
First draft prepared by
S.H. Henry1, T. Whitaker2, I. Rabbani2, J. Bowers2, D. Park2, W. Price2, F. X. Bosch3, J. Pennington4, P. Verger5, T. Yoshizawa6, H. van Egmond7, M.A Jonker7, R. Coker8
1Food and Drug Administration, Washington DC, USA
2
Agricultural Research Service, Department of Agriculture, Raleigh, North Carolina, USA3
Institute Catalan d’ Oncologia, L’Hospitalet del Llobregat, Barcelona, Spain; 4National Institutes of Health, Bethesda, Maryland, USA;5
Institut National de Recherche Agronome, Paris, France;6
Kagawa University, Kagawa, Japan;7
National Institute of Public Health and Environmental Protection, Bilthoven, The Netherlands;8
Natural Resources Institute, University of Greenwich, Kent, United Kingdom|
Intervention studies with vaccination against hepatitis B virus |
|
Dose–response relationship and estimation of carcinogenic risk |
The Expert Committee was requested by the Codex Committee on Food Additives and Contaminants at its Thirty-second Session (Codex Alimentarius, 2000) to ‘examine exposure to aflatoxin M1 and to conduct a quantitative risk assessment’ to compare the consequences of setting the maximum level in milk at 0.05 and 0.5 µg/kg.
Aflatoxins can be produced by three species of Aspergillus—A. flavus, A. parasiticus, and the rare A. nomius—which contaminate plants and plant products. A. flavus produces only B aflatoxins, while the other two species produce both B and G aflatoxins. Aflatoxins M1 and M2 are the hydroxylated metabolites of aflatoxins B1 and B2 and can be found in milk or milk products obtained from livestock that have ingested contaminated feed. The main sources of aflatoxins in feeds are peanut meal, maize and cottonseed meal.
Aflatoxins were evaluated by the Committee at its thirty-first, forty-sixth, and forty-ninth meetings (Annex 1, references 77, 122, and 131). At its forty-ninth meeting, the Committee considered estimates of the carcinogenic potency of aflatoxins and the potential risks associated with their intake. At that meeting, the Committee reviewed a wide range of studies conducted in animals and humans that provided qualitative and quantitative information on the hepatocarcinogenicity of aflatoxins. The Committee evaluated the potency of these contaminants, linked those potencies to estimates of intake, and discussed the potential impact of hypothetical standards on sample populations and their overall risk. In its evaluation, the Committee stated that the carcinogenic potency of aflatoxin M1 in sensitive species is about one order of magnitude less than that of aflatoxin B1. In particular, the Committee noted that the carcinogenic potency of aflatoxin B1 is substantially higher in carriers of hepatitis B virus (about 0.3 cancers per year per 100 000 persons per ng/kg bw per day), as determined by the presence in serum of the hepatitis B virus surface antigen (HBsAg+ individuals), than in HBsAg– individuals (about 0.01 cancers per year per 100 000 persons per ng/ kg bw per day). Populations with both a high prevalence of HBsAg+ and a high aflatoxin intake might benefit from reductions in aflatoxin intake. The Committee also noted that vaccination against hepatitis B virus would reduce the number of carriers of the virus, and thus reduce the potency of the aflatoxins in vaccinated populations, leading to a reduction in the risk for liver cancer.
The Committee at its forty-ninth meeting concluded that changing the hypothetical standard for aflatoxin B1 from 20 µg/kg to 10 µg/kg would not result in any observable difference in rates of liver cancer.
At its present meeting, the Committee reviewed studies published since its forty-ninth meeting, as well as other information, to elucidate further the carcinogenic potencies of aflatoxin M1 and aflatoxin B1 and the differences between animal species in their sensitivity to aflatoxins.
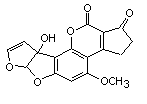
The chemical structure of aflatoxin M1 is shown in Figure 1. Aflatoxin M1 is the 4-hydroxy derivative of aflatoxin B1 and is secreted in the milk of mammals that consume aflatoxin B1. Aflatoxin M1 (CAS No. 6795-23-9) has a relative molecular mass of 328 Da and has the molecular formula C17H12O7.
A complete review of studies of the metabolism of aflatoxins conducted up to 1997 can be found in the report of the monograph on aflatoxins published after the forty-ninth meeting of the Committee (Annex 1, reference 132) and in a book by Eaton and Groopman (1994). The papers highlighted in this section address studies of matabolic differences among species in their sensitivity to aflatoxins, comparisons of the toxicity of aflatoxin M1 and aflatoxin B1, studies of the differences in the carcinogenic potency of aflatoxin M1 and aflatoxin B1, and papers published since the last report of the Committee.
The metabolism of aflatoxin B1 and the extent to which it binds to cell macro-molecules were compared in liver slices from humans and rats, as rats are more sensitive to the carcinogenicity of aflatoxin B1. Liver slices were prepared from three human liver samples and incubated with [3H]aflatoxin B1 at 0.5 µmol/L for 2 h. The rates of formation of oxidative metabolites and of specific binding to cell macromolecules showed significant interindividual variation. The rates of oxidative metabolism of aflatoxin B1 to aflatoxin Q1, aflatoxin P1, and aflatoxin M1 in the human liver samples were similar to those previously observed in rat liver slices. No aflatoxin B1–glutathione conjugate formation was detected in the human liver samples, and there was much less specific binding of aflatoxin B1 to cell macromolecules in the human than in the rat liver slices. For example, the level of binding between aflatoxin B1 and DNA ranged from 3 to 26% of that in control rats. These results suggest that humans do not form as much aflatoxin B1 8,9-epoxide as rats, but they also suggest that humans do not have glutathione-S-transferase (GST) isozymes with high specific activity towards this epoxide. Significant individual differences in aflatoxin B1 metabolism and binding suggest the presence of genetic and/or environmental factors that may result in large differences in susceptibility.
Aflatoxin M1 is usually considered to be a detoxication by-product of aflatoxin B1; it is also the metabolite present in the milk of nursing women who eat foods containing the toxin. Aflatoxin B1 epoxide has been shown to exist as two stereoisomers—endo- and exo-epoxides—the latter being the DNA-reactive form, and a similar situation may apply to aflatoxin M1 epoxide. In a study of the metabolism of aflatoxins M1 and B1 in vitro in human liver microsomes, they had a very limited capacity to catalyse epoxidation of aflatoxin M1. The small amount of aflatoxin M1 dihydrodiol formed from the epoxide also appeared to have a lower capacity to induce microsomal protein than did aflatoxin B1 dihydrodiol. GST catalysed conjugation of the epoxides of both aflatoxins with glutathione; GST activity was present in mouse cytosol but not in the human liver fraction. The authors concluded that the difference between the genotoxic potency of the two toxins in vivo correlates with their mutagenicity in vitro, metabolic activation and DNA binding (Neal et al., 1998). In rats, however, activation of aflatoxin M1 to the epoxide does not appear to be essential for its acute toxicity. Experiments in human cell lines indicated that cytochrome P450 (CYP) enzymes are involved in the cytotoxicity of aflatoxin B1 but not of aflatoxin M1. Studies of the toxicity of aflatoxin M1 in human lymphoblastoid cell lines expressing or not expressing human CYP enzymes showed a direct effect in the absence of metabolic activation, in contrast to aflatoxin B1. Aflatoxin M1 is therefore not strictly a detoxication product of aflatoxin B1 in biological responses in which cytotoxicity plays a significant role, such as immunotoxicity (Heinonen et al., 1996).
In studies of species sensitivity to the carcinogenicity of aflatoxin B1, mice were resistant because they constitutively express an alpha-GST, which is strongly active against aflatoxin B1 8,9-epoxide, whereas rats, which do not express such a GST, were sensitive. Human hepatic alpha-class GSTs have little capacity to detoxify aflatoxin B1 8,9-epoxide. The nonhuman primate Macaca fascicularis showed significant constitutive hepatic GST activity towards aflatoxin B1 8,9-epoxide. GSTs were purified from liver tissue from this species and characterized, and GST cDNAs were cloned by reverse transcriptase-coupled polymerase chain reaction (PCR). A protein, GSHA-GST, was purified by glutathione agarose affinity chromatography, which had stronger aflatoxin B1 8,9-epoxide-conjugating activity than other GST-containing peaks. The GSHA-GST was shown to belong to the µ class. The authors then showed that two distinct µ-class GST cDNAs have 97% and 98% homology with the human µ-class GSTs hGSTM4 and hGSTM2, respectively. µ-Class GSTs appear to be responsible for most of the conjugating activity of aflatoxin B1 8,9-epoxide in the liver of M. fascicularis. None of the known human µ-class GSTs acts preferentially on the ultimate genotoxic aflatoxin B1 metabolite exo-aflatoxin B1 8,9-epoxide, but large interindividual differences in the expression of GST isoforms have been shown in various tissues, and few human livers have been evaluated. The authors concluded that identification of a potential human homologue of GSHA-GST would be relevant to the design of chemointervention strategies to reduce aflatoxin B1-induced liver cancer in highly exposed populations. Nevertheless, induction of known human GSTs with little or no activity towards the epoxide of aflatoxin B1 might be ineffective in reducing the genotoxicity of aflatoxin B1 (Wang et al., 2000).
The extreme sensitivity of turkeys to aflatoxin B1 was studied by measuring microsomal activation of aflatoxin B1 to the 8,9-epoxide, the putative toxic intermediate, cytosolic GST-mediated detoxication of aflatoxin B1 8,9-epoxide, and hepatic phase I and phase II enzyme activities in 3-week-old male Oorlop turkeys. Liver microsomes prepared from these turkeys activated aflatoxin B1 in vitro with an apparent Km of 110 µmol/L and a Vmax of 1.25 nmol/mg per min. The involvement of CYP 1A2 and, to a lesser extent, 3A4 in the activation of aflatoxin B1 was assessed with specific mammalian CYP inhibitors. The possible presence of avian orthologues of these CYPs was indicated by activity towards ethoxyresorufin and nifedipine, as well as by western immunoblotting with antibodies to human CYPs. GST-mediated conjugation of 1-chloro-2,4-dinitrobenzene and 3,4-dichloronitrobenzene was demonstrated in cytosol prepared from the turkey livers, but the rate was much lower than that observed in other species. The presence of alpha- and µ-class GSTs and another aflatoxin B1 detoxifying enzyme, aflatoxin B1 aldehyde reductase, was shown by western immunoblotting. Quinone reductase activity was also present in the cytosol. Furthermore, the cytosol showed no measurable GST-mediated detoxication of microsomally activated aflatoxin B1. Thus, turkeys are deficient in the most crucial aflatoxin B1 detoxication pathway. The authors concluded that the extreme sensitivity of this species to aflatoxin B1 is due to a combination of efficient aflatoxin B1 activation and deficient detoxication by phase II enzymes such as GSTs (Klein et al., 2000).
The carcinogenicity of aflatoxin B1, aflatoxicol (aflatoxin L), aflatoxin M1, and aflatoxicol M1 (aflatoxin LM1) was compared in terms of their binding to target organ DNA in rainbow trout. Tritiated compounds were synthesized, dose–response curves for DNA binding were established, and liver DNA binding indices were calculated for the four aflatoxins after a 2-week dietary intake by trout fry. The adduct levels increased linearly with dietary concentration, with relative DNA binding indices of 21, 20, 2.4, and 2.2 x 103 (pmol/mg of DNA)/(pmol/g of diet) for aflatoxin M1, aflatoxin L, aflatoxin M1, and aflatoxin LM1, respectively.
In a similar protocol, over 7200 trout fry with an average initial body weight of 1.2 g were used to establish full carcinogen dose–response curves for each aflatoxin and an estimate of the DNA binding index after a single dose. Since trout are very sensitive, < 180 µg of each aflatoxin were required. Data analysed on logit incidence versus Ln dose coordinates generated four curves, which were modelled as parallel in slope over most or all the doses studied. In this analysis, the relative tumorigenic potencies were 1.0 for aflatoxin B1, 0.94 for aflatoxin L, 0.086 for aflatoxin M1, and 0.041 for aflatoxin LM1. When the data were plotted as logit incidence versus Ln adducts (effective dose received), dose–response relationships were found for all aflatoxin adducts, indicating that they are equally tumorigenic, except for aflatoxin LM1, which was two to three times less potent. Differences in the tumorigenicity of the four aflatoxins are largely or entirely accounted for by differences in uptake and metabolism leading to DNA adduction, rather than to any inherent difference in tumour initiating potency per DNA adduct (Bailey et al., 1998).
Since most people are exposed to carcinogens in food intermittently, the effects of intermittent intake of aflatoxin B1 on hepatic and testicular glutathione was studied in male Fischer 344 rats fed diets containing aflatoxin B1 at a concentration of 0, 0.01, 0.04, 0.4, or 1.6 ppm at 4-week intervals up to 20 weeks. The control animals were fed an aflatoxin B1-free NIH-31 diet. Rats eating diets containing 0.01 ppm aflatoxin B1 did not show induction of hepatic or testicular GST activity, but intermittent intake of concentrations of 0.04–1.6 ppm significantly increased GST activity. The increase in enzyme activity was proportional to the dose and the length of intake of aflatoxin B1 (Sahu et al., 2000).
Oltipraz is a competitive and perhaps irreversible inhibitor of CYP 1A2 and 3A4, and addition of oltipraz to rat liver microsomes or to cultured human hepatocytes blocks the oxidative metabolism of aflatoxin B1 to its 8,9-oxide and to the hydroxylated derivative aflatoxin M1. Inhibition of aflatoxin M1 excretion in urine during dietary intervention with oltipraz was examined in male Fischer 344 rats before, during, and after transient intervention. The animals were housed individually in glass metabolism cages and given 25 µg of [3H]aflatoxin B1 by gavage daily for 28 consecutive days. On days 6–16, half the rats were fed a diet supplemented with 0.075% oltipraz. Sequential 24-h urine samples were collected, and a subset was analysed for aflatoxin B1 metabolites. Aflatoxin M1 was the main metabolite in all samples, accounting for 2–6% of the administered dose. Its excretion was greatly reduced (by 77%) when oltipraz was added to the diet, but rapidly returned to control levels after cessation of the intervention. No such inhibition of aflatoxin M1 excretion was seen in animals given oltipraz by gavage 24 h before dosing with aflatoxin B1. These findings are consistent with the view that oltipraz or a short-lived metabolite inhibits CYP 1A2 in vivo (Scholl et al., 1996).
The effects of cancer preventive agents on the metabolism of aflatoxin B was examined in non-human primates in a study designed to complement a human chemointervention trial, in which oltipraz, an antischistosomal drug approved by the Food and Drug Administration of the USA, was used to modulate the metabolism of aflatoxin B in a human population naturally exposed to this toxin in the diet. This study is discussed in section 5.3. The hepatic metabolism of aflatoxin B1 was studied in macaque (M. nemestrina) and marmoset (Callithrix jacchus) monkeys and compared with that in humans. Thus, four adult male marmosets were used as controls, four were given oltipraz, and three received ethoxyquin. At time 0, each animal received a single dose of [3H]aflatoxin B at 100 µg/kg bw (0.5 mCi/kg) by gavage, and blood samples were drawn at 0, 2, 24, and 48 h. On days 16–28, the treated animals received the synthetic dithiolthione oltipraz at 18 mg/kg bw per day or the antioxidant ethoxyquin at 30 mg/kg bw per day in their diet, whereas the control animals received the vehicles only. On day 26, each animal received a second dose of [3H]aflatoxin B by gavage, and blood samples were drawn at 0, 2, 24, and 48 h. On day 28, the animals were killed, their livers were excised, and microsomal and cytosolic fractions were prepared. For comparison, livers from adult male macaques were obtained from the University of Washington Primate Center (USA). Twelve human liver samples were also obtained from the University of Washington, and the microsomal fractions were pooled. Microsomal oxidation of aflatoxin B1 and GST activity were measured, DNA adducts were isolated, and [3H]aflatoxin B-derived radioactivity in DNA fractions was quantified. [3H]Aflatoxin B1–albumin adducts in serum were determined.
The oxidative metabolism of aflatoxin B was quantitatively similar in the two monkey species and in humans. In contrast to macaques, humans and marmosets lacked aflatoxin B glutathione conjugating activity. As the metabolism of aflatoxin B in marmosets resembled that in humans more closely than that in macaques, the focus of the study was on marmosets. Both oltipraz and ethoxyquin induced aflatoxin B1–glutathione conjugating activity in the livers of some but not all marmosets. Oltipraz inhibited CYP-mediated activation of aflatoxin B to the ultimate carcinogenic metabolite, aflatoxin B1 8,9-epoxide, in vitro by up to 51%, and animals that received oltipraz in vivo showed a significant reduction (average, 53%) in aflatoxin B–DNA adduct formation in comparison with control animals.
The authors interpreted these findings as indicating that oltipraz and ethoxyquin induce modest aflatoxin B glutathione conjugating activity in the livers of some marmosets, most of the activity (about 70%) being directed against the exo isomer of aflatoxin B1 8,9-epoxide, which is by far the most potent DNA-reactive metabolite. Other workers have demonstrated aflatoxin B–mercapturic acid in the urine of marmosets exposed to aflatoxin B1. The hepatic GST activity towards aflatoxin B1 8,9-epoxide shown in this study in non-human primates was two orders of magnitude lower than that in mice, which are resistant to the carcinogenic effects of aflatoxin B1. The authors offered two explanations for the presence of DNA adducts and the decrease in steady state of exo-aflatoxin B1 8,9-epoxide shown in both treated groups: GST-mediated detoxication of exo-aflatoxin B1 8,9-epoxide and inhibition of the CYP(s) that form it. Consistent with the results of the chemointervention trial in a human population (section 5.3), administration of oltipraz and ethoxyquin would be likely to attenuate the adverse effects of aflatoxin B in primates (Bammler et al., 2000).
In a study of the inhibition of aflatoxin M1 production by bovine hepatocytes after intervention with oltipraz and another dithiolthione, 4-methyl-5-(2-pyrazinyl)-1,2-dithiole-3-thione, oltipraz inhibited the metabolism of aflatoxin B1, as neither aflatoxin M1 nor aflatoxin B dihydrodiol (the second metabolite found in bovine hepatocytes) was formed. The second dithiolthione did not significantly inhibit aflatoxin B1 metabolism. The authors suggested that the inhibition of aflatoxin B1 metabolism by oltipraz was due to inhibition of the activity of several CYP enzymes. Although the authors proposed that oltipraz could be administered to dairy cows that had accidentally received aflatoxin B1 in their feed, unmetabolized aflatoxin B1 would still reach the systemic circulation. These results obtained in vitro should be confirmed in vivo (Kuilman et al., 2000).
The roles of coumarin, benzyl isothiocyanate, and indole-3-carbinol, which are present in vegetable-enriched diets and are believed to protect against malignant disease, in regulating GST and aldo-keto reductase activity were examined in rat liver. The drugs butylated hydroxyanisole, diethyl maleate, ethoxyquin, beta-naphtho-flavone, oltipraz, phenobarbital, and trans-stilbene oxide were also investigated. In a complicated protocol, summarized briefly here, groups of three male and three female 10-week-old male and female Fischer 344 rats were given diets containing 0.75% butylated hydroxyanisole, 0.5% benzyl isothiocyanate, 0.5% coumarin, 0.5% ethoxyquin, 0.5% indole-3-carbinol, or 0.075% oltipraz for 2 weeks. Diethyl maleate at 0.5% was administered for 5 days in the food. trans-Stilbene oxide at 400 mg/kg bw was dissolved in 0.5 ml of peanut oil before daily intraperitoneal administration on 3 consecutive days, and beta-naphthoflavone at 200 mg/kg bw was dissolved in phosphate-buffered saline before daily intraperitoneal administration for 7 consecutive days. Phenobarbital was added to the drinking-water at a concentration of 0.1% for 7 days.
For the short-term intervention study of the effect of coumarin on the development of preneoplastic foci, six groups of eight 12-week-old male Fischer 344 rats were given one of the following experimental diets for 13 weeks: RM1 control maintenance diet throughout, 0.05% coumarin in RM1 diet throughout, 2 mg/kg of diet aflatoxin B1 in RM1 diet for 6 weeks followed by RM1 control diet for 7 weeks, 2 mg/kg of diet aflatoxin B1 in RM1 diet throughout, 0.05% coumarin in RM1 diet for 2 weeks followed by aflatoxin B1 at 2 mg/kg of RM1 diet containing 0.05% coumarin for 11 weeks, or 2 mg/kg of diet aflatoxin B1 in RM1 diet for 6 weeks followed by aflatoxin B1 in RM1 diet containing 0.05% coumarin for 7 weeks.
In a long-term intervention study with coumarin to study tumour formation, six groups of eight 12-week-old male Fischer 344 rats were given the same diets described above but were placed on diets containing coumarin and aflatoxin B1 for 24 weeks before being transferred to a control diet from week 25 until termination of the experiment at week 50. The animals were killed with CO2, and tissues were removed immediately. Microsomal and cytosolic fractions were prepared from fresh liver or from samples snap-frozen in liquid nitrogen.
Under these conditions, coumarin was the main inducer of aflatoxin B1 aldehyde reductase and the aflatoxin-conjugating µ-class GST A5 subunit in rat liver, increasing the concentrations of these proteins by 25–35 times. Coumarin caused similar increases in the concentration of pi-class GST P1 subunit and NAD(P)H:quinone oxidoreductase in rat liver.
To assess the biological significance of enzyme induction by dietary coumarin, two intervention studies were performed, in which the ability of benzopyrone to inhibit aflatoxin B1-initiated preneoplastic nodules (at 13 weeks) or aflatoxin B1-initiated liver tumours (at 50 weeks) was investigated. Pretreatment with coumarin for 2 weeks before administration of aflatoxin B1 and continued treatment during exposure to the carcinogen for a further 11 weeks protected the animals completely from development of hepatic preneoplastic lesions by 13 weeks. Treatment with coumarin in a longer-term dietary intervention, before and during exposure to aflatoxin B1 for 24 weeks, resulted in significant inhibition of the number and size of tumours that developed by 50 weeks. The authors concluded that consumption of a coumarin-containing diet provides substantial protection against the initiation of hepato-carcinogenesis by aflatoxin B1 in rats. The other phytochemicals and synthetic drugs tested in this study induced different zone- and sex-specific enzymes in the liver. The complexity of gene–environment interactions is emphasized by the fact that certain inducing agents can cause nuclear translocation of drug-metabolizing enzymes (Kelly et al., 2000).
The acute toxicity of aflatoxin M1 was reviewed by van Egmond (1994) and is summarized only briefly here. In the 1960s, newly hatched ducklings were shown by several investigators to be extremely sensitive to both aflatoxin B1 and aflatoxin M1, with LD50 values of 12–16 µg per bird. Histopathological examination showed liver lesions similar to those caused by aflatoxin B1 and necrosis of the renal tubules. Milk naturally contaminated with aflatoxin M1 produced fewer lesions than artificially contaminated milk, however, suggesting differences in the bioavailability of naturally and artificially occurring aflatoxin M1. Studies on the acute toxicity of aflatoxins in 1-day-old ducklings suggest that aflatoxin M1 and aflatoxin B1 act by a similar mechanism in causing acute toxicity and subcellular alterations, such as changes in liver parenchymal cells, dissociation of ribosomes from the rough endoplasmic reticulum, and proliferation of the smooth endoplasmic reticulum, and that only the naturally occurring isomer of each aflatoxin is biologically active.
van Egmond (1994) also summarized the results of long-term studies of toxicity. In a study by Sinnhuber in 1974, rainbow trout received diets containing aflatoxin B1 at 4 µg/kg or aflatoxin M1 at 0, 4, 16, 32, or 64 µg/kg for 12 months and then received a control diet. Selected groups were held for 20 months to determine the effect of maturation on tumour development, and some were fed aflatoxin M1 at 20 µg/kg of diet for 5–30 days to determine the effect of limited oral intake of this toxin. Female trout with aflatoxin M1-induced hepatomas had a significantly higher mortality rate at maturation (16–20 months) than males. The trout receiving aflatoxin M1 at 20 µg/kg of diet had a 3–12% incidence of hepatoma within 12 months. The author concluded that aflatoxin M1 is a potent liver carcinogen but less potent than aflatoxin B1.
Canton et al. in 1975 fed rainbow trout diets containing aflatoxin M1 at 0, 5.9, or 27 µg/kg and aflatoxin B1 at 5.8 µg/kg for 16 months. The fish were killed after 5, 9, and 12 months. Degeneration of the liver was seen in all three groups and in the control group, but no tumours or preneoplastic changes were found. At 15 months, however, the survivors fed the diet containing 5.8% aflatoxin B1 had a 13% incidence of hepatocellular carcinoma and a 23% incidence of hyperplastic nodules, and those fed the diet with 27.3 µg/kg aflatoxin M1 had a 2% incidence of hepatocellular carcinoma and a 6% incidence of hyperplastic nodules. The investigators concluded that differences in trout strain could have contributed to the differences between their results and those of Sinnhuber, but that aflatoxin M1 is less carcinogenic in trout than aflatoxin B1.
van Egmond (1994) summarized two further studies in rats. In a study in 1974, weanling Fischer rats were given 25 µg/day of synthetic aflatoxin M1 by intubation on 5 days/week for 8 consecutive weeks. A second group of rats was given natural aflatoxin B1 at the same concentration and under similar conditions. A control group was included. Only one rat (3%) given aflatoxin M1 developed a hepatocellular carcinoma, whereas 28% had liver lesions (preneoplastic lesions). All rats receiving aflatoxin B1 developed tumours, whereas controls showed no significant liver lesions. The carcinogenic potency of aflatoxin M1 was concluded to be much lower than that of aflatoxin B1.
In a second study, groups of Fischer rats were maintained on diets containing natural aflatoxin M1 at 0, 0.5, 5, or 50 µg/kg and were killed between 18 and 22 months. Hepatocellular carcinomas were detected in 5% and neoplastic nodules in 15% of rats fed diets containing aflatoxin M1 at 50 µg/kg between 19 and 20 months. No nodules or carcinomas were observed at the lower dose. Of rats fed the diet containing aflatoxin B1 at 50 µg/kg, 95% developed hepatocellular carcinomas. Only a few rats at 50 µg/kg of aflatoxin M1 developed intestinal carcinomas. The authors suggested that the greater polarity of aflatoxin M1 than aflatoxin B1 might be associated with the higher incidence of intestinal tumours. It was concluded that aflatoxin M1 was a hepatic carcinogen, but with a potency 2–10% that of aflatoxin B1 (Cullen et al., 1987, as described by van Egmond, 1994). This study is the one usually cited in comparisons of the carcinogenicity of aflatoxin B1 and aflatoxin M. van Egmond (1994) concluded that the toxicity of aflatoxin M1 is similar to or slightly lower than that of aflatoxin B1 in rats and ducklings, and the carcinogenicity of aflatoxin M1 is probably one to two orders of magnitude lower than that of aflatoxin B1 (see Table 1).
Table 1. Comparative toxicity of aflatoxin M1 and B1
|
Species, strain |
Sex |
Route |
Aflatoxin |
LD50 |
Reference |
|
Mouse, C57BL/6J, newborn |
M,F |
Intraperitoneal |
AFP1 |
< 5% that of AFB1 |
Buchi et al. (1973) |
|
Duck, Peking |
NR |
Oral |
AFM1, AFM2 |
Similar to AFB1, |
Purchase (1967) |
|
Peking duckling, |
M,F |
Oral |
AFB1 |
0.34 mg.kg bw |
Lijinsky & Butler (1966) |
|
Rainbow trout, |
M,F |
Intraperitoneal |
AFB1 |
0.81 mg/kg bw |
Bauer et al. (1969) |
|
Fischer rat, |
M |
Intraperitoneal |
AFB1 |
0.75 mg/kg bw |
McGuire (1969) |
|
CFW Swiss mouse, 30 days |
M |
Intraperitoneal |
AFB1 |
> 150 mg/kg bw |
McGuire (1969) |
|
Syrian hamster, 30 days |
M |
Oral |
AFB1 |
10 mg/kg bw |
Wogan (1966) |
Adapted from Roebuck & Maxuitenko (1994)
The potency of aflatoxin B1 and aflatoxin M1 in inducing DNA damage and genotoxicity was tested in Drosophila melanogaster in vivo in the mei-9a mei-41D5 DNA repair test and the mwh/flr3 wing spot test, respectively. In the repair test, larval stock consisting of meiotic recombination-deficient double-mutant mei-9a mei-41D5 males and repair-proficient females was exposed to the test agent, and preferential killing of the mutant larvae was taken as evidence of DNA damage. Aflatoxin M1 was found to be a DNA-damaging agent, with an activity about one-third that of aflatoxin B1. In the wing spot test, in which larval flies trans-heterozygous for the somatic cell markers mwh and flr3 were treated and the wings were inspected at adulthood for spots manifesting the phenotypes of the marker, the genotoxicity of aflatoxin M1 and aflatoxin B1was similar. The authors concluded that aflatoxin M1 is genotoxic in mammalian systems in vivo (Shibahara et al., 1995).
The tree shrew (Tupaia belangeri chinensis) is unique in that it can be infected with human hepatitis B virus (HBV) and is susceptible to aflatoxin B1-induced liver cancer; a synergistic interaction between HBV and aflatoxin B1 for liver cancer has been observed. In studies in which the tree shrew model was used to evaluate experimental chemoprevention strategies for populations at high risk for liver cancer, two groups of tree shrews were fed milk containing aflatoxin B1 at a concentration providing a dose of 400 µg/kg bw per day for 4 weeks. One week before administration of aflatoxin B1, one group also received oltipraz at 0.5 mmol/kg bw per day orally for 5 weeks. Samples of 1 ml of blood and 24-h urine were obtained from each animal at weekly intervals. Aflatoxin–albumin adducts in serum were identified by radioimmunological assay, and aflatoxin–N7-guanine adducts in urine were measured by high-performance liquid chromatography (HPLC). The concentration of aflatoxin–albumin adducts increased rapidly over 2 weeks, to reach a plateau at 20 pmol/mg of protein, and decreased after cessation of exposure to aflatoxin B1. Oltipraz significantly attenuated the overall burden of aflatoxin–albumin adducts throughout exposure, with a median reduction of 80%. As measured in a single cross-sectional analysis at the end of treatment with aflatoxin B1, oltipraz decreased the urinary aflatoxin–N7-guanine content by 93%. The authors concluded that oltipraz reduces risk biomarkers for aflatoxin B1 in the tree shrew, as it does in rodents and humans, and established a rationale to evaluate cancer chemoprevention by oltipraz in tree shrews infected with human HBV and exposed to aflatoxin B1.
The authors recalled that reductions of comparable magnitude in both aflatoxin–albumin adducts in serum and aflatoxin–N7-guanine adducts in urine were found in rats pretreated with oltipraz and exposed to aflatoxin B1. Tree shrews appear to be less susceptible to hepatocarcinogenesis than rats. The tree shrew model is useful and may allow determination of whether agents such as oltipraz sustain their chemopreventive effect against aflatoxin in the presence of chronic infection with HBV (Li et al., 2000).
The effects of methyl deficiency and dietary restriction on hepatic-cell proliferation and telomerase activity were studied in 5-week-old male Fischer 344 rats pretreated with aflatoxin B1 at 25 µg/rat per day by gavage on 5 days/week for 3 weeks or given solvent (100 µg of 75% dimethyl sulfoxide). The rats were then separated into groups fed a methyl-sufficient or -deficient diet ad libitum or with dietary restriction. When the rats were 15, 20, and 32 weeks of age, hepatic-cell proliferation, telomerase activity, and the number of GST-placental form (P)-positive foci were determined. Dietary restriction reduced hepatic-cell proliferation, while the methyl-deficient diet and aflatoxin B1 pretreatment increased cell proliferation. Telomerase activity was decreased by dietary restriction and increased by the methyl-deficient diet and aflatoxin B1 pretreatment. The same trend was observed for GST-P+ foci in aflatoxin B1-pretreated rats: methyl deficiency increased the number of foci, and dietary restriction decreased the number. These results are consistent with a role of telomerase in hepatocarcinogenesis, although the origin of the cells giving rise to the increase in telomerase activity was not determined (Chou et al., 2000).
In a study of the effect of ascorbic acid on the toxicity of aflatoxin B1, young guinea-pigs were either fed diets containing 0 or 25 mg/day of ascorbic acid or were given 300 mg/day by gavage for 21 days and the LD50 dose of aflatoxin B1 on day 22. Seven of 10 animals fed no ascorbic acid died within 73 h of administration of aflatoxin B1, and their livers showed massive regional necrosis and multilobular degeneration. None of the animals given 25 mg/day ascorbic acid died, but their livers showed changes similar to those seen in the group that received no ascorbic acid. The activities of serum alanine and aspartate aminotransferases were elevated.
None of the animals given 300 mg/day of ascorbic acid died or had pathological changes in the liver, and their alanine and aspartate aminotransferase activities were unaffected. Production of aflatoxin M1 by liver microsomes tended to be higher than that in the other two groups. Three animals receiving 300 mg/day of ascorbic acid were given a second intraperitoneal LD50 dose of aflatoxin B1 1 month after the first. One animal died, and the livers of all animals showed centrilobular degeneration and moderate necrosis in scattered hepatocytes. Hepatic microsomal CYP and cytosolic GST activities and aflatoxin M1 production were drastically reduced, and the activities of alanine and aspartate aminotransferase were increased. The results indicate that intake of 300 mg of ascorbic acid virtually protected the animals from the acute toxicity of aflatoxin B1 given by gavage but not when administered as a second dose intraperitoneally (Netke et al., 1997).
In a study of the effects of carotenoids on the initiation of liver carcinogenesis by aflatoxin B1, male weanling rats were fed beta-carotene, beta-apo-8´-carotenal, cantha-xanthin, astaxanthin, or lycopene at 300 mg/kg of diet; an excess of vitamin A (21 000 retinol equivalents per kg of diet); or 3-methylcholanthrene at 6 x 20 mg/kg bw intraperitoneally before and during treatment with aflatoxin B1 at 2 x 1 mg/kg bw. The rats were then treated with 2-acetylaminofluorene and partial hepatectomy, and GST-P+ liver foci were detected and quantified. Aflatoxin B1-induced hepatic DNA damage was evaluated as single-strand breaks and binding of [3H]aflatoxin B1 to liver DNA and plasma albumin in vivo. Modulation of aflatoxin B1 metabolism by carotenoids or by 3-methylcholanthrene was investigated by incubation in vitro of [14C]aflatoxin B1 with liver microsomes from rats that had been fed carotenoids or treated with 3-methylcholanthrene; the metabolites formed were analysed by HPLC.
Neither lycopene nor an excess of vitamin A had any effect, but beta-carotene, beta-apo-8´-carotenal, astaxanthin, and canthaxanthin decreased the metabolism of aflatoxin B1 to aflatoxin M1, a less genotoxic metabolite. The authors concluded that these carotenoids exert their protective effect by deviating aflatoxin B1 metabolism towards detoxication pathways. In contrast, beta-carotene did not protect hepatic DNA from aflatoxin B1-induced alterations and affected the metabolism of aflatoxin B1 to only a minor degree. Its protective effect against the initiation of liver preneoplastic foci by aflatoxin B1 appears to be mediated by other mechanisms (Gradelet et al., 1998).
The hepatotoxicity of aflatoxin B1 is augmented by bacterial endotoxin lipopoly-saccharide in rats. At intraperitoneal doses > 1 mg/kg bw, aflatoxin B1 caused pronounced injury to the periportal regions of the liver. Male Sprague-Dawley rats were given aflatoxin B1 at 1 mg/kg bw or the vehicle, 0.5% dimethyl sulfoxide and saline, and then Escherichia coli lipopolysaccharide (7.4 x 106 enzyme units per kg) or its saline vehicle 4 h later. Liver injury was assessed 6, 12, 24, 48, 72, or 96 h after administration of aflatoxin B1. Histological examination of liver sections and measurements of alanine and aspartate aminotransferase activity in serum were used to evaluate hepatic parenchymal-cell injury. Biliary-tract alterations were evaluated as increased concentration of serum bile acids and activities of gamma-glutamyl-transferase, alkaline phosphatase, and 5’-nucleotidase in serum.
No or little injury was seen in rats treated with aflatoxin B1 or lipopolysaccharide alone, but hepatic parenchymal-cell injury was pronounced by 24 h in the group treated with aflatoxin B1 and lipopolysaccharide, returning to control values by 72 h. The injury began in the periportal region and spread mid-zonally with time. Changes in serum markers indicative of biliary-tract alterations were evident by 12 h, but the values had returned to control levels by 72 h. The nature of the hepatic lesions suggested that lipopolysaccharide potentiated the effects of aflatoxin B1 on both parenchymal and bile-duct epithelial cells.
The authors suggested that the results of this study might partly explain the severity of human cases of acute aflatoxicosis. In addition, persons with hepatitis who have an inflammatory response may be predisposed to the carcinogenic effects of aflatoxin B1, as the results of this study suggest that inflammation accompanied by hepatic parenchymal-cell hyperplasia might contribute epigenetically to aflatoxin B-induced carcinogenesis by promoting tumour formation (Barton et al., 2000).
Aflatoxin M1, like other aflatoxins, is produced by fungi that grow naturally on plants in the field or on stored feeds. Aflatoxins are among the most toxic of the known mycotoxins and have been implicated in the deaths of humans and animals that have consumed mouldy food . While the liver is the target organ for aflatoxicosis, aflatoxins are also found in other animal tissues and products, such as meat, milk, and eggs. As mature animals modify and eliminate toxins effectively, however, the main concern is long-term intake of low concentrations of these toxins, which can lead to cancer and immunosuppression. Although intake of low doses of aflatoxins may not cause death or tissue damage, it may severely affect the cost-effectiveness of animal production.
Sensitivity to aflatoxins varies from one species to another, and, within the same species, the severity of toxicity depends on dose, duration of intake, age, and breed of the animals, and their dietary protein content. The results of toxicological studies in domestic animals are given in Table 2.
Table 2. Results of studies of the toxicity of aflatoxins in domestic animals
|
Species |
Sex |
Aflatoxin |
Dose |
Toxicological end-point |
Findings |
|
Mink |
Pregnant |
Mixed |
Diet,10 µg/kg |
Kit body weight at 3 weeks |
Decreased |
|
Kit mortality (birth to 3 weeks) |
Increased |
||||
|
Hamster |
Pregnant |
B1 |
4–6 mg/kg bw |
Hepatic, renal, and fetal lesions |
Increased |
|
Duck |
Male and female |
Mixed |
Diet, 33 µg/kg |
Thymus: |
|
|
Viable cells |
Decreased |
||||
|
Cells/g tissue |
Decreased |
||||
|
Spleen: |
|||||
|
Viable cells |
Decreased |
||||
|
Cells/g tissue |
Decreased |
||||
|
Bursa of Fabricius: |
|||||
|
Viable cells |
Decreased |
||||
|
Cells/g tissue |
Decreased |
||||
|
Chicken |
Male |
B1 |
Diet, 0.5–5.0 mg/kg, 5 weeks |
Weight loss, decreased weight gain, impaired blood coagulation, poor pigmentation, decreased bone strength, and hepatic lesions |
Positive |
|
Calves |
NR |
B1 |
Single s.c. dose |
Presence of aflatoxin B1, aflatoxin M1, aflatoxin L in tissues and urine |
|
|
0.8 mg/kg bw |
|
Positive |
|||
|
1.8 mg/kg bw |
|
Positive |
|||
|
Daily dose: 42 mg over 3 months |
|
Negative |
|||
|
Pig |
NR |
Mixed |
Diet: |
Weight loss, anorexia, haemorrhage. liver damage, and death |
Positive |
|
B1, B2, G1 |
2.3–4.5 mg/kg |
As above plus renal damage |
Positive |
||
|
Caprine |
NR |
B1, G1, M1 |
1.3–1.5 mg/day until death |
Anorexia, depression, jaundice, liver and kidney damage, dark urine, and nasal discharge |
Positive |
|
Rabbit |
NR |
B1 |
25–626 µg/kg bw for 24 days |
Hepatic lesions |
Positive |
|
Rat |
NR |
B1 |
7 mg/kg bw, once |
Hepatic carcinoma |
Positive |
|
0.5 mg/kg bw 4 days postnatally |
Gastrointestinal, urogenital, and hepatic carcinoma |
Positive |
|||
|
Guinea-pig |
NR |
B1 |
630 µg/kg bw once |
Hepatic lesions |
Positive |
|
Monkey |
NR |
B1 |
0.01–1.0 mg/day until death |
Hepatic lesions |
Positive |
Compiled from data reported by Aulerich et al. (1993), Miller & Wilson (1994), Shane (1994), Sabino et al. (1995), and Hurley et al. (1999)
NR, not reported; s.c., subcutaneous
In general, ingestion of aflatoxin results in a variety of clinical signs which depend on the amount consumed and the species and age of the animal. Aflatoxin may make an animal more susceptible to infectious diseases by impairing its immune system or potentiating a bacterial infection. Symptoms of secondary infection may obscure the symptoms of aflatoxicosis. Intake of aflatoxins during gestation may affect offspring as well as adults (Miller & Wilson, 1994).
The Food and Drug Administration (USA) set a tolerance limit of 20 µg/kg for aflatoxins in maize and 0.5 µg/kg of aflatoxin M1 in milk. The latter can be achieved by consuming a diet contaminated with < 30 µg/kg (Shane, 1994). The carry-over of aflatoxin from animal feed to milk and tissue is discussed in section 8.1.
Liver cancer has been related to dietary intake of aflatoxins. The most recent epidemiological studies tend to indicate that individuals who are carriers of persistent viral infection with HBV and who are exposed to aflatoxin in their diets are at increased risk for progression to liver cancer as compared with HBV carriers who are not exposed to aflatoxins. No similar interaction has been reported with chronic infection with hepatitis C virus (HCV). The epidemiological studies to date have focused on aflatoxin B1; ingestion of aflatoxin M1 with milk and milk products has not been directly related to liver cancer. Some of the epidemiological observations that implicate aflatoxins in the etiology of liver cancer derive from observations of unusual clusters of disease or unexplained trends in incidence. Additional indications are provided by the results of case–control and cohort studies based on adequate techniques and comprehensive evaluation of the risk factors in the etiology of liver cancer.
Increasing trends have been reported in the rates of hospitalization, incidence, and mortality attributable to liver cancer in the black and white populations of both sexes in the USA. The age-specific curves showed a shift towards liver cancer among persons aged 40–60. The authors discuss in detail and quite convincingly the factors that interfere in analyses of time trends for liver cancer, including the widespread introduction of new diagnostic means (ultrasound, a-fetoprotein analysis), improved registration practices (histological confirmation and coding), and the quality of analysis (trend and birth–cohort analyses). The authors attribute the trend to intravenous drug abuse in the relevant generations, HCV being the predominant causative factor (El Serag & Mason, 1999).
HCV was also shown to be related to the increased rate of death from liver cancer in Japan after a vaccination campaign against tuberculosis under non-sterile conditions (Okuda, 1991).
Also in Japan, a report on trends in death from liver cancer showed that alcohol was the agent primarily responsible. Birth cohort analyses showed small effects. The effect of alcohol may have been overestimated because the incidence of liver cancer was used as a surrogate measure of alcohol consumption by women, who are assumed to have low consumption of alcohol. Other surrogate measures used were the incidence of oesophageal cancer and the mortality rate from cirrhosis (Makimoto & Higuchi, 1999).
The role of chronic infection with HBV and HCV in the etiology of liver cancer is well established. Several epidemiological studies have examined the association between seropositivity for HBsAg and the risk for liver cancer. The risk estimates ranged from 3 to 30 in case–control studies and from 5.3 to 148 in cohort studies (IARC, 1994). A meta-analysis of studies published before 1998 gave an estimated relative risk of 17 for persons with antibodies to HCV who are HBsAg– (Donato et al., 1998). Table 3 shows current estimates of the attributable fractions for the main risk factors associated with liver cancer. Worldwide, 52% of liver cancer cases (230 000) have been attributed to chronic HBV infection, with 19 000 in developed countries and 210 000 in developing countries. The fraction of liver cancer cases attributable to HCV infection is 110 000 (25% of the world total), with 17 000 cases in developed countries and 93 000 in developing countries.
Table 3. Risk factors for liver cancer and estimates of attributable fractions (%)
|
Risk factor |
Low-risk countries in Europe and the USA |
Japan |
High-risk countries in Africa and Asia |
|||
|
Estimate |
Range |
Estimate |
Range |
Estimate |
Range |
|
|
Hepatitis B virus |
< 22 |
4–58 |
20 |
18–44 |
60 |
40–90 |
|
Hepatitis C virusa |
60 |
12–72 |
63 |
48–94 |
< 10 |
Not evaluated |
|
Aflatoxin |
Little exposure |
Little exposure |
Heavy exposureb |
|
|
|
|
Alcohol |
|
15–45 |
< 20 |
|
|
11–30 |
|
Tobacco |
< 12 |
|
40 |
38–51 |
22c |
Not evaluated |
|
Oral contraceptives |
|
10–50d |
Not evaluated |
8e |
Not evaluated |
|
|
Other |
< 5 |
|
|
|
< 5 |
|
From Bosch et al. (1999). The attributable fractions do not necessarily add up to 100% because of multiple exposures and possible interactions between risk factors.
a
Not including infection with both HBV and HCV; second-generation assays were used in few studiesb
Atrributable risk not quantifiedc
Estimates for HBsAg– black men > 50 years old (one study)d
Only in womene
Only in black women (one study)The presence of HBV DNA or HCV RNA serum and liver tumour tissue from patients with liver cancer, mostly in European countries, who were seronegative for antibodies to both viruses, was investigated in a collaborative multicentre study. Of the specimens, 33% contained HBV DNA and 7% contained HCV RNA (Brechot et al., 1998). The results have been confirmed. The trend suggests that, in countries where HBV is common, the presence of HBV DNA among HBsAg– patients with liver cancer is higher than the 33% found in Europe. These findings reinforce the strong relationship between HBV and HCV viral infections and liver cancer and suggest that the attributable fractions shown in Table 3 may be underestimates.
In a review of several studies in China (some of which were evaluated by the Committeeat its forty-ninth meeting), in which aflatoxin–albumin adducts were measured, a correlation was found between death from primary liver cancer and aflatoxin B1–albumin in serum from persons in Fusui but not in those from Shanghai. In Fusui County, primary liver cancer was correlated to intake of aflatoxin B1 but not aflatoxin, M1 and the decreasing trend in the aflatoxin–albumin adducts over time in Fusui were attributed to improved agricultural practices (Yu et al., 1998).
The mutation induced by aflatoxin B1 in exon 3 of the human HPRT gene in B lymphoblasts is a GC to TA transversion at base 209, occurring in 17% of aflatoxin B1-induced mutants. In an analysis of the HPRT mutation frequency in an area with heavy intake of aflatoxin B1, the residents of Qidong County, China, were studied to determine the combined contributions of aflatoxins and other risk factors to the high incidence rate of liver cancer in the region. The study cohort comprised 42 men and 65 women aged 40–65. Blood samples were analysed for mean aflatoxin B1 in albumin, HPRT mutations by a T-cell clonal assay, HBsAg status, serum alanine aminotransferase activity, leukocyte count, haemoglobin concentration, and platelet count. Subjects were categorized as having a low or a high intake of aflatoxin B1 and were dichotomized around the population mean of aflatoxin–albumin adducts.
A major assumption in this study was that an individual’s aflatoxin B1 content was representative of his or her intake of aflatoxin throughout life, even though aflatoxin B1–albumin adducts indicate recent intake. The amounts of aflatoxin B1 measured were comparable with those in previous year-long studies in this population. The typical mutation frequency in the HPRT gene in normal, healthy adults is 5–8 x 10–6 per cell, whereas the frequency in this population was 26 x 10–6. Thus, the population was exposed to environmental agents that damage DNA. The authors concluded that the aflatoxin-induced DNA damage in T lymphocytes, assessed as the validated marker, albumin adducts, led to an increased mutation frequency, reflected as the increase in HPRT gene mutations (Wang et al., 1999a).
The limitation of the study is that HBsAg seropositivity was presumed to indicate the presence of HBV; however, epidemiological studies that rely on HBsAg status instead of detection of HBV DNA (Brechot et al., 1998; Bosch et al., 1999) systematically underestimate the risk due to HBV. Furthermore, HCV status was not measured.
Studies on intake of aflatoxins and liver cancer published in 1997–2000 incorporated biomarkers of intake of aflatoxin in order to compare series of cases and controls. Some of these studies had the advantage of being nested in cohort studies, thus including data from biological specimens collected some time before the occurrence of liver cancer.
A small case–control study in the Sudan showed a relationship between grain storage conditions and the aflatoxin contamination of peanuts, and some association between storage conditions and the occurrence of liver cancer (Omer et al., 1998).
A 10-year follow-up study for hepatocellular carcinoma in Qidong, China, was reported in which 145 carriers of HBV provided eight monthly urine samples which were tested for aflatoxin M1 by a sensitive assay (3.6 ng/L). At the beginning of the study, 54% of the subjects had aflatoxin M1 in their urine; 22 subsequently developed hepatocellular carcinoma. The predictors of liver cancer among HBV carriers were found to be the presence of aflatoxin M1 in urine, antibodies to HCV, and a family history of hepatocellular carcinoma. The estimated relative risk associated with aflatoxin M1 was 3.6 (95% confidence interval, 1.3–9.9) . The authors compared this estimates with that for a cohort in Shanghai with exposure to both HBV and aflatoxins (relative risk, 59) and found no statistically significant difference, mainly because of the small number of cases in both studies. All four patients with hepatocellular carcinoma who had aflatoxin M1 in their urine and who were tested for mutations in the P53 oncogeneshowed the missense mutation in codon 249. The authors concluded that aflatoxin is a substantial risk factor for progression to hepatocellular carcinoma among carriers of HBV. The (unstable) estimated attributable fraction was 0.55 (0.09–0.94) (Sun et al., 1999).
In a prospective follow-up study of 737 HBV carriers and 699 with no HBV, aflatoxin–albumin adducts were measured in 30 HBsAg+ patients with liver cancer and 150 controls (HBV status unclear). A significantly larger proportion of patients had adducts (odds ratio [OR], 3.5), and they had a significantly higher overall level of adducts than controls. The authors concluded that aflatoxins are a significant co-factor with HBV in the induction of primary liver cancer (Lu et al., 1998).
Forty-three patients with hepatocellular carcinoma in Taiwan were compared with 86 matched controls for the urinary concentration of aflatoxin metabolites and aflatoxin B1–albumin adducts in specimens taken in 1988–92 and for GST activity. All but one of the patients were HBsAg+ and the other had antibodies to HCV. The levels of biomarkers of aflatoxin in urine differed somewhat between cases and controls. A trend to increased risk with urinary aflatoxin M1 was reported, but the numbers were too small to ensure proper power. A high risk was found for persons with detectable levels of aflatoxin–albumin adducts and aflatoxin B1–N7 guanine adducts (OR, 10; 1.6–61). The authors concluded that aflatoxin is a significant factor for primary liver cancer in HBV carriers and that there may be an interaction between the GSTM1 genotype and intake of aflatoxin B1 (Yu et al., 1997a).
A small case–control study was conducted among black Africans to determine the effect of iron overload and other environmental factors on the risk for hepato-cellular carcinoma. The OR associated with HBV seropositivity in 24 cases and 48 hospital controls was 33 (7.2–150) for HCV infection, 6.4 (0.30–130) for alcohol consumption, 2.0 (0.50–8.2) for iron overload, and 11 (1.5–77) for aflatoxin–albumin adducts. There was no association with hepatocellular carcinoma (median prevalence of adducts, 7.3 in cases and 22 in controls; OR not reported) (Mandishona et al., 1998).
Thus, three studies from Asia reported increased risks for persons infected with HBV and with either aflatoxin M1 metabolites in urine or aflatoxin–albumin adducts in serum. The ORs reported ranged from 3 to 10. The evidence is not entirely consistent, and the study from southern Africa found no significant association berween the presence of aflatoxin–albumin adducts in serum and liver cancer.
Other cohort studies gave conflicting results with regard to the role of aflatoxin in the etiology of liver cancer. A follow-up study was conducted to estimate the risk for primary liver cancer among male HBV carriers in areas with different intakes of aflatoxin: in Senegal, China, and persons of Asian origin resident in the USA. The cohorts were selected to examine why the estimated risk for liver cancer in some areas of China was significantly higher (two- to threefold) than that on the west coast of Africa. The prevalence of HBsAg was only moderately higher in Senegal (20% versus 16%), and the expected intake of aflatoxin was higher in the African setting. In an analysis of differences in host response to the viral infection in these populations, viral replication (HBV DNA detected by Southern blotting) was 25–30% in HBsAg carriers of all age groups in China, whereas in Senegal a strong decay in HBV DNA rates was seen with age, from 14% in persons aged 20–29 to 3% in those aged 30–49 and undetectable in persons > 50. Asian–American HBsAg carriers also showed a strong decline in HBV DNA with age, from 37% in the 20–29 age group to 5% in those over 50. Prolonged viral replication at a high titre correlated with several parameters of liver damage and may be a determinant in the high rate of mother-to-child transmission of HBV and in the incidence of liver cancer in Chinese populations .
In the first report of this study, the authors showed that the risk of an HBV carrier for progression to hepatocellular carcinoma was lower in Senegal (high risk for exposure to both HBV and aflatoxin) than in China (high risk for exposure to HBV but lower risk for intake of aflatoxin). This was contrary to expectations if a strong interaction between HBV and aflatoxins is the central determinant of liver cancer in these areas. In fact, active DNA replication throughout life seems to explain the higher progression rate in China and may be a consequence of the high rate of mother-to-child transmission of HBV and the high incidence of liver cancer in Chinese populations (Evans et al., 1998).
The metabolism of aflatoxins is not yet fully understood. It has been hypothesized that metabolic polymorphisms of the genes that regulate the metabolism of aflatoxins could explain the established interspecies differences in susceptibility to aflatoxin-induced carcinogenicity and the largely hypothetical differences in susceptibility among human groups. Some epidemiological studies have addressed the environmental factors that may modulate the natural history of aflatoxins and biomarkers of aflatoxins under various conditions of exposure.
The determinants of aflatoxin–albumin adducts in blood were investigated in 357 persons, including 181 chronic carriers of HBV, in The Gambia. Several environmental factors (season, place of residence, HBV status) and aspects of the metabolism of aflatoxin (the GST genotypes M1, T1, P1 and epoxide hydrolase) were recorded, and the ratio of 6beta-hydroxycortisol:cortisol as a marker of CYP 3A4 activity was measured in urine. The major determinants of the amounts of aflatoxin in blood were place of residence and season. The mean adduct levels were higher in persons without HBV infection and the GSTM1 null genotype. The authors concluded that environmental factors leading to food contamination are better determinants of intake than metabolic measures and are more amenable to intervention (Wild et al., 2000).
Understanding the natural history of the biomarkers used in epidemiological studies is an absolute requirement for proper interpretation of much of the available literature. In a study in Taiwan, aflatoxin B1–N7-guanine adducts were measured in urine as a function of the hormonal and nutritional parameters that may affect aflatoxin adduct formation. In a cross-sectional study of 42 male HBV carriers and 43 HBV-free men, adduct formation was detected in 42%. Significant determinants of adduct formation were HBV status, with higher levels in HBsAg carriers, and plasma concentrations of cholesterol, alpha-tocopherol and alpha- and beta-carotene. The associations were significant and dose-dependent. Lycopene concentrations were inversely related to adduct formation. This study is significant in that it indicates some of the environmental and host determinants of adduct formation for use in etiological studies (Yu et al., 1997b).
If HBV status is directly related to adduct formation, case–control studies will systematically show that the presence of adducts in urine is a risk factor for hepatocellular carcinoma (Sohn et al., 2000). Nutritional determinants of adduct formation, if validated, should be treated as confounders in epidemiological studies in which urinary adducts are used as a biomarker. The roles of folate and other nutrients in the natural history of HBV infection and liver cancer have been confirmed in a number of studies (e.g. McGlynn et al., 1999), and the role of nutrients in the mutagenicity and carcinogenicity of aflatoxins is corroborated by evidence from experiments in rats (Soni et al., 1997) and in studies summarized in section 2.3.4.
GST expression was found to be inversely related to the HBV status of patients with normal livers, suggesting that viral replication decreases the ability of liver cells to detoxify liver carcinogens such as aflatoxin efficiently. Tissue from liver tumours had less GST activity, and subjects with the null GSTM1 genotype had less GST alpha- and µ-isoenzymes, with some overexpression of pi. These results suggest that GST expression should be treated as a confounder in epidemiological studies in which urinary adducts are used as a biomarker (Zhou et al., 1997).
In a comparison of the sensitivity to mutagens of 28 cases of hepatocellular carcinoma and 110 controls, on the basis of the count of chromatid breaks induced by bleomycin or benzo[a]pyrene diol epoxide, the OR was 36. The tests show defects in predisposition to chromosome breakage or the capacity to repair chromatid breaks or both. The results suggest that individual susceptibility can be important in determining the outcome in exposed individuals. No epidemiological studies have been reported in which host factors determined by these methods were adjusted for (Wu et al., 1998).
A study in Taiwan evaluated by the Committee at its forty-ninth meeting showed that, in an area hyperendemic for infection and with moderate-to-high intake of aflatoxins, immunization against HBV had reduced the rate of HBV carriage in 6-year-old children from about 10% in 1981–86 to 0.8–0.9% in the period 1990–94. A more recent report noted that 15–20% of the population of Taiwan were estimated to be HBV carriers in the early 1980s. A programme of mass vaccination against HBV was launched in 1982, and, since 1986, all newborns and, progressively, preschool children, primary-school children, adolescents, young adults, and others have also been vaccinated. The coverage of newborns is over 90%, and 79% of pregnant women are screened for HBsAg. The proportion of babies born to highly infectious mothers who also became carriers decreased from 86–96% to 12–14%. The average annual incidence of hepatocellular carcinoma in children aged 6–14 decreased significantly from 0.7 per 100 000 in 1981–86 to 0.36 per 100 000 in 1990–94; and the annual incidence of hepatocellular carcinoma in children aged 6–9 declined from 0.52 per 100 000 for those born in 1974–84 to 0.13 per 100 000 in those born in 1986–88. Thus, the mass vaccination programme has been highly effective in controlling chronic HBV infection and in preventing liver cancer in Taiwan. If a vaccination coverage rate of 90% of all newborns against HBV can be maintained, the carriage rate in Taiwan can be expected to decline to 0.1% by 2010. The cost of the programme has been about US$ 100 million (Huang & Lin, 2000).
Recent reports from Taiwan and from other areas where massive HBV vaccination campaigns have been conducted have shown the presence of HBV mutants in the surface gene which induces chronic carriage among immunized children. The prognosis of these infected children is uncertain, but the observation should be considered in evaluating the occurrence of liver cancer in HBV-vaccinated populations (Hsu et al., 1999).
A study of vaccination against HBV was reported from the Republic of Korea, where the prevalence of HBV infection is among the highest in the world. In this prospective cohort study, 370 285 men over the age of 30 who were clinically free of liver disease and had not been vaccinated against HBV at the time of enrolment were included. About 5% of the cohort were HBsAg+, 78 094 had antibodies to the HBV surface marker, and 273 277 were negative for both. About 13% of the men had been vaccinated against HBV in 1985. Cases of liver cancer were ascertained by record linkage and from medical records covering 1986–89. A multivariate log-linear model was used to test for statistical significance and to estimate relative risks. The follow-up period represented 1 404 566 person-years (average, 3 years and 10 months), and 302 cases were ascertained, to give an overall incidence rate of liver cancer of 22 per 100 000 person-years. The relative risk for primary liver cancer was18% (95% confidence interval, 14–23) among chronically infected men, 0.34 (0.19–0.60) among unvaccinated infected men, and 0.58 (0.31–1.1) in the vaccinated group. The study suggests that vaccination against HBV, even in adulthood, reduces the risk for liver cancer (Lee et al., 1998).
A report on the results of vaccination programmes in China and The Gambia showed that the vaccine must be given as early as possible in life: vaccination within 48 h of birth reduced carriage by at least 70%. In China, some 40% of carriers of HBV were infected by perinatal transmission from their mothers. The effectiveness of vaccination has been reported to be 70% in some areas of China and as much as 90% in others. In Africa, introduction of the vaccine into the routine programme for infant vaccination reduced the carriage rate by 94%. In The Gambia, protection was shown to be maintained up to the age of 9 years, which is well past the age at which the risk of becoming a carrier is high; thus, these children effectively have lifelong protection against HBV-associated liver cancer (Wild & Hall, 2000).
Programmes to reduce the burden of liver cancer in developing countries should therefore give priority to HBV vaccination and to the prevention of HCV contamination. This implies reinforcing the control of blood and blood products and the use of sterile medical equipment. HBV carriers may benefit from reductions in intake of aflatoxins in their diets, and this may also offer some protection to HCV carriers. However, a reduction in intake of aflatoxin B1 or a reduction in the concentration of aflatoxin M1 in milk or milk products is unlikely to result in an observable reduction in the rate of liver cancer in most developed countries. In these populations, alcohol consumption may account for most cases of liver cancer without viral markers.
In spite of the effectiveness of hepatitis B vaccination in preventing chronic HBsAg carrier status in unexposed newborns, infants, and adults, a substantial number of persons (some 300 million worldwide) are HBsAg carriers. No effective treatment has been developed for these persons (Torresi & Locarnini, 2000).
Epidemiological studies suggest that dietary intake by chronic HBV carriers of aflatoxins may increase the rate of progression to hepatocellular carcinoma (Qian et al., 1994). Thus, it has been suggested that oltipraz, a drug that modifies the metabolism of aflatoxin and has a number of other biological properties (reviewed by Kensler et al., 1999) might be used as a chemopreventive agent. The experimental basis for this proposal is the demonstration of remarkable anticancer activity against aflatoxin B1-induced hepatocarcinogenesis in rats (see section 2.3.4). Oltipraz has also been evaluated as a chemopreventive agent for cancers of the colon, liver, bladder, and skin (reviewed by Kensler & Helzlsouer, 1985; Kensler et al., 1999).
In rats, continuous administration of oltipraz significantly reduced the formation of aflatoxin–albumin adducts and the occurrence of liver neoplasms (Kensler et al., 1997). Oltipraz affected the life cycle of HBV in vitro by blocking transcription in 2.2.15 cells, resulting in dose-related inhibition of HBV replication, perhaps mediated through induction of wild-type p53 (Chi et al., 1998).
Oltipraz has been tested in in phase I/II trials in China. The results of pilot studies for these trials showed reasonably good compliance with the regimen and mild toxicity, with no observed interaction with the HBV status of the individual (Jacobson et al., 1997). The results of another pilot trial showed that low daily doses of oltipraz induced phase-2 conjugation of aflatoxin, as measured by an increase in the urinary excretion of aflatoxin mercapturic acid, with no reduction in the concentration of aflatoxin M1. Intermittent high doses of oltipraz decreased the phase-1 metabolism of aflatoxin, leading to a significant reduction in excretion of aflatoxin M1, indicating that the metabolic pathways of aflatoxins in humans can be strongly modified by oltipraz (Wang et al., 1999b). The long-term effects of this chemopreventive treatment remain to be established.
Various biomarkers were studied in 23 cases of liver cancer in the USA. HBV markers including HBV DNA were found in 13 cases, HCV antibodies in sera in 5/22, overexpression of P53 in tissue in 5/23, and mutations in codon 249 in 0/5. Surprisingly, aflatoxin B1–DNA adducts were found in liver tumour tissue in 3/19 cases and aflatoxin B1–lysine adducts in sera in 5/5, none of which had concurrent overexpression of P53 or mutations at codon 249. Few cases were available for each test, and only the abstract has been published; however, the presence of markers of aflatoxin in cases of liver cancer in the USA is interesting because intake of aflatoxin in that country is expected to be low. HBV or HCV infection would also be expected to be infrequent, depending on the subpopulation sampled (Hoque et al., 1999).
Aflatoxin–albumin adducts were also identified in serum from 104 volunteers in the United Kingdom. There was no direct correlation with a particular food (Turner et al., 1998).
Mutations of P53 are a relevant marker in the molecular epidemiology of liver cancer, as some 20% of cases show mutations of this oncogene. Moreover, a mutation at the third base of codon 249 (a GC to TA transversion leading to a change from arginine to serine) has been described in geographical correlation studies of intake of aflatoxin B1.
The IARC and other databases on p53 clearly describe the ‘hot spot’ at 249 as the predominant mutation in liver cancer. The IARC database is heavily biased by publication and reporting selection, but 35–40% of cases of liver cancer in areas where there is high intake of aflatoxins show the presence of the 249 mutation, whereas there is a much lower prevalence (0–2%) in areas where there is low intake (Soussi et al., 2000).
The most recent reviews of ‘molecular fingerprints’ of carcinogens appear to converge in accepting a few for which some specificity can be claimed: GC to TA transversions in lung cancer associated with smoking; GC to TA transversions in codon 249 in liver cancer associated with aflatoxin B1; and CC:GG to TT:AA transversions in skin cancer associated with exposure to ultra-violet light (Hainaut & Vahakangas, 1997; Wang & Groopman, 1999).
Since the last evaluation by the Committee, additional studies have shown that a mutation in codon 249 of P53 is found regularly in a proportion of cases of liver cancer in certain countries and not in others. Several studies showed that these mutations are poorly correlated with another marker of aflatoxin intake, the presence of aflatoxin B1 adducts, in hepatocellular carcinoma tissue (Hsie et al., 1995; Soini et al., 1996; Lunn et al., 1997). Experimental data have also shown that the mutation can be induced in hepatocytes exposed to aflatoxin B1; aflatoxin metabolites bind to the third base in codon 249; and 249 ser p53 expression inhibits apoptosis and p53-mediated transcription and enhances liver cell growth in vitro (reviewed by Hussain & Harris, 2000).
Most of the studies reported below involved few cases, and even fewer cases with P53 mutations, and many of the comparisons and ORs calculated from them are therefore quite unstable. Many of the reports focus on mutations and not on the full spectrum of genetic alterations in P53 that characterizes hepatocellular carcinoma. Most of these mutations have been identified in individuals who are also HBV carriers.
Of 21 cases of hepatocellular carcinoma in India, three had mutations in P53 (two in 249 GT and one in a 250 CT transition). In investigations for HBV status, 59% of the cases were shown to have HBV DNA by dot blotting, 90% to have HBV DNA by polymer chain reaction, and 71% to be HBsAg+ by enzyme-linked immunosorbent assay (ELISA). The report indicated that intake of aflatoxin was common in that part of India (Katiyar et al., 2000).
In a report on 24 cases of liver cancer in Shanghai and Qidong, China, all specimens had integrated HBV DNA, and 63% had the null GSTM1 genotype; 95% had alterations in P53: 12 had mutations in P53, and 13 had overexpression. Loss of heterozygosity at 4q was found in 50%, at 1p in 46%, at 16q in 42%, and at 13q in 38%. Mutation at codon 249 was found in seven cases from Qidong (all those with a P53 mutation) and in three of five from Shanghai (Rashid et al., 1999).
Of 30 cases of hepatocellular carcinoma from Guangxi, China, an area of high risk for HBV and exposure to aflatoxin, 90% were HBsAg+, and 43% showed P53 expression and a linear response with the stage of tumour (Qin et al., 1997).
Seven of 21 samples of tissue from patients with hepatocellular carcinoma in Tongan, China, had point mutations at codon 249 resulting in a G to T transversion. Only one of the patients was HBV-negative (method not stated). The authors also reported another case of HBV-negative hepatocellular carcinoma with a mutation at codon 249 of P53 (Yang et al., 1997).
In a study in Taiwan of 110 cases of liver cancer and 37 controls, HBV status was assessed by assay for HBsAg, intake of aflatoxin by aflatoxin B1 adducts in liver tissue, P53 status by immunohistochemistry, and DNA mutations by single-stranded conformation polymorphism and sequencing. The main findings were elevated risks associated with HBsAg seropositivity (OR, 8.4) and aflatoxin B1 adducts (OR, 3.9), with an OR of 68 for both. P53 mutations were found in 29% of cases, and mutations at codon 249 in 13%. The presence of aflatoxin B1 adducts in liver tissue was related to the presence of P53 protein and DNA mutations (borderline significance). Mutations in codon 249 were found only in HBsAg+ subjects, suggesting that HBV is involved in the selection of these mutations. Because liver tissue was required for these comparisons, the controls were patients with liver or biliary-tract conditions, including hepatic metastases from other primary cancers (Lunn et al., 1997).
In a small correlation study, the P53 mutation patterns in 31 cases of hepato-cellular carcinoma from northern and southern Jiang-Su Province in China were compared. Mutations in codon 249 were found in 9/16 cases in the area with high intake of aflatoxin and 1/15 in the area with lower intake (Shimizu et al., 1999).
In a study in Spain, 120 paraffin blocks from hepatocellular carcinoma cases were studied by single-stranded conformation polymorphism and sequencing techniques. No mutation was found in P53, although P53 overexpression was found in 14 cases. The authors concluded that P53 mutations are not common in hepatocellular carcinoma induction or promotion in Spain (Boix-Ferrero et al., 1999).
An investigation was conducted of circulating DNA for P53 codon 249 mutations in a series of 53 hepatocellular carcinoma cases in The Gambia and in 13 patients with liver cirrhosis. There were 53 controls and a second control group of 60 French patients with a variety of liver conditions. The relevant mutation was found in 19 cases of hepatocellular carcinoma, two patients with cirrhosis and three controls. The OR for hepatocellular carcinoma was 16 (3–90). None of the patients in France had the 249 mutation (Kirk et al., 2000).
In a series of 62 hepatocellular carcinoma cases in Taiwan, P53 mutations were investigated by single-stranded conformation polymorphism and sequencing; 37 of the patients were HBsAg+ and 25 HBsAg–. Twenty patients had mutations, which were widely distributed along exons 5–8. Four patients, all HBsAg carriers, had a G to T mutation at codon 249 (Sheu, 1997).
A cluster of mutations was found at position 220 in P53 in patients with genetic haemochromatosis and liver cancer. Mutations in codon 249 exon 7 A/T were observed in one case. Although anecdotal, these observations suggest that a mutation at this locus may also be acquired in other contexts (Vautier et al., 1999).
A recent, unpublished meta-analysis on the relationship between HBV, aflatoxin, and mutation at codon 249 of P53 showed that the geographical relationship between intake of aflatoxins (broadly classified into three levels) and P53 mutations (any spot) was strongly correlated. The correlation was due almost entirely to the G to T mutation, but a significant (albeit unstable) trend with mutations at other codons remained. There was no indication that the presence of the 249 mutation varied with HBV status, although one study showed a significant interaction (Stern et al., 2001, personal communication).
Table 4 summarizes the results of studies on P53 mutations in cases of liver cancer. Intake of aflatoxins is expressed crudely in relation to the geographical source of the specimens, thus ignoring local and individual variation. Although P53 mutations occur at both the hot spot (249 G to T) and other spots, mutations at codon 249 predominated (92% versus 2%) in relation to the geographical classification. The presence of the mutation was affected only moderately by the concurrent presence of HBV.
Table 4. P53 mutations in cases of liver cancer according to exposure to aflatoxin
|
Extent of exposure to aflatoxins |
No. of cases of liver cancer |
G–T mutations at |
Total P53 mutations |
Mutations at codon 249/ total P53 mutations (%) |
||
|
% |
Range |
% |
Range |
|||
|
Higha |
259 |
49 |
30–83 |
56 |
45–69 |
92 |
|
Intermediateb |
495 |
10 |
0–30 |
28 |
13–50 |
27 |
|
Lowc |
651 |
2 |
0–11 |
24 |
0–35 |
2 |
From Lunn et al. (1997); Qin et al. (1997); Sheu (1997); Yang et al. (1997); Boix-Ferrero et al. (1999); Rashid et al. (1999); Shimizu et al. (1999); Katiyar et al. (2000)
a
China, rural sub-Saharan Africab
Urban China, urban sub-Saharan Africa, Thailandc
Europe, Japan, northern China, Singapore, USAThere is growing consensus that the mutational spectra of P53, and in particular codon 249, is a relevant biomarker of intake of aflatoxin B1 in relation to hepatocellular carcinoma. In order to make full use of this biomarker, the natural history of the mutation should be characterized, and its relationship to the dose of aflatoxin B1 and to better validated biomarkers, such as aflatoxin B1–albumin adducts in urine, should be established.
In epidemiological studies, the finding of this mutation in specimens of hepatocellular carcinoma can be interpreted as reflecting the involvement of aflatoxin B1, but its absence, particularly in cancers in people living in areas of heavy intake of aflatoxin B1 should be interpreted with caution, as aflatoxin B1 might induce hepatocellular carcinoma by mechanisms other than DNA damage.
Early detection of aflatoxin M1 and removal of small lots of contaminated milk can prevent contamination of much larger volumes. Screening methods are particularly useful if they can be carried out quickly, easily, and economically. They should allow detection of concentrations of aflatoxin M1 in milk as low as those detected by the ultimate quantitative methods (see section 3.2). Theoretically, screening methods should never give false-negative results.
The screening tests used for aflatoxin M1 in milk and milk products are usually immunochemical. For aflatoxin M1, both radioimmunoassays and enzyme immunoassays have been developed (Frémy & Chu, 1989), although enzyme immunoassays are used more often.
The protocol for radioimmunoassay of aflatoxin M1 usually includes simultaneous incubation of a test solution containing an unknown amount of aflatoxin M1 (or a standard solution of a known amount of aflatoxin M1 in phosphate buffer) with a constant amount of labelled aflatoxin and its specific antibody. Free and bound labelled aflatoxin are then separated, and the radiolabel on aflatoxin is determined. The aflatoxin M1 concentration of the test solution is determined by comparing the results to a standard curve, which is established by plotting the ratio of bound and the initial (total) amount of labelled aflatoxin multiplied by 100 (% binding) versus the concentration of aflatoxin M1 standard (Chu, 1984). Radioimmunoassay has found little use in routine investigations of aflatoxin M1 in milk. A recent exception is an investigation of samples of milk in Thailand (Saitanu, 1997). There have been no collaborative studies on radioimmunoassays for aflatoxin M1.
Most enzyme immunoassays for aflatoxin M1 are heterogeneous, with separation of the immunocomplex and the unreacted material. One of the commonest heterogeneous enzyme immunoassays, the ELISA, is generally used to determine aflatoxin M1. Several direct competitive ELISAs for aflatoxin M1 are available commercially. The 96-well microtitre plate assay is most commonly used for quantitative measurements. ELISAs for aflatoxin M1 are usually designed for rapid screening. For legal purposes, positive results in an ELISA require confirmation by an accepted reference method.
One successful use of a direct competitive ELISA, in which the results were confirmed by a validated HPLC method, involved ELISA for the determination of aflatoxin M1 in pasteurized milk, infant formula, powdered milk, and yoghurt (Kim et al., 2000). A valuable addition was comparison of the ELISA with the validated HPLC method of Ferguson-Foos & Warren (1984; see also section 3.2). Kim et al. concluded that the results for aflatoxin M1 obtained with ELISA were similar to those obtained by HPLC. In a study in which ELISA and HPLC with immunoaffinity purification were compared, the latter technique was found to be superior (Biancardi, 1997). An interesting development is a portable field test involving a patented, membrane-based flow-through enzyme immunoassay (Sibanda et al., 1999), which can be carried out on farms. The kit comprises a nylon membrane spotted with anti-mouse antibodies, a plastic snap-fit device, absorbent cotton wool, mouse monoclonal antibodies against aflatoxin M1, and aflatoxin B1–horse radish peroxidase conjugate. Clean-up is done on an immunoaffinity column (see section 3.2). No collaborative studies have been carried out, at least not under the auspices of international organizations, of ELISAs for aflatoxin M1.
Techniques other than immunochemical procedures can, in principle, be used for rapid analysis of milk and milk products for aflatoxin M1. One such technique involves electrochemical flow injection monitoring on filter-supported bilayer lipid membranes (Andreou & Nikolelis, 1998). The method has not been formally validated.
It has been recommended that extensive collaborative studies be conducted to validate the performance characteristics of immunoassays, including their reproducibility and repeatability, accuracy, sensitivity, limits of detection, specificity, and selectivity (Frémy & Chu, 1989). This recommendation was echoed in 2000 (van Egmond, 2000), as no such studies have been carried out by AOAC International, the organization responsible for collaborative studies of methods of analysis (International Union of Pure and Applied Chemistry, 1989). The International Dairy Federation (1999) has produced a guideline document that outlines the parameters necessary for the evaluation and validation of competitive enzyme immunoassays for quantitative determination of aflatoxin M1 in milk and milk products.
Quantitative analytical methods for aflatoxin M1 usually follow the general pattern for mycotoxin assays, i.e. extraction, clean-up, concentration, separation, detection, and quantification. A homogeneous sample of aflatoxin M1 in milk is easily obtained, because the toxin is distributed evenly in fluid milk. The initial problem encountered in analysing milk is the extraction step. Because milk is a complex natural product, aflatoxin M1 is not easily extracted and purified for final assay, owing (partly) to adsorption of aflatoxin M1 to casein proteins. A process is required to separate aflatoxin M1 from milk easily, efficiently, and economically. Once purified extracts are obtained, the concentration of aflatoxin M1 can be determined in one of several ways. Most quantitative methods involve thin-layer chromatography (TLC) or HPLC. Aflatoxin M1 is a weakly polar component and is extractable with solvents such as methanol, acetone, chloroform, or combinations of these solvents with water. In practice, the choice of solvent depends on the clean-up and the separation procedure.
The quantitative methods that have been developed and validated for aflatoxin M1 in milk and milk products were originally designed to analyse milk powder. Milk was spray-dried or lyophilized to preserve its shelf life and to reduce sample bulk. Various mixtures of methanol and water (Masri et al., 1968, 1969a; Fehr et al., 1971), acetone and water and acetone, chloroform, and water (Purchase & Steyn, 1967) were used to extract aflatoxin M1 from milk powder.
In the first effective method for the determination of aflatoxin M1 in fluid milk, methanol and water were used as the extraction solvents (Jacobson et al., 1971). This method was modified by others (McKinney, 1972; Stubblefield & Shannon, 1974a), leading to a method suitable for collaborative studies. The method of Stubblefield & Shannon (1974a) involved extraction with acetone and water, precipitation with lead acetate solution to deproteinize the milk, and a defatting step with hexane. TLC with fluorescence detection was used for ultimate separation, detection, and quantification. The collaborative study was successful (Stubblefield & Shannon, 1974b), and the method became an official method for aflatoxin M1 of the AOAC and IUPAC ( AOAC official method 974.17, surplussed in 1993; see Table 5 for performance characteristics).
Table 5. Performance characteristics of methods of analysis for aflatoxin M1 in milk and milk products that have been tested in laboratories providing acceptable results in formal collaborative studies by international organizations
|
Sample |
No. of laboratories |
Mean |
Recovery |
Relative SD (%) |
|
|
Repeatability |
Reproducibility |
||||
|
AOAC method 974.17 (Stubblefield & Shannon, 1974b), in accordance with Scott (1989) |
|||||
|
Naturally contaminated: |
|||||
|
Powdered skimmed milk |
14 |
1200 |
– |
– |
73 |
|
Powdered whole milka |
30 |
2800 |
– |
– |
51 |
|
Cheese (ricotta) |
12 |
850 |
– |
– |
48 |
|
Butter |
8 |
480 |
– |
– |
45 |
|
Spiked: |
|||||
|
Milk with 100 ng/kg |
14 |
140 |
136 |
– |
75 |
|
Cheddar cheese with 500 ng/kg |
11 |
270 |
54 |
– |
65 |
|
Uncontaminated: |
|||||
|
Milk |
14 |
0 |
– |
– |
– |
|
Blue cheese |
14 |
450b |
– |
– |
– |
|
Sample |
No. of laboratories |
Mean |
Recovery (%) |
Relative SD (%); visual densitometry |
|||
|
|
|
|
|
Repeatability |
Reproducibility |
||
|
AOAC method 980.21 (Stubblefield et al., 1980), in accordance with Scott (1989) |
|||||||
|
Naturally contaminated: |
|||||||
|
Powdered milkc |
32 |
3000 |
– |
36 |
23 |
36 |
38 |
|
Powdered milkc |
38 |
4800 |
– |
28 |
19 |
32 |
29 |
|
Cheesed |
48 |
340 |
– |
40 |
33 |
58 |
47 |
|
Cheesec |
37 |
980 |
– |
27 |
30 |
23 |
40 |
|
Cheesed |
53 |
1400 |
– |
19 |
20 |
33 |
42 |
|
Spiked: |
|||||||
|
Powdered milk with 1120 ng/kgc |
36 |
1000 |
91 |
32 |
21 |
71 |
34 |
|
Butter with 1000 ng/kg |
12 |
450 |
45 |
8.0e |
68 |
24e |
82 |
|
Uncontaminated: |
|||||||
|
Powdered milkc |
42 |
– |
– |
– |
|
– |
|
|
Cheesec |
38 |
– |
– |
– |
|
– |
|
|
Samplef |
No. of valuesg |
Mean |
Recovery |
Relative SD (%) |
|
|
Repeatabilityh |
Reproducibilityi |
||||
|
AOAC method 986.16 (Stubblefield & Kwolek, 1986), in accordance with Scott (1989) |
|||||
|
Spiked fluid milk |
|||||
|
At 770 ng/L |
20 (25) |
600 |
78 |
– |
– |
|
At 155 ng/L |
20 (26) |
140 |
92 |
– |
– |
|
At 67 ng/L |
18 (22) |
67 |
86 |
– |
– |
|
At 415 ng/L |
18 (23) |
420 |
88 |
– |
– |
|
At 1300 ng/L |
20 (26) |
1300 |
99 |
– |
– |
|
At 116 ng/L |
20 (26) |
120 |
97 |
– |
– |
|
Sample |
No. of laboratories |
Mean |
Recovery |
Relative SD (%) |
|
|
Repeatability |
Reproducibility |
||||
|
Dairy Federation standard 171 (Tuinstra et al., 1993) |
|||||
|
Naturally contaminated milk powder |
12 |
81 |
– |
9.9 |
23 |
|
14 |
150 |
– |
14 |
23 |
|
|
13 |
80 |
– |
6.8 |
18 |
|
|
11 |
200 |
– |
4.7 |
11 |
|
|
14 |
580 |
– |
12 |
19 |
|
|
Sample |
No. of laboratories |
Mean |
Recovery |
Relative SD (%) |
HORRAT |
|
|
Repeatability |
Reproducibility |
|||||
|
AOAC method 2000.08 (Dragacci et al., 2001) |
||||||
|
Naturally contaminated milk powder |
12 |
0.023 |
– |
17 |
27 |
0.33 |
|
12 |
0.046 |
– |
12 |
23 |
0.31 |
|
|
12 |
0.100 |
– |
8 |
21 |
0.33 |
|
|
Spiked milk powder with 0.05 ng/mL |
10 |
0.037 |
74 |
18 |
31 |
0.44 |
|
Uncontaminated milk powder |
12 |
– |
– |
– |
– |
– |
SD, standard deviation; Recovery, mean recovery from sample spiked with known amount of aflatoxin M1; HORRAT, ratio of relative SD for reproducibility in the trial to that predicted. A HORRAT of 1 indicates a relative SD for reproducibility corresponding exactly to the Horwitz equation (Horwitz, 1989); a HORRAT < 1.0 ± 0.5 indicates normal reproducibility; a HORRAT > 1.5 indicates that reproducibility is higher than expected, whereas a HORRAT > 2.0 indicates problematic reproducibility (AOAC International, 2000).
|
a |
Duplicate series of samples: no significant difference in means of two sets found by t test. |
|
b |
Fluorescent contaminant |
|
c |
Duplicate series of samples |
|
d |
Triplicate series of samples |
|
e |
Four samples instead of 12 |
|
f |
Duplicate samples |
|
g |
In parentheses, total number of values including normal-phase data used for estimates of precision |
|
h |
Individual values were reported, but it was not clear from the original publication whether they related to repeatability or reproducibility. Relative SD for repeatability for all samples combined, 28% |
|
i |
Individual values were reported, but it was not clear from the original publication whether they related to repeatability or reproducibility. Relative SD for reproducibility for all samples combined, 44% |
|
j |
Values based on reconstituted milk prepared from the milk powder |
In another method, liquid milk was partitioned with chloroform in a separating funnel and cleaned-up over a small silica gel column. Final separation was by TLC with fluorescence detection, and aflatoxin M1 spots were quantified by visual or densitometric estimation (Stubblefield, 1979). This method was further modified to allow determination of aflatoxin M1 in cheese, in which two-dimensional TLC was used to improve separation of the aflatoxin M1 spots from the background. The method was evaluated in an AOAC/IUPAC collaborative study (Stubblefield et al., 1980) and became an official AOAC method for aflatoxin M1 in milk and cheese (AOAC official method 980.21; see Table 5 for performance characteristics).
With advances in HPLC methods in the 1980s, laboratories moved away from TLC to HPLC determination. In addition, factory-prepared solid-phase extraction columns were introduced for the purification of milk extracts. A method that successfully combined these two developments was that of Ferguson-Foos & Warren (1984), originally developed for normal-phase HPLC. The method was modified for reversed-phase HPLC, with the preparation of trifluoroacetic acid derivatives of aflatoxins M1 and M2, and evaluated in an AOAC collaborative study (Stubblefield & Kwolek, 1986). The method became AOAC official method 986.16. for aflatoxin M1 in fluid milk (see Table 5 for performance characteristics).
A more recent advance in quantitative extraction of aflatoxin M1 and subsequent clean-up is use of immunoaffinity cartridges. These columns are composed of monoclonal antibodies specific for aflatoxin M1, which are immobilized on Sepharose® and packed into small cartridges (see Figure 2). The first published method for aflatoxin M1 with immunoaffinity columns was that of Mortimer et al. (1987).
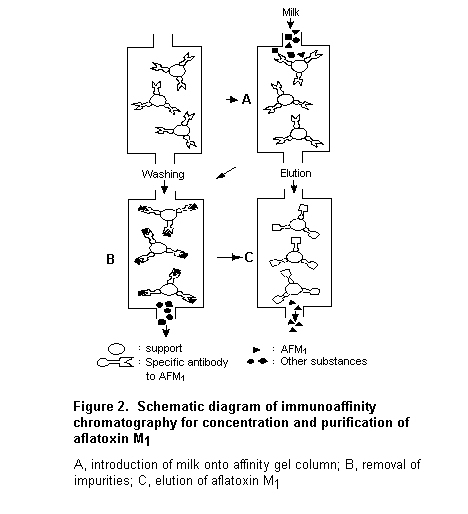
A milk sample containing aflatoxin M1 is loaded onto the affinity gel column, and the antigen aflatoxin M1 is selectively complexed by the specific antibodies on the solid support into an antibody–antigen complex. The column is then washed with water to remove all other matrix components of the sample. Aflatoxin M1 is eluted from the column with a small volume of pure acetonitrile, and the eluate is concentrated and analysed by HPLC coupled with fluorescence detection. The method can be applied to whole milk, skimmed milk, and low-fat milk.
Modifications of the immunoaffinity-based methods for aflatoxin M1 were subsequently published and studied collaboratively under the auspicies of the International Dairy Federation (Tuinstra et al., 1993) and AOAC International (Dragacci et al., 2001) by groups of mainly European laboratories that could determine aflatoxin M1 in milk at concentrations < 0.05 µg/L. The collaborative study of Tuinstra et al. led to International Dairy Federation Standard 171 (see Table 5 for performance characteristics). Another collaborative study resulted in approval as AOAC method 2000.08 (Dragacci et al., 2001; see Table 5 for performance characteristics).
The combination of immunoaffinity clean-up and liquid chromatography offers the best means for efficient purification and precise determination of low concen-trations of aflatoxin M1. The approach is widely used in some parts of the world where low limits for aflatoxin M1 in milk are in force, e.g. the European Union, where the method is successfully practised by the National Reference Laboratories for Milk and Milk Products (Dragacci et al., 2001). The method may, however, be too expensive for routine use in developing countries. An interesting lower-cost alternative is a method that combines immunoaffinity clean-up with TLC and a computer-based, low-cost densitometer. This method is being validated for detection of low concentrations of aflatoxin M1 in a formal collaborative study.
Ensuring that regulations on limits for aflatoxin M1 in milk and milk products are met requires validated methods of analysis. Several organizations (AOAC International, IUPAC, the International Dairy Federation, and the European Standardization Committee [Comité Européen de Normalisation]) have tested methods of analysis for aflatoxin M1 collaboratively in order to establish their performance characteristics. The practical characteristics include: cost of performance, time required, and level of training needed. The scientific characteristics include: accuracy, precision, specificity, and lower limit of detection. Although all these characteristics are important, the most important from the regulatory point of view are accuracy and interlaboratory variation (reproducibility).
Few collaboratively studied methods of analysis for aflatoxin M1 have been published in the scientific literature. Nearly all are based on TLC and HPLC. The performance characteristics of the methods described above, derived from collaborative studies, are summarized in Table 5.
Criteria for acceptance of collaboratively derived performance characteristics of methods for the determination of aflatoxin M1 were not formalized until 1999 in countries that had regulations with respect to aflatoxin M1. In 1999, European Union legislation for aflatoxin M1 came into force. The Directive (98/53/EC; Commission of the European Union, 1998) includes a section stating specific requirements for the methods of analysis used to determine concentrations of aflatoxins in certain foodstuffs, including aflatoxin M1. These requirements are based on report 13505 (Comité Européen de Normalisation, 1999). The recommended recovery of aflatoxin M1 present in milk is 60–120% at a concentration of 0.01–0.05 µg/L and 70–110% at a concentration > 0.05 µg/L. The recommended precision of the relative standard deviation for reproducibility for all concentrations of aflatoxin M1 in milk is that derived from the Horwitz (1989) equation, i.e. RSDR = 2 (1–0.5 logC), where RSDR is the relative standard deviation calculated from results generated under reproducible conditions [SR/x], and C is the concentration expressed as powers of 10 (e.g. 1 mg/kg (ppm) = 10–6). The maximum permitted value is twice the RSDR. The detection limits of the methods used are not stated, as the precision values are given at the concentrations of interest.
Most collaborative studies on aflatoxin M1 were carried out under the auspices of AOAC International, which did not require the reporting of recovery values or the limit of detection or determination in collaborative studies until 2000. In addition, reporting of HORRAT values was not mandatory, and no criteria were established for HORRAT values until 2000 (AOAC International, 2000). Thus, except for the collaborative study of Dragacci et al. (2001), there were no criteria to verify the performance of a method for aflatoxin M1 in collaborative studies.
The availability of collaboratively studied ‘official’ methods of analysis for aflatoxin M1, with acceptable performance characteristics, is no guarantee of accurate results. Check sample programmes for aflatoxins, including aflatoxin M1 in milk, organized by IARC (Friesen & Garren, 1982) have shown that there can be wide variation in results. In compliance with the principles of analytical quality assurance, measurements of the mycotoxin by different laboratories should be reliable and comparable. A quality assurance programme includes, when possible, use of (certified) reference materials and many other elements. Certified reference materials are stable, homogeneous products containing certified amounts of the analyte(s) of interest. Such materials for aflatoxin M1 have been developed in the past with the coordination of the European Union’s Community Bureau of Reference, now known as the Standards, Measurements and Testing Programme. Several full-cream milk powders certified for their aflatoxin M1 content and an aflatoxin M1 calibrating solution are available worldwide from the European Union Joint Research Centre, Institute for Reference Materials and Measurements in Geel, Belgium. Their characteristics are shown in Table 6 (Boenke, 1997). The supplies of certified milk powder reference materials are nearly exhausted, and the Institute is planning to produce and certify new batches in 2001.
Table 6. Certified reference materials for aflatoxin M1
|
Number |
Matrix |
Certified value (µg/kg) |
Uncertainty (µg/kg) |
|
CRM 282 |
Full-cream milk powder |
< 0.05 |
Not reported |
|
CRM 283 |
Full-cream milk powder |
0.09 |
+ 0.04; – 0.02 |
|
CRM 285 |
Full-cream milk powder |
0.76 |
± 0.05 |
|
RM 423 |
Chloroform |
Information value: 9.93 |
Not reported |
Another increasingly important quality assurance component is proficiency testing. The European Union’s Community Reference Laboratory for Milk and Milk Products, in Paris, France, conducted proficiency tests for national reference laboratories in 1996 (Dragacci et al., 1996) and 1998 (Grosso et al., 1999). The organizers concluded that, considering the very low concentrations of aflatoxin M1 in the distributed samples, the network of national reference laboratories had shown good analytical competency for the determination of aflatoxin M1 in milk at the permitted level in the European Union (0.05 µg/kg). It is foreseen that proficiency tests for aflatoxin M1 will be expanded to include national reference laboratories that are charged with detecting mycotoxins. Laboratories that do not belong to the network can take part in the food analysis performance assessment scheme based in the United Kingdom but with worldwide participation. The scheme has organized rounds of testing for aflatoxin M1 since 1999, and samples are sent out every 3–4 months (Ministry of Agriculture, Fisheries and Food, 1999, 2000a,b). Another possibility is participation in the laboratory proficiency programme of the American Oil Chemists (2000).
Many methods of analysis have become available for the determination of aflatoxin M1 in milk and milk products, both for screening and for quantitative estimates. Most were developed for the analysis of milk and milk products, but they can be used for other dairy products, with minor modifications. The limits of determination have decreased over the years, while the precision of the methods has improved, as demonstrated in formal collaborative studies of performance characteristics. With modern methods of analysis, aflatoxin M1 can be determined at concentrations well below 0.05 µg/kg of milk. The combination of immunoaffinity columns and liquid chromatography offers the best means for efficient clean-up and precise determination of low concentrations of aflatoxin M1. Cheaper alternatives based on immunoaffinity clean-up with TLC and computer-based densitometry have been developed and are being validated.
A value of 0.05 µg/kg of milk is currently the legal limit in those countries that have the most stringent regulations for aflatoxin M1. This value was proposed by the Codex Committee on Food Additives and Contaminants in 1992 (Codex Alimentarius, 1992), but it was still at the proposal stage in 2000, as several countries had reserved their positions (Codex Alimentarius, 2000). The availability of collaboratively studied methods (which usually involve TLC and HPLC) is, however, no guarantee of accurate results. The reliability and comparability of analytical results can be significantly improved by making use of reference materials and by taking part in proficiency studies. Certified milk powder reference materials for determination of aflatoxin M1 are available, and proficiency studies with worldwide participation are organized.
Two effective methods for controlling aflatoxin M1 in the food supply are to sample dairy feed for aflatoxin B1 or to sample the milk directly for aflatoxin M1. The performance of sampling plans for alfatoxin in granular feed products such as shelled maize (Park et al., 2000; Johansson et al., 2000a,b,c) and cottonseed (Whitaker et al., 1976) has been evaluated, but there has been little evaluation of sampling plans to detect aflatoxin M1 in milk. It might be difficult to design an effective programme to control aflatoxin in granular feed, particularly at low concentrations, because of its heterogeneous distribution in these commodities, which results in wide sampling variation.
As the distribution of aflatoxin M1 in liquid milk can be expected to be reasonably homogeneous, sampling of liquid milk for aflatoxin M1 will be more accurate than sampling of granular feed products. Most of the uncertainty in estimates of aflatoxin M1 in milk is probably associated with the analytical procedure.
The European Union, the South American Common Market (MERCOSUR), and the USA have designed plans for sampling aflatoxin M1. A Directive of the Commission of the European Union (1998) and a Decision (Commission of the European Union, 1991) specify that a minimum of 9.5 kg (or L) should be collected from a batch of milk mixed by manual or mechanical means and should be composed of at least five increments. The batch is accepted if the concentration of aflatoxin M1 does not exceed the permitted limit. In the USA, the Food & Drug Administration (1996) stipulates that samples should consist of at least 10 lbs (4.5 kg) of milk, composed of no fewer than 10 randomly selected portions.
In the absence of information on the efficacy of sampling plans for the determination of aflatoxin M1 in milk, it is recommended that the European model, in which a 500-g sample composed of five 100-g portions of milk is taken from a batch, be used for the minimum sample size and sample selection method.
As aflatoxin M1 occurs frequently in milk, two questions can be raised. What happens to the aflatoxin M1 when contaminated milk is processed normally in the dairy industry? What can be done to reduce or destroy aflatoxin M1 in contaminated milk and milk products? The many investigations performed to resolve these questions were reviewed in detail by Yousef & Marth (1989). The main studies and conclusions are summarized below.
Treatments that are common in the dairy industry can be separated into two distinct processes: those that do not involve separation of milk components, such as heat treatment, low-temperature storage, and yoghurt preparation; and processes that involve separation of milk components, such as concentration, drying, and cheese and butter production.
The stability of aflatoxin M1 during heat processing, such as pasteurization (Alcroft & Carnaghan, 1962) and heating milk directly on a fire for 3–4 h (Patel et al., 1981), has been studied. Although the results of the studies are not consistent, most indicate that such heat treatments do not change the amount of aflatoxin M1 in these products appreciably. Studies of the stability of aflatoxin M1 in milk during cool or frozen storage gave variable results (Yousef & Marth, 1989), but storage of frozen contaminated milk and other dairy products for a few months did not appear to affect the aflatoxin M1 content. The manufacture of cultured dairy products, such as kefir and yoghurt, also did not significantly decrease the aflatoxin M1 content (Wiseman & Marth, 1983).
The results of several investigations have been published in which the effects of removal of water on aflatoxin M1 content were studied. The processes involved both heat- (spray or roller drying) and freeze-drying. The studies were reviewed by Yousef & Marth (1989). Large losses of aflatoxin M1 were reported in some studies, whereas in others concentrating milk did not affect its aflatoxin M1 content substantially. The few studies that addressed partitioning of aflatoxin M1 during cream and butter processing confirmed that a small proportion of aflatoxin M1 is carried over to cream and a yet smaller proportion to butter. No loss of aflatoxin M1 occurred as the remainder was found in skim milk and buttermilk, respectively.
The manufacture of cheese involves several processes. Aflatoxin M1 does not appear to be degraded in the first phase, conversion of milk into pressed curd, as the amount in whey and curd is approximately the same as in the original milk (Yousef & Marth, 1989). Aflatoxin M1 seemed rather to occur predominantly with casein, so that cheese curd contained a higher concentration than whey. The association of aflatoxin M1 with casein is also manifested in a higher concentration in cheese than in the milk from which the cheese is made. Yousef & Marth (1989) expressed the concentration of aflatoxin M1 in milk divided by the concentration of aflatoxin M1 in cheese as the enrichment factor. On the basis of several studies, these researchers concluded that the enrichment factor is 2.5–3.3 in many soft cheeses and 3.9–5.8 in hard cheeses. During the second phase of cheese manufacture, ripening, some discrepancies were found in the stability of aflatoxin M1, but, in general, it did not appear to be degraded during ripening.
The observation that the processes described above do not generally lead to loss of aflatoxin M1 is of considerable practical importance. Several possibilities for eliminating or inactivating aflatoxin M1 in milk, involving chemical and physical treatment, have been investigated. The chemicals that have been studied for their ability to degrade aflatoxin M1 are limited to those that are permitted as food additives: sulfites, bisulfites, and hydrogen peroxide (Applebaum & Marth, 1982a,b). When raw milk naturally contaminated with aflatoxin M1 was treated with 0.4% potassium bisulfite at 25 °C for 5 h, the concentration decreased by 45%. A higher concentration of bisulfite was less effective. Aflatoxin M1 in naturally contaminated milk was not affected by the presence of 1% hydrogen peroxide at 30 °C for 30 min, but addition of hydrogen peroxide at a concentration of 0.05–0.1% with lactoperoxidase reduced the amount by 50%.
Physical processes that have been explored to remove aflatoxin M1 from milk include adsorption and radiation. Five per cent bentonite in milk adsorbed 89% of aflatoxin M1 (Applebaum & Marth, 1982a). In a study of the effects of ultra-violet radiation with and without hydrogen peroxide, the concentration of aflatoxin M1 was reduced by 3.6–100%, depending on the length of time the milk was exposed to radiation, the volume of treated milk, the presence of hydrogen peroxide, and other aspects of the experimental design (Yousef & Marth, 1985).
The chemical and physical treatments described are not readily applicable in the dairy industry, at least at present, as little is known about the biological safety, or the nutritional value of the treated products. Moreover, the costs of the processes may be considerable and prohibitive for large-scale application. If aflatoxin M1 cannot be destroyed or removed readily, it can be excluded from milk only by eliminating aflatoxin B1 from the diet of animals.
Data on contamination of milk and various milk products with aflatoxin M1 were submitted to FAO by Argentina, Canada, the European Union, Norway, the Dubai Municipality of the United Arab Emirates, and the USA. Although not formally submitted, unpublished data were also available from an Indonesian research institute. Other data were taken from literature published between 1985 and early 2000 and from a review by Galvano et al. (1996a). In addition to published data, information was presented by the Philippines, the Republic of Korea, and Thailand. Data on natural occurrence before the mid-1980s and mid-1990s were surveyed by van Egmond (1989) and Galvano et al. (1996a), respectively. Although the available data are incomplete, they are presented in Appendix A.
None of the samples examined in Argentina in 1999 (Centro de Referencia de Micología, 1999) and Uruguay in 1993–95 (Pineiro et al., 1996) contained aflatoxins at concentrations > 0.05 µg/kg, and the maximum concentration was 0.03 µg/kg. Data for Brazil were taken from Martins & Martins (1986), de Sylos et al. (1996), Correa et al. (1997), Oliveira et al. (1997), Souza et al. (1999), and Prado et al. (1999, 2000). None of the milk samples was contaminated with aflatoxin M1 at > 0.5 µg/kg. Four of 52 milk samples in 1992 contained > 0.05 µg/kg, with a maximum of 0.37 µg/kg (de Sylos et al., 1996), and 35 of 54 samples taken in 1992–93 contained 0.006–0.077 µg/kg (Prado et al., 1999, 2000). Of the samples of milk powder, 2% (6/300) of those taken in 1992–93 contained > 0.5 µg/kg, with a maximum of 1.0 µg/kg (Oliveira et al., 1997).
The data submitted by Health Canada for 1994, although limited, indicated that none of the processed milk samples contained > 0.5 µg/kg, and the mean was < 0.063 µg/kg. Data for 1997–98 (not shown in Appendix A), in which the number of samples was not stated, showed no aflatoxin M1 in any of six types of milk at the limit of detection (LOD; 0.015 µg/kg).
Extensive data collected between 1995 and early 2000 were submitted by the Center for Food Safety and Applied Nutrition (Food & Drug Administration, 2000) in Washington DC, USA. This submission contained some industry-generated data, for 1998–2000 from southwestern USA (n = 5801), the midwest (n = 438), and southeastern USA (n = 13 093), and data from three Food & Drug Administration (FDA) regional laboratories: laboratory A (n = 277), laboratory B (n = 380), and laboratory 27 (n = 4225). No data were provided from northern USA because aflatoxin M1 is not a problem under the climatic conditions that prevail.
In the surveys by FDA laboratory 27 for 1995–2000, 185 (4.6%) of 4000 raw milk samples were contaminated with aflatoxin M1 at a concentration > 0.05 µg/kg and five (0.13%) at > 0.5 µg/kg, only in 1996. Of the finished milk samples during the same period, 10 (4.4 %) of 225 samples contained 0.05–< 0.5 µg/kg. In midwestern and southwestern USA after 1998, 10 (2.3%) of 438 and 14 (0.11%) of 13 093 samples, respectively, contained > 0.5 µg/kg. The incidence of aflatoxin M1 contamination was higher in southwestern (industry data) and southern states (FDA laboratory B) than in other areas. In 1998–2000, milk samples contaminated with > 0.05 and > 0.5 µg/kg were found in 21% (1239/5801) and 0.78% (45/5801) of southwestern states and in 40% (153/380) and 18% (68/380) of southern states, respectively. The numbers of samples for southwestern and southern states were provided within two distributions (0.00–0.5 µg/kg and > 0.5 µg/kg), with no information on mean or maximum values.
In Asia, high incidences and levels of aflatoxin M1 contamination were found in Indonesia, the Philippines, and Thailand. Unpublished data for Indonesia in 1990–93 and 1999 were obtained from the Research Institute for Veterinary Science (2000) in Bogor. Of 342 milk samples, 199 samples (58%) contained aflatoxin M1 (limit of quantification [LOQ], 0.1 µg/kg), and 73 (21%) contained > 0.5 µg/kg, with mean values of 0.31–5.4 µg/kg and maximum values of 2.0–23 µg/kg.
Data for Thailand in 1990–93 and 1995–96 were obtained from the Department of Medical Sciences, Ministry of Public Health (Boriboon & Suprasert, 1994) and the literature (Saitanu, 1997). Of 310 liquid milk samples, more than 261 (> 84%) were contaminated with aflatoxin M1 at aconcentrations > 0.05 µg/kg, and 58 samples (19%) contained > 0.5 µg/kg, with a maximum of 6.6 µg/kg. In the Philippines, data from the Bureau of Animal Industry, Department of Agriculture, for 1997 (Begino, 1998) indicated that 88% and 18% of 91 milk samples were contaminated with aflatoxin M1 at > 0.05 µg/kg and > 0.5 µg/kg, respectively.
The data for the Republic of Korea in 1995 and 1997 were taken from the literature (Shon et al., 1996; Kim et al., 2000). Of 134 liquid milk samples, 50 (37%) contained aflatoxin M1 at a concentration > 0.05 µg/kg, with a maximum of 0.28 µg/kg. In the United Arab Emirates, data for 1998–99 from the Dubai Central Laboratory, Dubai Municipality, showed that 33 (56%) of 59 liquid milk samples (including 15 imported products) were contaminated with > 0.05 µg/kg, with a maximum of 0.31 µg/kg.
The report from the Commission of the European Union (1999) provided data on aflatoxin M1 concentrations in milk samples (total, 7573) in 1999 from 10 Member States: Austria (20 samples), Belgium (192), Finland (296), France (234), Germany (6537), Ireland (62), the Netherlands (30), Portugal (96), Sweden (11) and the United Kingdom (95). Of these, 7259 samples (96%) contained aflatoxin M1 at concentrations below the the LOQ or LOD of the method (0.001–0.03 µg/kg); 314 samples (4.2%) contained up to 0.05 µg/kg, and none of samples contained > 0.05 µg/kg.
The Commission of the European Union (1989–95) also provided the SCOOP report, which gives the concentrations of aflatoxin M1 in milk analysed between 1984 and 1995 in eight Member States (Austria, Belgium, Denmark, Finland, France, Italy, Spain, and the United Kingdom). Of the 8791 milk samples (including crude and heat-treated milk), 34% contained concentrations below the LOQ (0.01 µg/kg)/LOD (0.001–0.01 µg/kg), 44% had concentrations up to 0.05 µg/kg, 0.33% (23 French and six British samples) had 0.051–0.1 µg/kg, and three samples from the United Kingdom had > 0.1 µg/kg, with a maximum of 0.22 µg/kg.
Additional information on a total of 18 945 samples taken in 1994–99 and 2000 was provided by the Commission of the European Union. Of 1048 samples taken in Finland in 1995–98, only one contained < 0.05 µg/kg and the others contained < 0.005 µg/kg. All 251 samples taken in France in 1998 contained < 0.05 µg/kg, and four had > 0.03 µg/kg). In 17 181 samples taken in Germany in 1996–98 and 2000, the maximum concentration was 0.033 µg/kg. All 168 samples of reconstituted milk taken in the Netherlands in 1998 contained < 0.01 µg/kg. The maximum concentration in 42 samples taken in Spain in 1998 was 0.027 µg/kg, and all 255 samples taken in the United Kingdom in 1994–95 had < 0.05 µg/kg.
Data submitted by the Norwegian Food Control Authority showed that 51 (94%) of 54 samples contained aflatoxin M1 at a mean value of 0.0014 µg/kg and a maximum of 0.009 µg/kg. Data for Cyprus were derived from the literature (Ioannou-Kakouri et al., 1999). Of 112 samples, including 10 imported products, 11 were contaminated with aflatoxin M1 at a level of 0.01–0.04 µg/kg.
Appendix B shows the distribution of aflatoxin M1 contamination in various types of milk and milk products. Table 7 gives data calculated from the surveys of FDA laboratory B (1998–2000, 380 milk samples from southern USA), FDA laboratory 27 (1995–2000, 4225 raw and finished milk samples), and the Ministry of Public Health, Thailand (1990–93, 60 raw and pasteurized milk samples; Boriboon & Suprasert, 1994).
Table 7. Distribution of aflatoxin M1 in milk in samples fromThailand and the USA
|
Concentration |
Southern USA |
USA |
Thailand |
|||
|
No. of |
% |
No. of |
% |
No. of |
% |
|
|
0.05 |
20 |
12 |
1 |
0.5 |
|
|
|
0.05–< 0.10 |
18 |
10 |
144 |
74 |
0 |
0.0 |
|
0.10 – < 0.20 |
32 |
18 |
29 |
15 |
2 |
9.5 |
|
0.20–< 0.30 |
18 |
10 |
8 |
4.1 |
3 |
14 |
|
0.30–< 0.40 |
14 |
8.0 |
5 |
2.6 |
1 |
4.8 |
|
0.40–< 0.50 |
15 |
8.6 |
3 |
1.5 |
4 |
19 |
|
0.50–< 1.00 |
40 |
23 |
2 |
1.0 |
9 |
43 |
|
1.00–< 2.00 |
11 |
6.3 |
3 |
1.5 |
1 |
4.8 |
|
2.00–< 3.00 |
5 |
2.9 |
|
|
0 |
0.0 |
|
3.00–< 4.00 |
0 |
0.0 |
|
|
0 |
0.0 |
|
4.00–< 5.00 |
4 |
2.3 |
|
|
0 |
0.0 |
|
5.00–< 6.00 |
1 |
0.6 |
|
|
0 |
0.0 |
|
6.00–< 7.00 |
2 |
1.1 |
|
|
1 |
4.8 |
|
7.00–< 8.00 |
0 |
0.0 |
|
|
|
|
|
8.00–< 9.00 |
1 |
0.6 |
|
|
|
|
|
9.00–< 10.00 |
1 |
0.6 |
|
|
|
|
|
> 10.00 |
2 |
1.1 |
|
|
|
|
|
Total no. positive |
174 |
|
195 |
|
21 |
|
|
Total no. analysed |
380 |
|
4225 |
|
60 |
|
Limited information was available on annual variations in the concentration of aflatoxin M1 in milk. The most recent data are derived from the survey submitted by FDA Laboratory 27 for 1995–2000 (Table 8). The incidence of contamination with aflatoxin M1 at a concentration > 0.05 µg/kg varied from 1.9% to 3.4% in raw milk and from 0.0% to 3.8% in finished milk in a 6-year survey, except in 1996, when these concentrations were found in 25% of raw milk samples and 19% of finished milk samples. A concentration > 0.5 µg/kg was found in 1.3% of raw milk samples only in 1996.
Table 8. Annual variations in aflatoxin M1 contamination of milk in the USA, 1995-2000
|
Year |
Raw milk |
Finished milk |
||||||||
|
No. of samples |
> 0.05 µg/kg |
> 0.5 µg/kg |
No. of samples |
> 0.05 µg/kg |
> 0.5 µg/kg |
|||||
|
No . |
(%) |
No . |
(%) |
No . |
(%) |
No . |
(%) |
|||
|
1995 |
695 |
13 |
1.9 |
0 |
0 |
79 |
3 |
3.8 |
0 |
0 |
|
1996 |
381 |
94 |
25 |
5 |
1.3 |
21 |
4 |
19 |
0 |
0 |
|
1997 |
717 |
20 |
2.8 |
0 |
0 |
53 |
1 |
1.9 |
0 |
0 |
|
1998 |
855 |
16 |
1.9 |
0 |
0 |
|
|
|
|
|
|
1999 |
877 |
26 |
3.0 |
0 |
0 |
60 |
2 |
3.3 |
0 |
0 |
|
2000 |
475 |
16 |
3.4 |
0 |
0 |
12 |
0 |
0 |
0 |
0 |
In data for Germany in 1997–2000 submitted by the Commission of the European Union, 0.11–0.27% of samples were contaminated at > 0.005 µg/kg and 0.06–0.14% at > 0.01 µg/kg in 1997–98 and 2000, and 4.3% at 0.005–0.05 µg/kg and 0.45% at 0.01–0.05 µg/kg in 1999 (Appendix A).
Extensive data on annual variation is contained in the report of a 15-year survey (1978–92) of aflatoxin M1 in milk and bulk raw milk in France (Table 9; Dragacci & Frémy, 1993). For 1978, 6246 milk samples were classified as containing < 0.05 µg/kg, 0.05–0.5 µg/kg, and > 0.5 µg/kg. According to this classification, two periods of contamination occurred in France, 1978–84 and the winters of 1988–91; highly contaminated samples, some containing up to 5 µg/kg, were found during the winters of 1978–83. The seasonal trend in milk contamination was attributed to the fact that cows receive less concentrated feed in summer, when they are grazing. Few contaminated samples were found between 1984 and 1988 and after 1991.
Table 9. Annual variation in aflatoxin M1 contamination of milk in a 15-year survey (1978–92) in France
|
Year: month |
Milk |
Bulk raw milk |
||||||||
|
No. of samples |
> 0.05 µg/kg |
> 0.5 µg/kg |
No. of samples |
> 0.05 µg/kg |
> 0.5 µg/kg |
|||||
|
No . |
(%) |
No . |
(%) |
No . |
(%) |
No . |
(%) |
|||
|
1978:11–1979:5 |
49 |
29 |
59 |
1 |
2.0 |
|
|
|
|
|
|
1979:6–10 |
37 |
6 |
16 |
0 |
0 |
|
|
|
|
|
|
1979:11–1980:5 |
44 |
20 |
45 |
3 |
6.8 |
10 |
10 |
100 |
4 |
40 |
|
1980:6–10 |
58 |
12 |
21 |
0 |
0 |
26 |
23 |
89 |
5 |
19 |
|
1980:11–1981:5 |
104 |
62 |
60 |
1 |
0.96 |
67 |
50 |
75 |
5 |
7.5 |
|
1981:6–10 |
114 |
4 |
3.5 |
0 |
0 |
42 |
5 |
12 |
0 |
0 |
|
1981:11–1982:5 |
310 |
34 |
11 |
0 |
0 |
57 |
0 |
0 |
0 |
0 |
|
1982:6–10 |
|
|
|
|
|
33 |
1 |
3.0 |
0 |
0 |
|
1982:11–1983:5 |
238 |
10 |
4.2 |
0 |
0 |
54 |
1 |
1.8 |
0 |
0 |
|
1983:6–10 |
209 |
1 |
0.48 |
0 |
0 |
41 |
0 |
0 |
0 |
0 |
|
1983:11–1984:5 |
447 |
0 |
0 |
0 |
0 |
38 |
2 |
5.3 |
0 |
0 |
|
1984:6–10 |
|
|
|
|
|
18 |
0 |
0 |
0 |
0 |
|
1984:11–1985:5 |
494 |
7 |
1.4 |
0 |
0 |
37 |
5 |
14 |
0 |
0 |
|
1985:6–10 |
|
|
|
|
|
30 |
0 |
0 |
0 |
0 |
|
1985:11–1986:5 |
466 |
0 |
0 |
0 |
0 |
42 |
1 |
2.4 |
0 |
0 |
|
1986:6–10 |
265a |
0 |
0 |
0 |
0 |
26 |
0 |
0 |
0 |
0 |
|
1986:11–1987:5 |
|
|
|
|
|
24 |
0 |
0 |
0 |
0 |
|
1987:6–10 |
449b |
2 |
0.45 |
0 |
0 |
18 |
0 |
0 |
0 |
0 |
|
1987:11–1988:5 |
277c |
0 |
0 |
0 |
0 |
29 |
0 |
0 |
0 |
0 |
|
1988:6–10 |
|
|
|
|
|
27 |
0 |
0 |
0 |
0 |
|
1988:11–1989:5 |
549d |
2 |
0.36 |
0 |
0 |
8 |
0 |
0 |
0 |
0 |
|
1989:6–10 |
|
|
|
|
|
9 |
0 |
0 |
0 |
0 |
|
1989:11–1990:5 |
526d |
13 |
2.5 |
0 |
0 |
18 |
0 |
0 |
0 |
0 |
|
1990:6–10 |
|
|
|
|
|
19 |
0 |
0 |
0 |
0 |
|
1990:11–1991:5 |
550d |
3 |
0.55 |
0 |
0 |
35 |
0 |
0 |
0 |
0 |
|
1991:6–10 |
303e |
0 |
0 |
0 |
0 |
15 |
0 |
0 |
0 |
0 |
|
1991:11–1992:5 |
|
|
|
|
|
34 |
0 |
0 |
0 |
0 |
From Dragacci & Fremy (1993)
a
1986:9–11;b
1987:1–3, 9–11;c
1988:1–3;d
11–3;e
1991:9–11Dietary intake of aflatoxin M1 was estimated from data on the concentrations of aflatoxin M1 in milk submitted to FAO/WHO, from selected reports in the literature, and from data on milk consumption in the GEMS/Food Regional Diets (WHO, 1998).
Eight data sets providing information on aflatoxin M1 concentrations in milk were submitted for consideration by the Committee. Six of these were in the submissions from Argentina, Canada, the European Union, Norway, the United Arab Emirates, and the USA, and these provided the results of analyses for aflatoxin M1 in milk from government or university institutions. Experimental data were available from an Indonesian research institute. The other two submissions were summaries of published papers from laboratories in Brazil and Uruguay. The eighth data set was in the form of summaries of published papers on the concentrations of aflatoxin M1 in milk.
The data are discussed below and summarized in Appendix B, with separate entries for aflatoxin M1 in different samples of milk. The data in Appendix B for which the number of samples and the mean value were available are summarized in Appendix C by GEMS/Food regional diet.
Data for 1993–94, 1994–95, 1995–96, and 1997–98 were submitted by Health Canada (2000). The detection limit was 0.015 µg/kg, and the analytical method included immunoaffinity clean-up columns followed by liquid chromatography coupled with fluorimetric detection. In data for 1997–98, the mean concentrations and ranges of aflatoxin M1 in nine types of milk were all < 0.015 µg/kg (Table 10).
Table 10. Concentrations of aflatoxin M1 in milk in Canada
|
Years |
Type of sample |
No. of samples |
Concentration (µg/kg) |
|
|
Mean |
Range |
|||
|
1993-94 |
Whole milk |
15 |
< 0.069 |
< 0.015-0.21 |
|
|
2% fat milk |
7 |
< 0.043 |
< 0.015-0.16 |
|
|
1% fat milk |
1 |
< 0.015 |
|
|
|
Non-fat milk powder |
2 |
< 0.037 |
< 0.015-0.059 |
|
|
2% fat lactose-reduced milk |
1 |
< 0.047 |
< 0.015-0.090 |
|
|
non-fat lactose-reduced milk |
1 |
< 0.015 |
|
|
|
Evaporated milk |
1 |
0.060 |
|
|
1994-95 |
Whole milk |
23 |
< 0.013 |
< 0.01-< 0.015 |
|
|
2% fat milk |
24 |
< 0.012 |
< 0.01-< 0.015 |
|
|
Non-fat milk |
8 |
< 0.013 |
< 0.01-< 0.015 |
|
|
Whole-milk powder |
1 |
< 0.1 |
|
|
|
Non-fat milk powder |
6 |
< 0.032 |
< 0.01-< 0.015 |
|
|
Low-fat flavoured milk |
1 |
< 0.015 |
|
|
|
2% fat lactose-reduced milk |
1 |
< 0.015 |
|
|
1995-96 |
Whole milk |
43 |
< 0.012 |
< 0.01-< 0.015 |
|
|
2% fat milk |
37 |
< 0.013 |
< 0.01-< 0.015 |
|
|
1% fat milk |
5 |
< 0.012 |
< 0.01-< 0.015 |
|
|
Non-fat milk |
5 |
< 0.01 |
|
|
|
Whole-milk powder |
2 |
< 0.01 |
|
|
|
Non-fat milk powder |
9 |
< 0.1 |
|
|
|
2% fat lactose-reduced milk |
1 |
< 0.015 |
|
|
|
Whole goat milk |
1 |
< 0.015 |
|
|
1997-98 |
Whole milk |
23 |
< 0.015 |
|
|
|
2% fat milk |
35 |
< 0.015 |
|
|
|
1% fat milk |
3 |
< 0.015 |
|
|
|
Non-fat milk |
15 |
< 0.015 |
|
|
|
Whole-milk powder |
1 |
< 0.015 |
|
|
|
Non-fat milk powder |
16 |
< 0.015 |
|
|
|
Whole goat milk |
2 |
< 0.015 |
|
|
|
Non-fat lactose-reduced milk |
1 |
< 0.015 |
|
|
|
Chocolate milk |
2 |
< 0.015 |
|
Data on the aflatoxin M1 content of milk in Argentina were submitted by the Universidad National de Lujan (2000). Concentrations of 0.0, 0.4, 0.6, 0.6, 0.6, and 0.9 µg/L (roughly equal to µg/kg) were found in six samples of fluid milk from Buenos Aires that were analysed in 1998. In response to a request about the analytical method and its LOQ/LOD, the method was identified as ‘interchemistry’, which is similar to the AOAC method but with a different clean-up procedure, allowing for an LOQ of 0.026 µg/L. It was also indicated that the milk had come from cows with problematic milk production which had been given a feed not typically used in Argentina (< 12% of milk production obtained from cows given this type of feed). Hence, the aflatoxin M1 concentrations in the six milk samples may not be typical of those in milk in this country.
Data on aflatoxin M1 in milk in the USA were submitted by the FDA (2000), including some data provided by industry for 1998–2000 from the southwest (n = 5801), midwest (n = 438), and southeast (n = 13 093). The data from the southeast were provided as the number of samples within two distributions (0.00–0.5 ng/kg and > 0.5 ng/kg), with no information on mean or maximum values. The submission also included data from three FDA regional laboratories: Laboratory A provided data from southeastern states for 1999–2000 (n = 199) and for 1997–2000 (n = 78) within distributions of 0.00–0.5 ng/kg and > 0.5 ng/kg with no mean or maximum values. Laboratory B provided data from southern states for 1998 (n = 163), 1999 (n = 167), and 2000 (n = 49). Laboratory 27 provided two sets of data for 1995–2000, one set for raw milk (n = 3942) and one for finished milk (n = 273). As the data from laboratories B and 27 were provided as individual numbers, the mean values could be calculated, and values < 0.05 µg/kg and < 0.5 µg/kg, maximum values, and values at the 90th percentile of consumption were determined.
Most of the values (72–100%) were 0–0.5 µg/kg (see Appendix B). The values for the southern states were higher than those for other regions.
Data were submitted by the Norwegian Food Control Authority, with clarification of the mean values for all samples and for positive samples, the analytical method, and individual values for all samples. Aflatoxin M1 was determined in 54 samples of milk from 49 dairies in four areas of Norway in 1998. The LOD of the analytical method was 0.0001 µg/kg. Aflatoxin M1 was found in 51 (94%) of the samples, giving a mean value of 0.0014 µg/kg for all samples, a mean of 0.0014 µg/kg for positive samples, and a median value of 0.0009 µg/kg. The highest value was 0.009 µg/kg.
Data on the concentrations of aflatoxin M1 in eight types of milk product in 1998 and 1999 were submitted by the Dubai Municipality, United Arab Emirates (Dubai Central Laboratory, 2000). It was assumed that the milk described as from ‘local dairies’ was either raw or whole. The data are summarized in Table 11.
Table 11. Concentrations of aflatoxin M1 in milk and milk products in Dubai Municipality, United Arab Emirates
|
Year |
Type of sample |
No. of samples |
< LOQa |
Maximum |
Positive |
||
|
No. |
% |
No . |
% |
||||
|
1998 |
Whole, raw milk |
22 |
1 |
4.5 |
0.31 |
21 |
95 |
|
1999 |
Whole, raw milk |
22 |
14 |
64 |
0.35 |
8 |
36 |
|
1998 |
UHT milk |
11 |
11 |
100 |
0 |
0 |
0 |
|
1999 |
Imported milk |
15 |
12 |
80 |
0.08 |
3 |
20 |
|
1999 |
Low-fat milk |
11 |
2 |
18 |
0.24 |
9 |
82 |
|
1999 |
Non-fat milk |
9 |
2 |
22 |
0.14 |
7 |
78 |
|
Total |
|
90 |
42 |
65 |
0.35 |
48 |
53 |
a LOD, 0.005 µg/kg; LOQ, 0.01 µg/kg
Although the original report indicated that the LOD was 0.005 ng/kg and the LOQ was 0.01 ng/kg, the Committee assumed that this was an error and that the values should be in µg/kg. ‘< LOQ’ in Table 11 refers to the total number of samples less the number of samples designated as ‘positive’ in the report, as it was assumed that ‘positive’ values were those above the LOQ. These data are presented in Appendix B, but are not included in Appendix C because an overall mean for the samples was not provided and it could not be calculated because individual values were lacking.
The Commission of the European Union (1999) provided a summary of the concentrations of aflatoxin M1 detected in 7573 milk samples in 1999 from 10 Member States: Austria, Belgium, Finland, France, Germany, Ireland, the Netherlands, Portugal, Sweden and the United Kingdom. Concentrations of aflatoxin M1 below the LOD/LOQ (which differed among the countries) were found in 7259 (96%) of the samples. The concentrations in all 314 samples in which aflatoxin M1 was detected (4.2%) were < 0.05 µg/kg. Mean values were not reported.
The Commission of the European Union (1989–95) also provided a Scientific Cooperation (SCOOP) report, containing data for milk analysed between 1989 and 1995 in nine Member States. Of the 8791 samples, 3338 (38%) contained concentrations of aflatoxin M1 below the LOQ/LOD; 1017 samples (12%) contained concentrations < 0.05 µg/kg; six samples (0.07%) contained 0.05–0.1 µg/kg; and three samples (0.03%) contained > 0.1 µg/kg. The nine samples containing > 0.05 µg/kg were from the United Kingdom.
Appendix A gives unpublished data from the Research Institute for Veterinary Science in Bogor, Indonesia, on concentrations of aflatoxin M1 in milk. The data include the mean and maximum concentrations of aflatoxin M1 in 342 samples of milk collected during 1990–93 and 1999. The LOQ of the analytical method was 0.1 µg/kg. The concentration was below the LOQ in 143 samples, > 0.05 µg/kg in 199, and > 0.5 µg/kg in 73.
A submission from Brazil (Ministerio da Saude, 2000) provided a summary of data on the concentrations of aflatoxin M1 in samples of domestic cow milk from five references (Sabino et al., 1989; de Sylos et al., 1996; Souza et al., 1999; Prado et al., 1999, 2000). A second submission from Brazil (Rodriquez-Amaya, 2000) was a paper prepared for the Tenth International IUPAC Symposium on Mycotoxins and Phycotoxins, which summarized data on concentrations of aflatoxin M1 in milk and milk products in Brazil (de Sylos et al., 1996; Correa et al., 1997; Oliveira et al., 1997) and Uruguay (Pineiro et al., 1996). The information from the eight papers (converted to µg/kg) is summarized in Table 12.
Table 12. Concentrations of aflatoxin M1 in milk from Brazil and Uruguay in published reports
|
Food |
No. of samples |
Positive samples |
Concentration (µg/kg) |
|||
|
No. |
% |
All samples |
Positive samples |
|||
|
Mean |
Range |
|||||
|
de Sylos et al. (1996), Campinas, Brazil |
||||||
|
Milk, pasteurized, 1989 |
51 |
0 |
0 |
0 |
|
|
|
Milk, whole dry, 1989 |
20 |
0 |
0 |
0 |
|
|
|
Milk, non-fat dry, 1989 |
15 |
0 |
0 |
0 |
|
|
|
Yoghurt, 1990 |
30 |
0 |
0 |
0 |
|
|
|
Milk, pasteurized, 1992 |
52 |
4 |
8 |
0.012 |
0.16 |
0.073–0.37 |
|
Oliveira et al. (1997), Brazil |
||||||
|
Milk, whole, from powder |
300 |
33 |
11 |
0.030 |
0.27 |
0.10–1.0 |
|
Distribution: |
267 |
|
|
89 |
0.0 |
|
|
|
|
17 |
6 |
|
|
0.10–0.20 |
|
|
|
10 |
3 |
|
|
0.20–0.30 |
|
|
|
6 |
2 |
|
> 0.50 |
|
|
Sabino et al. (1989), Brazil |
||||||
|
Milk, market, 1979–81 |
100 |
1 |
1 |
0.2 |
0.2 |
|
|
Milk, farm |
50 |
9 |
18 |
0.086 |
0.48 |
0.10–1.7 |
|
Souza et al. (1999), Brazil |
||||||
|
Milk |
110 |
27 |
24 |
|
|
|
|
|
|
5 |
|
|
0.05 |
0.038–0.071 |
|
Prado et al. (1999), Brazil |
||||||
|
Milk, all samples |
61 |
50 |
82 |
0.020 |
0.025 |
0.006–0.077 |
|
Pasteurized |
18 |
15 |
83 |
0.027 |
0.032 |
0.008–0.077 |
|
Long-life |
21 |
16 |
76 |
0.018 |
0.023 |
0.006–0.070 |
|
Long-life, infant |
11 |
9 |
82 |
0.025 |
0.030 |
0.007–0.050 |
|
Powder |
6 |
6 |
100 |
0.012 |
0.012 |
0.006–0.038 |
|
Powder, infant |
5 |
4 |
80 |
0.010 |
0.013 |
0.007–0.021 |
|
Correa et al. (1997), Brazil |
||||||
|
Milk, raw |
144 |
0 |
0 |
0.0 |
|
|
|
Pineiro et al. (1996), Uruguay |
||||||
|
Milk |
22 |
7 |
32 |
|
|
< 0.2–> 0.5 |
|
Distribution: |
|
15 |
68 |
|
< 0.2 |
|
|
|
|
6 |
27 |
|
|
0.2–0.5 |
|
|
|
1 |
5 |
|
> 0.5 |
|
Additional information about concentrations of aflatoxin M1 in milk was sought in the literature for guidance on appropriate levels for estimating daily intake and for information for geographic areas not covered by the submitted data. In particular, there were no submissions from Africa and few from the Far East or Middle East. Pertinent data (converted to µg/kg, assuming that 1 L of milk weighs 1.02 kg) are shown in Table 13. Other data were evaluated but not included in Table 13, for several reasons. Data reported by Lemieszek-Chodorowska (1974) for Poland and by Brewington & Weihrauch (1970) for the USA were considered to be too old; Fukal & Brezina (1991) did not provide mean values for their data from the former Czechoslovakia; the data from Cyprus (Ioannou-Kakouri et al., 1999) were of doubtful quality because of a low per cent recovery; Smith et al. (1994) in the USA reported data for only nine samples of goat milk, which is not commonly consumed in the USA; and the data of Galvano et al. (1998) for Italy probably duplicated data submitted by the Commission of the European Union. Other reports that did not contain pertinent data were those of Masri et al. (1969b), Grant & Carlson (1971), Purchase et al. (1972), Jung & Hanssen (1974), Stoloff et al. (1975), Kiermeier & Mashaley (1977), Nikov et al. (1991), el-Nezami et al. (1995), Saad et al. (1995), and Navas et al. (2000).
Table 13. Concentrations of aflatoxin M1 in milk, three countries (published reports)
|
Food |
No. of samples |
Positive samples |
Concentration (µg/kg) |
|||
|
No . |
% |
All samples (mean) |
Positive samples |
|||
|
Mean |
Range |
|||||
|
Karaioannoglou et al. (1989), Greece |
||||||
|
Milk, raw |
99 |
4 |
4.0 |
0.005 |
0.12 |
0.10–0.13 |
|
Milk, pasteurized |
51 |
0 |
0.0 |
0.0 |
|
|
|
Markaki & Melissari (1997), Athens, Greece |
||||||
|
Milk, pasteurized |
81 |
72 |
89 |
0.008 |
0.009 |
0.0005–0.18 |
|
Distribution: |
|
9 |
11 |
|
0.0 |
|
|
|
|
31 |
38 |
|
|
0.0005–0.001 |
|
|
|
32 |
40 |
|
|
0.0025–0.005 |
|
|
|
9 |
11 |
|
|
0.010–0.18 |
|
Rajan et al. (1995), India |
||||||
|
Milk |
504 |
89 |
18 |
0.20 |
1.2 |
0.10–3.5 |
|
Kim et al. (2000), Seoul, Republic of Korea |
||||||
|
Milk, pasteurized |
70 |
53 |
76 |
0.014 |
0.018 |
0.002–0.037 |
|
Yoghurt |
60 |
50 |
83 |
0.023 |
0.029 |
0.017–0.12 |
|
Infant formula |
26 |
22 |
85 |
0.039 |
0.046 |
0.003–0.093 |
|
Milk, powdered |
24 |
18 |
75 |
0.15 |
0.200 |
0.026–0.33 |
Additional data from the literature, summarized in Appendix A, were considered. There is already considerable overlap in the data in Appendices A and B, and there may be some duplication, because the Commission of the European Union provided summaries of data from individual Member States without identifying the sources of the data within each country. The criteria for selecting data from Appendix A for addition to Appendix B were that the data were not already in Appendix B; they were relatively recent (1990 or later); they related to fluid milk or yoghurt, rather than dry milk or other dairy products; and the number of samples analysed and the mean concentration of aflatoxin M1 in the samples was given. The data from Appendix A added to Appendix B were those from Africa (El-Gohary, 1996), France (Dragacci & Frémy, 1993), the Philippines (Begino, 1998), Poland (Domagala et al., 1997), the Republic of Korea (Shon et al., 1996), Spain (Macho et al., 1992; Jalon et al., 1994), and Thailand (Boriboon & Suprasert, 1994; Saitanu, 1997). The data from France (Dragacci & Frémy, 1993) and Spain (Macho et al., 1992; Jalon et al., 1994) were assumed to be included in the summary from the Commission of the European Union and are therefore included in Appendix B but are not summarized in Appendix C, to avoid duplication.
The data in Appendix B are summarized in Appendix C for the five regional diets by aggregating milk types and/or periods from the same source or reference. The European regional diets are derived from data submitted from Canada, the Commission of the European Union and nine of its Member States, Norway, Poland, and the USA. Latin American diets are derived from data from Argentina, Brazil, and Uruguay. Far Eastern diets are represented by data from India, Indonesia, the Philippines, the Republic of Korea, and Thailand. Middle Eastern diets are represented by data from Greece and the United Arab Emirates, and African diets are represented by data from Egypt.
For the European regional diet, the mean value of aflatoxin M1 in milk ranged from about 0.004 to 0.40 µg/kg on the basis of data from the Commission of the European Union, Norway, and the USA. The calculated weighted mean was 0.023 µg/kg.
For the Latin American regional diet, the mean value ranged from 0 to 0.52 µg/kg. The sources were six published papers (Sabino et al., 1989; de Sylos et al., 1996; Correa et al., 1997; Oliveira et al., 1997; Prado et al., 1999; Souza et al., 1999) and a submission of laboratory data from Argentina. The weighted mean was calculated to be 0.022 µg/kg.
For the Far Eastern regional diet, the sources were five published papers (Boriboon & Suprasert, 1994; Rajan et al., 1995; Shon et al., 1996; Begino, 1998; Kim et al., 2000) and a submission from a laboratory in Indonesia. The weighted mean was calculated to be 0.36 µg/kg. The values from Indonesia were higher than those from other countries in both the Far Eastern region and other regions.
For the Middle Eastern regional diet, the sources were two published papers (Karaioannoglou et al., 1989; Markaki & Melissari, 1997). The weighted mean was 0.005 µg/kg.
Only one source, a published paper from Egypt (El-Gohary, 1996), was available for the African regional diet. This source provided an average value for aflatoxin M1 in powdered milk of 0.015 µg/kg, which would be equivalent to 0.0018 µg/kg in fluid milk, as 30 g of dried milk yields 244 g of fluid milk (the ratio of dried to fluid milk is 1:8.2, and 0.015/8.2 = 0.0018 µg/kg).
In the European, Latin American, Middle Eastern, and African diets, the weighted mean value for aflatoxin M1 in milk was below the proposed maximum levels of 0.05 µg/kg and 0.5 µg/kg. In the Far Eastern regional diet, the weighted mean value for aflatoxin M1 in milk (0.36 µg/kg) was greater than the proposed maximum level of 0.05 µg/kg but below 0.5 µg/kg.
The GEMS/Food regional diets (WHO, 1998) provide the dietary intakes of food commodities in five geographical areas. Table 14 shows the intake of all milk and milk products for the five regions. The major food class in which aflatoxin M1 was identified was milk, the term ‘milk’ being assumed to include the mammalian milks (buffalo, camel, cow, goat, and sheep) listed in the GEMS/Food regional diets and not cheese, butter, or other dairy products derived from milk. Because the GEMS/Food database does not include separate data on the consumption of fermented milks, it was assumed that consumption of yoghurt and other fermented milks was subsumed within the figures for milk.
Table 14. Regional consumption of milk and milk products (g/person per day)
|
Regional diet |
Milk and milk products |
All milk |
Cows’ milk |
|
Far Eastern |
33 |
32 |
23 |
|
African |
42 |
42 |
36 |
|
Middle Eastern |
130 |
120 |
80 |
|
Latin American |
170 |
160 |
160 |
|
European |
340 |
290 |
290 |
The figures for ‘all milks’ were used to estimate dietary intake, for two reasons. First, the group ‘milk and milk products’ includes butter, cheese, cream, and ghee, which were not specified to be of interest for this assignment. Second, aflatoxin M1 occurs in milks other than cows’ milk, and use of data only on consumption of cow milk might result in an underestimate of intake of aflatoxin M1. ‘All milks’ include milk (and presumably fermented milk) from buffalo, camels, cows, goats, and sheep. For all five geographical areas, cow milk was the major type of milk consumed.
On the basis of the weighted mean concentrations of aflatoxin M1 in milk in the five regions, the intake was 0.1 ng/day in the African diet, 0.6 ng/day in the Middle Eastern diet, 3.5 ng/day in the Latin American diet, 6.8 ng/day in the European diet, and 12 ng/day in the Far Eastern diet (Table15). When these intakes are expressed as nanograms of aflatoxin M1 per kg of body weight per day and assuming a body weight of 60 kg, the intakes are 0.002 for the African diet, 0.10 for the Middle Eastern diet, 0.058 for the Latin American diet, 0.11 for the European diet, and 0.20 for the Far Eastern diet.
Table 15. Daily intake of aflatoxin M1 in all milk in the five regional diets
|
Diet |
Milk intake |
Weighted mean |
||
|
Aflatoxin in milk (µg/kg) |
Aflatoxin intake |
|||
|
ng/person |
ng/kg bw |
|||
|
European |
0.29 |
0.023 |
6.8 |
0.11 |
|
Latin American |
0.16 |
0.022 |
3.5 |
0.058 |
|
Far Eastern |
0.032 |
0.36 |
12 |
0.20 |
|
Middle Eastern |
0.12 |
0.005 |
0.6 |
0.10 |
|
African |
0.042 |
0.002 |
0.1 |
0.002 |
One of the papers (Oliveira et al., 1997) provided an estimate of the aflatoxin M1 intake of a 4-month-old child in Brazil on the basis of a rather high value for this compound in milk prepared from powder (0.27 µg/kg; range, 0.10–1.0 µg/kg). The estimated daily intake was 0.0037 µg/kg bw per day. If it is assumed that a 4-month-old child weighs about 6 kg, this would represent a daily intake of 0.022 µg (22 ng) of aflatoxin M1 per day. No other literature or submitted national estimates were available on the intake of aflatoxin M1.
One approach to determining the effect of applying the proposed maximum levels of 0.5 and 0.05 µg/kg for aflatoxin M1 in milk on dietary intake is based on assessment of the mean aflatoxin M1 concentration in all samples of milk, in all samples containing 0.5 µg/kg, and in all samples containing 0.05 µg/kg for a given population. These three concentrations can then be multiplied by the milk consumption of the population to determine the intakes. The most recent data (1999) from some Member States of the European Union, comprising 7573 samples, showed concentrations 0.05 g/kg in all samples. The intake of aflatoxin M1 would not differ if the maximum level was 0.5 or 0.05 µg/kg, as none of the concentrations exceeded 0.05 µg/g. A similar situation is seen with the most recent data from Canada, comprising 81 samples reported in 1997–98, as all milk samples analysed contained < 0.015 µg/kg (the LOD). Data on 3620 samples taken in the USA between 1995 and 2000 show that the mean intake of aflatoxin M1 from all samples of milk was 0.0062 µg/kg, resulting in an intake of 1.8 ng/person per day. If all samples containing > 0.5 µg/kg (3615; 99%) were omitted, the mean concentration would be 0.0046 µg/kg, and the mean intake would be 1.3 ng/person per day. If all samples containing > 0.05 µg/kg (3491; 96%) were omitted, the mean concentration would be 0.00070 µg/kg, and the intake would be 0.20 ng/person per day. The estimates are based on milk consumption in the GEMS/Foods European diet of 0.29 kg/person per day. The intakes of aflatoxin M1 from milk are 1.8 ng/person per day for all samples, 1.4 ng/person per day when the maximum level is 0.5 µg/kg, and 0.21 ng/person per day when the maximum level is 0.05 µg/kg.
Another approach to determining the effect of the proposed maximum limits on dietary intake is to generate distribution curves for the concentrations of aflatoxin M1 in milk in the regional data (Figure 3). Log normality of the distribution was assumed, the weighted mean concentrations of aflatoxin M1 in the GEMS/Food regions were used, and the maximum values were reported. The distribution curve for the European regional diet shows that setting the maximum level at 0.05 or 0.5 µg/kg would have no effect on intake. The distribution curve for milk samples in the Middle Eastern region, for which there were relatively few data, is similar to that for the European region. For the Latin American diet, selection of a maximum level of 0.5 µg/kg would also have no effect; however, use of a maximum level of 0.05 µg/kg would probably reduce intake. In the Far Eastern region, where milk is more heavily contaminated, intake of aflatoxin M1 would be decreased at both proposed levels. It should be noted, however, that milk consumption is low in the Far East.
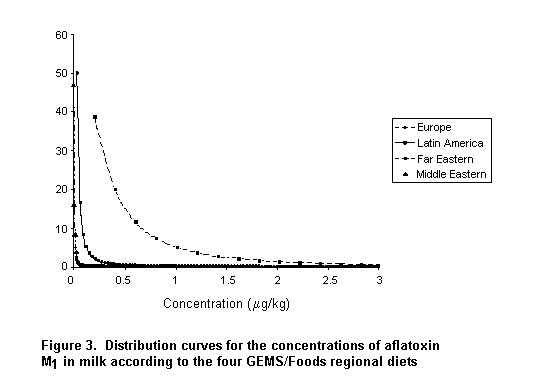
The weighted mean concentrations of aflatoxin M1 in milk in the regional diets are not the same in Appendix C and Figure 3 because generation of the distribution curves based on log normality required use of non-zero minimum values. Thus, the minimum values for aflatoxin M1 in milk were assumed to be one-half of the LOD/LOQ, resulting in slightly different weighted mean values than those shown in Appendix B, which were generated from the number of samples and the mean value from each submission within each geographical region.
An example of intake of aflatoxin M1 in milk was prepared from simulated distributions of contamination and of milk consumption by the French population (Figure 4). A probabilistic Monte-Carlo simulation was used to assess the intake of aflatoxin M1 in milk. The software used was @risk, and the formula used to derive the contamination curves was:
riskTlognorm (mean, SD, min, max).
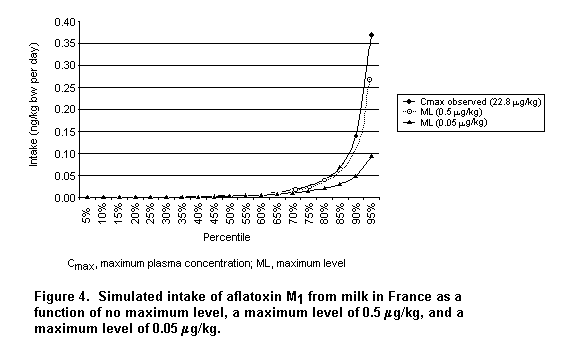
The data on food intake were taken from a French survey of individual food consumption conducted by the ‘Association Sucre–Produits sucrés, Communication, Consommation’ in 1994, and the results are those for participants who consumed milk. Of the 1161 persons surveyed, 79% consumed milk on the day of the survey. The mean consumption of milk was 27 g/week per kg bw, and the 95th percentile of consumption was 103 g/week per kg bw. The model is based on an assumption of log normality of the distribution and the mean, standard deviation, and minimum and maximum values for aflatoxin M1 in milk. The minimum value is assumed to be zero, and the maximum observed value was 23 µg/kg. Three simulations were made, on the basis of the maximum observed concentration of aflatoxin M1 in milk, the upper proposed maximum level (0.5 µg/kg), and the lower proposed maximum level (0.05 µg/kg).
The results show no difference between the curves when there is no maximum level and the upper proposed maximum level is up to the 90th percentile of consumption. The curves for the upper and the lower maximum levels begin to separate at the 70th percentile. The intakes at the 90th percentiles are 0.05, 0.12, and 0.14 ng/kg bw per day for a maximum level of 0.05 µg/kg or 0.5 µg/kg and no maximum level, respectively. The intake at the 95th percentile, is 0.09, 0.27, and 0.37 ng/kg bw per day for a maximum level of 0.05 µg/kg, a maximum level of 0.05 µg/kg, and no maximum level, respectively.
The validity and reliability of these estimates of intake of aflatoxin M1 from milk are limited by a number of factors. Insufficient data on the concentrations of aflatoxin M1 in milk were submitted for all five geographic regions and especially for the Middle East and Africa. As most of the submitted data were not accompanied by information on quality assurance, the validity or reliability of the resulting intake estimates cannot be assessed. Furthermore, little information was given about the sampling design or procedure used, and it is unlikely that the submitted data were representative of the area from which the samples were taken. Not all countries within a region submitted data, and the number of samples varied considerably. Very limited data were available from African and Middle Eastern countries. In addition, the concentration of aflatoxin M1 in milk could vary considerably within each geographic region.
The use of different analytical methods by various investigators probably affected the reported concentrations of aflatoxin M1 in milk, as some methods are more sensitive than others. The LOD/LOQ of the analytical method was less than the proposed maximum level of 0.05 µg/kg for the data from Indonesia, Thailand (Boriboon & Suprasert, 1994), and Uruguay. The lower values reported in these studies would not be accurate enough to determine the number of samples containing < 0.05 µg/kg. Comparison and aggregation of data from different laboratories is difficult not only because different analytical methods are used but also because the results are presented in different ways (distributions, means of positive values, values < LOD/LOQ, values below the maximum level) and in different units.
For studies in which a mean for all samples was not provided, the mean was calculated by assuming that values < LOQ/LOD were zero. In most cases, the number of samples at 0 and > 0 but < LOQ/LOD could not be determined. The assumption may have resulted in underestimation of the weighted mean concentrations of aflatoxin M1 in milk.
The effects of processing, season, climate, and other environmental variables on the aflatoxin M1 content of milk could not be ascertained, partly because these effects were not addressed in the various studies and reports, and also because various terms were used to describe milk and other dairy products. In most cases, the type of milk analysed was not specified.
For monitoring and evaluation of aflatoxin M1 in milk in the future, it would be useful to obtain more data, especially from African, Far Eastern, Middle Eastern, and Latin American countries, and detailed information on sampling plans and quality control of the analytical method in all submissions. In addition, individual data points should be provided for the construction of distribution curves and other statistical evaluations. The LOD and LOQ of the analytical methods should be provided. The samples that are analysed should be clearly described with regard to the type of milk, the extent of processing, geographical location, and whether or not the samples are representative of the entire country or region. Submissions should indicate whether values < LOD/LOQ are assigned a value of 0. They should provide information on the number of samples, the number of samples < LOD/LOQ, the number of samples containing aflatoxin M1 at < 0.05 µg/kg and < 0.5 µg/kg, the mean concentration in each type of milk (by time, if appropriate), the maximum value, and the 90th percentile value.
Since the observation that dairy cows consuming rations contaminated with aflatoxin B1 excrete aflatoxin M1 in their milk, various studies have been undertaken to establish the carry-over rates with consumption of high and low concentrations in feed. In studies conducted in the 1960s and 1970s, the aflatoxin B1 intake was relatively high, the milk yields relatively low, and the analytical methods not well developed. Varied results were obtained, the concentration of aflatoxin M1 in milk ranging from 0.2 to 4% of the concentration of aflatoxin B1 ingested with the feed (van Egmond, 1989; Pettersson, 1997). Kiermeier et al. (1975, 1977) indicated that concentrations of aflatoxin M1 in milk varied widely from animal to animal (even among animals in the same herd), from day to day, and from one milking to the next. The results of several researchers (van der Linde et al., 1964; Masri et al., 1969b; Kiermeier, 1973; McKinney et al., 1973; Polan et al., 1974) suggest that it takes 3–6 days of constant daily ingestion of aflatoxin B1 before steady-state excretion of aflatoxin M1 in milk is achieved, whereas aflatoxin M1 becomes undetectable 2–4 days after withdrawal of animals from the contaminated diet.
In the 1980s and 1990s, more information became available on the carry-over of aflatoxin B1 at concentrations in the range of the official limits. At the same time, the milk yields of cows increased considerably (up to 40 L/day), and analytical methods improved, became more reliable, and provided more precise results. The more recent studies widened the estimated range of concentrations of aflatoxin M1 in milk from 0.3 to 6.2%. The highest percentage was found in a carry-over experiment in Dutch dairy cows in early and late lactation stages that were fed diets naturally contaminated with low concentrations of aflatoxin B1 (Veldman et al., 1992). The authors assumed that the higher excretion ratios of cows with high milk production was a result of greater permeability of the cell membranes of the alveoli. The higher concentrations of aflatoxin M1 excreted by cows with mastitis is also believed to be due to increased permeability of the membranes. A linear relation between aflatoxin B1 intake and aflatoxin M1 content was found for aflatoxin B1 intakes of 5–80 µg:
aflatoxin M1 (ng/kg of milk) = 1.2 x aflatoxin B1 intake (µg/cow per day) + 1.9.
Thus, production of milk containing aflatoxin M1 at 0.05 µg/kg would require that the average intake of aflatoxin B1 by dairy cows be limited to approximately 40 µg/day per cow. On the basis of a daily consumption of 12 kg/cow of compound feed, the content of aflatoxin B1 in the feed would have to be 3.4 µg/kg in order to meet the proposed Codex limit. Carry-over studies conducted in the 1990s of cows producing 10–20 L of milk per day (Harvey et al., 1991; Galvano et al., 1996b) showed much less carry-over (0.5–0.6%).
The variations in carry-over are significant at both high and low levels of contamination of feed with aflatoxin B1. Pettersson (1997) gave a number of reasons for the considerable variation. Apart from problems of analysis and experimental technique, there are large individual differences in excretion; however, this is outweighed by the fact that the studies were performed on groups of cows. Any differences by breed that may exist are not clearly discernible in the available studies. Differences in milk production are important. The studies of Pettersson et al. (1990), Veldman et al. (1992), and Veldman (1992) of cows with high milk production showed the greatest carry-over, ranging from 2.6 to 6.2%. Tables 16 and 17 present data from Park & Pohland (1986) on the carry-over of aflatoxin from feed to edible tissue.
Table 16. Ratios of concentrations of aflatoxin in feed and in edible tissues
|
Animal |
Tissue |
Aflatoxin |
Feed:tissue ratioa |
|
Beef cattle |
Liver |
B1 |
14 000 |
|
Dairy cattle |
Milk |
M1 |
75 |
|
|
|
Aflatoxicol |
195 000 |
|
Pigs |
Liver |
B1 |
800 |
|
Laying hens |
Eggs |
B1 |
2 200 |
|
Broiler chickens |
Liver |
B1 |
1 200 |
From Park & Pohland (1986)
a
Concentration of aflatoxin B1 in feed divided by the concentration of the specified aflatoxin in the specified tissue
Table 17. Concentrations of aflatoxin B1 required in feed to result in 0.1 ng/g of residues of aflatoxins in edible tissues
|
Animal |
Level of contamination with aflatoxin B1 (ng/g) |
|||
|
Corn |
Peanut meal |
Cottonseed meal |
Cottonseed |
|
|
Beef cattle |
1800 |
14 000 |
12 725 |
14 000 |
|
Dairy cattle |
14 |
54 |
54 |
38 |
|
Pigs |
105 |
730 |
1 600 |
|
|
Laying hens |
325 |
1 835 |
2 445 |
|
|
Broiler chickens |
180 |
925 |
1 200 |
|
From Park & Pohland (1986)
The most effective way of controlling aflatoxin M1 in the food supply is to reduce contamination with aflatoxin B1 of raw materials and supplementary feedstuffs for dairy cattle. Specific regulations exist in many countries (FAO, 1997), and practical programmes are being developed; e.g. the Codex Committee on Food Additives and Contaminants has developed a code of practice for reducing aflatoxin B1 in raw materials (van Egmond et al., 1997). Reduction can be achieved by good manufac-turing practices and good storage practices. If preventive measures fail to reduce fungal growth and aflatoxin B1 formation in agricultural commodities intended for use as animal feeds, the last means for avoiding or reducing the occurrence of aflatoxins in feed is to eliminate (part of) the toxins. Feeds that have higher concentrations of aflatoxin B1 may be acceptable for feeding to dairy animals if they are blended with feed that has lower concentrations, provided that the resultant aflatoxin M1 concentration in milk does not exceed levels considered to be safe. In principle, aflatoxin-contaminated consignments of feeds can be decontaminated by removing the toxin (segregation) or by converting it to a non-toxic form (degradation). Degradation may be achieved by physical, chemical, or biological means.
Attempts have been made to degrade aflatoxins in feed by applying physical treatments such as heat, microwaves, gamma-rays, X-rays, ultra-violet light and adsorption (van Egmond & Speijers, 1999). Degradation of aflatoxin M1 has also been attempted by combined treatments, such as ultra-violet radiation followed by ultrafiltration. In most cases, neither heat treatment not irradiation is effective. Adsorption of aflatoxins from animal feed onto bentonite and hydrated sodium calcium aluminosilicate (Veldman, 1992; Galvano et al., 1996a) has been used in the feed industry to reduce the aflatoxin M1 content of milk (Harvey et al., 1991; Phillips et al., 1995).
A newer approach is use of oltipraz, a substituted dithiolthione that inhibits aflatoxin B1 metabolism by inhibiting the activity of several cytochrome P450 enzymes (Kuilman et al., 2000). No aflatoxin M1 formation was found in bovine hepatocytes incubated with aflatoxin B1 and oltipraz. The findings suggest that oltipraz is highly effective in inhibiting aflatoxin M1 contamination of milk from diary cows exposed to aflatoxin B1-contaminated feeds.
Some chemical procedures have been developed to degrade aflatoxins in animal feed, usually based on addition of oxidizing agents, aldehydes, acids, and bases. The most widely used chemical detoxication reagent is ammonia, as an anhydrous vapour or as an aqueous solution. Treatment of aflatoxin B1 with ammonia opens the lactone ring of the molecule. Ammoniation of agricultural commodities leads to decomposition of 95–98% of the aflatoxin B1 present. This process is used in various countries for the decomposition of animal feedstuffs; however, it has not been formally approved by the European Union or the Food and Drug Administration of the USA because of controversy about the safety of products from animals that have been fed diets that have undergone chemical decontamination.
Park and Price (2001) reviewed studies conducted over more than three decades on the development and safety evaluation of procedures for reducing risks associated with contamination of agricultural commodities by aflatoxins. Use of ammonia to alter the chemical structure of aflatoxins and thus reduce their toxic and mutagenic potential has gained acceptance in many countries, including Brazil, France, Mexico, Senegal, South Africa, the Sudan, and some states of the USA. Use of ammonia to treat cottonseed feed for lactating cows in Arizona, USA, and to treat peanut meal in France has kept the milk supply free from aflatoxin contamination for almost 20 years. In the aflatoxin decontamination programme, in which the products were tested for aflatoxin concentrations both before and after ammoniation, the process was shown to result usually in no detectable aflatoxin M1 residues in milk. The process and the treated products are accepted by the dairy industry.
Although several ammonia-based processes have been developed and studied, that in which ammonia (0.5–2.0%) is used under controlled conditions of moisture (12–16%), pressure (45–55 psi [3.2–3.9 kg/cm2]), and temperature (80–1000 °C) for 20–60 min, commonly called the ‘high-pressure, high-temperature method’, is the most efficient and produces the most reliably safe product. The procedure modifies the aflatoxin molecule chemically to compounds that are many orders of magnitude less toxic or mutagenic than the parent aflatoxin B1 or undetectable after exhaustive extraction, isolation, and purification. The aflatoxin–ammonia reaction products in cottonseed and maize consist of 12–14% volatile compounds, 20–24% compounds extractable with methylene chloride, and 6–13% compounds extractable with methanol. Treatment with acid, base, and proteolytic enzymes released an additional 19–22% of the compounds. After solvent extraction and enzymatic digestion, the remaining cottonseed or maize matrix contained ammonia reaction products representing only 37% of the original aflatoxin concentration.
Studies of metabolism and excretion showed that the feed-bound ammonia–aflatoxin products are excreted primarily in the faeces. Exhaustive studies showed that the reaction products have minimal if any effect on the health of animals receiving rations containing ammonia-treated aflatoxin-contaminated maize, peanut, or cottonseed meal. The ambient temperature method, which usually requires a 3–6-week treatment period, effectively reduces aflatoxin concentrations, but it requires close monitoring, and the safety of the ammoniated product has not been demonstrated unequivocally.
The effect of ammonia on feed composition and by-products is usually increased concentrations of total and non-protein nitrogen, crude protein, ash, and soluble solids, with reduced concentrations of sulfur-containing amino acids, available lysine, and reducing sugars. Production parameters such as milk and egg quality have been shown to be significantly improved or not adversely affected by the treatment. Long-term, short-term, relay, and multi-generation feeding studies showed no toxic effects or lesions related to the ammoniation procedure. It has been approved for use in the USA for increasing non-protein nitrogen in animal feeds.
Isolated aflatoxin–ammonia reaction products in maize and cottonseed or human foods derived from animals fed ammonia-treated aflatoxin-contaminated feeds had significantly reduced toxic and mutagenic potential. No tumours or neoplastic lesions were observed in trout fed rations containing milk obtained from lactating dairy cows fed ammonia-treated aflatoxin-contaminated feed. Trout are highly susceptible to tumorigenesis by aflatoxins. High mutagenic activity in milk from cows exposed to aflatoxin B1 was eliminated or reduced significantly by treating the feed with ammonia.
Thus, some decontamination reaction products are somewhat toxic, mutagenic, or have DNA or protein covalent binding potential. The effects are, however, many orders of magnitude less than those of the parent aflatoxin B1. These compounds usually represent < 1% of the original aflatoxin concentration in the feed matrix. A comparison of the equivalent toxicity of aflatoxin after ammonia treatment showed that the aflatoxin–ammonia reaction products do not contribute significant toxic or mutagenic potential to the residual aflatoxin B1 in ammoniated aflatoxin-contaminated feeds. Hence, the maximum level of aflatoxin in ammoniated aflatoxin-contaminated feeds would not have to be adjusted for the reaction products.
Alkaline heat treatment, or nixtamalization, which is used traditionally in the treatment of maize for the manufacture of tortillas, significantly reduced the concentration of aflatoxin. Subsequent studies showed, however, that much of the original aflatoxin was re-formed when the products were acidified (Lopez-Garcia & Park, 1998).
In principle, biological methods could also be used to eliminate aflatoxins. For instance, procedures have been developed to degrade aflatoxins in feedstuffs by exposing them to the bacterium Flavobacterium auranthiacum. These studies have not yet led to application.
In conclusion, there are several possibilities for preventing the presence of aflatoxin M1 in dairy products; each has its advantages and disadvantages. Although prevention of contamination of dairy cattle feed is the ideal, it may not be possible in practice. Various decontamination methods have a role to play in preventing and reducing the concentrations of aflatoxin M1 in dairy products.
At its forty-ninth meeting, the Committee concluded that there was ‘insufficient quantitative information available about competing aspects of metabolic activation and detoxication of aflatoxin B1 in vivo in various species to describe quantitatively a species-dependent effect of metabolism on aflatoxin B1 on carcinogenicity’. Further data have become available, however, to elucidate the effects of aflatoxin B1 and aflatoxin M1.
Studies of slices of human liver showed wide variation in the metabolism and activation of aflatoxins among individuals, and genetic and environmental factors may affect this process in vitro. For example, ascorbate and coumarin seem to protect against the toxic effects of aflatoxins, and epidemiological studies have also shown the importance of genetic and environmental factors in liver cancer associated with intake of aflatoxins.
Human hepatocytes appear to form less aflatoxin B-oxide than rat cells, and human cells appear not to have the important detoxifying GST enzymes with high specific activity against aflatoxin B1. Some workers have shown no GST activity in human liver cytosol fractions. Human liver enzymes have a limited capacity to form the toxic epoxide form from aflatoxin M1. Aflatoxin M1 is definitely cytotoxic, as is aflatoxin B1, in human hepatocytes, and this finding may have important implications for the effects of aflatoxin M1 on immunocompetence and growth.
Aflatoxins may potentiate liver damage caused by bacterial lipopolysaccharide, which may affect acute toxicity in humans and domestic animals. This potentiation of liver damage by aflatoxins may also play a role in the interaction between hepatitis viruses and liver carcinogenesis.
Sensitivity to aflatoxins varies among species. In a highly sensitive species like the turkey, the crucial aflatoxin B1-detoxifying enzyme pathway (GST-mediated) is deficient, while there is efficient oxidation of aflatoxin B1 to the epoxide. Hepatocytes from macaques, marmosets, and humans show a quantitatively similar ability to oxidatively metabolize aflatoxin B1. In one primate species (M. fascicularis), the µ class of GSTs is the most important in detoxifying aflatoxin B1, but human cells have not been shown to duplicate this pattern of detoxication.
Oltipraz inhibits cytochrome P450 1A2 activity in rats and humans in vivo and in tree shrew hepatocytes. Thus, oxidation of aflatoxin B1 to the 8,9-oxide and aflatoxin M1 is blocked. Oltipraz and ethoxyquin may also induce a modest increase in aflatoxin B glutathione conjugation activity.
The short-term toxicity of aflatoxin M1 is similar to that of aflatoxin B1, and it appears to act by the same mechanism. In ducklings and rats, the short-term toxicity of aflatoxin M1 was similar to or slightly less than that of aflatoxin B1. Aflatoxin M1 is a less potent carcinogen than aflatoxin B1, even in Fischer rats and rainbow trout, which are very sensitive. Aflatoxin M1 shows 2–10% of the carcinogenic activity of aflatoxin B1. In studies of genotoxicity, such as in Drosophila melanogaster, aflatoxin M1 was about 10-fold less mutagenic than aflatoxin B1.
The relationship between dose or exposure to aflatoxin M1, HBV, and/or HCV and the incidence of hepatocellular carcinoma and other risk factors has not been studied epidemiologically in studies suitable for modelling a dose–response relationship or the carcinogenic potency of aflatoxin M1. However, a number of conclusions can be drawn from the existing studies on the potency of aflatoxin M1 and aflatoxin B1 and risk factors for human liver cancer.
There is consistent, growing evidence that hepatitis viruses play an important role in the etiology of liver cancer. Progress has been made in the use of markers (based on the PCR), which show an increased fraction of cases with virus and, in some examples, an increased fraction of controls with virus. The risk estimates therefore differ little from previous ones, but the attributable fractions may be increased in relation to those shown in the table in the report of the Committee at its forty-ninth meeting (modified as Table 3 above) and in other reviews. That is, the attributable fractions due to hepatitis viruses might be increased and that due to aflatoxin thereby decreased.
Many epidemiological studies have involved use of biomarkers of aflatoxins. Many included biomarkers in the metabolic pathways in which aflatoxin is detoxified and eliminated. Evidence is available to show that aflatoxin–albumin adducts reflect both aflatoxin intake in the diet and aflatoxin–DNA adducts in liver cells. However, biomarkers of aflatoxins do not allow a quantitative measure of aflatoxin intake over the long term, as they are limited by the half-time of the urinary metabolites or serum protein adducts.
The risk for hepatocellular carcinoma in relation to urinary excretion of aflatoxin M1, aflatoxin adducts, or aflatoxin–albumin adducts has been estimated in many studies. The most consistent finding is that HBV carriers have an increased risk when some of these biomarkers of intake are present. The increase is usually modest (two- to threefold, with some exceptions), but many of the studies are based on small numbers of cases and wide confidence intervals. Some of these studies are particularly valuable because urine or serum samples were collected before liver cancer occurred. However, HBV carriers may have some degree of liver disease at any time. Either of these two factors can alter the natural history of the biomarker itself. In addition, as can be seen in Table 3, HCV appears to be an increasingly important risk factor for hepatocellular carcinoma, especially in developed countries.
P53 mutations may be of considerable value for identifying cases of liver cancer that can be related to aflatoxin intake. In areas of high risk, however, only 30–50% of people can be shown to harbour the relevant mutation (codon 249), even though the entire population would have been exposed to aflatoxins. The specificity of the marker is thus low, and the negative results are difficult to interpret. In order to make full use of this important biomarker, its natural history should be characterized, and the dose–response relationship between aflatoxin B1 and the mutation and the relationship between better validated biomarkers (aflatoxin B1–albumin adducts in urine) and the occurrence of P53 codon 249 mutations should be clarified. In epidemiological studies, the presence of mutations in specimens of hepatocellular carcinoma might be interpreted as reflecting the involvement of aflatoxin B1 or aflatoxin M1, but the absence of mutations, particularly in cancers from areas where there is high aflatoxin B1 intake (> 50% in most series), should be interpreted with caution, as aflatoxin B1 and aflatoxin M1 may induce hepatocellular carcinoma by other DNA damaging mechanisms.
Studies in which oltipraz is administered are likely to be informative if the proposed phase III trials are organized. In animals, olitpraz prevents aflatoxin-induced liver cancer. The results of studies in humans in China are awaited.
The Committee at its forty-ninth meeting had an extensive discussion of general modelling issues. That discussion is relevant and will not be repeated here; a few general points will be emphasized.
The main weakness of the risk assessment performed by the previous Committee was the lack of an ideal data set. "...the best data set to use for dose–response analysis would be a human study in which dose is accurately measured, response is determined without error and there are no confounding factors which are unexplained." In the case of aflatoxin M1, there is no epidemiological study that addresses these factors adequately. The dose of aflatoxin M1 or aflatoxin B1 has not been measured accurately over the long period generally associated with cancer induction. The additional risk factors for hepatocellular carcinoma that interact with aflatoxins, such as HBV and HCV infection, cannot be quantified completely. Species differences in sensitivity to aflatoxins are beginning to be elucidated, but they cannot be quantified precisely.
As stated by the Committee at its forty-ninth meeting , quantitative risk assessment involves four basic issues: (1) choice of data; (2) measure of exposure; (3) measure of response; and (4) choice of a mathematical relationship between dose and response for a given data set.
The carcinogenic potency of aflatoxins was estimated in studies in several animal species and in several epidemiological studies. Data were available generally only for aflatoxin B1, sometimes for mixtures of aflatoxins, and rarely for aflatoxin M1. A study by Yeh et al. (1989) was the main source used by the Committee at its forty-ninth meeting and by most other groups for estimating the carcinogenic potency of aflatoxin B1. This study addressed the roles of HBV and aflatoxin B1 in the development of primary hepatocellular carcinoma in a cohort of 7917 men aged 25–64 in southern Guangxi Province, China, where the incidence of this cancer is among the highest in the world. With 30 188 person-years accumulated, 149 deaths were observed, 76 (51%) of which were due to primary hepatocellular carcinoma. Of these patients, 69 (91%) were HBsAg+ at the time of enrolment into the study, in contrast to 23% of all members of the cohort. Aflatoxin intake was estimated for the population as a whole and not for individuals. When the estimated aflatoxin B1 intakes of subpopulations were plotted against the corresponding rates of death from primary hepatocellular carcinoma, a linear relationship was observed.
This study showed that HBV carriers in regions where HBV is highly prevalent and primary hepatocellular carcinoma is common are at high risk for this tumour. Further, the study indicated that the rate of death from this cancer is higher in an area of high aflatoxin B1 intake than in areas of lower intake. The limitations of the study, first, that intake of aflatoxin B1 was estimated from the availability of raw foodstuffs to the population and then attributed to individuals. Secondly, the correlation between the incidence of primary liver cancer and aflatoxin B1 intake was not adjusted for any of possible confounders, such as HCV infection, alcohol drinking, tobacco use, or nutritional status. Thirdly, exposure to HBV may have been underestimated since PCR was not used. The potency of aflatoxin in patients with primary liver cancer who were HBsAg– was estimated on the basis of only seven cases, some of which would have been shown to be HBsAg+ with PCR. Lastly, the prevalence of HBsAg was measured in a 25% sample of the cohort and attributed to the region.
The Committee at its forty-ninth meeting did not use the results of Campbell et al. (1990) in making quantitative estimates. This study, which showed no association between aflatoxin intake and the incidence of primary liver cancer in China, is more directly pertinent to determining the strength of the association between aflatoxin intake and the incidence of primary liver cancer (hazard assessment). A dose–response assessment, which is conducted after an assessment of the probability of an association on the basis of all the data, is guided by the selection of appropriate data, assuming that the association exists. A study with negative results is generally not suitable for this purpose.
In a cohort study in Shanghai, China (Ross et al., 1992; Qian et al., 1994), information from both measurements of biomarkers and dietary questionnaires was used to determine aflatoxin intake. The information on intake was, however, reported only as positive or negative, and the detailed data necessary for construction of a dose–response curve were not available. The study conducted by Wang et al. (1996) was the only one available to the Committee at its forty-ninth meeting in which HCV was considered, but the results were inconclusive.
In order to estimate the carcinogenic potency of aflatoxin M1 the Committee at its present meeting used a comparative approach, based on the results of studies in laboratory animals and in vitro, as shown in Figure 5. Thus, the relative carcinogenic potency of aflatoxin M1 and aflatoxin B1 in Fischer rats was estimated from the study of Cullen et al. (1987), which was considered the most relevant study for risk assessment, as it is the only 2-year study available, and, as noted above, there are no epidemiological studies in which the intake of both aflatoxin M1 and aflatoxin B1 was determined. Additional studies were considered in order to determine the robustness of this estimate of relative potency across species, by extrapolation. None of the studies considered here directly took into account HBV or HCV infection or other factors that might interact with and modify the carcinogenicity of aflatoxin M1.
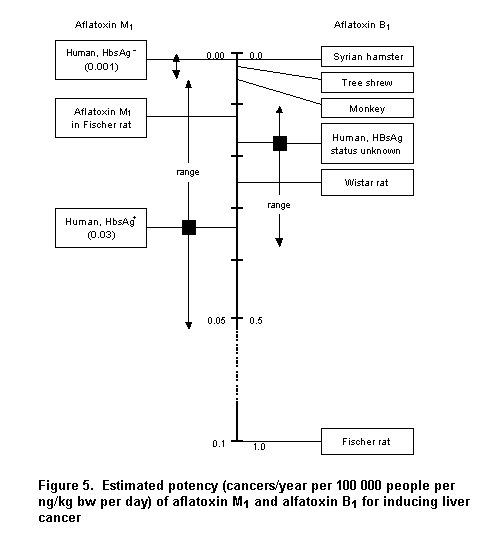
In all of the risk assessments, intake was expressed as the lifetime average intake in nanograms per kilogram of body weight per day.
Although aflatoxin M1 has been shown to be cytotoxic and to affect growth and the immune system, the focus of this risk assessment is its carcinogenicity.
The report of the forty-ninth meeting includes an abbreviated discussion of the mathematical models used in risk assessment. Since the data for aflatoxin M1 are so limited, a simple linear extrapolation was used here.
Figure 5 was modified from the report of the forty-ninth meeting of the Committee. It presents potencies estimated from the results of experimental and epidemiological studies. The carcinogenic potency of aflatoxin B1 in Fischer rats, a sensitive species, was arbitrarily set at 1; and the carcinogenic potencies of aflatoxin B1 in other species are represented on the line as a fraction of 1, i.e. relative to that in Fischer rats. The results of epidemiological studies in which the HBV infection status of the participants was unknown gave estimated potencies between those of HBV infected and uninfected persons. The potencies shown in Figure 5 do not apply directly to aflatoxin M1, since the estimates of intake given in many of the epidemiological studies did not include the contributions to total aflatoxin intake from milk and milk products. However, the potency of aflatoxin B1 provides some information about that of aflatoxin M1.
The significance of estimates of the potency of aflatoxin B1 relative to that of aflatoxin M1 derives from the fact that aflatoxin M1 is a metabolite of aflatoxin B1. At low intake, aflatoxin M1 is a potentially significant (~10–25%) component of the metabolic pathway of aflatoxin B1 in humans, as judged from studies of human liver slices and microsomes (Ramsdell & Eaton, 1990; Gallagher et al., 1996; Heinonen et al., 1996). Approximately 2–7% of ingested aflatoxin B1 has been estimated to be excreted in the form of urinary aflatoxin M1 (Groopman et al., 1992; Cheng et al., 1997). The difference in the percentage converted to aflatoxin M1 and that excreted suggests that at least some of the aflatoxin M1 derived from aflatoxin B1 is further metabolized to the DNA-reactive epoxide. Consequently, estimates of the potency of aflatoxin B1 provide some information about the potential risk posed by aflatoxin M1. Previous estimates of the carcinogenic potency of aflatoxin B1 based on the correlation with rates of primary liver cancer implicitly include the effects of aflatoxin M1 derived from metabolism of ingested aflatoxin B1. If exposure to aflatoxin M1 derived from metabolism of aflatoxin B1 is comparable in magnitude to the intake of aflatoxin M1 by ingestion, the potency of aflatoxin M1 can be no greater than that which has been estimated for aflatoxin B1, and is probably much less.
No epidemiological study equivalent to that of Yeh et al. (1989) for aflatoxin B1 has been reported that addressed the carcinogenicity of aflatoxin M1 in the presence and absence of HBV (and/or HCV). It was assumed that HBV and aflatoxin M1 interact in increasing the risk for liver cancer in a manner comparable to that seen in the study of Yeh et al. (1989).
As no epidemiological studies have been reported that bear on the relationship between risk for primary liver cancer and intake of aflatoxin M1 per se, the potency of aflatoxin M1 was estimated from a comparative perspective. For the purposes of risk assessment, the 2-year study in male Fischer rats (Cullen et al., 1987) was used. In this study, groups of 62 male Fischer rats received diets containing 0.5, 5.0, or 50 µg/kg of aflatoxin M1. An additional 63 animals received agar diet without aflatoxin M. A positive control received a diet containing 50 µg/kg of aflatoxin B1, and an additional 42 rats were fed rodent chow. All animals were offered food and water ad libitum. The total intake of aflatoxin was estimated on the basis of food consumption: rats fed diets containing aflatoxin M1 at 50 µg/kg ingested approximately 1 mg of aflatoxin M1, while those at 5.0 and 0.5 µg/kg groups ingested 0.1 and 0.01 mg, respectively. The group fed a diet containing aflatoxin B1 had received a total of 0.8 mg by the time they were killed. Rats were scheduled for killing at 3, 6, 10, 16, 17, 19, and 21 months, but all surviving rats receiving aflatoxin B1 were killed at 17 months.
In rats fed the diet containing aflatoxin M1 at 50 µg/kg, hepatocellular carcinomas were detected in two of 18 rats killed at 21 months, and neoplastic nodules were found in six of 37 killed between 19 and 21 months. No nodules or carcinomas were observed in groups receiving the lower doses. Nineteen of 20 rats fed the diet containing aflatoxin B1 had developed hepatocellular carcinomas by 17 months. From these results, it was estimated that the potency of aflatoxin M1 is 2–10% that of aflatoxin B1 (Hsieh et al., 1984). A conservative view of this result is that the potency of aflatoxin M1 is approximately one order of magnitude less than that of aflatoxin B1 in that species. Therefore, the carcinogenic potency of aflatoxin M1 in Fischer rats was placed at 0.1 the carcinogenic potency of aflatoxin B1 in Figure 5.
The potency of aflatoxin M1 in Fischer rats was calculated as follows:
2/18 risk / (1 mg/lifetime x 21 months/lifetime x 31 days/month) / 0.3 kg bw = 0.00057 mg/kg bw per day.
The corresponding calculation for aflatoxin B1 in Fischer rats is:
19/20 risk / (1 mg/lifetime x 17 months/lifetime x 31 days/month) / 0.3 kg bw = 0.006 mg/kg bw per day.
Thus, the potency of aflatoxin B1 is approximately 10 times that of aflatoxin M1. This is a conservative assessment of the carcinogenic potential of aflatoxin M1 relative to aflatoxin B1 because the tumour rate induced by aflatoxin B1 at 17 months was compared directly with that induced by aflatoxin M1 at 21 months. Although no hepatocellular carcinomas were observed in rats fed aflatoxin M1 and killed at 17 months, this may have been a consequence of the sample size. In any event, the true, as opposed to the observed, tumour rate induced by aflatoxin M1 at 17 months is certainly greater than 0 but lower than the rate at 21 months. This would imply a corresponding ratio greater than 10-fold.
Although the study of Cullen et al. (1987) was conducted with Fischer rats and did not include interaction with HBV or HCV, it is useful for estimating potency because the carcinogenicity of aflatoxin M1 and aflatoxin B1 is compared in the same experiment with the same control group. A limitation of this study for comparing the carcinogenic potency of aflatoxin M1 and aflatoxin B1 is that only one group received aflatoxin B1, and therefore a dose–response curve could not be constructed for aflatoxin B1 under these experimental conditions. Hence, the potency of aflatoxin B1 in this study was estimated from only one dose by linear extrapolation.
In the absence of epidemiological studies of aflatoxin M1, the relative risk associated with exposure to aflatoxin M1 versus aflatoxin B1 for human populations is assumed to be the same as that observed for Fischer rats. Use of this assumption is supported to some extent by studies of the relative potencies of the two toxins in vitro, as discussed below. The carcinogenic potency of aflatoxin M1 in HBsAg– individuals is placed in Figure 5 at 0.001 or 1/10 of the previously estimated carcinogenic potency of aflatoxin B1 in such persons. Similarly, the carcinogenic potency of aflatoxin M1 in HBsAg+ individuals is placed at 0.03, 1/10th of the previously estimated carcinogenic potency of aflatoxin B1. This approach, extrapolating potency, was adopted because of the apparent sensitivity of Fischer rats to aflatoxin B1.
As discussed in the report of the forty-ninth meeting, the customary three-quarters body weight scaling extrapolation of the potency of aflatoxin B1 in Fischer rats to humans results in an estimate of 37 cases per 100 000/year per ng/kg bw per day, which is orders of magnitude greater than the range of potency of aflatoxin B1 (0.002–0.036 per 100 000/year per ng of aflatoxin B1 per kg bw/day) determined from epidemiological studies of aflatoxin B1. Consequently, aflatoxin B1 is an extreme exception to the general pattern of three-quarters body weight and/or surface area scaling that has been observed to hold for many carcinogens. Aflatoxin M1 is also expected to be an exception to the general pattern.
A comparative study of aflatoxin M1 and aflatoxin B1 by Neal et al. (1998) suggested that cytotoxic effects in the MCL-5 lymphoblastoid cell line are generally absent at concentrations of aflatoxin B1 < 0.05 µg/ml and of aflatoxin M1 < 1 µg/ml. Although the susceptibility of liver cells to the cytotoxic effects of aflatoxins may differ from those of MCL-5 cells, the findings appear to be consistent with the conclusion that tumour induction in Fischer rats at the dietary concentration tested is likely to be a consequence of mutagenic effects of aflatoxin B1 and aflatoxin M1 rather than of increased liver cell proliferation due to cytotoxicity.
The relative potency of aflatoxin M1 and aflatoxin B1 in Fischer rats has been estimated in an animal model that is not infected with HBV, and no such studies in animals co-infected with HBV or any other virus (e.g. HCV) that causes significant toxicity, liver damage, and compensatory liver cell regeneration exist. Nevertheless, to the extent that the predominant mechanism of the carcinogenicity of aflatoxin M1 and aflatoxin B1 is likely to be genotoxic, at least at low levels of intake, it would appear reasonable to assume that the relative potency would be the same in populations with different background rates of liver cancer attributable to differences in general susceptibility to genotoxic carcinogens. Differences in the prevalence of HBV, which by itself is likely to cause liver cancer by a non-genotoxic process leading to enhanced cell proliferation, would appear to be represent an alteration in susceptibility to the effects of genotoxic agents.
The assumption that HBV affects the rates of primary liver cancer primarily by increasing and sustaining cell proliferation is supported by the study of Evans et al. (1998), discussed in section 2. Studies in woodchucks also seem to support the contention that the most significant mechanism of interaction between aflatoxins and hepatitis is increased cell proliferation, which fixes the adducts formed from the metabolism of aflatoxin B1 (Izzotti et al., 1995). This may reasonably be expected to be the case for aflatoxin M1 and HBV as well. Thus, in the absence of altered cell proliferation due to cytotoxic effects of either aflatoxin M1 or aflatoxin B1 per se, the induction of liver tumours by a genotoxic mechanism is generally correlated with adduct levels or other measures of genotoxic damage, irrespective of the background tumour rate (Poirier & Beland, 1994; Otteneder & Lutz, 1999; Wang & Groopman, 1999). This situation would appear to be likely for aflatoxin M1 and aflatoxin B1 at current levels of human intake.
Hepatitis may also significantly alter the metabolism of both aflatoxin B1 and aflatoxin M1, although there is little evidence to suggest that the metabolism of one would be altered preferentially. Consequently, for the purposes of this assessment and in the absence of specific data to the contrary, the potency of aflatoxin M1 and aflatoxin B1 is assumed to be the same in HBsAg+ and HBsAg– individuals.
The estimates of the potency of aflatoxin M1 were combined with estimates of intake for the European regional diet. The potency of aflatoxin M1 in a human population with an HBV prevalence rate P was projected to be the weighted combination of the potency estimates for HBsAg– individuals (0.001 cancers/100 000 per year per ng/kg bw per day) and HBsAg+ individuals (0.03 cancers/100 000 per year per ng/kg bw per day). Specifically, the average potency of aflatoxin M1 in a population of individuals with an HBV prevalence rate P was taken to be:
Potency = 0.001 x (1–P) + 0.03 x P.
Three rates of HBV prevalence were considered (1, 5, and 25%) in order to span the range of infection rates actually observed in various western and South-East Asian countries. The projected cancer risks were calculated as the product of the average potency and estimates of intake of aflatoxin M1 in a European diet, corresponding to relatively high intake of milk and milk products. The projected risks were calculated by assuming various standards of contamination, including the current standard in the USA of 0.5 µg/kg and the proposed standard of 0.05 µg/kg. Assuming that all products are contaminated at the standard results in a worst-case projection of the population risk. For a given standard, the average level of contami-nation is likely to be less than the standard, since producers seek to ensure that their product meets the standard. The calculations are show in Table 18.
Table 18. Projected risk for liver cancer attributed to intake of aflatoxin M1 in milk and comparison of effect of application of the proposed maximum levels
|
Concentration of aflatoxin M1 in milk (µg/kg) |
HBsAg+ |
Average potencya |
Aflatoxin M1 intakeb |
Prevalence of liver cancer attributable to aflatoxin M1 (cancer cases/ year per 106) |
|
|
ng/person per day |
ng/kg bw per dayc |
||||
|
Weighted mean of 0.023 |
1 |
0.0013 |
6.8 |
0.11 |
1.5 |
|
5 |
0.0025 |
6.8 |
0.11 |
2.8 |
|
|
25 |
0.0083 |
6.8 |
0.11 |
9.4 |
|
|
0.05 |
1 |
0.0013 |
15 |
0.25 |
3.2 |
|
5 |
0.0025 |
15 |
0.25 |
6 |
|
|
25 |
0.0083 |
15 |
0.25 |
20 |
|
|
0.5 |
1 |
0.0013 |
150 |
2.5 |
32 |
|
5 |
0.0025 |
150 |
2.5 |
60 |
|
|
25 |
0.0083 |
150 |
2.5 |
200 |
|
a
Potency in cases of cancer/year per 100 000 per ng/kg bw per dayb
Based on a European-type dietc
Assuming a body weight of 60 kgThe European diet was chosen from among the five regional diets because milk consumption is higher in that than in the other diets. It is obvious from the calculations that the additional risks for liver cancer predicted with the proposed maximum limit on aflatoxin M1 of 0.05 µg/kg or with the current limit of 0.5 µg/kg are very small. For example, in a population with an HBV prevalence rate of 1%, the additional number of cases of liver cancer associated with aflatoxin M1 contamination of milk at 0.5 µg/kg versus 0.05 µg/kg would be 0.0029 per 100 000 population/year (i.e. 0.0032–0.00032 cancers per 100 000 per year). Thus, in a population of 250 million with an HBV prevalence rate of 1%, the projected increase in risk would correspond to about seven additional cases per year at 0.5 µg/kg in comparison with the 0.05 µg/kg standard. This projected difference would not constitute a discernible reduction in the overall burden of liver cancer as measured in national cancer registries. Furthermore, the potency of aflatoxin M1 would appear to be so low in HBsAg– individuals that no carcinogenic effect of aflatoxin M1 intake in heavy consumers of milk and milk products could be demonstrated. The projected potency of aflatoxin M1 in HBsAg+ individuals is about 30-fold higher than that in HBsAg– individuals and comparable in magnitude to the estimated potency of aflatoxin B1.
Metabolism
In all species and tissues tested to date, the mutagenicity, carcinogenicity and DNA binding activity of aflatoxin B1 appear to result from its activation by cytochrome P450 enzymes to produce aflatoxin B1-8,9-epoxide. The metabolism of aflatoxin B1 to the epoxide and to aflatoxin M1 can be blocked in vitro (human hepatocytes) and in vivo (rats) by treatment with oltipraz, an antischistosomal drug, which blocks the formation of the epoxide and induces the major aflatoxin detoxication enzyme, glutathione-S-transferase (GST). Oltipraz is being tested in phase I and II clinical trials in China in the prevention of liver cancer; the results of these studies will be useful for clarifying the metabolism and mode of action of aflatoxins in humans.
Studies in human hepatocytes show wide variation among individuals in the metabolism and activation of aflatoxins. Human hepatocytes appear to form less of the epoxides of both aflatoxin B1 and M1 than rat hepatocytes. Conjugation of both epoxides with GST appears to occur more rapidly in mouse than in human hepatocytes. The details of the relationship between aflatoxin metabolism, activation. and detoxication in humans remains unclear.
Toxicological studies
Aflatoxin M1 is cytotoxic, as demonstrated in human hepatocytes in vitro and its acute toxicity in several species is similar to that of aflatoxin B1. In ducklings and rats, the acute and short-term toxicity of aflatoxin M1 was similar to or slightly less than that of aflatoxin B1. In studies of carcinogenicity, aflatoxin M1 was about one order of magnitude less potent than aflatoxin B1, even in sensitive species like the rainbow trout and the Fischer rat. The in vitro genotoxic potency of aflatoxin M1 was similar to that of aflatoxin B1 in some test systems and between one-half and one-sixth that of aflatoxin B1 in other test systems.
Observations in humans
No studies were available on the association between the dietary intake of aflatoxin M1 and the risk for liver cancer. The Committee reviewed the literature on this topic published since the previous evaluation to determine if the additional studies provided more accurate estimates of the dose–response relationships than those used in 1997. Studies in which the recently developed biomarkers of exposure (aflatoxin-albumin adducts in serum, aflatoxin–N7-guanine adducts in urine, aflatoxin M1 metabolites in urine or patterns of P53 mutations) were used did not provide additional evidence that would allow more accurate risk assessments. Studies with more sensitive markers of exposure to hepatitis B and/or C viruses in patients with liver cancer strongly suggested that the estimated fraction of cases of human liver cancer attributable to these viral infections is increasing. As a consequence, estimates of the potency of aflatoxin B1 estimated at the forty-ninth meeting in 1997 are likely to be overestimates. At its present meeting, the Committee made a conservative estimate of the potency of aflatoxin M1 on the basis of the estimates for aflatoxin B1.
Analytical methods
Screening tests for aflatoxin M1 in milk and milk products include radioimmuno-assay and enzyme-linked immunosorbent assay methods. The former have found little application in routine investigations of aflatoxin M1 in milk, whereas the latter are more often used. For regulatory purposes, positive results in enzyme-linked immunosorbent assays must be confirmed by an accepted reference method.
The quantitative analytical methods for aflatoxin M1 include thin-layer and liquid chromatography. Many of these methods were developed for the analysis of milk and milk powder but can often be used for other dairy products. Five such methods have been studied in formal collaborative studies, and their performance characteristics have been published. With the development of liquid chromatography in the 1980s, most laboratories abandoned use of thin-layer chromatography for the analysis of aflatoxin M1. Use of immunoaffinity cartridges for clean-up of milk extracts was introduced subsequently, and the combination of immunoaffinity and liquid chromatography now offers the best means for efficient clean-up and precise determination of low concentrations of aflatoxin M1. A method involving a combination of immunoaffinity column clean-up with thin-layer chromatography and a computer-based low-cost densitometer is being validated for low concentrations of aflatoxins in a formal collaborative study.
Sampling protocols
Sampling plans for aflatoxins in granular feed products have been evaluated, but there has been little work on the evaluation of sampling plans to detect aflatoxin M1 in milk. However, the European Union, the USA, and the Southern Common Market (Mercado Commun del Sur – MERCOSUR) have designed sampling plans for aflatoxin M1. A Directive of the Commission of the European Union specifies that from a batch of milk mixed by manual or mechanical means a minimum sample of 0.5 kg (or L) composed of at least five increments should be collected. The batch is accepted if the concentration of aflatoxin M1 does not exceed the permitted limit. In the USA, the Food and Drug Administration stipulates that samples should consist of at least 4.5 kg of milk, composed of no fewer than 10 randomly selected portions.
As the distribution of aflatoxin M1 in liquid milk can be expected to be reasonably homogeneous, sampling of liquid milk for aflatoxin M1 will involve less uncertainty than sampling of granular feed products for aflatoxins. Most of the uncertainty in estimating aflatoxin M1 in milk is probably associated with the analytical procedure.
Effects of processing
The results of the numerous studies on the effect of milk processing on the concentration of aflatoxin M1 are variable. The concentration is not appreciably reduced by heat treatment. Production of yoghurt, cheese, cream, milk powder, or butter does not lead to loss of aflatoxin M1, although it is redistributed differentially into the products resulting from these processes.
Aflatoxin M1 can be partially eliminated from milk by physical or chemical procedures, which include use of adsorbents, hydrogen peroxide, and ultra-violet radiation. These treatments are not readily applicable for the dairy industry, however, and their safety has not been tested; moreover, the costs may be prohibitive for large-scale application.
Food consumption/dietary intake assessment
Data on the concentrations of aflatoxin M1 in milk were submitted by Argentina, Brazil, Canada, Indonesia, Norway, the United Arab Emirates, the USA, and several Member States of the European Union; some data were also obtained from the literature. Some data may reflect biased or limited sampling designs and may not be representative of the area or nation in which the samples were obtained. Important information (number of samples, analytical quality assurance, mean value, individual sample values) was not available for some data sets. This does not imply that the data were not of good quality, but that they should be used with caution. All the data on aflatoxin M1 that were submitted were used in the evaluation.
The dietary intake of aflatoxin M1 was estimated from data on concentrations of aflatoxin M1 in milk and from data on milk consumption in the GEMS/Food regional diets. The weighted mean concentration of aflatoxin M1 in milk was 0.023 µg/kg in the European-type diet, 0.022 µg/kg in the Latin American diet, 0.36 µg/kg in the Far Eastern diet, 0.005 µg/kg in the Middle Eastern diet, and 0.002 µg/kg in the African diet. These mean concentrations were based on 10 778 milk samples for the European-type diet, 893 for the Latin American diet, 1191 for the Far Eastern diet, 231 for the Middle Eastern diet, and 15 for the African diet. The intake of aflatoxin M1 from milk was calculated to be 6.8 ng/person per day for the European-type diet, 3.5 ng/person per day for the Latin American diet, 12 ng/person per day for the Far Eastern diet, 0.7 ng/person per day for the Middle Eastern diet, and 0.1 ng/person per day for the African diet. Intake calculated from the European regional diet was used for the assessment of cancer risk because this diet had the highest milk consumption. If all milk consumed were contaminated with aflatoxin M1 at the proposed maximum levels of 0.05 µg/kg or 0.5 µg/kg, the intake of aflatoxin M1 from milk in the European regional diet would be 15 ng/person per day or 150 ng/person per day, respectively.
One approach for determining the potential effects on dietary intake of the two proposed maximum levels for aflatoxin M1 in milk involves estimating intake on the basis of mean aflatoxin M1 concentration in milk for all samples, for all samples containing less than 0.5 µg/kg, and for all samples containing less than 0.05 µg/kg for a given population. The three calculated concentrations are multiplied by the milk consumption of the population of interest to determine the intake of aflatoxin M1. The most recent data on aflatoxin M1 in milk in some European Union Member States (analyses of 7573 samples reported in 1999) indicated that all samples contained < 0.05 µg/kg, so the choice of either maximum level would not affect intake. Similarly, the data from Canada (81 samples reported in 1997-98) showed that all milk samples analysed contained < 0.015 µg/kg (the limit of detection) of aflatoxin M1 so intake would not be affected at either level. The USA submitted individual data for 3620 samples collected between 1995 and 2000. On the basis of these data, the intakes of aflatoxin M1 from milk were estimated to be 0.030 ng/kg of body weight per day for all samples, 0.023 ng/kg of body weight per day when a maximum level of 0.5 µg/kg was used, and 0.0035 ng/kg of body weight per day for a maximum level of 0.05 µg/kg. As other submissions and data from the literature did not include individual values for samples containing more than 0.5 µg/kg or more than 0.05 µg/kg aflatoxin M1 in milk, similar calculations could not be performed on any of the other data.
Another approach for determining the effect of the proposed maximum levels on dietary intake is to generate distribution curves for the regional data for the concentrations of aflatoxin M1 in milk. The distribution was constructed assuming log normality and with mean aflatoxin M1 concentrations in the GEMS/Food regions and the maximum values reported. The distribution curve for the European regional diet showed that if 0.05 and 0.5 µg/kg were set as the maximum levels, they would be at the extreme end of the distribution and consequently would have no effect on intake. The distribution curve for milk samples in the Middle Eastern diet, for which there were relatively few data, was similar to that for the European-type diet. For Latin American diets, selection of a maximum level of 0.5 µg/kg would also have no effect; however, use of a maximum level of 0.05 µg/kg would probably reduce intake. In the Far Eastern diet, in which the contamination of milk is higher, intake of aflatoxin M1 would be decreased at both proposed levels, but it should be noted that milk consumption is low in the Far Eastern diet.
Prevention and control
About 0.3–6.2 % of aflatoxin B1 in animal feed is transformed to aflatoxin M1 in milk. For contaminated feed, a linear relationship has been found between intakes of aflatoxin B1 in feed ranging from 5 to 80 µg/kg and the aflatoxin M1 content of milk, as follows: aflatoxin M1 (ng/kg milk) = [1.19 x aflatoxin B1 intake (µg/cow/day)] + 1.9. Thus, production of milk containing aflatoxin M1 at 0.05 µg/kg (the proposed Codex limit) would require that the average intake of aflatoxin B1 by dairy cows be limited to approximately 40 µg/day/cow. On the basis of a daily feed consumption of 12 kg/cow of compound feeds, the use of the limit of 40 µg aflatoxin B1 would mean that the content of aflatoxin B1 in the feed would have to be 3.4 µg/kg in order to meet the limit of 0.05 µg/kg for aflatoxin M1.
The most effective means for controlling aflatoxin M1 in the food supply is to control the amount of aflatoxin B1 in feed for dairy cows. Specific regulations exist in many countries to control aflatoxin B1 in the feed supply, but in parts of the world where cottonseed and maize are incorporated into animal feed, an effective aflatoxin control programme for feed may be difficult to design, particularly at low aflatoxin levels. The difficulty is related to the heterogeneous distribution of aflatoxin in these commodities, which results in a high degree of sampling variability.
The concentration of aflatoxin B1 in feed can be reduced by good manufacturing practice and good storage practices. If preventive measures fail, however, aflatoxin B1 can be reduced in feed by blending or by physical or chemical treatment. The physical treatments include heat, microwaves, gamma-rays, X-rays, ultra-violet light, and adsorption. Adsorption of aflatoxins onto hydrated sodium calcium alumino-silicate and other inert materials has been used in the animal feed industry in an attempt to reduce the aflatoxin M1 content of milk. The most successful chemical procedure for degrading aflatoxins in animal feed is treatment with ammonia. Ammoniation of agricultural commodities leads to decomposition of 95–98 % of the aflatoxin B1 present, and this procedure is used in various countries.
Since aflatoxin M1 is a metabolite of aflatoxin B1 and is presumed to induce liver cancer in rodents by a similar mechanism, estimates of the potency of aflatoxin B1 can be used for determining the risk due to intake of aflatoxin M1. No adequate epidemiological studies exist on the dose–response relationships between the intake of aflatoxin M1, exposure to hepatitis B or C virus, and liver cancer. The Committee therefore assumed that aflatoxin M1 acts similarly to aflatoxin B1 with hepatitis B (and possibly) C virus. The Committee used the comparative figure for carcinogenic potency derived at its forty-ninth meeting and assumed that the potency of aflatoxin M1 was one-tenth that of aflatoxin B1 in the Fischer rat. The carcinogenic potency of aflatoxin M1 was estimated to be 0.001/100 000 per year per ng/kg of body weight per day in HBsAg– individuals and 0.03/100 000 per year per ng/kg of body weight per day in HBsAg+ individuals.
The estimates of the potency of aflatoxin M1 were combined with estimates of intake from the GEMS/Food European regional diet. The potency of aflatoxin M1 in a human population with an HBsAg prevalence, P, was projected to be the weighted combination of the potency estimates for HBsAg– and HBsAg+ individuals. Specifi-cally, the average potency of aflatoxin M1 in a population of individuals with a hepatitis B virus prevalence rate P was taken to be: Potency = 0.001 x (1–P) + 0.03 x P.
Three rates of HBsAg+ prevalence (1, 5, and 25%) were considered to span the range of rates of infection with hepatitis B virus observed in various populations in western and South-East Asian countries. The risks for liver cancer were projected to be the product of the average potency values and estimates of intake of aflatoxin M1 from a European-type diet, corresponding to a relatively high intake of milk and milk products. The projected risks due to aflatoxin M1 were calculated for the two proposed maximum levels, 0.5 and 0.05 µg/kg. It was assumed that all products were contaminated at the two proposed maximum levels, giving worst-case projections of the population risk. Projected risks were also calculated for the weighted mean of 0.023 µg/kg of milk for the European regional diet (see Table 18).
The calculations show that, with worst-case assumptions, the projected risks for liver cancer at the proposed maximum levels of aflatoxin M1 of 0.05 and 0.5 µg/kg are very small. For example, in a population with a prevalence of hepatitis B virus infection of 1%, which is typical for the USA and western Europe, the additional numbers of liver cancer cases associated with contamination of all milk with aflatoxin M1 at 0.5 µg/kg versus 0.05 µg/kg would be 29 (i.e. 32 minus 3.2) cancers/1000 million persons per year. The potency of aflatoxin M1 appears to be so low in HBsAg– individuals that a carcinogenic effect of aflatoxin M1 intake in those who consume large quantities of milk and milk products in comparison with non-consumers of these products would be impossible to demonstrate.
Hepatitis B virus carriers might benefit from a reduction in the aflatoxin concentration in their diet, and the reduction might also offer some protection to hepatitis C virus carriers. Reduction of the current concentrations of aflatoxins in the diet in most developed countries is unlikely to produce an observable reduction in the rates of liver cancer.
At its present meeting, the Committee reiterated its previous conclusion that the liver cancer burden could best be reduced by giving priority to hepatitis B virus vaccination campaigns and to prevention of hepatitis C virus infection, which implies reinforcement of the control of blood and blood products and the use of sterile medical equipment.
Alcroft, R. & Carnaghan, R.B.A. (1962) Groundnut toxicity: Aspergillus flavus toxin (aflatoxin) in animal products: Preliminary communication. Vet. Rec., 74, 863–864.
American Oil Chemists’ Society (2000) Laboratory Proficiency Program, Pamphlet November 2000, AOCS Technical Services, Champaign, IL, USA.
Andreou, V.G. & Nikolelis, D.P. (1998) Flow injection monitoring of aflatoxin M1 in milk and milk preparations using filter-supported bilayer lipid membranes. Anal. Chem., 70, 2366–2371.
AOAC International (2000) Minutes of meeting official methods board meetings, Phoenix, AZ; Los Angeles, CA; Philadelphia, PA, USA. Arlington, VA, USA.
Applebaum, R.S. & Marth, E.H. (1982a) Inactivation of aflatoxin M1 in milk using hydrogen peroxide and hydrogen peroxide plus riboflavin or lactoperoxidase. J. Food Prot., 45, 557–560.
Applebaum, R.S. & Marth, E.H. (1982b) Use of sulfite or bentonite to eliminate aflatoxin M1 from naturally contaminated raw milk. Z. Lebensm. Unters. Forsch., 174, 303–305.
Association of Official Analytical Chemists (1984) Official Methods of Analysis of the AOAC, 14th Ed., Arlington, VA, sections 26.095–26.098.
Aulerich, R.J., Bursian, S.J. & Wetson, G.L. (1993) Effects of sublethal concentrations of aflatoxins on the reproductive performance of mink. Bull. Environ. Toxicol., 50, 750–756.
Bagni, A., Castagnetti, G.B., Chiavarti, C., Ferri, G., Losi, G. & Montanari, G. (1993) [Investigation of the presence of aflatoxins M1 and M2 in cows’ milk from sampling in the Province of Reggio Emilia.] Ind. Latte, 4, 55–66 (in Italian).
Bailey, G.S., Dashwood, R., Loveland, P.M., Pereira, C. & Hendricks, J.D. (1998) Molecular dosimetry in fish: Quantitative target organ DNA adduction and hepatocarcinogenicity for four aflatoxins by two exposure routes in rainbow trout. Mutat. Res., 339,233–244.
Bammler, T.K., Slone, D.H. & Eaton, D.L. (2000) Effects of dietary oltipraz and ethoxyquin on aflatoxin B1 biotransformation in non-human primates. Toxicol. Sci., 54, 30–41.
Barton, C.C., Hill, D.A., Yee, S.B., Barton, E.X., Ganey, P.E. & Roth, R.A. (2000) Bacterial lipopolysaccharide exposure augments aflatoxin B1-induced liver injury. Toxicol. Sci., 55, 444–452.
Bauer, D.H., Lee, D.J. & Sinnhuber, R.O. (1969) Acute toxicity of aflatoxins B1 and G1 in rainbow trout (Salmo garirdneri). Toxicol. Appl. Pharmacol., 15, 415–419.
Begino, E.T. (1998) Aflatoxin detection in milk in the Philippines. Poster presented at the 4th APFAN Conference, November 1998, Chiang Mai, Thailand. Data from the Bureau of Animal Industry, Department of Agriculture, Philippines.
Biancardi, A. (1997) Determination of aflatoxin M1 residues in milk: A comparative assessment of ELISA and IAC-HPLC methods. In. Aliment., 36, 870–876.
Boenke, A. (1997) The food and feed chain—A possible strategy for the production of certified reference materials (CRMs) in the area of mycotoxins? Food Chem., 60, 255–262.
Boix-Ferrero, J., Pellin, A., Blesa, R., Adrados, M. & Llombart-Bosch, A. (1999) Absence of p53 gene mutations in hepatocarcinomas from a Mediterranean area of Spain: A study of 129 archival tumor samples. Virchows Arch., 434, 497–501.
Boriboon, U. & Suprasert, D. (1994) Determination of aflatoxin M1 and M2 in milk and milk product. Minist. Public Health J. (Thailand), 13, 108–114.
Bosch, F.X., Ribes, J. & Borras, J. (1999) Epidemiology of primary liver cancer. Semin. Liver Dis., 19, 271.
Brechot, C., Jaffredo, F., Lagorce, D., Gerken, G., Buschenfelde, K.M., Papakonstontinou, A., Hadziyannis, S. Romeo, R., Colombo, M., Rodes, J., Bruix, J., Williams, R. & Naoumov, N. (1998) Impact of HBV, HCV and GBV-C/HGV on hepatocellular carcinomas in Europe: Results of a European concerted action. J. Hepatol., 29, 173–183.
Brewington, C.R. & Weihrauch, J.L. (1970) Survey of commercial milk samples for aflatoxin M. J. Dairy Sci., 53, 1509–1510.
Buchi, G., Spitzner, D., Paglialunga, S. & Wogan, G.N. (1973) Synthesis and toxicity evaluation of aflatoxin P1. Life Sci., 13, 1145.
Campbell, T.C., Chen, J., Liu, C., Li, J. & Parpia, B. (1990) Nonassociation of aflatoxin with primary liver cancer in a cross-sectional ecological survey in the People’s Republic of China. Cancer Res. 50, 6882-6893.
Centro de Referencia de Micología (1999) Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Argentina
Cheng, Z., Root, M., Pan, W., Chen, J. & Campbell, T.C. (1997) Use of an improved method for analysis of urinary aflatoxin M1 in a survey of mainland China and Taiwan. Cancer Epidemiol Biomarkers Prev., 6, 523–529.
Chi, W.J., Doong, S.L., Lin-Shiau, S.Y., Boone, C.W., Kelloff, G.J. & Lin, J.K. (1998) Oltipraz, a novel inhibitor of hepatitis B virus transcription through elevation of p53 protein. Carcinogenesis, 19, 2133–2138.
Chou, M.W., Mikhailova, M.V., Nichols, J., Poirier, L.A., Warbritton, A. & Beland, F.A. (2000) Interactive effects of methyl-deficiency and dietary restriction on liver cell proliferation and telomerase activity in Fischer 344 rats pretreated with aflatoxin B1. Cancer Lett, 152, 53–61.
Chu, F.S. (1984) Immunochemical studies on mycotoxins. In: Kurata, H. & Ueno, Y., eds, Toxigenic Fungi, Tokyo, Kodansha Ltd, pp. 234–244.
Codex Alimentarius (1992) Draft Report. Codex Committee on Food Additives and Contaminants, The Hague, 23–28 March.
Codex Alimentarius (2000) Codex Committee on Food Additives and Contaminants. CL 1999/13 GEN-CX 0016 FAC-Agenda item 16a. ALINORM 99/37, paras 103-105. Draft maximum level for aflatoxin M1 in milk, 20–24 March.
Comité Européen de Normalisation (1999) Food analysis—Biotoxins—Criteria of analytical methods of mycotoxins (CEN report CR 13505), Brussels.
Commission of the European Union (1989–95) Opinions of the Committee for Scientific Cooperation (SCOOP reports), Health & Consumer Protection Directorate.
Commission of the European Union (1991) Commission Decision 91/180/EEC of 14 February 1991 laying down certain methods of analysis and testing of raw milk and heat treated milk. Off- J. Eur. Commun., L93, 6–8.
Commission of the European Union (1998) Commission Directive 98/53/EC of 16 July 1998 laying down the sampling methods and the methods of analysis for the official control of the levels for certain contaminants in foodstuffs. Off. J. Eur. Commun., L201, 93–101.
Commission of the European Union (1999) Report.
Correa, B., Galhardo, M., Costa, E.O. & Sabino, M. (1997) Distribution of molds and aflatoxins in dairy cattle feeds and raw milk. Rev. Microbiol., 28, 279–283.
Cullen, J.M., Reubner, B.H., Hsieh, L.S., Hyde, D.M. & Hsieh, D.S.P. (1987) Carcinogenicity of dietary aflatoxin M1 in male Fisher rats compare to aflatoxin B1. Cancer Res., 47, 1913–1917.
Davoli, R., Tedeschi, M. & Russo, V. (1986) [Aflatoxin M1 content in pooled milk samples]. In: Proceedings of the 21st International Zootechnical Symposium, Milan, pp. 216-222 (in Italian).
De Natale, G., Fiorentino, A., Busco, V.P. & Pallamara, M. (1989) [Investigation on the presence of aflatoxin M1 in milk samples taken from some farms in Puglia.] Atti Soc. Ital. Sci. Vet., 43, 605–609 (in Italian).
Domagala, J., Kisza, J., Bluthgen, A. & Heescgen, W. (1997) Contamination of milk and aflatoxin M1 in Poland. Milchwissenschaft, 52, 631–633.
Donato, F., Boffetta, P. & Puoti, M. (1998) A meta-analysis of epidemiological studies on the combined effect of hepatitis B and C virus infections in causing hepatocellular carcinoma. Int. J. Cancer, 75, 347–354.
Dragacci, S. & Frémy, J. M. (1993) [Contamination of milk with aflatoxin M1]. Sci. Aliment., 13, 711–722 (in French).
Dragacci, S., Marchond, N. & Venant, A. (1996) First proficiency testing for aflatoxin M1 determination in dried milk in the European Union (CRL document, reference AFM1 06/96), Paris: CNEVA.
Dragacci, S., Grosso, F., Pfauwathel-Marchond, N., Frémy, J..M., Venant, A. & Lombard, B. (2001) Proficiency testing for the evaluation of the ability of European Union– national reference laboratories to determine aflatoxin M1 in milk at levels corresponding to the new European Union legislation. Food Addit. Contam. (in press).
Eaton, D.L. & Groopman, J.D., eds (1994) The Toxicology of Aflatoxins: Human Health, Veterinary, and Agricultural Significance, San Diego, CA, Academic Press.
van Egmond, H.P. (1989) Aflatoxin M1: Occurrence, toxicity and regulation. In: van Egmond, H.P., ed., Mycotoxins in Dairy Products, London, Elsevier Applied Science, pp. 11–55.
van Egmond, H.P. (1994) Aflatoxins in milk. In: Eaton, D.L. & Groopman, J.D., eds, The Toxicology of Aflatoxin: Human Health, Veterinary, and Agricultural Significance, San Diego, CA, Academic Press, pp. 365–381.
van Egmond, H.P. (2000) Associate referee report aflatoxin M. Submitted to AOAC International, June 2000.
van Egmond, H.P. & Speijers, G.J.A. (1999) Mycotoxins. In: van der Heyden, C.A., Younes, M., Fishbein, L. & Miller, S.,eds, International Food Safety Handbook, New York, Marcel Dekker, pp. 341–355.
van Egmond, H.P., Svensson, U.K. & Frémy, J.M. (1997) Mycotoxins. In: Residues and Contaminants in Milk and Milk Products (Special Issue 9701), Brussels, International Dairy Federation, pp. 17–88.
El-Gohary, A.H. (1996) Aflatoxin in some foodstuffs with special reference to public health hazard in Egypt. Indian J. Anim. Sci., 66, 468–473.
El Serag, H.B. & Mason, A.C. (1999) Rising incidence of hepatocellular carcinoma in the United States. New Engl. J. Med., 340, 745–750.
Evans, A.A., O’Connell, A.P., Puch, J.C., Mason, W.S., Shen F., Chen, G.-C., Lin, W.-Y., Dia, A., M-Boup, S., Drame´, B. & London, W.T. (1998) Geographic variations in viral load among hepatitis B carriers with differing risks of hepatocellular carcinoma. Cancer Epidemiol. Biomarkers Prev., 7, 559–565.
FAO (1997) Worldwide regulations for mycotoxins 1995. A compendium. FAO Food and Nutrition Paper 64, Rome, 43 pp.
Fehr, P.M., Bernage, L. & Vassilopoulis, V. (1971) [Effect of consumption of peanut oilcake polluted with Aspergillus flavus in lactating ruminants]. Lait, 48, 377–391 (in French).
Ferguson-Foos, J. & Warren, J.D. (1984) Improved clean-up for liquid chromatographic analysis and fluorescence detection of aflatoxins M1 and M2 in fluid milk products. J. Assoc. Off. Anal. Chem., 67, 1111–1114.
Food & Drug Administration (1996) Compliance Policy Guide (7106.10, Section 527.400 Whole Milk, Low Fat Milk, Skim Milk–Aflatoxin M1), Office of Regulatory Affairs, Washington DC, p. 219.
Food & Drug Administration (2000) Center for Food Safety and Applied Nutrition, Washington DC.
Fremy, J.M. & Boursier, B. (1981) Rapid determination of aflatoxin M1 in dairy products by reversed-phase high performance liquid chromatography. J. Chromatogr., 219, 156–161.
Frémy, J.M. & Chu, F.S. (1989) Immunochemical methods of analysis for aflatoxin M1. In: van Egmond, H.P, ed., Mycotoxins in Dairy Products, London, Elsevier Applied Science, pp. 97–125.
Friesen, M.D. & Garren, L. (1982) International mycotoxin check sample programme. Part II. Report on the performance of participating laboratories for the analysis of aflatoxin M1 in lyophilized milk. J. Assoc. Anal. Chem., 65, 864–868.
Fukal, L. (1988) Use of commercial RIA kit for detecting aflatoxin M1 contamination in milk. Prum. Protreavin, 39, 196–198 (In: Dairy Sci. Abstr., 51, 199).
Fukal, L. & Brezina, P. (1991) [Determination of aflatoxin M1 levels in milk in the production of baby and children’s food using immunoassay]. Nahrung, 35, 745–748 (in German).
Fukal, L., Brezina, P. & Marek, M. (1990) Immunochemical monitoring of aflatoxin M1 occurrence in milk produced in Czechoslovakia. Dtsch. Lebensm. Rundsch., 89, 289–291 (In: Dairy Sci. Abstr., 53, 287).
Gallagher, E.P., Kunze, K.L., Stapleton, P.l. & Eaton, D.L. (1996) The kinetics of alfatoxin B1 oxidation in human cDNA-expressed and human liver microsomal cytochromes P450 1A2 and 3A4. Toxicol. Appl. Pharmacol., 141, 595–606.
Galvano, F., Galofaro, V. & Galvano, G. (1996a) Occurrence and stability of aflatoxin M1 in milk and milk products: A worldwide review. J. Food Prot., 59, 1079–1090.
Galvano, F., Pietri, A., Bertuzzi, T., Fusconi, G., Galvano, M., Piva, A. & Piva, G. (1996b) Reduction of carryover of aflatoxin from cow feed to milk by addition of activated carbons. J. Food Prot., 59, 551–554.
Galvano, F., Galofaro, V., de Angelis, A., Galvano, M., Boganno, M. & Galvano, G. (1998) Survey of the occurrence of aflatoxin M1 in dairy products marketed in Italy. J. Food Prot., 61, 738–741.
Gelosa, L. (1986) [Presence of aflatoxin M1 in milk for consumption.] Latte, 11, 261–264 (in Italian).
Gilli, G., Bono, R. & Carraro, E. (1987) [Aflatoxins and human alimentation: First results on the contamination of milk.] Latte, 12, 229–234 (in Italian).
Gradelet, S., LeBon, A.-M., Berges, R., Suschetet, M. & Astorg, P. (1998) Dietary carotenoids inhibit aflatoxin B1-induced liver preneoplastic foci and DNA damage in the rat: Role of the modulation of aflatoxin B1 metabolism. Carcinogenesis, 19, 403–411.
Grant, D.W. & Carlson, F.W. (1971) Partitioning behavior of aflatoxin M in dairy products. Bull. Environ. Contam. Toxicol., 6, 521–524.
Groopman, J.D., Zhu, J., Donoahue, P.R., Pikul, A., Zhang, L., Chen, J.S. & Wogan, G.N. (1992) Molecular dosimetry of urinary aflatoxin–DNA adducts in people living in Guangxi Autonomous Region, People’s Republic of China. Cancer Res., 52, 45–52.
Grosso F., Dragacci S. & Lombard B. (1999) Second Community proficiency testing for the determination of aflatoxin M1 in milk (November 1998). Report aflatoxin SSA/TOMI/FG9901, May 1999, Paris.
Hainaut, P. & Vahakangas, K. (1997) p53 as a sensor of carcinogenic exposures: Mechanisms of p53 protein induction and lessons from p53 gene mutations. Pathol. Biol. (Paris), 45, 833–844.
Harvey, R.B., Phillips, T.D., Ellis, J.A., Kubena, L.F., Huff, W.E. & Petersen, H.D. (1991) Effects on aflatoxin M1 residues in milk by addition of hydrated sodium calcium aluminosilicate to aflatoxin-contaminated diets of dairy cows. Am. J. Vet. Res., 52, 1556–1559.
Heinonen, J.T., Fisher, R., Brendel, K. & Eaton, D.L. (1996) Determination of aflatoxin B1 biotransformation and binding to hepatic macromolecules in human precision liver slices. Toxicol. Appl. Pharmacol., 136, 1–7.
Hoque, A., Patt, Y.Z., Yoffe, B., Groopman, J.D., Greenblatt, M.S., Zhang, Y.-J. & Santella, R.M. (1999) Does aflatoxin B1 play a role in the etiology of hepatocellular carcinoma in the United States? Nutr. Cancer, 35, 27–33.
Horwitz, W. (1989) Guidelines for collaborative study of procedure to validate characteristics of a method of analysis. J. Assoc. Off. Anal. Chem., 72, 649–704.
Hsieh, D.S.P., Cullen, J.M. & Reubner, B.H. (1984) Comparative hepatocarcinogenicity of aflatoxins B1 and M1 in the rat. Food Chem. Toxicol., 22, 1027–1028.
Hsu, H.Y., Chang, M.H., Liaw, S.H., Ni, Y.H. & Chen, H.L. (1999) Changes of hepatitis B surface antigen variants in carrier children before and after universal vaccination in Taiwan. Hepatology, 30, 1312–1317.
Huang, K. & Lin, S. (2000) Nationwide vaccination: a success story in Taiwan. Vaccine, 18 (Suppl. 1), 35–38.
Hurley, D.J., Neiger, R.D., Higgins, K.F., Rottinghaus, GE. & Stahr, H. (1999) Short-term exposure to subacute doses of aflatoxin-induced mitogen responses in young mallard ducks. Avian Dis., 43, 649–655.
Hussain, S.P. & Harris, C.C. (2000) Molecular epidemiology and carcinogenesis: Endogenous and exogenous carcinogens. Mutat. Res., 462, 311–322.
IARC (1994) Hepatitis Viruses (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 59), Lyon, IARCPress.
International Dairy Federation (1982) (Provisional) International IDF Standard 111.
International Dairy Federation (1999) Guidelines for the standardized description of competitive enzyme immunoassays for the determination of aflatoxin M1: IDF group E501-organic contaminants. Bull. Int. Dairy Fed., 340, 24–26.
International Union of Pure and Applied Chemistry (1989) Guidelines for collaborative study procedure to validate characteristics of a method of analysis. J. Assoc. Off. Anal. Chem., 72, 694–705.
Ioannou-Kakouri, E., Christodoulidou, M., Christou, E. & Constandinidou, E. (1995) Immunoaffinity column/HPLC determination of aflatoxin M1 in milk. Food Agric. Immunol., 7, 131–137.
Ioannou-Kakouri, E., Aletrari, M., Christou, E., Hadjioannou-Ralli, A., Koliou, A. & Akkelidou, D. (1999) Surveillance and control of aflatoxins B1, B2, G1, G2, and M1 in foodstuffs in the Republic of Cyprus: 1992–1996. J. AOAC Int., 82, 883–892.
Izzotti, A., Scatolini, L., Lewtas, J., Walsh, D. & De Flora, S. (1995) Enhanced levels of DNA adducts in the liver of woodchucks infected with hepatitis virus. Chem.-Biol. Interactions, 97, 273–285.
Jacobson, W.C., Harmeyer, W.C. & Wiseman, H.G. (1971) Determination of aflatoxins B1 and M1 in milk. J.Dairy Sci., 54, 21–24.
Jacobson, L.P., Zhang, B.-C., Zhu, Y.R., Wang, J.-B., Wu, Y., Zhang, Q.-N., Yu, L.-Y., Qian, G.-S., Kuang, S.-Y., Li, Y.-F., Fang, X., Zarba, A., Chen, B., Enger, C., Davidson, N.E., Gorman, M.B., Gordon, G.B., Prochaska, H.J., Egner, P.A., Groopman, J.D., Munoz, A., Helzlsouer, K.J. & Kensler, T.W. (1997) Oltipraz chemoprevention trial in Qidon, People’s Republic of China: Study design and clinical outcomes. Cancer Epidemiol. Biomarkers Prev., 6, 257–265.
Jalon, M., Urieta, B.P. & Macho, M.L. (1994) [Surveillance of contamination with aflatoxin M1 of milk and milk products in the Autonomous Community of Vasca.] Alimentaria, 3, 25–29 (in Spanish).
Johansson, A.S., Whitaker, T.B., Hagler, W.M., Jr, Giesbrecht, F.G., Young, J.H. & Bowman, D.T. (2000a) Testing shelled corn for aflatoxin, Part I: Estimation of variance components. J. AOAC Int., 83, 1264–1269.
Johansson, A.S., Whitaker, T.B., Giesbrecht, F.G., Hagler, W.M., Jr & Young, J.H. (2000b) Testing shelled corn for aflatoxin, Part II: Modeling the observed distribution of aflatoxin test results. J. AOAC Int., 83, 1270–1278.
Johansson, A.S., Whitaker, T.B., Giesbrecht, F.G., Hagler, W.M., Jr & Young, J.H. (2000c) Testing shelled corn for aflatoxin, Part III: Evaluating the performance of aflatoxin sampling plans. J. AOAC Int., 83, 1279–1284.
Jung, M. & Hanssen, E. (1974) [Occurrence of aflatoxin M in dairy milk products.] Food Cosmet. Toxicol., 12, 131–138 (in German).
Karaioannoglou, P., Mantis, A., Koufidis, D., Koidis, P. & Triantafillou, J. (1989) Occurrence of aflatoxin M1 in raw and pasteurized milk and in feta and teleme cheese samples. Milchwissenschaft, 44,746–748.
Katiyar, S., Dash, B.C., Thakur, V., Guptan, R.C., Sarin, S.K. & Das, B.C.(2000) p53 tumor supressor gene mutations in hepatocellular carcinoma patients in India. Cancer, 88, 1565–1573.
Kawamura, O., Wang, D.S., Liang, Y.X., Hasegawa, A., Saga, C., Visconti, A. & Ueno, Y. (1994) Further survey of aflatoxin M1 in milk powders by ELISA. Food Agric. Immunol., 6, 465–467.
Kelly, V.P., Ellis, E.M., Manson, M.M., Chanas, S.A., Moffat, G.J., McLeod, R., Judah, D.J., Neal, G.E. & Hayes, J.D. (2000) Chemoprevention of aflatoxin B1 hepatocarcinogenesis by coumarin, a natural benzopyrone that is a potent inducer of aflatoxin B1-aldehyde reductase, the glutathione S-transferase A5 and P1 subunits and NAD(P)H:quinone oxidoreductase in rat liver. Cancer Res., 60, 957–969.
Kensler, T.W. & Helzlsouer, K.J. (1985) Oltipraz: Chemical opportunities for chemoprevention. J. Cell Biochem., 22S, 101–107.
Kensler, T.W., Gange, S.J., Egner, P.A., Donal, P.M., Munoz, A., Groopman, J.D., Rogers, A.E. & Roebuck, B.D. (1997) Predictive value of molecular dosimetry: Individual vs. group effects of oltipraz on aflatoxin–albumin adducts and risk of liver cancer. Cancer Epidemiol. Biomarkers Prev., 6, 603–610.
Kensler, T.W., He, X., Otieno, M., Egner, P.A., Jacobson, L.P., Chen, B.B., Wang, J.-S., Zhu, Y.-R., Zhang, B.-C., Wang, J.-B., Wu, Y., Zhang, Q.-N., Qian, G.-S., Kuang, S.-Y., Fang, X., Li, Y.-F., Yu, L.-Y., Prochaska, H.J., Davdson, N.E., Gordon, G.B., Gorman, M.B., Zarba, A., Enger, C., Munoz, A., Helzlsouer, K.J. & Groopman, J.D. (1998) Oltipraz chemoprevention trial in Qidong, People’s Republic of China: Modulation of serum alfatoxin albumin adduct biomarkers. Cancer Epidemiol. Biomarkers Prev., 7, 127–134.
Kensler, T.W., Groopman, J.D., Sutter, T.R., Curphey, T.J. & Roebuck, B.D. (1999) Development of cancer chemopreventive agents: Oltipraz as a paradigm. Chem. Res. Toxicol., 12, 113–136.
Kiermeier, F. (1973) [On the excretion of aflatoxin M into cows’ milk in relation to the intake of aflatoxin B1.] Michwissenschaft, 28, 683–685 (in German).
Kiermeier, F. & Mashaley, R. (1977) [Influence of raw milk processing on the aflatoxin M content of milk products.] Z. Lebensm. Unters. Forsch., 164, 183–187 (in German).
Kiermeier, F., Reinhardt, V. & Behringer, G. (1975) [Presence of aflatoxins in raw milk.] Dtsch. Lebensm.-Rdsch., 71, 35–38 (in German).
Kiermeier, F., Weiss, G., Behringer, G., Miller, M. & Ranfft, K. (1977) Presence and content of aflatoxin M1 in milk supplied to a dairy. Z. Lebensm. Unters. Forsch., 163, 71–74.
Kim, E.K., Shon, D.H., Ryu, D., Park, J.W., Hwang, H.J. & Kim, Y.B. (2000) Occurrence of aflatoxin M1 in Korean dairy products determined by ELISA and HPLC. Food Addit. Contam., 17, 59–64.
Kirk, G.D., Camus-Randon, A.-M., Mendy, M., Goedert, J.J., Merle, P., Trepo, C., Brechot, C., Hainaut, P. & Móntesano, R. (2000) Ser-249 p53 mutations in plasma DNA of patients with hepatocellular carcinoma from the Gambia. J. Natl Cancer Inst., 92, 148–153.
Klein, P.J., Buckner, R., Kelly, J. & Coulombe, R.A., Jr (2000) Biochemical basis for the extreme sensitivity of turkeys to aflatoxin B1. Toxicol. Appl. Pharmacol., 165, 45–52.
Kuilman, M.E., Maas, R.F., Woutersen-van Nijnanten, F.M. & Fink-Gremmels, J. (2000) Inhibition of aflatoxin M1 production by bovine hepatocytes after intervention with oltipraz. Vet. Q., 22, 30–35.
Lee, M.S., Kim, D.H., Kim, H., Lee, H.S., Kim, C.Y., Park, T.S., Yoo, K.Y., Park, B.J. & Ahn, Y.O. (1998) Hepatitis B vaccination and reduced risk of primary liver cancer among male adults: A cohort study in Korea. Int. J. Epidemiol., 27, 316–319.
Lemieszek-Chodorowska, K. (1974) [Detection and determination of aflatoxin M1 in milk]. Rocz. Panstw. Zakl. Hig., 25, 489–494 (in Polish).
Li, Y., Su, J.-J., Qin, L.-L., Egner, P.A., Wang, J.-S., Groopman, J.D., Kensler, T.W. & Roebuck, B.D. (2000) Reduction of aflatoxin B1 adduct biomarkers by oltipraz in the tree shrew (Tupaia belangeri chinensis). Cancer Lett., 154, 79–83.
Lijinsky, W. & Butler, W.H. (1966) Purification and toxicity of aflatoxin G1. Proc. Soc. Exp. Biol. Med., 123, 151–154.
van der Linde, J.A., Frens, A.M., de Longh, M. & Vles, R.O. (1964) Inspection of milk from cows fed aflatoxin-containing groundnut meal. Tijdschr. Diergeneesk., 89, 1082–1088.
Liu, X.-J., Li, X.-F. & Wen, S.-F. (1992) Application of direct enzyme-linked immunosorbent assay for determination of aflatoxin M1 in milk powder. Hyg. Res., 21, 251–253.
Lopez-Garcia, R. & Park, D.L. (1998) Effectiveness of postharvest procedures in management of mycotoxin hazards. In: Sinha, K.K. & Bhatnager, D., eds, Mycotoxins in Agriculture and Food Safety, New York, Marcel Dekker, pp. 407–433..
Lu, P., Kuang, S. & Wang, J. (1998) Hepatitis B virus and aflatoxin exposure in the development of primary liver cancer. Chung Hua I Hsueh Tsa Chih, 78, 340–342.
Lunn , R.M., Zhang, Y.-J., Wang, L.-Y., Chen, C.-J., Lee, P.-H., Le, C.-S., Tsai, W.-Y. & Santella, R.M. (1997) p53 mutations, chronic hepatitis B virus infection, and alfatoxin exposure in hepatocellular carcinoma in Taiwan. Cancer Res., 57, 3471–3477.
Macho, M.L., Munoz, P. & Eguileor, I. (1992) Aflatoxin M1 analysis of milk and dairy products of total diet in the Basque country. In: Morgan, M.R.A., Smith, C.J. & Williams, P.A., eds, Food Safety and Quality Assurance: Applications of Immunoassay Systems, London, Elsevier Scientific Publishers, pp. 115–118.
Makimoto, K. & Higuchi., S. (1999) Alcohol consumption as a major risk factor for the rise in liver cancer mortality rates in Japanese men. Int. J. Epidemiol., 28, 30–34.
Mandishona, E., MacPhail, A.P., Gordeuk, V.R., Kedda, M.-A., Paterson, A.C., Rouault, T.A. & Kew, M.C. (1998) Dietary iron overload as a risk factor for hepatocellular carcinoma in black Africans. Hepatology, 27, 1563–1566.
Markaki, P. & Melissari, E. (1997) Occurrence of aflatoxin M1 in commercial pasteurized milk determined with ELISA and HPLC. Food Addit. Contam., 14, 451–456.
Martins, J.L.S. & Martins, I.S. (1986) Aflatoxin M1 in type B milk sold in the city of Sao Paulo, SP (Brazil). Rev. Saude Publ., 20, 303–308.
Masri, M.S., Page, J.R. & Garcia, V.C. (1968) Analysis for aflatoxin M in milk. J. Assoc. Off. Anal. Chem., 51, 594–600.
Masri, M.S., Page, J.R. & Garcia, V.C. (1969a) Modification of method for aflatoxins in milk. J. Assoc. Off. Anal. Chem., 52, 641–643.
Masri, M.S., Garcia, V.C. & Page, J.R. (1969b) The aflatoxin M content of milk from cows fed known amounts of aflatoxin. Vet. Rec., 84, 146–147.
McGlynn, .K.A., Obroin, S., N’Dao, M., Chen, G., Buetow, K.H., Shen, F.M. & London, W.T. (1999) Plasma folate, chronic HBV infection and primary liver cancer. Proc. Am. Assoc. Cancer Res., 40, 993.
McGuire, R.A. (1969) Factors Affecting the Acute toxicity of Aflatoxin B1 in the Rat and Mouse. MSc Thesis, Massachusetts Institute of Technology, Cambridge, MA.
McKinney, J.D. (1972) Determination of aflatoxin M1 in raw milk: A modified Jacobson, Harmeyer and Wiseman method. J. Am. Oil Chem. Soc., 49, 444–445.
McKinney, J.D., Cavanagh, G.C., Bell, J.T., Hoversland, A.S., Nelson, D.M., Pearson, J. & Selkirk, R.J. (1973) Effects of ammoniation on aflatoxins in rations fed lactating cows. J. Am. Oil Chem. Soc., 50, 79–84.
Miller, D.M. & Wilson, D.M. (1994) Veterinary diseases related to aflatoxins. In: Eaton, D.L. & Groopman, J.D., eds, The Toxicology of Aflatoxin. Human Health, Veterinary and Agricultural Significance, New York, Academic Press, pp. 347–364.
Ministerio da Saude (2000) Agencia Nacional de Vigilancia Sanitaria, Diretoria de Alimentos e Toxicologia, Gerencia-Geral de Alimentos, Brasilia, Brazil.
Ministry of Agriculture, Fisheries and Food (1993) Mycotoxins: Third Report (Food Surveillance Paper No. 36), London, HMSO.
Ministry of Agriculture, Fisheries and Food (1999) Report to participants in the food analysis performance assessment scheme. Aflatoxins series IV, Round 23: January 1999. Report No. 0423, Norwich, Central Science Laboratory, 22 pp.
Ministry of Agriculture, Fisheries and Food (2000a) Report to participants in the food analysis performance assessment scheme. Aflatoxins series IV, Round 27: January 2000. Report No. 0427, Sand Hutton, York, Central Science Laboratory, 21 pp.
Ministry of Agriculture, Fisheries and Food (2000b) Report to participants in the food analysis performance assessment scheme. Aflatoxins series IV, Round 30: July 2000. Report No. 0430, Sand Hutton, York, Central Science Laboratory, 15 pp.
Mortimer, D.N., Gilbert, J. & Shepherd, M.J. (1987) Rapid and sensitive analysis of aflatoxin M1 in liquid and powdered milks using an affinity column clean-up. J. Chromatogr., 407, 393–398.
Mosso, C., Moda, G. & Barbarino, G. (1992) Study of some qualitative values affecting the hygiene of milk produced in the province of Turin. Ind. Alim., 31, 196–202.
Navas, S.A., Zorzetto, M.A.P. & Sabino, M. (2000) Determination of aflatoxin M1 in powdered milk using TCL (Abstract). X International IUPAC Symposium on Mycotoxins and Phycotoxins, 21–25 May 2000.
Neal, G.E., Eaton, D.L., Judah, D.J. & Verma, A. (1998) Metabolism and toxicity of aflatoxins M1 and B1 in human-derived in vitro systems. Toxicol. Appl. Pharmacol., 151, 152–158.
Netke, S.P., Roomi, M.W., Tsao, C. & Niedzwiecki, A. (1997) Ascorbic acid protects guinea pigs from acute aflatoxin toxicity. Toxicol. Appl. Pharmacol., 143, 429–435.
el-Nezami, H.S., Nicoletti, G., Neal, G.E., Donohue, D.C. & Ahokas, J.T. (1995) Aflatoxin M1 in human breast milk samples from Victoria, Australia and Thailand. Food Chem. Toxicol., 33, 173–179.
Nikov, P.S., Bukharbaeva, A.S., Baimbetova, A.M. & Amireeva, N.T. (1991) [Aflatoxin contamination of milk and milk products in south-eastern Kazakhstan.] Vopr. Pitan., 5, 72–74 (in Russian).
Okuda, K. (1991) Chapter 9. In: Tabor, E., DiBisceglie, A.M. & Purcell, R.l.H., eds, Etiology, Pathology and Treatment of Hepatocellular Carcinoma in North America, Houston, TX, Gulf Publishing Co., pp 119–126.
Oliveira, C.A., Germano, P.M., Bird, C. & Pinto, C.A. (1997) Immunochemical assessment of aflatoxin M1 in milk powder consumed by infants in Sao Paulo, Brazil. Food Addit. Contam., 14, 7–10.
Omer, R.E., Bakker, M.I., van’t Veer, P., Hoogenboom, R.L.A.P., Polman, T.H.G., Alink, G.M., Idris, M.O., Kadaru, A.M.Y. & Kok, F.J. (1998) Aflatoxin and liver cancer in Sudan. Nutr. Cancer, 32, 174–180.
Otteneder, M. & Lutz, W.K. (1999) Correlation of DNA adduct levels with tumor incidence: carcinogenic potency of DNA adducts. Mutat. Res., 424, 237–247.
Park, D.L. & Pohland, A.E. (1986) A rationale for the control of aflatoxin in animal feeds. In: Steyn, P. & Vleggaar, R., eds, Mycotoxins and Phycotoxins, Amsterdam, Elsevier Science Publishers, pp. 43–482.
Park, D.L. & Price, W.D. (2001) Reduction of aflatoxin hazards using ammoniation. Rev. Environ. Contam. Toxicol., 171 (in press).
Park, D.L., Whitaker, T.B., Giesbrecht, F.G. & Njapau, H. (2000) Performance of three pneumatic probe samplers and four analytical methods used to estimate aflatoxins in bulk cottonseed. J. AOAC Int., 83, 1247–1251.
Patel, P.M., Netke, S.P., Gupta, D.S. & Dabadghao, A.K. (1981) Note on the effect of processing milk into khoa on aflatoxin M1 content. Indian J. Anim. Sci., 51, 791–792.
Pettersson, H. (1997) Carry-over of aflatoxin from feedingstuffs to milk. Swedish derogations from EC legislation in the area of feedingstuffs. Report 2. Undesirable substances and products. Stockholm: Ministry of Agriculture, pp. 23–27.
Pettersson, H., Bertilsson, J. & Wennberg, O. (1990) Carry-over of aflatoxin from dairy cattle feed for milk. In: Proceedings of the World Association of Veterinary Food Hygienists Symposium, Stockholm, 1989, pp. 97–102.
Phillips, T.D., Sarr, A.B. & Grant, P.G. (1995) Selective chemisorption and detoxification of aflatoxins by phyllosilicate clay. Nat. Toxins, 3, 204–213.
Pineiro, M., Dawson, R. & Costarrica, M.L. (1996) Monitoring program for mycotoxin contamination in Uruguayan food and feeds. Nat. Toxins, 4, 242–245.
Poirier, M.C. & Beland, F.A. (1994) DNA adduct measurements and tumor incidence during chronic carcinogen exposure in rodents. Environ. Health Perspectives, 102 (Suppl. 6), 161–165.
Polan, C.E., Hayes, J.R. & Campbell, T.C. (1974) Consumption and fate of aflatoxin B1 by lactating cows. J. Agric. Food Chem., 22, 635–638.
Prado, G., Oliveira, M.S., Abrantes, F.M., Santos, L.G., Soares, C.R. & Veloso, T. (1999) [Occurrence of aflatoxin M1 in milk consumed in the City of Belo Horizonte, Minas Gerais, Brazil, August 1998 to April 1999.] Cienc. Tecnol. Aliment. Campinas, 19, 420–423 (in Portuguese).
Prado, G., de Oliveira, M.S., Abrantes, F.M., dos Santos, L.G., Barroso, R.E.S. & Veloso, T. (2000) Occurrence of aflatoxin M1 in milk marketed in the city of Belo Horizonte. Minas Gerais, Brazil—August/98 to April/99 (Abstract). X International IUPAC Symposium on Mycotoxins and Phycotoxins, 21–25 May 2000.
Purchase, I.F.H. (1967) Acute toxicity of aflatoxin M1 and M2 in one-day-old ducklings. Food Cosmet. Toxicol., 5, 339–342.
Purchase, I.F.H. & Steyn, M. (1967) Estimation of aflatoxin M1 in milk. J. Assoc. Off. Anal. Chem., 50, 363–364.
Purchase, I.F., Steyn, M., Rinsma, R. & Tustin, R.C. (1972) Reduction of the aflatoxin M content of milk by processing. Food Cosmet. Toxicol., 10, 383–387.
Qian, G.S., Yasei, P. & Young, G.C. (1984) Rapid extraction and detection of AFM1 in cow’s milk by HPLC and RIA. Anal. Chem., 56, 2079–2080.
Qian, G.-S., Ross, R.K., Yu, M.C., Yuan, J.-M., Gao, Y.-T., Henderson, B.E., Wogan, G.N. & Groopman, J.D. (1994) A followup study of urinary markers of aflatoxin exposures and liver cancer risk in Shanghai, People’s Republic of China. Cancer Epidemiol. Biomarkers Prev., 3, 3–10.
Qin, G., Su, J., NIng, Y., Duan, X., Luo, D. & Lotlikar, P.D. (1997) p53 protein expression in patients with hepatocellular carcinoma from the high incidence area of Guangxi, Southern China. Cancer Lett., 121, 303–310.
Rajan, A., Ismail, P.K. & Radhakrishnan, V. (1995) Survey of milk samples for aflatoxin M1 in Thrissur, Kerala. Indian J. Dairy Sci., 48, 302–305.
Ramsdell, H.S. & Eaton, D.L. (1990) Mouse liver glutathione S-transferase isoenzyme activity toward aflatoxin B1-8,9-epoxide and benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide. Toxicol. Appl. Pharmacol., 105, 216–225.
Rashid, A., Wang, J.S., Qian, G.S., Lu, B.X., Hamilton, S.R. & Groopman, J.D. (1999) Genetic alterations in hepatocellular carcinomas: Association between loss of chromosome 4q and p53 gene mutations. Br. J. Cancer, 80, 59–66.
Richard, J.L., Bennett, G.A., Ross, P.F. & Nelson, P.E. (1993) Analysis of naturally occurring mycotoxins in feedstuffs and food. J. Anim. Sci., 71, 2563–2574.
Roebuck, B.D. & Maxuitenko, Y.Y. (1994) Biochemical mechanisms and biological implications of the toxicity of aflatoxins as related to aflatoxin carcinogenesis. In: Eaton, D. & Groopman, J., eds, The Toxicology of Aflatoxins: Human Health, Veterinary and Agricultural Significance, New York: Academic Press, pp. 27–43.
Rodriguez-Amaya, D.B. (2000) Research on mycotoxins in Latin America. X International IUPAC Symposium on Mycotoxins and Phycotoxins. Submitted to WHO.
Ross, R.K., Yuan, J.-M., Yu, M.C., Wogan, G.N., Qian, G.-S., Tu, J.T., Groopman, J.D., Gao, Y.-T. & Henderson, B.E. (1992) Urinary aflatoxin biomarkers and risk of hepatocellular carcinoma. Lancet, 339, 943–946.
Saad, A.M., Abdelgadir, A.M. & Moss, M.O. (1995) Exposure of infants to aflatoxin M1 from mothers’ breast milk in Abu Dhabi, UAE. Food Addit. Contam., 12, 255–261.
Sabino, M., Purchio, A. & Zorzetto, M.A.P. (1989) Variations in the levels of aflatoxin in cows milk consumed in the city of Sao Paulo, Brazil. Food Addit. Contam., 6, 321–326.
Sabino, M., Purchio, A. & Milanez, T.V. (1995) Aflatoxin B1, M1 and aflatoxicol in tissues and urine of calves receiving aflatoxin. Food Addit. Contam., 12, 467–472.
Sahu, S.C., Chou, M.W., Sotomayor, R.E., Hinton, D.M., Barton, C.N. & O’Donnell, M.W. (2000) Effects of intermittent exposures of aflatoxin B1 on hepatic and testicular glutathione S-transferase in rats. J. Appl. Toxicol., 20, 215–219.
Saitanu, K. (1997) Incidence of aflatoxin M1 in Thai milk products. J. Food Prot., 60, 1010–1012.
Scholl, P., Musser, S.M., Kensler, T.W. & Groopman, J.D. (1996) Inhibition of aflatoxin M1 excretion in rat urine during sietary intervention with oltipraz. Carcinogenesis, 17, 1385–1388.
Scott, P. (1989) Methods for determination of aflatoxin M1 in milk and milk products—A review of performance characteristics. Food Addit. Contam., 6, 283–305.
Shane, S.M. (1994) Economic issues associated with aflatoxins. In: Eaton, D.L. & Groopman, J.D., eds, The Toxicology of Aflatoxin. Human Health, Veterinary, and Agricultural Significance, New York, Academic Press, pp. 513–527.
Sheu, J.C. (1997) Molecular mechanism of hepatocarcinogenesis. Gastroenterol. Hepatol., 12, 9–10.
Shibahara, T., Ogawa, H.I., Ryo, H. & Fujikawa, K. (1995) DNA-damaging potency and genotoxicity of aflatoxin M1 in somatic cells in vivo of Drosophila melanogaster. Mutagenesis, 10, 161–164.
Shimizu, Y., Zhu, J.-J., Han, F., Ishikawa, T. & Oda, H. (1999) Different frequencies of p53 codon-249 hot-spot mutations in hepatocellular carcinomas in Jiang-Su Province of China. Int. J. Cancer, 82, 187–190.
Shon, D.H., Lim, S.H. & Lee, Y.W. (1996) Detection of aflatoxin M1 in cow’s milk by an enzyme-linked immunosorbent assay. Korean J. Appl. Microbiol. Biotechnol., 24, 630–635.
Sibanda, L., De Saeger, S. & van Peteghem, C. (1999) Development of a portable field immunoassay for the detection of aflatoxin M1 in milk. Int. J. Food Microbiol., 48, 203–209.
Smith, E.E., Philips, T.D., Ellis, J.A., Harvey, R.B., Kubena, L.F., Thompson, J. & Newton, G. (1994) Dietary hydrated sodium calcium aluminosilicate reduction of aflatoxin M1 residue in dairy goat milk and effects on milk production and components. J. Anim. Sci., 72, 677–682.
Sohn, S. Jaitovitch-Groisman, I., Benlimame, N., Galipeau, J., Batist, G. & Alaoui-Jamali, M.A. (2000) Retroviral expression of the hepatitis B virus x gene promotes liver cell susceptibility to carcinogen-induced site specific mutagenesis. Mutat. Res., 460, 17–28.
Soini, Y., Chia, S.C., Bennett, W.P., Groopman, J.D., Wang, J.S., DeBenedetti, V.M., Cawley, H., Welsh, J.A., Hansen, C., Bergasa, N.V., Jones, E.A., DiBisceglie, A.M., Trivers, G.E., Sandoval, C.A., Calderon, I.E., Munoz Espinosa, L.E. & Harris, C.C. (1996) An aflatoxin-associated mutational hotspot at codon 249 in the p53 tumor suppressor gene occurs in hepatocellular carcinomas from Mexico. Carcinogenesis, 17, 1007–1012.
Soni, K.B., Lahiri, M., Chackradeo, P., Bhide, S.V. & Kuttan, R. (1997) Protective effect of food additives on aflatoxin-mutagenicity and hepatocarcinogenicity. Cancer Lett., 115, 129–133.
Soussi, T., Dehouche, K. & B’eroud, C. (2000) p53 website and analysis of p53 gene mutations in human cancer: Forging a link between epidemiology and carcinogenesis. Human Mutat., 15, 105–113.
Souza, S.V.C., Vargas, E.A. & Junqueira, P G. (1999) [Efficiency of an ELISA kit in the detection and quantification of aflatoxin M1 in milk and investigation of the occurrence in the State of Minas Gerais.] Cienc. Tecnol. Allment. Campinas, 19, 401–405 (in Portuguese).
Stoloff, L., Trucksess, M., Hardin, N., Francis, O.J., Hayes, J.R., Polan, C.E. & Campbell, T.C. (1975) Stability of aflatoxin M in milk. J. Dairy Sci., 58, 1789–1793.
Stubblefield, R.D. (1979) The rapid determination of aflatoxin M1 in dairy products. J. Am. Oil Chem. Soc., 56, 800–802.
Stubblefield, R.D. & Kwolek, W.F. (1986) Rapid liquid chromatographic determination of aflatoxins M1 and M2 in artificially contaminated fluid milks: Collaborative study. J. Assoc. Off. Anal. Chem., 69, 880–885.
Stubblefield, R.D. & Shannon, G.M. (1974a) Aflatoxin M1: Analysis in dairy production and distribution in dairy foods made from artificially contaminated milk. J. Assoc. Off. Anal. Chem., 57, 847–851.
Stubblefield, R.D. & Shannon, G.M. (1974b) Collaborative study of methods for the determination of aflatoxin M1 in dairy products. J. Assoc. Off. Anal. Chem., 57, 852–857.
Stubblefield, R.D., van Egmond, H.P., Paulsch, W.E. & Schuller, P.L. (1980) Determination and confirmation of identity of aflatoxin M1 in dairy products: Collaborative study. J. Assoc. Off. Anal. Chem., 63, 907–921.
Sun, Z., Lu, P., Gail, M.H., Pee, D., Zhang, Q., Ming, L., Wang, J., Wu, Y., Liu, G., Wu, Y. & Zhu, Y. (1999) Increased risk of hepatocellular carcinoma in male hepatitis B surface antigen carriers with chronic hepatitis who have detectable urinary alfatoxin metabolite M1. Hepatology, 30, 379–383.
de Sylos, C.M., Rodriguez-Amaya, D.B. & Carvalho, P.R. (1996) Occurrence of aflatoxin M1 in milk and dairy products commercialized in Campinas, Brazil. Food Addit. Contam., 13, 169–172.
Torresi, J. & Locarnini, S. (2000) Antiviral chemotherapy for the treatment of hepatitis B virus infections. Gastroenterology, 118, S83–S103.
Tuinstra, L.G.M.T., Roos, A.H. & van Trijp, J.M.P. (1993) Liquid chromatographic determination of aflatoxin M1 in milk powder using immunoaffinity columns for cleanup: Interlaboratory study. J. AOAC Int., 76, 1248–1254.
Turner, P.C., Dingley, K.H., Coxhead, J., Russell, S. & Garner, C.R. (1998) Detectable levels of serum aflatoxin B1–albumin adducts in the United Kingdom population: Implications for aflatoxin-B1 exposure in the United Kingdom. Cancer Epidemiol. Biomarkers Prev., 7, 441–447.
Universidad National de Lujan (2000) Centro de Investigacion en Micotoxinas, Lujan, Argentina.
Vautier, G., Bomford, A.B., Portmann, B.C., Metivier, E., Williams, R. & Ryder, S.C. (1999) p53 mutations in British patients with hepatocellular carcinoma: Clustering in genetic hemochromatosis. Gastroenterology, 117, 154–160.
Veldman, A. (1992) Effect of sorbentia on carry-over of aflatoxin from cow feed to milk. Milchwissenschaft, 47, 777–780.
Veldman, V.A., Meijs, J.A.C., Borggreve, G.J. & Heeres van der Tol, J.J. (1992) Carry-over of aflatoxin from cows’ food to milk. Anim. Prod., 55, 163–168.
Vittani, M. (1987) [Presence of aflatoxin M1 in pasteurized milk and low-fat milk.] Latte, 12, 1103–1107 (in Italian).
Wang, J.S. & Groopman, J.D. (1999) DNA damage by mycotoxins. Mutat. Res., 424, 167–181.
Wang, L.Y., Hatch, M., Chen, C.J., Levin, B., You, S.L., Lu, S.H., Wu, M.H., Wu, W.P., Wang, L.W., Wang, Q., Huang, G.T., Yang, P.M., Lee, H.S. & Santella, R.M. (1996) Aflatoxin exposure and risk of hepatocellular carcinoma in Taiwan. Int. J. Cancer, 67, 620–625
Wang, S.S., O’Neill, J.P., Qian, G.-S., Zhu, Y.-R., Wang, J.-B., Armenian, H., Zarba, A., Wang, J.-S., Kensler, T.W., Cariello, N.F., Groopman, J.D. & Swenberg, J.A. (1999a) Elevated HPRT mutation frequencies in aflatoxin-exposed residents of Daxin, Qidong County, People’s Republic of China. Carcinogenesis, 20, 2181–2184.
Wang, J.S., Shen, X., He, X., Zhu, Y.R., Zhang, B.C., Wang, J.B., Qian, G.S., Kuang, S.Y., Zarba, A., Egner, P.A., Jacobson, L.P., Munoz, A., Helzlsouer, K.J., Groopman, J.D. & Kensler, T.W. (1999b) Protective alterations in phase 1 and phase 2 metabolism of aflatoxin B1 by oltipraz in residents of Qidong, People’s Republic of China. J. Natl Cancer Inst., 91, 347–354.
Wang, C., Bammler, T.K., Guo, Y., Kelly, E.J. & Eaton, D.L. (2000) Mu-class GSTs are responsible for aflatoxin B1-8,9-epoxide-conjugating activity in the nonhuman primate Macaca fascicularis liver. Toxicol. Sci., 56, 26–36.
Whitaker, T.B., Dickens, J.W. & Monroe, R.J. (1976) Variability associated with testing cottonseed for aflatoxin. J. Am. Oil Chem. Soc., 53, 502–505.
WHO (1998) GEMS/Food regional diets. Regional per capita consumption of raw and semi-processed agricultural commodities (WHO/FSF/FOS/98.3), Geneva.
Wild, C.P. & Hall, A.J. (2000) Primary prevention of hepatocellular carcinoma in developing countries. Mutat. Res., 462, 381–393.
Wild, C.P., Yin, F., Turner, P.C., Chemin, I., Chapot, B., Mendy, M., Whittle, H., Kirk, G.D. & Hall, A.J. (2000) Environmental and genetic determinants of aflatoxin–albumin adducts in The Gambia. Int. J. Cancer, 86, 1–7.
Wiseman, D.W. & Marth, E.H. (1983) Behavior of aflatoxin M1 in yogurt, buttermilk and kefir. J. Food Prot., 46, 115–118.
Wogan, G.N. (1966) Chemical nature and biological effects of the aflatoxins. Bacteriol. Rev., 30, 460–470.
Wu, S., Gu, J., Patt, Y., Hassan, M., Spitz, M.R., Beasley, P.R. & Hwang, L.-Y. (1998) Mutagen sensitivity as a susceptibility marker for human hepatocellular carcinoma. Cancer Epidemiol. Biomarkers Prev., 17, 567–570.
Yang, M., Zhou, H., Kong, R.Y.C., Fong, W.F., Ren, L.-Q., Liao, X.-H., Wang, Y., Zhuang, W. & Yang, S. (1997) Mutations at codon 249 of p53 gene in human hepatocellular carcinomas from Tongan, China. Mutat. Res., 381, 25–29.
Yeh, F.-S., Yu, M.C., Mo, C.-C., Luo, S., Tong, M.J. & Henderson, B.E. (1989) Hepatitis B virus, aflatoxin, and hepatocellular carcinoma in Southern Guangxi, China. Cancer Res., 49, 2506–2509.
Yousef, A.E. & Marth, E.H. (1985) Degradation of aflatoxin M1 in milk by ultraviolet energy. J. Food Prot., 48, 697–698.
Yousef, A.E. & Marth, E.H. (1989) Stability and degradation of aflatoxin M1. In: van Egmond, H.P., ed., Mycotoxins in Dairy Products, London, Elsevier Applied Science, pp. 127–161.
Yu , M.-W., Lien, J.-P., Chiu, Y.-H., Santella, R.M., Liaw, Y.-F. & Chen, C.-J. (1997a) Effect of aflatoxin metabolism and DNA adduct formation on hepatocellular carcinoma among chronic hepatitis B carriers in Taiwan. J. Hepatol., 27, 320–330.
Yu, M.-W., Chiang, Y.-C., Lien, J-.P. & Chen, C.-J. (1997b) Plasma antioxidant vitamins, chronic hepatitis B virus infection and urinary aflatoxin B1–DNA adducts in healthy males. Carcinogenesis, 18, 1189–1194.
Yu, S.Z., Chen, G., Zhi, X.L. & Li, J. (1998) Primary liver cancer: Natural toxins and prevention in China. J. Toxicol. Sci., 23 (suppl. 2), 143–147.
Zhou, T., Evans, A.A., London, W.T., Xia, X., Zou, H., Shen, F.-M. & Clapper, M.L. (1997) Glutathione S-transferase expression in hepatitis B virus-associated human hepatocellular carcinogenesis. Cancer Res., 57, 2749–2753.
Results of surveys for aflatoxin M1, showing concentrations and distribution of
contamination in food commodities
|
Country/ Region |
Commodity |
Year/ Season |
No. of samples |
LOQ |
n < LOQ |
Mean/Max |
|
Americas |
||||||
|
Argentina |
Farm milk |
Mar–Sept 1999 |
56 |
0.01* |
50 |
0.0017/0.030 |
|
Powdered milk |
Mar–Sept 1999 |
5 |
0.01* |
1 |
0.010/0.014 |
|
|
Pasteurized fluid milk |
Mar–Sept 1999 |
16 |
0.01* |
8 |
0.0065/0.017 |
|
|
Brazil |
Milk |
NR |
224 |
NR |
220 |
Trace/0.002 |
|
Brazil |
Pasteurized milk |
1992 |
52 |
0.025 |
48 |
0.012/0.37 |
|
Brazil |
Raw milk |
1992–93 |
144 |
NR |
144 |
0 |
|
Brazil |
Powdered milk, pasteurized |
1989 |
86 |
0.15 |
86 |
0 |
|
Brazil |
Milk powder, reconstituted for infants |
Oct. 1992–Jan. 1993 |
300 |
0.1 |
267 |
0.030/1.0 |
|
Brazil |
Yoghurt |
1990 |
30 |
0.15 |
30 |
0 |
|
Brazil |
Milk |
1996–97 |
110 |
0.0063 |
83 |
NR/0.0071 |
|
Brazil |
Milk and milk products |
1998–99 |
61 |
0.006 |
11 |
0.020/0.077 |
|
Pasteurized milk |
|
18 |
0.006 |
3 |
0.027/0.077 |
|
|
Long–life milk |
1998–99 |
21 |
0.006 |
5 |
0.018/0.070 |
|
|
Long–life milk, infant |
|
5 |
0.006 |
1 |
0.010/0.021 |
|
|
Milk powder |
|
6 |
0.006 |
0 |
0.012/0.038 |
|
|
Milk powder, infant |
|
11 |
0.006 |
2 |
0.025/0.050 |
|
|
Canada |
Homogenized milk |
1994 |
15 |
0.015 |
|
< 0.069/0.21 |
|
2% partly skimmed milk |
1994 |
7 |
0.015 |
|
< 0.034/0.16 |
|
|
Skimmed milk powder |
1994 |
2 |
0.015 |
1 |
< 0.030/0.059 |
|
|
Partly skimmed milk, lactose reduced |
1994 |
2 |
0.015 |
1 |
< 0.053/0.090 |
|
|
1% partly skimmed milk |
1994 |
1 |
0.015 |
1 |
0 |
|
|
Evaporated milk |
1994 |
1 |
0.015 |
0 |
0.06 |
|
|
Partly skimmed milk, lactaid |
1994 |
1 |
0.015 |
0 |
0.038 |
|
|
Skimmed milk, lacteeze |
1994 |
1 |
0.015 |
1 |
0 |
|
|
Uruguay |
Milk |
1993–95 |
22 |
|
15 |
0.002/ > 0.02 |
|
USA |
Raw milk |
1995 |
695 |
|
682 |
0.0017/0.33 |
|
Raw milk |
1996 |
381 |
|
287 |
0.035/1.8 |
|
|
USA |
Raw milk |
1997 |
717 |
|
697 |
0.0023/0.41 |
|
Raw milk |
1998 |
855 |
|
839 |
0.0025/0.38 |
|
|
Raw milk |
1999 |
877 |
|
851 |
0.0024/0.24 |
|
|
Raw milk |
2000 |
475 |
|
459 |
0.0044/0.37 |
|
|
Fnished milk |
1995 |
79 |
|
76 |
0.0032/0.11 |
|
|
Finished milk |
1996 |
21 |
|
17 |
0.0110/0.08 |
|
|
Finished milk |
1997 |
53 |
|
52 |
0.0011/0.06 |
|
|
Finished milk |
1999 |
60 |
|
58 |
0.0038/0.17 |
|
|
Finished milk |
2000 |
12 |
|
12 |
0 |
|
|
Southwest |
Milk |
1998–2000 |
5 801 |
0.02*/ |
4 562 |
NR/> 0.5 |
|
|
|
|
0.05 |
|
|
|
|
Midwest |
Milk |
1998–2000 |
438 |
0.5 |
428 |
NR/> 0.5 |
|
Southeast |
Milk |
1998–2000 |
13 093 |
0.5 |
13 079 |
NR/> 0.5 |
|
Southern |
Milk |
1998 |
163 |
0.05 |
78 |
0.73/14.0 |
|
Southern |
Milk |
1999 |
168 |
0.05 |
96 |
0.17/5.2 |
|
Southern |
Milk |
2000 |
49 |
0.05 |
32 |
0.069/0.50 |
|
Southeast |
Milk |
Jun 1999–May 2000 |
199 |
0.1* |
196 |
NR/< 0.25 |
|
Southeast |
Milk |
Jul 1997–May 2000 |
78 |
0.1* |
49 |
NR/> 0.5 |
|
China, north |
Powdered milk |
NR |
NR |
0.08* |
2 |
NR |
|
China, south |
Powdered milk |
NR |
NR |
0.08* |
7 |
NR/4.2 |
|
China |
Powdered milk |
1992–93 |
27 |
0.015 |
1 |
0.10/0.46 |
|
Taiwan |
Fresh milk |
Mar–Sept 1986 |
56 |
0.1 |
56 |
0 |
|
Powdered milk |
Mar–Sept 1986 |
161 |
0.1 |
161 |
0 |
|
|
India |
Raw milk |
1992–93 |
504 |
|
415 |
0.20/3.50 |
|
Raw milk |
1993 |
325 |
|
289 |
0.12–1.0 |
|
|
Indonesia |
Milk |
Jan 1990 |
55 |
0.1 |
15 |
1.3/21 |
|
Milk |
Sept 1990 |
30 |
0.1 |
0 |
5.4/23 |
|
|
Milk |
July 1991 |
53 |
0.1 |
10 |
0.48/2.0 |
|
|
Milk |
Dec 1991 |
50 |
0.1 |
32 |
0.31/3.4 |
|
|
Milk |
Jan 1992 |
80 |
0.1 |
51 |
0.78/9.7 |
|
|
Java |
Milk |
Jan 1993 |
64 |
0.1 |
34 |
0.85/6.7 |
|
Indonesia |
Milk/factory |
Sept 1999 |
10 |
0.1 |
1 |
0.92/2.1 |
|
Ice cream/factory |
Aug 1999 |
5 |
0.1 |
0 |
0.31/0.48 |
|
|
Ice cream/factory |
Oct 1999 |
11 |
0.1 |
11 |
0 |
|
|
Korea, Republic of |
Raw milk |
Winter 1995 |
49 |
0.003* |
0 |
0.065/0.16 |
|
Market milk |
Winter 1995 |
15 |
0.003* |
0 |
0.13/0.28 |
|
|
Market milk |
Summer, winter 1997 |
70 |
0.01 |
31 |
0.014/0.052 |
|
|
|
0.002 |
17 |
0.014/0.037 |
|||
|
Korea, Republic of |
Infant formula |
Autumn 997 |
26 |
0.01 |
8 |
0.043/0.13 |
|
|
0.002 |
4 |
0.039/0.93 |
|||
|
Powdered milk |
Autumn 1997 |
24 |
0.01 |
7 |
0.16/0.34 |
|
|
|
0.002 |
6 |
0.15/0.33 |
|||
|
Yoghurt |
Autumn, winter, 1997 |
60 |
0.01 |
29 |
0.023/0.12 |
|
|
|
0.002 |
10 |
0.024/0.17 |
|||
|
Philippines |
Milk |
1997 |
91 |
0.05 |
11 |
0.13/NR |
|
Thailand |
Raw milk |
1990–93 |
45 |
0.1 |
33 |
0.11/0.80 |
|
Raw milk |
1995–96 |
67 |
0.025 |
1 |
NR |
|
|
Pasteurized milk |
1990–93 |
15 |
0.1 |
6 |
0.80/6.6 |
|
|
Pasteurized milk |
1995–96 |
63 |
0.025 |
0 |
NR |
|
|
UHT milk |
|
60 |
0.025 |
0 |
NR |
|
|
Sterilized milk |
|
60 |
0.025 |
0 |
NR |
|
|
Thailand |
Powdered milk |
1995–96 |
31 |
0.1 |
30 |
1.4 |
|
Powdered milk, imported |
1995–96 |
13 |
0.025 |
11 |
NR |
|
|
Pelleted milk |
1995–96 |
7 |
0.025 |
0 |
NR |
|
|
United Arab Emirates |
Fresh milk |
1998 |
22 |
0.01/0.005* |
1 |
NR/0.31 |
|
UHT milk |
1998 |
11 |
0.01/0.005* |
11 |
0 |
|
|
Milk/imported |
1999 |
15 |
0.01/0.005* |
12 |
NR/0.08 |
|
|
Low fat milk |
1999 |
11 |
0.01/0.005* |
2 |
NR/0.24 |
|
|
Milk powder |
1998 |
8 |
0.01/0.005* |
8 |
0 |
|
|
Milk powder |
1999 |
8 |
0.01/0.005* |
8 |
0 |
|
|
Full cream milk |
1999 |
22 |
0.01/0.005* |
14 |
NR/0.35 |
|
|
Skimmed milk |
1999 |
9 |
0.01/0.005* |
2 |
NR/0.14 |
|
|
Milk-based infant formula |
1998 |
11 |
0.01/0.005* |
11 |
0 |
|
|
Milk-based infant formula |
1999 |
3 |
0.01/0.005* |
3 |
0 |
|
|
Europe |
||||||
|
Austria |
Milk powder |
Feb 1983 |
65 |
0.03 |
65 |
0 |
|
Austria |
Milk |
1990–93 |
479 |
0.01 |
479 |
0.005/ < 0.01 |
|
Milk |
1999 |
20 |
0.005 |
20 |
0 |
|
|
Belgium |
Commercial blended milk |
Winter 1984–85 |
233 |
|
204 |
0.02–0.15 |
|
Winter 1985–86 |
89 |
0.03 |
89 |
0 |
||
|
Milk |
1993–94 |
153 |
0.01* |
98 |
0.01/0.037 |
|
|
Milk |
1999 |
192 |
0.005 |
168 |
NR/< 0.05 |
|
|
Denmark |
Milk |
1990–94 |
1664 |
0.01* |
1240 |
< 0.01/0.069 |
|
Cyprus |
Raw milk |
1993,1995– |
71 |
0.005 |
68 |
0.0015/0.04 |
|
Pasteurized milk, full |
96 |
19 |
0.01* |
14 |
0.0040/0.02 |
|
|
Pasteurized milk, light |
|
4 |
0.01* |
3 |
0.01 |
|
|
Pasteurized milk, skimmed |
|
8 |
0.01* |
5 |
0.0075/0.04 |
|
|
Baby milk, imported |
|
6 |
0.005 |
6 |
0 |
|
|
Evaporated milk, imported |
|
4 |
0.005 |
4 |
0 |
|
|
Czech Republic |
Raw milk |
1987–88 |
376 |
NR |
330 |
0.002–< 0.50 |
|
Raw milk |
1987–88 |
89 |
NR |
62 |
0.020–< 0.50 |
|
|
Milk |
NR |
191 |
NR |
166 |
0.050–0.10 |
|
|
Finland |
Milk powder |
1986 |
14 |
0.1 |
14 |
0 |
|
Finland |
Milk |
1992–15 |
122 |
0.01* |
122 |
0.005/< 0.01 |
|
Milk |
1995 |
59 |
0.005 |
59 |
0 |
|
|
Milk |
1996 |
112 |
0.005 |
112 |
0 |
|
|
Milk |
1997 |
378 |
0.005 |
377 |
< 0.0001/< 0.05 |
|
|
Milk |
1998 |
499 |
0.005 |
499 |
0 |
|
|
Milk |
1999 |
296 |
0.005 |
295 |
< 0.0002/< 0.05 |
|
|
France |
Milk |
Nov 1984– |
494 |
0.05 |
487 |
NR |
|
466 |
0.05 |
466 |
0 |
|||
|
Sept–Nov 1986 |
265 |
0.05 |
265 |
0 |
||
|
Jan–Mar, Sept–Nov 1987 |
449 |
0.05 |
447 |
NR |
||
|
Jan–Mar 1988 |
277 |
0.05 |
277 |
0 |
||
|
Nov 1988– |
549 |
0.05 |
547 |
NR |
||
|
Nov 1989– |
526 |
0.03 |
511 |
NR |
||
|
Nov 1990– |
550 |
0.03 |
544 |
NR |
||
|
Sept–Nov 1991 |
303 |
000 |
300 |
NR |
||
|
Bulk raw milk |
Nov 1984– |
37 |
0.05 |
32 |
NR |
|
|
June–Oct 1985 |
30 |
0.05 |
30 |
0 |
||
|
Bulk raw milk |
Nov 1985– |
42 |
0.05 |
41 |
NR |
|
|
June–Oct 1986 |
26 |
0.05 |
26 |
0 |
||
|
Nov 1986– |
24 |
000 |
24 |
0 |
||
|
June–Oct 1987 |
18 |
0.05 |
18 |
0 |
||
|
Nov 1987– |
29 |
0.05 |
29 |
0 |
||
|
May 1988 |
|
|
|
|
||
|
June–Oct 1988 |
27 |
0.05 |
27 |
0 |
||
|
Nov 1988– |
8 |
0.05 |
8 |
0 |
||
|
Bulk raw milk |
June–Oct 1989 |
9 |
0.05 |
9 |
0 |
|
|
Nov 1989– |
18 |
0.05 |
18 |
0 |
||
|
June–Oct 1990 |
19 |
0.05 |
19 |
0 |
||
|
Nov 1990– |
35 |
0.05 |
35 |
0 |
||
|
June–Oct 1991 |
15 |
0.05 |
15 |
0 |
||
|
Nov 1991– |
34 |
0.05 |
34 |
0 |
||
|
LOQ: 0.05, ND – < 0.05; 0.03, ND– < 0.03 |
|
|
||||
|
Cheese, milk |
1989–94 |
2670 |
0.01 |
2600 (< 0.05) |
0.016/0.37 |
|
|
Heat-treated milk |
1990–95 |
165 |
0.01 |
160 (< 0.05) |
0.015/NR |
|
|
Powdered milk |
1989–94 |
134 |
0.1 |
130 (< 0.05) |
0.15/< 0.3 |
|
|
Milk |
1998 |
251 |
0.03 |
247 |
NR/< 0.05 |
|
|
Milk |
1999 |
234 |
0.03 |
234 |
NR |
|
|
Germany |
Herd's bulk milk |
Nov 1984– |
135 |
|
NR |
0.003–0.080 |
|
Nov 1985– |
242 |
|
NR |
0.003–0.100 |
||
|
May–June 1986 |
74 |
|
NR |
0.003–0.020 |
||
|
Pasteurized commercial milk |
Nov 1984– |
132 |
|
NR |
0.003–0.060 |
|
|
Nov 1985– |
16 |
|
NR |
0.007–0.013 |
||
|
Nov 1985– |
473 |
|
NR |
0.004–0.010 |
||
|
Milk, butter, cream, cheese, ice cream |
1991–93 |
1853 |
0.01* |
1692 |
0.006/0.07 |
|
|
Baby food |
|
206 |
0.1* |
171 |
0.007/0.04 |
|
|
Cheese |
|
110 |
0.1* |
104 |
0.055/0.15 |
|
|
Milk |
1996 |
2822 |
|
NR |
NR/(0.033)* |
|
|
1997 |
4902 |
|
NR |
NR/(0.014)* |
||
|
1998 |
6150 |
|
NR |
NR/(0.036)* |
||
|
1998–2000 |
6537 |
0.005 |
4775 |
NR |
||
|
|
|
0.01 |
1550 |
NR |
||
|
2000 |
3307 |
|
NR |
NR (0.007, max stated value) |
||
|
|
|
|
|
|
||
|
Greece |
Raw milk |
Nov 1986– |
99 |
0.1* |
95 |
0.10–0.13 |
|
Pasteurized milk |
|
51 |
0.1* |
51 |
0 |
|
|
Pasteurized milk |
1995–96 |
81 |
0.005* |
72 |
0.0072/0.18 |
|
|
Ireland |
Milk |
1999 |
62 |
0.02 |
60 |
NR/< 0.05 |
|
Italy |
Milk |
NR |
59 |
|
13 |
Trace–0.38 |
|
Milk |
NR |
27 |
|
3 |
0.005–0.065 |
|
|
Milk |
NR |
107 |
|
102 |
0.024–0.094 |
|
|
Milk |
NR |
104 |
|
104 |
0 |
|
|
Milk, imported |
1984 |
313 |
0.001* |
271 |
0.001–0.050 |
|
|
Milk, dairy farm |
1985 |
276 |
0.001* |
206 |
0.001–0.20 |
|
|
Milk, dairy farm |
NR |
176 |
|
176 |
0 |
|
|
Milk, dairy farm |
NR |
107 |
|
41 |
0.006–0.10 |
|
|
Milk, dairy farm |
NR |
107 |
|
51 |
0.003–0.060 |
|
|
Milk, dairy farm |
1995 |
159 |
0.001 |
23 |
0.009/0.11 |
|
|
Dry milk |
NR |
10 |
|
0 |
0.015–0.10 |
|
|
Dry milk |
1995 |
97 |
0.001 |
16 |
0.018/0.10 |
|
|
Raw milk |
NR |
57 |
|
33 |
0.10–0.93 |
|
|
Raw milk |
NR |
60 |
|
57 |
0.10–0.28 |
|
|
Pasteurized milk |
NR |
68 |
|
7 |
0.005–0.050 |
|
|
Pasteurized milk |
NR |
30 |
|
3 |
0.003–0.022 |
|
|
Yoghurt |
1995 |
114 |
0.001 |
23 |
0.014/0.50 |
|
|
Italy |
Milk |
1984–95 |
997 |
0.001*–0.5* |
579 |
NR/< 0.05 |
|
Cheese |
1984–95 |
1593 |
0.001*–0.5* |
1270 |
NR/< 0.05 |
|
|
Netherlands |
Bulk milk and milk powder |
Jan–Apr 1985 |
209 |
0.010 |
40 |
0.010–0.070 |
|
May–Aug 1985 |
207 |
0.010 |
54 |
0.010–0.050 |
||
|
Sept–Dec 1985 |
207 |
0.010 |
19 |
0.010–0.090 |
||
|
Jan–Apr 1986 |
212 |
0.010 |
38 |
0.010–0.090 |
||
|
May–Aug 1986 |
204 |
0.010 |
89 |
0.010–0.070 |
||
|
Sept–Dec 1986 |
202 |
0.010 |
37 |
0.010–0.050 |
||
|
Milk |
1990–93 |
1903 |
NR |
331 |
0.009/0.02 |
|
|
Milk powder, home-produced |
1998 |
168 |
0.01 |
168 |
0 |
|
|
Milk powder, imported |
1998 |
39 |
0.01 |
NR |
NR |
|
|
Milk |
1999 |
30 |
0.005 |
25 |
NR/< 0.05 |
|
|
Norway |
Milk |
1998 |
54 |
0.0001 |
3 |
0.0014/0.009 |
|
Poland |
Raw milk |
1993–94 |
30 |
0.003* |
24 |
0.0036–0.0106 |
|
1993–94 |
157 |
0.003* |
120 |
< 0.010–0.025 |
||
|
Portugal |
Milk |
1999 |
96 |
NR |
28 |
NR/< 0.05 |
|
Spain |
Raw milk |
May–Nov 1990 |
61 |
0.01 |
52 |
0.010–< 0.050 |
|
UHT milk |
May–Nov 1990 |
33 |
0.01 |
28 |
0.010–0.025 |
|
|
Raw milk |
May–Nov 1990 |
61 |
0.01 |
49 |
0.010–0.040 |
|
|
29 |
0.01 |
28 |
0.020–0.040 |
|||
|
32 |
0.01 |
21 |
0.010–0.040 |
|||
|
Milk |
1989–92 |
307 |
0.005*–0.5* |
234 |
0.006/0.046 |
|
|
Butter, cream, cheese, and ice cream |
1989–92 |
221** |
0.005*–0.5* |
217 |
0.022/0.29 |
|
|
Baby food, powdered milk |
1989–92 |
4 |
0.005*–0.5* |
0 |
0.06/0.09 |
|
|
Confectionary |
1989–92 |
2 |
0.005*–0.5* |
0 |
0.033/0.05 |
|
|
Milk (34 UHT & 8 pastuerized) |
1998 |
42 |
NR |
NR |
NR/0.027 |
|
|
Sweden |
Milk |
Jan–Mar 1986 |
268 |
0.005 |
239 |
0.005–0.31 |
|
Milk |
Apr–June 1986 |
271 |
0.005 |
118 |
0.005–0.050 |
|
|
Milk powder |
Dec 1985 |
14 |
0.005 |
0 |
0.006–0.057 |
|
|
Milk |
1999 |
11 |
0.005 |
11 |
0 |
|
|
United Kingdom |
Milk, farm–gate |
1988 |
118 |
0.01 |
107 |
0.010–0.18 |
|
Milk, farm–gate |
1989 |
127 |
0.01 |
101 |
0.010–0.16 |
|
|
Milk |
1988–89,1995 |
331 |
0.01 |
255 |
0.01/0.22 |
|
|
United Kingdom |
Powdered milk |
1988–89, 1995 |
93 |
0.02 |
89 |
0.01/0.05 |
|
Yoghurt |
1988–89,1995 |
30 |
0.02 |
24 |
0.01/0.04 |
|
|
Cheese |
1988–89,1995 |
73 |
0.02 |
0 |
0.08/0.22 |
|
|
Retail milk |
1994–95 |
162 |
0.01 |
74 |
NR |
|
|
Dried milk |
1995 |
93 |
0.02 |
89 |
NR/0.05 |
|
|
Milk |
1999 |
95 |
0.001 |
93 |
NR/< 0.05 |
|
|
Country/ Region |
Commodity |
Year/ Season |
90th %ile |
n > 0.05(µg/kg) |
n > 0.5µg/kg) |
References |
Sampling procedure |
|
Americas |
|||||||
|
Argentina |
Farm milk |
Mar–Sept 1999 |
0 |
0 |
|
CEREMIC; A, ELISA |
|
|
Powdered milk |
Mar–Sept 1999 |
|
0 |
0 |
(Ridascreen) |
Whole and defatted |
|
|
Pasteurized fluid milk |
Mar–Sept 1999 |
|
0 |
0 |
(Ridascreen) |
Commercial samples |
|
|
Brazil |
Milk |
NR |
|
0 |
0 |
P, Martins & Martins (1986) |
|
|
Brazil |
Pasteurized milk |
1992 |
|
4 |
0 |
P,S, de Sylos et al. (1996); A (HPLC), AOAC 986.16 (1990) |
|
|
Brazil |
Raw milk |
1992–93 |
0 |
0 |
0 |
P, Correa et al. (1997) |
|
|
Brazil |
Powdered milk, pasteurized |
1989 |
0 |
NR |
0 |
P,S, de Sylos et al. (1996); A (TLC), AOAC 980.21 (1990) |
|
|
Brazil |
Milk powder, reconstituted for infants |
Oct. 1992–Jan. 1993 |
|
> 33 |
6 |
P,S, A (ELISA), Oliveira et al. (1997) |
|
|
Brazil |
Yoghurt |
1990 |
0 |
0 |
0 |
P,S, de Sylos et al. (1996); A (TLC), AOAC 980.21 (1990) |
|
|
Brazil |
Milk |
1996–97 |
|
0 |
0 |
P,S, Souza et al. (1999); A, ELISA |
|
|
Brazil |
Milk and milk products |
1998–99 |
|
0.006–0.077 |
0 |
P,S, Prado et al. (2000); A, HPLC |
|
|
Pasteurized milk |
|
|
0.008–0.077 |
0 |
|
||
|
Long–life milk |
1998–99 |
|
0.006–0.070 |
0 |
|
||
|
Long–life milk, infant |
|
|
0 |
0 |
|
||
|
Milk powder |
|
|
0 |
0 |
|
||
|
Milk powder, infant |
|
|
0.007–0.050 |
0 |
|
||
|
Canada |
Homogenized milk |
1994 |
|
NR |
0 |
USA (FDA); industry |
|
|
2% partly skimmed milk |
1994 |
|
NR |
0 |
Health Canada |
|
|
|
Skimmed milk powder |
1994 |
|
1 |
0 |
|
||
|
Partly skimmed milk, lactose reduced |
1994 |
|
1 |
0 |
|
||
|
1% partly skimmed milk |
1994 |
0 |
0 |
0 |
|
||
|
Evaporated milk |
1994 |
|
1 |
0 |
|
||
|
Partly skimmed milk, lactaid |
1994 |
|
0 |
0 |
|
||
|
Skimmed milk, lacteeze |
1994 |
0 |
0 |
0 |
|
||
|
Uruguay |
Milk |
1993–95 |
|
0 |
0 |
P, Pineiro et al. (1996) |
|
|
USA |
Raw milk |
1995 |
0.000 |
13 |
0 |
FDA laboratory 27 |
|
|
Raw milk |
1996 |
0.070 |
94 |
5 |
|
||
|
USA |
Raw milk |
1997 |
0.000 |
20 |
0 |
FDA laboratory 27 |
|
|
Raw milk |
1998 |
0.000 |
16 |
0 |
|
||
|
Raw milk |
1999 |
0.000 |
26 |
0 |
|
||
|
Raw milk |
2000 |
0.000 |
16 |
0 |
|
||
|
Fnished milk |
1995 |
0.000 |
3 |
0 |
|
||
|
Finished milk |
1996 |
0.060 |
4 |
0 |
|
||
|
Finished milk |
1997 |
0.000 |
1 |
0 |
|
||
|
Finished milk |
1999 |
0.000 |
2 |
0 |
|
||
|
Finished milk |
2000 |
0 |
0 |
0 |
|
||
|
Southwest |
Milk |
1998–2000 |
|
1239 |
45 |
FDA; industry |
|
|
|
639 |
|
|
||||
|
|
399 |
|
|
||||
|
|
134 |
|
|
||||
|
|
11 |
|
|
||||
|
|
11 |
|
|
||||
|
Midwest |
Milk |
1998–2000 |
|
NR |
10 |
FDA; industry |
|
|
Southeast |
Milk |
1998–2000 |
|
NR |
14 |
FDA; industry |
|
|
Southern |
Milk |
1998 |
1.4 |
82 |
47 |
FDA laboratory B |
|
|
Southern |
Milk |
1999 |
0.56 |
59 |
20 |
FDA laboratory B |
|
|
Southern |
Milk |
2000 |
0.28 |
13 |
1 |
FDA laboratory B |
|
|
Southeast |
Milk |
Jun 1999–May 2000 |
|
3 (0.1–< 0.25) |
0 |
FDA laboratory A |
|
|
Southeast |
Milk |
Jul 1997–May 2000 |
|
29 (< 0.1– 7 |
|
FDA; industry |
|
|
China, north |
Powdered milk |
NR |
|
> 17 |
3 |
P,S, Liu et al. (1992); A, ELISA |
From factories and markets in 12 provinces |
|
China, south |
Powdered milk |
NR |
|
>19 (< 0.5) |
33 |
||
|
China |
Powdered milk |
1992–93 |
|
16 |
0 |
P,S, Kawamura et al. (1994); |
|
|
Taiwan |
Fresh milk |
Mar–Sept 1986 |
|
NR |
0 |
P, van Egmond (1989) |
|
|
Powdered milk |
Mar–Sept 1986 |
|
NR |
0 |
|
||
|
India |
Raw milk |
1992–93 |
|
89 |
> 65 |
P,S, Rajan et al. (1995); A, AOAC (1990), HPLC |
|
|
Raw milk |
1993 |
|
36 |
3 |
|
||
|
Indonesia |
Milk |
Jan 1990 |
|
> 40 |
16 |
P,S, RIVS (2000; A, AOAC (1984) |
|
|
Milk |
Sept 1990 |
|
> 30 |
8 |
|
||
|
Milk |
July 1991 |
|
> 43 |
5 |
|
||
|
Milk |
Dec 1991 |
|
> 18 |
4 |
|
||
|
Milk |
Jan 1992 |
|
> 29 |
11 |
|
||
|
Java |
Milk |
Jan 1993 |
|
> 30 |
23 |
|
|
|
Indonesia |
Milk/factory |
Sept 1999 |
|
> 9 |
6 |
P,S, DL/RIVS (2000); A, AOAC (1984) |
|
|
Ice cream/factory |
Aug 1999 |
|
> 5 |
0 |
|
||
|
Ice cream/factory |
Oct 1999 |
|
NR |
0 |
|
||
|
Korea, Republic of |
Raw milk |
Winter 1995 |
0.120 |
29 |
0 |
P,S, and A (ELISA), Shon et al. (1996) |
|
|
Market milk |
Winter 1995 |
|
18 |
0 |
|
||
|
Market milk |
Summer, winter 1997 |
|
3 |
0 |
P,S,A (HPLC), Kim et al. (2000) |
|
|
|
|
0 |
0 |
|
||||
|
Korea, Republic of |
Infant formula |
Autumn 997 |
|
12 |
0 |
P,S,A (HPLC), Kim et al. (2000) |
|
|
|
9 |
0 |
|
||||
|
Powdered milk |
Autumn 1997 |
|
17 |
0 |
|
||
|
|
17 |
0 |
|
||||
|
Yoghurt |
Autumn, winter, 1997 |
|
9 |
0 |
|
||
|
|
10 |
0 |
|
||||
|
Philippines |
Milk |
1997 |
|
80 |
16 |
P, Begino (1998) |
|
|
Thailand |
Raw milk |
1990–93 |
0.42 |
> 12 |
4 |
P,S, Boriboon & Suprasert (1994); A, IDF (1981) |
|
|
Raw milk |
1995–96 |
|
57 |
17 |
P,S, Saitanu (1997); A, RIA |
~ 50 ml collected from 50-L farm milk can and tested within 3–4 days or stored at –15 °C until testing within 1 month |
|
|
Pasteurized milk |
1990–93 |
1.1 |
> 9 |
7 |
P,S, Boriboon & Suprasert (1994); A, IDF (1981) |
|
|
|
Pasteurized milk |
1995–96 |
|
63 |
20 |
P,S, Saitanu (1997); A, RIA |
One sample from each lot of factory-treated products tested on arrival or kept at 4 °C and tested within 2 days |
|
|
UHT milk |
|
|
60 |
7 |
|||
|
Sterilized milk |
|
|
60 |
3 |
|||
|
Thailand |
Powdered milk |
1995–96 |
|
> 1 |
1 |
P,S, Boriboon & Suprasert (1994); |
Tested within 2 weeks |
|
Powdered milk, imported |
1995–96 |
|
2 |
0 |
|||
|
Pelleted milk |
1995–96 |
|
7 |
1 |
|||
|
United Arab Emirates |
Fresh milk |
1998 |
|
21 |
0 |
Dubai Central Laboratory |
immuno-affinity column–HPLC/ fluorescence. |
|
UHT milk |
1998 |
0 |
0 |
0 |
|||
|
Milk/imported |
1999 |
|
NR |
0 |
|||
|
Low fat milk |
1999 |
|
NR |
0 |
|||
|
Milk powder |
1998 |
0 |
0 |
0 |
|||
|
Milk powder |
1999 |
0 |
0 |
0 |
|||
|
Full cream milk |
1999 |
|
NR |
0 |
|||
|
Skimmed milk |
1999 |
|
NR |
0 |
|||
|
Milk-based infant formula |
1998 |
0 |
0 |
0 |
|||
|
Milk-based infant formula |
1999 |
0 |
0 |
0 |
|||
|
Europe |
|||||||
|
Austria |
Milk powder |
Feb 1983 |
0 |
0 |
0 |
P, van Egmond (1989) |
|
|
Austria |
Milk |
1990–93 |
0 |
0 |
0 |
SCOOP Report (EU) |
|
|
Milk |
1999 |
0 |
0 |
0 |
|
||
|
Belgium |
Commercial blended milk |
Winter 1984–85 |
|
NR |
0 |
P, van Egmond (1989) |
|
|
Winter 1985–86 |
0 |
0 |
0 |
|
|||
|
Milk |
1993–94 |
|
0 |
0 |
SCOOP Report (EU) |
|
|
|
Milk |
1999 |
|
24 |
0 |
EU |
|
|
|
Denmark |
Milk |
1990–94 |
|
424 |
0 |
SCOOP Report (EU) |
|
|
Cyprus |
Raw milk |
1993,1995– |
|
3 |
0 |
P,S, Ioannou-Kakouri et al. |
|
|
Pasteurized milk, full |
96 |
|
0 |
0 |
(1999); A, Ioannou-Kakouri et al. (1995) |
|
|
|
Pasteurized milk, light |
|
|
0 |
0 |
|
||
|
Pasteurized milk, skimmed |
|
|
0 |
0 |
|
||
|
Baby milk, imported |
|
|
0 |
0 |
|
||
|
Evaporated milk, imported |
|
0 |
0 |
0 |
|
||
|
Czech Republic |
Raw milk |
1987–88 |
|
|
0 |
P, Fukal et al. (1990) |
|
|
Raw milk |
1987–88 |
|
|
0 |
|
||
|
Milk |
NR |
|
25 |
0 |
P, Fukal (1988) |
|
|
|
Finland |
Milk powder |
1986 |
0 |
0 |
0 |
P, van Egmond (1989) |
|
|
Finland |
Milk |
1992–15 |
0 |
0 |
0 |
SCOOP Report (EU) |
|
|
Milk |
1995 |
0 |
0 |
0 |
|
||
|
Milk |
1996 |
0 |
0 |
0 |
|
||
|
Milk |
1997 |
|
1(< 0.05) |
0 |
|
||
|
Milk |
1998 |
0 |
0 |
0 |
|
||
|
Milk |
1999 |
|
1(< 0.05) |
0 |
|
||
|
France |
Milk |
Nov 1984– |
|
7 |
0 |
P,S, Dragacci & Frémy (1993); A, Frémy & Boursier (1981) |
|
|
0 |
0 |
0 |
|
||||
|
Sept–Nov 1986 |
0 |
0 |
0 |
|
|||
|
Jan–Mar, Sept–Nov 1987 |
|
2 |
0 |
|
|||
|
Jan–Mar 1988 |
0 |
0 |
0 |
|
|||
|
Nov 1988– |
|
2 |
0 |
|
|||
|
Milk |
Nov 1989– |
|
13 |
0 |
P,S, Dragacci & Frémy (1993); A, Tuinstra et al. (1974) |
|
|
|
Nov 1990– |
|
3 |
0 |
|
|||
|
Sept–Nov 1991 |
|
0 |
0 |
|
|||
|
Bulk raw milk |
Nov 1984– |
|
5 |
0 |
P,S, Dragacci & Frémy (1993); A, Frémy & Boursier (1981) |
|
|
|
June–Oct 1985 |
0 |
0 |
0 |
|
|||
|
Bulk raw milk |
Nov 1985– |
|
1 |
0 |
P,S, Dragacci & Frémy (1993); A, Frémy & Boursier (1981) |
|
|
|
June–Oct 1986 |
0 |
0 |
0 |
|
|||
|
Nov 1986– |
0 |
0 |
0 |
|
|||
|
June–Oct 1987 |
0 |
0 |
0 |
|
|||
|
Nov 1987– |
0 |
0 |
0 |
|
|||
|
May 1988 |
|
|
|
|
|||
|
June–Oct 1988 |
0 |
0 |
0 |
|
|||
|
Nov 1988– |
0 |
0 |
0 |
|
|||
|
Bulk raw milk |
June–Oct 1989 |
0 |
0 |
0 |
P,S, Dragacci & Frémy (1993); A, Tuinstra et al. (1974) |
|
|
|
Nov 1989– |
0 |
0 |
0 |
|
|||
|
June–Oct 1990 |
0 |
0 |
0 |
|
|||
|
Nov 1990– |
0 |
0 |
0 |
|
|||
|
June–Oct 1991 |
0 |
0 |
0 |
|
|||
|
Nov 1991– |
0 |
0 |
0 |
|
|||
|
|
|
|
|
|
|||
|
Cheese, milk |
1989–94 |
|
21 |
0 |
SCOOP Report (EU) |
|
|
|
Heat-treated milk |
1990–95 |
|
2 |
0 |
|
||
|
Powdered milk |
1989–94 |
0 |
0 |
0 |
|
||
|
Milk |
1998 |
|
3 (0.03–0.05) |
|
|
||
|
Milk |
1999 |
0 |
0 |
0 |
|
||
|
Germany |
Herd's bulk milk |
Nov 1984– |
|
NR |
0 |
P, van Egmond (1989) |
|
|
Nov 1985– |
|
NR |
0 |
|
|||
|
May–June 1986 |
|
0 |
0 |
|
|||
|
Pasteurized commercial milk |
Nov 1984– |
|
NR |
0 |
|
||
|
Nov 1985– |
|
0 |
0 |
|
|||
|
Nov 1985– |
|
0 |
0 |
|
|||
|
Milk, butter, cream, cheese, ice cream |
1991–93 |
|
159 (< 0.05), 2(0.051–0.10) |
0 |
SCOOP Report (EU) |
|
|
|
Baby food |
|
|
35 (< 0.05) |
0 |
|
||
|
Cheese |
|
|
1 (< 0.05), |
0 |
|
||
|
Milk |
1996 |
|
0 |
0 |
|
||
|
1997 |
|
11 (> 0.005), |
|
|
|||
|
1998 |
|
7 (> 0.005), |
|
|
|||
|
1998–2000 |
|
205 (< 0.05) |
0 |
|
|||
|
|
|
7 (< 0.05) |
0 |
|
|||
|
2000 |
9 (> 0.005), |
NR |
|
|
|||
|
|
|
2 (> 0.01) |
|
|
|||
|
Greece |
Raw milk |
Nov 1986– |
|
> 4 |
0 |
P,S, Karaioannoglou et al. (1989); A, IDF (1982) |
|
|
Pasteurized milk |
|
0 |
NR |
0 |
|
||
|
Pasteurized milk |
1995–96 |
|
3 |
0 |
P,S, and A, Markaki & Melissari (1997); A, Qian et al. (1994) |
|
|
|
Ireland |
Milk |
1999 |
|
2 (< 0.05) |
|
EU |
|
|
Italy |
Milk |
NR |
|
NR |
0 |
P, Davoli et al. (1986) |
|
|
Milk |
NR |
|
NR |
0 |
P, Gelosa (1986) |
|
|
|
Milk |
NR |
|
NR |
0 |
P, Gilli et al. (1987) |
|
|
|
Milk |
NR |
0 |
0 |
0 |
P, Oliviero et al. (1987) |
|
|
|
Milk, imported |
1984 |
|
0 |
0 |
P,S, Piva et al. (1987); |
|
|
|
Milk, dairy farm |
1985 |
|
7 |
0 |
A (HPLC), AOAC (1984) |
|
|
|
Milk, dairy farm |
NR |
|
0 |
0 |
P, Mosso et al. (1992) |
|
|
|
Milk, dairy farm |
NR |
|
0 |
0 |
P, Bagni et al. (1992) |
|
|
|
Milk, dairy farm |
NR |
|
NR |
0 |
P, Bagni et al. (1992) |
|
|
|
Milk, dairy farm |
1995 |
|
2 |
0 |
P,S, Galvano et al. (1998); A, Mortimer et al. (1987) |
|
|
|
Dry milk |
NR |
|
NR |
0 |
P, Vittani (1987) |
|
|
|
Dry milk |
1995 |
|
10 |
0 |
P,S, Galvano et al. (1998); A, Mortimer et al. (1987) |
|
|
|
Raw milk |
NR |
|
NR |
NR |
P, De Natale et al. (1989) |
|
|
|
Raw milk |
NR |
|
NR |
0 |
|
||
|
Pasteurized milk |
NR |
|
NR |
0 |
P, Gelosa (1986) |
|
|
|
Pasteurized milk |
NR |
|
0 |
0 |
P, Vittani (1987) |
|
|
|
Yoghurt |
1995 |
|
2 |
0 |
P,S, Galvano et al. (1998); A, Richard et al. (1993) |
|
|
|
Italy |
Milk |
1984–95 |
|
398 |
0 |
SCOOP Report (EU); A, Immunoaffinity– |
|
|
Cheese |
1984–95 |
|
323 |
0 |
HPLC/ fluorescence |
|
|
|
Netherlands |
Bulk milk and milk powder |
Jan–Apr 1985 |
|
NR |
0 |
P, van Egmond (1989) |
|
|
May–Aug 1985 |
|
NR |
0 |
|
|||
|
Sept–Dec 1985 |
|
NR |
0 |
|
|||
|
Jan–Apr 1986 |
|
NR |
0 |
|
|||
|
May–Aug 1986 |
|
NR |
0 |
|
|||
|
Sept–Dec 1986 |
|
NR |
0 |
|
|||
|
Milk |
1990–93 |
|
1572 |
|
SCOOP Report (EU); A, HPLC/ fluorescence |
|
|
|
Milk powder, home-produced |
1998 |
0 |
0 |
0 |
EU |
|
|
|
Milk powder, imported |
1998 |
|
NR |
NR |
EU |
|
|
|
Milk |
1999 |
|
5 (< 0.05) |
0 |
EU |
|
|
|
Norway |
Milk |
1998 |
|
0 |
0 |
Norwegian Food Control Authority |
|
|
Poland |
Raw milk |
1993–94 |
|
0 |
0 |
P,S, Domagala et al. (1997) |
|
|
1993–94 |
|
0 |
0 |
|
|||
|
Portugal |
Milk |
1999 |
|
68 (< 0.05) |
0 |
EU |
|
|
Spain |
Raw milk |
May–Nov 1990 |
|
0 |
0 |
P,S, Macho et al. |
|
|
UHT milk |
May–Nov 1990 |
|
0 |
0 |
(1992); A (HPLC), Mortimer et al. (1987) |
|
|
|
Raw milk |
May–Nov 1990 |
|
0 |
0 |
P, Jalon et al. (1994) |
|
|
|
|
0 |
0 |
|
||||
|
|
0 |
0 |
|
||||
|
Milk |
1989–92 |
|
73 (< 0.05) |
0 |
SCOOP Report (EU); |
|
|
|
Butter, cream, cheese, and ice cream |
1989–92 |
|
5** (< 0.05) |
0 |
A, HPLC |
**Excluding rejected data |
|
|
Baby food, powdered milk |
1989–92 |
|
4 (< 0.05) |
0 |
|
||
|
Confectionary |
1989–92 |
|
2 (< 0.05) |
0 |
|
||
|
Milk (34 UHT & 8 pastuerized) |
1998 |
|
0 |
NR |
EU |
|
|
|
Sweden |
Milk |
Jan–Mar 1986 |
|
NR |
0 |
P, van Egmond (1989) |
|
|
Milk |
Apr–June 1986 |
|
NR |
0 |
|
||
|
Milk powder |
Dec 1985 |
|
NR |
0 |
|
||
|
Milk |
1999 |
0 |
0 |
0 |
EU |
|
|
|
United Kingdom |
Milk, farm–gate |
1988 |
|
2 |
0 |
P,S, MAFF (1993) |
|
|
Milk, farm–gate |
1989 |
|
3 |
0 |
|
||
|
Milk |
1988–89,1995 |
|
67 (< 0.05), |
0 |
SCOOP Report (EU); A, HPLC/ fluorescence |
|
|
|
United Kingdom |
Powdered milk |
1988–89, 1995 |
4 (< 0.05) |
|
0 |
SCOOP Report (EU) |
|
|
Yoghurt |
1988–89,1995 |
|
6 (< 0.05) |
0 |
|
||
|
Cheese |
1988–89,1995 |
|
24 (< 0.05), |
0 |
|
||
|
Retail milk |
1994–95 |
|
88 (< 0.05) |
0 |
EU |
|
|
|
Dried milk |
1995 |
|
4 (0.05) |
0 |
EU |
31 dried milk and 62 infant formula dried milk; 4 infant formula samples had max of 0.05, i.e. < 0.05 when reconstituted |
|
|
Milk |
1999 |
|
2 (< 0.05) |
0 |
EU |
|
|
When the mean value for a positive samples was quoted and used (or its use implied), the data were recalculated, so all data represent the true mean for all samples, those below the limit of detection being considered 0. When possible, the numbers of samples with values > 0.05 and 0.5 µg/kg are included, as these values represent the legislative limits in many countries.
LOQ, limit of quantification; mean, true mean (for n analytical values; the true mean is the sum Xi / n, where Xi is the value of each analytical result); Max, maximum level
References: P, parent reference; S, sampling method; A, analytical method
*, limit of detection
ND, not determined; NR, not reported; TLC, thin-layer chromatography; ELISA, enzyme-linked immunosorbent assay; HPLC, high-performance liquid chromatography; RIA, radioimmunoassay
CEREMIC, Centro de Referencisa de Micología (Argentina); EU, European Union; FDA, Food and Drug Administration (USA); IDF, International Dairy Federation; MAFF, Ministry of Agriculture, Fisheries and Food (United Kingdom); RIVS, Research Institute for Veterinary Science (Indonesia)
Concentrations of aflatoxin M1 in milk
|
Country |
Commodity |
Date |
No. of samples |
LOQ/LOQ µg/kg |
n (%) |
|||
|
< LOQ/LOQ |
< 0.05 µg/kg |
0.5 µg/kg |
1 µg/kg |
|||||
|
Submitted laboratory data |
||||||||
|
Argentina |
Milk |
1998 |
6 |
NR |
1 (17)a |
2 (33) |
2 (33) |
0 (0.0) |
|
Austria |
Milk |
1990–93 |
479 |
NR/0.01 |
479 (100) |
479 (100) |
479 (100) |
0 (0.0) |
|
Belgium |
Milk |
1993–94 |
153 |
NR/0.01 |
98 (64) |
153 (100) |
153 (100) |
0 (0.0) |
|
Canada |
Milk, 7 types |
1997–98 |
81 |
0.015/NR |
81 (100.0) |
81 (100) |
81 (100) |
0 (0.0) |
|
Milk, 6 types |
1995-96 |
92 |
0.015/NR |
92 (100) |
92 (100) |
92 (100) |
0 (0.0) |
|
|
Milk, 5 types |
1994–95 |
57 |
0.015/NR |
57 (100) |
57 (100) |
57 (100) |
0 (0.0) |
|
|
Milk, 5 types |
1993–94 |
27 |
0.015/NR |
13 (48) |
17 (63) |
27 (100) |
0 (0.0) |
|
|
Denmark |
Milk |
1990–94 |
1664 |
0.01/NR |
1240 (74) |
1664 (100) |
1664 (100) |
0 (0.0) |
|
European Union |
Milk |
1999 |
7573 |
Variable |
7259 (96) |
7573(100) |
7573(100) |
0 (0.0) |
|
Finland |
Milk |
1992–95 |
122 |
0.01/NR |
122 (100) |
122 (100) |
122 (100) |
0 (0.0) |
|
France |
Milk, raw |
1989–94 |
2670 |
NR/0.01 |
0 (0.0) |
2649 (99) |
2670 (100) |
0 (0.0) |
|
Milk, pasteurized |
1990–95 |
165 |
NR/0.01 |
0 (0.0) |
163 (99) |
165 (100) |
0 (0.0) |
|
|
Indonesia |
Milk |
1990–93, 1999 |
342 |
NR/0.1 |
143 (42) |
NA |
269 (79) |
NR |
|
Italy |
Milk |
1984–95 |
997 |
0.001/NR |
579 (58) |
977 (98) |
997 (100) |
0 (0.0) |
|
Netherlands |
Milk |
1990–93 |
1903 |
NR |
331 (17) |
1572 (83) |
1903 (100) |
0 (0.0) |
|
Norway |
Milk, dairy |
1998 |
54 |
0.0001/NR |
3 (5.6) |
54 (100) |
54 (100) |
0 (0.0) |
|
Spain |
Milk |
1989–92 |
307 |
0.005/NR |
234 (76) |
307 (100) |
307 (100) |
0 (0.0) |
|
United Arab Emirates |
Milk, raw |
1998 |
22 |
0.005/0.01 |
1 (4.5) |
1 (4.5) |
22 (100) |
0 (0.0) |
|
Milk, whole |
1999 |
22 |
0.005/0.01 |
14 (64) |
NR |
22 (100) |
0 (0.0) |
|
|
Milk, UHT |
1998 |
11 |
0.005/0.01 |
11 (100) |
11 (100) |
11 (100) |
0 (0.0) |
|
|
Milk, imported |
1999 |
15 |
0.005/0.01 |
12 (80) |
NR |
15 (100) |
0 (0.0) |
|
|
Milk, low-fat |
1999 |
11 |
0.005/0.01 |
2 (18) |
NR |
11 (100) |
0 (0.0) |
|
|
Milk, non-fat |
1999 |
9 |
0.005/0.01 |
2 (22) |
NR |
9 (100) |
0 (0.0) |
|
|
United Kingdom |
Milk |
1988–89 |
331 |
NR/0.01 |
255 (77) |
322 (97) |
331 (100) |
0 (0.0) |
|
USA |
||||||||
|
Southwest |
Milk |
1998–2000 |
5801 |
0.02/0.05 |
4562 (79) |
4562 (79) |
5756 (99) |
NR |
|
Midwest |
Milk |
1998–2000 |
438 |
0.5/NQ |
10 (2.3) |
10 (2.3) |
428 (98) |
NR |
|
Southeast |
Milk |
1998–2000 |
13 093 |
0.5/NQ |
NR |
NR |
13 079 (100) |
NR |
|
Southeast |
Milk |
1999–2000 |
199 |
0.02/0.05 |
196 (98)a |
NR |
199 (100) |
0 (0.0) |
|
Southeast |
Milk |
1997–2000 |
78 |
0.02/0.05 |
49 (63)a |
NR |
71 (91) |
NR |
|
Southern |
Milk |
1998 |
163 |
0.02/0.05 |
81 (50) |
81 (50) |
117 (72) |
21 (13) |
|
Southern |
Milk |
1999 |
167 |
0.02/0.05 |
108 (65) |
108 (65) |
147 (88) |
2 (1.2) |
|
Southern |
Milk |
2000 |
49 |
0.02/0.05 |
36 (74) |
36 (74) |
49 (100) |
0 (0.0) |
|
USA |
Milk, raw |
1995 |
755 |
0.02/0.05 |
742 (98) |
742 (98) |
755 (100) |
0 (0.0) |
|
Milk, raw |
1996 |
381 |
0.02/0.05 |
289 (76) |
289 (76) |
376 (99) |
3 (0.8) |
|
|
Milk, raw |
1997 |
597 |
0.02/0.05 |
589 (99) |
589 (99) |
597 (100) |
0 (0.0) |
|
|
Milk, raw |
1998 |
855 |
0.02/0.05 |
843 (99) |
843 (99) |
855 (100) |
0 (0.0) |
|
|
Milk, raw |
1999 |
877 |
0.02/0.05 |
853 (97) |
853 (97) |
877 (100) |
0 (0.0) |
|
|
Milk, raw |
2000 |
477 |
0.02/0.05 |
462 (97) |
462 (97) |
477 (100) |
0 (0.0) |
|
|
Milk, finished |
1995 |
79 |
0.02/0.05 |
76 (96) |
76 (96) |
79 (100) |
0 (0.0) |
|
|
Milk, finished |
1996 |
21 |
0.02/0.05 |
17 (81) |
17 (81) |
21 (100) |
0 (0.0) |
|
|
Milk, finished |
1997 |
53 |
0.02/0.05 |
52 (98) |
52 (98) |
53 (100) |
0 (0.0) |
|
|
Milk, finished |
1998 |
48 |
0.02/0.05 |
44 (92) |
44 (92) |
48 (100) |
0 (0.0) |
|
|
Milk, finished |
1999 |
60 |
0.02/0.05 |
58 (97) |
58 (97) |
60 (100) |
0 (0.0) |
|
|
Milk, finished |
2000 |
12 |
0.02/0.05 |
12 (100) |
12 (100) |
12 (100) |
0 (0.0) |
|
|
Literature data |
||||||||
|
Brazil |
Milk, market |
1979–81 |
100 |
NR |
99 (99)a |
99 (99) |
100 (100) |
0 (0.0) |
|
Milk, farm |
1979–81 |
50 |
NR |
41 (82) |
41 (82) |
46 (92) |
1 (2.0) |
|
|
Milk |
1996–97 |
110 |
NR/0.0063 |
83 (76) |
110 (100) |
110 (100) |
0 (0.0) |
|
|
Milk, fluid |
1998–99 |
50 |
NR/0.006 |
10 (20)a |
46 (92) |
50 (100) |
0 (0.0) |
|
|
Brazil |
Milk, reconstituted |
1997 |
300 |
NR |
267 (89) |
267 (89) |
294 (98) |
0 (0.0) |
|
Milk, raw |
1992–93 |
144 |
0.5/NR |
144 (100) |
144 (0.0) |
144 (100) |
0 (0.0) |
|
|
Milk, powder |
1989 |
35 |
NR |
35 (100)a |
35 (100) |
35 (100) |
0 (0.0) |
|
|
Milk, past |
1989 |
51 |
NR |
51 (100)a |
51 (100) |
51 (100) |
0 (0.0) |
|
|
Milk, past |
1992 |
52 |
NR |
48 (92)a |
48 (92) |
52 (100) |
0 (0.0) |
|
|
Yoghurt |
1990 |
30 |
NR |
30 (100) |
30 (100) |
30 (100) |
0 (0.0) |
|
|
Egypt |
Milk, reconstituted |
1995 |
15 |
NR |
14 (93) |
15 (100) |
15 (100) |
0 (0.0) |
|
France |
Milk |
1990–91 |
853 |
NR/0.05 |
844 (99) |
850 (100) |
853 (100) |
0 (0.0) |
|
Milk, raw |
1990–92 |
103 |
NR/0.05 |
103 (100) |
103 (100) |
103 (100) |
0 (0.0) |
|
|
Greece |
Milk, past |
1997 |
81 |
NR/0.005 |
72 (89) |
78 (96) |
81 (100) |
0 (0.0) |
|
Milk, raw |
1986–87 |
99 |
NR/0.1 |
95 (96) |
NA |
99 (100) |
0 (0.0) |
|
|
Milk, past |
1986–87 |
51 |
NR/0.1 |
51 (100) |
NA |
51 (100) |
0 (0.0) |
|
|
India |
Milk, raw |
1992–93 |
504 |
NR/0.1 |
415 (82) |
415 (82) |
NR |
NR |
|
Italy |
Milk |
1995 |
159 |
0.001 |
23 (14) |
NR |
136 (100) |
0 (0.0) |
|
Yoghurt |
1995 |
114 |
0.001 |
23 (20) |
NR |
114 (100) |
0 (0.0) |
|
|
Philippines |
Milk |
1997 |
91 |
NR/0.05 |
11 (12) |
11 (12) |
75 (82) |
NR |
|
Poland |
Milk, raw |
1993–94 |
187 |
NR/0.003 |
144 (77) |
187 (100) |
187 (100) |
0 (0.0) |
|
Republic of Korea |
Yoghurt |
2000 |
60 |
NR/0.01 |
29 (48) |
51 (85) |
60 (100) |
0 (0.0) |
|
Milk, past |
2000 |
70 |
NR/0.01 |
31 (44) |
67 (96) |
70 (100) |
0 (0.0) |
|
|
Milk, raw |
1995 |
49 |
NR/0.003 |
0 (0.0) |
20 (41) |
49 (100) |
0 (0.0) |
|
|
Milk, market |
1995 |
15 |
NR/0.003 |
0 (0.0) |
0 (0.0) |
15 (100) |
0 (0.0) |
|
|
Spain |
Milk, raw |
1990 |
61 |
NR/0.01 |
52 (85) |
61 (100) |
61 (100) |
0 (0.0) |
|
Milk, UHT |
1990 |
33 |
NR/0.01 |
28 (85) |
33 (100) |
33 (100) |
0 (0.0) |
|
|
Milk, raw |
1990 |
122 |
NR/0.01 |
98 (80) |
122 (100) |
122 (100) |
0 (0.0) |
|
|
Thailand |
Milk, raw |
1990–93 |
45 |
NR/0.1 |
33 (73) |
NA |
41 (91) |
0 (0.0) |
|
Milk, market |
1995–96 |
183 |
NR/0.025 |
0 (0.0) |
0 (0.0) |
153 (84) |
NR |
|
|
Uruguay |
Milk |
1993–95 |
22 |
0.2/NR |
15 (68.2) |
NA |
21 (96) |
NR |
|
Country |
Commodity |
Date |
No. of samples |
LOQ/LOQ µg/kg |
Mean |
Maximum |
90th %ile |
References |
|
Submitted laboratory data |
||||||||
|
Argentina |
Milk |
1998 |
6 |
NR |
0.52 |
0.9 |
NR |
Universidad de Lujan |
|
Austria |
Milk |
1990–93 |
479 |
NR/0.01 |
0.005 |
< 0.01 |
NR |
SCOOP Report |
|
Belgium |
Milk |
1993–94 |
153 |
NR/0.01 |
0.01 |
0.037 |
NR |
SCOOP Report |
|
Canada |
Milk, 7 types |
1997–98 |
81 |
0.015/NR |
< 0.015 |
< 0.015 |
NR |
Health Canada (2000) |
|
Milk, 6 types |
1995-96 |
92 |
0.015/NR |
< 0.015 |
< 0.015 |
NR |
||
|
Milk, 5 types |
1994–95 |
57 |
0.015/NR |
< 0.015 |
< 0.015 |
NR |
||
|
Milk, 5 types |
1993–94 |
27 |
0.015/NR |
< 0.056 |
0.21 |
NR |
||
|
Denmark |
Milk |
1990–94 |
1664 |
0.01/NR |
< 0.01 |
0.069 |
NR |
SCOOP Report |
|
European Union |
Milk |
1999 |
7573 |
Variable |
NR |
NR |
NR |
European Union |
|
Finland |
Milk |
1992–95 |
122 |
0.01/NR |
0.005 |
< 0.01 |
NR |
SCOOP Report |
|
France |
Milk, raw |
1989–94 |
2670 |
NR/0.01 |
0.016 |
0.37 |
NR |
SCOOP Report |
|
Milk, pasteurized |
1990–95 |
165 |
NR/0.01 |
0.015 |
NR |
NR |
SCOOP Report |
|
|
Indonesia |
Milk |
1990–93, 1999 |
342 |
NR/0.1 |
0.86 |
23 |
NR |
Research Institute for Veterinary Science |
|
Italy |
Milk |
1984–95 |
997 |
0.001/NR |
NR |
NR |
NR |
SCOOP Report |
|
Netherlands |
Milk |
1990–93 |
1903 |
NR |
0.009 |
0.02 |
NR |
SCOOP Report |
|
Norway |
Milk, dairy |
1998 |
54 |
0.0001/NR |
0.0014 |
0.009 |
NR |
Food Control Authority |
|
Spain |
Milk |
1989–92 |
307 |
0.005/NR |
0.006 |
0.046 |
NR |
SCOOP Report |
|
United Arab Emirates |
Milk, raw |
1998 |
22 |
0.005/0.01 |
NR |
0.31 |
NR |
Dubai Central Laboratory |
|
Milk, whole |
1999 |
22 |
0.005/0.01 |
NR |
0.35 |
NR |
||
|
Milk, UHT |
1998 |
11 |
0.005/0.01 |
0 |
0 |
0 |
||
|
Milk, imported |
1999 |
15 |
0.005/0.01 |
NR |
0.08 |
NR |
||
|
Milk, low-fat |
1999 |
11 |
0.005/0.01 |
NR |
0.24 |
NR |
||
|
Milk, non-fat |
1999 |
9 |
0.005/0.01 |
NR |
0.14 |
NR |
||
|
United Kingdom |
Milk |
1988–89 |
331 |
NR/0.01 |
0.01 |
0.22 |
NR |
SCOOP Report |
|
USA |
||||||||
|
Southwest |
Milk |
1998–2000 |
5801 |
0.02/0.05 |
NR |
> 0.5 |
NR |
Industry |
|
Midwest |
Milk |
1998–2000 |
438 |
0.5/NQ |
NR |
> 0.5 |
NR |
Industry |
|
Southeast |
Milk |
1998–2000 |
13 093 |
0.5/NQ |
NR |
> 0.5 |
NR |
Industry |
|
Southeast |
Milk |
1999–2000 |
199 |
0.02/0.05 |
NR |
< 0.5 |
NR |
FDA Laboratory A |
|
Southeast |
Milk |
1997–2000 |
78 |
0.02/0.05 |
NR |
> 0.5 |
NR |
FDA Laboratory A |
|
Southern |
Milk |
1998 |
163 |
0.02/0.05 |
0.73 |
14 |
1.5 |
FDA Laboratory B |
|
Southern |
Milk |
1999 |
167 |
0.02/0.05 |
0.17 |
5.2 |
0.58 |
FDA Laboratory B |
|
Southern |
Milk |
2000 |
49 |
0.02/0.05 |
0.07 |
0.5 |
0.28 |
FDA Laboratory B |
|
USA |
Milk, raw |
1995 |
755 |
0.02/0.05 |
0.002 |
0.33 |
0 |
FDA Laboratory 27 |
|
Milk, raw |
1996 |
381 |
0.02/0.05 |
0.03 |
1.8 |
0.07 |
||
|
Milk, raw |
1997 |
597 |
0.02/0.05 |
0.001 |
0.41 |
0 |
||
|
Milk, raw |
1998 |
855 |
0.02/0.05 |
0.002 |
0.38 |
0 |
||
|
Milk, raw |
1999 |
877 |
0.02/0.05 |
0.002 |
0.24 |
0 |
||
|
Milk, raw |
2000 |
477 |
0.02/0.05 |
0.004 |
0.37 |
0 |
||
|
Milk, finished |
1995 |
79 |
0.02/0.05 |
0.003 |
0.11 |
0 |
||
|
Milk, finished |
1996 |
21 |
0.02/0.05 |
0.011 |
0.06 |
0.06 |
||
|
Milk, finished |
1997 |
53 |
0.02/0.05 |
0.001 |
0.06 |
0 |
||
|
Milk, finished |
1998 |
48 |
0.02/0.05 |
0.005 |
0.06 |
0 |
||
|
Milk, finished |
1999 |
60 |
0.02/0.05 |
0.004 |
0.17 |
0 |
||
|
Milk, finished |
2000 |
12 |
0.02/0.05 |
0 |
0 |
0 |
||
|
Literature data |
||||||||
|
Brazil |
Milk, market |
1979–81 |
100 |
NR |
0.002 |
0.2 |
NR |
Sabino et al. (1989) |
|
Milk, farm |
1979–81 |
50 |
NR |
0.087 |
1.7 |
NR |
||
|
Milk |
1996–97 |
110 |
NR/0.0063 |
0.012 |
0.071 |
NR |
Souza et al. (1999) |
|
|
Milk, fluid |
1998–99 |
50 |
NR/0.006 |
0.028 |
0.077 |
NR |
Prado et al. (1999) |
|
|
Brazil |
Milk, reconstituted |
1997 |
300 |
NR |
0.03 |
1 |
NR |
Oliveira et al. (1997) |
|
Milk, raw |
1992–93 |
144 |
0.5/NR |
0 |
0 |
0 |
Correa et al. (1997) |
|
|
Milk, powder |
1989 |
35 |
NR |
0 |
0 |
0 |
de Sylos et al. (1996) |
|
|
Milk, past |
1989 |
51 |
NR |
0 |
0 |
0 |
||
|
Milk, past |
1992 |
52 |
NR |
0.012 |
0.37 |
NR |
||
|
Yoghurt |
1990 |
30 |
NR |
0 |
0 |
0 |
||
|
Egypt |
Milk, reconstituted |
1995 |
15 |
NR |
0.0018b |
NR |
NR |
El-Gohary (1995) |
|
France |
Milk |
1990–91 |
853 |
NR/0.05 |
NR |
NR |
NR |
Dragacci & Fremy (1993) |
|
Milk, raw |
1990–92 |
103 |
NR/0.05 |
0 |
0 |
0 |
||
|
Greece |
Milk, past |
1997 |
81 |
NR/0.005 |
0.008 |
0.18 |
NR |
Markaki & Melissari (1997) |
|
Milk, raw |
1986–87 |
99 |
NR/0.1 |
0.005 |
0.13 |
NR |
Karaioannoglou et al. (1989) |
|
|
Milk, past |
1986–87 |
51 |
NR/0.1 |
0 |
0 |
0 |
||
|
India |
Milk, raw |
1992–93 |
504 |
NR/0.1 |
0.20 |
3.5 |
NR |
Rajan et al. (1995) |
|
Italy |
Milk |
1995 |
159 |
0.001 |
0.0086 |
0.11 |
NR |
Galvano et al. (1998) |
|
Yoghurt |
1995 |
114 |
0.001 |
0.014 |
0.50 |
NR |
||
|
Philippines |
Milk |
1997 |
91 |
NR/0.05 |
0.13 |
NR |
NR |
Begino (1998) |
|
Poland |
Milk, raw |
1993–94 |
187 |
NR/0.003 |
NR |
0.025 |
NR |
Domagala et al. (1997) |
|
Republic of Korea |
Yoghurt |
2000 |
60 |
NR/0.01 |
0.024 |
0.12 |
NR |
Kim et al. (2000) |
|
Milk, past |
2000 |
70 |
NR/0.01 |
0.014 |
0.052 |
NR |
||
|
Milk, raw |
1995 |
49 |
NR/0.003 |
0.065 |
0.16 |
0.12 |
Shon et al. (1996) |
|
|
Milk, market |
1995 |
15 |
NR/0.003 |
0.13 |
0.28 |
NR |
||
|
Spain |
Milk, raw |
1990 |
61 |
NR/0.01 |
NR |
< 0.050 |
NR |
Macho et al. (1992) |
|
Milk, UHT |
1990 |
33 |
NR/0.01 |
NR |
0.025 |
NR |
||
|
Milk, raw |
1990 |
122 |
NR/0.01 |
NR |
0.04 |
NR |
Jalon et al. (1994) |
|
|
Thailand |
Milk, raw |
1990–93 |
45 |
NR/0.1 |
0.112 |
0.8 |
NR |
Boriboon & Suprasert (1994) |
|
Milk, market |
1995–96 |
183 |
NR/0.025 |
NR |
NR |
NR |
Saitanu (1997) |
|
|
Uruguay |
Milk |
1993–95 |
22 |
0.2/NR |
NR |
20 |
NR |
Pineiro et al. (1996) |
NA, not applicable; NR, not reported; NQ, not quantified; past, pasteurized
a No samples; number of samples < LOD/LOQ may be higher
b Converted from 0.018 µg/kg of dry milk
Concentrations of aflatoxin M1 in milk (µg/kg) by regional diet
|
Diet |
Commodity |
Date |
No. of samples |
No. (%) |
Mean |
Maximum |
Reference |
||
|
< 0.05 µg/kg |
< 0.5 µg/kg |
> 0.5 µg/kg |
|||||||
|
European |
|||||||||
|
Austria |
Milk |
1990–93 |
479 |
479 (100) |
479 (100) |
0 (0.0) |
0.005 |
< 0.01 |
SCOOP Report |
|
Belgium |
Milk |
1993–94 |
153 |
153 (100) |
153 (100) |
0 (0.0) |
0.01 |
0.037 |
SCOOP Report |
|
Finland |
Milk |
1992–95 |
122 |
122 (100) |
122 (100) |
0 (0.0) |
0.005 |
< 0.01 |
SCOOP Report |
|
France |
Milk, 2 types |
1990–95 |
2 835 |
2812 (99) |
2835 (100) |
0 (0.0) |
0.016 |
0.37 |
SCOOP Report |
|
Netherlands |
Milk |
1990–93 |
1 903 |
1572 (83) |
1903 (100) |
0 (0.0) |
0.009 |
0.02 |
SCOOP Report |
|
Norway |
Milk, dairy |
1998 |
54 |
54 (100) |
54 (100) |
0 (0.0) |
0.0014 |
0.009 |
Food Control Authority |
|
Spain |
Milk |
1989–92 |
307 |
307 (100) |
307 (100) |
0 (0.0) |
0.006 |
0.046 |
SCOOP Report |
|
United Kingdom |
Milk |
1988–89 |
331 |
322 (97) |
331 (100) |
0 (0.0) |
0.01 |
0.22 |
SCOOP Report |
|
Southern USA |
Milk |
1998–2000 |
379 |
225 (59) |
313 (83) |
66 (17) |
0.40 |
14 |
FDA Laboratory B |
|
USA |
Milk, raw |
1995–2000 |
3 942 |
3778 (96) |
3937 (100) |
5 (0.1) |
0.0048 |
1.8 |
FDA Laboratory 27 |
|
USA |
Milk, finished |
1995–2000 |
273 |
259 (95) |
273 (100) |
0 (0.0) |
0.0037 |
0.17 |
FDA Laboratory 27 |
|
|
|
|
10 778 |
|
Weighted mean = |
|
0.023 |
|
|
|
Latin American |
|||||||||
|
Argentina |
Milk |
1998 |
6 |
2 (33) |
2 (33) |
4 (67) |
0.52 |
0.9 |
Universidad Nacional de Lujan (2000) |
|
Brazil |
Milk, 2 types |
1979–81 |
150 |
140 (93) |
146 (97) |
4 (2.7) |
0.03 |
1.7 |
Sabino et al. (1989) |
|
Brazil |
Milk, reconst. |
1997 |
300 |
267 (89) |
294 (98) |
6 (2.0) |
0.03 |
1 |
Oliveira et al. (1997) |
|
Brazil |
Milk and yoghurt |
1989, 1990, 1992 |
133 |
129 (97) |
133 (100) |
0 (0.0) |
0.0047 |
0.37 |
de Sylos et al. (1996) |
|
Brazil |
Milk |
1996–97 |
110 |
110 (100) |
110 (100) |
0 (0.0) |
0.012 |
0.071 |
Souza et al. (1999) |
|
Brazil |
Milk, various |
1998–99 |
50 |
46 (92) |
50 (100) |
0 (0.0) |
0.028 |
0.077 |
Prado et al. (1999) |
|
Brazil |
Milk, raw |
1992–93 |
144 |
144 (100) |
144 (100) |
0 (0.0) |
0 |
0 |
Correa et al. (1997) |
|
|
|
|
893 |
|
Weighted mean = |
0.022 |
|
|
|
|
Far Eastern |
|||||||||
|
India |
Milk, raw |
1992–93 |
504 |
415 (82) |
NR |
> 65 (> 13) |
0.20 |
3.5 |
Rajan et al. (1995) |
|
Indonesia |
Milk |
1990–93, 1999 |
342 |
> 199 (> 58) |
269 (79) |
73 (21) |
0.86 |
23 |
Research Institute for Veterinary Science (2000) |
|
Philippines |
Milk |
1997 |
91 |
|
75 (82) |
16 (18) |
0.13 |
NR |
Begino (1998) |
|
Republic of Korea |
Milk and yoghurt |
2000 |
130 |
|
130 (100) |
0 (0.0) |
0.019 |
0.124 |
Kim et al. (2000) |
|
Republic of Korea |
Milk, 2 types |
1995 |
64 |
|
64 (100) |
0 (0.0) |
0.080 |
0.28 |
Shon et al. (1996) |
|
Thailand |
Milk, 2 types |
1990–93 |
60 |
|
49 (82) |
11 (18) |
0.28 |
6.6 |
Boriboon & Suprasert (1994) |
|
|
|
|
1191 |
|
Weighted mean = |
|
0.36 |
|
|
|
Middle Eastern |
|||||||||
|
Greece |
Milk, past |
1997 |
81 |
78 (89) |
81 (100) |
0 (0.0) |
0.008 |
0.18 |
Markaki & Melissari (1997) |
|
Greece |
Milk, 2 types |
1986–87 |
150 |
146 (97) |
150 (100) |
0 (0.0) |
0.003 |
0.13 |
Karaioannoglou et al. (1989) |
|
|
|
|
231 |
|
Weighted mean = |
0.005 |
|
|
|
|
African |
|||||||||
|
Egypt |
Milk, reconstituted |
1995 |
15 |
15 (100) |
15 (100) |
0 (0.0) |
0.0018** |
NR |
El-Gohary (1995) |
* Converted from 0.015 µg/kg of dried milk
See Also:
Toxicological Abbreviations
AFLATOXIN M1 (JECFA Evaluation)