.3.2.1 Breast cancer
More than 10 cohort and 50 case-control studies have assessed the
relationship between use of combined (estrogen plus progestogen) oral
contraceptives and the risk for breast cancer (IARC, 1999). Individual
data on 53 297 women with breast cancer and 100 239 women without
breast cancer from 54 studies conducted in 25 countries were
collected, checked, and analysed centrally by the Collaborative Group
on Hormonal Factors in Breast Cancer (1996). The studies included in
the collaboration represented about 90% of the epidemiological
information on the topic. The results provided strong evidence of a
small increase in the relative risk for a diagnosis of breast cancer
among women who are taking combined oral contraceptives and during the
10 years after stopping: RR = 1.2; 95% CI, 1.12-1.3 for current users;
1.2; 95% CI, 1.1-1.2 one to four years after stopping; 1.1; 95% CI,
1-1.1 five to nine years after stopping. The results also showed no
significance excess risk of having breast cancer diagnosed 10 or more
years after stopping use (RR = 1.01; 95% CI, 0.96-1.05; Figure 2).
There was no pronounced variation in the results for recent use
among women with different background risks for breast cancer,
including women from different countries and ethnic groups, women with
different reproductive histories, and those with or without a family
history of breast cancer.
Other features of hormonal contraceptive use such as duration of
use, age at first use, and the dose and type of hormone in the
contraceptives had little additional effect on risk once recent use
had been taken into account. Women who began use before the age of 20
had higher relative risks for breast cancer being diagnosed while they
were using combined oral contraceptives and in the five years after
stopping than women who began use when older, but the higher relative
risks apply at ages when breast cancer is rare and for a given
duration of use earlier use does not result in more cancers being
diagnosed than use begun later.
Because the incidence of breast cancer rises steeply with age,
the estimated excess number of cancers diagnosed between starting use
and 10 years after stopping increases with age at last use: for
example, among 10 000 women in Europe and North America who used oral
contraceptives between the ages of 16 and 19, 20 and 24, and 25 and
29, the estimated excess numbers of cancers diagnosed up to 10 years
after stopping use were 0.5 (95% CI, 0.3-0.7), 1.5 (0.7-2.3), and 4.7
(2.7-6.7), respectively. Up to 20 years after cessation of use, the
difference between women who have ever used oral contraceptives and
those who have never used them is not so much in the total number of
cancers diagnosed but in their clinical presentation: breast cancers
diagnosed in women with any use are less advanced clinically than
those diagnosed in women with no use, the relative risk for tumours
that had spread beyond the breast compared with localized tumours
being 0.88 (95% CI, 0.81-0.95). The relationship observed between the
risk for breast cancer and use of hormones is unusual, and it is not
possible to infer from these data whether it is due to earlier
diagnosis of breast cancer in women who have used oral contraceptives,
the biological effects of hormonal contraceptives, or a combination
(Collaborative Group on Hormonal Factors in Breast Cancer, 1996).
Data on injectable progestogen-only contraceptives were available
from two case-control studies and a pooled analysis of original data,
overall involving about 350 women with breast cancer who had used
these drugs (IARC, 1999). Data on oral progestogen-only contraceptives
were available from a pooled analysis of original data on 725 women
with breast cancer. Overall, there is no evidence of an increased risk
for breast cancer in users of progestogen-only contraceptives
(Collaborative Group on Hormonal Factors in Breast Cancer, 1996).
4.3.2.2 Endometrial cancer
Three cohort and 16 case-control studies addressed the
relationship between use of combined oral contraceptives and risk for
endometrial cancer (IARC, 1999). The results of these studies
consistently show that the risk for endometrial cancer of women who
have taken these pills is approximately halved. The reduction in risk
is generally stronger the longer the oral contraceptives have been
used and persists for at least 10 years after cessation of use. Few
data are available for the more recent, low-dose formulations. Use of
sequential oral contraceptives which were removed from the consumer
market in the 1970s was associated with an increased risk for
endometrial cancer. One case-control study addressed the relationship
between use of oral progestogen-only contraceptives and risk for
endometrial cancer. Less than 2% of the control women had used these
preparations, and women with endometrial cancer were less likely to
have used oral progestogen-only contraceptives than control women but
not significantly so.
The effects of use of depot medroxyprogesterone acetate on the
risk for endometrial cancer have been evaluated in one cohort and one
case-control study. No reduction in risk was seen in the cohort study,
whereas a strong reduction was observed in the case-control study.
Although the evidence is based on small numbers of women, the results
of these studies suggest that women who use progestogen-only
contraceptives have a reduced risk for endometrial cancer.
In biological terms, the favourable effect of oral contraceptive
use with regard to endometrial cancer is attributed to the daily
presence of progestogens, whereas in the normal cycle they are present
for only 14 days.
Hydroxyestrogens may be further modified by the action of
catechol-O-methyltransferase (COMT). High activity of COMT is found in
many tissues, including liver and kidneys, blood cells, endometrium,
and breast. A genetic polymorphism for this enzyme results in a
trimodal distribution of activity, but epidemological studies of the
polymorphism in relation to breast cancer risk have yielded
conflicting results (Lavigne et al., 1997; Millikan et al., 1998;
Thompson et al., 1998). The methylation of catecholestrogens
effectively prevents these compunds from entering the redox cycling
pathway, and 2-methoxyestradiol may be an antitumour agent (Zhu &
Conney, 1998a,b).
Methylation of 4-hydroxyestradiol by COMT is inhibited by
2-hydroxy-estradiol (Roy et al., 1990). Interestingly, tissues which
develop estradiol-induced tumours (rat pituitary, male Syrian hamster
kidney and mouse uterus) have very high concentrations of endogenous
catecholamines (up to 50-fold relative to other strains or species and
non-target tissues). Catecholamines in target tissues may inhibit or
compete for COMT-catalysed methylation, thus leading to increased
concentrations of hydroxylated metabolites of estradiol (Zhu and
Conney, 1998a).
1.2.2.2 Conjugation
Studies of the conjugation of estrogens with glucuronic acid or
sulfate have been reviewed in detail (IARC, 1979). Lysosomes from male
Syrian hamster livers and kidneys can catalyse the deconjugation of
estradiol and estrone glucuronides. The rates of deconjugation of
estrogen glucuronides were higher in kidney than in liver, by 56% for
estrone and 34% for estradiol. Treatment of hamsters for nine days
with subcutaneous implants containing 25 mg estradiol (releasing 61
µg/day) increased lysosomal estrone and estradiol 3ß-glucuronidase
activity in kidney by 15 and 25%, respectively, and by about 100% in
liver. Estradiol was deconjugated at negligible rates in both liver
and kidney (Zhu et al., 1996). Human liver microsomal sulfatases
convert estrone sulfate to estrone before 16 alpha-hydroxylation
(Huang et al., 1998). Estrone sulfate, the most abundant estrogen in
blood, and other estrogen conjugates may serve as a circulating
reservoir of estradiol, and regulation of deconjugation reactions may
affect intracellular estradiol concentrations.
Demethylation of catechol estrogens has also been reported. The
rates of demethylation of 2-and 4-methoxyestradiol were about equal in
kidney microsomes, but the rate of 2-methoxyestradiol demethylation in
liver was fivefold higher than that of 4-methoxyestradiol. Estradiol
treatment decreased hepatic 2-methoxyestradiol demethylation by about
20% relative to controls with little effect on 4-methoxyestradiol
demethylation, whereas the opposite was observed in kidney (Zhu
et al., 1996).
In the absence of conjugation, a pathway for further catalysis of
catechol estrogens has been suggested. Redox cycling of catechol
(hydroxyquinone) to quinone through semiquinone intermediates is
catalysed by oxidation of catechol estrogens by peroxidases or CYP1A1
lipid hydroperoxide cofactors. Reduction of the quinone to the
hydroquinone is catalysed by NADPH-dependent P450 reductase and other
enzymes. Oxygen radicals formed in this redox process may increase the
carbonyl content of proteins, formation of DNA 8-hydroxydeoxyguanosine
adducts, and lipid peroxidation. The authors concluded that redox
cycling is a critical step in estrogen-mediated carcinogenesis (Wang &
Liehr, 1994; Yager & Liehr, 1996).
1.2.3 Biochemical parameters
1.2.3.1 Synthesis
In mammals, estradiol, estrone, and estriol are synthesized from
steroid precursors in the gonads, adrenal cortex, and placenta or
through peripheral conversion of androgens in other tissues of
mammals. Cholesterol, obtained primarily from circulating low-density
lipoprotein, serves as the precursor for steroid biosynthesis,
although steroidogenic cells are capable of local cholesterol
synthesis de novo. In non-pregnant premenopausal women, the
principal estrogens found in the blood are estradiol and estrone.
Estradiol is synthesized and secreted primarily from ovarian granulosa
cells, whereas most estrone (approximately 40% of total estrogens) is
formed peripherally from estradiol and androstenedione. Estradiol and
estrone may be intra converted through the action of the enzyme
17ß-hydroxysteroid dehydrogenase. Estrone may be further metabolized
to estriol, primarily in the liver. Estriol is also formed in the
fetal liver and in the placenta from
16alpha-hydroxydehydroepiandrosterone sulfate, which is secreted from
the fetal adrenal and circulating dehydroepiandrosterone sulfate. At
least 90% of urinary estriol is derived from fetal sources (Carr,
1992). In men and postmenopausal women, the source of serum estradiol
is peripheral conversion of androgens by the enzyme aromatase. In men,
approximately 0.3% of plasma testosterone is aromatized to estradiol;
an additional contribution of approximately 25% of the total estradiol
may be due to testicular secretion (Griffin & Wilson, 1998). Estrone
is the predominant circulating estrogen in postmenopausal women,
formed by peripheral conversion of adrenal androgens in adipose
tissue.
Gonadal synthesis of estradiol is regulated by luteinizing and
follicle stimulating hormones secreted by the anterior pituitary
gland. The secretion of these two hormones is regulated by
gonadotropin-releasing hormone secreted by the hypothalamus, steroid
hormones, and other factors in a complex feedback loop which
effectively regulates the serum concentrations of hormone within a
physiological concentration range, which is particularly variable in
premenopausal women. The feedback loop is controlled by the dominant
circulating hormone (estradiol and progesterone in women, testosterone
in men). Feedback control for estradiol in men and for testosterone in
women is therefore not operative (Wilson et al., 1998). There is
evidence that this feedback loop exists in prepubertal children but is
quiescent.
In humans, plasma estradiol concentrations generally remain low
during the first 12 years of life. Around the time of menarche, rising
plasma concentrations of gonadotropins from the anterior pituitary
stimulate the ovary to produce estradiol. During a normal menstrual
cycle, plasma estradiol concentrations change very little throughout
the first half of the follicular phase but increase as the follicles
develop, reaching serum concentrations that are up to nine times
greater than the basal concentrations near mid-cycle. After the
mid-cycle surge, the estradiol concentrations fall precipitously.
During the luteal phase, the serum estradiol concentrations rise to a
plateau for 8-10 days, before declining. Should fertilization occur,
the corpus luteum formed from the dominant follicle after ovulation
remains active as the principal source of estradiol for the first 6-8
weeks of pregnancy. The corpus luteum is later supplanted by the
placenta as the site of estrogen synthesis. As the placenta lacks
17 alpha-hydroxylase, fetal and maternal circulating androgens are
necessary for placental estrogen synthesis. During pregnancy, the
feto-placental unit secretes a large quantity of estriol into the
maternal circulation, which is ultimately excreted in the urine.
The concentrations of circulating estrogens, their daily
production, and their metabolic clearance rates can be found in
previous reviews (IARC, 1979, specifically 'General remarks on sex
hormones') and in most textbooks of endocrinology (e.g. Braunstein,
1994; Goldfien & Monroe, 1994; Carr, 1998; Griffin & Wilson, 1998; see
also textbook appendices for summary tables). They are summarized in
Table 1. Somewhat different values can be calculated for the basal
production rate of estradiol in prepubertal boys and girls
(Angsusingha et al., 1974; IARC, 1979) when measurements of hormone in
urine or plasma are used as the basis for the calculation (4 or 12
mg/day).
1.2.3.2 Mechanism of action
The conventional view of the action of steroid sex hormones
involves interaction of the sex hormone with specific intracellular
receptors, which subsequently bind tightly to specific DNA sequences
in the genome (Malayer & Gorski, 1993). This tight nuclear binding
initiates transcription of specific genes, which ultimately leads to
physiological events. These include include development of
reproductive tissues, maturation of the ovarian follicle, development
of the uterus and vagina, and ductal development in the breast.
Estrogen withdrawal results in menstruation. In non-reproductive
tissues, estrogens may affect bone growth and prevent bone resorption,
and effects on the plasma lipid profile through action in the liver.
Estrogens typically promote cell growth or cell proliferation in
responsive cells in culture.
Table 1. Levels of circulating estrogens, metabolic clearance rates, and
daily production
Sex Age or phase Serum Metabolic Total daily
concentration clearance production
(pg/ml) (L/day) (mg/day)
Male 1400
Prepubertal < 10 < 0.014
12-16 years < 23 < 0.031
> 16 years 20-50 0.027-0.068
Female 1400
< 8 years < 7 < 0.01
2-12 8-18 0.01-0.024
12-14 16-34 0.02-0.09
14-16 20-68 0.03-0.09
Early follicular 20-100 0.03-0.14
Preovulatory 100-350 0.14-0.47
Luteal 100-350 0.14-0.47
Late pregnancy 18 000 24
Postmenopausal 10-30 0.01-0.04
It has been proposed that the carcinogenicity of estrogens is
distinct from its hormonal properties; however, since that hypothesis
originated, information on estrogen-receptor and ligand interactions
has been re-evualuated in the light of recent identification of
additonal estrogen receptor proteins. Three specific receptors have
now been identified for the endogenous ligand estradiol: the classical
receptor ER alpha, ERß, and ERß2. Analyses of ER alpha and ERß RNA
indicate that ER alpha is widely distributed and that ER ß is
prominent in prostate, ovary (localized to the granulosa cells of the
maturing ovary), epididymis, urinary bladder, uterus, lung, thymus,
colon, small intestine, vessel walls, pituitary glan, hypothalamus,
cerebellum, and brain cortex (Couse et al., 1997a; Kuiper et al.,
1998). These receptor isoforms differ in their agonist and antagonist
reactions to agents such as tamoxifen and to classes of agents
variously referred to as 'endocrine disruptors' or 'xenoestrogens' and
to endogenous estrogen metabolites such as 2-hydroxyestradiol.
Moreover, alpha-estrogen receptor knockout (alpha ERKO; Couse et al.,
1997a) and ERß-/- female mice (Krege et al., 1998) have very different
phenotypes. Alpha ERKO females lack the ability to complete
folliculogenesis, are infertile, and have multiple cystic,
haemorrhagic, and atretic follicles, whereas ERß-/- female mice
develop normally and are indistinguishable from their wild-type
littermates. These mice are fertile, but their fertility is
compromised, as demonstrated by reductions in litter size. Mammary
gland development is normal in ERß-/- mice, in contrast to the absence
of breast development beyond that of prepubertal females in alpha ERKO
mice. Male mice lacking ERß are also fertile (alpha ERKO males are
infertile) but develop prostate and bladder hyperplasia as they age.
These ERKO mice and the receptor-binding properties of various ligands
demonstrate that ER alpha and ERß have different functional
responsibilities.
Abundant evidence exists that hormonal carcinogenicity is linked
to the relative balance of various estrogens. Proliferation of mammary
glands and other reproductive (i.e. target) tissues is inextricable
linked to hormonal changes during the menstrual cycle, pregnancy, and
the initiation or cessation of cyclic menstruation. The cyclic
proliferation of tissues is correlated to the cellular content of the
estrogen receptor. In cultured cells, the growth of cells with
estrogen receptors (e.g. MCF-7 cells) but not those without estrogen
receptors (e.g. MDA-MB-231 cells) is dependent on the estradiol
concentration in serum. Molecular genetic studies of human cancer
indicate that the progression from a normal to malignant phenotype
requires activation of one or several oncogenes and/or inactivation of
tumour suppressor genes. These events require cell division (reviewed
by Bernstein & Ross, 1993; Russo & Russo, 1996; Tsai et al., 1998).
A functional role for catechol estrogens distinct from that for
estradiol has been suggested (reviewed by Zhu & Conney, 1998a,b).
Because of the extremely short half-lives of these compounds
(Schneider et al., 1984), they are unlikely candidates for circulating
hormones. Locally, 2-hydroxyestradiol reportedly stimulates
progesterone production in ovarian granulosa cells (Spicer et al.,
1990), and 2-hydroxyestrone has been shown to inhibit MCF-7 cell
proliferation in the presence of quinalizarin, a potent inhibitor of
COMT. This challenge can be overcome with physiological concentrations
of estradiol. No effects on cell growth were observed with
concentrations of 10-9 to 10-6 mol/L of 2-hydroxyestradiol in the
absence of methyltransferase inhibition (Schneider et al., 1984).
The endogenous metabolite 2-methoxyestradiol has been suggested
to be an antiangiogenic factor and tumour suppressor (Fotis et al.,
1994; Zhu & Conney, 1998a); however, the concentrations required to
induce apoptotis in cultured cells are about 10 times greater than
those observed in humans (Yue et al., 1997). Those authors suggested
that unless the concentration of 2-methoxyestradiol in the lipid phase
(i.e. membranes) exceeds that in the aqueous phase, as reported for
some lipophilic calcium blockers, the antiangiogenic properties of
2-methoxyestradiol are of little physiological relevance.
A role for catechol estrogens in implantation in the mouse uterus
has been suggested on the basis of the observation of increased
activity of 4-hydroxylase activity on day 4 of pregnancy (Paria et
al., 1990). 2-and 4-Hydroxyestradiol bind to the estrogen receptor
with reduced relative affinities of 23 and 26%, respectively (Schultze
et al., 1994). Additionally, a distinct signaling pathway separate
from the estrogen receptor may exist for 4-hydroxyestradiol. In
alpha ERKO mice, 4-hydroxyestradiol stimulated up-regulation of
lactoferrin mRNA and water imbibition (Das et al., 1997). In these
mice, the effects of 4-hydroxyestradiol could not be mimicked by
estradiol, nor could the effects be blocked by the anti-estrogen ICI
182,780; however, it has been postulated that the estrogenic effects
in the uterus in alpha ERKO mice are mediated through ERß (Krege et
al., 1998).
The physiological effects of estradiol that are reported not to
be receptor-mediated (i.e. not mediated via the classical receptor
mechanism) include those on myometrial, neuronal, pituitary, maturing
oocyte, and granulosa cells (Wehling, 1997). Estradiol may protect
against oxidative damage in neuronal cells (Behl et al., 1995), but
very high concentrations of estradiol were required for this effect:
10-5 but not 10-7 mol/L was effective in preventing cell death. At
physiological concentrations in blood, the antioxidant properties of
estradiol protect against oxidation of low-density lipoprotein (Rifici
& Kachadurian, 1992; Hoogerbrugge et al., 1998). Novel so-called
'scavestrogens', structurally related to 17 alpha-estradiol, have
antioxidant properties that protect against the radical-mediated death
of cultured cells (Blum-Degen et al., 1998). Other studies indicate
that the pro-oxidant and antioxidant properties of estrogens may be
dependent on structure and concentration. Estradiol, estriol, and
methoxyestrogen metabolites had only antioxidant properties. Catechol
estrogens showed pro-oxidant properties at low concentrations (5
pmol/L to 100 nmol/L), but antioxidant properties dominated at high
concentrations (0.5-50 µmol/L) (Markides et al., 1998). In addition to
oxidant and antioxidant effects, a rapid, non-genomic effect of
estradiol, possibly mediated by cAMP, Ca++, or ion channel gating,
has been postulated (reviewed by Moss et al., 1997).
1.3 Toxicological studies
1.3.1 Acute toxicity
The therapeutic dose of fine-particle estrogen given orally is
0.5-2 mg/day. No adverse effects were reported in children after
accidental ingestion of large doses of estrogen-containing oral
contraceptives (Physician's Desk Reference, 1999). An
electroencephalogram of a young woman who took 160 mg of estradiol
valerate (80 tablets of 2 mg), which is converted to estradiol-ß
in vivo, showed traces typical of subcortical disturbance on the
first day; however, the recording was normal one week later (Punnenon
& Salmi, 1983).
1.3.2 Short-term studies of toxicity
Estradiol was administered in the diet to female Crl:CD BR rats
at doses equal to 0. 0.003, 0.17, 0.69, or 4.1 mg/kg bw per day for 90
days. The end-points were chosen to evaluate both short-term and
reproductive toxicity and several mechanistic and biochemical
parameters. Administration of doses > 0.17 produced dose-dependent
increases in body weight, food consumption, and feed efficiency. At
0.69 and 4.1 mg/kg bw per day, minimal to mild non-regenerative
anaemia, lymphopenia, decreased serum cholesterol (at the high dose
only), and altered splenic lymphocyte subtypes were observed. Changes
in the weights of several organs were noted. Evidence of ovarian
malfunction (reduced corpora lutea and large antral folicles) was
found at doses > 0.17 mg/kg bw per day. Pathological changes in
males and females fed 0.69 or 4.1 mg/kg bw per day included
centrilobular hepatocellular hypertrophy, diffuse hyperplasia of the
pituitary gland, feminization of the male mammary gland, mammary gland
hyperplasia in females, cystic ovarian follicles, hypertrophy of the
endometrium and endometrial glands in the uterus, degeneration of the
seminiferous epithelium, and atrophy of the testes and acessory sex
glands (Biegel et al., 1998b).
1.3.3 Long-term studies of toxicity and carcinogenicity
The toxicity of estrogens, including estradiol and related
esters, has been reviewed extensively by working groups convened by
the IARC (1979, 1987). After reviewing studies of estradiol
administered orally to mice and by subcutaneous injection or
implantation in mice, rats, hamsters, guinea-pigs, and monkeys, the
group concluded that there is sufficient evidence for the
carcinogenicity of estradiol in experimental animals, noting that:
"Administration to mice increased the incidences of mammary,
pituitary, uterine, cervical, vaginal, testicular, lymphoid and bone
tumours. In rats, there was an increased incidence of mammary and/or
pituitary tumours. estradiol-17b produced a statistically
nonsignificant increase in the incidence of foci of altered
hepatocytes and hepatic nodules induced by partial hepatectomy and
administration of N-nitrosodiethylamine in rats. In hamsters, a high
incidence of malignant kidney tumours occurred in intact and castrated
males and in ovariectomized females, but not in intact females. In
guinea-pigs, diffuse fibromyomatous uterine and abdominal lesions were
observed.' The IARC working group concluded that the carcinogenic
effects of estrogens and progestogens were inextricably linked to the
hormonal milieu and to dose-effect relationships (IARC, 1987).
Hormonal effects on non-cancer end-points were not evaluated by the
groups.
The induction of renal tumours by various steroidal and
non-steroidal estrogens was examined in castrated male Syrian hamsters
treated subcutaneously for nine months with a capsule that released an
average of 110 µg of the hormone per day. The tumour incidences
associated with the hormonal activity of the substances tested are
presented in Table 2. These data demonstrate a good correlation among
the hormonal parameters progesterone receptor induction and serum
prolactin and relative estrogenic potency (estrogen receptor binding)
in hamster kidney. All animals trested with estrone, equilin plus
d-equilin, or Premarin(R) developed renal tumours, with combined
numbers of tumour foci in both kidneys of 15, 18, and 16, respectively
(Li et al., 1995).
Castrated adult male Syrian hamsters were treated for eight
months with subcutaneous pellets containing 20 mg estrogen or
anti-estrogen; the release rates in µg/day were as follows:
diethylstilbestrol, 156; ethinyl-estradiol, 215; estradiol, 96;
estradiol-17 alpha, 104; 17 alpha-ethinyl-11ß-methoxy-estradiol
(moxestrol), 104; and tamoxifen, 183. Treatment with ethinylestradiol
resulted in progressive dysplasia but no renal tumours, but dysplasia
was observed in the proximal tubules of the renal cortex, which was
uncommon in animals treated with diethylstilbestrol or estradiol.
Animals treated with estradiol, diethylstilbestrol, or moxestrol had a
tumour incidence of 100%, which was completely abolished by
concurrrent treatment with ethinylestradiol (Table 3). Simultaneous
administration of diethylstilbestrol and estradiol-17ß or estradiol-17
alpha (a noncarcinogenic estrogen) did not mitigate the
carcinogenicity of diethylstilbestrol (Li et al., 1998).
The carcinogenicity of estradiol and its metabolites was
investigated in groups of 20-35 B6C3F1 mice of each sex given a
single daily intraperitoneal injection of the compound on four
consecutive days starting at 12 days of age. They were then maintained
for approximately 18 months and killed. Of the catechol estrogens and
their quinones tested, only estrone-3,4-quinone was significantly
carcinogenic in the livers of male mice (Table 4). It was also highly
toxic, as most of the mice died from unknown causes shortly after
treatment. Estrone was protective against liver tumour formation in
this system, and few tumours were induced in female mice (Cavalieri et
al.,1997).
The tumour formation in the kidneys of male Syrian hamsters given
25-mg pellets containing estrogen or catechol estrogen:cholesterol
(90:10) by subcutaneous implantation and killed 175 days later was
studied histologically. Estradiol and 4-hydroxyestradiol each induced
renal tumours in four of five animals, whereas neither
2-hydroxyestradiol nor 2-methoxystradiol was carcinogenic. The lack of
carcinogenicity of 2-hydroxyestradiol was not due to failure of the
hormone to stimulate cell growth in vivo, as estradiol,
4-hydroxyestradiol, and 2-hydroxyestradiol supported the growth of
estrogen-dependent H-301 cells injected into male hamsters.
2-Methoxyestradiol was not effective in stimulating these cells. The
authors note that none of the three compounds was mutagenic in
Salmonella typhimurium TA100 strain (see below; Liehr et al., 1986).
The role of estrogen and its metabolites in tumour formation was
examined in castrated male Syrian hamsters implanted for 9-10 months
with pellets containing various estrogens (doses not given). The
results are shown in Table 5. The authors suggested that the
neoplastic changes seen in hamster kidney after continuous exposure to
estrogens are due to synergistic action between hormonal and
carcinogenic factors (Li & Li, 1989).
Castrated male Syrian hamsters received subcutaneously implanted
pellets containing diethylstilbestrol, alpha-dienestrol, hexestrol,
diethylstilbestrol 3,4-oxide, estradiol, estrone, ethinylestradiol,
equilin or (+)-equilenin, which released 100-210 µg/day, for a total
of nine months. The tumour incidence was 75-100%, except with
ethinylestradiol (20%) and (+)-equilenin (0%). The ability of
estrogens to cause renal tumours correlated well with their ability to
compete for estrogen receptor binding, with the notable exception of
ethinylestradiol (Li et al., 1983). The tumours induced by
ethinylestradiol are of a different origin than other hormone-induced
renal tumours (Oberley et al., 1991). The presence of estrogen
receptors, probably ER alpha, in interstitial cells of control and
estrogen-treated hamsters was confirmed by immuno-histochemical
staining and northern blotting. Receptors were also found in renal
corpuscles, arterial cells, and interstitial capillaries but not in
the tubular epithelia of the cortex, further indicating that the
tumours have an interstitial or mesenchymal origin (Bhat et al.,
1993). The presence of ERß, which was identified after the study, has
not been investigated.
Virgin female C3H/HeJ mice with a high titre of antibodies to the
mouse mammary tumour virus were fed diets that provided doses
equivalent to 0.015, 0.15, or 0.75 mg/kg bw per day estradiol from
week 6 to week 110 of age (Highman et al., 1978). The microscopic
findings in animals sacrificed after 52 weeks of feeding are shown in
Table 6. In a continuation of this study, the authors reported the
preneoplastic and neoplastic findings in mice sacrificed after up to
104 weeks on the estrogenic diets (Highman et al., 1980). The results
are shown in Tables 7 and 8. Uterine cervical adenosis may be a
precursor of cervical adenocarcinoma in C3H/HeJ mice and may therefore
serve as an early indicator of the uterine carcinogenicity of a
compound. In these experiments, high doses of estradiol increased the
incidence of adenosis but did not affect the incidence of ovarian
tubular adenomas. After 66-91 weeks of treatment, high doses of
estradiol also increased the incidence of mammary gland hyperplastic
alveolar nodules but not mammary adenocarcinomas. The authors reported
the occurrence of several other sporadic tumours at different sites in
both control and treated animals. They concluded that the incidence of
lesions in mice given estradiol was generally dose-dependent,
indicating that this compound either induces or facilitates the
development of these lesions (Highman et al., 1980)
Table 2. Estrogenicity and carcinogenicity of various steroidal and stilbene estrogens in Syrian hamster kidney
Estrogen % of competitive Induction of Serum % of animals No. of tumour
binding to estrogen progesterone prolactin with tumours foci in both
receptors due to receptors in kidney (ng/ml) kidneys
renal tumours (fmol/mg protein)
Steroidal
Estradiol-17ß 55 48 390 100 17
11ß-Methoxy ethinyloestradiol 52 60 330 100 22
16 alpha-Hydroxyestrone 48 45 390 38 3
11ß-Methoxyestradiol 30 35 390 25 2
11ß-Methylestradiol 14 18 150 0 0
Estradiol-17 alpha 34 6 130 0 0
Deoxestrone 14 8 94 0 0
Stilbene
Diethylstilbestrol 46 50 450 100 19
Indenestrol B 46 49 280 100 11
Indanestrol 10 29 100 0 0
From Li et al. (1995)
Table 3. Prevention of the carcinogenicity of estrogens by ethinylestradiol
Estrogen % tumour induction No. of tumour
nodules in both
kidneys
Diethylstilbestrol 100 15
Estradiol-17ß 100 13
Ethinylestradiol 10 2
Moxestrol 100 18
Diethylstilbestrol + ethinylestradiol 0 0
17ß-Estradiol + ethinylestradiol 0 0
Moxestrol + ethinylestradiol 0 0
Diethylstilbestrol + diethylstilbestrol 100 12
Diethylstilbestrol + 17ß-estradiol 100 12
Diethylstilbestrol + 17 alpha-estradiol 100 9
From Li et al. (1998)
Intact and ovariectomized or hysterectomized nulliparous female
C3H/HeJ mice, five months of age, received either estradiol at a dose
of 0.5 mg/L in drinking-water for one year or estradiol plus daily
injections of 0.1 mg 2-bromo-alpha-ergocryptine, an effective
suppressor of prolactin secretion. All mice were examined weekly for
mammary tumours. One year after the onset of treatment, all surviving
mice were sacrificed. The formation of mammary hyperplastic nodules
was highly significantly suppressed in mice that received both
estradiol and 2-bromo-alpha-ergocryptine, and the mammary tumour
incidence was slightly but significantly reduced in comparison with
that in animals receiving estradiol alone. The tumour incidence in
concurrent controls was not reported. In a separate study, nulliparous
30-day-old mice received estradiol in the drinking-water with or
without a daily injection of 0.1 mg 2-bromo-alpha-ergocryptine for
19-20 months. The effects on the mammary gland are shown in Table 9.
The authors concluded that at least a portion of the oncogenic
activity of estrogenic steroids on the mammary gland in rodents is
manifested through a stimulatory effect on prolactin secretion
(Welsch, 1976).
The results of short-and long-term studies of the carcinogenicity
of estrogens are summarized in Table 10.
Table 4. Carcinogenicity of estradiol and its metabolites in male
B6C3F1 mice
Compound Dose No. of mice with tumours/
(µmol/kg bw) total no. of animals (%)
Estradiol-17ß 30 6/20 (30)
2-Hydroxy-17ß-estradiol 30 10/28 (36)
4-Hydroxy-17ß-estradiol 30 10/24 (42)
17ß-Estradiol-2,3-quinone 7.5 8/26 (31)
17ß-Estradiol-3,4-quinone 7.5 4/21 (19)
Estrone 30 3/32 (9.4)
2-Hydroxyestrone 30 9/30 (30)
4-Hydroxyestrone 30 8/33 (24)
Estrone-2,3-quinone 7.5 12/25 (48)
Estrone-3,4-quinone 3.7 6/10 (60)
Benzo[a]pyrene 60 12/12 (100)
Solvent 7/19 (37)
Untreated 11/33 (33)
From Cavalieri et al. (1997)
1.3.4 Genotoxicity
A working group convened by IARC evaluated the results of tests
for genetic toxicity conducted with estradiol and concluded:
"Oestradiol-17ß did not induce chromosomal aberrations in bone-marrow
cells of mice treated in vivo. Unusual nucleotides were found in
kidney DNA of treated hamsters. It induced micronuclei but not
aneuploidy, chromosomal aberrations or sister chromatid exchanges in
human cells in vitro. In rodent cells in vitro, it induced
aneuploidy and unscheduled DNA synthesis but was not mutagenic and did
not induce DNA strand breaks or sister chromatid exchanges.
Oestradiol-17ß was not mutagenic to bacteria." The group also
concluded that the limited data available on estrone and estriol were
indicative of genotoxicity (IARC, 1987).
A mechanism has been proposed by which catechol estrogens
interact with DNA. Nonmethylated catechol estrogens can be oxidized to
a quinone which can bind to DNA; thus, 2-and 4-hydroxy estradiol
produce 2,3-and 3,4-quinones, respectively. Reaction of the
3,4-quinone of estrone or estradiol with deoxyguanosine (dG) at N7
resulted in loss of the deoxyribose moiety and thus induced
depurinating adducts. No adducts were observed after reaction of the
3,4-quinone with deoxyadenosine (dA). Reaction of estrone-2,3-quinone
with dG and dA produced a stable N2-dG or N6-dA adduct, the
deoxyribose group remaining intact. Formation of depurinating and
stable adducts in calf thymus DNA by activated catechol estrogens and
in mammary glands of female Sprague-Dawley rats injected with 200 nmol
4-hydroxy-estrone was confirmed by the 32P-postlabelling technique.
The authors suggested that the formation of depurinating adducts via
3,4-quinone followed by misreplication of unrepaired apurinic sites
are the critical steps in initiation of cancer by estrogens (Cavalieri
et al., 1997; Stack et al., 1998).
Studies of the genetic toxicology of estrogens are summarized in
Table 11.
The mutagenicity of estradiol and its metabolites was assessed in
Salmonella typhimurium strain TA100, but the authors concluded that
none of the compounds was mutagenic in this assay (Liehr et al.,
1986). Estradiol was evaluated in several short-term tests for
genotoxic potentiol and at 1, 10, or 100 µg/ml was found to cause
chromosomal aberrations and sister chromatid exchange in cultured
human lymphocytes with and without metabolic activation. In the
absence of metabolic activation, the lowest dose caused aberrations
after 72 h of treatment but not after 24 or 48 h; the intermediate
dose caused aberrations after 48 and 72 h but not after 24 h of
treatment. With a 6-h treatment, aberrations were observed at 10 and
100 µg/ml in the presence of metabolic activation but not in its
absence. Estradiol caused sister chromatid exchange at most doses with
or without metabolic activation (Dhillon & Dhillon, 1995).
Table 5. Carcinogenicity of estradiol and its metabolites in castrated
male Syrian hamsters
Compound No. of mice with tumours/
total no. of animals (%)
Estradiol-17ß 6/6 (100)
Estrone 8/10 (80)
Estriol 4/7 (57)
2-Hydroxy-17ß-estradiol 0/6 (0)
2-Hydroxyestrone 0/6 (0)
4-Hydroxy-17ß-estradiol 5/5 (100)
4-Hydroxyestrone 2/6 (33)
Ethinylestradiol 3/15 (20)
Equilin 6/8 (75)
(+)-Equilenin 0/9 (0)
From Li & Li (1989)
Table 6. Percent incidence of pathological changes in female mice given estradiol for 52 weeks
Dose No. of Effects in the Effects in the Effects in the
(mg/kg bw animals cervix uterine horn mammary gland
per day)
Adenosis in Adenosis in Glandular Hyperplastic Adeno- Osseus
upper third upper and hyperplasia alveolar carcinoma hyperplasia
lower thirds nodules
0 47 11 0 26 0 4 0
0.015 35 17 0 24 0 0 0
0.15 36 15 0 62 3 6 18
0.75 48 38 36 96 9 8 82
From Highman et al. (1977)
Table 7. Incidences (and%) of uterine cervical adenosis and ovarian tubular
adenomas in mice fed estradiol
Dose Week
(mg/kg bw
per day) 26 52 78 104
Adenosis Adenosis Adenosis Tubular Adenosis Tubular
adenoma adenoma
0 2/37 (5) 5/46 (11) 1/14 (7) 2/14 (14) 13/19 (16) 12/24 (50)
0.015 1/37 (3) 5/29 (17) 1/5 (20) 0/5 (0) 3/24 (12) 11/25 (44)
0.15 5/45 (110) 5/35 (14) 3/14 (21) 2/16 (12) 8/20 (40) 12/20 (60)
0.75 25/42 (60) 33/45 (73) 4/7 (57) 0/7 (0) 3/6 (50) 3/7 (43)
Table 8. Incidences (and%) of pathological changes in the mammary gland of female mice fed estradiol
Dose Hyperplastic alveolar nodules Adenocarcinomas
(mg/kg bw
per day) Weeks 0-39 Weeks 40-65 Weeks 66-91 Weeks 92-105 Weeks 0-39 Weeks 40-65 Weeks 66-91 Weeks 92-105
0 0/91 (0) 0/57 (0) 3/29 (10) 6/50 (12) 4/91 (4) 15/57 (26) 13/29 (45) 19/50 (38)
0.015 0/89 (0) 0/63 (0) 0/19 (0) 4/31 (13) 0/89 (0) 28/63 (44) 8/19 (42) 13/31 (42)
0.15 0/94 (0) 2/56 (4) 8/29 (28) 7/21 (33) 4/94 (4) 21/56 (37) 14/24 (58) 6/21 (29)
0.75 0/93 (0) 5/78 (6) 5/19 (26) 6/17 (35) 5/93 (5) 34/78 (44) 11/19 (58) 8/17 (47)
From Highman et al. (1980)
Table 9. Effects of treatment with estradiol with or without 2-bromo-alpha-ergocryptine on the number of mammary hyperplastic
nodules and mammary tumours in young nulliparous C3H/HeJ mice
Treatment No. of mice at No. of mice at No. of hyperplastic nodules No. of mice with
start of study end of study in inguinal mammary glands mammary tumours
Controls 100 16 3.1 11
Estradiol 100 12 4.8 27
Estradiol + 2-bromo-alpha-ergocryptine 100 28 2.8 9
Table 10. Summary of results of short-term and long-term studies
of the carcinogenicity of estrogens
Species Dose Findings Reference
Short-term study
Rats 0.003-4.12 mg/kg bw Histopathological changes, particularly at Biegel et al. (1998b)
per day for 90 days intermediate and high doses
Long-term studies
Mice Increased incidences of mammary, pituitary, IARC (1979)
uterine, vaginal, testicular, lymphoid, and
bone tumours
Rats Mammary and pituitary tumours; statistically IARC (1979)
nonsignificant increase in incidence of foci
of altered hepatocytes and hepatic nodules
after partial hepatotectomy and adminsitration
of N-nitrosodiethylamine
Hamsters Malignant renal tumours in intact and IARC (1979)
castrated males and in ovariectomized
but not intact females
Castrated male 111 µg/day 100% renal tumour incidence Li et al. (1995)
Syrian hamsters,
9 months
Castrated male 96 µg/day 100% incidence of renal tumours; modulated by Li et al. (1998)
Syrian hamsters, ethinylestradiol
8 months
B6C3F1 mice 4 daily Estrone was protective; estradiol-17ß did not Cavalieri et al.
intraperitoneal increase incidence over control (1997)
injections
(30 µmol/kg bw)
Table 10. (continued)
Species Dose Findings Reference
Male Syrian 25 mg subcutaneously, Estradiol-17ß and 4-hydroxy-17ß-estradiol Liehr et al. (1986)
hamsters 175 days produced tumours, but 2-hydroxy-17ß-estradiol
did not
Castrated male Not reported; Estradiol-17ß and 4-hydroxy-17ß-estradiol Li & Li (1989)
Syrian hamsters 9-10 months produced tumours, but 2-hydroxy-17ß-estradiol
did not
Castrated male 100-200 µg/day Ethinylestradiol produced a lower incidence of Li et al. (1983)
Syrian hamsters tumours than estradiol-17ß
Virgin C3H/HeJ 0.015-0.75 mg/kg bw Estradiol-17ß caused a dose-dependent increase Highman et al.
mice per day in feed in tumour incidence (1977, 1980)
C3H/HeJ mice 0.5 mg/l in Estradiol-17ß caused tumours Welsch (1976)
drinking-water
When Swiss albino mice were given a single intraperitoneal
injection of 100, 1000, or 10 000 µg/kg bw, the highest dose increased
the number of micronucleated polychromatic erythrocytes and the
frequency of sister chromatid exchange, although the
polychromatic:normochromatic erythrocyte ratio did not appear to be
affected. Estradiol did not cause reverse mutation in Salmonella
strains TA100, TA1535, TA97a or TA98, at concentrations of 1-10 000
µg/plate in the absence of metabolic activation and 1-1000 µg/plate in
its presence. In a host-mediated assay in which mice were given 100,
1000, or 10 000 µg/kg bw followed 2 h later by injection of
S. typhimurium, no change in the number of His+ revertants per
plate was observed (Dhillon & Dhillon, 1995).
Estradiol was evaluated in five tests for the induction of
micronuclei in bone marrow in vivo in female rats given three daily
subcutaneous doses of 20 µg/kg bw and in mice given a single
intraperitoneal injection of 10-150 mg/kg bw. The authors concluded
that estradiol was not genotoxic to the bone marrow of rodents (Ashby
et al., 1997).
In male B6C3F1 mice and male Fischer 344 rats that received
estradiol at 310, 620, or 1250 mg/kg bw in three injections, there was
no increase in the frequency of polychromatic erythrocytes. In male
and female B6C3F1 mice treated with various numbers of injections,
solvents, routes of administration, and killing schedules, no
significant increase in the frequency of micronuclei was observed
(Shelby et al., 1997).
The onset of genomic rearrangements was tested at 10-5 mol/L
estradiol in two X-ray-transformed cell lines (X-ray-9 and F-17a) and
two untransformed cell lines (10T1/2b and 10T1/2c). Genomic
rearrangements (deletions or additions in minisatellites) were
observed in the transformed but not the untransformed lines. No new
rearrangements were observed after withdrawal of estradiol (Paquette,
1996).
The ability of estradiol to induce morphological transformation,
gene mutations, chromosomal aberrations, sister chromotid exchange,
unscheduled DNA synthesis and other chromosomal changes was assessed
in Syrian hamster embryo cells. Cell growth was completely inhibited
at 10-30 µg/ml but was not affected at a concentration < 3 µg/ml.
Treatment of cells with 0.3-6 µg/ml did not affect their
colony-forming efficiency, but at 10 µg/ml colony formation was 53%
that of controls. Incubation with estradiol at 0.3-10 µg/ml for 48 h
induced a dose-dependent increase in the frequency of morphological
changes. Estradiol in this dose range also induced numerical changes
(chromosome gains and loses). The majority of these cells (94%) were
diploid. Estradiol had no other effects in this assay (Tsutsui et al.,
1987). No exogenous metabolic activation was used in these
experiments.
Table 11. Genetic toxicity of estrogens
End-point Test system Dose Result Reference
In vitro
Reverse mutation S. typhimurium TA100 50-1500 µg/plate Negative Liehr et al.
(1986)
Reverse mutation S. typhimurium TA100, 1-10 000 µg/plate -S9 Negative Dhillon & Dhillon
TA1535, TA97a, TA98 1-1000 µg/plate +S9 Negative (1995)
Chromosomal aberration, Cultured human 1-100 µg/ml Positive Dhillon & Dhillon
sister chromatid lymphocytes (1995)
exchange
Micronucleus formation Human cells Positive IARC (1987)
Aneuploidy, chromosomal Human cells Negative IARC (1987)
aberrations, sister
chromatid exchange
Aneuploidy, unscheduled Rodent cells Positive IARC (1987)
DNA synthesis
Mutagenicity, DNA damage, Rodent cells Negative IARC (1987)
sister chromatid
exchange
Cell transformation, Syrian hamster 0-10 µg/ml Positive Tsutsui et al.
numerical chromosomal embryo cells (-S9) (1987)
changes
Gene mutation, Syrian hamster 0-10 µg/ml Negative Tsutsui et al.
chromosomal aberration, embryo cells (-S9) (1987)
sister chromatid
exchange, unscheduled
DNA synthesis
Table 11. (continued)
End-point Test system Dose Result Reference
Numerical chromosomal Cultured human 0.05-75 µmol/l Positive Schuler et al.(1998)
changes lymphocytes
Chromosomal breakage Cultured human 0.05-75 µmol/l Negative Schuler et al.(1998)
lymphocytes
DNA damage pBR322 (-S9) 0.01-0.1 mmol/l Single-strand Yoshie & Ohshima
breaks with 2- (1998)
and 4-hydroxy
estradiol and
estrone; negative
with estradiol
and estrone
Microtubule disruption Chinese hamster 0-100 µmol/l EC50, 10 µmol/l Aizu-Yokota et al.
V79 cells (1995)
Adduct formation
Syrian hamster embryo cells 1 µg/ml (-S9) Increase with Hayashi et al.
estradiol and (1996)
2- and 4-hydroxy
estradiol
Syrian hamsters 2-150 mg/kg bw Increase in Han & Liehr
intraperitoneally kidney at 50 (1994a)
mg/kg bw; no
increase in liver
Male Syrian hamsters 50 mg/kg bw Increase in Han & Liehr
intraperitoneally kidney; no (1994a)
time dependence
Table 11. (continued)
End-point Test system Dose Result Reference
Male Syrian hamsters 100 mg/kg bw Increase in liver Han & Liehr
intraperitoneally 1-2 h after (1994a)
dosing but not
later
Male Syrian hamsters 25 mg Increase in Han & Liehr
subcutaneous implant kidney on day 3 (1994a)
but not day 6;
no hepatic
adducts;
substantial
differences in
adduct levels in
controls between
days 3 and 6
Male Syrian hamsters 100 mg/animal per Negative for Han & Liehr
day intraperitoneally kidney with (1994a)
for 3 days estradiol and
2- and 4-hydroxy
estradiol
NBL rats Dose not reported; Unidentified Han et al.
serum level 14 times adduct after 16 (1995)
that of control but not 8 weeks
of treatment
Mongrel dogs Dose not reported Decrease in Winter & Liehr
prostate adduct (1996)
level; increase
in carbonyl
content
Table 11. (continued)
End-point Test system Dose Result Reference
Human liver 2 mmol/l Negative Seraj et al.
(1996)
Rat liver 2 mg/kg bw per day Adducts in male Feser et al. (1996)
by gavage but not for female liver
14 days
Human breast tissue Positive Musarrat et al.
correlation (1996)
Human breast tissue No correlation Nagashima et al.
(1995)
In vivo
Micronucleus formation, Mouse bone marrow 100-10 000 µg/kg Positive at Dhillon & Dhillon
sister chromatid exchange bw as single highest dose (1995)
intraperitoneal
injection
Micronucleus formation Rat bone marrow 20 µg/kg bw as Negative Ashby et al.
three daily (1997)
subcutaneous
injections
Micronucleus formation Mouse bone marrow 10-10 mg/kg bw Negative Ashby et al.
as single (1997)
intraperitoneal
injection
Micronucleus formation Mouse and rat bone 0.1-10 mg/kg bw Negative Shelby et al.
marrow intraperitoneally (1997)
Table 11. (continued)
End-point Test system Dose Result Reference
Frequency of Male and female mice 310-1250 mg/kg bw Negative Shelby et al.
polychromatic as three (1997)
erythrocytes injections
Male Syrian hamster 5-150 mg/kg bw Negative Han & Liehr
intraperitoneally (1994a)
DNA damage Male Syrian hamster kidney 25 mg subcutaneously Single-strand Han & Liehr
and liver every two weeks breaks in kidney (1994a)
but not liver
DNA damage Male Syrian hamster 250 µg/animal per Single-strand Han & Liehr
day for 7 days by breaks with (1994a)
infusion estradiol and
4-hydroxy but
not 2-hydroxy
Chromosomal aberration Male Syrian hamster 20 mg via Positive in Banerjee et al.
subcutaneous capsule kidney (1994)
DNA damage NBL rat Subcutaneous capsules, Single-strand Ho & Roy (1994)
16 weeks; dose breaks in
not reported prostate with
estradiol +
testosterone
The effect of estradiol at 0.05-75 µmol/L was examined in
cultured human lymphocytes by multicolour fluorescence in-situ
hybridization. DNA probes for the centromere and adjacent
heterochromatin regions of chromosomes 1, 9, and 16 were used to
detect hyperdiploidy, polyploidy, and chromosomal breakage. Nonlinear
increases in hyperdiploidy but no chromosomal breakage was observed.
The authors concluded that induction of numerical changes in
chromosomes by estradiol followed a sublinear dose-response
relationship, probably with a threshold concentration. Binding of
estradiol to microtubules or saturation of detoxification mechanisms
are possible explanations for the observation (Schuler et al., 1998).
Male Syrian hamsters were treated with intraperitoneal injections
of 5, 15, or 150 mg/kg bw estradiol; subcutaneous implants containing
25 mg estradiol for two weeks; or continuous infusion of estradiol or
2-or 4-hydroxy-estradiol at 250 mg/animal per day for seven days. A
single intraperitoneal injection of estradiol had no effect on DNA
single-strand breaks in liver or kidney DNA, but the subcutaneous
implants increased the number of renal single-strand breaks by 10%; no
effect was seen in liver. Infusion of estradiol or 4-hydroxyestradiol,
but not 2-hydroxyestradiol, for one week resulted in a 9% increase in
the number of single-strand breaks relative to untreated controls. The
authors suggested that estrogen-induced carcinogenesis is mediated by
free-radical damage (Han & Liehr, 1994a).
Male Syrian hamsters received capsules containing 20 mg
diethyl-stilbestrol, estradiol, moxestrol, 17 alpha-estradiol, or
ß-dienestrol, and between 94 (dienestrol) and 140 (diethylstilbestrol)
µg/day were obsorbed daily from the pellet. Animals were sacrificed at
0.5, 1, 2, 3, 4, or 5 months (diethylstil-bestrol) or at 5 months (all
other treatments). Chromosomal aberrations but not exchanges in
hamster kidney DNA were cumulative with continued exposure to
diethylstilbestrol. The kidneys of estradiol-and moxestrol-treated
animals had chromosomal aberrations at frequencies similar to those
seen with diethylstilbestrol, whereas the frequency of chromosomal
aberrations in animals treated with the weaker estrogens were similar
to those of controls. The authors suggested that estrogen-induced
chromosomal aberrations are involved in tumorigenesis but that the
process does not involve metabolic activation, since moxestrol, which
is poorly metabolized, did induce chromosomal aberrations (Banerjee et
al., 1994).
2-Catechol estradiol and 4-catechol estrone at 0.01-0.1 mmol/L
induced strand breaks in the pBR322 plasmid, and the level was greatly
enhanced by a nitric oxide-releasing compound. The strand breaks could
be inhibited by antioxidants such as N-acetylcysteine and ascorbate
and by superoxide dismutase. Estradiol, estrone, O-methylcatechol
estrogens, and diethylstilbestrol did not induce strand breaks. The
authors suggest that NO mediates conversion of catechol estrogens to
quinones, and the oxygen radicals produced by the quinone/hydroquinone
redox system react with NO to form peroxynitrite, which causes strand
breaks (Yoshie & Ohshima, 1998).
Natural estrogens and their derivatives were tested for the
ability to induce microtubule disruption in Chinese hamster V79 cells
(which lack cytochrome P450 enzymes) at concentrations of 1-100
µmol/L. The EC50 values were 10 µmol/L for estradiol and 9 µmol/L for
17 alpha-estradiol. The most potent disrupting agent tested was
2-methoxyestradiol (EC50, 2 µmol/L). Preincubation of cells with 1
µmol/L taxol for 2 h protected them against microtubule disruption by
estradiol at doses up to 50 µmol/L. The authors concluded that some
natural estrogens cause microtubule disruption in a nongenomic manner
(Aizu-Yokota et al., 1995).
An increase in the frequency of DNA strand breakage and
accumulation of lipid peroxidation products in the dorsolateral but
not the ventral prostate were seen in four NBL rats treated
subcutaneously with testosterone plus estradiol, relative to control
rats. Treatment of castrated rats with testosterone resulted in a
slightly lower rate of strand breaks than in untreated controls. The
authors concluded that estradiol was responsible for the single-strand
breaks in these animals (Ho & Roy, 1994).
Studies of adducts
Male Syrian hamsters were injected intraperitoneally with 2, 10,
50, or 150 mg/kg bw estradiol and sacrificed 4 h later; with 50 mg/kg
bw and sacrificed 1-8 h later; or with 100 mg/kg bw and sacrificed 1-8
h later. Their livers and kidneys were examined for
8-hydroxy-2'-deoxyguanosine (8-OHdG) as a marker of hydroxy radical
interaction with DNA. Four hours after dosing with 50 mg/kg bw, the
renal 8-OHdG levels were double those of controls; adducts were not
determined in kidneys from animals treated at 150 mg/kg bw. No
dose-dependence was observed. The levels of hepatic DNA adducts in
treated animals were similar to those in controls. In hamsters treated
with 50 mg/kg estradiol and killed 1-8 h later, the level of renal DNA
adducts was greater than that in controls at 4 h but not at 1, 2, or 8
h after dosing; hepatic DNA adducts were not determined. In animals
injected with 100 mg/kg estradiol, the number of hepatic DNA adducts
was increased 1 and 2 h after dosing. Treatment of hamsters with
subcutaneous implants containing 25 mg estradiol for three or six days
increased the renal levels of 8-OHdG by 50% over that in controls by
day 3, but the levels were no different from those of controls in
animals implanted with estradiol for six days. No effect was observed
on the background level of liver DNA adducts at either time. Treatment
of hamsters for three days by intraperitoneal injection with 100
µg/animal per day estradiol or 2-or 4-hydroxyestradiol also had no
effect on renal DNA 8-OHdG levels. The authors concluded that the
mechanism of the carcinogenic action of estrogen occurs through
generation of free radicals via redox cycling of catechol estrogen
metabolites (Han & Liehr, 1994b). Substantial differences were
observed in the levels of adducts in untreated animals after three and
six days in the implant experiment. While no statistical analysis was
performed, the differences were sufficient to indicated a substantial
variation in the background level of adducts. The lack of dose-and
time-dependence of adduct formation in these experiments is
inconsistent with the hypothesis that estradiol is a genotoxic
carcinogen.
An unidentified adduct was observed in DNA isolated from the
dorsolateral prostate of NBL rats treated by subcutaneous
administration of estradiol and testosterone for 16 weeks but not 8
weeks. The circulating estradiol concentrations were increased
approximately 14 times over those of controls, while normal plasma
testosterone concentrations were maintained. The appearance of the
adduct correlated with the appearance of dysplastic lesions (Han
et al., 1995).
Incubation of Syrian hamster embryo cells with 1 µg/ml estradiol
or 2-or 4-hydroxy estradiol for 24 h induced DNA adduct formation in
parallel with cell transformation. The level of DNA adduct formation
was greatest with 4-hydroxy estradiol and then with 2-hydroxy
estradiol and estradiol. No exogenous metabolic activation was used in
these experiments. In later experiments, diethylstilbestrol increased
adduct formation in the absence but not in the presence of metabolic
activation (Hayashi et al., 1996).
Mongrel dogs were treated with capsules containing 5
alpha-dihydro-testosterone (DHT) and/or estradiol for 60 days. The
capsules were of uniform length (7 cm), but the quantity of hormone
used was not described. Blood sampoles were obtained for the
measurement of hormone. The 8-OHdG adduct levels in DNA from prostate
were reduced in all dogs that received DHT, whereas treatment with
estradiol or DHT plus estradiol had no effect. Free radical-induced
damage (carbonyl content) of proteins was observed in prostate tissue,
and the authors concluded that the damage was consistent with injury
by estrogen metabolites followed by DHT-stimulated growth of altered
prostatic cells (Winter & Liehr, 1996).
Sterol-initiated DNA adduct formation was examined in vitro by
32P-postlabelling. After exposure of human liver DNA to 2 mmol/L
steroid, several steroids but not estradiol, estrone, or estriol
formed DNA adducts. The presence of a carbonyl group at C17 (which
estradiol lacks) was strongly associated with DNA binding (Seraj et
al., 1996).
When three male and three female Han:Wistar rats given estradiol
at 2 mg/kg bw per day intragastrically as an aqueous microcrystalline
suspension for 14 days, an estrogen-specific DNA adduct was observed
by 32P-postlabelling in the livers of male but not female rats (Feser
et al., 1996).
To assess the hypothesis that estrogen-induced adduct formation
is related to estrogen-induced tumorigenesis in humans, DNA from
normal human breast tissue, benign tumours, and malignant tissue with
invasive ductal carcinoma was examined for the presence of 8-OHdG
adducts by a novel solid-phase immunoslot-blot assay with
adduct-specific antibodies. The amounts of 8-OHdG found in the three
tissues were 0.25, 0.98, and 2.4 pmol/µg DNA, respectively; 13 times
more endogenous formation of 8-OHdG was observed in MCF-7 cells which
undergo hormone-dependent cell growth and have estrogen receptor s
than in normal cultured human mammary epithelial cells, but no
difference in adduct levels was observed between normal cells and
MDA-MB 231 cells, which undergo receptor-independent growth and lack
estrogen receptors. 8-OHdG adduct levels also correlated to the
estrogen receptor status of the tissue, with higher adduct levels in
malignant tissue with estrogen receptors than in those without. Age
and smoking status did not correlate to the 8-OHdG content of DNA. The
authors concluded that accumulation of 8-OhdG adducts in DNA is
predictive of the risk for breast cancer and may be a major
contributor to the development of breast neoplasia (Musarrat
et al., 1996).
No difference in 8-OhdG adduct levels was found by
high-performance liquid chromatography-electrochemical detection in
breast cancer tissue and adjacent non-cancerous tissue, and no
correlation was found with expression of estrogen or progesterone
receptors or with clinical stage or histological grade (Nagashima et
al., 1995).
1.3.5 Reproductive toxicity
The embryotoxicity and teratogenicity of estradiol were reviewed
by a working group convened by IARC (1979), which concluded that
"Oestradiol-17ß has teratogenic actions on the genital tract and
possibly on other organs and impairs fertility."
Estradiol was administered in the feed to female Crl:CD BR rats
at doses equal to 0, 0.003, 0.17, 0.69, or 4.1 mg/kg bw per day in a
90-day, one-generation study. As no pups were born to dams at the two
highest doses, only three dose groups of the F1 generation were
assessed. The mean daily intakes of the F1 females were 0, 0.005 and
0.27 mg/kg bw per day, respectively. The F0 rats were 49 days of age
on test day 0, and serum hormones were evaluated after 7, 28, and 90
days of feeding; they were evaluated on postnatal day 98 for the F1
generation. In the F0 generation, estradiol at doses > 0.17 mg/kg bw
per day produced a dose-dependent increase in serum estradiol
concentration, and all doses produced a dose-dependent decrease in
serum progesterone concentration on test day 90, which correlated with
ovarian atrophy and lack of corpora lutea. The serum concentration of
luteinizing hormone was decreased at all times at > 0.69 mg/kg bw
per day and at 0.17 mg/kg bw per day on test day 90. Little change was
observed in the serum concentrations of follicle-stimulating hormone.
The serum concentration of prolactin was increased at 4.1 mg/kg bw per
day on test day 90. In the F1 generation on postnatal day 28, the
serum estradiol concentration was increased and that of progesterone
decreased at 0.27 mg/kg bw per day. No change in serum prolactin,
follicle-stimulating hormone, or luteinizing hormone concentration was
noted. Dietary estradiol caused marked effects on the estrus cycle at
0.17 mg/kg bw per day (F0) and 0.27 mg/kg bw per day (F1) and at
0.69 and 4.1 mg/kg bw per day (F0 generation) (Biegel et al., 1998b).
Information on the pups in this study was presented elsewhere.
The groups at the two highest doses produced no pups, and the weights
of the pups in the two remaining groups decreased relative to that of
controls; the weights of pups of the F0 generation at 0.003 mg/kg bw
per day (0.005 mg/kg bw per day for the F1 generation) recovered
after birth and remained similar to those of controls throughout the
study. The mean length of gestation in this dose group was
statistically nonsignificantly decreased, which the authors suggested
contributed to the decreased birth weight. The body weights of animals
at 0.27 mg/kg bw per day remained below control levels throughout the
study. Parenteral administration of estradiol did not affect the
anogenital distance in male or female pups. Onset of sexual maturity,
as measured by prepubertal separation in males and vaginal opening in
females, was delayed. Some of the histopathological findings were more
severe in the F1 generation than in the parent generation. The
authors concluded that additional studies were needed to define the
dose-response curve more accurately (Biegel et al., 1998a).
The average litter size of transgenic 'knockout' female mice
deficient in steroid 5 alpha-reductase type I (SRD5 alpha 1-/-,
wild-type C57Bl/6J/129Sv) is reduced in comparison with wild-type
controls (2.7 vs. 8 pups, respectively). In reductase-deficient
animals, the maternal serum estrogen concentrations were chronically
increased by two-to threefold relative to control animals. In control
animals, spikes in DHT and to a much smaller extent testosterone
concentrations occurred in maternal plasma on day 9 of gestation. In
the 5a-reductase-deficient animals, the androgen peaks at day 9 were
reversed. Oogenesis, fertilization, implantation, and placental
morphology appeared normal in reductase-deficient animals. Fetal loss
occurred between gestation days 10.75 and 11, commensurate with a
surge in placental androgen production. Minimal fetal loss was
observed on gestation day 10.5. To test the hypothesis that steroid
hormones contribute to fetal loss in reductase-deficient animals,
pregnant animals were treated with pellets containing various amounts
of steroid hormone. Table 12 shows the effect on mean embryo survival.
Bleeding was observed grossly in the uteri of control and experimental
animals. Administration of estrogen receptor antagonists or inhibitors
of aromatase prevented the excess fetal loss observed in the
reductase-deficient mice. Testosterone was mildly protective against
fetal loss in the knockout mice. The authors concluded that
5a-reductase guards against the toxic effects of estrogen during
pregnancy. They also noted that the human placenta, unlike the rodent
placenta, has high levels of aromatase, resulting in very high
concentrations of estrogens in the amniotic fluid. They speculated
that the human fetoplacental unit has developed a mechanism to protect
itself against estrogens (Mahendroo et al., 1997).
Pregnant female CF-1 mice were treated subcutaneously on
gestation day 13 with Silastic capsules containing 0, 25, 100, 200, or
300 µg estradiol. Male fetuses positioned in utero between a male and
female (MF males) were examined for treatment-related prostatic
effects. In some experiments, MF males were obtained from pregnant
dams killed on gestation day 19 and reared by foster dams. At the age
of seven months, MF males were castrated, implanted subcutaneously
with capsules containing 500 µg testosterone, and killed three weeks
later. The total concentration of estradiol in serum was increased in
a dose-dependent manner in treated MF fetuses collected on day 18,
with 94, 150, 230, 360, and 530 pg/ml in the animals at the five
doses, respectively. A 40% increase in the number of prostatic
glandular epithelial buds was found in MF males from dams treated with
25 µg estradiol, relative to controls. An increase in prostate size
was also noted, but the size of the individual buds was not changed;
prostate weight was increased in MF males at the low dose sacrificed
at eight months but was decreased at the high dose, resulting in an
inverted-U dose-response curve. The authors concluded that increased
fetal serum estrogen concentrations affect androgen regulation of
prostate differentiation, resulting in a permanent increase in the
number of prostatic androgen receptors and in prostate size (vom Saal
et al., 1997). The doses used in this study were below the NOEL.
Under conditions associated with reduced estrogen synthesis in
humans (aromatase deficiency, placental sulfatase deficiency, fetal
anencephaly), estrogen production and concentrations may be reduced by
80-90%. However, progesterone production and fetal development remain
normal, indicating that considerably more estrogen is produced during
normal pregnancy than is necessary (Fisher, 1998).
The embryotoxicity of steroidal and nonsteroidal estrogens was
examined in cultured whole embryos obtained from Sprague-Dawley rats.
Preliminary experiments resulted in steep dose-response curves for all
estrogens examined at doses ranging from 0.05 to 0.5 mmol/L. For
example, diethylstilbestrol had no effect at concentrations < 0.15
mmol/L but was lethal to 100% of cultured embryos at doses > 0.25
mmol/L. The concentrations tested resulted in low embryolethality
(2-20%). Estrogens had dysmorpho-genic effects at concentrations of
0.1-0.2 mmol/L. The commonest effect observed was hypoplasia of the
prosencephalon. Estradiol and estrone were markedly and statistically
significantly more toxic in the presence of metabolic activation (from
livers of pregnant and non-pregant females and Aroclor 1254-treated
adult male rats), but metabolic activation attenuated the embryotoxic
effects of ethinylestradiol, tamoxifen, and erythrohexestrol and had
no effect on other estrogens. In this system, estradiol was more
efficiently converted to catechol estrogens in male liver, but
Table 12. Effects of steroid hormones in 21-day release pellets on embryo survival
Pellet Dose Control females Srd5a1-/- females
treatment
No. Live Dead No. Live Dead
litters embryos embryos litters embryos embryos
None 4 8.0 0.4 8 3.2 5.1
Placebo 3 9.6 0.67 3 4.3 5.0
Androstenedione 0.5 4 3.3 4.5 5 1.0 7.8
Testosterone 0.5 2 8.8 1.5 5 6.2 3.2
Estradiol 0.5 2 0 7.0 5 0 8.4
0.08 5 0 7.4 6 0 7.8
0.02 2 0 5.0 6 0.3 6.3
0.01 2 0 11 6 0 6.8
0.005 2 0 8.5 5 0 5.0
0.0025 2 4.5 4.0 4 2.8 5.5
ethinylestradiol was converted to catechol estrogens approximately
three times more effectively than estradiol when metabolic activation
systems from pregnant and non-pregnant animals was used. The authors
concluded that the effects observed are independent of steroid
structure or estrogen activity and are strongly dependent on the
pathways and rates of biotransformation of some (but not all) of the
parent compounds (Beyer et al., 1989).
Ten mg of estradiol or testosterone were implanted subcutaneously
into groups of five and seven female Sprague-Dawley rats on day 10 of
pregnancy, whereas control pregnant rats were given dextran by the
same method. Implantation with estradiol or testosterone resulted in
complete resorption of embryos in all treated animals (Sarkar et al.,
1986).
A review of the birth certificates and hospital records of 7723
infants whose mothers had reported using oral contraceptives indicated
that these compounds present no major teratogenic hazard (Rothman &
Louik, 1978).
Studies of reproductive toxicity are summarized in Table 13.
1.3.6 Special studies of mechanisms of action
Estrogen-induced tumorigenesis has been the subject of two lines
of investigation: receptor-based effects and redox cycling and DNA
adduct formation leading to genetic damage. During the past decade,
considerable attention has been focused on understanding the molecular
basis of hormone receptor biology. Recently, transgenic animals that
overexpress or lack estrogen receptors (Couse et al., 1997b) or
aromatase (Fisher et al., 1998) have been developed. The role of
estrogens and other hormones in mammary neoplasia in rodents and their
relevance to human risk has been reviewed (Russo & Russo, 1996), and
it was noted that rodent models mimic some but not all of the complex
external and endogenous factors involved in initiation, promotion, and
progression of carcinogenesis. Tumour type and incidence are
influenced by the age, reproductive history, and the endocrine milieu
of the host at the time of exposure. The spontaneous incidence of
tumours differs in different strains of rats and mice. In rats, most
spontaneously developed neoplasias, with the exception of leukaemia,
are of endocrine organs or organs under endocrine control. Russo &
Russo (1996) concluded that mechanism-based toxicology is not yet
sufficient for human risk assessment, and the approach should be
coupled to and validated by traditional long-term bioassays.
The estrogen-responsive male Syrian hamster kidney model has been
widely used to study the carcinogenicity of estrogens in vivo.
Separation of carcinogenic from hormonal effects in male and
ovariectomized female Syrian hamster kidney has been reviewed (Yager &
Liehr, 1996). In hamsters treated chronically with relatively high
doses by subcutaneous implantation, certain potent synthetic estrogens
such as ethinylestradiol result in < 10% tumour incidence in kidney,
whereas treatment with other estrogens result in renal tumour
development in nearly all animals. Ethinylestradiol also acts at
different sites from other estrogens in the hamster kidney. Thus, the
estrogenicity of a compound is essential but not sufficient for renal
carcinogenesis in hamsters. Other factors that have been suggested to
contribute to carcinogenicity include cell-type-specific uptake and
differential estrogen metabolism (such as high 4-hydroxylation rates)
leading to estrogen-induced damage to cell macro-molecules (DNA
and protein). Similarly, the progesterone metabolite
5 alpha-pregnane-3,20-dione successfully competes with progesterone
for receptor binding and biological effectiveness in some tissues but
not others (Tsai et al., 1998). Since a second estrogen receptor
isoform (ERb) has recently been identified, the results described
below should be interpreted with caution.
The estrogen metabolite 4-hydroxyestradiol, but not
2-hydroxyestradiol, was tumorigenic in male hamster kidneys (Yager &
Liehr, 1996), the proposed mechanism of action being redox cycling
resulting in oxygen radical formation and subsequent damage to cell
macromolecules. It is not certain that this pathway is relevant
in vivo at physiological concentrations of estradiol. For example,
micromolar concentrations of estradiol are necessary to cause
microtubular disruption in Chinese hamster V79 cells, and these are
greatly in excess of the picomolar to nanomolar concentrations
normally found in serum. At higher concentrations, the lipophilicity
of estradiol and some metabolites (such as methoxy derivatives) and
their ability to intercalate into DNA and lipid membranes may be more
important from a toxicological perspective than the estrogenic
properties.
The hormonal (i.e. receptor-mediated) and carcinogenic (i.e.
genotoxic) properties of synthetic hormones were differentiated by
measuring the rates of catechol estrogen and methyl ester formation by
a weak carcinogen, 17a-ethinylestradiol, and by a strong hormonal
carcinogen, moxestrol. The rates of hydroxylation in comparison with
that for estradiol were 40-50% for moxestrol and 25-35% for
ethinylestradiol, the differences being apparent at longer reaction
times (i.e. 20 min but not 10 min). 2-Hydroxymoxestrol was a poor
substrate for COMT, proceeding at a rate of about 3% of the
methylation of 2-hydroxyestradiol. In hamster kidney cytosolic protein
extracts, 10 nmol/L progesterone decreased binding of 2 nmol/L
3H-progesterone by 78%, and 10 nmol/L ethinylestradiol inhibited it
by 35%. Interpretation of this result is complicated, as the assay was
performed with insufficient excess progesterone. Estradiol and
moxestrol had no effect. The authors suggested that the decreased
capacity of ethinylestradiol to form catechol metabolites and its
progestogen antagonist activity contribute to the low tumour incidence
seen with this compound (Zhu et al., 1993).
Table 13. Studies of the reproductive toxicity of estrogens
Species Dose Findings Reference
Mice, rats 0.1-35 mg/day Teratogenic IARC (1979)
subcutaneously
Female rats 0.003-4.1 mg/kg bw No NOEL identified Biegel et al.
per day orally for (1998b)
90 days
Transgenic mice Estrogen No NOEL; effects Mahendroo et
concentrations observed between days al. (1997)
reduced two- 10.75 and 11
to threefold
Mice 0-300 µg/animal No NOEL for effects on vom Saal et
fetal prostate al. (1997)
Cultured whole 0.5-0.5 mmol/l Dysmorphogenic effects Beyer et al.
embryos of at 0.1-0,2 mmol/L (1989)
Sprague-Dawley
rats
Sprague-Dawley 10-mg implant Embryo resorption Sarkar et al.
rats (1986)
Humans Acidental exposure No effect reported Rothman & Louk
(1978)
Several steroidal estrogens were tested at doses of 0.1-100
nmol/L for their ability to increase proliferation of primary renal
proximal tubular cells in culture. Most of the estrogens tested
(including 4-hydroxyestradiol and estrone and, to a lesser extent,
2-hydroxyestradio) increased cell proliferation at a concentration of
0.1-10 nmol/L but inhibited it at 100 nmol/L. The authors concluded
that the ability to induce cell proliferation is a more accurate
predictor of carcinogenicity in this system than estrogen-responsive
end-points or the amount of catechol metabolites generated (Li et al.,
1995).
To determine the role of estrogens in tubular renal damage and
the subsequent reparative cell proliferation, castrated adult male
Syrian hamsters were given subcutaneous pellets that released hormones
at the following rates (µg/day): diethylstilbestrol, 145; estradiol,
134, estrone, 104, ethinylestradiol, 154; tamoxifen, 141;
progesterone, 147; and DHT, 121. Diethylstilbestrol was administered
for one to nine months, while the other compounds were administered
for five months. The severity of tubular damage increased with
progressive estrogen treatment, with a prominent rise in the number of
secondary and tertiary lysosomes. The concentration of cathepsin D was
increased in estrogen-treated kidneys (by approximately 2.7-and
3.5-fold at four and five months, respectively) and paralleled the
rise in estrogen receptor content. Progesterone and DHT alone had no
effect, and concomitant treatment of animals with estrogen and either
tamoxifen or DHT mitigated the estrogenic effects. The primary form of
cathepsin D found in the kidneys of control and estrogen-treated
animals was the 52-kDa isoform, considered to be the inactive form of
the protein. The 31-and 27-kDa isoforms, believed to be the active
forms, were found in significant amounts only in the kidneys of
estrogen-treated animals, primary renal tumours, and their metastases.
The authors suggested that cathepsin D mediates renal tubular damage
as a first step in reparative cell proliferation (Li et al., 1997).
Estradiol- or diethylstilbestrol-induced growth of cultured
proximal renal tubular cells could be inhibited by ethinylestradiol.
Expression of estrogen-responsive protooncogene (c- myc, c- fos, and
c- jun) RNA and protein in kidneys was reduced in animals treated
concomitantly relative to that found in animals treated with estradiol
or diethylstilbestrol. The authors concluded that ethinylestradiol
interferes with estrogen receptor-mediated mitogenic pathways,
preventing gene dysregulation and tumour development. This effect does
not appear to be due to differential binding to estrogen receptors by
estrogenic substances (Li et al., 1998).
Other hormones, notably progesterone, testosterone, and
deoxycortico-sterone, and the antiestrogen tamoxifen prevent or
inhibit the growth of estrogen-induced renal tumours in Syrian
hamsters (reviewed by Yager & Liehr, 1996). Progesterone and tamoxifen
exert a protective effect on mammary carcinogenesis (Inoh et al.,
1985). A review of clinical data indicated that adjuvant progestogen
therapy for treatment of patients with metastatic renal-cell carcinoma
is not effective, indicating that carcinogenesis in the Syrian hamster
model is not representative of human renal carcinogenesis (Linehan et
al., 1997).
Rodent tissues that form estrogen-induced tumours have high
concentrations of the caetcholamine noradrenaline. In a study to test
the hypothesis that hydrogen peroxide formed by monoamine oxidase
deamination of catecholamines provides a source of free radicals, in
addition to that postulated to be provided by metabolic redox cycling
of catechol estrogen intermediates, Syrian hamsters and Sprague-Dawley
rats received 25 mg estradiol in a subcutaneous capsule for two weeks.
Treatment increased monoamine oxidase activity in hamster kidney but
not liver and had no effect on monoamine oxidase activity in rat liver
or kidney. The induction of hamster kidney monoamine oxidase activity
could be prevented by tamoxifen. The authors concluded that
receptor-mediated induction of monoamine oxidase, which deaminates
catecholamines, may increase production of hydrogen peroxide and
hydroxyl radicals, thus contributing to tumour initiation (Sarabia &
Liehr, 1998).
Male Syrian hamsters were treated with quercetin, an inhibitor of
COMT, in order to assess the potentiating effects of this compound on
renal tumorigenesis. All six animals treated with subcutaneous pellets
that released estradiol at 61 µg/day developed kidney tumours, but no
tumours were seen in hamsters treated with quercetin at 0.3 or 3% in
the diet for 5.7 or 6.5 months, respectively. Concomitant
administration of estradiol and quercetin increased the number of
large tumours and the incidence of metastases over that seen with
hormone treatment alone. Quercetin inhibited 2 and 4-catechol estrogen
methylation by 34 and 22%, respectively. The rates of redox cycling in
liver and kidney were not affected by treatment with quercetin or
estradiol (Zhu & Liehr, 1994).
Male Syrian hamsters received subcutaneously implanted pellets
containing 25 mg estradiol (which were replaced every three months)
for seven months. Dietary supplementation with 1% vitamin C decreased
estrogen-induced renal carcinogenesis by 50% in a small number of male
Syrian hamsters. In related experiments, the effect of estradiol
and/or vitamin C was examined on the renal activity of the detoxifying
enzymes quinone reductase, catalase, superoxide dismutase, glutathione
peroxidase, glutathione reductase, glucose-6-phosphate dehydrogenase,
and gamma-glutamyl transpeptidase. Glutathione peroxidase activity was
increased in the kidneys of hamsters treated with estradiol for 1
month (141% of control). Quinone reductase activity was reduced in the
kidneys of estradiol-treated animals (18% of control), but the
activity was partially restored by dietary supplementation with
vitamin C for one month (32% of control); in liver, concomitant
treatment with vitamin C and estradiol reduced the activity of this
enzyme (6% of control), and estradiol treatment alone caused a smaller
decrease in activity (68% of control). Differences in catalase
activities were observed after one month but not by seven months of
treatment. Vitamin C had no effect on the intensity or specificity of
estrogen-related kidney DNA adducts. The authors concluded that the
enzymatic changes observed in estradiol-and/or vitamin C-treated
animals were insufficient to account for the differences in renal
tumour incidence. The authors concluded that vitamin C inhibits
estrogen-induced carcinogenesis by reducing the concentration of
estrogen quinone metabolites (Liehr et al., 1989).
COMT is present in the epithelial cells of the proximal
convoluted tubules of the kidney, predominantly in the juxtamedullary
region, where estrogen-induced tumours arise. Treatment of male Syrian
hamsters with estradiol or ethinylestradiol for two or four weeks
altered the intensity, distribution, and subcellular location of
immunoreactivity to COMT. Staining for this enzyme in control animals
was largely of the soluble cytoplasm and nuclear membrane-bound forms,
whereas staining for the soluble form of nuclear COMT was observed in
estrogen-treated animals. No differences were observed between the two
estrogens or between animals treated with estrogen for two or four
weeks; no difference in nuclear location was observed between treated
and control animals. Estradiol-induced renal tumours did not stain for
COMT, and the nuclear signal present in human cells was lacking in
hamster kidney. The authors suggested that a change in the subcellular
distribution of COMT is a protective response to catechol estrogen
metabolic damage to the genome (Weiss et al., 1998).
Male Noble rats were treated with subcutaneous Silastic implants
containing testosterone and/or estradiol or diethylstilbestrol for 16
weeks. In estradiol-treated animals, the plasma testosterone
concentration, determined after three weeks of treatment, was
decreased more than 10-fold (from 4.8 ng/ml to < 0.3 ng/ml), whereas
the estradiol concentration was increased 4.5-fold (from 16 pg/ml to
75 pg/ml). Diethylstilbestrol but not estradiol caused a statistically
significant decrease in body weight, and both hormones decreased the
relative weight of the dorsolateral and ventral prostate and seminal
vesicles plus coagulating glands. The body weights of animals treated
for 16 weeks with testosterone and estradiol were significantly lower
than those of controls, the relative weights of the dorsolateral and
ventral prostate and seminal vesicles plus coagulating glands were
increased, and the testicular weight was decreased approximately
twofold. Multifocal epithelial dysplasia and marked inflammatory
changes were observed in the lateral prostate. No changes were seen in
the morphology of the ventral prostate seminal vesicle, coagulating
gland (anterior prostate), or ampullary gland. Implants of
testosterone plus diethylstilbestrol induced widespread dysplasia in
the ventral prostate and lesser or no ventral prostatic dysplasia. In
explant cultures, animals treated with testosterone plus
diethylstilbestrol or testosterone plus estradiol showed a reduced
ability to convert the 5 alpha,3ß-hydroxysteroid derivative of 3H-DHT
to the more polar 6 alpha-and 7 alpha-hydroxylated derivatives,
resulting in accumulation of 3ß-androstanediol. These metabolic
changes resulted in a threefold (testosterone plus estradiol) or
eightfold (testosterone plus diethylstilbestrol) increase in
accumulation of 3ß-androstanediol in the dysplastic ventral prostate;
no accumulation was observed in the explanted dorsolateral prostate.
In animals treated with testosterone plus diethylstilbestrol, the
ratio of estrone:estradiol was reversed in the ventral prostate,
whereas in animals treated with testosterone plus estradiol, estradiol
metabolism was decreased in the dysplastic dorsolateral prostate but
not in the ventral prostate. The authors concluded that differences
between target tissues in the bioavailability of the estrogen
component determines in which lobe prostate dysplasia develops (Ofner
et al., 1992).
Male Noble rats were treated with Silastic implants containing
testosterone and estradiol for 16 weeks since it had been reported
previously that such implants increase the plasma estradiol
concentration threefold while maintaining testosterone at
physiological concentrations. This treatment regimen produced
dysplasia in the dorsolateral prostate, without liver dysplasia.
Microsomes were prepared from the liver, ventral prostate, and
dorsolateral prostate of control and treated animals to determine
metabolic conversion of estradiol to catechol estrogens. Catechol
estrogen formation was observed at high levels in liver microsomal
incubates and low levels in prostate incubates. Treatment failed to
alter the extent or profile of hepatic estradiol metabolism, except
for a significant reduction in estriol production relative to
controls. A nonsignificant reduction in 2-hydroxyestradiol formation
was also observed. The authors concluded that catechol estrogen
formation is not a mediating step in estrogen-induced tumorigenesis
(Lane et al., 1997).
Mongrel dogs were treated for 60 days subcutaneously with DHT
and/or estradiol; however, the quantity of hormone in the implant and
the resulting plasma concentrations were not measured. Previous
studies had indicated that such implants maintain plasma hormone
concentrations at physiological levels. The activities of aryl
hydrocarbon hydroxylase, 7-ethoxycoumarin O-deethylase, and
estradiol 2-and 4-hydroxylase were elevated in the prostate glands of
animals treated with either hormone or their combination and were
either decreased or unchanged in liver and kidney. The increase
observed in estradiol-treated animals was substantially modified by
concomitant treatment with DHT. The activity of estrogen 2-hydroxylase
was increased tenfold and fourfold in animals given estradiol and
estradiol plus DHT, respectively. The activities of
7-ethoxycoumarin- O-deethylase, aryl hydrocarbon hydroxylase,
glutathione peroxidase I, and catalase were also increased in the
prostate. Hormones had variable effects on these enzymes in liver and
kidney. The free radical-generated carbonyl content of the prostate
increased 2.5-fold after treatment with estradiol and twofold with
treatment with estradiol plus DHT. No hormone-related effects on
carbonyl content were seen in kidney proteins, whereas DHT and the
combination with estradiol increased the carbonyl content by 60 and
150% over the control level. Treatment with estradiol alone resulted
in a substantial but nonsignificant decrease in the hepatic protein
carbonyl content relative to controls. In DNA hydrolysates, a 45%
decrease was observed in the 8-OHdG content in DNA from the prostate
of DHT-treated animals relative to control DNA; no changes were
observed in DNA from other tissues or in that from animals treated
with estradiol or estradiol plus DHT. The authors concluded that the
hormone-induced changes in the activities of catechol estrogen
synthetase and other enzymes were consistent with the postulated
generation of free radicals by redox cycling of catechol estrogen
specifically in the prostate. The protein damage observed is
consistent with induction of benign prostatic hyperplasia injury by
estrogen metabolites followed by growth stimulation of altered foci by
DHT (Winter & Liehr, 1996).
Adult male Noble rats received subpannicularimplantations of
pellets that released a daily mean average of 120 µg of testosterone
propionate and 110 µg of estradiol. Mammary gland ductal
adenocarcinomas were induced in 100% of the rats after eight to nine
months of treatment, whereas neither hormone alone was effective in
inducing adenocarcinomas. Testosterone propionate disrupted mammary
gland ducts and caused proliferation of stromal tissue, while
estradiol induced ductal epithelial growth and nodular atypical
hyperplasia. The authors concluded that androgens interact with
androgen receptors or progesterone receptors, or perhaps both, in
mammary glands induced by estradiol and testosterone propionate to
cause tumours, reduce the latency, enhance tumour size, and increase
the incidence to 100% (Liao et al., 1998).
1.4 Observations in humans
1.4.1 Therapeutic use
Estrogens, sometimes in conjunction with progestogens, are used
for contraception and for hormone replacement. They are among the most
commonly prescribed drugs and include the parent molecule, its esters,
and other synthetic steroidal and non-steroidal derivatives. Estrogens
can be delivered orally, by intramuscular injection, or
percutaneously. The main concern associated with use of estrogens for
these purposes is a possibly increased risk for cancer; other
side-effects may include hypertension, thromboembolic and other
vascular diseases, breakthrough bleeding, gall-bladder disorders,
nausea, migraine, and mood changes. The progestational component of
combined hormonal therapy may be responsible for some of these effects
(Williams & Stancel, 1996; Carr & Griffin, 1998).
The effects of various dosages of orally administered conjugated
estrogens, mestranol, and ethinylestradiol on the binding capacity of
serum corticosteroid-binding globulin (CBG)-binding capacity was
assessed in three healthy postmenopausal women, each of whom received
Premarin(R) orally at 0.3, 0.62, 1.2, or 2.5 mg/day for 14 days.
Each dose increased serum CBG-binding capacity, but the increase was
not significant at 0.3 mg/day. The serum concentration was
approximately doubled with a doubling of the dose. The authors
concluded that there may be a threshold concentration of exogenous
estrogens below which there is no associated increase in serum
concentrations of CBG and above which there is a dose-response
relationship between the exogenous estrogen and serum CBG-binding
capacity (Moore et al., 1978).
Twenty-three healthy postmenopausal women received one or more
two-week courses of estrogens orally, including fine-particle
estradiol (1, 2, or 10 mg/day) and conjugated estrogens (Premarin(R)
at 0.3, 0.62, 1.2, or 2.5 mg/day). The other estrogens that were
assessed included piperizine estrone sulfate, ethinylestradiol, and
diethylstilbestrol. Each dose was ingested by three women, and the
pre-treatment and post-treatment concentrations of
follicle-stimulating hormone, luteinizing hormone, CBG-binding
capacity, SHBG-binding capacity, angiotensinogen, estrone, and
estradiol were determined. The relative potency of the estrogen
preparations were assessed. Conjugated estrogens were 3.2 times more
potent than fine-particle estradiol in increasing SHBG-binding
capacity, and fine-particle estradiol was 1.9 times more potent than
piperizine estrone sulfate in inducing CBG-binding capacity, whereas
conjugated estrogens were 2.5 times more potent than piperizine
estrone sulfate. Doses < 1.7 mg equivalents of piperizine estrone
sulfate failed to increase CBG-binding capacity significaqntly.
Fine-particle estrogens and piperizine estrone sulfate were equipotent
in more potent than piperizine estrone sulfate. The angiotensinogen
response exceeded the baseline range only after administration of 2.2
mg equivalents piperizine estrone sulfate. The natural estrogen
preparations, piperizine estrone sulfate, fine-particle estradiol, and
conjugated estrogens were nearly equipotent on affecting serum
follicle-stimulating hormone concentrations. No dose-response
relationship for serum luteinizing hormone could be demonstrated with
any estrogen preparation (Mashchak et al., 1982).
See also section 4, 'Epidemiological studies of women exposed to
postmenopausal estrogen therapy and hormonal contraceptives'.
1.4.2 Estradiol-related genetic markers of carcinogenicity
The A2 allele of CYP17, the cytochrome P450 that catalyses the
rate-limiting step in androgen biosynthesis, has been suggested to be
a genetic marker for hormone-dependent disease such as polycystic
ovarian syndrome, anovulatory infertility, hirsutism, and breast
cancer. In a nested case-control study, no association was found
between higher basal transcription of the A2 allele and subsequent
development of breast cancer, stage of disease at diagnosis, or age at
menarche. No racial differences in genotype were observed among
Caucasian, African-American, Asian, and Hispanic women (Helzlsouer et
al., 1998).
The data on the effect of COMT polymorphism on human risk for
hormone-induced cancer are conflicting and are discussed elsewhere in
this report.
2. PROGESTERONE
2.1 Explanation
Progesterone (100 or 200 mg) in combination with estradiol
benzoate (10 mg or 20 mg) is administered to cattle as an ear implant
to increase the rate of weight gain (i.e. growth promotion) and
improve feed efficiency. Progesterone is also administered on an
intravaginal sponge to lactating and non-lactating dairy cows and
goats to synchronize estrus. Progesterone is considered to be an
endogenous substance, as exogenously administered progesterone is
structurally identical to the progesterone produced in animals and
humans.
2.2 Biological data
2.2.1 Absorption, distribution, and excretion
Progesterone is largely inactive when administered by the oral
route because of its low systemic bioavailability. High doses of
certain fine-particle forms have been studied for possible use in
contraception or in hormone replacement therapy, and the results are
discussed below.
The pharmacokinetics of orally administered fine-particle
progesterone have been reviewed (Sitruk-Ware et al., 1987). Two 100-mg
capsules of fine-particle progesterone given to women in the early
follicular phase resulted in a peak plasma concentration at the second
hour of 13 ng/ml. When compared with crystallized compound, the same
dose of fine-particle progesterone resulted in twice as the peak serum
concentration. The plasma profiles of the three major metabolites,
pregnanediol 3a-glucuronide, 17-hydroprogesterone, and
20a-dihydroprogesterone, mimicked that of progesterone.
Six fasting women received 100 mg of fine-particle progesterone
vaginally or orally in the early follicular phase of the menstrual
cycle. The maximal plasma concentration was reached 2 h after oral
administration and had returned to baseline by 6 h. Progesterone was
metabolized predominantly to 5a derivatives, regardless of the route
of administration; 5a-pregnanolone was found after oral but not
vaginal administration. The serum concentrations of
deoxycorticosterone and deoxycorticosterone sulfate were also
increased after oral administration of progesterone. The authors
concluded that ingested progesterone is metabolized mainly in the
intestine (Nahoul et al., 1993).
Seven subjects consumed several oral formulations containing 200
mg progesterone, and blood samples were collected after 0.5, 1, 2, 3,
4, and 6 h. A mean peak serum concentration of 30 ng/ml progesterone
was achieved with a fine-particle formulation in oil 2 h after
administration; the mean peak concentrations achieved with the other
formulations were 9.6 ng/ml at 4 h with plain-milled, 13 ng/ml at 3.2
h with fine-particle, 11 ng/ml at 4 h with plain-milled in oil, and 11
ng/ml at 4.1 h with fine-particle enteric coated capsules (Hargrove et
al., 1989).
Five postmenopausal women received 100 mg progesterone per day
orally for five days. The peak plasma progesterone concentrations,
7-11 ng/ml, were reached within 4 h after the last dose, and the
concentrations remained raised for 96 h. The concentration of the
metabolite 20a-dihydro-progesterone mimicked that of progesterone
closely, at 3-5.1 ng/ml. Other metabolites present in plasma included
pregnanediol-3alpha-glucuronide (550-1000 ng/ml) and
17-hydroxyprogesterone (1.4-3.2 ng/ml) (Whitehead et al., 1980).
The effect of food on progesterone absorption was tested in 15
normal postmenopausal women who received 200 mg fine-particle
progesterone for five days or placebo under fasting and non-fasting
conditions; 100, 200, or 300 mg fine-particle progesterone for five
days under fasting conditions; or 200 mg fine-particle progesterone or
50 mg progesterone in oil intramuscularly for two days. Ingestion of
food increased the integrated area under the curve of plasma
concentration-time and maximal plasma concentration but not the time
to the maximal concentration, and increased the bioavailability of
progesterone twofold. Absorption and elimination were independent of
dose over the range tested. The bioavailability of orally administered
progesterone was 8.6% that of intramuscularly administered compound
(Simon et al., 1993).
The bioavailability of 50-200 mg fine-particle progesterone was
studied in normally menstruating women treated by sublingual, oral
(capsule and tablet), vaginal, or rectal administration during the
follicular phase. Increased serum concentrations persisted for over 8
h in women given > 100 mg progesterone vaginally or rectally and over
24 h in those given 200-mg tablets orally. The authors concluded that
clinically relevant concentrations of progesterone can be reached with
fine-particle oral formulations (Maxson & Hargrove, 1985).
Progesterone is bound in serum to CBG and to albumin. Albumin has
a high capacity but low affinity for progesterone, while CBG has a
higher affinity but a lower capacity. Approximately 17% of serum
progesterone is bound to CBG and 80% to albumin, and 2.5% is in the
free state. CBG is synthesized and secreted by the liver and other
tissues, and fetal hepatocytes can synthesize it. A receptor for
unliganded CBG exists on the surface of target cell membranes, which,
when bound to steroid, can activate adenyl cyclase (Orth & Kovacs,
1998).
Progesterone is eliminated via the urine after conversion in the
liver to pregnane-3 alpha,20 alpha-diol (preganediol) and conjugation
to glucuronic acid at C3. Pregnanediol is the major urinary metabolite
of progesterone and serves as an index of progesterone secretion. The
metabolic clearance rate for progesterone is approximately 2200 l/day
(Goldfien & Monroe, 1994).
Elimination of progesterone after intravenous administration of
500 mg/kg bw to ovariectomized rats was described by a two-compartment
model, with distribution and elimination half-lives of 0.13 ± 0.024
(mean ± SD) and 1.2 ± 0.21 h, respectively, and a volume of
distribution of 2.4 l/kg bw (Gangrade et al., 1992). The plasma
half-life of progesterone has also been reported as 5 min (Williams &
Stancel, 1996).
In five ovariectomized gilts given 47.4 nmol progesterone,
bi-exponential clearance was observed, with mean half-lives of 2.5 and
34 min. The authors concluded that the half-life of the first
compartment indicated that the liver is the principal organ of
clearance. In two gilts given 44.7 or 64.6 nmol progesterone, a
secondary increase in the concentration of progesterone in portal and
mesenteric plasma was observed, indicating that progesterone may
undergo enterohepatic circulation (Symonds et al., 1994).
Six adult anestrous bitches were given progesterone
intramuscularly at 2 mg/kg bw, and the progesterone concentrations in
serum were monitored for 72 h. A peak serum concentration of 34 ng/ml
was reached 1.8 h after dosing, but by 72 h the serum concentrations
had reached the preinjection concentration of 0.9 ng/ml. The mean
half-life of absorption was 0.5 h and the mean half-life of
elimination 12 h. The authors used computer modelling to establish
that a dose of 3 mg/kg bw intramuscularly once a day would maintain
the serum concentration at > 10 ng/ml (Scott-Moncrieff et al., 1990).
2.2.2 Biotransformation
Progesterone is irreversibly converted for subsequent elimination
by hydroxylation to more polar and less active compounds, particularly
at C21, C6, and C16. Progesterone hydroxylation is catalysed by
members of various families of cytochromes P450 and other enzymes
found in hepatic and extrahepatic tissues. Human 6ß-hydroxylase
activity is catalysed most efficiently by CYP3A4 and to a lesser
extent by CYP2D6, whereas 16 alpha-hydroxylation is catalysed by
CYP3A4, 1A1, and 2D6 (Niwa et al., 1998).
Tissue and species differ in the metabolism of progesterone. For
example, human but not rat kidneys possess 6ß-hydroxylase activity.
Steroid 21-hydroxylase in liver is provided primarily by CYP2C5 in
rabbits (Kedderis & Mugford, 1998) and by CYP2C6 in rats (Endoh et
al., 1995), which are P450 isozymes that have not been found in human
liver. Rabbit liver CYP2C3 catalyses the 6ß-and 16 alpha-hydroxylation
of progesterone (Hsu et al., 1993), but this enzyme has also not been
found in human liver.
It is well recognized that progesterone can serve as a precursor
for other bioactive steroids, including hydroxylated progestogens,
corticosteroids, androgens, and estrogens. Progesterone in plasma may
be 21-hydroxylated to deoxycorticosterone enzymatically, independently
of the adrenal steroid 21-hydroxylase (Mellon & Miller, 1989).
Alternatively, progesterone may be reduced to 5
alpha-dihydroprogesterone (5 alpha-pregnane-3,20-dione) and further to
5 alpha-pregnan-3 alpha-ol-20-one, which may bind to the GABAA
receptor on cell surface membranes (reviewed by Baulieu, 1997; see
also below). Both of these compounds are secreted by the ovary and
both are also produced by progesterone metabolism in the hypothalamus
and pituitary (Mahesh et al., 1996).
Progesterone can be metabolized to 17 alpha-hydroxyprogesterone
by P450 17 alpha-hydroxylase and further to androstenedione. Cells
that do not express this enzyme cannot metabolize progesterone to
androgenic or estrogenic hormones. In human ovarian thecal cells,
17 alpha-hydroxypro-gesterone was not further metabolized to
androstenedione or to any other product, whereas pregnenolone and
17 alpha-hydroxypregnenolone were converted to dihydroepiandrosterone
and androsterone (McAllister et al., 1989). Thus, the conversion of
progesterone to androgen is a minor pathway in some tissues or during
particular stages of follicular development.
A substantial increase was noted in the ability of the rat uterus
to metabolize progesterone to 5 alpha-pregnane-3,20-dione and
3 alpha-hydroxy-5 alpha-pregnan-20-one (allopregnanolone) between days
11 and 21 of gestation when serum progesterone concentrations are
high. While the former compound actively binds progesterone receptors
in some tissues, this ability is apparently lacking in rat uterus
(Tsai et al., 1998). Some indirect evidence exists that uterine
contractility is mediated, at least in part, through GABAA receptors
(Mahesh et al., 1996).
2.2.3 Biochemical parameters
2.2.3.1 Synthesis
Progesterone is synthesized from steroid precursors in the
gonads, adrenal cortex, and placenta of mammals. Cholesterol obtained
primarily from circulating low-density lipoprotein serves as the
precursor for steroid biosynthesis, although steroidogenic cells are
capable of local de novo cholesterol synthesis. In non-pregnant women,
progesterone in the bloodstream is derived principally from secretion
by the granulosa and thecal cells of the ovary. Gonadal synthesis of
progesterone is regulated by luteinizing and follicle-stimulating
hormones secreted by the anterior pituitary. The secretion of these
hormones, in turn, is regulated by gonadotropin-releasing hormone,
steroid hormones, and other regulating factors in a complex feedback
loop which is not completely understood. During the follicular phase,
about half of the progesterone is of ovarian origin and the other half
of adrenal origin. During the luteal phase, production shifts
predominantly to progesterone of ovarian origin.
The concentrations of circulating progestogens, daily production,
and metabolic clearance rates have been reported in previous reviews
(IARC, 1979, specifically 'General remarks on sex hormones') and in
most textbooks of endocrinology (e.g. Braunstein, 1994; Goldfien &
Monroe, 1994; Carr, 1998; Griffin & Wilson, 1998).
2.2.3.2 Mechanism of action
In the conventional view of the action of steroid sex hormones,
they diffuse passively into the cell, interact with specific
intranuclear receptors, and bind to specific DNA sequence on the
genome (O'Malley et al., 1991). Ligand-receptor binding in conjunction
with interactions with other nuclear transcription factors initiates
transcription of a specific set of genes which are ultimately
responsible for the translation of hormonal action into physiological
events. Intracellular receptors for progesterone, a superfamily of
ligand-activated transcriptional factors, mediate most of its effects.
In humans, three specific progesterone-binding receptors have been
defined: the 116-kDa B-receptor, an N-terminal truncated 94-kDa
A-receptor, and the 60-kDa C-receptor; the last lacks the first zinc
finger but contains, as do isoforms A and B, the hormone-binding
domain (Leighton & Wei, 1998). Progesterone receptors, like other
steroid hormone receptors, form dimers when a hormone binds. The
various progesterone receptor isoforms have similar binding affinities
for progesterone but may modulate regulation of a defined set of
hormone-dependent genes. In addition, receptors for progesterone,
glucocorticoids, mineralocorticoids, and androgens bind to overlapping
DNA response elements and can thus regulate the same genes, adding
complexity to the biological function of the steroid (Tsai et al.,
1998). The specificity depends on the type or number of receptors
present in the cell and other DNA sequences outside the
steroid-binding domain.
Progesterone and estrogen interact to control the growth and
development of organ systems, primarily the female reproductive tract.
Progesterone inhibits estrogen-stimulated endometrial proliferation
and prepares the endometrium for implantation of the blastocytst. In
this process, the endometrium is converted from a proliferative to a
secretative phase, with accompanying morphological and biochemical
changes. Although progesterone stimulates formation of secretory
alveoli in the mammary gland, particularly during pregnancy,
progesterone itself has minimal effect in the absence of estrogen
priming, as in the uterus. Of the sex steroid hormones, progesterone
has the smallest identified distribution of receptor tissue (Williams
& Stancel, 1996) and perhaps the smallest cohort of transcriptionally
responsive genes. Receptor levels and hormone responsiveness vary
throughout the estrus cycle in all species: in the follicular phase,
the levels are low but increase in response to serum estrogen; the
levels decrease during the luteal phase in response to output of
progesterone by the corpus luteum. In rats, the progesterone receptor
levels increase throughout gestation despite high circulating
concentrations of progesterone, whereas in other species little change
or a decrease (guinea-pig) is noted (Tsai et al., 1998). An
estrogen-stimulated increase in progesterone receptors is essential
for progestogen activity in the uterus. Estrogen is required for the
maintenance of intracellular progesterone receptor levels, and
progesterone, in turn, decreases the estrogen receptor concentration
and the levels of progesterone receptor.
The metabolic effects of progesterone in humans include increased
plasma concentrations of cholesterol and low-density lipoprotein and
decreased high-density lipoprotein, and transient decreases in sodium
excretion due to its capacity to antagonize aldosterone. Progesterone
has well-known effects on basal body temperature. Hydroxylated
metabolites of progesterone, namely the 3 alpha-and 3 alpha,5 alpha
(allopregnanolone) derivatives, are suspected to act as
neuropharmacological agents through the GABAA receptor (Mahesh et
al., 1996; Wiebe et al., 1997; Concas et al., 1998; Smith et al.,
1998). Synthesis and accumulation of the progestogen-type
neuropharmacological agents are not related to plasma progesterone
concentrations (Concas et al., 1998).
2.3 Toxicological studies
The toxicological studies reviewed below were reported in the
scientific literature.
2.3.1 Acute toxicity
The therapeutic dose of fine-partcile progesterone is 400 mg/day
for 10 days 5Physicians' Desk Reference, 1999). A dose of 300 mg/day
taken by women at bedtime for 10 days per month was well tolerated,
the only specific side-effect being mild, transient drowsiness (de
Lignieres).
2.3.2 Short-term studies of toxicity
In order to clarify the nature of the vaginal response to
neonatal treatment with progesterone and to record the responses of
other target organs, including the mammary gland, to such treatment,
291 female BALB/cfC3H/Crgl mice bearing the mouse mammary tumour virus
received daily subcutaneous injections of 5 or 20 µg estradiol or 100
µg progesterone; 5 µg estradiol and 100 µg progesterone; or 20 µg
estradiol and 100 µg progesterone for five days beginning 36 h after
birth. One half of the mice were ovariectomized on day 40. Vaginal
smears were taken for 25 days when the mice were 40-50 days old. The
mice were killed and autopsied at the time of onset of mammary tumours
or at one year of age. The results are shown in Table 14. Mice
receiving progesterone alone showed ovary-dependent, persistent
vaginal cornification. When progesterone and estradiol were given
concurrently, the occurrence of persistent vaginal cornification was
significantly lower than in mice receiving estradiol alone.
Progesterone alone produced hyperplastic downgrowths and lesions of
both vaginal and cervical epithelia but to a lesser degree than in
mice treated neonatally with estrogen. When progesterone was given
concurrently with estradiol, the incidence of lesions was lower but
their severity was greater. The low doses of estradiol and
progesterone each resulted in an earlier age of onset and a higher
incidence of mammary tumours; this also occurred after both combined
estrogen plus progesterone treatments. In treated mice ovariectomized
on day 40, normal mammary development did not occur and mammary
tumours failed to appear, regardless of neonatal treatment. The
authors concluded that progesterone causes ovary-dependent, persistent
vaginal cornification and hyperplastic changes in the vaginal and
cervical epithelium, affects the incidence of these lesions in mice
treated simultaneously with estradiol, and significantly increases the
incidence of mammary tumours in mammary tumour-virus bearing mice
(Jones & Bern, 1977).
2.3.3 Long-term studies of toxicity and carcinogenicity
The toxicology of progesterone administered by subcutaneous or
intramuscular injection to mice, rabbits, and dogs or by subcutaneous
implantation in mice has been extensively reviewed (IARC, 1979).
Progesterone increased the incidences of ovarian, uterine, and mammary
tumours in mice and mammary gland tumours in dogs. Neonatal treatment
increased the occurrence of precancerous and cancerous lesions of the
genital tract and mammary tumorigenesis in female mice. The IARC
Working Group concluded that the carcinogenic effects of estrogens and
progestogens were inextricably linked to the hormonal milieu and to
dose-effect relationships (IARC, 1979, 1987)
The role of hormones, including progesterone, in mammary
neoplasia in rodents and their relevance to human risk assessment have
been reviewed (Russo & Russo, 1996), and it was noted that rodent
models mimic some but not all of the complex external and endogenous
factors involved in initiation, promotion, and progression of
carcinogenesis. Tumour type and incidence are influenced by the age,
reproductive history, and endocrine milieu of the host at the time of
exposure. The spontaneous incidence of tumours varies from strain to
strain of rats and mice. In rats, most spontaneously developed
neoplasias, with the exception of leukemia, are of endocrine organs or
organs under endocrine control. Russo & Russo (1996) concluded that
mechanism-based toxicology is not yet sufficient for human risk
assessment, and the approach should be coupled to and validated by
traditional long-term bioassays.
Table 14. Incidences of mammary tumours and nodules in mice treated neonatally with progesterone
Neonatal treatment No. bearing tumours/ Age at No. bearing nodules/
no. examined (%) appearance no. examined (%)
Estradiol Progesterone (months)
0 100 23/32 (72) 9.3 27/32 (84)
5 0 17/19 (89) 9.2 19/19 (100)
5 100 20/32 (63) 9.3 27/32 (84)
20 0 4/11 (30) 9.8 6/11 (55)
20 100 33/44 (75) 9.6 40/44 (91)
0 0 5/17 (29) 11.3 1/17 (6)
Female rabbits were treated with large doses of either
progesterone or testosterone for up to 763 days, and
hydroxyprogesterone caproate was given intramuscularly every other
week at an average dose of 13 mg/rabbit to 19 animals; testosterone
ethanate was given intramuscularly every other week at an average dose
of 15 mg to 21 animals. Rabbits treated with progesterone developed
numerous cysts of the endometrium, sometimes associated with atypical
hyperplasia. Active mammary secretion also occurred. After large doses
of testosterone, one animal developed two adenomatous polyps of the
endometrium, but there were no other noteworthy endometrial changes.
One control animal developed similar polyps. Neither hormone
significantly altered other tissues, including the ovary, adrenal,
thyroid, and pituitary glands. No precancerous endometrial changes or
cancers were found (Meissner & Sommers, 1966).
2.3.4 Genotoxicity
Genetic toxicity was not studied to support registration of
progesterone for use in cattle. A working group convened by IARC
examined the available information and concluded that: 'Progesterone
did not induce dominant lethal mutations in mice or chromosomal
aberrations in rats treated in vivo. It did not induce chromosomal
aberrations or sister chromatid exchanges in cultured human cells, nor
chromosomal aberrations or DNA strand breaks in rodent cells. Studies
on transformation of rodent cells in vitro were inconclusive: a
clearly positive result was obtained for rat embryo cells, a weakly
positive result for mouse cells and a negative result for Syrian
hamster embryo cells. Progesterone was not mutagenic to bacteria.'
(IARC, 1987).
Several studies on the genetic toxicity of progesterone have been
reported in the scientific literature since the IARC evaluation. After
exposure of DNA to 2 mmol/L progesterone, no DNA adducts were formed,
as determined by 32P-postlabelling. The presence of a carbonyl group
at C17, which progesterone lacks, was strongly associated with DNA
binding by other sterols (Seraj et al., 1996).
No significant increase in the frequency of structural or
numerical chromosomal aberrations was seen in Syrian hamster embryo
cells after treatment for 24 h with 3-30 µg/ml progesterone.
Progesterone was able to transform cells only at the highest dose
tested (30 µg/ml), but even at this dose, the frequency of
transformation was one half that found with 1 µg/ml benzo[ a]pyrene
(Tsutsui et al., 1995).
Progesterone was administered by gavage to female rats as a
single dose of 100 mg/kg bw three days before a two-thirds
hepatectomy, and the rats were killed for cell sampling two days after
the hepatectomy. The frequencies of micronucleated and binucleated
hepatocytes were determined in a suspension of liver cells isolated by
collagenase perfusion. The frequency of micronucleated hepatocytes was
3.5-fold higher in progesterone-treated animals than in controls, but
no increase in the frequency of binucleated hepatocytes was observed.
The increase in micronucleus frequency may be related to chromosomal
breakage or to loss of whole chromosomes. A dose of 100 mg/kg bw
progesterone administered by gavage weekly for six weeks, followed by
partial hepatocetomy, did not increase the number of
gamma-glutamyltranspeptidase-positive foci in liver. The authors noted
that similar findings were made under similar experimental conditions
with the non-genotoxic liver mitogen 4-acetylaminofluorene,
progesterone did not form DNA adducts in rat liver, and equivocal
responses were observed for DNA repair in primary rat heptocytes.
In vitro, progesterone was not mutagenic in bacteria, did not induce
chromosomal aberrations or sister chromosome exchange in cultured
human cells or chromosomal aberrations or DNA strand breaks in rodent
cells, inconclusive results were obtained with regard to
transformation of rodent cells and in the L5178Y TK+/- forward
mutation assay, and a slight but statistically significant increase in
the frqueny of sister chromatid exchange was observed in lymphocytes
from children. In vivo, progesterone did not induce dominant lethal
mutations in mice or chromosomal aberrations in rat bone marrow
(Martelli et al., 1998).
In three male and three female Han:Wistar rats given progesterone
at 100 mg/kg bw per day intragastrically as an aqueous
microcrystalline suspension for 14 days, no progesterone-specific
adducts were observed in the liver, as determined by 32P-postlabelling
(Feser et al., 1996).
2.3.5 Reproductive toxicity
Progesterone at doses of 5-200 mg was administered daily by
subcutaneous injection to 22 pregnant Long-Evans rats on days 15-20 of
gestation. The pups were surgically removed from the dams on day 22
and killed after 20 days. No difference in the anogenital distance was
observed between treated and control rats, and sex was easily
determined, whereas animals treated with concentrations of 1.5-2.5 mg
synthetic progestogens were all masculinized (Revesz et al., 1960).
Six pregnant rhesus monkeys were treated with progesterone in oil
intramuscularly at a dose of 50 mg daily for five days each week.
Treatment was begun on days 24-28 of gestation and was maintained
until delivery. The newborns were weighed, killed, and completely
autopsied, with appropriate histological sections. The offspring of
animals treated with progesterone (three males and thee females) were
delivered at about term, and no abnormalities were noted. In contrast,
most of the offspring of monkeys treated with synthetic progestogens
were born prematurely and were not viable; furthermore, all of the
females were masculinized. The authors concluded that progesterone in
comparatively large doses produces no abnormalities and does not
interfere with the normal conclusion of a pregnancy in rhesus monkeys
(Wharton & Scott, 1964).
2.4 Observations in humans
The commonest uses of progestogens are for contraception and for
hormone replacement therapy. For these purposes, synthetic
progestational agents are usually administered, which differ in
structure and function from natural progesterone (Williams & Stancel,
1996).
Changes in endometrial morphology was investigated in 43 patients
without ovaries who were given progesterone intravaginally,
intramuscularly, or orally. The response to intramuscular
administration in oil was heterogeneous, whereas the endometrial
morphology seen after intravaginal administration of fine-particle
progesterone closely matched that observed in the natural cycle, and
pregnancies were supported in two women. Orally administered
progesterone did not affect endometrial morphology (Bourgain et al.,
1990).
Sixty women with oligomenorrhea or amenorrhoea received a 10-day
course of 200 or 300 mg fine-particle progesterone orally. Withdrawal
bleeding occurred in 90% of the women receiving 300 mg progesterone,
58% receiving 200 mg, and 29% of those given placebo. Their lipid
concentrations were unchanged (Shangold et al., 1991).
Seven women with luteal phase defects, treatment with 200 mg
fine-particle progesterone orally three times a day was effective
(Frishman et al., 1995).
The side-effects occurring in women receiving fine-particle
progesterone orally as part of hormone replacement therapy have been
reviewed. They include minor changes in plasma lipoprotein profile in
some but not all studies but no effect on haemostatic parameters (such
as platelet aggregation and fibronolytic capacity) (Sitruk-Ware et
al., 1987).
Two groups of six healthy postmenopausal women were given 300 mg
of fine-particle progesterone once or twice a day on days 1-14 after
estrogen priming for 30 days. Endometrial biopsy samples were then
studied for effects on histological appearance, glycogen content,
ribosomal RNA, and nuclear estrogen receptors in glands, surface
epithelial, and stroma, and the samples from treated women were
compared with biopsy samples obtained before therapy. The group given
progesterone once a day showed incomplete secretory conversion,
whereas the other group showed full secretory conversion with
suppression of mitotic activity and the presence of a predecidual
reaction. Staining for glycogen was significantly increased in both
groups, by 124% at 300 mg/day and 291% at 600 mg/day. Ribosomal RNA
was decreased by 50 and 73% in the two groups, respectively. The
nuclear receptor content decreased in every region of the endometrium
in both groups; although the decrease in nuclear estrogen receptor
content in the stroma of the group given 300 mg/day did not reach
significance, this finding was attributed to insufficient samples (Kim
et al., 1996).
Postmenopausal women were treated for 21 of 28 days for five
years with percutaneous estradiol at a dose of 1.5 mg/day. Within the
first six months, some women received a dose of 3 mg/day estradiol for
25 of 28 days. The initial dose of progesterone was 200 mg/day orally
for the last 14 days of estradiol treatment. This dose was increased
to 300 mg/day for women in whom the desired clinical effect was not
achieved at the lower dose (i.e. withdrawal bleeding). The baseline
plasma concentration was 18 pg/ml, which increased to an average of
68-89 pg/ml at various times during the treatment period. During the
last 12 months of treatment, 2% of the women had episodes of irregular
bleeding, 21% had regular cyclic bleeding, and 77% had no bleeding.
The highest incidence of cyclic bleeding was observed with the
high-estradiol plus high-progesterone regimen (3 mg estradiol and 300
mg progesterone), and the highest incidence of amenorrhoea was seen in
women given the low-estradiol plus low-progesterone regimen. There was
no evidence of endometrial hyperplasia after five years of treatment
with estradiol and progesterone (Moyer et al., 1993).
See also section 4, 'Epidemiological studies of women exposed to
postmenopausal estrogen therapy and hormonal contraceptives'.
3. TESTOSTERONE
3.1 Explanation
Testosterone propionate (200 mg) in combination with estradiol
benzoate (20 mg) is administered to cattle as an ear implant to
increase the rate of weight gain (growth promotion) and to improve
feed efficiency. Testosterone propionate is rapidly cleaved to
testosterone in vivo and is thus considered an 'endogenous
substance', as the residues are structurally identical to testosterone
produced in animals and humans.
3.2 Biological data
3.2.1 Absorption, distribution, and excretion
Testosterone is generally considered to be inactive when given by
the oral route owing to gastrointestinal and/or hepatic inactivation.
Maintenance of physiological concentrations after injection of
testosterone is also difficult because of its rapid clearance (Griffin
& Wilson, 1998). There is little clinical interest in oral forms of
testosterone (i.e. fine-particle formulations), as the high doses
required to overcome first-pass metabolism in the liver result in a
high hepatic load (WHO, 1992).
Absorption of the ester testosterone undecanoate has been studied
for its possible use in testosterone replacement therapy. The plasma
concentrations of testosterone were measured by radioimmunoassay after
oral administration of 25 mg testosterone or 40 mg testosterone
undecanoate (approximately 25 mg testosterone) to young women, and
were compared with that in subjects receiving testosterone at 1.5
µg/kg bw by intravenous administration. Bioavailable testosterone
represented 3.6% of administered testosterone and 6.8% of administered
testosterone undecanoate. The low bioavailability of orally
aministered testosterone has been attributed to its high metabolic
clearance rate, 25 ml/min per kg. The estimated bioavailability
provides justification for the high dose (120-140 mg/day) considered
to be necessary to replace daily production (5-7 mg) (Tauber et al.,
1986). The long aliphatic side-chain of testosterone undecanoate is
thought to facilitate its lymphatic absorption and thus to avoid
first-pass metabolism (Butler et al., 1992; Griffin & Wilson, 1998).
In normal men, circulating androgens are bound to SHBG and to a
lesser extent to serum albumin. Only 1-2% of the testosterone in
circulation is unbound: 44% is bound to SHBG and 54% is bound to
albumin and other proteins (Griffin & Wilson, 1998). Testosterone
binds to albumin with lower affinity than to SHBG. Plasma SHBG is
secreted from the liver, but adult rodent livers do not produce the
secretory form of SHBG (Reventos et al., 1993). The plasma
concentration of SHBG is increased 5-10-fold by estrogens and
decreased twofold by testosterone (Griffin & Wilson, 1998). A
non-secretory form of SHBG is present in many tissues, including
reproductive tissues and the brain. A SHBG receptor on the surface of
some cell membranes binds unliganded SHBG; steroid hormone then binds
SHBG, which can then not bind receptor (Hryb et al., 1990). The
SHBG-receptor complex present on membranes of target tissues may be
responsible for the interaction between the steroid hormone and cAMP
pathways (Rosner, 1991). The intracellular form of SHBG may also
sequester or direct hormones to their target tissue.
When radiolabelled testosterone is administered intragastrically
to rats, about one fourth appears in bile within 12 h as glucuronide
and sulfate conjugates, indicating enterohepatic circulation (IARC,
1979). When testosterone is used as a substrate, hepatic
glycuronsyltransferase and sulfotransferase activities are higher in
men than in women and show extensive individual variation (Pacifici et
al., 1997). Individual but not sex differences were observed in the
activities of the human jejunal estrogen and dehydroepiandrosterone
sulfotransferases (Her et al., 1996).
About 90% of an administered dose of radiolabelled testosterone
is found in the urine, and about 6% undergoes enterohepatic
circulation and appears in the feces (Wilson, 1996). About 1% of
testosterone is secreted in urine as glucuronide, while the remainder
is secreted primarily as 17-ketosteroids (androsterone and
etiocholanolone). Smaller amounts of glucuronide and sulfate
conjugates of androstanediol and estrogens are found.
The plasma half-life of testosterone after intravenous
administration was 10 min (Tauber et al., 1986).
3.2.2 Biotransformation
The metabolism of testosterone has been reviewed (IARC, 1979;
Goldfien & Monroe, 1994; Wilson, 1996; Griffin & Wilson, 1998).
17ß-Hydroxysteroid dehydrogenase catalyses the reversible
oxido-reduction of androstenedione to testosterone, and of estrone to
estradiol and dehydroepi-androsterone to D5-androstenediol. Further
reductions in this pathway (5 alpha and 5 beta followed by 3 keto)
lead to the formation of androsterone and etiocholanolone, the
principal urinary products of testosterone.
Significant differences are found between the metabolic pathways
of testosterone in rodents and humans. Sex-specific regulation of
cytochrome P450s has not been found in human liver, although sex
differences in the metabolism of xenobiotics exist (Kedderis &
Mugford, 1998). Hepatic rat testosterone 7 alpha-hydroxylase (P450
2A1) and mouse testosterone 15 alpha-hydroxylase (P450 2A4) are not
found in human liver. Testosterone is converted to its 6ß form by
human liver CYP3A4 (Lacroix et al., 1997). The major pathways for
inactivation of testosterone in female mouse liver are hydroxylation
at the 16ß-, 6 alpha-, and 16 alpha-positions followed by conjugation
(Wilson & LeBlanc, 1998). Metabolism of testosterone in rat liver,
used as a diagnostic tool for P450 isozyme activity, produced
7 alpha-, 6ß-, 16 alpha-, and 2 alpha-hydroxytestosterones (Swales et
al., 1996). Testosterone 6ß-and 16 alpha-hydroxylation is carried out
in rat liver by CYP3A18 and by other members of the 3A family (Nagata
et al., 1996). No 16ß-hydroxylase (CYP2B1) activity was detected in
rat liver (Swales et al., 1996).
Extensive reductive metabolism of testosterone occurs in the
liver and extrahepatic tissues. The rat small intestine is capable of
oxidizing testosterone, and the enzyme is present in homogenates of
rat gastric and duodenal mucosa at significantly higher levels than in
jejunum; it was also found in the ileum and colon. The major portal
vein metabolite was androstenedione, indicaing that the gut rather
than the liver is primarily responsible for the metabolism of orally
administered testosterone (Farthing et al., 1982)
Testosterone is a precursor of other physiologically relevant
steroids. The active metabolite DHT, formed through the action of
5 alpha-reductase, is an important physiological substrate for the
androgen receptor. DHT binds with higher affinity to the androgen
receptor than testosterone and is considered to be the main cellular
mediator of androgen activity in some tissues. DHT is metabolized to
androsterone, androstanedione, and 3 alpha- and 3ß-androstanediol.
3ß-Hydroxysteroid dehydrogenase also catalyses the conversion of
dehydroepiandrosterone to androstenedione in the testosterone
biosynthetic pathway. Estradiol is formed by aromatization of the A
ring of testosterone. DHT cannot be converted to estrogen.
3.2.3 Biochemical parameters
3.2.3.1 Synthesis
Testosterone is synthesized in testicular Leydig cells, ovarian
thecal cells, and the adrenal cortex. The principal regulator of
gonadal steroidogenic synthesis is luteinizing hormone, secreted by
the anterior pituitary. DHT, derived from reduction of testosterone in
peripheral tissues, and D5-androstenediol are also physiologically
relevant androgens (Griffin & Wilson, 1998; Miyamoto et al., 1998).
Testosterone synthesis begins in the fetus during the first trimester
of pregnancy after stimulation by maternal chorionic gonadotropin. In
males, the plasma concentrations of testosterone are high during the
prenatal period, peak at around three months of age, and then remain
very low until about 11 years of age. The testosterone concentrations
reach a plateau at around 17 years of age, remain elevated for
decades, and then gradually decline (see Table 15). The blood
concentrations of testosterone oscillate in response to the pulsatile
secretion of luteinizing hormone throughout the day. Testosterone
decreases the pulsatile release of gonadotropin-releasing hormone from
the hypothalamus and luteinizing hormone from the pituitary by
negative feedback (Griffin & Wilson, 1998). A diurnal trend in the
blood concentrations of testosterone is found as well, the
concentrations in plasma being approximately 25% higher in the early
morning than in the afternoon (Orth & Kovacs, 1998).
Plasma testosterone concentrations also increase in females
during puberty, although to a lesser extent than in males. In adult
premenopausal women, 70-80% of circulating testosterone is derived
from conversion of dehydroxyepiandrosterone and androstenedione from
the adrenals and ovaries. In postmenopausal women, ovarian secretion
may account for up to 50% of testosterone production, as bilateral
oophorectomy results in a significant reduction in circulating
androgens. Women in their 40s have about 50% less circulating
androgens than women in their 20s, probably because of declining
adrenal dehydroxyepiandrosterone secretion (reviewed by Gelfand &
Wiita, 1997).
The concentrations of circulating androgens, their daily
production and metabolic clearance rates are given in reviews (e.g.
IARC, 1979, specifically 'General remarks on sex hormones') and in
most textbooks of endocrinology (e.g. Braunstein, 1994; Goldfien &
Monroe, 1994; Carr, 1998; Griffin & Wilson, 1998).
3.2.3.2 Mechanism of action
Testosterone and DHT readily diffuse into target cells and bind
with high affinity to the androgen receptor. Only one intracellular
receptor type has so far been identified, and molecular and genetic
analyses indicate that multiple androgen receptor subtypes are
unlikely to exist. The receptor-hormone complex binds to specific
target DNA sequences and initiates transcription of androgen-regulated
genes, translating exposure to hormones into physiological events
(Wilson, 1996).
Testosterone is necessary during pubertal development for the
initiation and maintenance of spermatogenesis and for the libido.
Androgens interact with other growth factors to regulate the growth
and differentiation of the prostate. Androgens also stimulate
erythropoietin production in the kidney, stimulate stem cells in the
haematopoietic system, increase nitrogen retention, and regulate male
hair growth, including balding in genetically susceptible individuals.
Androgens in conjunction with growth hormone may accelerate linear
growth during puberty. Androgens decrease serum SHBG concentrations
and shift the high-or low-density lipoprotein:cholesterol ratio
(Griffin & Wilson, 1998; WHO, 1992)
Testosterone and DHT produce different biological effects in
reproductive and non-reproductive organs and during development. In
tissues such as bone, muscle, and testis, testosterone is thought to
have a direct androgenic action, while conversion to DHT is essential
for androgenic action in other target tissues. In the fetus,
testosterone is responsible for development of the seminal vesicles
and epididymis and for virilization of the mesonephric ducts, whereas
DHT is required for external virilization.
3.3 Toxicological studies
The toxicological studies reviewed below were reported in the
scientific literature.
3.3.1 Acute toxicity
Fine-particle orally administered testosterone has not been
approved for clinical use. In studies with 400 mg of fine-particle
testosterone administered orally for 21 days to six healthy male
volunteers, testosterone was well tolerated, with few effects on
lmiver enzyme systems, but induction of the heaptic drug-metabolizing
system, including testosterone metabolism, was reported in all six
subjects (Johnsen et al., 1976).
In one report of an adverse event, a hypogonadal 21-year-old man
began treatment with 200-mg intramuscular injections of testosterone
enanthate every two weeks. He had a cerebral accident which was
attributed to treatment. His plasma testosterone concentration was 11
400 ng/dL, whereas the range in normal men is 200-1000 ng/dL. The
authors concluded that cerebral infarct is a potential complication of
over-zealous andrigen replacement (Nagelberg et al., 1986).
Table 15. Concentrations, metabolic clearance rates, and daily production of circulating androgens
Person Serum concentration Metabolic clearance Total daily production
(ng/ml) (l/day) (mg/day)
Male 690
Prepubertal 0.08-0.14 0.06-0.1
Pubertal 0.84-1.8 0.58-1.2
Adult 3-10 2.1-6.9
Female 690
Prepubertal 0.05-0.13 0.03-0.09
Pubertal 0.09-0.24 0.06-0.17
Adult 0.3-0.7 0.21-0.48
3.3.2 Short-term studies of toxicity
Six intact adult male baboons received weekly intramuscular
injections of 200 mg testosterone enanthate (equivalent to 8 mg/kg bw)
for up to 28 weeks, while two control animals received weekly
injections of the vehicle only. Quantitative increases in the weight
and volume of both prostatic lobes were seen after 15 weeks of
treatment, and by week 28 there was an increase in stromal tissue with
papillary ingrowth or invagination of glandular epithelium in the
caudal lobe of the prostate. The serum concentrations of testosterone
and DHT were significantly elevated, from 10 and 2-3 ng/ml to 30-40
and 5-6 ng/ml, respectively. The androstenedione concentrations were
increased by three to four times and that of estradiol from 20 to
80-90 pg/ml. The authors concluded that these steroids play a direct
role in inducing early benign prostate hypertrophy in baboons and that
their observations were similar to those in human benign prostate
hypertrophy (Karr et al., 1984).
3.3.3 Long-term studies of toxicity and carcinogenicity
The effects of subcutaneously injected or implanted testosterone
and its esters have been reviewed extensively (IARC, 1979). The
working group convened by IARC concluded that: "There is sufficient
evidence for the carcinogenicity of testosterone in experimental
animals. In the absence of adequate data in humans, it is reasonable,
for practical purposes, to regard testosterone as if it presented a
carcinogenic risk to humans."
The relevance of animal models to human prostate disorders has
been reviewed (WHO, 1992). Besides humans, dogs are the only animals
that develop prostatic cancer and benign prostatic hyperplasia at a
high frequency. In this model, long-term treatment with androgens and
estrogens is required to produce hyperplasia, although such synergism
is not observed in other species. ACI rats spontaneously develop
histologically evident prostatic cancer which does not progress to
clinically relevant disease when pharmaco-logically relevant amounts
of exogenous androgen are administered. Prostate cancer has been
induced only in the Noble and Lobund-Wistar strains of rat.
The role of hormones, including androgens, in the development of
mammary neoplasia in rodents and their relevance to human risk
assessment have been reviewed (Russo & Russo, 1996). Endogenous
androgens are necessary for mammary development in rodents, and it was
noted that rodent models mimic some but not all of the complex
external and endogenous factors involved in initiation, promotion, and
progression of carcinogenesis. Tumour type and incidence are
influenced by the age, reproductive history, and the endocrine milieu
of the host at the time of exposure. The spontaneous incidence of
tumours differs in different strains of rats and mice. In rats, most
spontaneous neoplasias, with the exception of leukaemia, are of
endocrine organs or organs under endocrine control. Russo & Russo
(1996) concluded that mechanism-based toxicology is not yet sufficient
for human risk assessment, and the approach should be coupled to and
validated by traditional long-term bioassays.
Fischer 344 rats were given 3,2'-dimethyl-4-aminobiphenyl (a
prostate carcinogen) at 50 mg/kg bw 10 times at two-week intervals,
and then, from week 20, testosterone propionate and/or
diethylstilbestrol by subcutaneous Silastic implant for 40 weeks, as
seven cycles of 30 days' treatment and 10 days' withdrawal.
Intermittent administration of testosterone resulted in suppression of
the development of ventral prostate adenocarcinomas and slight
(non-significant) increases in the incidences of invasive carcinomas
of the lateral prostate and seminal vesicles. Diethylstilbestrol
completely suppressed tumorigenesis, and the combination with
testosterone propionate inhibited prostate tumour development (Cui
et al., 1998).
Hydroxyprogesterone caproate was given intramuscularly every
other week at an average dose of 13 mg to 19 female rabbits, and
testosterone ethanate was given intramuscularly every other week at an
average dose of 15 mg to 21 animals; both treatments were given for up
to 763 days. Rabbits treated with progesterone developed numerous
endometrial cysts, sometimes associated with atypical hyperplasia;
active mammary secretion was also seen. Treatment with testosterone
induced two adenomatous polyps of the endometrium in one animal, but
no other noteworthy endometrial changes were found and one control
animal developed similar polps. Neither significantly altered other
tissues such as the ovary, adrenal, thyroid, or pituitary gland. No
precancerous endometrial changes or cancers were found (Meissner &
Sommers, 1966).
3.3.4 Genotoxicity
No studies of genetic toxicity were available to the IARC working
group (IARC, 1979). Summaries of more recent studies from the
scientific literature are presented below.
Testosterone at concentrations of 1-100 µg/ml had weak
transforming activity, but with no dose-response relationship, in
Syrian hamster embryo cells. It inhibited the transforming potential
of benzo[ a]pyrene when incubated concomitantly or sequentially. The
transforming effect of testosterone was amplified by phorbol acetate
but was completely inhibited by dexamethasone (Lasne et al., 1990).
The growth of Syrian hamster embryo cells was reduced by
incubation with testosterone or testosterone propionate at 10-30 µg/ml
in a dose-dependent manner, and the two compounds were equipotent in
producing morphological transformation. The propionate induced
dose-dependent cell transformation at concentrations of 1-30 µg/ml,
while testosterone was effective only at 30 µg/ml. At that
concentration, the transformation frequency induced by testosterone
and the propionate was one-half that induced by 1 µg/ml
benzo[a]pyrene. Neither steroid significantly increased the frequency
of chromosomal aberrations or aneuploidy. Testosterone did not induce
gene mutations at the hprt or Na+/K+ ATPase locus (Tsutsui et
al., 1995).
When testosterone and other steroids were added at a final
concentration of 2 mmol/L to DNA obtained from human surgical
resections, rat liver, HepG2 cells, and calf thymus, testosterone did
not form adducts with naked DNA. Furthermore, no adducts were observed
in DNA isolated from HepG2 cells incubated with 10-100 µmol/L
testosterone for 24 h. The presence of a carbonyl group at C17 (which
testosterone lacks) was predictive of DNA reactivity (Seraj et al.,
1996). DNA strand breaks were observed in the dorsolateral prostate of
Nobel rats treated with a combination of testosterone and estradiol
but not in those treated with testosterone alone (Ho & Roy, 1994).
NBL/Cr rats received two Silastic implants subcutaneously, one of
which contained testosterone and the other estradiol, at the age of
eight to nine weeks, and were killed 8, 16, or 24 weeks after the
start of treatment. The quantity of steroid in the implant was not
described, but the release rate was predicted to increase the
estradiol concentrations by approximately 14-fold while maintaining
normal plasma testosterone concentrations. Adduct levels were
determined by 32P-postlabelling. The prostate glands were found to
contain two major endogenous adducts and several minor spots,
irrespective of treatment, but in treated animals the relative levels
of the two adducts were decreased from 1 and 10 × 109 to 0.5 and 3 ×
109 in the ventral but not in the dorsolateral or anterior prostate.
A major adduct spot was observed in the dorsolateral prostate of
animals treated with both hormones for 16 and 24 weeks but not in
those treated for 8 weeks or in control animals. The presence of this
adduct coincided with the occurrence of dysplastic lesions in the
dorsolateral prostate. It appears to be different from the indirect
estrogen-DNA adducts found in the kidneys of male hamsters treated
with estrogen implants and from I-compounds. The authors suggested
that testosterone enhances prostatic carcinogenesis, perhaps by
generating estradiol through peripheral conversion (Han et al., 1995).
3.3.5 Reproductive toxicity
Complete resorption of embryos was seen in female Sprague-Dawley
rats that received 10 mg estradiol (five rats) or testosterone (seven
rats) as a subcutaneous implant on day 10 of gestation. No effect was
seen in control pregnant rats given dextran by the same route (Sarkar
et al., 1986).
3.4 Observations in humans
Androgens are used therapeutically in men with deficient
testicular function to replace normal testosterone. In women, an
estrogen-androgen combination is used in postmenopausal hormone
replacement therapy (Sands & Studd, 1995; Wilson, 1996). The effects
of excess androgens particularly in young boys and women include
virilizing effects (such as hair loss, deepened voice, acne, growth of
facial hair, and menstrual irregularities); estrogenic effects (such
as gynecomastia in men), and toxic effects (such as oedema and
hepatotoxic effects, especially with 17 alpha substitutions). The
serious side-effects of orally administered androgens are particularly
common in the liver in the treatment of aplastic anaemia but are also
observed during treatment for other conditions, including
hypogonadism. The complications include blood-filled cysts and
heaptomas. Increasing the plasma concentration of testosterone to
above physiological levels results in decreased secreation of
follicle-stimulating and luteinizing hormones and decreased testicular
volume and sperm production (Wilson, 1996; Griffin & Wilson, 1998).
In postmenopausal women treated with androgen replacement therapy
at doses up to 10 mg/day for more than six months, the virilizing
effects of orally administered alkylated andrigens such as
methyltestosterone were found to be deoendent on dose and duration.
The masculinizing effects abated when exposure to the andrigens was
decreased or eliminated (reviewed by Gelfand & Wiita, 1997).
Methylation of testosterone at the 17a position effectively reduces
its first-pass metabolism in the liver, thus increasing its
bioavailability. It can thus reach the systemic circulation when given
at therapeutic concentrations (Friffin & Wilson, 1998).
Testosterone undecanotate, the only oral testosterone ester
preparation available, has been used successfully for the management
of delayed puberty in boys by administration of 40 mg daily for 15-21
months. No side-effects were observed, but progression of
virilization, testicular growth, and acceleration of growth associated
with puberty were observed (Butler et al., 1992).
Subcutaneously implanted pellets containing 100 mg crystalline
testosterone release the compound at a rate of 1 mg/day, which falls
to 0.5 mg by three months and 0.35 mg at six months. In postmenopausal
women, such implants raised the serum testosterone concentration
threefold over that of controls. Development of downy facial hair,
hirsutism, acne, and, rarely, voice changes and clitoromegaly have
been reported as adverse effects. Estrogen and estrogen plus
testosterone implants lowered low-density lipoprotein cholesterol and
raised high-density lipoprotein cholesterol in the same proportion
(Sands & Studd, 1995).
The role of endogenous androgens in the development of benign
prostatic hyperplasia and prostate cancer and the effectiveness of
androgen ablation have been reviewed (WHO, 1992; Osterling et al.,
1997). These disorders arise from different parts of the prostate but
are highly associated statistically. The prevalence of histologically
discernible prostate cancer appears to be similar in various
geographic locations, but progression to clinical prostate cancer is
associated with environmental and genetic factors. These disorders
develop later in life only in men who had normal concentrations of
androgens throughout puberty and possibly up to 40 years. Differences
in serum testosterone concentrations have been studied as a possible
cause for the geographical differences in the occurrence of prostate
cancer. Dutch men were found to have a higher average plasma
testosterone concentration than Japanese men, which was statistically
associated with body weight. Nevertheless, the serum testosterone
concentrations of Japanese men with prostate cancer were not different
than those in men without this disease (WHO, 1992).
Epidemiological studies of the relationship between serum
concentrations of androgens and estrogens and prostate cancer have
yielded inconsistent but largely negative results (for references, see
Dorgan et al., 1998; Ross et al., 1998). Since a possible association
with markers of androgen metabolism (androstanediol glucuronide and
DHT) has been observed in some studies, a complex, androgen-dependent,
multi-gene model involving both androgen biosynthesis and metabolic
pathways has been suggested to be critical for susceptibility to
prostatic cancer (Ross et al., 1998).
The effectiveness of orally administered testosterone was
assessed in five eunuchs in a trial in which controls received a
placebo and neither the participants nor the investigators was aware
of which group received treatment. Initial studies indicated that a
dose of 25 mg testosterone had no effect on any clinical parameter,
including sexual desire, erection, ejaculation, and general
well-being. In a second study, a dose of 100 mg/day did not alter the
clinical parameters, whereas 400 mg/day (100 mg four times a day) was
fully effective (Johnsen et al., 1974). Oral administration of
testosterone at 30 mg/day did not restore sexual function to
castrates, while 100 mg/day had a slight effect; 20 mg of testosterone
propionate administered twice a week by injection had moderate
potency. Thus, testosterone is more than 16 times as active by
injection as it is orally (Foss & Camb, 1939).
4. EPIDEMIOLOGICAL STUDIES OF WOMEN EXPOSED TO POSTMENOPAUSAL
ESTROGEN THERAPY AND HORMONAL CONTRACEPTIVES
4.1 Methods
Although no large epidemiological studies on estradiol,
progesterone, or testosterone alone are available, millions of women
have been exposed, chiefly since the 1960s, to a range of exogenous
estrogens and/or progestogens in the course of postmenopausal estrogen
therapy or hormonal contraception. A few extensive reviews on these
compounds have been published recently (WHO, 1998; IARC, 1999) which
provide many more details of the studies summarized here. The findings
of a large number of case-control and cohort studies and clinical
trials in which the relationship between use of exogenous estrogens
and progestogens and the risk for disease was evaluated are reviewed.
Each study design has its strengths and weaknesses: cohort studies are
considered to be less susceptible to bias in recall or information,
but case-control studies are useful for investigating relatively rare
conditions. Whenever possible, the results of pooled analyses of
original data or meta-analyses of published results are given, in
order to provide the best estimate of the relationship between hormone
use and cancer risk. The strength of an association between exposure
to exogenous hormones and the development of disease is given, as is
customary in epidemiological studies, in terms of the relative risk
(RR), which is the ratio of the incidence rate of the disease among
subjects exposed to the factor to that among unexposed subjects. A
relative risk of 1.0 indicates no association, whereas a relative risk
significantly greater than 1.0 implies a positive association between
exposure to the factor and the development of disease; conversely, a
relative risk of less than 1.0 implies that exposure to the factor may
have a protective effect. In order to provide some indication of the
variation of relative risks, 95% confidence intervals (CIs) are
usually calculated. If the interval does not include 1.0, the finding
is considered to be statistically significant. In a few instances,
such as for breast cancer, the estimated excess number of breast
cancer among 10 000 women who had used hormones is also given.
4.2 Postmenopausal estrogen therapy
4.2.1 Exposure
The numbers of women who have used postmenopausal estrogen
therapy varies among countries and within regions of countries (IARC,
1999). The prevalence of use has been greater in the United States
than in most other countries, and use of estrogen therapy after the
menopause is rare in developing countries, although it is increasing.
Conjugated equine estrogens at a standard dose of 0.625 mg daily are
the most widely prescribed preparation for estrogen therapy in women
in the United States, but estradiol and its esters such as estradiol
valerate (1-2 mg daily) are more widely used in most of Europe (Table
16). Conjugated equine estrogens and estradiol compounds yield similar
plasma concentrations of estradiol and estrone. Progestogens such as
medroxyprogesterone acetate, levonorgestrel, and norethindrone acetate
(Table 17) are added to estrogens, typically for 10 days of each
21-day cycle, in combined regimens. Oral administration is the most
popular route, although percutaneous methods are becoming commoner;
use of injections, the first form of postmenopausal estrogen therapy,
has been declining.
4.2.2 Human carcinogenicity
4.2.2.1 Breast cancer
The relationship between estrogen use and breast cancer risk is
of paramount importance because of the frequency of this neoplasm, of
which there are approximately 800 000 new cases per year worldwide,
representing 21% of all cancers in women (Parkin et al., 1999).
Information on the relationship between postmenopausal estrogen
therapy and the risk for breast cancer is available from many
epidemiological studies (Colditz, 1998; IARC, 1999). A pooled analysis
of the original data for 52 705 women with breast cancer and 108 411
women without breast cancer from 51 studies in 21 countries showed a
small increase in risk in current and recent users with longer
duration of use (five years or more) (Collaborative Group on Hormonal
Factors in Breast Cancer, 1997).
The relative risk for having breast cancer was 1.02 (95% CI,
1.01-1.04) for each year of use among current users or those who had
ceased use one to four years previously and 1.4 (95% CI, 1.2-1.5) for
women who had used postmenopausal estrogen therapy for five years or
longer (average duration, 11 years). This increase is comparable with
the effect on risk for breast cancer of delaying menopause, since the
risk for women who have never used such preparations is 1.03 (95% CI,
1.02-1.03) for each additional year at menopause. Five or more years
after cessation of estrogen use there was no significant excess risk
for breast cancer overall or in relation to duration of use.
Of the many factors examined that might affect the relationship
between risk for breast cancer and estrogen use, only a woman's weight
and body-mass index had an effect: the increase in the risk for breast
cancer of current and recent users associated with long duration of
use was greater for women of lower weight than of higher weight or
higher body-mass index. There was no marked variation in the results
according to hormonal type or dose, but little information was
available about long duration of use of any specific preparation.
Cancers diagnosed in women who had ever used postmenopausal estrogen
therapy tended to be less advanced clinically than those diagnosed in
women who had never used them.
Table 16. Routes of administration and dosages of commonly used estrogens
Estrogen Route Dosage
Natural and equine estrogens
Conjugated equine estrogens Oral 0.3-2.5 mg/day
Intramuscular 25 mg/day
Intravenous 25 mg/day
Vaginal 2-4 g/day
Piperazine estrone sulfate Oral 0.625-2.5 mg/day
Vaginal 2-4 g/day
Estradiol Patch 0.05-0.1 mg every
3-4 days
Vaginal 1-4 g/day
Micronized, valerate Oral 1-2 mg/day
Valerate Intramuscular 10 mg/month
Cypionate Intramuscular 1-5 mg/month
Synthetic estrogens
Ethinylestradiol Oral 20-50 µg/day
Mestranol Oral 50-100 µg/day
Diethylstilbestrol Oral 1-5 mg/day
Quinestrol Oral 0.1 mg/day
Table 17. Routes of administration and dosages of commonly used progestogens
Progestogen Route Dosage
Progesterone
Suppositories Vaginal 25-200 mg/day
Rectal 25-200 mg/day
In oil Intramuscular 50-200 mg/day
Micronized Oral 100-300 mg/day
17 alpha-Hydroxy derivatives
Medroxyprogesterone acetate Oral 2.5-10 mg/day
Intramuscular 250-1000 mg/day or week
Megestrol acetate Oral 20-320 mg/day
17 alpha-Hydroxyprogesterone Intramuscular 125-250 mg/week
caproate
Table 17. (continued)
Progestogen Route Dosage
19-Nortestosterone derivatives
Ethynodiol diacetate Oral 1 mg/day
Norethindrone Oral 0.35-10 mg/day
Norethindrone acetate Oral 1-10 mg/day
Norethyndrol Oral 2.5-5 mg/day
Norgestrel Oral 0.3-0.5 mg/day
Levonorgestrel Oral 0.075-0.5 mg/day
Halogenated progesterone
Cyproterone acetate Oral 10-50 mg/day
Retroprogesterone
Dydrogesterone Oral 5-20 mg/day
In Europe and North America, the cumulative incidence of breast
cancer among women aged 50-70 who have never used postmenopausal
estrogen therapy is about 45 per 1000. The cumulative excess numbers
of breast cancers diagnosed per 1000 women of these ages who began use
of hormones at age 50 and used them for 5, 10, or 15 years were
estimated to be 2 (95% CI, 1-3), 6 (3-9), and 12 (5-20), respectively
(Collaborative Group on Hormonal Factors in Breast Cancer, 1997).
4.2.2.2 Endometrial cancer
The association with use of postmenopausal estrogen therapy is
most consistent for endometrial cancer (IARC, 1999). In the United
States, the incidence of endometrial cancer began to rise in the
1960s, reached a peak in the mid-1970s, and then declined until the
1990s. The increased incidence occurred primarily among postmenopausal
women and followed and then paralleled an increase in use of
postmenopausal estrogen therapy.
At least eight cohort and 30 case-control studies have been
conducted to investigate the impact of postmenopausal estrogen therapy
on the occurrence of endometrial cancer. Virtually all (6/8 cohort
studies and 29/30 case-control studies) reported elevated risks
associated with any use of postmenopausal estrogen therapy, and the
excess was statistically significant in the majority (IARC, 1999). The
published results from 30 such studies have been summarised by
meta-analytic methods (Table 18; Grady et al., 1995). The summary
relative risk for an increased incidence of this cancer was 2.3 (95%
CI, 2.1-2.5) for estrogen users when compared to non-users, and a much
higher relative risk was associated with prolonged duration of use
(RR = 9.5 for 10 or more years' use); the risk remained elevated five
or more years after discontinuation of use of unopposed (without the
addition of progestogen) estrogen therapy (RR = 2.3). Interruption of
estrogen use for five to seven days per month did not result in a
lower risk than daily use. Users of unopposed conjugated equine
estrogens had a greater increase in the relative risk of developing
endometrial cancer than users of synthetic estrogens. The risk for
dying from endometrial cancer was increased among users of unopposed
estrogens (RR = 2.7; 95% CI, 0.9-8). Cohort studies showed a decreased
risk for endometrial cancer among users of estrogen plus
progestogen(RR = 0.4), whereas case-control studies showed a small
increase (RR = 1.8). Information on the risk for endometrial cancer
among users of estrogen plus progestogen is, however, still limited
(Grady et al., 1995; IARC, 1999).
In conclusion, the risk for endometrial cancer increases
substantially with long duration of use of unopposed estrogens, and
the increased risk persists for several years after discontinuation of
use. Although not statistically significant, the risk of users of
unopposed estrogens of dying from endometrial cancer is also
increased, so that the association with use of estrogens cannot be
attributed exclusively to greater medical attention for users.
4.2.2.3 Cervical cancer
Only one cohort and two case-control studies are available on the
relationship between use of postmenopausal estrogen therapy and the
risk for invasive cervical cancer (IARC, 1999); in none of them were
the possible confounding effects of oncogenic human papillomaviruses
(the most important cause of cervical cancer, IARC, 1995) considered.
On balance, the limited evidence available suggests that
postmenopausal estrogen therapy is not associated with an increased
risk for invasive cervical carcinoma. The results provide some
suggestion that it is associated with a reduced risk for cervical
cancer, but that may be due to more active screening for preinvasive
disease among women who have received postmenopausal estrogen therapy.
4.2.2.4 Ovarian cancer
The four cohort and 12 case-control studies that addressed the
risk for ovarian cancer (largely epithelial) among women undergoing
postmenopausal estrogen therapy gave somewhat variable results (IARC,
1999). One cohort study and one large case-control study showed a
significant excess risk for ovarian cancer among women who used this
therapy. A pooled analysis of individual data from 12 studies in the
United States based on 2197 cases of invasive epithelial ovarian
cancer among white women and 8893 white controls (Whittemore et al.,
1992) gave pooled multivariate relative risks for invasive ovarian
cancer among women who had ever used postmenopausal estrogen therapy
for more than three months of 0.9 (95% CI, 0.7-1.3) in comparison with
hospital-based controls and 1.1 (95% CI, 0.9-1.4) in comparison with
population-based controls; no consistent duration-risk relationship
was seen. The relative risk for use for > 15 years was 0.5 (95% CI,
0.2-1.3) in the hospital-based and 1.5 (95% CI, 0.8-3.1) in the
population-based studies. The overall trend per year of use was 1.0
for both types of study, and both risk estimates were nonsignificant.
Allowance was made in the analysis for age, study, parity, and use of
combined oral contraceptives. The relative risk in a similar pooled
analysis of individual data for 327 cases of epithelial ovarian
tumours of borderline malignancy associated with any use of
postmenopausal estrogen therapy was 1.1 (95% CI, 0.7-2) (Harris et
al., 1992).
The original data from the four available European studies on
ovarian cancer (from Greece, Italy, and the United Kingdom) were also
reassessed. For a total of 1470 ovarian cancer cases and 327 controls,
a pooled relative risk of 1.7 (95% CI, 1.3-2.2) was found (Negri et
al., in press). Although this relative risk appears to be higher than
those reported from North America, it is not incompatible.
Table 18. Pooled relative risks (RRs) and 95% confidence intervals (CIs)
derived in a meta-analysis of studies of post-menopausal estrogen therapy
and endometrial cancer
Analysis RR 95% CI No. of studies
Any use of estrogens
All eligible studies 2.3a 2.1-2.5 29
Cohort studies 1.7a 1.3-2.1 4
Case-control studies 2.4a 2.2-2.6 25
Hospital controls 2.2 2.0-2.5 10
Gynaecologcial controls 3.3b 2,7-4.0 6
Community controlsc 2.4a 2.0-2.9 10
Dose of conjugated
estrogens (mg)
0.3 3,9 1.6-9.5 3
0.625 3.4 2.0-5.6 4
> 1.25 5.8 4.5-7.5 9
Duration of use (years)
< 1 1.4 1.0-1.8 9
1-5 2.8 2.3-3.5 12
5-10 5.9 4.7-7.5 10
> 10 9.5b 7.4-12 10
Table 18. (continued)
Analysis RR 95% CI No. of studies
Regimen
Intermittent and cyclic 3.0b 2.4-3.8 8
Continuous 2.9b 2.2-3.8 8
Type of estrogen
Conjugated 2.5d 2.1-2.9 9
Synthetice 1.3b 1.1-1.6 7
Time since last use
(years)
< 1 4.1b 2.9-5.7 3
1-4 3.7 2.5-5.5 3
> 5 2.3 1.8-3.1 5
Stage of tumour
0-1 4.2 3.1-5.7 3
2-4 1.4 0.8-2.4 3
Not invasive 6.2 4.5-8.4 4
Invasive 3.8b 2.9-5.1 6
Death from endometrial 2,7 0.9-8.0 3
cancer
a p for homogeneity, < 0.0001
b p for homogeneity, < 0.01
c Residential-, neighbourhood- and population-based controls
d p for homogeneity, < 0.005
e Primarily ethinylestradiol, estradiol valerate, estriol,
and other, unspecified estrogens; diethylstilbestrol and
estrogens combined with androgen were excluded, except
when such use was included with use of all synthetic estrogens
4.2.2.5 Cancers of the liver and biliary tract
Two cohort and two case-control studies that addressed the
association between use of postmenopausal estrogen therapy and the
risk for cancers of the liver or biliary tract showed no alteration in
risk.
4.2.2.6 Colorectal cancer
Seven cohort and 12 case-control studies provide information on
use of postmenopausal estrogen therapy and the risk for colorectal
cancer (Franceschi & La Vecchia, 1998a; IARC, 1999). The risk was not
increased and appeared to be reduced in half of the studies. In a
meta-analysis of the 20 independent estimates of the association
between any use of postmenopausal hormones and colorectal cancer
published up to December 1996 (Herbert-Croteau, 1998), the summary
relative risk was 0.85 (95% CI, 0.73-0.99), although there was
substantial heterogeneity among the studies. The protective effect of
hormones was found to be stronger in more recent publications. The
relative risks were lower for current or recent users (RR = 0.69; 95%
CI,0.52-0.91) and among users of > 5 years (RR = 0.73; 95% CI,
0.53-1) than for short-term users (RR = 0.88; 95% CI, 0.64-1.2). These
data suggest that use of postmenopausal estrogen therapy has a
protective effect against colorectal carcinogenesis. Inadequate
assessment of exposure, poor control of confounding factors such as
lifestyle, and changing patterns of use over time possibly contributed
to the results of studies conducted so far.
4.2.2.7 Cutaneous malignant melanoma
One cohort and nine case-control studies addressed the risk for
cutaneous malignant melanoma in relation to use of postmenopausal
estrogen therapy. Most suggested no alteration (IARC, 1999).
4.2.2.8 Thyroid cancer
Seven case-control studies on thyroid cancer and use of
postmeno-pausal estrogen therapy showed no clear effect on risk (IARC,
1999). La Vecchia et al. (1999) carried out a pooled analysis of the
original data from eight studies on estrogens and thyroid cancer,
comprising a total of 1305 cases and 2300 controls: 110 (8%) cases and
205 (9%) controls reported ever having used postmenopausal estrogen
therapy (RR = 0.78; 95% CI, 0.58-1.0).
4.2.2.9 Summary and conclusions
The working group convened by IARC (1999) concluded that there is
sufficient evidence in humans for the carcinogenicity of
postmenopausal estrogen therapy, largely based on the clear evidence
and substantial strength of the association with risk for endometrial
cancer. The association with breast cancer is weak but consistent with
biological mechanisms such as the adverse effects of delay in age at
menopause and obesity in postmenopausal women. Users of postmenopausal
estrogen therapy had no excess risk for cancers at other sites. The
evidence for the carcinogenicity of postmenopausal combined
estrogen-progestogen therapy was deemed to be limited.
4.2.3 Cardiovascular disease
Many cohort (Grodstein et al., 1996) and case-control (Rosenberg
et al., 1993) studies and three meta-analyses (Stampfer & Colditz,
1991; Grady et al., 1992, US Congress, 1995) have concluded that
postmenopausal estrogen therapy decreases the risk for coronary heart
disease by 35-50%. The predicted increase in the life expectancy of
hormone users, based on estimates from observational studies, is two
to three years (Grady et al., 1992). In a few studies of the outcomes
of estrogen therapy, with or without the addition of a progestogen, in
women with established coronary disease, lower risks were found among
users for a second infarct, death related to coronary heart disease,
and a second coronary artery stenosis, due to a reduction in lipids
and increased fibrinolysis (Petitti, 1998).
A pooled analysis (Hemminki & McPherson, 1997) of 22 randomized
trials involving 4124 women who received oestrogen therapy, placebo,
no therapy, or vitamins and minerals did not support the favourable
findings of the observational studies. The relative risk for women
taking hormones compared with those not taking them was 1.4 (95% CI,
0.48-4) for cardiovascular events without pulmonary embolus and deep
vein thrombosis and 1.6 (95% CI, 0.65-4.2) for cardiovascular events
with these aspects. The largest trial so far on the effect of
postmenopausal therapy with estrogen plus progestoge on the risk for
coronary heart disease involved 2763 women under the age of 80 (mean
age, 67) with established coronary disease (Hulley et al., 1998).
During an average follow-up period of 4.1 years, hormonal treatment
did not reduce the overall rate of coronary heart disease (RR = 0.99;
95% CI, 0.80-1.2) but did increase the frequency of thromboembolic
events and gall-bladder disease. The difference between the findings
of the observational studies and the trials may reflect some selection
bias in the former (Petitti, 1998). Trials, however, reflect
exclusively the short-term effects of postmenopausal estrogen therapy
on cardiovascular disease, while the long-term effects, reflected in
observational studies may be substantially different. An early
thrombogenic effect of estrogen may be counterbalanced by an
anti-atherogenic effect that occurs with longer duration of use
(Rosenberg et al., 1993).
4.2.4 Osteoporosis
Osteoporosis, a systemic skeletal disease characterized by low
bone mass and microarchitectural deterioration with a consequent
increase in bone fragility and susceptibility to fractures, affects an
estimated 75 million people in developed countries. Evidence that
postmenopausal estrogen therapy prevents fractures comes from cohort
and case-control studies, the typical relative risk for non-spinal
fracture being about 0.7 for women currently taking hormones. The
beneficial effect of postmenopausal estrogen therapy was more marked
in women who began therapy within five years after the menopause, and
it appears to be unaffected by age and concomitant progestogen
therapy. Evidence from randomized, controlled clinical trials of a
long-term favourable effect of postmenopausal estrogen therapy is
scanty (Eastell, 1998).
4.2.5 Overall mortality
The estimates of the excess numbers of cases of endometrial
cancer and, possibly, breast cancer diagnosed in women who used
postmenopausal estrogen therapy should be considered in the context of
other effects of such therapy on health (IARC, 1999). Use of
postmenopausal estrogens affects organs other than the breast and may
well decrease the incidences of coronary heart disease and
osteoporotic fractures, although it increases the incidence of venous
thromboembolism (Petitti, 1998).
A few studies of mortality in women showed an inverse association
with postmenopausal estrogen therapy use, most of the relative risks
ranging from 0.4 to 0.8 (Folsom et al., 1995; Sturgeon et al., 1995;
Grodstein et al., 1997; Schairer et al., 1997). In a prospective study
of nurses' health in the United States, current users of hormones had
a lower risk for death (RR = 0.63; 95% CI, 0.56-0.7) than subjects who
had never taken them, after adjustment for confounding variables;
however, the apparent benefit decreased with long-term use (RR = 0.8;
95% CI, 0.67-0.96 after > 10 years) because of an increase in the
rate of mortality from breast cancer. Current hormone users with
coronary risk factors (69% of the women) had the greatest reduction in
mortality (RR = 0.51; 95% CI, 0.45-0.57), and those at low risk had
substantially less benefit (RR = 0.89; 95% CI, 0.62-1.3). The rate of
mortality among women who use postmenopausal hormones is thus lower
than that of non-users, but the survival benefit diminishes with
longer duration of use and is lower for women at low risk of coronary
disease (Grodstein et al., 1997). Since women who use hormones may be
basically healthier than women who do not (Sturgeon et al., 1995), the
benefits attributed to use of estrogens in non-randomized studies of
postmenopausal estrogen users may be overestimated.
4.3 Hormonal contraceptives
4.3.1 Exposure
Oral contraceptives have been used since the early 1960s and are
now used by about 90 million women worldwide (IARC, 1979, 1999 and
related references). 'The pill' is given as a combination of an
estrogen and progestogen or, formerly, as sequential therapy.
Progestogen-only contraceptives have been available for over 20 years
(IARC, 1999) and are administered primarily by intramuscular depot
injections and subcutaneous implants in developing countries, where
there is widest use. Oral progestogen-only 'mini pills' are used
primarily in Europe and North America, but fewer women use these
preparations than parenterally administered progestogens and combined
oral contraceptives. The hormone content of combined preparations has
decreased over time, multiphase formulations have been marketed, and
various progestogens have been introduced (WHO, 1998). Formulations
containing 30-50 µg of estrogen, called 'low-dose' pills, are
available. Oral contraceptives may be used for emergency post-coital
contraception, and the components of oral contraceptives are used to
treat peri-and postmenopausal symptoms and number of other conditions.
Oral contraceptive preparations containing 50 µg of ethinylestradiol
contain 1, 2.5, 3, or 4 mg of norethisterone, while preparations
containing 30 µg of ethinylestradiol contain 150 or 250 µg of
levonorgestrel. The scientific validity of classifying different oral
contraceptive formulations by 'potency' is a controversial issue.
Combined oral contraceptives tend to have progestogenic, estrogenic,
and anti-oetrogenic effects which vary according to the target organ
and the hormonal content of the formulation (WHO, 1998). The estrogen
component of combined oral contraceptives is either ethinylestradiol
or mestranol and the progestogens used are chlormadinone acetate,
cyproterone acetate, desogestrel, ethynodiol diacetate, gestodene,
levonorgestrel, lynestrenol, megestrol, norethisterone, norethisterone
acetate, norethynodrel, and norgestrel. The estrogen used most
commonly is ethinylestradiol, and the most commonly used progestogens
are desogestrel, levonorgestrel, and norethisterone.
Use of oral contraceptives varies widely (IARC, 1999). They were
already being used extensively in the Netherlands, Sweden, the United
Kingdom, and the United States in the 1960s; extensive use of oral
contraceptives by adolescents was documented in Sweden and the United
Kingdom as early as 1964. In contrast, very little use of oral
contraceptives is reported in Japan, the countries of the former
Soviet Union, and most developing countries. The type of oral
contraceptives prescribed also differs among countries, and both the
type of oral contraceptive and the doses of estrogens and progestogens
have changed between and within countries over time.
It is important to stress that use of oral contraceptives is a
relatively recent human activity, and the health benefits and adverse
effects in women have not yet been followed over a complete
generation, even though these preparations are some of the most widely
used and best studied drugs in the world. Women who began using oral
contraceptives before the age of 20 in the 1960s are only now reaching
the ages (50-60 years) at which the incidences of most malignancies
begin to increase.
Estrogens and progestogens belonging to the same chemical groups
may have different estrogenic, androgenic, and progestogenic effects.
Little is known about the long-term health risks and potential
protective effects of the individual components. The effects become
increasingly complex as women grow older and may be exposed to
different types and doses of hormones, starting with oral
contraceptives and progressing to postmenopausal hormone therapy.
4.3.2 Human carcinogenicity
4.3.2.1 Breast cancer
More than 10 cohort and 50 case-control studies have assessed the
relationship between use of combined (estrogen plus progestogen) oral
contraceptives and the risk for breast cancer (IARC, 1999). Individual
data on 53 297 women with breast cancer and 100 239 women without
breast cancer from 54 studies conducted in 25 countries were
collected, checked, and analysed centrally by the Collaborative Group
on Hormonal Factors in Breast Cancer (1996). The studies included in
the collaboration represented about 90% of the epidemiological
information on the topic. The results provided strong evidence of a
small increase in the relative risk for a diagnosis of breast cancer
among women who are taking combined oral contraceptives and during the
10 years after stopping: RR = 1.2; 95% CI, 1.12-1.3 for current users;
1.2; 95% CI, 1.1-1.2 one to four years after stopping; 1.1; 95% CI,
1-1.1 five to nine years after stopping. The results also showed no
significance excess risk of having breast cancer diagnosed 10 or more
years after stopping use (RR = 1.01; 95% CI, 0.96-1.05; Figure 2).
There was no pronounced variation in the results for recent use
among women with different background risks for breast cancer,
including women from different countries and ethnic groups, women with
different reproductive histories, and those with or without a family
history of breast cancer.
Other features of hormonal contraceptive use such as duration of
use, age at first use, and the dose and type of hormone in the
contraceptives had little additional effect on risk once recent use
had been taken into account. Women who began use before the age of 20
had higher relative risks for breast cancer being diagnosed while they
were using combined oral contraceptives and in the five years after
stopping than women who began use when older, but the higher relative
risks apply at ages when breast cancer is rare and for a given
duration of use earlier use does not result in more cancers being
diagnosed than use begun later.
Because the incidence of breast cancer rises steeply with age,
the estimated excess number of cancers diagnosed between starting use
and 10 years after stopping increases with age at last use: for
example, among 10 000 women in Europe and North America who used oral
contraceptives between the ages of 16 and 19, 20 and 24, and 25 and
29, the estimated excess numbers of cancers diagnosed up to 10 years
after stopping use were 0.5 (95% CI, 0.3-0.7), 1.5 (0.7-2.3), and 4.7
(2.7-6.7), respectively. Up to 20 years after cessation of use, the
difference between women who have ever used oral contraceptives and
those who have never used them is not so much in the total number of
cancers diagnosed but in their clinical presentation: breast cancers
diagnosed in women with any use are less advanced clinically than
those diagnosed in women with no use, the relative risk for tumours
that had spread beyond the breast compared with localized tumours
being 0.88 (95% CI, 0.81-0.95). The relationship observed between the
risk for breast cancer and use of hormones is unusual, and it is not
possible to infer from these data whether it is due to earlier
diagnosis of breast cancer in women who have used oral contraceptives,
the biological effects of hormonal contraceptives, or a combination
(Collaborative Group on Hormonal Factors in Breast Cancer, 1996).
Data on injectable progestogen-only contraceptives were available
from two case-control studies and a pooled analysis of original data,
overall involving about 350 women with breast cancer who had used
these drugs (IARC, 1999). Data on oral progestogen-only contraceptives
were available from a pooled analysis of original data on 725 women
with breast cancer. Overall, there is no evidence of an increased risk
for breast cancer in users of progestogen-only contraceptives
(Collaborative Group on Hormonal Factors in Breast Cancer, 1996).
4.3.2.2 Endometrial cancer
Three cohort and 16 case-control studies addressed the
relationship between use of combined oral contraceptives and risk for
endometrial cancer (IARC, 1999). The results of these studies
consistently show that the risk for endometrial cancer of women who
have taken these pills is approximately halved. The reduction in risk
is generally stronger the longer the oral contraceptives have been
used and persists for at least 10 years after cessation of use. Few
data are available for the more recent, low-dose formulations. Use of
sequential oral contraceptives which were removed from the consumer
market in the 1970s was associated with an increased risk for
endometrial cancer. One case-control study addressed the relationship
between use of oral progestogen-only contraceptives and risk for
endometrial cancer. Less than 2% of the control women had used these
preparations, and women with endometrial cancer were less likely to
have used oral progestogen-only contraceptives than control women but
not significantly so.
The effects of use of depot medroxyprogesterone acetate on the
risk for endometrial cancer have been evaluated in one cohort and one
case-control study. No reduction in risk was seen in the cohort study,
whereas a strong reduction was observed in the case-control study.
Although the evidence is based on small numbers of women, the results
of these studies suggest that women who use progestogen-only
contraceptives have a reduced risk for endometrial cancer.
In biological terms, the favourable effect of oral contraceptive
use with regard to endometrial cancer is attributed to the daily
presence of progestogens, whereas in the normal cycle they are present
for only 14 days.
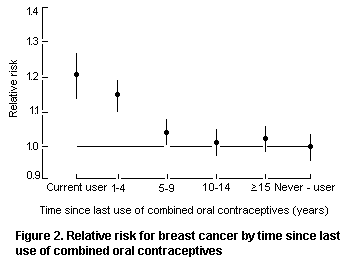 Risk (and 95% confidence interval) relative to that of women who have
never used combined oral contraceptives, stratified by study, age at
diagnosis, parity, age at birth of first child, and age at which risk
for conception ceased
4.3.2.3 Cervical cancer
Two cohort and 16 case-control studies of use of combined oral
contraceptives and invasive cervical cancer have been published (IARC,
1999); these consistently show relative risks of approximately 1.5-2
associated with long duration of use. Similar associations were seen
in four studies in which some analyses were restricted to cases and
controls who had human papillomavirus infection, the most important
cause of cervical cancer (IARC, 1995). Biases related to differences
in sexual behaviour, screening practices, and other factors between
users and non-users of oral contraceptives cannot be ruled out as
possible explanations for the observed associations. There is little
evidence that use of depot medroxyprogesterone acetate or other
progestational injectable contraceptives alters the risk for either
squamous-cell carcinoma or adenocarcinoma of the uterine cervix.
4.3.2.4 Ovarian cancer
Four cohort and 21 case-control studies addressed the
relationship between ovarian cancer and use of combined oral
contraceptives (IARC, 1999). Most of the information available on the
topic has been summarized in two pooled analyses (Table 19) of studies
involving 971 cases and 2258 controls in three European countries
(Franceschi et al., 1991) and 2197 cases and 8893 controls in 12
studies in the United States (Whittemore et al., 1992), giving a total
of over 3100 cases and 11 000 controls. In the pooled analysis of
original data from three hospital-based European studies, the relative
risk was 0.6 (95% CI, 0.4-0.8) for any use and 0.4 (0.2-0.7) for
longest (> 5 years) use. Allowance was made in the analysis for age
and other sociodemographic factors, menopausal status, and parity. The
protection persisted for at least 15 years after use ceased. In the
pooled analysis of original data from studies in the United States,
adjustment was made for age, study, and parity, and the corresponding
values were 0.7 (95% CI, 0.6-0.8) for any use and 0.3 (0.2-0.4) for
use for more than six years. Similar results were obtained in
population-based and hospital-based studies considered separately,
with a relative risk of 0.7 in both types of study for any use of
combined oral contraceptives, 0.6 in hospital-based studies and 0.3 in
population-based ones for use for more than six years, and 0.95
(nonsignificant) and 0.90 (p < 0.001), respectively, per additional
year of use.
An inverse association was also observed in a further analysis of
seven studies of 110 black cases and 251 black controls from the
United States pooled analysis, with a relative risk of 0.7 for any use
and 0.6 for longest use (John et al., 1993). In an analysis of data on
327 epithelial ovarian neoplasms of borderline malignancy in white
women, the relative risks were 0.8 (95% CI, 0.6-1) for any use of
combined oral contraceptives and 0.6 (0.4-0.9) for more than five
years of use (Harris et al., 1992).
Table 19. Results of pooled analysis of the association between use of oral contraceptives
and ovarian cancer
Reference, Type of study No. of cases Relative risk (95% CI) Comments
country (age)
Any use Longest use Duration
(years)
Franceschi et al. Three 971 (< 65) 0.6 (0.4-0.8) 0.4 (0.2-0.7) > 5 Protection still present
(1991), Greece, hospital-based > 15 years after use
Italy, United ceased (odds ratio, 0.5)
Kingdom
Whittemore et al. Pooled analysis 2197 (all ages) 0.7 (0.6-0.87) 0.3 (0.2-0.4 > 6 Invasive epithelial
(1992), of 12 neoplasms in white women;
United States population- and protection seen in both
hospital-based types of study
case-control
studies
Harris et al. Same as above 327 0.8 (0.6-1.1) 0.6 (0.4-0.9) > 5 Epithelial tumours of low
(1992), malignant potential in
United States white women
The most convincing aspect of the inverse relationship found
between use of combined oral contraceptives and risk for ovarian
cancer is the consistency of the results, independently of the type of
study, geographical area (Australia, Europe, North America, and
developing countries), or type of analysis, although the covariates
differed from study to study. Likewise, the inverse relationship was
observed for most formulations considered, including low-dose ones
(Rosenberg et al., 1994), even though relatively few data are
available on the more recent formulations.
One case-control study addressed use of progestogen-only oral
contraceptives and one case-control study specifically addressed any
use of depot medroxyprogesterone acetate. Neither showed any
alteration in risk, either overall or in relation to duration of use.
In biological terms, the favourable effect of oral contraceptives
with regard to the risk for ovarian cancer can be interpreted in the
context of the 'incessant ovulation theory'. Thus, any event such as
pregnancy or eaqrly menopause that diminishes the number of ovulations
over a lifetime reduces the risk for ovarian cancer.
4.3.2.5 Cancers of the liver and biliary tract
Three cohort studies showed no significant association between
use of combined oral contraceptives and the incidence of or mortality
from liver cancer, but the expected numbers of cases were very small,
resulting in low statistical power. The association between use of
combined oral contraceptives and liver cancer was evaluated in 11
case-control studies (IARC, 1999). Three European and two North
American studies showed relative risks that were significantly
increased.
The two largest investigations on the topic were carried out in
countries at intermediate to high risk for liver cancer, i.e.
developing countries and southern Europe, where the relative risks for
use of oral contraceptives for two years or more were 0.2 and 0.8,
respectively. Use of oral contraceptives does not therefore enhance
the risk for liver cancer in populations where the prevalence of
hepatitis B and C is high, but they may be a rare cause of cancer in
developed countries where the prevalence of hepatitis is low (IARC,
1999). Positive associations have been reported chiefly with use of
high-dose formulations.
Two case-control studies addressed the association between risk
for liver cancer and use of injectable progestogen-only
contraceptives. In neither study did the risk for liver cancer differ
significantly between women who had ever or never used these
contraceptives. Both studies were conducted in areas endemic for
hepatitis viruses.
4.3.2.6 Colorectal cancer
Four cohort and 10 case-control studies provided information on
use of combined oral contraceptives and the risk for colorectal cancer
(IARC, 1999; Franceschi & La Vecchia, 1998b). None showed
significantly elevated risks in women who used these preparations for
any duration. Relative risks below 1.0 were found in nine studies, and
the risk was significantly reduced in two.
4.3.2.7 Cutaneous malignant melanoma
Four cohort and 18 case-control studies provided information on
use of combined oral contraceptives and the risk for cutaneous
malignant melanoma (IARC, 1999). The relative risks were generally
close to 1.0 and not related to duration of use. A meta-analysis of
published data from 18 case-control studies on melanoma, involving
3796 cases and 9442 controls, yielded a relative risk for any use of
oral contraceptives of 0.95 (95% CI, 0.87-1). Further analyses in
different subgroups defined by personal characteristics and study
design did not materially alter this finding (Gefeller et al., 1997).
One case-control study of cutaneous malignant melanoma showed no
increase in risk among users of progestogen-only contraceptives.
4.3.2.8 Thyroid cancer
Ten case-control studies published information on use of combined
oral contraceptives and risk for cancer of the thyroid gland. In
general there was no elevation in risk associated with oral
contraceptive use Original data from 13 studies from North America,
Asia and Europe were reanalysed (La Vecchia et al., 1999). Based on
2,132 cases of thyroid cancer and 3,301 controls, relative risk for
ever use of oral contraceptives was 1.15 (95% CI, 0.97-1.37). There
was no relationship with duration of use, or age at first use, or use
before first birth. The relative risk was significantly increased for
current oral contraceptive users (RR = 1.45; 95% CI, 1.02-2.07), but
declined with increasing time since stopping.
4.3.2.9 Summary and conclusions
The working group convened by IARC in 1998 (IARC 1999) concluded
that there was sufficient evidence in humans for the carcinogenicity
of combined oral contraceptives. Conversely, the evidence for the
carcino-genicity of progestogen-only contraceptives was deemed
inadequate. It is impossible to infer whether the association between
breast cancer and use of combined oral contraceptives is due to
earlier diagnosis of breast cancer in users, to the biological effects
of contraceptives, or to a combination.
4.3.3 Cardiovascular disease
4.3.3.1 Acute myocardial infarct
Myocardial infarct is uncommon in women of reproductive age (WHO,
1998 and related references). Age, cigarette smoking, diabetes,
hypertension, and raised cholesterol concentration in blood are
important risk factors for myocardial infarct in young women.
After the first reports of a roughly threefold increase in risk
for myocardial infarct in current users of combined oral
contraceptives, three cohort and six case-control studies were
reported which included data collected after 1980. The relative risks
varied widely, from 0.9 to 5 (WHO, 1998), but six of the studies
reported elevated relative risks, three of which were statistically
significant. None of the studies found a relationship between
myocardial infarct and duration of current use. in nine studies that
included data on past use of combined oral contraceptives, there was
no evidence of either an increase or a decrease in the risk for
myocardial infarct among previous users when compared with women who
had never used oral contraceptives.
The available data do not allow an evaluation of the effect of
dose of estrogen on the relative risk for myocardial infarct
independently of the type and dose of progestogen. There are
insufficient data to assess whether the risk of users of low-dose
combined oral contraceptives is modified by the type of progestogen,
and the suggestion that users of low-dose combined oral contraceptives
containing gestodene or desogestrel have a lower risk for myocardial
infarct than users of low-dose formulations containing levonorgestrel
remains to be substantiated.
4.3.3.2 Stroke
Strokes can be divided into two broad categories, infarcts and
haemorrhages, according to the nature of the cerebral lesion. The
relationship between use of combined oral contraceptives and ischaemic
stroke has been demonstrated in a number of epidemiological studies,
from as early as 1968 (WHO, 1998). Two cohort and 11 case-control
studies show a fairly consistent pattern of association, especially if
greater weight is given to larger studies conducted in areas where the
prevalence of use of oral contraceptives among women in the control
group was more than 10%. Most of the studies found that current use of
combined oral contraceptives was associated with an overall increase
in the risk for ischaemic stroke that was approximately 3.4 times that
of non-users. An association with transient ischaemic attacks was also
reported.
None of the studies of the relationship between risk for stroke
and duration of use of contraceptives provided strong evidence for an
association. Four studies found no significantly elevated risk among
past users, and two others found that the relative risk for ischaemic
stroke among former users of combined oral contraceptives was lower
than that among women who had never used them (WHO, 1998). The risk
for ischaemic stroke among women who do not smoke, who have their
blood pressure checked regularly, and who do not have hypertension is
increased by about 1.5-fold with current (albeit not past) use of
low-dose combined oral contraceptives when compared with non-users.
Two recent cohort and four case-control studies examined the risk
for haemorrhagic stroke in current users and found relative risks of
1-2. The incidence of fatal and non-fatal haemorrhagic stroke was not
increased in current users of combined and oral contraceptives who did
not smoke and did not have hypertension, but the incidence of
haemorrhagic stroke increases with age, smoking, and hypertension, and
current use of oral contraceptives appears to magnify these effects.
Use of progestogen-only oral and injectable contraceptives does
not appear to increase the relative risk for all types of stroke.
4.3.3.3 Venous thromboembolism
Thrombosis arises from the interaction of disturbances of the
wall of the blood vessels, of the components of blood, and of the
dynamics of blood flow (stasis). The risk factors for venous
thromboembolism include pregnancy and puerperium, recent surgery,
immobilization, obesity, and various chronic diseases. Venous
thromboembolic disease is the most common cardiovascular event among
users of oral contraceptives, but it is associated with low rates of
mortality and long-term disability. Three cohort and six case-control
studies which included data collected after 1980 found that current
users of combined oral contraceptives had a risk for thromboembolic
disease that was three to four times higher than that of women who
were not using oral contraceptives (WHO, 1998). The risk of current
users is probably highest in the first year of use but falls to that
of non-users within three months of stopping use of oral
contraceptives.
4.3.4 Overall mortality
Since use of oral contraceptives probably increases the risks for
cancers of the breast, cervix uteri, and liver but decreases those for
cancers of the ovary and endometrium, it is important to evaluate the
net effect of use of oral contraceptives. This obviously depends on a
woman's age and the background frequencies of cancers affected by oral
contraceptives at various sites.
The risks for developing cancers of the breast, cervix,
endometrium, ovary, and liver among women in the United States aged
20-54 and using oral contraceptives was evaluated in a meta-analysis
of 79 relevant studies (Schlesselman, 1995). Women who had used oral
contraceptives for eight years were estimated to have a small net
benefit, with 73 fewer cancer cases (approximately 2%) than women who
had never used these preparations.
In a 25-year follow-up of 46 000 British women, half of whom had
used oral contraceptives in 1968-69 (Beral et al., 1999), the risks
for dying from all cancer were 1.0 (95% CI, 0.8-1.1) for any use and
1.3 (1.0-1.6) for use for > 10 years. The relative risk for death
from all causes combined, adjusted for age, parity, social class, and
smoking, was 1.0 (95% CI, 0.9-1.1) for any use and 1.1 (95% CI,
0.9-1.3) for long-term users. Women who had stopped using oral
contraceptives > 10 years previously had no significant excess or
deficits of deaths from any specific cause or from all causes combined
(Beral et al., 1999).
5. COMMENTS AND EVALUATION
5.1 Estradiol-17ß
The Committee considered data in the published literature from
studies on the bioavailability, metabolism, short-term and long-term
toxicity, reproductive toxicity, geneotoxicity, and carcinogenicity of
exogenous estrogens administered orally. Numerous reports on studies
of the use of exogenous estrogens in women were considered, as were
studies in experimental animals on the mechanisms of action of
estradiol. The extensive database derived from the results of
epidemiological studies of women taking oral contraceptive
preparations containing estrogens or postmenopausal estrogen
replacement therapy was also used to evaluate the safety of estradiol.
Estradiol is an 18-carbon steroid and the most potent of the
natural estrogens. It exerts its biological effects largely by
receptor-mediated mechanisms, as it binds with high affinity and high
specificity to intracellular receptors. Binding of estradiol directly
affects the growth and development of the reproductive tract and
breast and the appearance of secondary sex characteristics. In
non-pregnant females, estradiol acts synergistically with progesterone
during the luteal phase of the menstrual cycle to initiate events
leading to a new cycle. Continued estradiol production is essential
for the normal growth and development of the fetus. In addition to its
effects on reproductive tissues, estradiol is an important metabolic
hormone, particularly because of its effects on the cardiovascular,
skeletal, and gastrointestinal systems.
In general, estradiol is inactive when given orally because it is
inactivated in the gastrointestinal tract and liver, although
fine-particle formulations of estradiol are effective when given
orally and are used therapeutically. The bioavailability of a single
4-mg dose of fine-particle estradiol administered orally to 14 young
women was 5% of that of a dose administered intravenously. At least
60% of the absorbed dose appeared in the serum as estrone and estrone
sulfate and was available as part of the endogenous pool.
Estradiol shows little toxicity when given as a single oral dose.
Few conventional short-and long-term studies of the systemic toxicity
of estradiol in animals treated orally were available, but there is
sufficient information to demonstrate that the adverse effects of
estradiol seen in animals are associated with estrogenic activity.
Because of the specificity and affinity with which estradiol binds to
its receptors, the hormonal effects occur at much lower doses than
other toxicological responses and hence are the most appropriate for
use in evaluating the safety of the compound.
In studies of developmental toxicity in rats, all embryos were
resorbed when estradiol was administered subcutaneously at a dose
equivalent to 25 mg/kg bw on day 10 of gestation. In an interim report
of a multigeneration study of reproductive toxicity in female rats,
estradiol was administered in the feed a a dose of 0.003, 0.17, 0.69,
or 4.1 mg/kg bw per day. No viable pups were observed at the two
higher doses. As the concen tration of progesterone was altered at
various times in the treated groups, a NOEL was not identified in this
study.
The Committee reviewed studies of the genotoxic potential of
estradiol. The compound did not cause gene mutations in vitro. In
some other assays, sporadic but unconfirmed positive results were
obtained. There was more consistent evidence for the induction of
micronuclei in vitro, aneuploidy in vitro, cell transformation
in vitro, oxidative damage to DNA in vivo, and DNA single-strand
breakage in vivo by estradiol. The Committee concluded that
estradiol has genotoxic potential.
A normal biological function of estrogens is to increase the
number of proliferating cells in the endometrium and breast. This
effect is exerted by binding with high affinity to estrogen receptors.
These receptors, of which there are several forms, are found in many
tissues. In cultured human breast cancer cells containing estrogen
receptors, estradiol stimulated growth when added at concentrations of
10-12 mol/L and above, with a maximal response at about 10-10 mol/L.
Estradiol does not stimulate the growth of cultured human breast
cancer cells that do not contain estrogen receptors. At higher
concentrations, estrogens also stimulate cell proliferation in rat
liver in vivo and in vitro. Any factor that increases mitotic
activity reduces the time available for repair of DNA damage before
the next cell division. An agent that causes cell proliferation may
not, however, induce the mutagenic events that are required for
neoplasia. If receptor-mediated stimulation of cell growth is an
important mechanism in the induction of neoplasia by estradiol,
late-stage carcinogenic activity would be expected to predominate.
Experimental studies in rodents in which estradiol was administered in
conjunction with known carcinogens support this mechanism, as do
observations of an increased incidence of cancer among women taking
postmenopausal hormone replacement therapy. In long-term studies of
carcinogenicity in animals, revieiwed at the thirty-second meeting,
oral and parenteral administration of estradiol increased the
incidences of tumours only in hormone-dependent tissues, including the
kidneys of male Syrian hamsters. The Committee concluded that the
carcinogenicity of estradiol is most probably a result of its
interaction with hormonal receptors.
The commonest uses of estrogens in humans are for oral
contraception and postmenopausal hormone replacement therapy. For oral
contraception, the xenobiotic estrogen ethinylestradiol is usually
used. Fine-particle estradiol and conjugated equine estrogen
preparations are commonly used in postmenopausal estrogen replacement
therapy. When healthy postmeno-pausal women were given four courses of
0.3, 0.62, 1.2, or 2.5 mg/day of conjugated equine estrogens for two
weeks followed by no medication for three weeks, the NOEL was 0.3
mg/day on the basis of changes in the serum concentrations of
corticosteroid-binding globulin (CBG). These results indicate that
there is a threshold concentration of estrogen administered orally,
below which there is no increase in serum concentrations of CBG. In a
study involving 23 healthy postmenopausal women receiving various
estrogen preparations orally, 0.3 mg/day of conjugated equine
estrogens or fine-particle estradiol had no effect on the serum
concentrations of follicle-stimulating hormone, angiotensinogen, sex
hormone-binding globulin, or CBG. Thus, 0.3 mg/day, equivalent to 5
µg/kg bw per day, was the NOEL for these hormonal effects of
estradiol. A further indication that this is the NOEL is that 0.3
mg/day of estradiol administered orally to women did not relieve
symptoms of the menopause.
A study of approximately 7700 infants whose mothers had reported
taking oral contraceptives while pregnant with them showed no evidence
that estrogens present a teratogenic hazard.
Because of differences in the pharmacokinetics and
pharmaco-dynamics of natural and xenobiotic estrogen preparations, the
Committee concluded that data on the use of estrogens for
postmenopausal hormone replacement therapy are more appropriate for
evaluating the safety of estradiol than data on their use for oral
contraception. Epidemiological studies on women who took estrogens,
either alone or in combination with progestogens and androgens, showed
that the risks for cancers at most sites were unaffected; the risks
for cancers of the endometrium and breast were increased. In a
meta-analysis of 30 epidemiological studies of women who had ever used
postmenopausal estrogen-only therapy, the relative risk for
endometrial cancer was 2.3 (95% confidence interval, 2.1-2.5). The
relative risk of women who had taken postmenopausal estrogen-only
therapy for more than 10 years was 9.5 (95% confidence interval,
7.4-12). The addition of progestogens to postmenopausal estrogen
therapy reduces the excess risk substantially, althoug it may not be
completely eliminated. In a review of 51 epidemiological studies of
women taking hormonal replacement therapy, the relative risk for
breast cancer was increased by a factor of 1.023 (95% confidence
interval, 1.011-1.036) for each year of use. These estimates of
relative risk are based on the results of studies of women who used
postmenopausal estrogen replacement therapy preparations containing
either conjugated equine estrogens (average dose, 0.625 mg/day) or
estradiol (1-2 mg/day). Overall, the available data suggest that the
increased incidences of cancers of the breast and endometrium observed
among women receiving postmenopausal estrogen replacement therapy is
due to the hormonal effects of estrogens.
See also section 4, 'Epidemiological studies of women exposed to
postmenopausal estrogen therapy and hormonal contraceptives'.
5.2 Progesterone
The Committee considered published data from studies on the
bioavailability, metabolism, short-term toxicity, reproductive
toxicity, genotoxicity, and the long-term toxicity and carcinogenicity
of orally administered progesterone. Numerous reports of studies on
progesterone in humans were considered. In addition, the extensive
database derived from studies of women taking progestogens as a
component of oral contraception, as injectable progestogen-only
contraception, and in postmenopausal hormone replacement therapy was
used to support the safety evaluation.
Progesterone is a 21-carbon steroid that is the only important
natural progestogen. Its normal role is to prepare the uterus for
implantation and to maintain pregnancy. Continued production of
progesterone is necessary to maintain pregnancy. The production of
progesterone by the corpus luteum is controlled by release of
luteinizing hormone from the pituitary gland. In non-pregnant females,
an elevated concentration of progesterone inhibits the cyclic release
of luteinizing hormone, and higher levels inhibit production of
follicle-stimulating hormone. Progesterone opposes some of the effects
of estrogens, but prior stimulation with estrogens is essential for
progesterone to elicit biological responses. Progesterone binds with
high afinity and high specificity to an intracellular receptor
protein; binding of progesterone activates the receptor, resulting in
activation of specific genes.
Less than 10% of progesterone is bioavailable after oral
administration, as it is inactivated in the gastroinetestinal tract
and/or the liver. Progesterone has little toxicity when given as a
single oral dose.
No conventional studies of toxicity in animals treated with
progesterone orally were available, and few other studies were found.
Because progesterone binds specifically and with high affinity to its
receptor, the hormonal effects are the most sensitive toxicological
end-points in these studies. The results of studies of toxicity after
administration by other routes suggest that the effects seen in
animals are associated with hormonal activity. Although equivocal
results have been reported for the induction of single-strand DNA
breaks and DNA adducts have been seen in vitro and in vivo in some
studies, progesterone was not mutagenic. The Committee concluded that,
on balance, progesterone has no genotoxic potential.
Mouse pups given subcutaneous injections of 100 µg of
progesterone on five consecutive days (equivalent to 200 mg/kg bw per
day), beginning 36 h after birth and observed for up to one year had
an increased incidence of mammary gland tumours. Female rabbits given
an average dose of 8 mg/kg bw intramuscularly every second week for
two years developed endometrial cysts, which were sometimes associated
with atypical hyperplasia, but no significant changes were observed in
other tissues.
No multigeneration study of reproductive toxicity with
progesterone was available. Developmental toxicity was not seen in
studies in rats and rhesus monkeys. Rats dosed at 5-25 mg/kg bw per
day on days 14-19 of gestation delivered pups that showed no evidence
of masculinization. Rhesus monkeys given progesterone at 5 mg/kg bw
per day intramuscularly on five days per week beginning after one
month of gestation and continuing to delivery had healthy offspring
with no evident abnormalities. The Committee noted that exogenous
progesterone has been used to maintain pregnancy, with no evidence of
toxicity and with no effect on the normal outcome of pregnancy.
The commonest uses of progesterone by humans are in contraception
and in postmenopausal hormone replacement therapy in which synthetic
progestogens are used either alone or in combination with estrogens.
In a study designed to explore anti-proliferatory and secretory
end-points in the endometrium, women received fine-particle
progesterone at 300 or 600 mg/day (300 mg twice a day) orally for two
weeks after pretreatment with estrogens for 30 days. The group
receiving 300 mg/day showed incomplete conversion of the uterus to
full secretory activity, while the group receiving 600 mg/day showed
full secretory conversion of the uterus with suppression of mitotic
activity. In studies in which women were given 200 or 300 mg of
progesterone orally for one or five years, there was no evidence of
endometrial hyperplasia or carcinoma. Oral administration of a single
dose of 200 mg of fine-particle progesterone (equivalent to 3.3 mg/kg
bw) to women provided concentrations in blood similar to those found
during the luteal phase of the ovulatory cycle. This dose was
considered to be the lowest-observed-effect level (LOEL) in humans.
There is extensive literature on the use of synthetic
progestogens in combination with estrogens for oral contraception.
While synthetic progestogens differ from natural products in their
pharmacokinetics, pharmacodynamics, tissue specificity, and potency, a
prominent feature of the synthetic agents is a protective effect
against the untoward effects of estrogens. No increase in the risk for
cancer at any site was found in women taking only progesterone or
other progestogens orally for contraception. When used in combination
with estrogens in postmenopausal replacement therapy, progesterone
reduced the excess risk for endometrial cancer found with estrogen
alone but did not alter the increased risk for breast cancer.
See also section 4, 'Epidemiological studies of women exposed to
postmenopausal estrogen therapy and hormonal contraceptives'.
5.3 Testosterone
The Committee considered published data from experimental studies
on the bioavailability, metabolism, short-term toxicity, reproductive
toxicity, genotoxicity, and the long-term toxicity and carcinogenicity
of testosterone administered orally. Reports of studies of human use
were also considered.
Testosterone is a 19-carbon steroid which has potent androgenic
properties, including maintenance of testicular function and growth
and differentiation of secondary sex characteristics. It exerts its
biological effects through receptor-mediated mechanisms, as it binds
with high affinity and high specificity to an intracellular receptor
protein, the androgen receptor, which is consequently activated,
resulting in activation of specific genes. In certain target tissues,
testosterone is metabolized to 5 alpha-dihydrotestosterone, which has
greater binding affinity for the androgen receptor.
Androgens have marked anabolic effects which include increased
protein synthesis in muscle and bone. This results in an increased
rate of body growth. In females, androgens have actions in the breast,
uterus, and vagina that are similar to those of progestogens.
Luteinizing hormone and follicle-stimulating hormone from the
pituitary gland control the production of testosterone by the testes.
Testosterone in turn modulates the concentration of
follicle-stimulating hormone and luteinizing hormone, thus controlling
the circulating levels of testosterone through a feedback mechanism.
Little testosterone is bioavailable after oral administration, as
it is inactivated in the gastrointestinal tract and the liver. After
oral administration of 25 mg of testosterone to young women,
approximately 4% of the dose was found to be bioavailable.
Testosterone has very little toxicity when given as a single oral
dose. Few studies have been conducted of the toxicity of testosterone
in animals treated by the oral route. Short-and long-term studies in
animals demonstrate that its adverse effects are due to its hormonal
activity. Therefore, the most sensitive toxicological targets are
hormone-sensitive tissues such as the prostate. Six adult male baboons
received intramuscular injections of testosterone enanthate to provide
a dose equivalent to 8 mg/kg bw each week for up to 28 weeks. At the
end of the study, histological evidence of non-neoplastic alterations
was found in the prostate. Female rabbits given an average dose of
testosterone equivalent to 6 mg/kg bw intramuscularly every second
week for two years developed endometrial cysts and secretions from the
mammary gland. No significant changes were found in tissues from
organs other than those of the reproductive system.
In studies of developmental toxicity, testosterone was
embryotoxic; in rats, a dose of testosterone equivalent to 25 mg/kg bw
administered as subcutaneous implants on day 10 of gestation resulted
in resorption of all embryos. No multigeneration studies of
reproductive toxicity have been conducted with testosterone, since
while normal circulating concentrations are required for normal
reproductive function in males eleveated concentrations of
testosterone interfere with normal reproductive function in both males
and females.
In mammalian cells, no chromosomal aberrations, mutations, or DNA
adducts were found with testosterone alone, and the Committee
concluded that testosterone has no genotoxic potential. No studies to
investigate the carcinogenicity of testosterone in experimental
animals had become available since the previous evaluation. The
Committee re-affirmed that the increased rate of prostatic cancer
detected in rats is consistent with the hormonally mediated effects of
testosterone and its metabolites.
In men, the physiological concentrations of circulating
testosterone are 3-10 ng/ml. In women, the value is less than 1 ng/ml,
and most of the circulating testosterone is derived from the
conversion of dehydroepiandrosterone and androsterone from adrenal and
ovarian sources. Androgens are used therapeutically in men with
deficient testicular function to restore normal testosterone levels.
The effects of excess androgens, particularly in young boys, include
deepened voice, acne, and growth of facial hair, while in women hair
loss and menstrual irregularities may be seen. In a clinical trial
involving five eunuchs, an oral dose of 100 mg/day of a fine-particle
formulation of testosterone had no effect on sexual function, while an
oral dose of 400 mg/day was effective in restoring full sexual
function. Thus, oral administration of 100 mg/day (equivalent to 1.7
mg/kg bw per day) was the NOEL in this study. In studies in which
postmenopausal women received the testosterone analogue
methyltestosterone, alone or in combination with estrogens, a dose of
10 mg/day induced virilizing signs such as acne and hirsutism in a
sizeable proportion of the women. The effects were dose-and
time-dependent. The Committee noted that methyltestosterone is more
potent than testosterone when given orally.
No epidemiological studies of long-term treatment of humans were
available. Therapeutic doses of testosterone given for the treatment
of aplastic anaemia or hypogonadism have resulted in the induction of
liver cysts and hepatomas.
See also section 4, 'Epidemiological studies of women exposed to
postmenopausal estrogen therapy and hormonal contraceptives'.
6 EVALUATION
6.1 Estradiol-17ß
The Committee established an ADI of 0-50 ng/kg bw on the basis of
the NOEL of 0.3 mg/day (equivelent to 5 µg/kg bw per day) in studies
of changes in sev eral hormone-dependent parameters in postmenopausal
women. A safety factor of 10 was used to account for normal variation
among individuals, and an additional factor of 10 was added to protect
sensitive populations.
6.2 Progesterone
The Committee established an ADI of 0-30 µg/kg bw for
progesterone on the basis of the LOEL of 200 mg/day (equivalent to 3.3
mg/kg bw) for changes in the uterus. A safety factor of 100 was used
to allow for extrapolation from a LOEL to a NOEL and to account for
normal variation among individuals.
6.3 Testosterone
The Committee established an ADI of 0-2 µg/kg bw for testosterone
on the basis of the NOEL of 100 mg/day (equivalent to 1.7 mg/kg bw per
day) in the study of eunuchs and a safety factor of 1000. The large
safety factor was used in order to protect sensitive populations and
because of the small number of subjects in the study from which the
NOEL was identified.
7. REFERENCES
Aizu-Yokota, E., Susaki, A. & Sato, Y. (1995) Natural estrogens induce
modulation of microtubules in Chinese hamster V79 cells in culture.
Cancer Res., 55, 1863-1868.
Aldercreutz, H., Gorbach, S.L., Goldin, B.R., Woods, M.N., Dwyer, J.T.
& Hamalainen, E. (1994) Estrogen metabolism and excretion in oriental
Caucasian women. J. Natl Cancer Inst., 86, 1076-1082.
Angsusingha, K., Kenny, F.M., Nankin,H.R. & Taylor, F.H. (1974)
Unconjugated estrone, estradiol and FSH and LH in prepubertal and
pubertal males and females. J. Clin. Endocrinol. Metab., 39,
63-68.
Ashby, J., Fletcher, K., Williams, C., Odum, J. & Tinwell, H. (1997)
Lack of activity of estradiol in rodent bone marrow micronucleus
assays. Mutat. Res., 395, 83-88.
Banerjee, S.K., Banerjee, S., Li, S.A. & Li, J.J. (1994) Induction of
chromosome aberrations in Syrian hamster renal cortical cells by
various estrogens. Mutat. Res., 311, 191-197.
Baulieu, E.E. (1997) Neurosteroids: Of the nervous system, by the
nervous system, for the nervous system. Recent Prog. Horm. Res.,
52, 1-32.
Behl, C., Widmann, M., Trapp, T. & Holsboer, F. (1995) 17ß-Estradiol
protects neurons from oxidative stress-induced cell death. Biochem.
Biophys. Res. Commun., 216, 473-482.
Beral, V., Hernon, C., Kay, C., Hannaford, P., Darby, S. & Reeves, G.
(1999) Mortality associated with oral contraceptive use: 25 year
follow up of cohort or 46000 women from Royal College of General
Practitioners' oral contraception study. Br. Med. J., 318, 96-100.
Bernstein, L. & Ross, R.K. (1993) Endogenous hormones and breast
cancer risk. Epidemiol. Rev., 15, 48-65.
Beyer, B.K., Stark, K.L., Fantel, A.G. & Juchau, M.R. (1989)
Biotransformation, estrogenicity, and steroid structure as
determinants of dysmorphogenic and generalized embryotoxic effects of
steroidal and nonsteroidal estrogens. Toxicol. Appl. Pharmacol.,
98, 113-127.
Bhat, H.K., Hacker, H.J., Bannasch, P., Thompson, E.A. & Liehr, J.G.
(1993) Localization of estrogen receptors in interstitial cells of
hamster kidney and in estradiol-induced renal tumors as evidence of
the mesenchymal origin of this neoplasm. Cancer Res., 53,
5447-5451.
Biegel, L.B., Flaws, J.A., Hirshfield, A.N., O'Connor, J.C., Elliot,
G.S., Ladics, G.S., Silbergold, E.K., Van Pelt, C.S., Hurtt, M.E.,
Cook, J.C. & Frame, S.R. (1998a) 90-day feeding study and
one-generation reproduction study in Crl:CD BR rats with 17
alpha-estradiol. Toxicol. Sci., 44, 116-142.
Biegel, L.B., Cook, J.C., Hurtt, M.E. & O'Connor, J.C. (1998b) Effects
of 17 beta-estradiol on serum hormone concentrations and estrous cycle
in female Crl:CD BR rats: Effects on parental and first generation
rats. Toxicol. Sci., 44, 143-154.
Blum-Degen, D., Haas, M., Pohli, S., Harth, R., Romer, W., Oettel, M.,
Riederer, P. & Gotz, M.E. (1998) Scavestrogens protect IMR 32 cells
form oxidative stress-induced cell death. Toxicol. Appl. Pharmacol.,
152, 49-55.
Bourgain, C., Devroey, P., Van Waesverghe, L., Smitz, J. & van
Steirteghem, A.C. (1990) Effects of natural progesterone on the
morphology of the endometrium in patients with primary ovarian
failure. Hum. Reprod., 5, 537-543.
Braunstein, G.D. (1994) Testes. In: Greenspan, F.S. & Baxter, J.D.,
eds, Basic and Clinical Endocrinology, 4th Ed., Norwalk,
Connecticut, Appleton & Lange, pp. 391-418.
Butler, G.E., Sellar, R.E., Walker, R.F., Kelnar, C.J.H. & Wu, F.C.W.
(1992) Oral testosterone undecanoate in the management of delayed
puberty in boys: Pharmacokinetics and effect on sexual maturation.
J. Clin. Endocrinol. Metab., 75, 37-44.
Butterworth, M., Lau, S.S. & Monks, T.J. (1996) 17ß-Estradiol
metabolism by hamster hepatic microsomes: Implications for the
catechol-O-methyltransferase-mediated detoxication of catechol
estrogens. Drug. Metab. Disposition, 24, 588-594.
Carr, B.R. (1992) Fertilization, implantation, and endocrinology of
pregnancy. In: Griffin, J.E. & Ojeda, S.R., eds, Textbook of
Endocrine Physiology, 2nd Ed., New York, Oxford University Press,
pp.189-209.
Carr, B.R. (1998) Disorders of the ovary and female reproductive
tract. In: Wilson, J.D., Foster, D.W., Kronenberg, H.M. & Larsen,
P.R., eds, Williams Textbook of Endocrinology, 9th Ed.,
Philadelphia, W.B. Saunders Co., pp. 751-817.
Carr, B.R. & Griffin, J.E. (1998) Fertility control and its
implications. In: Wilson, J.D., Foster, D.W., Kronenberg, H.M. &
Larsen, P.R., eds, Williams Textbook of Endocrinology, 9th Ed.,
Philadelphia, W.B. Saunders Co., pp. 901-925.
Cavalieri, E.L., Stack, D.E., Devanesan, P.D., Todorovic, R., Dwivedy,
I., Higginbotham, S., Johansson, S.L., Patil, K.D., Gross, M.L.,
Gooden, J.K., Ramanathan, R., Cerny, R.L. & Rogan, E.G. (1997)
Molecular origin of cancer: Catechol estrogen-3,4-quinones as
endogenous tumor initiators. Proc. Natl Acad. Sci. USA, 94,
10937-10942.
Colditz, G.A. (1998) Relationship between estrogen levels, use of
hormone replacement therapy, and breast cancer. J. Natl Cancer
Inst., 90, 814-823.
Collaborative Group on Hormonal Factors in Breast Cancer (1996) Breast
cancer and hormonal contraceptive reanalysis of individual data on 53
297 women with breast cancer and 100 239 women without breast cancer
from 54 epidemiological studies. Lancet, 347, 1713-1727.
Collaborative Group on Hormonal Factors in Breast Cancer (1997) Breast
cancer and hormone replacement therapy: Collaborative reanalysis of
data from 51 epidemiological studies of 52 705 women with breast
cancer and 108 411 women without breast cancer. Lancet, 350,
1047-1059.
Concas, A., Mostallino, M.C., Porcu, P., Follesa, P., Barbaccia, M.L.,
Trabucchi, M., Purdy, R.H., Grisenti, P. & Biggio, G. (1998) Role of
brain allopregnanolone in the plasticity of gamma-aminobutyric acid
type A receptor in rat brain during pregnancy and after delivery.
Proc. Natl Acad. Sci. USA, 95, 13248-13289.
Coppoc, G.L., Bottoms, G.D., Monk, E., Moore, A.B. & Roesel, O.F.
(1982) Metabolism of estrogens in the gastrointestinal tract of swine.
II. Orally administered gestadiol-17ß-D-glucuronide. J. Anim. Sci.,
55, 135-144.
Couse, J.F., Lindzey, J., Grandien, K., Gustafsson, J.A. & Korach,
K.S. (1997a) Tissue distribution and quantitative analysis of estrogen
receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger
ribonucleic acid in the wild-type ERalpha-knockout mouse.
Endocrinology, 138, 4613-4621.
Couse, J.F., Davis, V.L. & Korach, K.S. (1997b) Physiological findings
from transgenic mouse models with altered levels of estrogen receptor
expression. In: Pavlik, E.J., ed., Estrogens, Progestogens and Their
Antagonists, Boston, Birkhauser, Vol. 2, pp. 69-98.
Cui, L., Mori, T., Takahashi, S., Imaida, K., Akagi, K., Yada, H.,
Yaono, M. & Shirai, T. (1998) Slight promotion effects of intermittent
administration of testosterone propionate and/or diethylstilbestrol on
3,3'-dimethyl-4-aminobiphenyl-initiated rat prostate carcinogenesis.
Cancer Lett., 9, 195-199.
Das, S.K., Talor, J.A., Korach, K.S., Paria, B.C., Dey, S.K. & Lubahn,
D.B. (1997) Estrogenic responses in estrogen receptor-a deficient mice
reveal a distinct estrogen signaling pathway. Proc. Natl Acad. Sci.
USA, 94, 12786-12791.
Dhillon, V.S. & Dhillon, I.K. (1995) Genotoxicity evaluation of
estradiol. Mutat. Res., 345, 87-95.
Dorgan, J.F., Albanes, D., Virtamo, J., Heinonen, O.P., Chandler,
D.W., Galmarini, M., McShane, L.M., Barrett, M.J., Tangrea, J. &
Taylor, P.R. (1998) Relationship of serum androgens and estrogens to
prostate cancer risk: Results from a prospective study in Finland.
Cancer Epidemiol. Biomarkers Prev., 7, 1069-1074.
Eastell, R. (1998) Treatment of postmenopausal osteoporosis.
N. Engl. J. Med., 338, 736-746
Endoh, A., Natsume, H. & Igarashi, Y. (1995) Dual regulation of
21-hydroxylase activity by sex steroid hormones in rat hepatocytes.
J. Steroid Biochem. Mol. Biol., 54, 163-165.
Farthing, M.J.G., Vinson, G.P., Edwards, C.R.W. & Dawson, A.M. (1982)
Testosterone metabolism by the rat gastrointestinal tract, in vitro
and in vivo. Gut, 23, 226-234.
Feser, W., Kerdar, R.S., Blode, H. & Reimann, R. (1996) Formation of
DNA-adducts by selected sex steroids in rat liver. Hum. Exp.
Toxicol., 15, 556-562.
Fisher, D.A. (1998) Endocrinology of fetal development. In: Wilson,
J.D., Foster, D.W., Kronenberg, H.M. & Larsen, P.R., eds, Williams
Textbook of Endocrinology, 9th Ed., Philadelphia, W.B. Saunders Co.,
pp. 1273-1301.
Fisher, C.R., Graves, K.H., Parlow, A.F. & Simpsor, E.R. (1998)
Characterization of mice deficient in aromatase (ArKO) because of
targeted disruption in cyp19 gene. Proc. Natl Acad. Sci. USA, 95,
6965-6970.
Folsom, A.R., Mink, P.J., Sellers, T.A., Hong, C.P., Zheng, W. &
Potter, J.D. (1995) Hormonal replacement therapy and morbidity and
mortality in a prospective study of postmenopausal women. Am. J.
Public Health, 85, 1128-1132.
Foss, G.L. & Camb, M.B. (1939) Clinical administration of androgens.
Lancet, i,502-504.
Fotis, T., Zhang, Y., Pepper, M.S., Aldercreutz, H., Montesano, R.,
Nawroth, P.P. & Schwelgerer, L. (1994) The endogenous estrogen
metabolite 2-methoxyestradiol inhibits angiogenesis and suppresses
tumor growth. Nature, 368, 237-239.
Franceschi, S. & La Vecchia, C. (1998a) Colorectal cancer and hormone
replacement therapy: An unexpected finding. Eur. J. Cancer Prev.,
7, 427-438.
Franceschi, S. & La Vecchia, C. (1998b) Oral contraceptives and
colorectal tumors. Contraception, 58, 335-343.
Franceschi, S., Parazzini, F., Negri, E., Booth, M., La Vecchia, C.,
Beral, V., Tzonou, A. & Trichopoulos, D. (1991) Pooled analysis of 3
Euopean case-control studies of epithelial ovarian cancer. III. Oral
contraceptives use. Int. J. Cancer, 49, 61-65.
Frishman, G.N., Klock, S.C., Luciano, A.A. & Nulsen, J.C. (1995)
Efficacy of oral micronized progesterone in the treatment of luteal
phase defects. J. Reprod. Med., 40, 521-524.
Gangrade, N.K., Boudinot, F.D. & Price, J.C. (1992) Pharmacokinetics
of progesterone in ovariectomized rats after single dose intravenous
administration. Biopharm. Drug Disposition, 13, 703-709.
Gefeller, O., Hassan, K. & Wille, L. (1997) A meta-analysis on the
relationship between oral contraceptives and melanoma: Results and
methodological aspects. J. Epidemiol. Biostat., 2, 225-235.
Gelfand, M.M. & Wiita, B. (1997) Androgen and estrogen-androgen
hormone replacement therapy: A review of the safety literature,
1941-1996. Clin. Ther., 19, 383-404.
Goldfien, A. & Monroe, S.E. (1994) Ovaries. In: Greenspan, F.S. &
Baxter, J.D., eds, Basic and Clinical Endocrinology, 4th Ed.,
Norwalk, Connecticut, Appleton & Lange, pp. 419-470.
Grady, D., Rubin, S.M., Petitti, D.B., Fox, C.S., Black, D., Ettinger,
B., Ernster, V.L. & Cummings, S.R. (1992) Hormone therapy to prevent
disease and prolong life in postmenopausal women. Ann. Intern. Med.,
117, 1016-1037.
Grady, D., Gebretasadik, T., Kerlikowske, K., Ernster, V. & Petitti,
D. (1995) Hormone replacement therapy and endometrial cancer risk: A
meta-analysis. Obstet. Gynecol., 85, 304-313.
Griffin, J.E. & Wilson, J.D. (1998) Disorders of the testes and the
male reproductive tract. In: Wilson, J.D., Foster, D.W., Kronenberg,
H.M. & Larsen, P.R., eds, Williams Textbook of Endocrinology, 9th
Ed., Philadelphia, W.B. Saunders Co., pp. 819-875.
Grodstein, F., Stampfer, M.J., Manson, J.-A.E., Colditz, G.A.,
Willett, W.C., Rosner, B., Speizer, F.E. & Hennekens, C.H. (1996)
Postmenopausal estrogen and progestin use and the risk of
cardiovascular disease. New Engl. J. Med., 335, 453-461.
Grodstein, F., Stampfer, M.J., Colditz, G.A., Willett, W.C., Manson,
J.-A.E., Joffe, M., Rosner, B., Fuchs, C., Hankinson, S.E., Hunter,
D.J., Hennekens, C.H. & Speizer, F.E. (1997) Postmenopausal hormone
therapy and mortality. New Engl. J. Med., 336, 1769-1775.
Grumbach, M.M. & Styne, D.M. (1998) Puberty: Ontogeny,
neuroendocrinology, physiology and disorders. In: Wilson, J.D.,
Foster, D.W., Kronenberg, H.M. & Larsen, P.R., eds, Williams
Textbook of Endocrinology, 9th Ed., Philadelphia, W.B. Saunders
Co., pp. 1509-1625.
Hammond, D.K., Zhu, B.T., Wang, M.Y., Ricci, M.J. & Liehr, J.G. (1997)
Cytochrome P540 metabolism of estradiol in hamster liver and kidney.
Toxicol. Appl. Pharmacol., 145, 54-60.
Han, X. & Liehr, J.G. (1994a) DNA single strand breaks in the kidneys
of Syrian hamsters treated with steroidal estrogens: Hormone-induced
free radical damage preceeding renal malignancy. Carcinogenesis,
15, 997-1000.
Han, X. & Liehr, J.G. (1994b) 8-Hydroxylation of guanine bases in
kidney and liver DNA of hamsters trested with estradiol: Role of free
radicals in estrogen-induced carcinogenesis. Cancer Res., 54,
5515-5517.
Han, X., Liehr, J.G. & Bosland, M.C. (1995) Induction of a DNA adduct
detectable by 32P-postlabeling in the dorsolateral prostate of NBL/Cr
rats treated with estradiol-17b and testosterone. Carcinogenesis,
16, 951-954.
Hargrove, J.T., Maxson, W.S. & Wentz, A.C. (1989) Absorption of oral
progesterone is influenced by vehicle and particle size. Am. J.
Obstet. Gynecol., 161, 948-951.
Harris, R., Whittemore, A.S., Intyre, J. & the Collaborative Ovarian
Cancer Group (1992) Characteristic relating to ovarian cancer risk:
Collaborative analysis of 12 US case-control studies. Am. J.
Epidemiol., 136, 1204-12011.
Hayashi, N., Hasegawa, K., Komine, A., Tanaka, Y., McLachian, J.A.,
Barrett, J.C. & Tsutsui, T. (1996) Estrogen-induced cell
transformation and DNA adduct formation in cultured Syrian hamster
embryo cells. Mol. Carcinog., 16, 149-156.
Helzlsouer, K.J., Huang, H.Y., Strickland, P.T., Hoffman, S., Alberg,
A.J., Comstock, G.W. & Bell, D.A. (1998) Association between CYP17
polymorphisms and the development of breast cancer. Cancer
Epidemiol. Biomarkers Prev., 7, 945-949.
Hemminki, E. & McPherson, K. (1997) Impact of postmenopausal hormone
therapy on cardiovascular events and cancer: Pooled data from clinical
trials. Br. Med. J., 315, 149-153.
Her, C., Szumlanski, C., Aksoy, I.A. & Weinshilboum, R.M. (1996) Human
jejunal estrogen sulfotransferase and dehydroepiandrosterone
sulfotrans-ferase. Drug Metab Disposition, 24, 1328-1335.
Herbert-Croteau, N. (1998) A meta-analysis of hormone replacement
therapy and colon cancer among women. Am. J. Epidemiol., 147, 87.
Highman, B., Norvell, M.J. & Shellenberger, T.E. (1978) Pathological
changes in female C3H mice continuously fed diets containing
diethylstilbestrol or 17beta-estradiol. J. Environ. Pathol.
Toxicol., 1, 1-30.
Highman, B., Greenman, D.L., Norvell, M.J., Farmer, J. &
Shellenberger, T.E. (1980) Neoplastic and preneoplastic lesions
induced in female C3H mice by diets containing diethylstilbestrol or
17beta-estradiol. J. Environ. Pathol. Toxicol., 4, 81-95.
Ho, S.M. & Roy, D. (1994) Sex hormone-induced nuclear DNA damage and
lipid peroxidation in the dorsolateral prostates of Noble rats.
Cancer Lett., 84, 155-162.
Hoogerbrugge, N., Zillikens, M.C., Jansen, H., Meeter, K., Deckers,
J.W. & Birkenhager, J.C. (1998) Estrogen replacement decreases the
level of antibodies against oxidized low-density lipoprotein in
postmenopausal women with coronary heart disease. Metabolism, 47,
675-680.
Hryb, D.J., Khan, M.S., Romas, N.A. & Rosner, W. (1990) The control of
the interaction of sex hormone-binding globulin with its receptor by
steroid hormones. J. Biol. Chem., 265, 6048-6054.
Hsu, M.H., Griffin, K.J., Wang, Y., Kemper, B. & Johnson, E.F. (1993)
A single amino acid substitution confers progesterone 6
beta-hydroxylase activity to rabbit cytochrome P4502C2. J. Biol.
Chem., 268, 6939-6944.
Huang, Z., Guengerich, F.P. & Kaminsky, L.S. (1998)
16-alpha-Hydroxylation of estrone by human cytochrome P4503A4/5.
Carcinogenesis, 19, 867-872.
Hulley, S., Grady, D., Bush, T., Furberg, C., Herrington, D., Riggs,
B. & Vittinghoff, E. for the Heart and Estrogen/Progestin Replacement
Study (HERS) Research Group. (1998) Randomized trial of estrogen plus
progestin for secondary prevention of coronary heart disease in
postmenopausal women. J. Am. Med. Assoc., 280, 605-613.
IARC (1979) IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Humans, Vol. 21, Sex Hormones (II), Lyon,
IARCPress, pp. 35-82, 139-547.
IARC (1987) IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Humans, Suppl. 7, Overall Evaluations of
Carcinogenicity: An Updating of IARC Monographs Volumes 1 to 42,
Lyon, IARCPress, pp. 272-310, 395-400.
IARC (1995) IARC Monographs on the Evaluation of Carcinogenic Risks
to Humans, Vol. 64, Human Papillomaviruses, Lyon, IARCPress.
IARC (1999) IARC Monographs on the Evaluation of Carcinogenic risks
to Humans, Vol. 72, Hormonal Contraception and Postmenopausal
Hormone Therapy Lyon, IARCPress.
Inoh, A., Kamiya, K., Fujii, Y. & Yokoro, K. (1985) Protective effects
of progesterone and tamoxifen in estrogen-induced mammary
carcinogenesis in ovariectomized W/Fu rats. Jpn. J. Cancer Res.,
76, 699-704.
John, E.M., Whittermore, A.S., Harris, R., Intyre, J. & the
Collaborative Ovarian Cancer Group (1993) Characteristics relating to
ovarian cancer risk: Collaborative analysis of seven US case-control
studies. Epithelial ovarian cancer in black women. J. Natl Cancer
Inst., 85, 142-147.
Johnsen, S.G., Bennett, E.P. & Jensen, V.G. (1974) Therapeutic
effectiveness of oral testosterone. Lancet, ii, 1473-1475.
Johnsen, S.V., Kampmann, J.P., Bennett, E.P. & Jorgensen, F.S. (1976)
Enzyme induction by oral testosterone. Clin. Ther., 20, 233-237.
Jones, L.A. & Bern, H.A. (1977) Long-term effects of neonatal
treatment with progesterone alone and in combination with estrogen, on
the mammary gland and reproductive tract of female BALB/cfc3H mice.
Cancer Res., 37, 67-75.
Karr, J.P., Kim, U., Resko, J.A., Schneider, S., Chai, L.S., Murphy,
G.P. & Sandburg, A.A. (1984) Induction of benign prostatic hypertrophy
in baboons. Urology, 3, 276-289.
Kedderis, G.L. & Mugford, C.A. (1998) Sex-dependent metabolism of
xenobiotics. CIIT Activities, 18, 1-7.
Kim, S., Korhonen, M., Wilborn, W., Foldesy, R., Snipes, W., Hodgen,
G.D. & Anderson, F.D. (1996) Antiproliferative effects of low-dose
micronized progesterone. Fertil. Steril., 65, 323-331.
Krege, J.H., Hodgin, J.B., Couse, J.F., Enmark, E., Warner, M.,
Mahler, J.F., Sar, M., Korach, K.S., Gustafsson, J.A. & Smithies, O.
(1998) Generation and reproductive phenotypes of mice lacking estrogen
receptor ß. Proc. Natl Acad. Sci. USA, 95, 15677-15682.
Kuhnz, W., Gansau, C. & Mahler, M. (1993) Pharmacokinetics of
estradiol, free and total estrone, in young women following single
intravenous and oral administration of 17beta-estradiol.
Arzeimittelforschung, 43, 966-973.
Kuiper, G.G.J.M., Lemmen, J.G., Carlsson, B., Corton, J.C., Safe,
S.H., van der Saag, P.T., van der Burg, B. & Gustafsson, J.-A. (1998)
Interaction of estrogenic chemicals and phytoestrogens with estrogen
receptor ß. Endocrinology, 139, 4252-4263.
Lacroix, D., Sonnier, M., Moncion, A., Cheron, G. & Cresteil, T.
(1997) Expression of CYP3A in the human liver--Evidence that the shift
between CYP3A7 and CYP3A4 occurs immediately after birth. Eur. J.
Biochem., 247, 625-634.
Lane, K.E., Ricci, M.J. & Ho, S.M. (1997) Effect of combined
testosterone and estradiol-17ß treatment on the metabolism of E2 in
the prostate and liver of Noble rats. Prostate, 30, 256-262.
Larsen, M.C., Angus, W.G.R., Brake, P.B., Eltom, S.E., Sukow, K.A. &
Jefcoate, C.R. (1998) Characterization of CYP1B1 and CYP1A1 expression
in human mammary epithelial cells: Role of the aryl hydrocarbon
receptor in polycyclic aromatic hydrocarbon metabolism. Cancer Res.,
58, 2366-2374.
Lasne, C., Lu, Y.P., Orfila, L., Ventura, L. & Chouroulinkov, I.
(1990) Study of various transforming effects of the anabolic agents
trenbolone and testosterone on Syrian hamster embryo cells.
Carcinogenesis, 11, 541-547.
La Vecchia, C., Ron, E., Franceschi, S., Dal Maso, L., Mark, S.D.,
Chatenoud, L., Braga, C., Preston-Martin, S., McTiernan, A., Kolonel,
L., Mabuchi, K., Yin, F., Wingren, G., Galanti, M.R., Hallquist, H.,
Lund., E., Levi, F., Linos, D. & Negri, E. (1999) A pooled analysis of
case-control studies of thyroid cancer. III. Oral contraceptives,
hormonal replacement therapy and other female hormones. Cancer
Causes Control, 10, 157-166.
Lavigne, J.A., Helzlsouer, K.J., Huang, H.Y., Strickland, P.T., Bell,
D.A., Selmin, O., Watson, M.A., Hoffman, S., Comstock, G.W. & Yager,
J.D. (1997) An association between the allele coding for a low
activity variant of catechol-O-methyltransferase and the risk of
breast cancer. Cancer Res., 57, 5493-5497.
Leighton, J.K. & Wei, L.L. (1998) Progesterone and development. In:
Dickson, R.B. & Salomon, D.S., eds, Hormones and Growth Factors in
Development and Neoplasia, New York, Wiley-Liss, pp. 177-190.
Lewis, S.J., Heaton, K.W., Oakey, R.E. & McGarrigle, H.H. (1997) Lower
serum oestrogen concentrations associated with faster intestinal
transit. Br. J. Cancer, 76, 395-400.
Lewis, S.J., Oakey, R.E. & Heaton, K.W. (1998) Intestinal absorption
of oestrogen: The effect of altering transit-time. Eur. J.
Gastroenterol. Hepatol., 10, 33-39.
Li, J.J. & Li, S.A. (1989) Estrogen carcinogenesis in Syrian hamster
tissues: Role of metabolism. Fed. Proc., 46, 1858-1863.
Li, J.J., Li, S.A., Klicka, J.K., Parsons, J.A. & Lam, L.K.T. (1983)
Relative carcinogenic activity of various synthetic and natural
estrogens in the Syrian hamster kidney. Cancer Res., 43,
5200-5204.
Li, J.J., Li, S.A., Oberly, T.D. & Parsons, J.A. (1995) Carcinogenic
activities of various steroidal and nonsteroidal estrogens in the
hamster kidney: Relation to hormonal activity and cell proliferation.
Cancer Res., 55, 4347-4351.
Li, S.A., Liao, D.Z.J., Yazlovitskaya, E.M., Pantazis, C.G. & Li, J.J.
(1997) Induction of cathepsin D protein during estrogen
carcinogenesis: Possible role in estrogen-mediated kidney tubular cell
damage. Carcinogenesis, 18, 1375-1380.
Li, J.J., Hou, X., Bentel, J., Yaslovitskaya, E.M. & Li, S.A. (1998)
Prevention of estrogen carcinogenesis in the hamster kidney by ethinyl
estradiol: Some unique properties of a synthetic estrogen.
Carcinogenesis, 19, 471-477.
Liao, D.Z., Pantazis, C.G., Hou, X. & Li, S.A. (1998) Promotion of
estrogen-induced mammary gland carcinogenesis by androgen in the male
Noble rat: Probable mediation by steroid receptors. Carcinogenesis,
19, 2173-2180.
Liehr, J.G. & Ricci, M.J. (1996) 4-Hydroxylation of estrogens as
marker of human mammary tumors. Proc. Natl Acad. Sci. USA, 93,
3294-3296.
Liehr, J.G., Fang, W.F., Sirbasku, D.A. & Ari-Ulubelen, A. (1986)
Carcinogenicity of catecholestrogens in Syrian hamsters. J. Steroid
Biochem., 24, 353-356.
Liehr, J.G., Roy, D. & Gladek, A. (1989) Mechanism of inhibition of
estrogen-induced renal carcinogenesis in male Syrian hamsters by
vitamin C. Carcinogenesis, 10, 1983-1988.
Liehr, J.G., Ricci, M.J., Jefcoate, C.R., Hannigan, E.V., Hokanson,
J.A. & Zhu, B.T. (1995) 4-Hydroxylation of estradiol by human uterine
myometrium and myoma microsomes: Implications for the mechanism of
uterine tumorigenesis. Proc. Natl Acad. Sci. USA, 92, 9220-9224.
de Lignieres, B. (1999) Oral micronized progesterone. Clin. Ther.,
21, 41-60.
Linehan, W.M., Shipley, W.U. & Parkinson, D.R. (1997) Cancer of the
kidney and ureter. In: DeVita, V.T., Jr, Hellman, S. & Rosenberg,
S.A., eds, Cancer: Principles and Practice of Oncology, 5th Ed.,
Philadelphia, Lippincott-Raven, pp. 1271-1300.
Mahendroo, M.S., Cala, K.M., Landrum, D.P. & Russell, D.W. (1997)
Fetal death in mice lacking 5alpha-reductase type I caused by estrogen
excess. Mol. Endocrinol., 11, 917-927.
Mahesh, V.B., Brann, D.W. & Hendry, L.B. (1996) Diverse modes of
action of progesterone and its metabolites. J. Steroid Biochem. Mol.
Biol., 56, 209-219.
Malayer, J.R. & Gorski, J. (1993) An integrated model of estrogen
receptor action. Domest. Anim. Endocrinol., 10, 159-177.
Markides, C.S.A., Roy, D. & Liehr, J.G. (1998) Concentration
dependence of prooxidant and antioxidant properties of
catecholestrogens. Arch. Biochem. Biophys., 360, 105-112.
Martelli, A., Mereto, E., Ghia, M., Orsi, P., Allavena, A., De
Pascalis, C.R. & Brambilla, G. (1998) Induction of micronucli and
enzyme-altered foci in liver of female rats exposed to progesterone
and three synthetic progestins. Mutat. Res., 9, 33-41.
Mashchak, C.A., Lobo, R.A., Dozono-Takano, R., Eggena, P., Nakamura,
R.M., Brenner, P.F. & Mishell, D.R., Jr (1982) Comparison of
pharmaco-dynamic properties of various estrogens. Am. J. Obstet.
Gynecol., 144, 511-518.
Maxson, W.S. & Hargrove, J.T. (1985) Bioavailability of oral
micronized progesterone. Fertil. Steril., 44, 622-626.
McAllister, J.M., Kerin, J.F.P., Trant, J.M., Estabrook, R.W., Mason,
J.I., Waterman, M.R. & Simpson, E.R. (1989) Regulation of cholesterol
side-chain cleavage and 17 alpha-hydroxylase/Lyase activities in
proliferating human theca interna cells in long term monolayer
culture. Endocrinology, 125, 1959-1966.
Meissner, W.A. & Sommers, S.C. (1966) Endometrial changes after
prolonged progesterone and testosterone administration to rabbits.
Cancer Res., 26, 474-478.
Mellon, S.H. & Miller, W.L. (1989) Extraadrenal steroid
21-hydroxylation is not mediated by P450c21. J. Clin. Invest., 84,
1497-1502.
Michnovicz, J.J. & Bradlow, H.L. (1990) Induction of estradiol
metabolism by dietary indole-3-carbinol in humans. J. Natl Cancer
Inst., 82, 947-949.
Michnovicz, J.J., Hershcopf, R.J., Haley, N.J., Bradlow, H.L. &
Fishman, J. (1989) Cigarette smoking alters hepatic estrogen
metabolism in men: Implications for athersclerosis. Metabolism,
38, 537-541.
Millikan, R.C., Pittman, G.S., Tse, C.K., Duell, E., Newman, B.,
Savitz, D., Moorman, P.G., Boissy, R.J. & Bell, D.A. (1998)
Catechol-O-methyltransfe-rase and breast cancer risk.
Carcinogenesis, 19, 1943-1947.
Miyamoto, H., Yeh, S., Lardy, H., Messing, E. & Chang, C. (1998)
Delta5-androstenediol is a natural hormone with androgenic activity in
human prostate cells. Proc. Natl Acad. Sci. USA, 15, 11083-11088.
Moore, D.E., Kawagoe, S., Davajan, V., Nakamura, R.M. & Mishell, D.R.
(1978) An in vivo system in man for quantitation of estrogenicity.
II. Pharmacologic changes in binding capacity of serum
corticosteroid-binding globulin induced by conjugated estrogens,
mestranol, and ethinyl estradiol. Am. J. Obstet. Gynecol., 130,
482-486.
Moore, A.B., Bottoms, G.D., Coppoc, R.C., Pohland, R.C. & Roesel, O.F.
(1982) Metabolism of estrogens in the gastrointestinal tract of swine.
1. Instilled estradiol. J. Anim. Sci., 55, 124-134.
Moss, R.L., Gu, Q. & Wong, M. (1997) Estrogen: Nontranscriptional
signaling pathway. Recent Prog. Horm. Res., 52, 33-69.
Moyer, D.L., de Lingnieres, B., Driguez, P. & Pez, J.P. (1993)
Prevention of endometrial hyperplasia by progesterone during long-term
estradiol replacement: Influence of bleeding pattern and secretory
changes. Fertil. Steril., 59, 992-997.
Musarrat, J., Arezina-Wilson, J. & Wani, J.J. (1996) Prognostic and
aetiolo-gical relevance of 8-hydroxyguanosine in human breast
carcinogenesis. Eur. J. Cancer, 32A, 1209-1214.
Nagashima, M., Tsuda, H., Takenoshita, S., Nagamachi, Y., Hirohashi,
S., Yokota, J. & Kasai, H. (1995) 8-Hydroxydeoxyguanosine levels in
DNA of human breast cancers are not significantly different from those
of non-cancerous breast tissues by the HPLC-ECD method. Cancer
Lett., 90, 157-162.
Nagata, K., Murayama, N., Miyata, M., Shimada, M., Urahashi, A.,
Yamozoe, Y. & Kato, R. (1996) Isolation and characterization of a new
rat P450 (CYP3A18) cDNA encoding P450(6)beta-2 catalyzing testosterone
6 beta-and 16 alpha-hydroxylations. Pharmacogenetics, 6, 103-111.
Nagelberg, S.B., Laue, L., Loriaux, D.L., Liu, L. & Sherins, R.J.
(1986) Cerebrovascular accident associated with testosterone therapy
in a 21-year-old hypogonadal man. New Engl. J. Med., 314, 649-650.
Nahoul, K., Dehennin, L., Jondet, M. & Roger, M. (1993) Profiles of
plasma estrogens, progesterone and their metabolites after oral or
vaginal administration of estradiol or progesterone. Maturitas,
16, 185-202.
Negri, E., Tzonou, A., Beral, V., Lagiou, P., Trichopoulos, D.,
Parazzini, F., Franceschi S., Booth, M. & La Vecchia C. (in press)
Hormonal therapy for menopause and ovarian cancer in a collaborative
re-analysis of European studies. Int. J. Cancer
Niwa, T., Yabusaki, Y., Honma, K., Matsuo, N., Tatsuta, K., Ishibashi,
F. & Katagiri, M. (1998) Contribution of human hepatic cytochrome P450
isoforms to regioselective hydroxylation of steroid hormones.
Xenobiotica, 28, 539-547.
Oberley, T.D., Gonzalez, A., Lauchner, L.J., Oberley, L.W. & Li, J.J.
(1991) Characterization of early kidney lesions in estrogen-induced
tumors of the Syrian hamster. Cancer Res., 51, 1922-1929.
Ofner, P., Bosland, M.C. & Vena, R.L. (1992) Differential effects of
diethylstilbestrol and estradiol-17ß in combination with testosterone
on rat prostate lobes. Toxicol. Appl. Pharmacol., 112, 300-309.
O'Malley, B.W., Tsai, S.Y., Bagchi, M., Weigel, N.L., Schrader, W.T. &
Tsai, M.J. (1991) Molecular mechanisms of action of a steroid hormone
receptor. Recent Prog. Horm. Res., 47, 1-26.
Orth, D.N. & Kovacs, W.J. (1998) The adrenal cortex. In: Wilson, J.D.,
Foster, D.W., Kronenberg, H.M. & Larsen, P.R., eds, Williams
Textbook of Endocrinology, 9th Ed., Philadelphia, W.B. Saunders
Co., pp. 517-664.
Osterling, J., Fuks, Z., Lee, C.T. & Scher, H.I. (1997) Cancer of the
prostate. In: DeVita, V.T., Jr, Hellman, S. & Rosenberg, S.A., eds,
Cancer: Principles and Practice of Oncology, 5th Ed., Philadelphia,
Lippincott-Raven, pp.1322-1386.
Pacifici, G.M., Gucci, A. & Giuliani, L. (1997) Testosterone
sulphation and glucuronidation in the human liver: Interindividual
variability. Eur. J. Drug Metab. Pharmacokinet., 22, 253-258.
Paquette, B. (1996) Enhancement of genomic instability by
17beta-estradiol in minisatellite sequences of X-ray-transformed mouse
10T1/2 cells. Carcinogenesis, 17, 1221-1225.
Paria, B.C., Chakraborty, C. & Dey, S.K. (1990) Catechol estrogen
formation in the mouse uterus and its role in implantation. Mol.
Cell Endocrinol., 69, 25-32.
Parkin, D.M., Pisani, P. & Ferlay, J. (1999) Estimates of the
worldwide incidence of twenty-five major cancers in 1990. Int. J.
Cancer
Petitti, D.B. (1998) Hormone replacement therapy and heart disease
prevention. Experimentation trumps observation. J. Am. Med. Assoc.,
280, 650-651.
Philip, A. & Murphy, B.E. (1986) Relative binding of certain steroids
of low polarity to human sex-hormone binding globulin: Strong binding
of 2-methoxyestrone, a steroid lacking the 17beta-OH group.
Steroids, 47, 373-379.
Physicians' Desk Reference (1999), 53rd Ed., pp. 830-833, 3124-3126.
Pohland, R.C., Coppoc, G.L., Bottoms, G.D. & Moore, A.B. (1982)
Metabolism of estrogens in the gastrointestinal tract of swine. III.
Estradiol-17ß-D-glucuronide instilled into sections of intestine.
J. Anim. Sci., 55, 145-152.
Punnenon, R. & Salmi, T. (1983) Effects of a massive single oral dose
of oestradiol valerate in a young woman. Ann. Clin. Res., 15,
134-136.
Renoir, J.-M., Mercier-Bodard, C. & Baulieu, E.-E. (1980) Hormonal and
immunological aspects of the phylogeny of sex steroid binding plasma
protein. Proc. Natl Acad. Sci. USA, 77, 4578-4582.
Reventos, J., Sullivan, P.M. Josheh, D.R. & Gordon, J.W. (1993)
Tissue-specific expression of the rat androgen-binding protein/sex
hormone-binding globulin gene in transgenic mice. Mol. Cell
Endocrinol., 96, 69-73.
Revesz, C., Chappel, C.I. & Gaudry, R. (1959) Masculinization of
female fetuses in the rat by progestational compounds.
Endocrinology, 66, 140-144.
Rifici, V.A. & Kachadurian, A.K. (1992) The inhibition of low-density
lipoprotein oxidation by 17-ß estradiol. Metabolism, 41,
1110-1114.
Rosenberg, L., Palmer, J.R. & Shapiro, S. (1993) A case-control study
of myocardial infacton in relation to use of estrogen supplements.
Am. J. Epidemiol., 137, 54-63.
Rosenberg, L., Palmer, J.R., Zauber, A.G., Washauer, M.E., Lewis,
J.L., Jr, Strom, B.L., Harlap, S. & Shapiro, S. (1994) A case-control
study of oral contraceptive use and invasive epithelial ovarian
cancer. Am. J. Epidemiol., 139, 654-661.
Rosner, W. (1991) Plasma steroid-binding proteins. Endocrinol.
Metab. Clin. North Am., 20, 697-720.
Ross, R.K., Pike, M.C., Coetzee, G.A., Reichardt, J.K.V., Yu, M.C.,
Feigelson, H., Stanczyk, F.Z., Kolonel, L.N. & Henderson, B.E. (1998)
Androgen metabolism and prostate cancer: Establishing a model of
genetic susceptibility. Cancer Res., 58, 4497-4505.
Rothman, K.J. & Louik, C. (1978) Oral contraceptives and birth
defects. New Engl. J. Med., 210, 522-524.
Roy, D., Weisz, J. & Liehr, J.G. (1990) The O-methylation of
4-hydroxyestradiol is inhibited by 2-hydroxyestradiol: Implications
for estrogen-induced carcinogenesis. Carcinogenesis, 11, 459-462.
Ruoff, W.L. & Dziuk, P.J. (1994) Absorption and metabolism of
estrogens from the stomach and duodenum of pigs. Domest. Anim.
Endocrinol., 11, 197-208.
Russo, I.H. & Russo, J. (1996) Mammary gland neoplasia in long-term
rodent studies. Environ. Health Perspectives, 104, 938-967.
vom Saal, F.S., Timms, B.G., Montano, M.M., Palanza, P., Thayer, K.A.,
Nagel, S.C., Dhar, M.D., Ganjam, V.K., Parmigiani, S. & Welshons, W.V.
(1997) Prostate enlargement in mice due to fetal exposure to low doses
of estradiol or diethylstilbestrol and opposite effects at high doses.
Proc. Natl Acad. Sci. USA, 94, 2056-2061.
Sands, R. & Studd, J. (1995) Exogenous androgens in postmenopausal
women. Am. J. Med., 98 (Suppl. 1A), 76S-79S.
Sarabia, S.F. & Liehr, J.G. (1998) Induction of monoamine oxidase B by
17beta-estradiol in the hamster kidney preceding carcinogenesis.
Arch. Biochem. Biophys., 355, 249-253.
Sarabia, S.F., Zhu, B.T., Kurosawa, T., Tohma, M. & Liehr, J.G. (1997)
Mechanism of P450-catalyzed aromatic hydroxylation of estrogens.
Chem. Res. Toxicol., 10, 767-771.
Sarkar, K., Kinson, G.A. & Rowsell, H.C. (1986) Embryo resorption
following administration of steroidal compounds to rats in
mid-pregnancy. Can. J. Vet. Res., 50, 433-437.
Savas, U., Carstens, C.P. & Jefcoate, C.R. (1997) Biological
oxidations and P450 reactions. Recombinant mouse CYP1B1 expressed in
Escherichia coli exhibits selective binding by polycyclic hydrocarbons
and metabolism which parallels C3H10T1/2 cell microsomes, but differs
from human recombinant CYP1B1. Arch. Biochem. Biophys., 347,
181-192.
Schairer, C., Adami, H.-O., Hoover, R. & Persson, I. (1997)
Cause-specific mortality in women receiving hormone replacement
therapy. Epidemiology, 8, 59-65.
Schleicher, F., Tauber, U., Louton, T. & Schunack, W. (1998) Tissue
distribution of sex steroids: Concentration of 17ß-oestradiol and
cyproterone acetate in selected organs of female Wistar rats.
Pharmacol. Toxicol., 82, 34-39.
Schlesselman, J.J. (1995) Net effect of oral contraceptive use on the
risk of cancer in women in the United States. Obstet. Gynecol.,
85, 793-801.
Schneider, J., Huh, M.M., Bradlow, H.L. & Fishman, J. (1984)
Antiestrogen action of 2-hydroxy estrone on MCF-7 breast cancer cells.
J. Biol. Chem., 259, 4840-4845.
Schuler, M., Hasegawa, L., Parks, R., Metzler, M. & Eastmond, D.A.
(1998) Dose-response studies of the induction of hyperdiploidy and
polyploidy by diethylstilbestrol and 17ß-estradiol in cultured human
lymphocytes using multicolor fluorescense in situ hybridization.
Environ. Mol. Mutag., 31, 263-273.
Schultze, N., Vollmer, G., Wunsche, W., Grote, A., Feit, B. & Knuppen,
R. (1994) Binding of 2-hydroxyestradiol and 4-hydroxyestradiol to the
estrogen receptor of MCF-7 cells in cytosolic extracts and in nuclei
of intact cells. Exp. Clin. Endocrinol., 102, 399-405.
Scott-Moncrieff, J.C., Nelson, R.W., Bill, R.L., Natlock, C.L. &
Bottoms, G.D. (1990) Serum disposition of exogenous progesterone after
intramuscular administration in bitches. Am. J. Vet. Res., 51,
893-895.
Seraj, M.J., Umemoto, A., Tanaka, M., Kajikawa, A., Hamada, K. &
Monden, Y. (1996) DNA adduct formation by hormonal steroids in
vitro. Mutat. Res., 370, 49-59.
Shangold, M.M., Tomai, T.P., Cook, J.D., Jacobs, S.L., Zinaman, M.J.,
Chin, S.Y. & Simon, J.A. (1991) Factors associated with withdrawal
bleeding after administration of oral micronized progesterone in women
with secondary amenorrhea. Fertil. Steril., 56, 1040-1047.
Shelby, M.D., Tice, R.R. & Witt, K.L. (1997) 17b-Estradiol fails to
induce micronucli in the bone marrow cells of rodents. Mutat. Res.,
395, 89-90.
Shou, M., Korzekwa, K., Brooks, E.N., Krausz, K.W., Gonzalez, F.J. &
Gelboin, H.V. (1997) Role of human hepatic cytochrome P450 1A2 and 3A4
in the metabolic activation of estrone. Carcinogenesis, 18,
207-214.
Simon, J.A., Robinson, D.E., Andrews, M.C., Hildebrand, J.R., III,
Rocci, M.L., Jr, Blake, R.E. & Hodgen, G.D. (1993) The absorption of
oral micronized progesterone: The effect of food, dose
proportionality, and comparison with intramuscular progesterone.
Fertil. Steril., 60, 26-33.
Sitruk-Ware, R., Bricaire, C., de Lignieres, B., Yaneva, H. &
Mauvais-Jarvis, P. (1987) Oral micronized progesterone.
Contraception, 36, 373-402.
Smith, S.S., Gong, Q.H., Hsu, F.C., Markowitz, R.S., ffrench-Mullen,
J.M.H. & Li, X. (1998) GABAA receptor alpha 4subunit suppression
prevents withdrawal properties of an endogenous steroid. Nature,
302, 926-930.
Spicer, L.J., Kao, L.-C., Strauss, J.F., III & Hammond, J.M. (1990)
2-Hydroxyestradiol enhanced progesterone production by porcine
granulosa cells: Dependence on de novo cholesterol synthesis and
stimulation of cholesterol side-chain cleavage activity and cytochrome
P450scc messenger ribonucleic acid levels. Endocrinology, 127,
2736-2770.
Spink, D.C., Spink, B.C., Cao, J.Q., Gierthy, J.F., Hays, C.L., Li, Y.
& Sutter, T.R. (1997) Induction of cytochrome P450 1B1 and catechol
estrogen metabolism in ACHN human renal adenocarcinoma cells.
J. Steroid Biochem. Mol. Biol., 62, 223-232.
Stack, D.E., Cavalieri, E.L. & Rogan, E.G. (1998) Catecholestrogens as
procarcinogens: Depurinating adducts and tumor initiation.
Adv. Pharmacol., 42, 833-836.
Stampfer, M.J. & Colditz, G.A. (1991) Estrogen replacement therapy and
coronary heart disease: A quantitative assessment of the epidemiologic
evidence. Prev. Med., 20, 47-63.
Sturgeon, S.R., Schairer, C., Brinton, L.A., Pearson, T. & Hoover,
R.N. (1995) Evidence of a healthy estrogen user survivor effect.
Epidemiology, 6, 227-231.
Swales, N.J., Johnson, T. & Caldwell, J. (1996) Cryopreservation of
rat and mouse hepatocytes. Drug Metab. Disposition, 24, 1224-1230.
Symonds, H.W., Prime, G.R. & Pullar, R.A. (1994) Preliminary evidence
for the enterohepatic circulation of progesterone in the pig. Br.
Vet. J., 150, 585-593.
Tauber, U., Schroder, K., Dusterberg, B. & Matthes, H. (1986) Absolute
bioavailability of testosterone after oral administration of
testosterone-undecanoate and testosterone. Eur. J. Drug Metab.
Pharmacokinet., 11, 145-149.
Thompson, P.A., Shields, P.G., Freudenheim, J.L., Stone, A., Vena,
J.E., Marshall, J.R., Grahm, S., Laughlin, R., Nemoto, T., Kadlubar,
F.F. & Ambrosone, C.B. (1998) Genetic polymorphisms in
catechol-O-methyltransferase, menopausal status and breast cancer
risk. Cancer Res., 58, 2107-2110.
Tsai, M.J., Clark, J.H., Schrader, W.T. & O'Malley, B.W. (1998)
Hormones that act as transcription-regulatory factors. In: Wilson,
J.D., Foster, D.W., Kronenberg, H.M. & Larsen, P.R., eds, Williams
Textbook of Endocrinology, 9th Ed., Philadelphia, W.B. Saunders Co.,
pp. 55-94.
Tsutsui, T., Suzuki, N., Fukuda, S., Sato, M., Maizumi, H., McLachlan,
J.A. & Barrett, J. (1987) 17ß-Estradiol-induced cell transformation
and aneuploidy of Syrian hamster embryo cells in culture.
Carcinogenesis, 11, 1715-1719.
Tsutsui, T., Konine, A., Huff, J. & Barrett, J.C. (1995) Effects of
testosterone, testosterone propionate, 17beta-trenbolone and
progesterone on cell transformation and mutagenesis in Syrian hamster
embryo cells. Carcinogenesis, 16, 1329-1333.
Ursin, G., London, S., Stanczyk, F.Z., Gentzschein, E., Paganini-Hill,
A., Ross, R.K. & Pike, M.C. (1997) A pilot study of urinary estrogen
metabolites (16alpha-OHE1 and 2-OHE1) in postmenopausal women with and
without breast cancer. Environ. Health Perspectives, 105 (Suppl
3), 601-605.
US Congress (1995) Effectiveness and Costs of Osteoporosis Screening
and Hormone Replacement Therapy, Vol. II, Evidence of Benefits,
Risks, and Costs (Publication OTA-BP-H-144), Washington DC, Office
of Technology Assessment.
Wang, M.Y. & Liehr, J.G. (1994) Identification of fatty acid
hydroperoxide cofactors in the cytochrome P450-mediated oxidation of
estrogens to quinone metabolites. J. Biol. Chem., 269, 284-291.
Wehling, M. (1997) Specific, nongenomic actions of steroid hormones.
Annu. Rev. Physiol., 59, 365-393.
Weiss, J., Fritz-Wolz, G., Clawson, G.A., Benedict, C.M., Abendroth,
C. & Creveling, C.R. (1998) Induction of nuclear
catechol-O-methyltransferase by estrogens in hamster kidney:
Implications for estrogen-induced renal cancer. Carcinogenesis,
19, 1307-1312.
Weisz, J., Bui, Q.D., Roy, D. & Leihr, J.G. (1992) Elevated
4-hydroxylation of estradiol by hamster kidney microsomes: A potential
pathway of metabolic activation of estrogens. Endocrinology, 131,
655-661.
Welsch, C.W. (1976) Interaction of estrogen and prolactin in
spontaneous mammary tumorigenesis of the mouse. J. Toxicol. Environ.
Health, Suppl. 1, 161-175.
Wharton, L.R. & Scott, R.B. (1964) Experimental production of genital
lesions with norethindrone. Am. J. Obstet. Gynecol., 89, 701-715.
White, C.M., Ferraro-Borgida, M.J., Fossati, A.T., McGill, C.C.,
Ahlberg, A.W., Feng, Y.J., Heller, G.V. & Chow, M.S. (1998) The
pharmacokinetics of intravenous estradiol-A preliminary study.
Pharmacotherapy, 18, 1343-1346.
Whitehead, M.I., Townsend, P.T., Gill, D.K., Collins, W.P. & Campbell,
S. (1980) Absorption and metabolism of oral progesterone. Br. Med.
J., 280, 825-827.
Whittemore, A.S, Harris, R., Intyre, J. & the Collaborative Ovarian
Cancer Group (1992) Characteristics relating to ovarian cancer risk:
Collaborative analysis of 12 US case-control studies. II. Invasive
epithelial ovarian cancers in white women. Am. J. Epidemiol., 136,
1184-1203.
WHO (1992) Guidelines for the Use of Androgens in Men
(WHO/HPR/MALE/92), Geneva, Special Programme of Research, Development
and Research Training in Human Reproduction.
WHO (1998) Cardiovascular Disease and Steroid Hormone Contraception.
Report of a WHO Scientific Group (WHO Technical Report Series 877),
Geneva.
Wiebe, J.P., Boushy, D. & Wolfe, M. (1997) Synthesis, metabolism and
levels of the neuroactive steroid, 3alpha-hydroxy-4-pregnen-20-one
(3alpha HP), in rat pituitaries. Brain Res., 764, 158-166.
Williams, C.L. & Stancel, G.M. (1996) Estrogens and progestins. In:
Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddon, R.W. & Gilman,
A.G., eds, Goodman and Gilman's The Pharmacological Basis of
Therapeutics, 9th Ed., pp. 1411-1440.
Wilson, J.D. (1996) Androgens. In: Hardman, J.G., Limbird, L.E.,
Molinoff, P.B., Ruddon, R.W. & Gilman, A.G., eds, Goodman and
Gilman's The Pharmacological Basis of Therapeutics, 9th Ed.,
pp.1441-1457.
Wilson, V.S. & LeBlanc, G.A. (1998) Endosulfan elevated testosterone
biotransformation and clearance in CD-1 mice. Toxicol. Appl.
Pharmacol., 148, 158-168.
Wilson, J.D., Foster, D.W., Kronenberg, H. & Larsen, P.R. (1998)
Principles of endocrinology. In: Wilson, J.D., Foster, D.W.,
Kronenberg, H.M. & Larsen, P.R., eds, Williams Textbook of
Endocrinology, 9th Ed., Philadelphia, W.B. Saunders Co., pp. 1-10..
Wingard, L.M., Brody, T.M., Larner, J. & Schwartz, A. (1991)
Estrogens, progestins, and oral contraceptives. In: Human
Pharmacology: Molecular to Clinical, St Louis, Mosby-YearBook, pp.
494-514.
Winter, M.L. & Liehr, J.G. (1996) Possible mechanism of induction of
benign prostatic hyperplasia by estradiol and dihydrotestosterone in
dogs. Toxicol. Appl. Pharmacol., 136, 211-219.
World Cancer Research Fund in association with the American Institute
for Cancer Research (1997) Food, Nutrition and the Prevention of
Cancer: A Global Perspective, Washington DC.
Yager, J.D. & Liehr, J.G. (1996) Molecular mechanisms of estrogen
carcinogenesis. Annu. Rev. Pharmacol. Toxicol., 36, 203-232.
Yamazaki, H., Shaw, P.M., Guengerich, F.P. & Shimada, T. (1998) Roles
of cytochromes P450 1A2 and 3A4 in the oxidation of estradiol and
estrone in human liver microsomes. Chem. Res. Toxicol., 11,
659-665.
Yoshie, Y. & Ohshima, H. (1998). Synergistic induction of DNA strand
breakage by catechol-estrogens and nitrous oxide: Implications for
hormonal carcinogenesis. Free Radicals Biol. Med., 15, 341-348.
Yue, T.L., Wang, X., Louden, C.S., Gupta, S., Pillarisetti, K., Gu,
J.L., Hart, T.K., Lysko, P.G. & Feuerstein, G.Z. (1997)
2-Methoxyestradiol, an endogenous estrogen metabolite, induces
apoptosis in endothelian cells and inhibits angiogenesis: Possible
role for stress-activated protein kinase signaling pathway and Fas
expression. Mol. Pharmacol., 51,951-962.
Zhu, B.T. & Conney, A.H. (1998a) Functional role of estrogen
metabolism in target cells: Review and perspectives. Carcinogenesis,
19, 1-27.
Zhu, B.T. & Conney, A.H. (1998b) Is 2-methoxyestradiol an endogenous
estrogen metabolite that inhibits mammary carcinogenesis? Cancer
Res., 58, 2269-2297.
Zhu, B.T. & Liehr, J.G. (1994) Quercitin increases the severity of
estradiol-induced tumorigenesis in hamster kidney. Toxicol. Appl.
Pharmacol., 125, 149-158.
Zhu, B.T., Roy, D. & Liehr, J.G. (1993) The carcinogenic activity of
ethinyl estrogens is determined by both their hormonal characteristics
and their conversion to catechol metabolites. Endocrinology, 132,
577-583.
Zhu, B.T., Evaristus, E.N., Antoniak, S.K., Sarabia, S.F., Ricci, M.J.
& Liehr, J.G. (1996) Metabolic deglucuronidation and demethylation of
estrogen conjugates as a source of parent estrogens and
catecholestrogen metabolites in Syrian hamster kidney, a target organ
of estrogen-induced tumorignesis. Toxicol. Appl. Pharmacol., 136,
186-193.
Risk (and 95% confidence interval) relative to that of women who have
never used combined oral contraceptives, stratified by study, age at
diagnosis, parity, age at birth of first child, and age at which risk
for conception ceased
4.3.2.3 Cervical cancer
Two cohort and 16 case-control studies of use of combined oral
contraceptives and invasive cervical cancer have been published (IARC,
1999); these consistently show relative risks of approximately 1.5-2
associated with long duration of use. Similar associations were seen
in four studies in which some analyses were restricted to cases and
controls who had human papillomavirus infection, the most important
cause of cervical cancer (IARC, 1995). Biases related to differences
in sexual behaviour, screening practices, and other factors between
users and non-users of oral contraceptives cannot be ruled out as
possible explanations for the observed associations. There is little
evidence that use of depot medroxyprogesterone acetate or other
progestational injectable contraceptives alters the risk for either
squamous-cell carcinoma or adenocarcinoma of the uterine cervix.
4.3.2.4 Ovarian cancer
Four cohort and 21 case-control studies addressed the
relationship between ovarian cancer and use of combined oral
contraceptives (IARC, 1999). Most of the information available on the
topic has been summarized in two pooled analyses (Table 19) of studies
involving 971 cases and 2258 controls in three European countries
(Franceschi et al., 1991) and 2197 cases and 8893 controls in 12
studies in the United States (Whittemore et al., 1992), giving a total
of over 3100 cases and 11 000 controls. In the pooled analysis of
original data from three hospital-based European studies, the relative
risk was 0.6 (95% CI, 0.4-0.8) for any use and 0.4 (0.2-0.7) for
longest (> 5 years) use. Allowance was made in the analysis for age
and other sociodemographic factors, menopausal status, and parity. The
protection persisted for at least 15 years after use ceased. In the
pooled analysis of original data from studies in the United States,
adjustment was made for age, study, and parity, and the corresponding
values were 0.7 (95% CI, 0.6-0.8) for any use and 0.3 (0.2-0.4) for
use for more than six years. Similar results were obtained in
population-based and hospital-based studies considered separately,
with a relative risk of 0.7 in both types of study for any use of
combined oral contraceptives, 0.6 in hospital-based studies and 0.3 in
population-based ones for use for more than six years, and 0.95
(nonsignificant) and 0.90 (p < 0.001), respectively, per additional
year of use.
An inverse association was also observed in a further analysis of
seven studies of 110 black cases and 251 black controls from the
United States pooled analysis, with a relative risk of 0.7 for any use
and 0.6 for longest use (John et al., 1993). In an analysis of data on
327 epithelial ovarian neoplasms of borderline malignancy in white
women, the relative risks were 0.8 (95% CI, 0.6-1) for any use of
combined oral contraceptives and 0.6 (0.4-0.9) for more than five
years of use (Harris et al., 1992).
Table 19. Results of pooled analysis of the association between use of oral contraceptives
and ovarian cancer
Reference, Type of study No. of cases Relative risk (95% CI) Comments
country (age)
Any use Longest use Duration
(years)
Franceschi et al. Three 971 (< 65) 0.6 (0.4-0.8) 0.4 (0.2-0.7) > 5 Protection still present
(1991), Greece, hospital-based > 15 years after use
Italy, United ceased (odds ratio, 0.5)
Kingdom
Whittemore et al. Pooled analysis 2197 (all ages) 0.7 (0.6-0.87) 0.3 (0.2-0.4 > 6 Invasive epithelial
(1992), of 12 neoplasms in white women;
United States population- and protection seen in both
hospital-based types of study
case-control
studies
Harris et al. Same as above 327 0.8 (0.6-1.1) 0.6 (0.4-0.9) > 5 Epithelial tumours of low
(1992), malignant potential in
United States white women
The most convincing aspect of the inverse relationship found
between use of combined oral contraceptives and risk for ovarian
cancer is the consistency of the results, independently of the type of
study, geographical area (Australia, Europe, North America, and
developing countries), or type of analysis, although the covariates
differed from study to study. Likewise, the inverse relationship was
observed for most formulations considered, including low-dose ones
(Rosenberg et al., 1994), even though relatively few data are
available on the more recent formulations.
One case-control study addressed use of progestogen-only oral
contraceptives and one case-control study specifically addressed any
use of depot medroxyprogesterone acetate. Neither showed any
alteration in risk, either overall or in relation to duration of use.
In biological terms, the favourable effect of oral contraceptives
with regard to the risk for ovarian cancer can be interpreted in the
context of the 'incessant ovulation theory'. Thus, any event such as
pregnancy or eaqrly menopause that diminishes the number of ovulations
over a lifetime reduces the risk for ovarian cancer.
4.3.2.5 Cancers of the liver and biliary tract
Three cohort studies showed no significant association between
use of combined oral contraceptives and the incidence of or mortality
from liver cancer, but the expected numbers of cases were very small,
resulting in low statistical power. The association between use of
combined oral contraceptives and liver cancer was evaluated in 11
case-control studies (IARC, 1999). Three European and two North
American studies showed relative risks that were significantly
increased.
The two largest investigations on the topic were carried out in
countries at intermediate to high risk for liver cancer, i.e.
developing countries and southern Europe, where the relative risks for
use of oral contraceptives for two years or more were 0.2 and 0.8,
respectively. Use of oral contraceptives does not therefore enhance
the risk for liver cancer in populations where the prevalence of
hepatitis B and C is high, but they may be a rare cause of cancer in
developed countries where the prevalence of hepatitis is low (IARC,
1999). Positive associations have been reported chiefly with use of
high-dose formulations.
Two case-control studies addressed the association between risk
for liver cancer and use of injectable progestogen-only
contraceptives. In neither study did the risk for liver cancer differ
significantly between women who had ever or never used these
contraceptives. Both studies were conducted in areas endemic for
hepatitis viruses.
4.3.2.6 Colorectal cancer
Four cohort and 10 case-control studies provided information on
use of combined oral contraceptives and the risk for colorectal cancer
(IARC, 1999; Franceschi & La Vecchia, 1998b). None showed
significantly elevated risks in women who used these preparations for
any duration. Relative risks below 1.0 were found in nine studies, and
the risk was significantly reduced in two.
4.3.2.7 Cutaneous malignant melanoma
Four cohort and 18 case-control studies provided information on
use of combined oral contraceptives and the risk for cutaneous
malignant melanoma (IARC, 1999). The relative risks were generally
close to 1.0 and not related to duration of use. A meta-analysis of
published data from 18 case-control studies on melanoma, involving
3796 cases and 9442 controls, yielded a relative risk for any use of
oral contraceptives of 0.95 (95% CI, 0.87-1). Further analyses in
different subgroups defined by personal characteristics and study
design did not materially alter this finding (Gefeller et al., 1997).
One case-control study of cutaneous malignant melanoma showed no
increase in risk among users of progestogen-only contraceptives.
4.3.2.8 Thyroid cancer
Ten case-control studies published information on use of combined
oral contraceptives and risk for cancer of the thyroid gland. In
general there was no elevation in risk associated with oral
contraceptive use Original data from 13 studies from North America,
Asia and Europe were reanalysed (La Vecchia et al., 1999). Based on
2,132 cases of thyroid cancer and 3,301 controls, relative risk for
ever use of oral contraceptives was 1.15 (95% CI, 0.97-1.37). There
was no relationship with duration of use, or age at first use, or use
before first birth. The relative risk was significantly increased for
current oral contraceptive users (RR = 1.45; 95% CI, 1.02-2.07), but
declined with increasing time since stopping.
4.3.2.9 Summary and conclusions
The working group convened by IARC in 1998 (IARC 1999) concluded
that there was sufficient evidence in humans for the carcinogenicity
of combined oral contraceptives. Conversely, the evidence for the
carcino-genicity of progestogen-only contraceptives was deemed
inadequate. It is impossible to infer whether the association between
breast cancer and use of combined oral contraceptives is due to
earlier diagnosis of breast cancer in users, to the biological effects
of contraceptives, or to a combination.
4.3.3 Cardiovascular disease
4.3.3.1 Acute myocardial infarct
Myocardial infarct is uncommon in women of reproductive age (WHO,
1998 and related references). Age, cigarette smoking, diabetes,
hypertension, and raised cholesterol concentration in blood are
important risk factors for myocardial infarct in young women.
After the first reports of a roughly threefold increase in risk
for myocardial infarct in current users of combined oral
contraceptives, three cohort and six case-control studies were
reported which included data collected after 1980. The relative risks
varied widely, from 0.9 to 5 (WHO, 1998), but six of the studies
reported elevated relative risks, three of which were statistically
significant. None of the studies found a relationship between
myocardial infarct and duration of current use. in nine studies that
included data on past use of combined oral contraceptives, there was
no evidence of either an increase or a decrease in the risk for
myocardial infarct among previous users when compared with women who
had never used oral contraceptives.
The available data do not allow an evaluation of the effect of
dose of estrogen on the relative risk for myocardial infarct
independently of the type and dose of progestogen. There are
insufficient data to assess whether the risk of users of low-dose
combined oral contraceptives is modified by the type of progestogen,
and the suggestion that users of low-dose combined oral contraceptives
containing gestodene or desogestrel have a lower risk for myocardial
infarct than users of low-dose formulations containing levonorgestrel
remains to be substantiated.
4.3.3.2 Stroke
Strokes can be divided into two broad categories, infarcts and
haemorrhages, according to the nature of the cerebral lesion. The
relationship between use of combined oral contraceptives and ischaemic
stroke has been demonstrated in a number of epidemiological studies,
from as early as 1968 (WHO, 1998). Two cohort and 11 case-control
studies show a fairly consistent pattern of association, especially if
greater weight is given to larger studies conducted in areas where the
prevalence of use of oral contraceptives among women in the control
group was more than 10%. Most of the studies found that current use of
combined oral contraceptives was associated with an overall increase
in the risk for ischaemic stroke that was approximately 3.4 times that
of non-users. An association with transient ischaemic attacks was also
reported.
None of the studies of the relationship between risk for stroke
and duration of use of contraceptives provided strong evidence for an
association. Four studies found no significantly elevated risk among
past users, and two others found that the relative risk for ischaemic
stroke among former users of combined oral contraceptives was lower
than that among women who had never used them (WHO, 1998). The risk
for ischaemic stroke among women who do not smoke, who have their
blood pressure checked regularly, and who do not have hypertension is
increased by about 1.5-fold with current (albeit not past) use of
low-dose combined oral contraceptives when compared with non-users.
Two recent cohort and four case-control studies examined the risk
for haemorrhagic stroke in current users and found relative risks of
1-2. The incidence of fatal and non-fatal haemorrhagic stroke was not
increased in current users of combined and oral contraceptives who did
not smoke and did not have hypertension, but the incidence of
haemorrhagic stroke increases with age, smoking, and hypertension, and
current use of oral contraceptives appears to magnify these effects.
Use of progestogen-only oral and injectable contraceptives does
not appear to increase the relative risk for all types of stroke.
4.3.3.3 Venous thromboembolism
Thrombosis arises from the interaction of disturbances of the
wall of the blood vessels, of the components of blood, and of the
dynamics of blood flow (stasis). The risk factors for venous
thromboembolism include pregnancy and puerperium, recent surgery,
immobilization, obesity, and various chronic diseases. Venous
thromboembolic disease is the most common cardiovascular event among
users of oral contraceptives, but it is associated with low rates of
mortality and long-term disability. Three cohort and six case-control
studies which included data collected after 1980 found that current
users of combined oral contraceptives had a risk for thromboembolic
disease that was three to four times higher than that of women who
were not using oral contraceptives (WHO, 1998). The risk of current
users is probably highest in the first year of use but falls to that
of non-users within three months of stopping use of oral
contraceptives.
4.3.4 Overall mortality
Since use of oral contraceptives probably increases the risks for
cancers of the breast, cervix uteri, and liver but decreases those for
cancers of the ovary and endometrium, it is important to evaluate the
net effect of use of oral contraceptives. This obviously depends on a
woman's age and the background frequencies of cancers affected by oral
contraceptives at various sites.
The risks for developing cancers of the breast, cervix,
endometrium, ovary, and liver among women in the United States aged
20-54 and using oral contraceptives was evaluated in a meta-analysis
of 79 relevant studies (Schlesselman, 1995). Women who had used oral
contraceptives for eight years were estimated to have a small net
benefit, with 73 fewer cancer cases (approximately 2%) than women who
had never used these preparations.
In a 25-year follow-up of 46 000 British women, half of whom had
used oral contraceptives in 1968-69 (Beral et al., 1999), the risks
for dying from all cancer were 1.0 (95% CI, 0.8-1.1) for any use and
1.3 (1.0-1.6) for use for > 10 years. The relative risk for death
from all causes combined, adjusted for age, parity, social class, and
smoking, was 1.0 (95% CI, 0.9-1.1) for any use and 1.1 (95% CI,
0.9-1.3) for long-term users. Women who had stopped using oral
contraceptives > 10 years previously had no significant excess or
deficits of deaths from any specific cause or from all causes combined
(Beral et al., 1999).
5. COMMENTS AND EVALUATION
5.1 Estradiol-17ß
The Committee considered data in the published literature from
studies on the bioavailability, metabolism, short-term and long-term
toxicity, reproductive toxicity, geneotoxicity, and carcinogenicity of
exogenous estrogens administered orally. Numerous reports on studies
of the use of exogenous estrogens in women were considered, as were
studies in experimental animals on the mechanisms of action of
estradiol. The extensive database derived from the results of
epidemiological studies of women taking oral contraceptive
preparations containing estrogens or postmenopausal estrogen
replacement therapy was also used to evaluate the safety of estradiol.
Estradiol is an 18-carbon steroid and the most potent of the
natural estrogens. It exerts its biological effects largely by
receptor-mediated mechanisms, as it binds with high affinity and high
specificity to intracellular receptors. Binding of estradiol directly
affects the growth and development of the reproductive tract and
breast and the appearance of secondary sex characteristics. In
non-pregnant females, estradiol acts synergistically with progesterone
during the luteal phase of the menstrual cycle to initiate events
leading to a new cycle. Continued estradiol production is essential
for the normal growth and development of the fetus. In addition to its
effects on reproductive tissues, estradiol is an important metabolic
hormone, particularly because of its effects on the cardiovascular,
skeletal, and gastrointestinal systems.
In general, estradiol is inactive when given orally because it is
inactivated in the gastrointestinal tract and liver, although
fine-particle formulations of estradiol are effective when given
orally and are used therapeutically. The bioavailability of a single
4-mg dose of fine-particle estradiol administered orally to 14 young
women was 5% of that of a dose administered intravenously. At least
60% of the absorbed dose appeared in the serum as estrone and estrone
sulfate and was available as part of the endogenous pool.
Estradiol shows little toxicity when given as a single oral dose.
Few conventional short-and long-term studies of the systemic toxicity
of estradiol in animals treated orally were available, but there is
sufficient information to demonstrate that the adverse effects of
estradiol seen in animals are associated with estrogenic activity.
Because of the specificity and affinity with which estradiol binds to
its receptors, the hormonal effects occur at much lower doses than
other toxicological responses and hence are the most appropriate for
use in evaluating the safety of the compound.
In studies of developmental toxicity in rats, all embryos were
resorbed when estradiol was administered subcutaneously at a dose
equivalent to 25 mg/kg bw on day 10 of gestation. In an interim report
of a multigeneration study of reproductive toxicity in female rats,
estradiol was administered in the feed a a dose of 0.003, 0.17, 0.69,
or 4.1 mg/kg bw per day. No viable pups were observed at the two
higher doses. As the concen tration of progesterone was altered at
various times in the treated groups, a NOEL was not identified in this
study.
The Committee reviewed studies of the genotoxic potential of
estradiol. The compound did not cause gene mutations in vitro. In
some other assays, sporadic but unconfirmed positive results were
obtained. There was more consistent evidence for the induction of
micronuclei in vitro, aneuploidy in vitro, cell transformation
in vitro, oxidative damage to DNA in vivo, and DNA single-strand
breakage in vivo by estradiol. The Committee concluded that
estradiol has genotoxic potential.
A normal biological function of estrogens is to increase the
number of proliferating cells in the endometrium and breast. This
effect is exerted by binding with high affinity to estrogen receptors.
These receptors, of which there are several forms, are found in many
tissues. In cultured human breast cancer cells containing estrogen
receptors, estradiol stimulated growth when added at concentrations of
10-12 mol/L and above, with a maximal response at about 10-10 mol/L.
Estradiol does not stimulate the growth of cultured human breast
cancer cells that do not contain estrogen receptors. At higher
concentrations, estrogens also stimulate cell proliferation in rat
liver in vivo and in vitro. Any factor that increases mitotic
activity reduces the time available for repair of DNA damage before
the next cell division. An agent that causes cell proliferation may
not, however, induce the mutagenic events that are required for
neoplasia. If receptor-mediated stimulation of cell growth is an
important mechanism in the induction of neoplasia by estradiol,
late-stage carcinogenic activity would be expected to predominate.
Experimental studies in rodents in which estradiol was administered in
conjunction with known carcinogens support this mechanism, as do
observations of an increased incidence of cancer among women taking
postmenopausal hormone replacement therapy. In long-term studies of
carcinogenicity in animals, revieiwed at the thirty-second meeting,
oral and parenteral administration of estradiol increased the
incidences of tumours only in hormone-dependent tissues, including the
kidneys of male Syrian hamsters. The Committee concluded that the
carcinogenicity of estradiol is most probably a result of its
interaction with hormonal receptors.
The commonest uses of estrogens in humans are for oral
contraception and postmenopausal hormone replacement therapy. For oral
contraception, the xenobiotic estrogen ethinylestradiol is usually
used. Fine-particle estradiol and conjugated equine estrogen
preparations are commonly used in postmenopausal estrogen replacement
therapy. When healthy postmeno-pausal women were given four courses of
0.3, 0.62, 1.2, or 2.5 mg/day of conjugated equine estrogens for two
weeks followed by no medication for three weeks, the NOEL was 0.3
mg/day on the basis of changes in the serum concentrations of
corticosteroid-binding globulin (CBG). These results indicate that
there is a threshold concentration of estrogen administered orally,
below which there is no increase in serum concentrations of CBG. In a
study involving 23 healthy postmenopausal women receiving various
estrogen preparations orally, 0.3 mg/day of conjugated equine
estrogens or fine-particle estradiol had no effect on the serum
concentrations of follicle-stimulating hormone, angiotensinogen, sex
hormone-binding globulin, or CBG. Thus, 0.3 mg/day, equivalent to 5
µg/kg bw per day, was the NOEL for these hormonal effects of
estradiol. A further indication that this is the NOEL is that 0.3
mg/day of estradiol administered orally to women did not relieve
symptoms of the menopause.
A study of approximately 7700 infants whose mothers had reported
taking oral contraceptives while pregnant with them showed no evidence
that estrogens present a teratogenic hazard.
Because of differences in the pharmacokinetics and
pharmaco-dynamics of natural and xenobiotic estrogen preparations, the
Committee concluded that data on the use of estrogens for
postmenopausal hormone replacement therapy are more appropriate for
evaluating the safety of estradiol than data on their use for oral
contraception. Epidemiological studies on women who took estrogens,
either alone or in combination with progestogens and androgens, showed
that the risks for cancers at most sites were unaffected; the risks
for cancers of the endometrium and breast were increased. In a
meta-analysis of 30 epidemiological studies of women who had ever used
postmenopausal estrogen-only therapy, the relative risk for
endometrial cancer was 2.3 (95% confidence interval, 2.1-2.5). The
relative risk of women who had taken postmenopausal estrogen-only
therapy for more than 10 years was 9.5 (95% confidence interval,
7.4-12). The addition of progestogens to postmenopausal estrogen
therapy reduces the excess risk substantially, althoug it may not be
completely eliminated. In a review of 51 epidemiological studies of
women taking hormonal replacement therapy, the relative risk for
breast cancer was increased by a factor of 1.023 (95% confidence
interval, 1.011-1.036) for each year of use. These estimates of
relative risk are based on the results of studies of women who used
postmenopausal estrogen replacement therapy preparations containing
either conjugated equine estrogens (average dose, 0.625 mg/day) or
estradiol (1-2 mg/day). Overall, the available data suggest that the
increased incidences of cancers of the breast and endometrium observed
among women receiving postmenopausal estrogen replacement therapy is
due to the hormonal effects of estrogens.
See also section 4, 'Epidemiological studies of women exposed to
postmenopausal estrogen therapy and hormonal contraceptives'.
5.2 Progesterone
The Committee considered published data from studies on the
bioavailability, metabolism, short-term toxicity, reproductive
toxicity, genotoxicity, and the long-term toxicity and carcinogenicity
of orally administered progesterone. Numerous reports of studies on
progesterone in humans were considered. In addition, the extensive
database derived from studies of women taking progestogens as a
component of oral contraception, as injectable progestogen-only
contraception, and in postmenopausal hormone replacement therapy was
used to support the safety evaluation.
Progesterone is a 21-carbon steroid that is the only important
natural progestogen. Its normal role is to prepare the uterus for
implantation and to maintain pregnancy. Continued production of
progesterone is necessary to maintain pregnancy. The production of
progesterone by the corpus luteum is controlled by release of
luteinizing hormone from the pituitary gland. In non-pregnant females,
an elevated concentration of progesterone inhibits the cyclic release
of luteinizing hormone, and higher levels inhibit production of
follicle-stimulating hormone. Progesterone opposes some of the effects
of estrogens, but prior stimulation with estrogens is essential for
progesterone to elicit biological responses. Progesterone binds with
high afinity and high specificity to an intracellular receptor
protein; binding of progesterone activates the receptor, resulting in
activation of specific genes.
Less than 10% of progesterone is bioavailable after oral
administration, as it is inactivated in the gastroinetestinal tract
and/or the liver. Progesterone has little toxicity when given as a
single oral dose.
No conventional studies of toxicity in animals treated with
progesterone orally were available, and few other studies were found.
Because progesterone binds specifically and with high affinity to its
receptor, the hormonal effects are the most sensitive toxicological
end-points in these studies. The results of studies of toxicity after
administration by other routes suggest that the effects seen in
animals are associated with hormonal activity. Although equivocal
results have been reported for the induction of single-strand DNA
breaks and DNA adducts have been seen in vitro and in vivo in some
studies, progesterone was not mutagenic. The Committee concluded that,
on balance, progesterone has no genotoxic potential.
Mouse pups given subcutaneous injections of 100 µg of
progesterone on five consecutive days (equivalent to 200 mg/kg bw per
day), beginning 36 h after birth and observed for up to one year had
an increased incidence of mammary gland tumours. Female rabbits given
an average dose of 8 mg/kg bw intramuscularly every second week for
two years developed endometrial cysts, which were sometimes associated
with atypical hyperplasia, but no significant changes were observed in
other tissues.
No multigeneration study of reproductive toxicity with
progesterone was available. Developmental toxicity was not seen in
studies in rats and rhesus monkeys. Rats dosed at 5-25 mg/kg bw per
day on days 14-19 of gestation delivered pups that showed no evidence
of masculinization. Rhesus monkeys given progesterone at 5 mg/kg bw
per day intramuscularly on five days per week beginning after one
month of gestation and continuing to delivery had healthy offspring
with no evident abnormalities. The Committee noted that exogenous
progesterone has been used to maintain pregnancy, with no evidence of
toxicity and with no effect on the normal outcome of pregnancy.
The commonest uses of progesterone by humans are in contraception
and in postmenopausal hormone replacement therapy in which synthetic
progestogens are used either alone or in combination with estrogens.
In a study designed to explore anti-proliferatory and secretory
end-points in the endometrium, women received fine-particle
progesterone at 300 or 600 mg/day (300 mg twice a day) orally for two
weeks after pretreatment with estrogens for 30 days. The group
receiving 300 mg/day showed incomplete conversion of the uterus to
full secretory activity, while the group receiving 600 mg/day showed
full secretory conversion of the uterus with suppression of mitotic
activity. In studies in which women were given 200 or 300 mg of
progesterone orally for one or five years, there was no evidence of
endometrial hyperplasia or carcinoma. Oral administration of a single
dose of 200 mg of fine-particle progesterone (equivalent to 3.3 mg/kg
bw) to women provided concentrations in blood similar to those found
during the luteal phase of the ovulatory cycle. This dose was
considered to be the lowest-observed-effect level (LOEL) in humans.
There is extensive literature on the use of synthetic
progestogens in combination with estrogens for oral contraception.
While synthetic progestogens differ from natural products in their
pharmacokinetics, pharmacodynamics, tissue specificity, and potency, a
prominent feature of the synthetic agents is a protective effect
against the untoward effects of estrogens. No increase in the risk for
cancer at any site was found in women taking only progesterone or
other progestogens orally for contraception. When used in combination
with estrogens in postmenopausal replacement therapy, progesterone
reduced the excess risk for endometrial cancer found with estrogen
alone but did not alter the increased risk for breast cancer.
See also section 4, 'Epidemiological studies of women exposed to
postmenopausal estrogen therapy and hormonal contraceptives'.
5.3 Testosterone
The Committee considered published data from experimental studies
on the bioavailability, metabolism, short-term toxicity, reproductive
toxicity, genotoxicity, and the long-term toxicity and carcinogenicity
of testosterone administered orally. Reports of studies of human use
were also considered.
Testosterone is a 19-carbon steroid which has potent androgenic
properties, including maintenance of testicular function and growth
and differentiation of secondary sex characteristics. It exerts its
biological effects through receptor-mediated mechanisms, as it binds
with high affinity and high specificity to an intracellular receptor
protein, the androgen receptor, which is consequently activated,
resulting in activation of specific genes. In certain target tissues,
testosterone is metabolized to 5 alpha-dihydrotestosterone, which has
greater binding affinity for the androgen receptor.
Androgens have marked anabolic effects which include increased
protein synthesis in muscle and bone. This results in an increased
rate of body growth. In females, androgens have actions in the breast,
uterus, and vagina that are similar to those of progestogens.
Luteinizing hormone and follicle-stimulating hormone from the
pituitary gland control the production of testosterone by the testes.
Testosterone in turn modulates the concentration of
follicle-stimulating hormone and luteinizing hormone, thus controlling
the circulating levels of testosterone through a feedback mechanism.
Little testosterone is bioavailable after oral administration, as
it is inactivated in the gastrointestinal tract and the liver. After
oral administration of 25 mg of testosterone to young women,
approximately 4% of the dose was found to be bioavailable.
Testosterone has very little toxicity when given as a single oral
dose. Few studies have been conducted of the toxicity of testosterone
in animals treated by the oral route. Short-and long-term studies in
animals demonstrate that its adverse effects are due to its hormonal
activity. Therefore, the most sensitive toxicological targets are
hormone-sensitive tissues such as the prostate. Six adult male baboons
received intramuscular injections of testosterone enanthate to provide
a dose equivalent to 8 mg/kg bw each week for up to 28 weeks. At the
end of the study, histological evidence of non-neoplastic alterations
was found in the prostate. Female rabbits given an average dose of
testosterone equivalent to 6 mg/kg bw intramuscularly every second
week for two years developed endometrial cysts and secretions from the
mammary gland. No significant changes were found in tissues from
organs other than those of the reproductive system.
In studies of developmental toxicity, testosterone was
embryotoxic; in rats, a dose of testosterone equivalent to 25 mg/kg bw
administered as subcutaneous implants on day 10 of gestation resulted
in resorption of all embryos. No multigeneration studies of
reproductive toxicity have been conducted with testosterone, since
while normal circulating concentrations are required for normal
reproductive function in males eleveated concentrations of
testosterone interfere with normal reproductive function in both males
and females.
In mammalian cells, no chromosomal aberrations, mutations, or DNA
adducts were found with testosterone alone, and the Committee
concluded that testosterone has no genotoxic potential. No studies to
investigate the carcinogenicity of testosterone in experimental
animals had become available since the previous evaluation. The
Committee re-affirmed that the increased rate of prostatic cancer
detected in rats is consistent with the hormonally mediated effects of
testosterone and its metabolites.
In men, the physiological concentrations of circulating
testosterone are 3-10 ng/ml. In women, the value is less than 1 ng/ml,
and most of the circulating testosterone is derived from the
conversion of dehydroepiandrosterone and androsterone from adrenal and
ovarian sources. Androgens are used therapeutically in men with
deficient testicular function to restore normal testosterone levels.
The effects of excess androgens, particularly in young boys, include
deepened voice, acne, and growth of facial hair, while in women hair
loss and menstrual irregularities may be seen. In a clinical trial
involving five eunuchs, an oral dose of 100 mg/day of a fine-particle
formulation of testosterone had no effect on sexual function, while an
oral dose of 400 mg/day was effective in restoring full sexual
function. Thus, oral administration of 100 mg/day (equivalent to 1.7
mg/kg bw per day) was the NOEL in this study. In studies in which
postmenopausal women received the testosterone analogue
methyltestosterone, alone or in combination with estrogens, a dose of
10 mg/day induced virilizing signs such as acne and hirsutism in a
sizeable proportion of the women. The effects were dose-and
time-dependent. The Committee noted that methyltestosterone is more
potent than testosterone when given orally.
No epidemiological studies of long-term treatment of humans were
available. Therapeutic doses of testosterone given for the treatment
of aplastic anaemia or hypogonadism have resulted in the induction of
liver cysts and hepatomas.
See also section 4, 'Epidemiological studies of women exposed to
postmenopausal estrogen therapy and hormonal contraceptives'.
6 EVALUATION
6.1 Estradiol-17ß
The Committee established an ADI of 0-50 ng/kg bw on the basis of
the NOEL of 0.3 mg/day (equivelent to 5 µg/kg bw per day) in studies
of changes in sev eral hormone-dependent parameters in postmenopausal
women. A safety factor of 10 was used to account for normal variation
among individuals, and an additional factor of 10 was added to protect
sensitive populations.
6.2 Progesterone
The Committee established an ADI of 0-30 µg/kg bw for
progesterone on the basis of the LOEL of 200 mg/day (equivalent to 3.3
mg/kg bw) for changes in the uterus. A safety factor of 100 was used
to allow for extrapolation from a LOEL to a NOEL and to account for
normal variation among individuals.
6.3 Testosterone
The Committee established an ADI of 0-2 µg/kg bw for testosterone
on the basis of the NOEL of 100 mg/day (equivalent to 1.7 mg/kg bw per
day) in the study of eunuchs and a safety factor of 1000. The large
safety factor was used in order to protect sensitive populations and
because of the small number of subjects in the study from which the
NOEL was identified.
7. REFERENCES
Aizu-Yokota, E., Susaki, A. & Sato, Y. (1995) Natural estrogens induce
modulation of microtubules in Chinese hamster V79 cells in culture.
Cancer Res., 55, 1863-1868.
Aldercreutz, H., Gorbach, S.L., Goldin, B.R., Woods, M.N., Dwyer, J.T.
& Hamalainen, E. (1994) Estrogen metabolism and excretion in oriental
Caucasian women. J. Natl Cancer Inst., 86, 1076-1082.
Angsusingha, K., Kenny, F.M., Nankin,H.R. & Taylor, F.H. (1974)
Unconjugated estrone, estradiol and FSH and LH in prepubertal and
pubertal males and females. J. Clin. Endocrinol. Metab., 39,
63-68.
Ashby, J., Fletcher, K., Williams, C., Odum, J. & Tinwell, H. (1997)
Lack of activity of estradiol in rodent bone marrow micronucleus
assays. Mutat. Res., 395, 83-88.
Banerjee, S.K., Banerjee, S., Li, S.A. & Li, J.J. (1994) Induction of
chromosome aberrations in Syrian hamster renal cortical cells by
various estrogens. Mutat. Res., 311, 191-197.
Baulieu, E.E. (1997) Neurosteroids: Of the nervous system, by the
nervous system, for the nervous system. Recent Prog. Horm. Res.,
52, 1-32.
Behl, C., Widmann, M., Trapp, T. & Holsboer, F. (1995) 17ß-Estradiol
protects neurons from oxidative stress-induced cell death. Biochem.
Biophys. Res. Commun., 216, 473-482.
Beral, V., Hernon, C., Kay, C., Hannaford, P., Darby, S. & Reeves, G.
(1999) Mortality associated with oral contraceptive use: 25 year
follow up of cohort or 46000 women from Royal College of General
Practitioners' oral contraception study. Br. Med. J., 318, 96-100.
Bernstein, L. & Ross, R.K. (1993) Endogenous hormones and breast
cancer risk. Epidemiol. Rev., 15, 48-65.
Beyer, B.K., Stark, K.L., Fantel, A.G. & Juchau, M.R. (1989)
Biotransformation, estrogenicity, and steroid structure as
determinants of dysmorphogenic and generalized embryotoxic effects of
steroidal and nonsteroidal estrogens. Toxicol. Appl. Pharmacol.,
98, 113-127.
Bhat, H.K., Hacker, H.J., Bannasch, P., Thompson, E.A. & Liehr, J.G.
(1993) Localization of estrogen receptors in interstitial cells of
hamster kidney and in estradiol-induced renal tumors as evidence of
the mesenchymal origin of this neoplasm. Cancer Res., 53,
5447-5451.
Biegel, L.B., Flaws, J.A., Hirshfield, A.N., O'Connor, J.C., Elliot,
G.S., Ladics, G.S., Silbergold, E.K., Van Pelt, C.S., Hurtt, M.E.,
Cook, J.C. & Frame, S.R. (1998a) 90-day feeding study and
one-generation reproduction study in Crl:CD BR rats with 17
alpha-estradiol. Toxicol. Sci., 44, 116-142.
Biegel, L.B., Cook, J.C., Hurtt, M.E. & O'Connor, J.C. (1998b) Effects
of 17 beta-estradiol on serum hormone concentrations and estrous cycle
in female Crl:CD BR rats: Effects on parental and first generation
rats. Toxicol. Sci., 44, 143-154.
Blum-Degen, D., Haas, M., Pohli, S., Harth, R., Romer, W., Oettel, M.,
Riederer, P. & Gotz, M.E. (1998) Scavestrogens protect IMR 32 cells
form oxidative stress-induced cell death. Toxicol. Appl. Pharmacol.,
152, 49-55.
Bourgain, C., Devroey, P., Van Waesverghe, L., Smitz, J. & van
Steirteghem, A.C. (1990) Effects of natural progesterone on the
morphology of the endometrium in patients with primary ovarian
failure. Hum. Reprod., 5, 537-543.
Braunstein, G.D. (1994) Testes. In: Greenspan, F.S. & Baxter, J.D.,
eds, Basic and Clinical Endocrinology, 4th Ed., Norwalk,
Connecticut, Appleton & Lange, pp. 391-418.
Butler, G.E., Sellar, R.E., Walker, R.F., Kelnar, C.J.H. & Wu, F.C.W.
(1992) Oral testosterone undecanoate in the management of delayed
puberty in boys: Pharmacokinetics and effect on sexual maturation.
J. Clin. Endocrinol. Metab., 75, 37-44.
Butterworth, M., Lau, S.S. & Monks, T.J. (1996) 17ß-Estradiol
metabolism by hamster hepatic microsomes: Implications for the
catechol-O-methyltransferase-mediated detoxication of catechol
estrogens. Drug. Metab. Disposition, 24, 588-594.
Carr, B.R. (1992) Fertilization, implantation, and endocrinology of
pregnancy. In: Griffin, J.E. & Ojeda, S.R., eds, Textbook of
Endocrine Physiology, 2nd Ed., New York, Oxford University Press,
pp.189-209.
Carr, B.R. (1998) Disorders of the ovary and female reproductive
tract. In: Wilson, J.D., Foster, D.W., Kronenberg, H.M. & Larsen,
P.R., eds, Williams Textbook of Endocrinology, 9th Ed.,
Philadelphia, W.B. Saunders Co., pp. 751-817.
Carr, B.R. & Griffin, J.E. (1998) Fertility control and its
implications. In: Wilson, J.D., Foster, D.W., Kronenberg, H.M. &
Larsen, P.R., eds, Williams Textbook of Endocrinology, 9th Ed.,
Philadelphia, W.B. Saunders Co., pp. 901-925.
Cavalieri, E.L., Stack, D.E., Devanesan, P.D., Todorovic, R., Dwivedy,
I., Higginbotham, S., Johansson, S.L., Patil, K.D., Gross, M.L.,
Gooden, J.K., Ramanathan, R., Cerny, R.L. & Rogan, E.G. (1997)
Molecular origin of cancer: Catechol estrogen-3,4-quinones as
endogenous tumor initiators. Proc. Natl Acad. Sci. USA, 94,
10937-10942.
Colditz, G.A. (1998) Relationship between estrogen levels, use of
hormone replacement therapy, and breast cancer. J. Natl Cancer
Inst., 90, 814-823.
Collaborative Group on Hormonal Factors in Breast Cancer (1996) Breast
cancer and hormonal contraceptive reanalysis of individual data on 53
297 women with breast cancer and 100 239 women without breast cancer
from 54 epidemiological studies. Lancet, 347, 1713-1727.
Collaborative Group on Hormonal Factors in Breast Cancer (1997) Breast
cancer and hormone replacement therapy: Collaborative reanalysis of
data from 51 epidemiological studies of 52 705 women with breast
cancer and 108 411 women without breast cancer. Lancet, 350,
1047-1059.
Concas, A., Mostallino, M.C., Porcu, P., Follesa, P., Barbaccia, M.L.,
Trabucchi, M., Purdy, R.H., Grisenti, P. & Biggio, G. (1998) Role of
brain allopregnanolone in the plasticity of gamma-aminobutyric acid
type A receptor in rat brain during pregnancy and after delivery.
Proc. Natl Acad. Sci. USA, 95, 13248-13289.
Coppoc, G.L., Bottoms, G.D., Monk, E., Moore, A.B. & Roesel, O.F.
(1982) Metabolism of estrogens in the gastrointestinal tract of swine.
II. Orally administered gestadiol-17ß-D-glucuronide. J. Anim. Sci.,
55, 135-144.
Couse, J.F., Lindzey, J., Grandien, K., Gustafsson, J.A. & Korach,
K.S. (1997a) Tissue distribution and quantitative analysis of estrogen
receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger
ribonucleic acid in the wild-type ERalpha-knockout mouse.
Endocrinology, 138, 4613-4621.
Couse, J.F., Davis, V.L. & Korach, K.S. (1997b) Physiological findings
from transgenic mouse models with altered levels of estrogen receptor
expression. In: Pavlik, E.J., ed., Estrogens, Progestogens and Their
Antagonists, Boston, Birkhauser, Vol. 2, pp. 69-98.
Cui, L., Mori, T., Takahashi, S., Imaida, K., Akagi, K., Yada, H.,
Yaono, M. & Shirai, T. (1998) Slight promotion effects of intermittent
administration of testosterone propionate and/or diethylstilbestrol on
3,3'-dimethyl-4-aminobiphenyl-initiated rat prostate carcinogenesis.
Cancer Lett., 9, 195-199.
Das, S.K., Talor, J.A., Korach, K.S., Paria, B.C., Dey, S.K. & Lubahn,
D.B. (1997) Estrogenic responses in estrogen receptor-a deficient mice
reveal a distinct estrogen signaling pathway. Proc. Natl Acad. Sci.
USA, 94, 12786-12791.
Dhillon, V.S. & Dhillon, I.K. (1995) Genotoxicity evaluation of
estradiol. Mutat. Res., 345, 87-95.
Dorgan, J.F., Albanes, D., Virtamo, J., Heinonen, O.P., Chandler,
D.W., Galmarini, M., McShane, L.M., Barrett, M.J., Tangrea, J. &
Taylor, P.R. (1998) Relationship of serum androgens and estrogens to
prostate cancer risk: Results from a prospective study in Finland.
Cancer Epidemiol. Biomarkers Prev., 7, 1069-1074.
Eastell, R. (1998) Treatment of postmenopausal osteoporosis.
N. Engl. J. Med., 338, 736-746
Endoh, A., Natsume, H. & Igarashi, Y. (1995) Dual regulation of
21-hydroxylase activity by sex steroid hormones in rat hepatocytes.
J. Steroid Biochem. Mol. Biol., 54, 163-165.
Farthing, M.J.G., Vinson, G.P., Edwards, C.R.W. & Dawson, A.M. (1982)
Testosterone metabolism by the rat gastrointestinal tract, in vitro
and in vivo. Gut, 23, 226-234.
Feser, W., Kerdar, R.S., Blode, H. & Reimann, R. (1996) Formation of
DNA-adducts by selected sex steroids in rat liver. Hum. Exp.
Toxicol., 15, 556-562.
Fisher, D.A. (1998) Endocrinology of fetal development. In: Wilson,
J.D., Foster, D.W., Kronenberg, H.M. & Larsen, P.R., eds, Williams
Textbook of Endocrinology, 9th Ed., Philadelphia, W.B. Saunders Co.,
pp. 1273-1301.
Fisher, C.R., Graves, K.H., Parlow, A.F. & Simpsor, E.R. (1998)
Characterization of mice deficient in aromatase (ArKO) because of
targeted disruption in cyp19 gene. Proc. Natl Acad. Sci. USA, 95,
6965-6970.
Folsom, A.R., Mink, P.J., Sellers, T.A., Hong, C.P., Zheng, W. &
Potter, J.D. (1995) Hormonal replacement therapy and morbidity and
mortality in a prospective study of postmenopausal women. Am. J.
Public Health, 85, 1128-1132.
Foss, G.L. & Camb, M.B. (1939) Clinical administration of androgens.
Lancet, i,502-504.
Fotis, T., Zhang, Y., Pepper, M.S., Aldercreutz, H., Montesano, R.,
Nawroth, P.P. & Schwelgerer, L. (1994) The endogenous estrogen
metabolite 2-methoxyestradiol inhibits angiogenesis and suppresses
tumor growth. Nature, 368, 237-239.
Franceschi, S. & La Vecchia, C. (1998a) Colorectal cancer and hormone
replacement therapy: An unexpected finding. Eur. J. Cancer Prev.,
7, 427-438.
Franceschi, S. & La Vecchia, C. (1998b) Oral contraceptives and
colorectal tumors. Contraception, 58, 335-343.
Franceschi, S., Parazzini, F., Negri, E., Booth, M., La Vecchia, C.,
Beral, V., Tzonou, A. & Trichopoulos, D. (1991) Pooled analysis of 3
Euopean case-control studies of epithelial ovarian cancer. III. Oral
contraceptives use. Int. J. Cancer, 49, 61-65.
Frishman, G.N., Klock, S.C., Luciano, A.A. & Nulsen, J.C. (1995)
Efficacy of oral micronized progesterone in the treatment of luteal
phase defects. J. Reprod. Med., 40, 521-524.
Gangrade, N.K., Boudinot, F.D. & Price, J.C. (1992) Pharmacokinetics
of progesterone in ovariectomized rats after single dose intravenous
administration. Biopharm. Drug Disposition, 13, 703-709.
Gefeller, O., Hassan, K. & Wille, L. (1997) A meta-analysis on the
relationship between oral contraceptives and melanoma: Results and
methodological aspects. J. Epidemiol. Biostat., 2, 225-235.
Gelfand, M.M. & Wiita, B. (1997) Androgen and estrogen-androgen
hormone replacement therapy: A review of the safety literature,
1941-1996. Clin. Ther., 19, 383-404.
Goldfien, A. & Monroe, S.E. (1994) Ovaries. In: Greenspan, F.S. &
Baxter, J.D., eds, Basic and Clinical Endocrinology, 4th Ed.,
Norwalk, Connecticut, Appleton & Lange, pp. 419-470.
Grady, D., Rubin, S.M., Petitti, D.B., Fox, C.S., Black, D., Ettinger,
B., Ernster, V.L. & Cummings, S.R. (1992) Hormone therapy to prevent
disease and prolong life in postmenopausal women. Ann. Intern. Med.,
117, 1016-1037.
Grady, D., Gebretasadik, T., Kerlikowske, K., Ernster, V. & Petitti,
D. (1995) Hormone replacement therapy and endometrial cancer risk: A
meta-analysis. Obstet. Gynecol., 85, 304-313.
Griffin, J.E. & Wilson, J.D. (1998) Disorders of the testes and the
male reproductive tract. In: Wilson, J.D., Foster, D.W., Kronenberg,
H.M. & Larsen, P.R., eds, Williams Textbook of Endocrinology, 9th
Ed., Philadelphia, W.B. Saunders Co., pp. 819-875.
Grodstein, F., Stampfer, M.J., Manson, J.-A.E., Colditz, G.A.,
Willett, W.C., Rosner, B., Speizer, F.E. & Hennekens, C.H. (1996)
Postmenopausal estrogen and progestin use and the risk of
cardiovascular disease. New Engl. J. Med., 335, 453-461.
Grodstein, F., Stampfer, M.J., Colditz, G.A., Willett, W.C., Manson,
J.-A.E., Joffe, M., Rosner, B., Fuchs, C., Hankinson, S.E., Hunter,
D.J., Hennekens, C.H. & Speizer, F.E. (1997) Postmenopausal hormone
therapy and mortality. New Engl. J. Med., 336, 1769-1775.
Grumbach, M.M. & Styne, D.M. (1998) Puberty: Ontogeny,
neuroendocrinology, physiology and disorders. In: Wilson, J.D.,
Foster, D.W., Kronenberg, H.M. & Larsen, P.R., eds, Williams
Textbook of Endocrinology, 9th Ed., Philadelphia, W.B. Saunders
Co., pp. 1509-1625.
Hammond, D.K., Zhu, B.T., Wang, M.Y., Ricci, M.J. & Liehr, J.G. (1997)
Cytochrome P540 metabolism of estradiol in hamster liver and kidney.
Toxicol. Appl. Pharmacol., 145, 54-60.
Han, X. & Liehr, J.G. (1994a) DNA single strand breaks in the kidneys
of Syrian hamsters treated with steroidal estrogens: Hormone-induced
free radical damage preceeding renal malignancy. Carcinogenesis,
15, 997-1000.
Han, X. & Liehr, J.G. (1994b) 8-Hydroxylation of guanine bases in
kidney and liver DNA of hamsters trested with estradiol: Role of free
radicals in estrogen-induced carcinogenesis. Cancer Res., 54,
5515-5517.
Han, X., Liehr, J.G. & Bosland, M.C. (1995) Induction of a DNA adduct
detectable by 32P-postlabeling in the dorsolateral prostate of NBL/Cr
rats treated with estradiol-17b and testosterone. Carcinogenesis,
16, 951-954.
Hargrove, J.T., Maxson, W.S. & Wentz, A.C. (1989) Absorption of oral
progesterone is influenced by vehicle and particle size. Am. J.
Obstet. Gynecol., 161, 948-951.
Harris, R., Whittemore, A.S., Intyre, J. & the Collaborative Ovarian
Cancer Group (1992) Characteristic relating to ovarian cancer risk:
Collaborative analysis of 12 US case-control studies. Am. J.
Epidemiol., 136, 1204-12011.
Hayashi, N., Hasegawa, K., Komine, A., Tanaka, Y., McLachian, J.A.,
Barrett, J.C. & Tsutsui, T. (1996) Estrogen-induced cell
transformation and DNA adduct formation in cultured Syrian hamster
embryo cells. Mol. Carcinog., 16, 149-156.
Helzlsouer, K.J., Huang, H.Y., Strickland, P.T., Hoffman, S., Alberg,
A.J., Comstock, G.W. & Bell, D.A. (1998) Association between CYP17
polymorphisms and the development of breast cancer. Cancer
Epidemiol. Biomarkers Prev., 7, 945-949.
Hemminki, E. & McPherson, K. (1997) Impact of postmenopausal hormone
therapy on cardiovascular events and cancer: Pooled data from clinical
trials. Br. Med. J., 315, 149-153.
Her, C., Szumlanski, C., Aksoy, I.A. & Weinshilboum, R.M. (1996) Human
jejunal estrogen sulfotransferase and dehydroepiandrosterone
sulfotrans-ferase. Drug Metab Disposition, 24, 1328-1335.
Herbert-Croteau, N. (1998) A meta-analysis of hormone replacement
therapy and colon cancer among women. Am. J. Epidemiol., 147, 87.
Highman, B., Norvell, M.J. & Shellenberger, T.E. (1978) Pathological
changes in female C3H mice continuously fed diets containing
diethylstilbestrol or 17beta-estradiol. J. Environ. Pathol.
Toxicol., 1, 1-30.
Highman, B., Greenman, D.L., Norvell, M.J., Farmer, J. &
Shellenberger, T.E. (1980) Neoplastic and preneoplastic lesions
induced in female C3H mice by diets containing diethylstilbestrol or
17beta-estradiol. J. Environ. Pathol. Toxicol., 4, 81-95.
Ho, S.M. & Roy, D. (1994) Sex hormone-induced nuclear DNA damage and
lipid peroxidation in the dorsolateral prostates of Noble rats.
Cancer Lett., 84, 155-162.
Hoogerbrugge, N., Zillikens, M.C., Jansen, H., Meeter, K., Deckers,
J.W. & Birkenhager, J.C. (1998) Estrogen replacement decreases the
level of antibodies against oxidized low-density lipoprotein in
postmenopausal women with coronary heart disease. Metabolism, 47,
675-680.
Hryb, D.J., Khan, M.S., Romas, N.A. & Rosner, W. (1990) The control of
the interaction of sex hormone-binding globulin with its receptor by
steroid hormones. J. Biol. Chem., 265, 6048-6054.
Hsu, M.H., Griffin, K.J., Wang, Y., Kemper, B. & Johnson, E.F. (1993)
A single amino acid substitution confers progesterone 6
beta-hydroxylase activity to rabbit cytochrome P4502C2. J. Biol.
Chem., 268, 6939-6944.
Huang, Z., Guengerich, F.P. & Kaminsky, L.S. (1998)
16-alpha-Hydroxylation of estrone by human cytochrome P4503A4/5.
Carcinogenesis, 19, 867-872.
Hulley, S., Grady, D., Bush, T., Furberg, C., Herrington, D., Riggs,
B. & Vittinghoff, E. for the Heart and Estrogen/Progestin Replacement
Study (HERS) Research Group. (1998) Randomized trial of estrogen plus
progestin for secondary prevention of coronary heart disease in
postmenopausal women. J. Am. Med. Assoc., 280, 605-613.
IARC (1979) IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Humans, Vol. 21, Sex Hormones (II), Lyon,
IARCPress, pp. 35-82, 139-547.
IARC (1987) IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Humans, Suppl. 7, Overall Evaluations of
Carcinogenicity: An Updating of IARC Monographs Volumes 1 to 42,
Lyon, IARCPress, pp. 272-310, 395-400.
IARC (1995) IARC Monographs on the Evaluation of Carcinogenic Risks
to Humans, Vol. 64, Human Papillomaviruses, Lyon, IARCPress.
IARC (1999) IARC Monographs on the Evaluation of Carcinogenic risks
to Humans, Vol. 72, Hormonal Contraception and Postmenopausal
Hormone Therapy Lyon, IARCPress.
Inoh, A., Kamiya, K., Fujii, Y. & Yokoro, K. (1985) Protective effects
of progesterone and tamoxifen in estrogen-induced mammary
carcinogenesis in ovariectomized W/Fu rats. Jpn. J. Cancer Res.,
76, 699-704.
John, E.M., Whittermore, A.S., Harris, R., Intyre, J. & the
Collaborative Ovarian Cancer Group (1993) Characteristics relating to
ovarian cancer risk: Collaborative analysis of seven US case-control
studies. Epithelial ovarian cancer in black women. J. Natl Cancer
Inst., 85, 142-147.
Johnsen, S.G., Bennett, E.P. & Jensen, V.G. (1974) Therapeutic
effectiveness of oral testosterone. Lancet, ii, 1473-1475.
Johnsen, S.V., Kampmann, J.P., Bennett, E.P. & Jorgensen, F.S. (1976)
Enzyme induction by oral testosterone. Clin. Ther., 20, 233-237.
Jones, L.A. & Bern, H.A. (1977) Long-term effects of neonatal
treatment with progesterone alone and in combination with estrogen, on
the mammary gland and reproductive tract of female BALB/cfc3H mice.
Cancer Res., 37, 67-75.
Karr, J.P., Kim, U., Resko, J.A., Schneider, S., Chai, L.S., Murphy,
G.P. & Sandburg, A.A. (1984) Induction of benign prostatic hypertrophy
in baboons. Urology, 3, 276-289.
Kedderis, G.L. & Mugford, C.A. (1998) Sex-dependent metabolism of
xenobiotics. CIIT Activities, 18, 1-7.
Kim, S., Korhonen, M., Wilborn, W., Foldesy, R., Snipes, W., Hodgen,
G.D. & Anderson, F.D. (1996) Antiproliferative effects of low-dose
micronized progesterone. Fertil. Steril., 65, 323-331.
Krege, J.H., Hodgin, J.B., Couse, J.F., Enmark, E., Warner, M.,
Mahler, J.F., Sar, M., Korach, K.S., Gustafsson, J.A. & Smithies, O.
(1998) Generation and reproductive phenotypes of mice lacking estrogen
receptor ß. Proc. Natl Acad. Sci. USA, 95, 15677-15682.
Kuhnz, W., Gansau, C. & Mahler, M. (1993) Pharmacokinetics of
estradiol, free and total estrone, in young women following single
intravenous and oral administration of 17beta-estradiol.
Arzeimittelforschung, 43, 966-973.
Kuiper, G.G.J.M., Lemmen, J.G., Carlsson, B., Corton, J.C., Safe,
S.H., van der Saag, P.T., van der Burg, B. & Gustafsson, J.-A. (1998)
Interaction of estrogenic chemicals and phytoestrogens with estrogen
receptor ß. Endocrinology, 139, 4252-4263.
Lacroix, D., Sonnier, M., Moncion, A., Cheron, G. & Cresteil, T.
(1997) Expression of CYP3A in the human liver--Evidence that the shift
between CYP3A7 and CYP3A4 occurs immediately after birth. Eur. J.
Biochem., 247, 625-634.
Lane, K.E., Ricci, M.J. & Ho, S.M. (1997) Effect of combined
testosterone and estradiol-17ß treatment on the metabolism of E2 in
the prostate and liver of Noble rats. Prostate, 30, 256-262.
Larsen, M.C., Angus, W.G.R., Brake, P.B., Eltom, S.E., Sukow, K.A. &
Jefcoate, C.R. (1998) Characterization of CYP1B1 and CYP1A1 expression
in human mammary epithelial cells: Role of the aryl hydrocarbon
receptor in polycyclic aromatic hydrocarbon metabolism. Cancer Res.,
58, 2366-2374.
Lasne, C., Lu, Y.P., Orfila, L., Ventura, L. & Chouroulinkov, I.
(1990) Study of various transforming effects of the anabolic agents
trenbolone and testosterone on Syrian hamster embryo cells.
Carcinogenesis, 11, 541-547.
La Vecchia, C., Ron, E., Franceschi, S., Dal Maso, L., Mark, S.D.,
Chatenoud, L., Braga, C., Preston-Martin, S., McTiernan, A., Kolonel,
L., Mabuchi, K., Yin, F., Wingren, G., Galanti, M.R., Hallquist, H.,
Lund., E., Levi, F., Linos, D. & Negri, E. (1999) A pooled analysis of
case-control studies of thyroid cancer. III. Oral contraceptives,
hormonal replacement therapy and other female hormones. Cancer
Causes Control, 10, 157-166.
Lavigne, J.A., Helzlsouer, K.J., Huang, H.Y., Strickland, P.T., Bell,
D.A., Selmin, O., Watson, M.A., Hoffman, S., Comstock, G.W. & Yager,
J.D. (1997) An association between the allele coding for a low
activity variant of catechol-O-methyltransferase and the risk of
breast cancer. Cancer Res., 57, 5493-5497.
Leighton, J.K. & Wei, L.L. (1998) Progesterone and development. In:
Dickson, R.B. & Salomon, D.S., eds, Hormones and Growth Factors in
Development and Neoplasia, New York, Wiley-Liss, pp. 177-190.
Lewis, S.J., Heaton, K.W., Oakey, R.E. & McGarrigle, H.H. (1997) Lower
serum oestrogen concentrations associated with faster intestinal
transit. Br. J. Cancer, 76, 395-400.
Lewis, S.J., Oakey, R.E. & Heaton, K.W. (1998) Intestinal absorption
of oestrogen: The effect of altering transit-time. Eur. J.
Gastroenterol. Hepatol., 10, 33-39.
Li, J.J. & Li, S.A. (1989) Estrogen carcinogenesis in Syrian hamster
tissues: Role of metabolism. Fed. Proc., 46, 1858-1863.
Li, J.J., Li, S.A., Klicka, J.K., Parsons, J.A. & Lam, L.K.T. (1983)
Relative carcinogenic activity of various synthetic and natural
estrogens in the Syrian hamster kidney. Cancer Res., 43,
5200-5204.
Li, J.J., Li, S.A., Oberly, T.D. & Parsons, J.A. (1995) Carcinogenic
activities of various steroidal and nonsteroidal estrogens in the
hamster kidney: Relation to hormonal activity and cell proliferation.
Cancer Res., 55, 4347-4351.
Li, S.A., Liao, D.Z.J., Yazlovitskaya, E.M., Pantazis, C.G. & Li, J.J.
(1997) Induction of cathepsin D protein during estrogen
carcinogenesis: Possible role in estrogen-mediated kidney tubular cell
damage. Carcinogenesis, 18, 1375-1380.
Li, J.J., Hou, X., Bentel, J., Yaslovitskaya, E.M. & Li, S.A. (1998)
Prevention of estrogen carcinogenesis in the hamster kidney by ethinyl
estradiol: Some unique properties of a synthetic estrogen.
Carcinogenesis, 19, 471-477.
Liao, D.Z., Pantazis, C.G., Hou, X. & Li, S.A. (1998) Promotion of
estrogen-induced mammary gland carcinogenesis by androgen in the male
Noble rat: Probable mediation by steroid receptors. Carcinogenesis,
19, 2173-2180.
Liehr, J.G. & Ricci, M.J. (1996) 4-Hydroxylation of estrogens as
marker of human mammary tumors. Proc. Natl Acad. Sci. USA, 93,
3294-3296.
Liehr, J.G., Fang, W.F., Sirbasku, D.A. & Ari-Ulubelen, A. (1986)
Carcinogenicity of catecholestrogens in Syrian hamsters. J. Steroid
Biochem., 24, 353-356.
Liehr, J.G., Roy, D. & Gladek, A. (1989) Mechanism of inhibition of
estrogen-induced renal carcinogenesis in male Syrian hamsters by
vitamin C. Carcinogenesis, 10, 1983-1988.
Liehr, J.G., Ricci, M.J., Jefcoate, C.R., Hannigan, E.V., Hokanson,
J.A. & Zhu, B.T. (1995) 4-Hydroxylation of estradiol by human uterine
myometrium and myoma microsomes: Implications for the mechanism of
uterine tumorigenesis. Proc. Natl Acad. Sci. USA, 92, 9220-9224.
de Lignieres, B. (1999) Oral micronized progesterone. Clin. Ther.,
21, 41-60.
Linehan, W.M., Shipley, W.U. & Parkinson, D.R. (1997) Cancer of the
kidney and ureter. In: DeVita, V.T., Jr, Hellman, S. & Rosenberg,
S.A., eds, Cancer: Principles and Practice of Oncology, 5th Ed.,
Philadelphia, Lippincott-Raven, pp. 1271-1300.
Mahendroo, M.S., Cala, K.M., Landrum, D.P. & Russell, D.W. (1997)
Fetal death in mice lacking 5alpha-reductase type I caused by estrogen
excess. Mol. Endocrinol., 11, 917-927.
Mahesh, V.B., Brann, D.W. & Hendry, L.B. (1996) Diverse modes of
action of progesterone and its metabolites. J. Steroid Biochem. Mol.
Biol., 56, 209-219.
Malayer, J.R. & Gorski, J. (1993) An integrated model of estrogen
receptor action. Domest. Anim. Endocrinol., 10, 159-177.
Markides, C.S.A., Roy, D. & Liehr, J.G. (1998) Concentration
dependence of prooxidant and antioxidant properties of
catecholestrogens. Arch. Biochem. Biophys., 360, 105-112.
Martelli, A., Mereto, E., Ghia, M., Orsi, P., Allavena, A., De
Pascalis, C.R. & Brambilla, G. (1998) Induction of micronucli and
enzyme-altered foci in liver of female rats exposed to progesterone
and three synthetic progestins. Mutat. Res., 9, 33-41.
Mashchak, C.A., Lobo, R.A., Dozono-Takano, R., Eggena, P., Nakamura,
R.M., Brenner, P.F. & Mishell, D.R., Jr (1982) Comparison of
pharmaco-dynamic properties of various estrogens. Am. J. Obstet.
Gynecol., 144, 511-518.
Maxson, W.S. & Hargrove, J.T. (1985) Bioavailability of oral
micronized progesterone. Fertil. Steril., 44, 622-626.
McAllister, J.M., Kerin, J.F.P., Trant, J.M., Estabrook, R.W., Mason,
J.I., Waterman, M.R. & Simpson, E.R. (1989) Regulation of cholesterol
side-chain cleavage and 17 alpha-hydroxylase/Lyase activities in
proliferating human theca interna cells in long term monolayer
culture. Endocrinology, 125, 1959-1966.
Meissner, W.A. & Sommers, S.C. (1966) Endometrial changes after
prolonged progesterone and testosterone administration to rabbits.
Cancer Res., 26, 474-478.
Mellon, S.H. & Miller, W.L. (1989) Extraadrenal steroid
21-hydroxylation is not mediated by P450c21. J. Clin. Invest., 84,
1497-1502.
Michnovicz, J.J. & Bradlow, H.L. (1990) Induction of estradiol
metabolism by dietary indole-3-carbinol in humans. J. Natl Cancer
Inst., 82, 947-949.
Michnovicz, J.J., Hershcopf, R.J., Haley, N.J., Bradlow, H.L. &
Fishman, J. (1989) Cigarette smoking alters hepatic estrogen
metabolism in men: Implications for athersclerosis. Metabolism,
38, 537-541.
Millikan, R.C., Pittman, G.S., Tse, C.K., Duell, E., Newman, B.,
Savitz, D., Moorman, P.G., Boissy, R.J. & Bell, D.A. (1998)
Catechol-O-methyltransfe-rase and breast cancer risk.
Carcinogenesis, 19, 1943-1947.
Miyamoto, H., Yeh, S., Lardy, H., Messing, E. & Chang, C. (1998)
Delta5-androstenediol is a natural hormone with androgenic activity in
human prostate cells. Proc. Natl Acad. Sci. USA, 15, 11083-11088.
Moore, D.E., Kawagoe, S., Davajan, V., Nakamura, R.M. & Mishell, D.R.
(1978) An in vivo system in man for quantitation of estrogenicity.
II. Pharmacologic changes in binding capacity of serum
corticosteroid-binding globulin induced by conjugated estrogens,
mestranol, and ethinyl estradiol. Am. J. Obstet. Gynecol., 130,
482-486.
Moore, A.B., Bottoms, G.D., Coppoc, R.C., Pohland, R.C. & Roesel, O.F.
(1982) Metabolism of estrogens in the gastrointestinal tract of swine.
1. Instilled estradiol. J. Anim. Sci., 55, 124-134.
Moss, R.L., Gu, Q. & Wong, M. (1997) Estrogen: Nontranscriptional
signaling pathway. Recent Prog. Horm. Res., 52, 33-69.
Moyer, D.L., de Lingnieres, B., Driguez, P. & Pez, J.P. (1993)
Prevention of endometrial hyperplasia by progesterone during long-term
estradiol replacement: Influence of bleeding pattern and secretory
changes. Fertil. Steril., 59, 992-997.
Musarrat, J., Arezina-Wilson, J. & Wani, J.J. (1996) Prognostic and
aetiolo-gical relevance of 8-hydroxyguanosine in human breast
carcinogenesis. Eur. J. Cancer, 32A, 1209-1214.
Nagashima, M., Tsuda, H., Takenoshita, S., Nagamachi, Y., Hirohashi,
S., Yokota, J. & Kasai, H. (1995) 8-Hydroxydeoxyguanosine levels in
DNA of human breast cancers are not significantly different from those
of non-cancerous breast tissues by the HPLC-ECD method. Cancer
Lett., 90, 157-162.
Nagata, K., Murayama, N., Miyata, M., Shimada, M., Urahashi, A.,
Yamozoe, Y. & Kato, R. (1996) Isolation and characterization of a new
rat P450 (CYP3A18) cDNA encoding P450(6)beta-2 catalyzing testosterone
6 beta-and 16 alpha-hydroxylations. Pharmacogenetics, 6, 103-111.
Nagelberg, S.B., Laue, L., Loriaux, D.L., Liu, L. & Sherins, R.J.
(1986) Cerebrovascular accident associated with testosterone therapy
in a 21-year-old hypogonadal man. New Engl. J. Med., 314, 649-650.
Nahoul, K., Dehennin, L., Jondet, M. & Roger, M. (1993) Profiles of
plasma estrogens, progesterone and their metabolites after oral or
vaginal administration of estradiol or progesterone. Maturitas,
16, 185-202.
Negri, E., Tzonou, A., Beral, V., Lagiou, P., Trichopoulos, D.,
Parazzini, F., Franceschi S., Booth, M. & La Vecchia C. (in press)
Hormonal therapy for menopause and ovarian cancer in a collaborative
re-analysis of European studies. Int. J. Cancer
Niwa, T., Yabusaki, Y., Honma, K., Matsuo, N., Tatsuta, K., Ishibashi,
F. & Katagiri, M. (1998) Contribution of human hepatic cytochrome P450
isoforms to regioselective hydroxylation of steroid hormones.
Xenobiotica, 28, 539-547.
Oberley, T.D., Gonzalez, A., Lauchner, L.J., Oberley, L.W. & Li, J.J.
(1991) Characterization of early kidney lesions in estrogen-induced
tumors of the Syrian hamster. Cancer Res., 51, 1922-1929.
Ofner, P., Bosland, M.C. & Vena, R.L. (1992) Differential effects of
diethylstilbestrol and estradiol-17ß in combination with testosterone
on rat prostate lobes. Toxicol. Appl. Pharmacol., 112, 300-309.
O'Malley, B.W., Tsai, S.Y., Bagchi, M., Weigel, N.L., Schrader, W.T. &
Tsai, M.J. (1991) Molecular mechanisms of action of a steroid hormone
receptor. Recent Prog. Horm. Res., 47, 1-26.
Orth, D.N. & Kovacs, W.J. (1998) The adrenal cortex. In: Wilson, J.D.,
Foster, D.W., Kronenberg, H.M. & Larsen, P.R., eds, Williams
Textbook of Endocrinology, 9th Ed., Philadelphia, W.B. Saunders
Co., pp. 517-664.
Osterling, J., Fuks, Z., Lee, C.T. & Scher, H.I. (1997) Cancer of the
prostate. In: DeVita, V.T., Jr, Hellman, S. & Rosenberg, S.A., eds,
Cancer: Principles and Practice of Oncology, 5th Ed., Philadelphia,
Lippincott-Raven, pp.1322-1386.
Pacifici, G.M., Gucci, A. & Giuliani, L. (1997) Testosterone
sulphation and glucuronidation in the human liver: Interindividual
variability. Eur. J. Drug Metab. Pharmacokinet., 22, 253-258.
Paquette, B. (1996) Enhancement of genomic instability by
17beta-estradiol in minisatellite sequences of X-ray-transformed mouse
10T1/2 cells. Carcinogenesis, 17, 1221-1225.
Paria, B.C., Chakraborty, C. & Dey, S.K. (1990) Catechol estrogen
formation in the mouse uterus and its role in implantation. Mol.
Cell Endocrinol., 69, 25-32.
Parkin, D.M., Pisani, P. & Ferlay, J. (1999) Estimates of the
worldwide incidence of twenty-five major cancers in 1990. Int. J.
Cancer
Petitti, D.B. (1998) Hormone replacement therapy and heart disease
prevention. Experimentation trumps observation. J. Am. Med. Assoc.,
280, 650-651.
Philip, A. & Murphy, B.E. (1986) Relative binding of certain steroids
of low polarity to human sex-hormone binding globulin: Strong binding
of 2-methoxyestrone, a steroid lacking the 17beta-OH group.
Steroids, 47, 373-379.
Physicians' Desk Reference (1999), 53rd Ed., pp. 830-833, 3124-3126.
Pohland, R.C., Coppoc, G.L., Bottoms, G.D. & Moore, A.B. (1982)
Metabolism of estrogens in the gastrointestinal tract of swine. III.
Estradiol-17ß-D-glucuronide instilled into sections of intestine.
J. Anim. Sci., 55, 145-152.
Punnenon, R. & Salmi, T. (1983) Effects of a massive single oral dose
of oestradiol valerate in a young woman. Ann. Clin. Res., 15,
134-136.
Renoir, J.-M., Mercier-Bodard, C. & Baulieu, E.-E. (1980) Hormonal and
immunological aspects of the phylogeny of sex steroid binding plasma
protein. Proc. Natl Acad. Sci. USA, 77, 4578-4582.
Reventos, J., Sullivan, P.M. Josheh, D.R. & Gordon, J.W. (1993)
Tissue-specific expression of the rat androgen-binding protein/sex
hormone-binding globulin gene in transgenic mice. Mol. Cell
Endocrinol., 96, 69-73.
Revesz, C., Chappel, C.I. & Gaudry, R. (1959) Masculinization of
female fetuses in the rat by progestational compounds.
Endocrinology, 66, 140-144.
Rifici, V.A. & Kachadurian, A.K. (1992) The inhibition of low-density
lipoprotein oxidation by 17-ß estradiol. Metabolism, 41,
1110-1114.
Rosenberg, L., Palmer, J.R. & Shapiro, S. (1993) A case-control study
of myocardial infacton in relation to use of estrogen supplements.
Am. J. Epidemiol., 137, 54-63.
Rosenberg, L., Palmer, J.R., Zauber, A.G., Washauer, M.E., Lewis,
J.L., Jr, Strom, B.L., Harlap, S. & Shapiro, S. (1994) A case-control
study of oral contraceptive use and invasive epithelial ovarian
cancer. Am. J. Epidemiol., 139, 654-661.
Rosner, W. (1991) Plasma steroid-binding proteins. Endocrinol.
Metab. Clin. North Am., 20, 697-720.
Ross, R.K., Pike, M.C., Coetzee, G.A., Reichardt, J.K.V., Yu, M.C.,
Feigelson, H., Stanczyk, F.Z., Kolonel, L.N. & Henderson, B.E. (1998)
Androgen metabolism and prostate cancer: Establishing a model of
genetic susceptibility. Cancer Res., 58, 4497-4505.
Rothman, K.J. & Louik, C. (1978) Oral contraceptives and birth
defects. New Engl. J. Med., 210, 522-524.
Roy, D., Weisz, J. & Liehr, J.G. (1990) The O-methylation of
4-hydroxyestradiol is inhibited by 2-hydroxyestradiol: Implications
for estrogen-induced carcinogenesis. Carcinogenesis, 11, 459-462.
Ruoff, W.L. & Dziuk, P.J. (1994) Absorption and metabolism of
estrogens from the stomach and duodenum of pigs. Domest. Anim.
Endocrinol., 11, 197-208.
Russo, I.H. & Russo, J. (1996) Mammary gland neoplasia in long-term
rodent studies. Environ. Health Perspectives, 104, 938-967.
vom Saal, F.S., Timms, B.G., Montano, M.M., Palanza, P., Thayer, K.A.,
Nagel, S.C., Dhar, M.D., Ganjam, V.K., Parmigiani, S. & Welshons, W.V.
(1997) Prostate enlargement in mice due to fetal exposure to low doses
of estradiol or diethylstilbestrol and opposite effects at high doses.
Proc. Natl Acad. Sci. USA, 94, 2056-2061.
Sands, R. & Studd, J. (1995) Exogenous androgens in postmenopausal
women. Am. J. Med., 98 (Suppl. 1A), 76S-79S.
Sarabia, S.F. & Liehr, J.G. (1998) Induction of monoamine oxidase B by
17beta-estradiol in the hamster kidney preceding carcinogenesis.
Arch. Biochem. Biophys., 355, 249-253.
Sarabia, S.F., Zhu, B.T., Kurosawa, T., Tohma, M. & Liehr, J.G. (1997)
Mechanism of P450-catalyzed aromatic hydroxylation of estrogens.
Chem. Res. Toxicol., 10, 767-771.
Sarkar, K., Kinson, G.A. & Rowsell, H.C. (1986) Embryo resorption
following administration of steroidal compounds to rats in
mid-pregnancy. Can. J. Vet. Res., 50, 433-437.
Savas, U., Carstens, C.P. & Jefcoate, C.R. (1997) Biological
oxidations and P450 reactions. Recombinant mouse CYP1B1 expressed in
Escherichia coli exhibits selective binding by polycyclic hydrocarbons
and metabolism which parallels C3H10T1/2 cell microsomes, but differs
from human recombinant CYP1B1. Arch. Biochem. Biophys., 347,
181-192.
Schairer, C., Adami, H.-O., Hoover, R. & Persson, I. (1997)
Cause-specific mortality in women receiving hormone replacement
therapy. Epidemiology, 8, 59-65.
Schleicher, F., Tauber, U., Louton, T. & Schunack, W. (1998) Tissue
distribution of sex steroids: Concentration of 17ß-oestradiol and
cyproterone acetate in selected organs of female Wistar rats.
Pharmacol. Toxicol., 82, 34-39.
Schlesselman, J.J. (1995) Net effect of oral contraceptive use on the
risk of cancer in women in the United States. Obstet. Gynecol.,
85, 793-801.
Schneider, J., Huh, M.M., Bradlow, H.L. & Fishman, J. (1984)
Antiestrogen action of 2-hydroxy estrone on MCF-7 breast cancer cells.
J. Biol. Chem., 259, 4840-4845.
Schuler, M., Hasegawa, L., Parks, R., Metzler, M. & Eastmond, D.A.
(1998) Dose-response studies of the induction of hyperdiploidy and
polyploidy by diethylstilbestrol and 17ß-estradiol in cultured human
lymphocytes using multicolor fluorescense in situ hybridization.
Environ. Mol. Mutag., 31, 263-273.
Schultze, N., Vollmer, G., Wunsche, W., Grote, A., Feit, B. & Knuppen,
R. (1994) Binding of 2-hydroxyestradiol and 4-hydroxyestradiol to the
estrogen receptor of MCF-7 cells in cytosolic extracts and in nuclei
of intact cells. Exp. Clin. Endocrinol., 102, 399-405.
Scott-Moncrieff, J.C., Nelson, R.W., Bill, R.L., Natlock, C.L. &
Bottoms, G.D. (1990) Serum disposition of exogenous progesterone after
intramuscular administration in bitches. Am. J. Vet. Res., 51,
893-895.
Seraj, M.J., Umemoto, A., Tanaka, M., Kajikawa, A., Hamada, K. &
Monden, Y. (1996) DNA adduct formation by hormonal steroids in
vitro. Mutat. Res., 370, 49-59.
Shangold, M.M., Tomai, T.P., Cook, J.D., Jacobs, S.L., Zinaman, M.J.,
Chin, S.Y. & Simon, J.A. (1991) Factors associated with withdrawal
bleeding after administration of oral micronized progesterone in women
with secondary amenorrhea. Fertil. Steril., 56, 1040-1047.
Shelby, M.D., Tice, R.R. & Witt, K.L. (1997) 17b-Estradiol fails to
induce micronucli in the bone marrow cells of rodents. Mutat. Res.,
395, 89-90.
Shou, M., Korzekwa, K., Brooks, E.N., Krausz, K.W., Gonzalez, F.J. &
Gelboin, H.V. (1997) Role of human hepatic cytochrome P450 1A2 and 3A4
in the metabolic activation of estrone. Carcinogenesis, 18,
207-214.
Simon, J.A., Robinson, D.E., Andrews, M.C., Hildebrand, J.R., III,
Rocci, M.L., Jr, Blake, R.E. & Hodgen, G.D. (1993) The absorption of
oral micronized progesterone: The effect of food, dose
proportionality, and comparison with intramuscular progesterone.
Fertil. Steril., 60, 26-33.
Sitruk-Ware, R., Bricaire, C., de Lignieres, B., Yaneva, H. &
Mauvais-Jarvis, P. (1987) Oral micronized progesterone.
Contraception, 36, 373-402.
Smith, S.S., Gong, Q.H., Hsu, F.C., Markowitz, R.S., ffrench-Mullen,
J.M.H. & Li, X. (1998) GABAA receptor alpha 4subunit suppression
prevents withdrawal properties of an endogenous steroid. Nature,
302, 926-930.
Spicer, L.J., Kao, L.-C., Strauss, J.F., III & Hammond, J.M. (1990)
2-Hydroxyestradiol enhanced progesterone production by porcine
granulosa cells: Dependence on de novo cholesterol synthesis and
stimulation of cholesterol side-chain cleavage activity and cytochrome
P450scc messenger ribonucleic acid levels. Endocrinology, 127,
2736-2770.
Spink, D.C., Spink, B.C., Cao, J.Q., Gierthy, J.F., Hays, C.L., Li, Y.
& Sutter, T.R. (1997) Induction of cytochrome P450 1B1 and catechol
estrogen metabolism in ACHN human renal adenocarcinoma cells.
J. Steroid Biochem. Mol. Biol., 62, 223-232.
Stack, D.E., Cavalieri, E.L. & Rogan, E.G. (1998) Catecholestrogens as
procarcinogens: Depurinating adducts and tumor initiation.
Adv. Pharmacol., 42, 833-836.
Stampfer, M.J. & Colditz, G.A. (1991) Estrogen replacement therapy and
coronary heart disease: A quantitative assessment of the epidemiologic
evidence. Prev. Med., 20, 47-63.
Sturgeon, S.R., Schairer, C., Brinton, L.A., Pearson, T. & Hoover,
R.N. (1995) Evidence of a healthy estrogen user survivor effect.
Epidemiology, 6, 227-231.
Swales, N.J., Johnson, T. & Caldwell, J. (1996) Cryopreservation of
rat and mouse hepatocytes. Drug Metab. Disposition, 24, 1224-1230.
Symonds, H.W., Prime, G.R. & Pullar, R.A. (1994) Preliminary evidence
for the enterohepatic circulation of progesterone in the pig. Br.
Vet. J., 150, 585-593.
Tauber, U., Schroder, K., Dusterberg, B. & Matthes, H. (1986) Absolute
bioavailability of testosterone after oral administration of
testosterone-undecanoate and testosterone. Eur. J. Drug Metab.
Pharmacokinet., 11, 145-149.
Thompson, P.A., Shields, P.G., Freudenheim, J.L., Stone, A., Vena,
J.E., Marshall, J.R., Grahm, S., Laughlin, R., Nemoto, T., Kadlubar,
F.F. & Ambrosone, C.B. (1998) Genetic polymorphisms in
catechol-O-methyltransferase, menopausal status and breast cancer
risk. Cancer Res., 58, 2107-2110.
Tsai, M.J., Clark, J.H., Schrader, W.T. & O'Malley, B.W. (1998)
Hormones that act as transcription-regulatory factors. In: Wilson,
J.D., Foster, D.W., Kronenberg, H.M. & Larsen, P.R., eds, Williams
Textbook of Endocrinology, 9th Ed., Philadelphia, W.B. Saunders Co.,
pp. 55-94.
Tsutsui, T., Suzuki, N., Fukuda, S., Sato, M., Maizumi, H., McLachlan,
J.A. & Barrett, J. (1987) 17ß-Estradiol-induced cell transformation
and aneuploidy of Syrian hamster embryo cells in culture.
Carcinogenesis, 11, 1715-1719.
Tsutsui, T., Konine, A., Huff, J. & Barrett, J.C. (1995) Effects of
testosterone, testosterone propionate, 17beta-trenbolone and
progesterone on cell transformation and mutagenesis in Syrian hamster
embryo cells. Carcinogenesis, 16, 1329-1333.
Ursin, G., London, S., Stanczyk, F.Z., Gentzschein, E., Paganini-Hill,
A., Ross, R.K. & Pike, M.C. (1997) A pilot study of urinary estrogen
metabolites (16alpha-OHE1 and 2-OHE1) in postmenopausal women with and
without breast cancer. Environ. Health Perspectives, 105 (Suppl
3), 601-605.
US Congress (1995) Effectiveness and Costs of Osteoporosis Screening
and Hormone Replacement Therapy, Vol. II, Evidence of Benefits,
Risks, and Costs (Publication OTA-BP-H-144), Washington DC, Office
of Technology Assessment.
Wang, M.Y. & Liehr, J.G. (1994) Identification of fatty acid
hydroperoxide cofactors in the cytochrome P450-mediated oxidation of
estrogens to quinone metabolites. J. Biol. Chem., 269, 284-291.
Wehling, M. (1997) Specific, nongenomic actions of steroid hormones.
Annu. Rev. Physiol., 59, 365-393.
Weiss, J., Fritz-Wolz, G., Clawson, G.A., Benedict, C.M., Abendroth,
C. & Creveling, C.R. (1998) Induction of nuclear
catechol-O-methyltransferase by estrogens in hamster kidney:
Implications for estrogen-induced renal cancer. Carcinogenesis,
19, 1307-1312.
Weisz, J., Bui, Q.D., Roy, D. & Leihr, J.G. (1992) Elevated
4-hydroxylation of estradiol by hamster kidney microsomes: A potential
pathway of metabolic activation of estrogens. Endocrinology, 131,
655-661.
Welsch, C.W. (1976) Interaction of estrogen and prolactin in
spontaneous mammary tumorigenesis of the mouse. J. Toxicol. Environ.
Health, Suppl. 1, 161-175.
Wharton, L.R. & Scott, R.B. (1964) Experimental production of genital
lesions with norethindrone. Am. J. Obstet. Gynecol., 89, 701-715.
White, C.M., Ferraro-Borgida, M.J., Fossati, A.T., McGill, C.C.,
Ahlberg, A.W., Feng, Y.J., Heller, G.V. & Chow, M.S. (1998) The
pharmacokinetics of intravenous estradiol-A preliminary study.
Pharmacotherapy, 18, 1343-1346.
Whitehead, M.I., Townsend, P.T., Gill, D.K., Collins, W.P. & Campbell,
S. (1980) Absorption and metabolism of oral progesterone. Br. Med.
J., 280, 825-827.
Whittemore, A.S, Harris, R., Intyre, J. & the Collaborative Ovarian
Cancer Group (1992) Characteristics relating to ovarian cancer risk:
Collaborative analysis of 12 US case-control studies. II. Invasive
epithelial ovarian cancers in white women. Am. J. Epidemiol., 136,
1184-1203.
WHO (1992) Guidelines for the Use of Androgens in Men
(WHO/HPR/MALE/92), Geneva, Special Programme of Research, Development
and Research Training in Human Reproduction.
WHO (1998) Cardiovascular Disease and Steroid Hormone Contraception.
Report of a WHO Scientific Group (WHO Technical Report Series 877),
Geneva.
Wiebe, J.P., Boushy, D. & Wolfe, M. (1997) Synthesis, metabolism and
levels of the neuroactive steroid, 3alpha-hydroxy-4-pregnen-20-one
(3alpha HP), in rat pituitaries. Brain Res., 764, 158-166.
Williams, C.L. & Stancel, G.M. (1996) Estrogens and progestins. In:
Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddon, R.W. & Gilman,
A.G., eds, Goodman and Gilman's The Pharmacological Basis of
Therapeutics, 9th Ed., pp. 1411-1440.
Wilson, J.D. (1996) Androgens. In: Hardman, J.G., Limbird, L.E.,
Molinoff, P.B., Ruddon, R.W. & Gilman, A.G., eds, Goodman and
Gilman's The Pharmacological Basis of Therapeutics, 9th Ed.,
pp.1441-1457.
Wilson, V.S. & LeBlanc, G.A. (1998) Endosulfan elevated testosterone
biotransformation and clearance in CD-1 mice. Toxicol. Appl.
Pharmacol., 148, 158-168.
Wilson, J.D., Foster, D.W., Kronenberg, H. & Larsen, P.R. (1998)
Principles of endocrinology. In: Wilson, J.D., Foster, D.W.,
Kronenberg, H.M. & Larsen, P.R., eds, Williams Textbook of
Endocrinology, 9th Ed., Philadelphia, W.B. Saunders Co., pp. 1-10..
Wingard, L.M., Brody, T.M., Larner, J. & Schwartz, A. (1991)
Estrogens, progestins, and oral contraceptives. In: Human
Pharmacology: Molecular to Clinical, St Louis, Mosby-YearBook, pp.
494-514.
Winter, M.L. & Liehr, J.G. (1996) Possible mechanism of induction of
benign prostatic hyperplasia by estradiol and dihydrotestosterone in
dogs. Toxicol. Appl. Pharmacol., 136, 211-219.
World Cancer Research Fund in association with the American Institute
for Cancer Research (1997) Food, Nutrition and the Prevention of
Cancer: A Global Perspective, Washington DC.
Yager, J.D. & Liehr, J.G. (1996) Molecular mechanisms of estrogen
carcinogenesis. Annu. Rev. Pharmacol. Toxicol., 36, 203-232.
Yamazaki, H., Shaw, P.M., Guengerich, F.P. & Shimada, T. (1998) Roles
of cytochromes P450 1A2 and 3A4 in the oxidation of estradiol and
estrone in human liver microsomes. Chem. Res. Toxicol., 11,
659-665.
Yoshie, Y. & Ohshima, H. (1998). Synergistic induction of DNA strand
breakage by catechol-estrogens and nitrous oxide: Implications for
hormonal carcinogenesis. Free Radicals Biol. Med., 15, 341-348.
Yue, T.L., Wang, X., Louden, C.S., Gupta, S., Pillarisetti, K., Gu,
J.L., Hart, T.K., Lysko, P.G. & Feuerstein, G.Z. (1997)
2-Methoxyestradiol, an endogenous estrogen metabolite, induces
apoptosis in endothelian cells and inhibits angiogenesis: Possible
role for stress-activated protein kinase signaling pathway and Fas
expression. Mol. Pharmacol., 51,951-962.
Zhu, B.T. & Conney, A.H. (1998a) Functional role of estrogen
metabolism in target cells: Review and perspectives. Carcinogenesis,
19, 1-27.
Zhu, B.T. & Conney, A.H. (1998b) Is 2-methoxyestradiol an endogenous
estrogen metabolite that inhibits mammary carcinogenesis? Cancer
Res., 58, 2269-2297.
Zhu, B.T. & Liehr, J.G. (1994) Quercitin increases the severity of
estradiol-induced tumorigenesis in hamster kidney. Toxicol. Appl.
Pharmacol., 125, 149-158.
Zhu, B.T., Roy, D. & Liehr, J.G. (1993) The carcinogenic activity of
ethinyl estrogens is determined by both their hormonal characteristics
and their conversion to catechol metabolites. Endocrinology, 132,
577-583.
Zhu, B.T., Evaristus, E.N., Antoniak, S.K., Sarabia, S.F., Ricci, M.J.
& Liehr, J.G. (1996) Metabolic deglucuronidation and demethylation of
estrogen conjugates as a source of parent estrogens and
catecholestrogen metabolites in Syrian hamster kidney, a target organ
of estrogen-induced tumorignesis. Toxicol. Appl. Pharmacol., 136,
186-193.