
WHO FOOD ADDITIVES SERIES: 53
First draft prepared by
Professor Fritz R. Ungemach
Institute of Pharmacology, Pharmacy and Toxicology Veterinary Faculty, University of Leipzig, Leipzig, Germany
|
Pharmacodynamic effects: beta-adrenergic receptor selectivity for stereoisomers of ractopamine |
Ractopamine hydrochloride, a beta-adrenoceptor agonist, is a phenethanolamine salt approved for use as a feed additive. The formulated products, which contain four stereoisomers of the compound, are recommended for use in finishing pigs, at a dose of 5-20 mg/kg of feed to improve feed efficiency and to increase weight gain, or at 10-20 mg/kg of feed to improve carcass leanness. The recommended dose for finishing cattle is 10-30 mg/kg of feed to improve feed efficiency and to increase weight gain and carcass leanness.
Ractopamine was previously evaluated by the Committee at its fortieth meeting (Annex 1, reference 105). At that time, the Committee concluded that residues of ractopamine appeared to have little toxic potential and the effects recorded were mainly those to be expected from a beta-adrenoceptor agonist. It might, therefore, be appropriate to assess ractopamine on the basis of a no-observed-effect level (NOEL) for pharmacological effects. However, because such a NOEL could not be determined, the Committee was not able to establish an acceptable daily intake (ADI) at that time. Before reviewing the compound again, the Committee wished to see further evidence and arguments pertaining to the perceived gaps in data on genotoxicity, carcinogenicity, pharmacology, and data in humans.
The present Committee considered additional data on the toxicity of ractopamine, including the results of studies of long-term toxicity, genotoxicity and carcinogenicity. Results of unpublished and published studies on pharmacodynamic and pharmacokinetic properties of ractopamine and other beta-adrenoceptor agonists in animals and humans were also considered. All the pivotal studies were carried out according to appropriate standards for protocol and conduct.
Ractopamine has been approved for use in swine in 22 countries. In 2003, ractopamine was in approved for use in cattle in the USA. In the European Union, use of beta-adrenoceptor agonists in food-producing animals has been banned since 1996, except for tocolysis in mares and cows and bronchodilation in horses.
The chemical structure of ractopamine hydrochloride is shown in Figure 1.
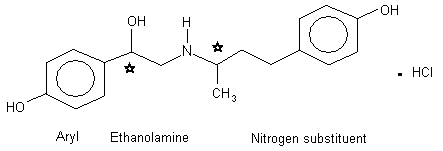
Figure 1. Chemical structure of ractopamine hydrochloride
Chiral carbons are denoted by asterisks
Ractopamine hydrochloride exists in two diastereomeric forms resulting from the presence of two chiral carbons. The commercial preparation is a racemic mixture of the four stereoisomers RR, RS, SR, and SS with a minimal purity of 96%. The RR isomer (butopamine) is a potent cardiostimulant in humans and is likely the functional and most active stereoisomer mediating the growth response in pigs, particularly through the beta2ARbeta2-adrenoceptor (Mills et al., 2003a, 2003b).
At its fortieth meeting (Annex 1, reference 104, 105), the Committee considered studies in several animal species (rats, dogs, swine), which indicated that [14C]ractopamine was rapidly absorbed after oral administration. Peak plasma concentrations were reached 0.5-2 h after dosing. The elimination half-life was about 6-7 h.
In a bioavailability study that complied with good laboratory practice (GLP), groups of five male and five female rats were given [14C]ractopamine as a single oral dose at 0.5, 2.0, or 20 mg/kg bw by gavage. The amount of radiolabel was quantified in samples of plasma and whole blood collected for 24 h after dosing. Comparison of the area under the curve (AUC) of concentration-time for plasma and whole blood indicated that the bioavailability of [14C]ractopamine was proportional to dose for males and females at doses up to 2.0 mg/kg bw. Increasing the dose to 20 mg/kg bw resulted in an increase in AUC versus dose in males and, to a more pronounced degree, in females. The absolute bioavailability of [14C]ractopamine in rats cannot be determined from the results of this study since [14C]ractopamine was not administered intravenously for comparison of oral and intravenous AUC values (Williams et al., 1987).
In studies in rats, the mean recovery of an orally administered dose of [14C]ractopamine at 2.85 or 7.7 mg/kg bw was 29% and 59% in the urine and bile, respectively, during the 24 after dosing, indicating an absorption of about 88%. Absorption and excretion of radioactivity was rapid; 55% of the administered radiolabel was excreted into bile during the first 8-h collection period (Smith & Paulson, 1994; Smith et al., 1995).
The bioavailability of ractopamine can be estimated by the urinary excretion of radioactivity determined in balance-excretion studies in dogs, monkeys, pigs, and cattle given [14C]ractopamine. After a single oral dose of [14C]ractopamine at 0.125 mg/kg bw, a total of 79.4% and 69.8% of the radiolabel was recovered, with an average of 55.2% and 45.2% in the urine and 24.2% and 24.6% in the faeces in dogs and monkeys, respectively, during the 72-h collection period after dosing (Williams, 1987a). When pigs fed unlabelled ractopamine hydrochloride at a dose of 20 mg/kg of feed were given a single oral dose of [14C]ractopamine, around 88% of the radiolabel was excreted in the urine within 7 days. A further 9% was recovered in the faeces over this time (Dalidowicz & Babbit, 1986; Dalidowicz et al., 1986). In a balance-excretion study, two crossbred steers were fed for 8 days with unlabelled ractopamine at a concentration of 30 mg/kg of feed and then given [14C]ractopamine in a single oral dose at 0.67 mg/kg bw. The administered dose of radiolabelled ractopamine was excreted almost quantitatively, with about 46% being recovered in the urine and 52% in the faeces within 10 days (Dalidowicz & Thomson, 1989).
The data on urinary excretion of radiolabel after oral administration of [14C]ractopamine suggest that this beta-agonist is rapidly and well absorbed from gastrointestinal tract, with a fraction of the administered dose of at least 45% (cattle, monkeys) and up to >85% (swine) reaching the systemic circulation.
Ractopamine is rapidly excreted in dogs, monkeys, and pigs given a single oral dose of [14C]ractopamine. The bulk of the radiolabel (85 - > 95% of the excreted dose) was cleared in the first 24 h after dosing, with urine being the major route of excretion. In rats, 59% of the administered dose was excreted in the bile and only 29% in the urine (Dalidowicz & Babbit, 1986; Dalidowicz et al., 1986, Smith, 1998; Williams, 1987a). In cattle, the bulk of the administered dose of radiolabelled ractopamine was excreted in the first 4 days (93%), while 74% was eliminated during the first 2 days after dosing (Dalidowicz & Thomson, 1989). In another study, four bull calves were given feed containing unlabelled ractopamine hydrochloride at a concentration providing a daily dose of 0.1 mg/kg bw for 17 days. Urine samples were collected during the treatment period and 14 days after drug withdrawal. An enzyme-linked immunosorbent assay (ELISA) with confirmation by liquid chromatography-mass spectrometry-mass spectrometry (LC-MS/MS) was used to analyse the urine before and after deconjugation. Ractopamine residues were excreted mainly in the form of glucuronides. High concentrations of ractopamine residues were found throughout the treatment period (44-473ng/ml; LC-MS/MS data) and remained present for several days after removal of the drug from the diet. Ractopamine residues were no longer detectable (limit of detection, 2 ng/ml) at 2 weeks after withdrawal (Elliot at al., 1998).
The tissue distribution of ractopamine is extensive and occurs rapidly, as judged by studies using radiolabelled ractopamine in swine and cattle. Three groups of three swine were fed [14C]ractopamine at a concentration of 30 mg/kg of feed for 4, 7, or 10 days. About 12 h after the last feeding, total concentrations of radioactive residues were measured in muscle, fat, liver, and kidney. No detectable residues were measured in the fat, suggesting that the drug is of low lipophilicity. Statistical analyses showed that total residues reached steady-state levels 4 days after initiation of feeding with ractopamine. Ranges of concentrations found in tissues during days 4-10 of feeding were: liver, 0.254-0.424 mg of ractopamine equivalents/kg; kidneys, 0.466-0.655 mg of ractopamine equivalents/kg; and muscle, 0.019-0.024mg of ractopamine equivalents/kg. Non-extractable residues also reached steady-state levels after 4 days and represented 26-29% and 15-16% of the total radioactive residue in liver and kidney, respectively (Dalidowicz et al., 1985). Upon withdrawal of feed containing ractopamine at a concentration of 20 mg/kg of feed, after the 7-day feeding regimen, residue concentrations in swine declined from 0.106 mg/kg to 0.056 mg/kg in liver, and from 0.116 mg/kg to 0.036 mg/kg in kidney after 24 and 72h, respectively. Simultaneously, the percentage of parent substance decreased from 14.1% to 3.6% of total residue in liver and from 27.5% to 3% in kidney (for review see Smith, 1998).
The length of time required for radioactive residues to reach steady state was determined in groups of three cattle receiving [14C]ractopamine at 1.01 mg/kg bw per day for 4,7, and 10 days via rumen fistula. The concentrations of 14C-labelled residue were determined in liver, kidney, muscle, and fat 12 h after the last dose. No detectable residues were measured in the muscle or fat. Steady-state concentrations were reached in 4 days in kidney (0.4-0.5 mg of ractopamine equivalents/kg) and 7 days in liver (0.59-0.75 mg of ractopamine equivalents/kg) after initiating administration of [14C]ractopamine. Non-extractable residues also reached steady-state levels after 4 days and represented 4.3-5.1% and 3.6-8.6% of the total radioactive residue in liver and kidney, respectively. The amount of parent ractopamine represented a mean of 12.7% and 14.2% of total radioactivity in liver and kidney, respectively, and was statistically the same for all dosing periods (Dalidowicz & Thomson, 1988).
Studies previously evaluated by the Committee revealed three major metabolites, identified as monoglucuronides of ractopamine (Annex 1, reference 105; Dalidowicz, 1986a, 1986b; Dalidowicz, 1987).
In studies in rats, dogs, pigs, and cattle fed [14C]ractopamine, a fourth metabolite was identified as a glucuronic acid diconjugate. The conjugation of the hydroxyl groups in both the aromatic ring attached to the carbinol and the phenol attached to the nitrogen substituent was not stereospecific.
After a withdrawal of 6 h (rats, dogs) or 12h (swine, cattle), unchanged ractopamine represented 40, 14, 52, and 13-16% of the total extractable and identifiable residues in the rat, dog, pig, and cattle livers, respectively, and 21, 29, 28-30, and 14% in the kidneys, respectively. After a withdrawal of 24 h and 72 h, parent ractopamine represented 14.1% and 3.6% in liver, and 27.5% and 3% of total residues in kidney, respectively, in swine. The remaining residue was found to comprise conjugates of ractopamine. The chromatographic profiles of the 14C-labelled residue extracts of rat, dog, pig, and cattle liver were qualitatively similar. The laboratory animals had generally a higher percentage of metabolites as residues. Studies in rats and dogs showed that urine from animals dosed with [14C]ractopamine contained the same four glucuronide metabolites of ractopamine as in pigs. It is concluded that the dogs and rats used in the toxicological studies were exposed to the same metabolites as those found in the edible tissues of pigs and cattle (Dalidowicz, 1986a, 1986b; Dalidowicz & Babbit, 1986; Dalidowicz, 1987; Dalidowicz, 1989; Smith, 1998).
In the bile of rats dosed orally with [14C]ractopamine, at least seven different crude metabolite fractions were partitioned chromatographically. Four of the crude metabolite fractions representing 76% of biliary radioactivity were isolated and identified with a sulfate-ester/glucuronic acid diconjugate of ractopamine as the main metabolite (46% of total biliary radioactivity). A further 6% of radioactivity was identified as a monosulfate conjugate and 25% as monoglucuronides of ractopamine. The site of sulfation was established at the C-10’ phenol (aromatic ring attached to carbinol). The sulfate conjugation was not stereospecific. The major site of glucuronidation was the C-10 phenol (phenol attached to the nitrogen substituent) (Smith et al., 1995).
In urine, only a minor fraction of radioactivity recovered was parent ractopamine. Swine excreted about 4-16% of the parent compound in the urine after a single oral dose of ractopamine. After repeated doses, the amount of unchanged drug increased to 36-85% of total radioactivity in the urine collected on day 4 of a 4-day dosing regimen. In rats injected with [14C]ractopamine at 9 mg/kg bw intraperitoneally, parent drug represented 22.6% of total urinary radioactivity while only 1.9% of radioactivity was associated with unchanged ractopamine after an oral dose of 9.9 mg/kg bw. The greater proportion of parent drug in the urine after parenteral administration than after oral administration suggests that liver and intestine play an important role in the biotransformation of ractopamine after oral administration. Therefore, although well absorbed from the gastrointestinal tract, the systemic availability of parent ractopamine is reduced, owing to a significant first-pass metabolism (for review, see Smith, 1998).
In a study to define the pharmacological response of humans to ractopamine, the pharmacokinetics of ractopamine were determined in six healthy male human volunteers given a single oral dose of 40 mg of ractopamine hydrochloride. Blood plasma and urine samples were collected 24 after dosing and analysed by high-performance liquid chromatography (HPLC) with electrochemical detection. Ractopamine was rapidly absorbed, with the mean peak plasma concentration of 41.2 ng/ml occurring after an average of 0.6 h. The mean half-life was 3.94 h. Ractopamine was no longer detected in plasma 24 h after dosing, except at very low concentrations in one volunteer. Only about 2% of the total administered dose was excreted in the urine as unchanged ractopamine by 24 after dosing. After treatment of urine samples with beta-glucuronidase and sulfatase for hydrolysis of ractopamine conjugates, urine excreted 0-24 h after dosing contained an average of 45.7% of the administered dose as free ractopamine, indicating that <5% of total ractopamine excreted represented the parent drug. The urinary metabolites were monoglucuronide and monosulfate conjugates, with ractopamine monosulfate being the major metabolite present. Of the total administered dose that was excreted in the urine, about 72% was excreted within 6 h after dosing. The results confirmed that ractopamine was extensively and rapidly absorbed, with an oral bioavailability of a minimum of 45.7% of the administered dose. Owing to rapid metabolism, the orally administered dose of 40mg produced low systemic concentrations of parent ractopamine, suggesting significant first-pass metabolism of the drug. The available data strongly suggest that disposition, metabolism, and excretion of ractopamine in humans are consistent with the pharmacokinetics and biotransformation observed in animals and for other phenolic, catecholic, and resorcinolic phenethanolamine beta-adrenergic agents (Hunt, 1994; Smith & Rodewald, 1994; Smith, 1998).
Phenethanolamine beta-adrenergic agonists share common structural characteristics that modulate their biological activity at beta-adrenoceptors, and their pharmacokinetic properties. For a beta-agonist to elicit biological activity, it must have a six-membered aromatic ring attached to the beta-carbon of the carbinol ("aryl") with substituents (hydroxyl groups or halogens), an hydroxyl group bonded to the beta-carbon in the levorotatory form, a positively-charged nitrogen in the ethylamine side-chain and a bulky substituent at the aliphatic nitrogen to confer specificity at the beta-adrenoceptor (Figure 1).
According to their substitutions at the aryl ring, the beta-agonists can be assigned to five different groups: the hydroxylated compounds (1) catecholamines (norepinephrine, epinephrine, isoproterenol, dobutamine); (2) resorcinols (fenoterol, terbutaline); (3) saligenins (salbutamol, salmeterol); and (4) simple phenols (ractopamine, ritodrine); and (5) the halogenated haloanilines (clenbuterol, mabuterol). Free bases of beta-agonists are lipophilic, with halogen-substituted compounds having increased lipophilicity of the aromatic portion in the free base form relative to hydroxylated beta-agonists. Because aliphatic amines are present on beta-agonists, these compounds have an alkaline pKa ranging from 8.5 (fenoterol) to 10.1 (terbutaline). The pKa for ractopamine is 9.4. At physiological pH, the vast majority of molecules of the free (unprotonated) base form of all beta-agonists will become ionized by protonation (e.g. about 99% of ractopamine will be positively charged at pH 7.4). Thus, the dissociation constant of the aliphatic nitrogen common to all beta-agonists dictates that these molecules are generally not lipophilic in the organism, with the notable exception of salmeterol. At the receptor only, the ionized form of beta-agonists would have biological activity (Smith, 1998).
(b) Absorption, distribution and excretion
beta-Agonists other than catecholamines are generally rapidly and extensively absorbed from the gastrointestinal tract. Peak plasma concentrations occur within 1 to 3 h after oral administration in humans, and a similar pattern is observed after oral administration to laboratory animals and farm animals. The apparent extent of absorption of beta-agonists, as assessed by urinary excretion of parent substance and metabolites, is shown in Table 1.
Table 1. Absorption and excretion of beta-agonists in various species
|
Compound |
Species |
Excretion and absortiona |
Bioavailability (% of parent drug) |
Half-lives in plasma (h) |
||
|
Time (h) |
Percentage of dose (%) |
Plasmab |
Urinec |
|||
|
Clenbuterol |
Human |
96 |
80 (U) |
75 (oral) |
66.4 (oral) |
30-33.9 |
|
Dog |
72 |
74 (U) |
33 (oral) |
41.1 (oral) |
— |
|
|
Rabbit |
96 |
92 (U) |
39 (oral) |
67 (oral) |
9 |
|
|
Rat |
72 |
86 (U + F) |
45 (oral) |
70 (oral) |
25.4-30 |
|
|
Ractopamine |
Human |
24 |
>46 (U) |
— |
2 (oral) |
3.9 |
|
Dog |
72 |
76 (U + F) |
— |
— |
6 |
|
|
Rat |
24 |
88 (U + B) |
— |
1.9 (oral) |
7 |
|
|
23 (intraperitoneal) |
||||||
|
Salbutamol |
Human |
72 |
76 (U) |
20 (oral) |
<30 (oral) |
2-3.9 |
|
>50 (intravenous) |
50-64 (intravenous) |
|||||
|
Dog |
24 |
72 (U) |
— |
— |
3 |
|
|
Rabbit |
48 |
63 (U) |
— |
— |
0.68-1.5 |
|
|
Rat |
48 |
56 (U) |
— |
— |
— |
|
|
Terbutaline |
Human |
72 |
40 (U) |
15 (oral) |
6 (oral) |
11-18 |
|
>50 (intravenous) |
60 (intravenous) |
|||||
|
Dog |
96 |
75 (U) |
50 (oral) |
72 (oral) |
— |
|
|
Rat |
72 |
44 (U) |
— |
8 (oral) |
— |
|
|
48 (intravenous ) |
||||||
|
From Morgan (1990); Smith (1998); Annex 1, references 105, 126. |
|
|
a |
Apparent absorption expressed as a percentage of the orally administered dose excreted in urine (U), bile (B) or faeces (F) at the indicated time (h) after dosing. |
|
b |
Percentage of radioactivity in plasma represented by the parent drug. Generally measured at peak concentration in the plasma or serum. |
|
c |
Percentage of radioactivity in urine represented by the parent drug. |
Although most beta-agonists are well absorbed, the unchanged molecules are not equally available to target tissues after absorption. Published data indicate greatest bioavailability of unchanged drug for halogenated phenethanolamines. In general, the quantity of parent clenbuterol in plasma or urine is 40-75% after oral administration, while for resorcinols, saligenins, and phenols concentrations of unchanged drug account for 30% or less of the urinary concentrations, with the exception of terbutaline in dogs (Table 1). In contrast, the proportion of parent drug present in the urine of animals after intravenous administration is greater than that after oral administration, regardless of the compound. These data suggest that there is significant first-pass metabolism of beta-agonists in the intestine and liver after oral administration.
Apart from the lipid-soluble agents, such as clenbuterol, the binding of beta-agonists to plasma proteins is low or negligible. All beta-agonists are widely distributed to the tissues. Radioactive residues are generally greatest in excretory organs. At physiological pH, the vast majority of molecules of any given beta-agonist exist in a protonated form that will not partition into adipose tissue. The higher lipophilicity of the free base of clenbuterol will thus not cause a higher accumulation in fat relative to hydroxylated beta-agonists. Residues of ractopamine in liver and kidney of pigs declined more rapidly when compared with residue depletion of clenbuterol from liver and kidneys of calves. After 48 h of withdrawal for both compounds, the total residues in livers of clenbuterol-treated calves were 72 times greater than the total residues in ractopamine-treated swine, and 123 times greater than in kidney. The second difference is the greater percentage of parent compound remaining in each tissue. After 48 h of withdrawal, unchanged clenbuterol represented 44% and 63% of the total radioactive residues in liver and kidneys of calves, respectively, while the amount of parent ractopamine remaining in livers and kidneys of swine was 5.5 and 16.7%, respectively. Laboratory animals generally had a higher percentage of beta-agonist metabolites as residues when compared with farm animals.
For most compounds and species, excretion of radioactivity after intravenous or oral administration of radiolabelled beta-agonist was predominantly via the urine and nearing completion by 48 h. Biliary excretion of a beta-agonist seems to be species-specific for any given compound. In all species, with the exception of rabbits, halogenated beta-agonists have significantly longer half-lives in plasma than do the beta-agonists bearing hydroxyl groups at the aromatic ring (Table 1).
Not all beta-agonists are metabolized in the same manner. The pattern of chemical substitution at the aromatic ring is the major factor in determining the route of metabolism and greatly influences the longevity of beta-agonists within mammalian tissues and their efficacy at the receptor (Table 2).
Table 2. Aromatic substitutions and biotransformation pathways of beta-agonists
|
Group |
Example |
Aromatic Substitutions |
COMTa |
Conjugationb |
Oxidation |
|
Catecholamines |
Isoproterenol |
3-,4-OH |
Yes |
Yes |
Yes |
|
Resorcinols |
Terbutaline |
3-,5-OH |
No |
Yes |
No |
|
Saligenins |
Salbutamol |
4-OH; |
No |
Yes |
No |
|
Phenols |
Ractopamine |
4-OH |
No |
Yes |
No |
|
Haloanilines |
Clenbuterol |
3-,5-Cl; |
No |
Yes |
Yes |
Modified from Smith (1998).
a COMT, Catechol O-methyltransferase.
b Glucuronidation and/or sulfation.
Catechols are rapidly deactivated by methylation of the 3-hydroxyl group of the aromatic ring by catechol O-methyltransferase (COMT) severely limiting their effectiveness after oral administration. Other patterns of aromatic hydroxylation are resistant to deactivation by COMT. Thus, resorcinolic, saligenic, and phenolic beta-agonists are not substrates for COMT. However, these beta-agonists are rapidly deactivated in liver and intestine by conjugative biotransformation enzymes. In laboratory and farm animals as well as in humans, the pathways of biotransformation of these hydroxylated compounds are exclusively through glucuronidation and sulfation. Studies with laboratory animals have established the involvement of intestine and liver in the presystemic elimination of hydroxylated beta-agonists such as fenoterol, ritodrine, salbutamol and terbutaline with a higher extraction ratio by the intestine when compared with liver. Oxidative metabolism of resorcinols, phenols, and saligenins other than salmeterol has not been confirmed in any species.
beta-Agonists with an aromatic ring containing halogen atoms substituting for hydroxyl groups (e.g. clenbuterol or mabuterol) are resistant to rapid metabolic degradation by conjugative biotransformation enzymes. Clenbuterol is metabolized through oxidative pathways, forming a range of oxidative metabolites with 4-amino-3,5-dichloromandelic acid, 3-amino-3,5-dichlorobenzoic acid, and 4-amino-3,5-dichlorohippuric acid being the major components in laboratory animals, cattle, and humans. The conjugation pathways reported for parent clenbuterol with glucuronidation of the beta-hydroxyl group and the aliphatic nitrogen, sulfation of the aromatic amine, and unusual ethylation of the benzylic hydroxyl seem to be unique for clenbuterol.
Because beta-agonists exist as racemates of stereoisomers, they are susceptible to stereospecific or stereoselective biotransformation. Stereospecific metabolism has been demonstrated for glucuronidation of only the RR and RS stereoisomers of ractopamine at the phenol attached to the beta-carbon. Stereoselective biotransformation, which is more common than stereospecific biotransformation, has not been extensively studied for beta-agonists other than terbutaline and salbutamol.
Overall, the data suggest that the metabolism of any given beta-agonist is similar in all the species studied, and that the major differences are quantitative rather than qualitative. In general, beta-agonists with halogenated aromatic ring systems (clenbuterol) are metabolized by oxidative and conjugative pathways and have long plasma half-lives, while beta-agonists with hydroxylated aromatic rings (ractopamine) are metabolized solely by conjugation and have relatively short half-lives (Table 1) (Smith, 1998; Annex 1, references 105, 126).
The differences in the pharmacokinetics of beta-agonists suggest that residues of halogenated phenethanolamines contain greater amounts of parent substance, higher oral bioavailability, relatively slow rates of elimination, and higher oral potencies in humans when compared with residues of hydroxylated beta-agonists.
A series of short-term studies of toxicity with orally administered ractopamine in mice, rats, dogs, and monkeys was evaluated by the Committee at its fortieth meeting (Annex 1, reference 105). These studies are summarized in Table 3.
Table 3. Summary of previously evaluated short-term studies of toxicity with ractopamine
|
Species (strain) |
Duration (route) |
Dose (mg/kg bw per day) |
NOEL (mg/kg bw per day) |
Critical effects |
|
Mouse (B6C3F1), 10 of each sex per group |
3 months (oral) |
0, 25, 125, or 1250 |
Not identified |
Significant increase in weight gain for male at the intermediate and highest doses. Males and females at the highest dose showed increased heart weight. Decreased testicular weight was seen in males at all doses. |
|
Rat (Fisher 344), 20 of each sex per group |
90-days (oral) |
Males: |
Males |
Decrease in uterine and spleen weight, and slight to moderate cytoplasmic changes in brown fat at intermediate and highest doses. Increased food consumption and decreased food utilization at highest dose. |
|
Females: |
Females |
|||
|
Monkeys (rhesus), two of each sex per group |
6 weeks (oral) |
0, 0.25, 0.5, or 4 |
Not established due to ongoing evaluation of heart and lung beta-adrenergic receptor binding |
Daily tachycardia, maximal at 30 min after dosing, remaining elevated for entire dosing period, without the nocturnal bradycardia seen in all other groups |
|
Monkeys (rhesus), three of each sex per group |
90-days (oral) |
0 or 0.125 |
0.125 |
No treatment-related effects reported |
|
Monkey (rhesus), Two of each sex per group |
18 days (inhalation) |
0, 0.38, 1.69, or 23.8 ractopamine hydrochloride/m3 |
Not identified |
Increased day and night heart rate in all groups |
From Annex 1, reference 105.
In addition, a study of target animal safety and drug tolerance with ractopamine in swine was submitted. Groups of four male and four female crossbred swine were given access ad libitum to a complete diet containing ractopamine hydrochloride at 0, 0.002, 0.01, or 0.05%, equal to time-weighted average daily doses (both sexes combined) of about 0, 0.6, 3, or 15 mg/kg bw for 56 days during the finishing period. Daily clinical observations, observations for mortality, haematological measurements, body weight, feed consumption, gross necroscopy, and histopathology were evaluated to assess potential toxicity in swine.
Pigs tolerated ractopamine at dietary concentrations equal to 15 mg/kg bw per day without physical signs of toxicity. A mild decrease in erythrocyte number, haemoglobin concentration, erythrocyte volume fraction, and serum urea nitrogen was observed at the intermediate and highest doses. A slight increase in serum concentration of creatinine occurred in all treated groups and was probably associated with a treatment-related increase in carcass muscle mass. No compound-related gross lesions or effects on organ weights were observed, except a decrease in the relative organ weight of the thyroid. Microscopically, the only treatment-related alteration was an increase in glycogen in periportal hepatocytes of a few pigs at the intermediate and highest doses. A NOEL was not identified in this study (Williams, 1987c).
Additional studies submitted on short-term toxicity of ractopamine in fish, other aquatic organisms (Williams et al., 1986d-f) and birds (Williams et al., 1986a-c), on ocular irritancy in rabbits (Williams et al., 1984b), and sensitizing potential of compound injected intradermally in albino Hartley guinea-pigs (Mercier, 1992) were considered to be not appropriate for the assessment of the safety of residues of ractopamine.
Mice
In a lifespan study that complied with GLP, the oncogenic potential of ractopamine administered in the diet was assessed. Groups of 60 male and 60 female CD-1 mice were given diets containing 0, 0.02%, 0.1% or 0.6% ractopamine hydrochloride, approximately equal to an average daily dose of 0, 25, 130, or 840 mg/kg bw per day in males, and 0, 35, 175, or 1085 mg/kg bw per day in females, for 21 months.
Data on plasma analysis confirmed that dietary administration resulted in systemic exposure to ractopamine that was roughly proportional to dose, and that was similar for males and females and similar after 6, 12 and 18 months of treatment. Mean total concentrations of ractopamine ranged up to 17.8 µg/ml for the group receiving ractopamine at 0.6% in the diet. Parent ractopamine represented <1% of total ractopamine at any time-point.
The mice were observed daily for survival and a detailed clinical evaluation was performed at weekly intervals; body weight and food consumption were measured every 2 weeks. Blood samples for clinical chemistry and haematology were collected from mice killed in a moribund condition at interim and from all survivors at termination, before necropsy. Gross and microscopic anatomical appearance was evaluated for all animals. All tissues were examined histologically.
Survival to termination was 62%, 75%, 63%, and 25% for males, and 72%, 58%, 63%, and 32% forfemales at 0, 0.02%, 0.1%, and 0.6%, respectively. These results indicate that the highest dietary concentration exceeded the maximum tolerated for mice of both sexes. The increased mortality at this dose was attributable to an enhanced severity of cardiomyopathy. In both males and females at 0.1%, the histopathological severity of cardiomyopathy was also slightly increased. Dose-related decreased body weight, body-weight gain, and efficiency of food utilization, coupled with increased food consumption were most apparent in males at the highest dose, and were expected findings after exposure to high levels of a sympathomimetic amine with thermogenic properties. No further clinical abnormalities were reported that were unique to treatment with ractopamine. Slight differences in haematology (slight dose-related increases in erythrocyte counts, haemoglobin, and erythrocyte volume fraction) and clinical chemistry parameters (mild dose-related increases in urea nitrogen) in the treated groups had no clinical or morphological correlate and were not clinically important. No trends in incidences of neoplasms in either sex were observed, except for a dose-dependent increase in the incidence of uterine leiomyomas in 1 out of 60, 5 out of 60, 8 out of 60, and 10 out of 60 mice at 0, 0.02%, 0.1%, and 0.6%, respectively. Treatment-related non-neoplastic proliferative lesions involved the smooth muscle of the female genital tract, with non-dose-related hyperplasia of the smooth muscle tunic of the uterus and smooth muscle proliferation at the costo-uterine ligament in some ovarian sections. All treatment-related effects of long-term toxicity, including tumorigenicity, observed were attributable to the beta-adrenergic potency of ractopamine. The NOEL was 25 mg/kg bw per day for males on the basis of body weight and food consumption changes (Williams, 1998a). However, owing to the dose-dependent increased incidence of uterine leiomyomas observed at all doses, no NOEL could be identified for females. The NOEL was approached by calculation of a benchmark dose to define a point of departure from the background incidence of leiomyoma. Given a benchmark dose of 320 mg/kg bw per day based on an excess leiomyoma incidence of 10% above control group, the incidence observed was still considered to be an effect level and an increased safety factor of 300 was applied for establishment of an acute reference dose (RfD) equivalent to an ADI (Food & Drug Administration, 1999). A benchmark dose was calculated as 201 mg/kg bw per day based on an excess incidence of leiomyoma of 5% above that in controls and a 95% confidence limit.
Rats
In a GLP-compliant study to assess the oncogenic potential of ractopamine, groups of 60 male and 60 female Fischer 344 rats were fed diets containing ractopamine for 24 months. The time-weighted average doses of ractopamine hydrochloride were 0, 2, 60, or 200 mg/kg bw per day for males and 0, 2, 60, 200, or 400 mg/kg bw per day for females.
Data on plasma analysis confirmed that dietary administration resulted in systemic exposure to ractopamine that was proportional to dose, was similar for males and females and was similar after 6, 12 and 18 months of treatment. Mean total concentrations of ractopamine ranged up to 7.8 µg/ml at 200 mg/kg bw per day (both sexes combined) and 19.1 µg/ml for the females at 400 mg/kg bw per day. Parent ractopamine represented <1% of total ractopamine at any time-point.
The rats were observed daily for survival and a detailed clinical evaluation was performed at weekly intervals; body weight and food consumption were measured every 2 weeks. Blood samples for clinical chemistry and haematology were collected from rats killed in a moribund condition at interim and from all survivors at termination. Necropsies were performed on all rats killed at interim and sacrificed at the end of the study, and tissues were submitted for histological examination.
Survival was significantly increased for the males and females at the highest dose tested. Survival to termination was 23%, 30%, 18%, and 68% for males, and 52%, 63%, 75%, and 63% for females at 0, 2, 60, and 200 mg/kg bw per day, respectively, and 78% for females at 400 mg/kg bw per day. Increased survival at the highest doses was associated with fewer deaths from chronic nephropathy in males and from common neoplasms in both sexes, and decreased mean body weight (which was about 20% less than that of controls). Decreased body weight and efficiency of food utilization, coupled with increased food consumption in these groups were expected findings after exposure to high levels of a beta-adrenergic agonist with thermogenic and repartitioning properties. No further clinical abnormalities were reported that were unique to treatment with ractopamine. No adverse compound-related adverse clinical observations were reported. Slight differences in haematology (increase in total erythrocyte count, haemoglobin, erythrocyte volume fraction) and clinical chemistry parameters (increases in concentrations of potassium, triglycerides, and cholesterol, decreases in urea nitrogen) at the highest dose were consistent with previous observations in rats treated with ractopamine and were not clinically important. Treatment-related morphological changes included an increased incidence in slight to moderate cardiomyopathy in females at the highest dose and in males at 60 and 200 mg/kg bw per day, with increased fatalities being limited to males at 60 mg/kg bw per day. Mucous acinar hypertrophy of the submandibular salivary gland was noted in males at the highest dose and in females at 200 and 400 mg/kg bw per day. Dose-related hyperplasia of the costo-uterine smooth muscle was observed in females at 0 (0 out of 60), 2 (0 out of 60), 60 (3 out of 60), 200 (17 out of 60), and 400 mg/kg bw per day (25 out of 60). No significant trends for incidences of neoplasms in males were observed, except the slightly increased incidence of testicular interstitial cell tumours. As Leydig cell tumours are common in Fischer 344 rats, the incidence of these tumours was not significantly increased in males treated with ractopamine. The only tumour with increased incidence in females was the pharmacologically mediated costo-uterine leiomyoma in 6 and 27 out of 60 rats at 200 and 400 mg/kg per day, respectively, while no such tumours were noted at lower doses or in controls. The NOEL for costo-uterine leiomyoma formation was 60 mg/kg bw per day. The NOEL for enhanced cardiomyopathy with increased fatality in males was 2 mg/kg bw per day (Williams, 1998b).
The Committee concluded that all treatment-related effects observed in the long-term studies of toxicity in mice and rats, including severe cardiomyopathy and tumorigenicity, were attributable to the beta-adrenergic potency of ractopamine.
The induction of mesovarian leiomyomas in mice and rats also appears to be a general feature of beta-adrenergic stimulants, as shown for various other beta2-agonists such as salbutamol and terbutaline. Evidence for the induction of this rare benign tumour in mice and rats as a function of prolonged stimulation of beta2-adrenergic receptors in mesovarian smooth muscle was provided by the fact that co-administration of the beta-adrenergic blocker propranolol prevented their development, while in control animals propranolol did not block the spontaneous occurrence of these tumours. Studies of carcinogenicity with other beta-agonists also reported no tumorigenic effects in mice, rats, and hamsters, except formation of benign mesovarian and uterine leiomyomas and, in the case of metaproterenol in mice, benign hepatic adenomas (Cantox, 2000; Jack et al., 1983; Gibson et al., 1987; Gopinath & Gibson, 1987). Ractopamine, like other beta-adrenerigic sympathomimetics, is therefore not a direct carcinogen and the induction of leiomyomas is considered to be a non-genotoxic event with a threshold, similar to other toxicological end-points (Food & Drug Administration, 1999; Gibson et al., 1987).
Monkeys
In a 1-year study, which complied with GLP, the toxicity of ractopamine was evaluated in a species (not rodents) whose cardiovascular responsiveness to beta-adrenoceptor beta-agonists approximates that of humans. Groups of four male and four female rhesus monkeys (there were only three males at the lowest dose owing to injury of one monkey before the beginning of treatment) were given ractopamine hydrochloride at doses of 0, 0.125, 0.5, or 4 mg/kg bw dissolved in purified water in a volume of 1 ml/kg bw once daily by nasogastric gavage.
Toxicokinetic analysis of plasma concentrations confirmed that nasogastric administration resulted in systemic exposure to ractopamine that was similar for males and females and in similar daily plasma concentration profiles after 0.5, 3, 6, 9, and 12 months of treatment. At 4 mg/kg bw per day, the time to maximum plasma concentration was about 1 h.
Peak plasma concentrations ranged from 44 ng/ml on day 190 to 59 ng/ml on day 15. At 0.125 and 0.5 mg/kg bw per day, the plasma concentrations of ractopamine were below the limit of detection (<5 ng/ml) of the assay.
All monkeys survived the treatment period. The treatment had no significant toxicological effect on food consumption, ophthalmic and daily physical examination, haematology, clinical chemistry, urine analysis, and gross and microscopic pathology. A significant 15-20% increase in body weight occurred in the animals at the highest dose and was attributed to the known repartitioning effect of this class of compounds.
The most striking effect of treatment with ractopamine was cardiostimulation, an expected pharmacological effect of a beta-adrenergic agonist. The degree and extent of cardiostimulation occurred in a dose-dependent manner and was significantly increased at 0.5 and 4 mg/kg bw per day compared with controls. The increased heart rate was maximal during the first 4 h after dosing, and in weeks 5-52 ranged from 185 to 199 beats per min for monkeys at 4 mg/kg bw per day compared with 120 to 133 beats per min for the control animals. At 0.5 mg/kg bw per day, heart rates were an average of about 20 beats per min above those of the controls. A mean increase of 8 beats per min at the lowest dose was not significantly different to means for the controls. Resting or night-time heart rates were also significantly increased compared with those for the controls. There was no accommodation to the increased heart rates during the 1-year period of treatment. The electrocardiogram wave forms showed decreased P-R and Q-T intervals. At 0.125 mg/kg bw per day, there was no significant effect on cardiac rate, conduction or repolarization. The observed decrease in heart weight relative to body weight in females at the intermediate and highest doses and a similar trend in males at the corresponding doses was not expected under conditions of prolonged increased heart rates. No treatment-related changes occurred in haematology, clinical chemistry and urine analysis parameters (Williams, 1993).
In conclusion, significant treatment-related effects on heart weight and heart rate were observed in monkeys treated with ractopamine at 0.5 and 4.0 mg/kg bw per day. The increase in body weight was present only in monkeys at 4 mg/kg bw per day. The effect on heart rate occurred in the absence of cutaneous erythema and other clinical signs or microscopic lesions of cardiotoxicity, thus suggesting that cardiovascular response in monkeys differs from that in dogs, with the dog being more sensitive to vasodilation, hypotension, and reflex cardiostimulation (Williams 1987a; Sarazan etal., 1993; Annex 1, reference 105). However, no direct measurements of inotropic response, peripheral vascular resistance or plasma catecholamine concentrations were conducted in this study; data on these parameters would have allowed direct assessment of ractopamine-mediated peripheral vasodilation through beta2-adrenoceptors and consecutive reflex sympathetic chronotropic and inotropic activation of the heart. In another study on acute cardiovascular effects of ractopamine in monkeys, intravenous infusion of ractopamine at 35 µg/kg bw gradually decreased total peripheral vascular resistance by about 30% during infusion (Williams & Stone, 1987c) (section 2.2.5(b)).
beta-Adrenoceptor binding assays were conducted using the nonselective radioligand [3H]dihydroalprenolol in membranes prepared from heart (left ventricle) and lung tissue samples taken at necropsy. The receptor density (Bmax) and affinity (Kd) were determined by Rosenthal-Scatchard analysis using linear regression. Non-specific radioligand binding was assessed by displacement studies with (-)-propranolol. The number and affinity (mean concentration for all doses: 0.61 nmol/l) and number of heart beta-adrenoceptors were not affected by prolonged treatment with ractopamine at any dose. The lung beta-adrenoceptor density significantly decreased at the highest dose in both sexes, being reduced by 23% when compared with controls (mean of 319.9 fmol/mg of protein) while receptor affinity (mean concentration for all doses: 0.188 nmol/l) remained unchanged. No significant changes were observed at lower doses.
Overall, the NOEL in the 1-year study in rhesus monkeys was 0.125 mg/kg bw per day (Williams, 1993).
The evaluation of ligand-binding kinetics of beta-adrenoceptors in heart and lung tissue of monkeys would address concerns that chronic exposure to residues of ractopamine in edible tissues may lead to desensitization of beta-adrenoceptors and decreased responsiveness to therapy among patients who require therapy with beta-adrenergic drugs. The reduced number of beta-adrenoceptors in lung tissue observed at the highest dose of 4 mg/kg bw per day indicates a receptor down-regulation after prolonged exposure to ractopamine. In a pilot study to find the dose range, groups of two male and two female rhesus monkeys were treated with ractopamine at a dose of 0.25, 0.5, or4.0 mg/kg bw per day by nasogastric gavage for 6 weeks; radioligand studies with [3H]dihydroalprenolol also revealed a statistically significant decrease in the density (Bmax) of lung beta-adrenoceptors at these lower doses of ractopamine. The number of receptors was reduced by 27.8% and 32.1% at the intermediate and highest doses, respectively. No significant down-regulation was observed at the lowest dose of 0.25 mg/kg bw per day. There was no treatment-related effect on the affinity of the beta-adrenoceptors for the radioligand used at any of the doses tested. In this study, the NOEL was 0.25 mg/kg bw per day on the basis of the decreased density of lung beta-adrenoceptors (Williams & Shoufler, 1993). In membranes of lung tissue, beta2-adrenoceptors are the predominant subtype of beta-adrenergic receptor, while in heart tissue beta1adrenoceptors are predominant (Abraham et al., 2003; Lohse et al., 1996). The lack of effects on beta-adrenoceptor density in heart membranes suggests a selective ractopamine-induced desensitization confined to beta2-adrenoceptors, which was almost at maximum at a dose of 0.5 mg/kg bw per day after 6 weeks. The validity of the results of the radioligand studies is weakened and interpretation of the data is complicated by the use of crude membrane preparations and [3H]dihydroalprenolol as a ligand. The higher non-specific binding of this radioligand complicates the interpretation of the results when compared with other ligands (Staehlin et al., 1983). beta-Adrenoceptors are biologically active only when exposed at the cell membrane surface, while internalized receptors are desensitized. Thus, the exact number of biologically active receptors is best determined in intact cells using impermeable ligands. In crude cell membrane preparations, the actual number of membrane-exposed beta-adrenoceptors may be overestimated as a result of binding of the radioligand to internalized receptors contaminating the membrane preparations. Desensitization of beta-adrenoceptors is a complex process that may involve both the down-regulation of the receptor expression at the cell surface and uncoupling of the receptors and G-proteins by phosphorylation resulting in a loss of receptor function. Receptor phosphorylation seems to be most important at fairly high concentrations of agonist. The simultaneous occurrence of both types of receptor desensitization by long-term exposure to beta-agonists and catecholamines has been shown for beta2-adrenoceptors of mast cells, equine lymphocytes, lung tissue of rats, and for beta-adrenoceptors of failing hearts (Lohse et al., 1996; Finney et al. 2000; Abraham et al., 2003; Scola et al., 2004). Thus, the true extent and degree of beta-adrenoceptor desensitization by exposure to a beta-agonist will be largely underestimated by only measuring the receptor density in crude membrane preparations. The available database on the effects of ractopamine on beta-adrenoceptor binding kinetics and functional coupling does not adequately address the concerns of receptor desensitization. However, since it can be supposed that beta-adrenoceptor desensitization only occurs upon activation of beta-adrenoceptors, the concerns may indirectly be addressed by identifying a NOEL for ractopamine at which beta1- and beta2-adrenergic activity is virtually absent (Williams, 1993).
At its fortieth meeting, the Committee considered a limited series of studies of genotoxicity in vitro and in vivo (in vitro: unscheduled DNA synthesis in rat hepatocytes, Ames tests, assay for forward mutation in mouse lymphoma Tk+/ cells; in vivo: sister chromatid exchange in bone-marrow cells of hamsters), the results of which were all reported to be negative (Annex 1, reference 105).
The results of additional studies of genotoxicity with ractopamine, including studies in vivo, are summarized in Table 4. The studies complied with GLP and the validity of the tests was checked with solvent controls and substances of known genotoxicity that served adequately as positive controls in the respective test system.
Table 4. Results of studies of genotoxicity with ractopamine
|
End-point |
Test object |
Concentration/dose |
Resultsa |
Reference |
|
In vitro |
||||
|
Reverse mutation |
S. typhimurium strains TA1535, TA1537, TA98, and TA100, and E. coli strain WP2 uvrA |
312.5-5000 µg/plate ±S9 |
Negative |
Garriott & Rexroat (1996) |
|
Chromosomal aberration |
Cultured human lymphocytes |
First test: 50, 156.3, and 250 µg/ml -S9; 300, 600, and 1000 µg/ml +S9 |
First test: negative -S9; positive at 1000 µg/ml +S9 |
Akhurst (1995) |
|
Second test: 25, 100, and 200 µg/ml -S9; 75, 150, and 300 µg/ml +S9 |
Second test: positive at 100 and 200 µg/ml -S9; positive at 300 µg/ml +S9 |
|||
|
Chromosomal aberration |
Human lymphocytes (from whole blood) |
First test: 150, 200, 250, and 300 µg/ml -S9; 1200, 1500, 1990, and 2490 µg/ml +S9 |
First test: negative -S9; positive at 2490 µg/ml +S9b |
Murli (1996a) |
|
Second test: 700, 800, 900, 1000 and 1100 µg/ml -S9 |
Second test: positive at 800, 900, 1000 and 1100 µg/mlb |
|||
|
Chromosomal aberration |
Chinese hamster ovary cells |
400, 800, 900, or 1000 µg/ml for 4 h, -S9; 850, 875, or 900 µg/ml for 4 h,+S9; 65, 100, or 250 µg/ml for 19 h, -S9 |
Negative |
Jackson & Garriott (1996) |
|
Forward mutation |
Mouse lymphoma L5178Y cells |
300, 325, and 350 µg/ml, -S9; 200, 225, 250, and 275 µg/ml, +S9 |
Positive |
Garriott & Yount (1996) |
|
Forward mutation |
Mouse lymphoma L5178Y cells |
50-225 µg/ml, -S9; 75-275 µg/ml,+S9 |
Negative -S9; positive +S9 |
Cifone (1996) |
|
In vivo |
||||
|
Chromosomal aberration/ micronucleus formation |
Mouse bone-marrow cells |
300, 600, and 1200 mg/kg bw, single oral dosec |
Negative |
Murli (1996b) |
|
Chromosomal aberration/ micronucleus formation |
Mouse bone-marrow cells |
200, 400, and 800 mg/kg bw daily for 14 daysc |
Negative |
Murli (1996c) |
|
Micronucleus formation |
Mouse bone-marrow cells |
185.75, 371.5, or 743 mg/kg bw daily for 2 daysc |
Negative |
Garriott et al. (1993) |
|
Micronucleus formation |
Rat bone-marrow cells |
50, 100, 200, 400, 600 mg/kg bw daily for 14 days4 |
Equivocal: increases in MPCEd at doses of 50-200 mg/kg but not at 400-600 mg/kg |
Garriott et al. (1996) |
a With and without S9, 9000 × g supernatant of induced rodent liver.
b No increase in polyploidy or endoreduplication was observed at the concentrations analysed.
c Cyclophosphamide was used in the positive control groups.
d MPCE, micronucleated polychromatic erythrocytes.
Two studies were conducted to determine evidence for exposure to ractopamine in the bone marrow. Groups of four male ICR mice or four Fischer 344 rats, respectively, were given a single oral dose of [14C]ractopamine hydrochloride at 748 mg/kg bw in mice and 200 mg/kg bw in rats. At four time-points from 0.5 to 6 h after treatment, the concentration of radioactivity in plasma and bone marrow was determined. The results of the studies confirmed that the bone-marrow compartment of mice and rats was exposed to ractopamine and/or its metabolites at peak concentrations of 6.36 µg equivalents/g in mice and 3.85 µg equivalents/g in rats, respectively (Chay & Pohland, 1996a, 1996b).
Ractopamine, which does not contain a structural alert, gave negative results in a battery of assays in prokaryotic and eukaryotic cells in vitro and test systems in mice and rats in vivo. Inconsistent positive effects were reported in assays for cyogenetic effects in vitro with cultured human whole blood lymphocytes. In one study, ractopamine showed evidence of clastogenic activity in both the absence and presence of metabolic activation (Akhurst, 1995). In a study of induction of chromosomal abberations, it was considered that ractopamine gave negative results without metabolic activation with an 18 h treatment, positive results with an incubation of 3 h at higher concentrations, and weakly positive results at a single highly toxic dose in the presence of metabolic activation. No induction of polyploidy or endoreduplication was observed with and without metabolic activation at the concentrations analysed (Murli, 1996a). In vivo, however, the compound did not induce a genotoxic response in bone-marrow cells of mice with respect to chromosome aberrations and formation of micronuclei (Garriot et al., 1993; Murli, 1996b, 1996c). The positive trend in increased micronucleation of polychromatic erythrocytes in rat bone marrow was within the range for historical controls. After expanding the test to include higher, but not overly toxic, doses, neither the individual group data, nor the trend analysis indicated a statistically significant increase in the frequency of micronucleation at a maximum tolerated dose (Garriot at al., 1996). Ractopamine was weakly mutagenic in the assay for forward mutation at the thymidine kinase (Tk) locus in mouse lymphoma L5178Y cells with and without metabolic activation (Garriot & Yount, 1996). In another study in the same test system, ractopamine was reported to give negative results without metabolic activation and was only weakly mutagenic with metabolic activation at similar concentrations (Cifone, 1996), while in a previous study, complying with GLP, that was evaluated by the Committee at its fortieth meeting, ractopamine did not show induce forward mutation at the Tk locus of mouse lymphoma L5178Y cells (Williams et al., 1984a).
In order to provide an explanation for the weak genotoxic effects of ractopamine in the assay in mouse lymphoma cells, special studies were conducted to partially test the hypothesis that ractopamine induces a clastogenic effect through a secondary mechanism in vitro, after undergoing oxidation at the 3 (meta-) carbon of the beta-carbon-attached aromatic ring producing ractopamine-catechol. The proposed mechanism by which catecholamines such as epinephrine cause mutagenesis is based on oxidative injury caused by auto-oxidation from a catechol to a quinone. This mechanism involves the formation of reactive intermediates and reactive oxygen species. In a study that did not comply with GLP, the formation of ractopamine-catechol in cultured mouse lymphoma cells was tested with and without metabolic activation and with co-incubation of ractopamine with the nucleophilic antioxidant and glutathione precursor Nacetyl-L-cystein (NAC) and/or S-adenosyl-L-methionine plus COMT for methylating potentially unstable catechols. Ractopamine and its catechol were analysed by LC-MS/MS using product ion scans for qualitative mass spectral confirmation and multiple reaction monitoring for semi-quantitative determination. Considerable accumulation of ractopamine-catechol occurred only in the presence of metabolic activation and in cultures treated with NAC, while the minor amounts observed under the other test conditions appeared to be insignificant with respect to any catechol-mediated genotoxicity in mouse lymphoma cells (Murphy & Gillespie, 1996). In a subsequent mechanistic study, which complied with GLP, the potential of various chemical agents to eliminate block, reduce or increase the genotoxic effect of ractopamine in the assay in L5178Y Tk+/ mouse lymphoma cells was investigated. The chemicals were chosen on the basis of their ability to alter the oxidative state of the cell. Ascorbic acid or N,N’-diphenyl-1,4-phenylenediamine as antioxidants, NAC as antioxidant and glutathione precursor, superoxidedismutase/catalase and copper(II) 3,5 diisopropylsalicylate hydrate to reduce superoxide/hydrogen peroxide and their subsequent dismutation to hydroxyl radicals were chosen to reduce the oxidative stress to the cells. L-Buthionine-[S,R]-sulfoximine reduces the cellular capacity for radical scavenging by diminishing glutathione. The studies were performed without metabolic activation, except in the case of superoxide dismutase/catalase, which was tested with and without metabolic activation. Ethyl-methanesulfonate and 3-methylcholanthrene served as positive controls and their mutagenicity was unaffected by the presence of the blocking agents. NAC and catalase (without metabolic activation) were the only agents that effectively blocked the mutagenic effect of ractopamine, while buthionine-[S,R]-sulfoximine failed to enhance the mutagenic acitivity of the compound. The results of the studies with ractopamine were compared with the results for epinephrine in the assay in mouse lymphoma cells. Among the chemicals tested, NAC and ascorbic acid blocked epinephrine-induced cytotoxicity and genotoxicity by reducing epinephrine oxidation and thus decreasing oxidative stress in the cell. It was considered to be likely that the slight difference from the results obtained with ractopamine is caused by dissimilar redox potentials for the ractopamine-catechol versus epinephrine (Oberly & Yount, 1996). Together with analytical evidence that ractopamine auto-oxidizes to a catechol in cell culture media alone, these results suggest that ractopamine produces genotoxicity through secondary oxidative mechanisms in vitro and that ractopamine, therefore, may be considered to be not intrinsically genotoxic in the assay in mouse lymphoma cells. Whether this effect can mechanistically define the genotoxic response of mouse lymphoma cells to ractopamine remains to be elucidated.
On the basis of the results of studies of genotoxicity, ractopamine was considered to be a weak mutagen and clastogen in human lymphocytes and mouse lymphoma cells in vitro, while assays for chromosomal aberration in bone-marrow cells and for micronucleus formation in mice and rats indicated a lack of genotoxic effects in vivo. This conclusion is supported by the lack of genotoxically-induced tumours in studies of oncogenicity in rodents (Williams, 1998a, 1998b).
The Committee therefore considered that ractopamine is not genotoxic.
At its fortieth meeting, the Committee considered a two-generation study of reproductive and developmental performance in Sprague-Dawley rats treated with ractopamine. Significant embryotoxic effects and a minor teratogenic response were observed only at the highest dose of 150 mg/kg bw per day, at which maternal toxicity was also noted. The NOEL was 15 mg/kg bw per day (Williams & Hoyt, 1987; Annex 1, reference 105).
A single-generation study of reproductive toxicity in crossbred swine was conducted to identify the adverse effects of feeding gilts with diets containing ractopamine during the finishing period. After withdrawal of ractopamine from the diet, the gilts were bred and allowed to farrow, and to nurse their litters. It was concluded that with ractopamine at the tested dietary concentrations of 20 and 60 mg/kg of feed, the reproductive performance of gilts would not be adversely affected after the drug was withdrawn (Williams, 1989). The study did not comply with appropriate standards for protocol and was therefore not considered to be suitable for the assessment of the safety of residues of ractopamine.
(a) Pharmacodynamic effects: beta-adrenergic receptor selectivity for stereoisomers of ractopamine
Ractopamine is reported to be active at both beta1 and beta2-adrenoceptors. Results of previous studies suggest that racemic ractopamine is about 20 times more selective for beta1- versus beta2-adrenoceptors, as shown by values for binding affinity in rat glioma cells. This selectivity contrasts with the selectivity for beta2-adrenoceptors reported for other beta-agonists that are effective in growth modification in rodents, such as clenbuterol or salbutamol. Furthermore, racemic ractopamine was shown to be almost equally effective with respect to the effective dose (ED50) values reported for beta1adrenergic cardiac effects and beta2-adrenergic bronchodilation and relaxation of costo-uterine muscle in vitro. The effects were submaximal in heart preparations and maximal in tracheal and costo-uterine muscle preparations when compared with the effects of isoproterenol (Williams et al., 1989; Anderson et al., 1993). Results from recently published studies indicate that the biologically most active RR isomer (butopamine) of ractopamine binds non-selectively to cloned porcine beta1- and beta2-adrenoceptors expressed in Chinese hamster ovary cells, but that adenylyl cyclase activation is more efficacious through the beta2-adrenoceptor. Among the ractopamine stereoisomers, butopamine has the highest affinity for each receptor subtype and has nearly equivalent binding affinity (about 25nmol/l). Butopamine stimulated the accumulation of adenosine 3’,5’-cyclic phosphate with equal efficacy to isoproterenol through the cloned porcine beta2-adrenoceptor, but was only 35% as efficacious through the cloned porcine 1adrenoceptor. The other isomers exhibited preferential binding to the beta2-adrenoceptors, but had a much lower affinity, in the rank order RS>SR>SS, and Kd values ranged from 78 to 16 600 nmol/l. Signalling through adenlyl cyclase appears to be restricted to the RR and RS isomer. A similar finding was noted in rats.
The relative contributions of beta1- and beta2-adrenoceptor activation to the spectrum of effects produced by ractopamine may also differ by the different ratio of the beta-adrenoceptor subtypes in tissues and species. The beta1- and beta2-adrenoceptor are co-expressed in most tissues of the body, but the ratio of these receptor subtypes can vary such that the beta1-adrenoceptor is predominant in heart (70-80% in humans, 72% in pigs) and adipose tissue (75% in rats, 80% in pigs), while skeletal muscle (60% in pigs), uterus (80% in humans) and lung (65% in pigs, 80% in humans or horses) have mostly beta2-adrenoceptors. The distribution of beta1- and beta2-adrenoceptors in the cardiovascular system and their contribution to cardiovascular effects of ractopamine are described in section 2.2.5(b).
Collectively, the RR isomer is most likely the functional stereoisomer of ractopamine, but its effectiveness may be limited by the presence of competing isomers, in particular the RS isomer in the racemic mixture. The RR isomer is a non-selective ligand at the beta1- and beta2-adrenoceptor, but signal transduction is more efficiently coupled through the beta2-adrenoceptor than the beta1-adrenoceptor. Therefore, the RR isomer of ractopamine is considered to be a full agonist at the beta2-adrenoceptor and a partial agonist at the beta1-adrenoceptor (Smith, 1998; Brodde, 1991; Mills, 2002; Abraham et al., 2003; Mills et al., 2003a, 2003b).
A series of special studies of cardiovascular effects in dogs, monkeys, and humans was conducted to characterize the comparative cardiostimulatory effects of ractopamine hydrochloride.
Dogs
At its fortieth meeting, the Committee considered a 1-year study of toxicity in beagle dogs given ractopamine orally at a dose of 0.112, 0.224, or 5.68 mg/bw per day. Clinical signs of cardiovascular effects recorded were mild nocturnal bradycardia at all doses tested and transient cutaneous erythema in the dogs at the highest dose. A clear NOEL was not observed in this study. In a pilot study, a single oral dose of ractopamine at 1.5 mg/kg bw produced near maximal cardiostimulation at 2 h after dosing. An oral dose of 0.05 mg/kg bw was associated with skin and oral mucosal reddening; the NOEL was 0.035 mg/kg bw (Williams, 1987b; Annex 1, reference 105).
In a study complying with GLP, the acute cardiovascular effects or ractopamine hydrochloride were evaluated in two male and two female beagle dogs anaesthetized with pentobarbital at a dose of 30 mg/kg bw). The animals were prepared for haemodynamic monitoring and the response of pulsatile arterial pressures, pulsatile aortic flow wave forms, and left ventricular pressure to a 10-min intravenous infusion of ractopamine at 35 µg/kg bw was determined.
Heart rate increased by approximately 65% during the 10-min infusion and remained elevated by at least 50% throughout the following 30-min monitoring period. Mean arterial pressure fell sharply to about 50% of its control level during the infusion and subsequently returned towards the control level, but remained depressed by approximately 25% at 40min after infusion. The fall in mean arterial pressure was accompanied by an increase in pulse pressure as a result of the relatively greater reduction in diastolic as compared with systolic pressure.
Cardiac output increased by approximately 50% during the infusion, and then stabilized at a level about 40% higher than that of controls for the next 30min. The increase in cardiac output was caused primarily by an increase in heart rate, since stroke volume only transiently increased by approximately 12% at the start of infusion and later decreased to about 82% of the control level. Total peripheral resistance fell sharply to about 35% of the control level immediately after the onset of infusion. Resistance subsequently returned to about 50% of the control level for the next 30min.
Peak aortic flow increased by approximately 90% immediately after the onset of infusion and subsequently stabilized at about 70%. The aortic flow ejection period shortened by approximately -27% during the infusion and remained shorter for the next 30min. Changes in the time to peak flow were small and variable.
The cardiac arrhythmia observed in one of the four animals could not be clearly ascribed to treatment with ractopamine or any other particular cause.
The systemic cardiovascular effects included tachycardia and peripheral vasodilatation. Despite concomitant and potent augmentation of cardiac output, the magnitude of the peripheral vasodilatation was sufficient to cause a dramatic fall in mean arterial pressure.
Cardiac output increased as a result of the pronounced increase in heart rate, and in spite of the decrease in left ventricular stroke volume. The decrease in stroke volume was accompanied by other changes in the aortic flow wave form consistent with an increase in left ventricular contractility. These included an increase in peak aortic flow and a reduction in the duration of the ejection period (Williams & Stone, 1987b).
Another study in conscious dogs was conducted to provide data on acute cardiovascular toxicity of oral doses of ractopamine. Groups of four male and four female beagle dogs (aged 10-19 months) were given ractopamine orally at a dose of 0, 2, 50, or 125 µg/kg bw. The lowest dose of 2 µg/kg bw was selected to be higher than the human exposure anticipated from eating 125 g of pig kidney from an animal that had not been removed from treatment with ractopamine before slaughter (zero time withdrawal). The intermediate dose of 50 µg/kg was selected because this dose had produced skin erythema without any statistically significant increase in heart rate in a previous study in dogs. The highest dose was selected because it was known to increase heart rate in this study and corresponded to the NOEL previously identified in a long-term study in monkeys (section 2.2.2). The study was designed as a double Latin square that allowed testing for residual effects and was certified for compliance with GLP and quality assurance.
Left ventricular pressure, aortic blood pressure, heart rate and electrocardiograms were recorded to provide data on the effects of an oral dose of ractopamine on left ventricular function and systemic blood pressure. The peak value of the first derivative of left ventricular pressure (dP/dtmax) was used as an index of left ventricular inotropic state. Systolic, diastolic, mean aortic, and aortic pulse pressures were derived by the data acquisition system from the aortic pressure signal. Heart rate and left ventricular end-diastolic pressure were obtained from the ventricular pressure signals.
All dogs survived the treatment. There was no residual carry-over effect from one treatment to the next in the Latin square design. Ractopamine caused statistically significant dose-dependent increases in heart rate and left ventricular inotropic state at 50 and 125 µg/kg bw. Maximum effects occurred approximately 2 h after dosing with an increased heart rate of 40 and 80 beats per min at the intermediate and highest doses, respectively. Increases in left ventricular inotropy were recorded during the 6-h period after dosing. No significant change in heart rate or left ventricular inotropic state was observed at 2 µg/kg. A drop in blood pressure was evident in both the systolic and diastolic (and therefore the mean) pressures in response to treatment at 50 and 125 µg/kg during the 6-h period immediately after dosing. Treatment at 125 µg/kg caused a decrease in aortic pulse pressure. Analysis of electrocardiograms did not indicate any treatment-related effects. Two dogs at 50 µg/kg and seven dogs at 125 µg/kg had a slight pinking of the abdominal skin (erythema). No other clinical signs were observed in the study. At 2 µg/kg, there was no significant effect on any of the parameters measured.
In conclusion, treatment with ractopamine at 50 and 125 µg/kg bw caused tachycardia, an increased left ventricular inotropic state, and a fall in systemic blood pressure in dogs. The increase in heart rate was consistent with a reflex tachycardia since the increased heart rate was always accompanied by a fall in systemic in blood pressure. The results of this study are consistent with expected pharmacological effects associated with vascular beta2-adrenoceptor stimulation and subsequent vasodilation. It is possible that the increased left tachycardia and ventricular inotropy were the result of some direct effect on cardiac beta1- and beta2-adrenoceptors in the dogs. The NOEL was 2 µg/kg bw in this study (Sarazan et al., 1993).
Monkeys
At its fortieth meeting, the Committee considered studies on cardiovascular effects of ractopamine in rhesus monkeys. The animals were given the test compound by nasogastric gavage for 6 weeks or 90 days. Monkeys given ractopmaine at 4 mg/kg bw per day developed tachycardia without changes of the electrocardiogram wave forms or cardiac histopathological appearance. The NOEL for haemodynamic effects was 0.125 mg/kg bw per day (Williams & Stone, 1987a; (Annex 1, reference 105)).
In a one-year study of toxicity in rhesus monkeys given ractopamine by nasogastric gavage at daily doses of 0.125, 0.5, and 4 mg/kg bw, significant increase of heart rate and characteristic changes of the electrocardiogram wave forms were recorded. The NOEL for cardiovascular effects was determined to be 0.125 mg/kg bw per day (section 2.2.5(b)).
The first of two primate experiments, which were compliant with GLP regulations, was performed to determine the appropriateness of a primate model for safety assessment. Two male and two female anaesthetized rhesus monkeys (body weight, 2.5-3.5 kg; given pentobarbital at 30 mg/kg bw) were prepared for haemodynamic monitoring. The response of pulsatile arterial pressures, pulsatile aortic flow waveforms, and left ventricular pressure to a 10-min intravenous infusion of ractopamine at 35 µg/kg bw was determined.
All animals survived the experimental phase of this terminal study, and no signs of toxicity were observed. Heart rate increased approximately 20% during the infusion and remained elevated throughout the 40-min monitoring period. Mean arterial pressure was maintained throughout the experiment while there was a slight augmentation of systolic pressure during the infusion. Cardiac output increased approximately 35% during the infusion and decreased steadily throughout the monitoring period. Total peripheral resistance decreased gradually to approximately -30% of its initial level during the infusion and then slowly returned towards control. Stroke volume increased approximately 14% during the infusion and exhibited a gradual return to baseline during the monitoring period. Peak aortic flow increased approximately 80% during the infusion and gradually returned toward baseline levels. The aortic flow ejection period shortened by approximately -18% during the infusion and remained so during the monitoring period. Changes in the time to peak flow were small and variable (Williams & Stone, 1987c).
The second of two experiments in primates was performed to further characterize the response to infusion of ractopamine by assessing the influence of anaesthesia. Eight male rhesus monkeys (body weight, about 6 kg) were prepared for haemodynamic monitoring, and their response to a 10 min intravenous infusion of ractopamine at 35 µg/kg bw, as compared with an infusion of sodium chloride (NaCl), was determined in both the awake, and in the anaesthetized states. Heart rates, systolic, diastolic, and mean arterial pressures were determined from the arterial pressure wave forms.
All animals survived the study, and no signs of toxicity were observed. There were no apparent effects on the haemodynamic parameters measured in response to the control infusion. In response to the infusion of ractopamine, the haemodynamic changes described below were recorded.
During the conscious state, pulse pressure was increased owing to the increase in systolic pressure, which appeared virtually immediately with the onset of infusion. After the peak response approximately 8 min later, systolic pressure declined slowly to the control level over the next 90 min and was stable throughout the subsequent 6-h monitoring period.
In the anaesthetized state, pulse pressure was increased by virtue of both an increase in systolic, and a decrease in diastolic pressure. The increase in systolic pressure appeared virtually immediately with the onset of infusion and continued to increase throughout the 40-min monitoring period. The decrease in diastolic pressure was shorter and less pronounced than the increase in systolic pressure.
In the conscious state, heart rate increased from its control level of approximately 120 beats per min to a maximum of almost 190 beats per min by the end of the 10-min infusion. Thereafter, heart rate declined rapidly with the cessation of infusion.
In the anaesthetized state, despite elevated heart rate (about 150 beats per min) present before ractopamine infusion, heart rate was further increased by ractopamine to approximately 214 beats per min by the end of the 10-min infusion period. Thereafter, heart rate declined, yet it remained elevated for the duration of the 40-min monitoring period.
In conclusion, the changes in heart rate and arterial pressures observed under anaesthetized conditions were qualitatively and quantitatively similar to those previously documented for this species. These included tachycardia and slightly increased pulse and mean arterial pressure. In the conscious state, the haemodynamic responses to ractopamine infusion also resulted in tachycardia and slightly increased pulse and mean arterial pressures. Barbiturate anaesthesia appears to play an insignificant role in the modulation of haemodynamic responses to a 10-min infusion of ractopamine at 35 µg/kg bw and does not alter the qualitative acute haemodynamic response in rhesus monkeys (Williams & Stone, 1987d).
Overall, data from these studies have characterized haemodynamic responses to a 10-min infusion of ractopamine at 35 µg/kg bw in anaesthetized and conscious rhesus monkeys. The cardiovascular effects are consistent with expected beta2-adrenoceptor-mediated peripheral vasodilation and modest reflex tachycardia associated with some possible direct cardiac beta-adrenoceptor stimulation.
It was demonstrated in studies of acute and long-term effects that dogs are more sensitive to the cardiovascular effects of ractopamine than are monkeys (Table 5 and Table 6).
Table 5. Acute cardiovascular effects of ractopamine administered by infusionb in anaesthetized monkeys and dogs
|
Cardiovascular parametera |
Species |
|
|
Rhesus monkeys |
Beagle dogs |
|
|
Heart rate |
+20% |
+65% |
|
Cardiac output |
+35% |
+50% |
|
Stroke volume |
+14% |
+12% to-18% |
|
Aortic flow ejection period |
-18% |
-27% |
|
Peak aortic flow |
+80% |
+70 to +90% |
|
Peripheral resistance |
-30% |
-67% |
|
From Williams & Stone (1987b, 1987c). |
|
|
a |
Values are expressed as percentage changes relative to the baseline values before infusion of ractopamine. |
|
b |
10-Min intravenous infusion of ractopamine hydrochloride at 35 mg/kg bw. |
Table 6. NOELs for cardiovascular effects of ractopamine in dogs and monkeys
|
Species |
Study type, route |
Dose |
NOEL |
Reference |
|
Dog |
Single oral dose |
0, 0.002, 0.05, 0.125 |
0.002 |
Sarazan et al. (1993) |
|
Dog |
1-year, oral |
0, 0.112, 0.224, 5.68 |
Not identified |
Annex 1, reference 105 |
|
Monkey |
90-day, gavage |
0, 0.125 |
0.125 |
Annex 1, reference 105 |
|
Monkey |
6-week, gavage |
0, 0.25, 0.5, 4.0 |
0.25 |
Williams & Shoufler (1993) |
|
Monkey |
1-year, oral |
0, 0.125, 0.5, 4.0 |
0.125 |
Williams (1993) |
The ractopamine-induced decrease of peripheral vascular resistance is mediated through beta2-adrenoceptor-stimulated vasodilation. In contrast, the effects on the heart occur primarily via beta1-adrenoceptors. It is uncertain to what extent the increase in heart rate is also caused by activation of cardiac beta2-adrenoceptors or to reflex sympathetic effects that stem from beta2-adrenoceptor-mediated peripheral vasodilation. No information was available on the relative contributions of either beta1- or beta2-adrenoceptors activation to the cardiostimulatory effects produced by ractopamine (e.g. studies with co-administration of the beta1-selective antagonist atenolol).
The traditional notion that only beta1-adrenoceptors modulate cardiac contractile function has been challenged by studies providing compelling evidence that both beta1- and beta2-adrenoceptors functionally coexist in cardiomyocytes of many mammalian species. For cardiac beta-adrenoceptor subtypes, differences in behaviour within species and great diversity in beta2-adrenoceptor-mediated cardiac responses between species were reported. The ratio of pr to beta2-adrenoceptors in the human heart is approximately 70:30 to 80:20. Studies in vivo and in vitro have shown that positive inotropic effects in heart atria and ventricles can be stimulated through both beta-adrenoceptor subtypes. In contrast to beta1adrenoceptors, stimulation of beta2-adrenoceptors induces maximal positive inotropic effects only in atria, but submaximal responses in ventricles. Studies in vitro have shown that beta1- and beta2-adrenoceptors are equally effective in stimulating positive chronotropic effects. There is further evidence for cardiac differential regulation and functionality of beta-adrenoceptors in healthy and diseased hearts, with the number of beta1 adrenoceptors being selectively reduced without alteration in the density of b2-adrenoceptors in chronic heart failure. Thus, extrapolation of cardiac responses to treatment with ractopamine from animals to humans as well as from healthy human volunteers to patients with chronic heart failure would be complicated (Brodde, 1991; Newton et al., 1999; Xiao et al, 1999).
Ractopamine hydrochloride was tested for antimicrobial activity against a variety of Gram-positive and Gram-negative aerobic (strains of Staphlycocci, Streptococci, Haemophilus, E. coli, Klebsiella, Enterobacter, Salmonella, Pseudomonas, Serratia, Shigella, Proteus, Citrobacter, and Acinetobacter) and anaerobic microorganisms (strains of Clostridia, Eubacterium, Peptococci, Propionobacterium, Bacteroides, and Fusobacteria) using tests with agar dilution in vitro. Ractopamine hydrochloride and compounds serving as positive controls were incorporated into agar plates at a range of concentrations from 0.5 to 128 µg/ml for the aerobic screen and from 0.008 to 256 µg/ml for the anaerobic screen. A total of 55 microorganisms (36 Gram-positive and Gram-negative aerobic pathogens and 19 Gram-positive and Gram-negative anaerobes) were then inoculated onto the surface of the plates. After incubation, the minimum inhibitory concentration (MIC) was determined by the presence or absence of bacterial growth at the inoculation site.
The MICs were >128 µg/ml for all the aerobes and >256 µg/ml for all the anaerobes tested, except Bacteroides vulgatus for which the MIC was 128 µg/ml. These high MICs indicate that ractopamine hydrochloride has no antimicrobial activity against the microorganisms tested (Lewis, 1985).
At its fortieth meeting, the Committee requested a survey of all non-therapeutic effects that follow long-term use of beta-adrenoceptor agonists in humans.
Adverse effects of prolonged therapeutic use of beta-agonists including tachycardia, vasodilation, skeletal muscle tremor, nervousness, metabolic disturbances, and beta-adrenoceptor desensitization are pharmacologically predictable, dose-related and potency-related. Non-pharmacological effects include airway hyper-responsiveness and increased airway inflammation. The incidence and severity of adverse reactions may vary for any given compound. The impact of the R- and S-enantiomers of beta-agonists on adverse effects remains unclear (Hoffman, 2001; Sears, 2002).
In pilot clinical trials, the bronchodilator effects of ractopamine were evaluated. There was little evidence for a bronchodilator activity in four patients (Annex 1, reference 105).
Tachycardia, decrease in blood pressure, and occasional palpitations are common adverse effect of systemically administered beta-agonists that are considered to be clinically most relevant. In patients without cardiac disease, beta-agonists rarely cause significant arrhythmias or myocardial ischaemia. However, patients with underlying coronary artery disease or pre-existing arrhythmias are at a much greater risk. Rhythm disturbances may occur more frequently with fenoterol than albuterol. Terbutaline use resulted in ectopic activity, but not frank arrhythmias. In single case reports and reports from small case series, cardiac ischaemia, heart failure, and cardiomyopathy were recorded with use of isoproterenol and albuterol at high doses. The risk of adverse cardiovascular effects is also increased in patients receiving monoamine oxidase (MAO) inhibitors or tricyclic antidepressants. The concern that beta-agonists at high doses increase asthma mortality rates through cardiac adverse effects has not been fully disproved. Tolerance develops rapidly to the cardiac-stimulating effects of beta2-agonists. (Hoffman, 2001; Sears, 2002).
The inotropic effects of ractopamine were evaluated in pilot clinical trials. Four patients showed little evidence for an increase in pulse rate. Two patients showed a mild elevation of blood pressure lasting for about 1 h. An infusion study showed inotropic and chronotropic enhancement in both healthy volunteers and heart patients (Annex 1, reference 105).
A study was conducted in humans to evaluate the effects of increasing doses of ractopamine hydrochloride on the indices of cardiovascular function and safety in normal human subjects and thereby define a dose that would not produce a detectable effect. The purpose of this study in humans was also to determine whether the dog or non-human primate provides a better model for estimating the acute cardiovascular and other potential toxic effects of ractopamine in the human diet. It was demonstrated in previous studies of acute and long-term effects in laboratory animals that dogs were more sensitive to the cardiovascular effects of ractopamine than were rodents and primates (sections 2.2.2 and 2.2.5(b); Annex 1, reference 105). The secondary objective was to describe the pharmacokinetics of single oral doses of 40 mg of ractopamine hydrochloride in normal human subjects (section 2.1.3). The protocol for this study was jointly developed with the cooperation and concurrence of the Food & Drug Administration/Center for Veterinary Medicine (CVM), and the sponsor (Elanco Animal Health).
The dose-response effect of ractopamine on the human cardiovascular system was studied in a single-blind, placebo-controlled, ascending single-dose protocol. The study was conducted with six healthy male volunteers (body weight: range, 67.8-79.6 kg; mean, 75.5 kg), given oral placebo plus five oral doses of 5, 10, 15, 25, and 40 mg of ractopamine, with a washout period of 48 h between doses. On a body-weight basis, the doses ranged from 0.063 to 0.590 mg/kg bw. Using standard and echocardiographic methods, measurements for 14 cardiovascular variables were obtained at nine hourly time-points in each subject.
No serious adverse events were reported. At doses of 15, 25, and 50 mg, sensations of increase in heart rate were reported for two, three, and four men, and sensation of heart pounding in one, three, and one man, respectively. The adverse effects were considered to be treatment-related and to be either mild or moderate in severity. One man was withdrawn from the study after the 25 mg dose because of adverse cardiac effects. Dose-dependent changes of cardiac variables appeared within the first hour after administration of ractopamine and gradually returned to baseline values before treatment. At a dose of 5 mg, there was apparently no cardiovascular response, and at 10 mg only minor effects were reported. At 15, 25, and 40 mg, the heart rate was elevated about 20, 30, and 50 beats per min above control and the cardiac output increased by approximately 35%, 55%, and 90%, respectively. At the same doses, the electromechanical systole was shortened by about 10%, 14%, and 19%, respectively. The systolic blood pressure increased in a dose-dependent manner. In contrast to the vasodilative effects recorded in monkeys and dogs, ractopamine did not change or even increase the diastolic pressure. The observed NOELs for different cardiovascular parameters in this study are shown in Table 7.
Table 7. NOELs for cardiovascular effects in healthy human volunteers after oral administration of ractopamine
|
Parameter |
NOEL (mg/person) |
NOEL( mg/kg bw)a |
|
Electromechanical systole |
5 |
67 |
|
Left ventricular ejection time |
5 |
67 |
|
Maximum velocity of circumferential fiber shortening |
5 |
67 |
|
Heart rate |
10 |
133 |
|
End systolic volume |
10 |
133 |
|
Systolic blood pressure |
10 |
133 |
|
Cardiac output |
15 |
200 |
|
End diastolic volume |
25 |
333 |
|
Diastolic blood pressure |
25 |
333 |
|
Corrected QT-interval |
40 |
597 |
|
Pre-ejection period |
40 |
597 |
From Hunt (1994) and Food & Drug Administration (1999).
a Based on a mean body weight of 75 kg for the persons tested.
Using a population modelling strategy, estimates of NOEL and confidence intervals (95%) were calculated for three salient parameters of cardiac function and for a composite over all variables (Table 8).
Table 8. Estimated NOELs and 95% confidence intervals for main parameters of cardiac function in healthy human volunteers after oral administration of ractopamine
|
Parameter |
NOEL (µg/kg bw) |
Confidence interval |
|
|
Lower |
Upper |
||
|
Electromechanical systole |
96 |
35 |
156 |
|
Heart rate |
114 |
58 |
171 |
|
Cardiac output |
84 |
41 |
128 |
|
Composite over all variables |
99 |
77 |
120 |
From Hunt (1994) and Food & Drug Administration (1999).
No estimates of NOEL were calculated for blood pressure variables using the linear extrapolation model.
The oral bioavailability of ractopamine was estimated in this study by measuring the amount of free and conjugated ractopamine in the urine of the six subjects during the 24 after the oral dosing. Forty-six percent of the administered dose was collected in the urine in 24 . This compared favourably with excretion rate in dogs and monkeys (section 2.1.1).
It was concluded that the primate model is more predictive of the human acute toxicity response after oral exposure to ractopamine than is the canine model. Appropriate timings for observations were used to reveal the onset, time to peak, and duration of cardiac effects induced by ractopamine. The results indicate a comparable time course of cardiostimulatory effects of ractopamine in humans, monkeys, and dogs (Hunt, 1994; Food & Drug Administration, 1999).
Skeletal muscle tremor is the most common adverse effect of beta2-selective adrenergic agonists and is more likely to be seen after oral administration than after inhalation. Tremor results from an imbalance between fast- and low-twitch muscle groups of the extremities, and its severity varies greatly between individuals. The more potent full agonist fenoterol is more likely to produce tremors than albuterol or terbutaline. Tolerance to this effect generally develops within weeks of the start of the treatment in the majority of patients. It is not clear whether this tolerance reflects desensitization of beta2-adrenoceptors of skeletal muscle or adaptation within the central nervous system. This adverse effect is minimized at low oral doses of beta-agonists. No such effects were recorded at the NOEL determined in the toxicological studies conducted in laboratory animals given ractopamine or in the study in humans on cardiovascular effects of ractopamine (Hunt, 1994; Hoffman, 2001; Sears, 2002).
Feelings of restlessness, apprehension, and anxiety were reported side-effects after the use of various beta-agonists, particularly after oral or parenteral treatment. In pilot clinical trials with ractopamine, four patients showed little evidence for central nervous system stimulation. It is unclear whether long-term treatment with these drugs results in the development of tolerance to these adverse effects (Annex 1, reference 105; Hoffman, 2001).
When given orally or parenterally, beta-agonists may increase the concentrations of glucose, lactate, and free fatty acids and decrease the concentration of potassium in plasma. These changes were reported in clinical trials and during treatment with beta-agonists. Hyperglycaemia and hypokalaemia represent expected pharmacological effects of these class of drugs through beta2-adrenoceptor-mediated stimulation of gluconeogenesis and glycogenolysis and activation of Na+,K+-ATPase with an intracellular shift in potassium. The hypokalaemic effect is more profound with fenoterol than with albuterol, formoterol orterbutaline. The decrease in serum potassium concentration may be especially important in patients with cardiac disease, particularly those taking cardiac glycosides, and is aggravated by the concomitant use of diuretics or glucocorticoids. In some diabetic patients, hyperglycaemia may be made worse by beta-agonists, and higher doses of insulin may be required. The effects on plasma glucose and potassium concentrations show tolerance with repeated use. In laboratory animals, similar changes in serum chemistry parameters were occasionally recorded with ractopamine at higher doses, exceeding the NOEL established in toxicological studies (Hoffman, 2001; Annex 1, reference 105).
Desensitization of beta-adrenoceptors in some tissues and decreased pharmacological responses have been demonstrated in patients with asthma upon long-term systemic administration of beta-agonists. In monkeys, long-term administration of ractopamine induced a down-regulation of beta-adrenoceptor density in membrane preparations of lung tissue. The study authors considered that the NOEL was 0.25 mg/kg bw per day; however, the range of mechanisms of beta-adrenoceptor desensitization was not fully explored in this study, precluding the identification of a NOEL (Hoffman, 2001).
The results of a number of epidemiological studies have suggested a possible adverse connection between prolonged use of beta-agonists and severe asthmatic sensations. There is some evidence suggesting that regular use of beta2-selective agonists may cause increased bronchial hyperreactivity and deterioration in disease control. Long-acting beta-agonists have not been shown to increase airway hyperresponsiveness in adults. Findings in asthmatic subjects suggest that regular beta2-agonist use increased allergen responses (early and late) with increased mediator release, implying that these drugs may potentiate allergic inflammation in the airway (Hoffman, 2001; Sears, 2002).
beta-Adrenergic stimulants have been extensively used for decades in humans and a search of the published literature did not reveal any studies on tumour development in humans as a result of long-term administration of beta-agonists (Cantox, 2000). There was no evidence of any increased incidence in smooth muscle tumours such as leiomyomas among users of these drugs (Gopinath & Gibson, 1987). In the non-pregnant human uterus, little or no relaxant response to beta2-agonists can be demonstrated. Thus, the response of these tissues to beta2-adrenoceptor agonists may represent a species-specific effect and it is considered unlikely that administration of beta2-agonists in humans would result in uterine leiomyomas as it does in mice and rats (Jack et al., 1983; Gibson et al., 1987).
In summary, the non-therapeutic effects that follow long-term beta-adrenoceptor agonist use in humans are pharmacologically predictable and dose-related, with cardiovascular effects being the most common adverse reactions. Observations in humans treated with beta-agonists suggest that the adverse effects generally are not problematic at therapeutic doses, except perhaps in the presence of hypoxia or co-morbidity (e.g. heart failure, cardiac arrhythmias, coronary disease, diabetes). In humans, all beta-agonists may induce a similar profile of adverse effects with long-acting beta-agonists (e.g. formoterol, salmeterol) showing less pronounced adverse effects compared with short-acting agents (e.g. isoproterenol, fenoterol, salbutamol, terbutaline). Tolerance to pharmacologically predictable non-therapeutic effects occurs readily. The potential adverse effects of beta-agonists were adequately addressed in the toxicological studies with ractopamine in laboratory animals, allowing prediction of the consequences of the long-term intake of residues of ractopamine by consumers of meat.
Ractopamine is well absorbed in a number of animal species and rapidly excreted, with urine as the major route of excretion. Biliary excretion of ractopamine varies with species, with the highest percentage of the administered dose found in the bile of rats. In laboratory animals, the plasma half-life ranged from about 4 to 7 h. Absorption of orally administered radiolabelled ractopamine was >80% of the administered dose, as estimated by recovery of radioactivity in the urine and faeces of rats, dogs, monkeys, pigs and cattle over a collection period of up to 10 days. Only a minor fraction of radioactivity in the urine represented parent ractopamine. More parent drug was detected in rat urine after parenteral administration of [14C]ractopamine than after oral administration, suggesting significant first-pass metabolism in the intestine and liver after oral administration.
In addition to the three major monoglucuronide metabolites of ractopamine, designated as metabolites A, B, and C, a fourth metabolite, D, was identified as a glucuronic acid diconjugate of ractopamine in the urine, liver and kidney of rats, dogs, pigs and cattle. In metabolite D, the glucuronic acid is attached to both hydroxyphenyl rings. Two minor metabolites, designated E and F, also found in all species, were not identified. The chromatographic profiles of radioactive extracts of urine and liver from rats, dogs, pigs and cattle given [14C]ractopamine were qualitatively, but not quantitatively, similar for the same four glucuronide metabolites of ractopamine. A monosulfate conjugate and a sulfate/glucuronic acid diconjugate were identified as the major metabolites in the bile of rats. While the metabolites generally represented a greater fraction of the concentrations of residues in laboratory animals, it was concluded that dogs and rats used in the studies of toxicity were exposed to the same metabolites as those found in the edible tissues of pigs and cattle.
The results of a study in six healthy male human volunteers receiving a single oral dose of 40 mg of ractopamine hydrochloride indicate a similar profile of pharmacokinetics and biotransformation in humans and animals. Orally administered ractopamine was extensively and rapidly absorbed and low systemic concentrations of parent drug were found. The urinary metabolites were monoglucuronide and monosulfate conjugates. The half-life in plasma was about 4 h.
The Committee conducted a review of the literature and submitted data on the pharmacokinetics of beta-adrenoceptor agonists in humans and in laboratory species, including those studies relevant to oral administration. The data suggest, for most compounds and species, rapid and extensive absorption from the gastrointestinal tract, significant first-pass metabolism, wide distribution to the tissues, and predominantly urinary excretion. Most beta-adrenoceptor agonists have low lipophilicity at physiological pH. The metabolism of any given beta-adrenoceptor agonist is similar in all the species studied and the major differences are quantitative rather than qualitative. In general, beta-adrenoceptor agonists with halogenated aromatic ring systems (e.g. clenbuterol) are metabolized by oxidative and conjugative pathways and have long half-lives in plasma. beta-Adrenoceptor agonists with hydroxylated aromatic rings (e.g. ractopamine) are metabolized solely by conjugation and have relatively short half-lives. The pharmacokinetic data suggest that halogenated phenethanolamine beta-adrenoceptor agonist residues comprising greater amounts of parent substance have a higher oral bioavailability and relatively slower rate of elimination and thus a greater oral potency than residues of hydroxylated beta-adrenoceptor agonists.
In a long-term study of toxicity and carcinogenicity, CD-1 mice were given diets containing ractopamine at a concentration equal to 0, 25, 130, or 840 mg/kg bw per day for males and 0, 35, 175, or 1085 mg/kg bw per day for females for 21 months. A significantly increased rate of mortality at the highest dose was attributed to the more severe cardiomyopathy observed in groups receiving the two higher doses. Dose-related decreases in body weight, body-weight gain, and food efficiency relative to controls could be anticipated after exposure to a beta-adrenoceptor agonist with thermogenic activity. There was a dose-dependent increase in the incidence of uterine leiomyomas in female mice at 0 (1 out of 60 mice), 35 (5 out of 60), 175 (8 out of 60), and 1085 (10 out of 60) mg/kg bw per day. Treatment-related non-neoplastic proliferative lesions of the female genital tract smooth muscle were not strictly dose-dependent. The NOEL was 25 mg/kg bw per day in male mice on the basis of changes in body weight and food consumption. No NOEL could be identified for the formation of uterine leiomyomas in females. A benchmark dose was estimated as an alternative approach to a NOEL in order to define the point at which the incidence of leiomyoma in treated animals was higher than that in controls. The benchmark dose was calculated as 201 mg/kg bw per day on the basis of an excess incidence of leiomyoma of 5% higher than in controls, and a 95% lower confidence limit.
In a 2-year study of toxicity and carcinogenicity, groups of 60 male and 60 female Fischer 344 rats received diets containing ractopamine at a dose equal to about 0, 2, 60, 200, or 400 (in females only) mg/kg bw per day. Survival was significantly increased in both sexes at the highest doses. Dose-related decreases relative to control in body weight, body-weight gain, and food efficiency were consistent with increased thermogenesis after exposure to a beta-adrenoceptor agonist. Treatment-related morphological changes included an increased incidence of slight to moderate cardiomyopathy in females at the highest dose and in males at the two higher doses. Dose-related hyperplasia of the costo-uterine smooth muscle was observed in females at 0 (0 out of 60 mice), 2 (0 out of 60), 60 (3 out of 60), 200 (17 out of 60), and 400 (25 out of 60) mg/kg bw per day. No significant trends for neoplasm incidences in males were recorded. The only tumour that occurred at an increased incidence in females was the costo-uterine leiomyoma at 200 (6 out of 60) and 400 (27 out of 60) mg/kg bw per day; these tumours were not observed in other treated groups. The NOEL was 60 mg/kg bw per day in female rats on the basis of formation of costo-uterine leiomyomas. The NOEL was 2 mg/kg bw per day in male rats on the basis of enhanced cardiomyopathy.
The Committee noted that the induction of benign leiomyomas in mice and rats appears to be a general feature of beta-adrenoceptor agonists, as shown by the prevention of the development of these tumours by co-administration of the beta-adrenoceptor blocker propranolol in studies with other beta-adrenoceptor agonists. The Committee considered, therefore, that ractopamine is not a direct carcinogen and the induction of leiomyomas is a non-genotoxic event with a threshold and concluded that all treatment-related effects observed in the long-term studies of toxicity in mice and rats were attributable to the beta-adrenergic activity of ractopamine.
A 1-year study of toxicity in rhesus monkeys given ractopamine at a dose of 0, 0.125, 0.5, or 4 mg/kg bw by gavage once daily. No significant toxicological effects were recorded. A significant increase in body weight occurred in the animals at the highest dose. Monkeys treated with ractopamine developed tachycardia, with no other changes in the electrocardiogram. The heart rate was significantly increased in the groups given the two highest doses compared with controls; the maximum increase occurred during the first 4 h after dosing, and no significant slowing of the resting and nocturnal heart rates was seen. The heart weight relative to body weight was decreased at the two highest doses. beta-Adrenoceptor binding assays were conducted using the nonselective radioligand [3H]dihydroalprenolol in membrane preparations made from heart and lung tissue. The affinity and number of beta-adrenoceptors in the heart were not affected by ractopamine treatment, whereas there was a statistically significant decrease (23.8%) in the density of beta-adrenoceptors in the lung in the group given the highest dose. The NOEL in this study in rhesus monkeys was 0.125 mg/kg bw per day on the basis of tachycardia.
In another study in which rhesus monkeys were treated with ractopamine at a dose of 0.25, 0.5, or 4.0 mg/kg bw per day by gavage for 6 weeks, assays with the radioligand [3H]dihydroalprenolol at termination also revealed statistically significant decreases of 27.8% and 32.1%, respectively, in the density of beta-adrenoceptors in the lung at the intermediate and the highest dose. No treatment-related effects on the affinity of beta-adrenoceptors for the radioligand used were observed at any dose tested. The NOEL was 0.25 mg/kg bw per day on the basis of the decreased density of beta-adrenoceptors in the lung.
The Committee noted that the two studies in monkeys could have underestimated ractopamine-induced beta-adrenoceptor desensitization as measured by radioligand binding in preparations of tissue membranes. The Committee concluded, however, that beta-adrenoceptor desensitization would not be induced at a NOEL at which beta-adrenergic activity was virtually absent. Therefore, the NOEL for ractopamine was considered to be 0.125 mg/kg bw per day in monkeys.
The Committee further noted that ractopamine-induced cardiostimulation in monkeys occurred in the absence of cutaneous erythema and in the absence of clinical signs or microscopic lesions indicative of cardiotoxicity, thus suggesting that the cardiovascular response in monkeys differs from that in dogs. Dogs appear to be more sensitive than monkeys to vasodilation, hypotension, and reflex cardiostimulation, although the vasodilatory effects of ractopamine after oral administration have not been directly assessed in monkeys.
Ractopamine has been tested in a wide range of assays for genotoxicity, and gave negative results in a battery of prokaryotic and eukaryotic assays in vitro and test systems in vivo. Positive results were reported with and without metabolic activation in cytogenetic assays with cultured human whole blood lymphocytes in vitro, but not in a cell line derived from Chinese hamster ovary. Positive results were also obtained in the L5178Y Tk assay in lymphoma cells of mice in vitro, although ractopamine had given negative results in this assay in a previous study evaluated by the Committee at its fortieth meeting. It was suggested that the weak genotoxic effects observed in the assay in lymphoma cells of mice were mediated by oxidative stress induced by auto-oxidation of ractopamine to ractopamine-catechol and its further oxidation to a quinone in vitro. Co-incubation of antioxidants blocked the mutagenic effect of ractopamine and that of the positive control, epinephrine. Ractopamine did not induce aberrations in chromosomes or micronuclei of bone marrow cells when administered to mice and rats in vivo. The Committee concluded that ractopamine was not intrinsically genotoxic in vitro or in vivo.
Recently published studies indicate that the RR-isomer (butopamine) is the stereoisomer with the most activity at the beta-adrenoceptor. Butopamine was shown to be a non-selective ligand at the beta1 and beta2-adrenoceptors, but signal transduction is more efficiently coupled through the b2-adrenoceptor than the beta1 adrenoceptor. Therefore, the RR-isomer of ractopamine is considered to be a full agonist at the beta2-adrenoceptor and a partial agonist at the beta1adrenoceptor. These results are consistent with the pharmacological characterization of racemic ractopamine in isolated cardiac (atria) and smooth muscle (costo-uterine, vas deferens, trachea), which shows a maximal response at beta2- and a submaximal response at beta1adrenoceptors when compared with the full beta1 and beta2-adrenoceptor agonist isoproterenol.
In a study of acute cardiovascular effects, anaesthetized dogs were given a 10-min infusion of ractopamine at 35 µg/kg bw. All animals developed tachycardia (65% increases in heart rate) and decreased peripheral vascular resistance (67% decrease) with a fall in mean arterial pressure (50% decrease) during the infusion. Cardiac output increased by 50% as a result of the increase in heart rate, in spite of a decrease in left ventricular stroke volume. The decrease in stroke volume was accompanied by changes in the aortic flow waveform, consistent with an increase in left ventricular contractility. Upon cessation of infusion, the cardiovascular parameters returned towards control levels, but remained changed during the 30 min observation period.
In another study of acute cardiovascular toxicity, conscious dogs were given ractopamine as a single oral dose of 0, 2, 50, or 125 µg/kg bw. At the two higher doses, ractopamine caused dose-dependent increases in heart rate and left ventricular contractility, with maximum effects at about 2 h after dosing. There was a drop in both systolic and diastolic blood pressures during the 6 h period immediately after dosing. No significant changes were observed at the lowest dose. Dogs receiving the two highest doses demonstrated a slight abdominal skin erythema. The NOEL for this study was 2 µg/kg bw.
The acute cardiovascular effects of ractopamine were monitored in rhesus monkeys during a 10 min intravenous infusion of ractopamine at 35 mg/kg bw and a subsequent 30-min observation period. Heart rate increased about 20% during infusion and remained elevated throughout the monitoring period. Cardiac output, stroke volume and peak aortic flow increased by 35%, 14% and 80%, respectively, during the infusion and decreased steadily throughout the monitoring period. The aortic flow ejection period was shortened by 18% during the infusion and the monitoring period. Total peripheral resistance gradually decreased to approximately 70% of its initial value during the infusion and then slowly returned towards control values. In a second experiment in anaesthetized and conscious rhesus monkeys receiving this treatment, haemodynamic responses to ractopamine were not significantly affected by barbiturate anaesthesia.
The Committee noted that in studies of acute and short-term toxicity, dogs were more sensitive than monkeys to the cardiovascular effects of ractopamine. In both species, however, the relative contribution of direct stimulation of cardiac beta1 and beta2-adrenoceptors, and of reflex-mediated sympathetic effects reflecting the stimulation of peripheral beta-adrenoceptors, to the overall cardiovascular effects of ractopamine is uncertain.
The dose-dependent effects of ractopamine on the human cardiovascular system were studied in a limited number of human volunteers (six persons) given ascending single oral doses equal to 67, 133, 200, 333, and 597 µg/kg bw, with an interval of 48 h between doses. Occasional mild to moderate sensations of increase in heart rate and heart pounding were reported at doses of 200, 333, and 597 µg/kg bw. Dose-dependent increases in heart rate and cardiac output, and shortened electromechanical systole, as measured by echocardiography, were observed. The changes appeared within the first hour after the administration of ractopamine and values gradually returned to those before treatment. The systolic blood pressure increased in a dose-dependent manner. Unlike in monkeys and dogs, ractopamine had little effect on diastolic blood pressure in humans. Only minor cardiovascular effects were observed at 133 µg/kg bw. The NOELs for the relevant cardiac variables were 67 µg/kg bw for electromechanical systole, ventricular ejection time, and maximum velocity of circumferential fibre shortening, 133 µg/kg bw for heart rate and 200 µg/kg bw for cardiac output.
The Committee concluded that the response in monkeys is more predictive of the acute cardiovascular response in humans exposed to dietary ractopamine than is that in dogs. Monitoring in the studies in animals and humans was appropriately timed to reveal the onset, time-to-peak, and duration of ractopamine-induced cardiac effects. The time course of the cardiostimulatory effects of ractopamine was comparable in humans, monkeys, and dogs.
The Committee reviewed publicly available literature on non-therapeutic effects in humans after long-term use of beta-adrenoceptor agonists. The reported side-effects of prolonged therapeutic use of beta-adrenoceptor agonists include tachycardia, vasodilation, skeletal muscle tremor, nervousness, metabolic disturbances (hyperglycaemia and hypokalaemia), and beta-adrenoceptor desensitization. These effects are pharmacologically predictable, dose-related and potency-related, with cardiovascular effects being the most commonly reported side-effects. Non-pharmacological effects include airway hyper-responsiveness and increased airway inflammation. The incidence and severity of side-effects vary for any given compound. Tolerance to pharmacologically predictable, non-therapeutic effects occurs readily. There is no evidence for any increased incidence of smooth muscle tumours such as leiomyomas, or of any other tumours, among human users of these drugs. Little or no relaxant response to beta2-adrenoceptor agonists has been reported for the non-pregnant human uterus. The potential side-effects of beta-adrenoceptor agonists were adequately addressed in studies of toxicity with ractopamine in laboratory animals and in humans, in order to predict the consequences of the intake of residues of ractopamine by consumers.
After reviewing the additional data on toxicology and pharmacology, the Committee reaffirmed the evaluation made by the Committee at its fortieth meeting, which stated that it was appropriate to assess ractopamine on the basis of a NOEL for pharmacological effects that are relevant to its ingestion by humans as a residue in meats.
The Committee concluded that the acute cardiac responses to ractopamine in humans were the most appropriate end-points for the calculation of an ADI. A combined NOEL of 67 µg/kg was determined on the basis of changes in electromechanical systole, left ventricular ejection time, and maximum velocity of circumferential fibre shortening. A safety factor of 10 was used to account for individual variability and an additional safety factor of 5 was considered appropriate to protect sensitive individuals and in view of the small sample size in the human volunteer study, thus resulting in a combined safety factor of 50. This approach provides a margin of safety of at least 20 000 times with respect to the formation of leiomyomas in mice and rats.
The Committee established an ADI for ractopamine of 0-1 µg/kg bw per day based on the NOEL of 67 µg/kg bw in the study in human volunteers and the application of a safety factor of 50, rounded to one significant figure.
Abraham, G., Kottke, C., Dhein, S. & Ungemach, F.R. (2003) Pharmacological and biochemical characterization of beta-adrenergic signal transduction pathway in different segments of the respiratory tract. Biochem. Pharmacol., 66, 1067-1081.
Akhurst, L.C. (1995) Ractopamine hydrochloride metaphase chromosome analysis of human lymphocytes cultured in vitro. Unpublished report on study No. DTA 5/941656 from Huntington Research Centre, Ltd., Huntington, Cambridgeshire, England. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Anderson, D.B., Veenhuizen, E.L., Smith, C.K., Dalidowicz, J.E., Donoho, A.L., Williams, G.D., Jones, D.J., Schroeder, A.L., Turberg, M.P., Guerrero, R.J. & Guneratne, R.J. (1993) Ractopamine hydrochloride: a unique PLE (phenethanolamine lean enhancer) for swine. In: Proceedings of the 11th International Symposium of WAVFH, October 24-29, pp. 96-102.
Brodde, O.E. (1991) beta1- and beta2adrenoceptors in the human heart: properties, function, and alterations in chronic heart failure. Pharmacol. Rev., 43, 203-242.
Cantox (2000) Responses to commentary regarding ractopamine hydrochloride by the Joint Expert Committee on Food Additives. Unpublished report from Cantox Health Sciences International, Mississauga, Ontario, Canada. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Chay, S.H. & Pohland, R.C. (1996a) Radiocarbon concentrations in bone marrow and plasma of male ICR mice after a single oral 748 mg/kg dose of 14C-ractopamine hydrochloride (Compound 031537). Unpublished report on study No. M05895 from Toxicology Research Laboratories, Lilly Research Laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Chay, S.H. & Pohland, R.C. (1996b) Radiocarbon concentrations in bone marrow and plasma of male Fischer 344 rats after a single oral 200 mg/kg dose (free base) of 14C-ractopamine hydrochloride. Unpublished report on study No. R13696 from Toxicology Research Laboratories, Lilly Research Laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Cifone, M.A. (1996) Mutagenicity test on ractopamine hydrochloride in the L5178Y TK+/-mouse lymphoma forward mutation assay. Unpublished report on study No. 17282-0-431 from Corning Hazleton Inc. (CHV), 9200 Leesburg Pike, Vienna, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Dalidowicz, JE (1986a) Metabolism of 14C-ractopamine HCl in rats. Unpublished study No. ABC-0285 from Agricultural Biochemistry, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Dalidowicz, J.E. (1986b) Metabolism of 14C-ractopamine HCl in swine. Unpublished report on study No. ABC-0301 from Agricultural Biochemistry, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Dalidowicz, J.E. (1987) Comparative metabolism of 14C-ractopamine HCl in swine, dogs and rats. Unpublished report on study No. ABC-0369 from Agricultural Biochemistry, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Dalidowicz, J.E. (1989) Comparative metabolism of 14C-ractopamine HCl in cattle, dogs, and rats. Unpublished report on study No. ABC-0387 from Animal Science Chemical Research, Lilly Research laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Dalidowicz, J.E. & Babbitt, M.S. (1986) Characterization of 14C-residues in tissues and excreta from swine fed 14C-ractopamine HCl. Unpublished report on study No.ABC-0355 Agricultural Biochemistry, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Dalidowicz, J.E. & Thomson, T.D. (1988) 14C-Ractopamine HCl tissue residue steady-state study in cattle. Unpublished report on study No. ABC-0398 from Agricultural Biochemistry, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Dalidowicz, J.E. & Thomson, T.D. (1989) 14C-Ractopamine HCl balance-excretion study in cattle. Unpublished report on study No. ABC-0422 from Agricultural Biochemistry, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Dalidowicz, J.E., Thomson, T.D. & Herberg, R.J. (1985) 14C-EL-737 steady-state residue study with swine fed the highest anticipated dose. Unpublished report on study No. ABC-0273 from Agricultural Biochemistry, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Dalidowicz, J.E., Thomson, T.D. & Herberg, R.J. (1986) 14C-Ractopamine HCl balance-excretion study in swine. Unpublished report on study No. ABC-0330 from Agricultural Biochemistry, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Elliott, C.T., Thompson, C.S., Arts, J.M., Crooks, S.R., van Baak, M.J., Verheij, E.R. & Baxter, G.A. (1998) Screening and confirmatory determination of ractopamine residues in calves treated with growth promoting doses of the beta-agonist. Analyst, 123, 1103-1107.
Food & Drug Administration (1999). Ractopamine hydrochloride (Paylean®) type A medicated article in the feed of swine. Freedom of Information Summary, NADA Number 140-0863. United States Food & Drug Administration, Washington, DC, USA. Available by written request from the United States Food and Drug Administration; Office of Management Programs; Division of Freedom of Information (HFI-35);5600 Fishers Lane; Rockville, MD 20857 (http://www.fda.gov/cvm/efoi/foidocs.htm). Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Food & Drug Administration (2003). Ractopamine hydrochloride (OptaflexxTM45®) Type A medicated article for beef cattle. Freedom of Information Summary, NADA Number 141-0221. United States Food and Drug Administration, Washington, D.C.
Finney, P.A., Belvisi, M.G., Donnelly, L.E., Chuang, T.T., Mak, J.C.W., Scorer, C., Barnes, P.J., Adcock, I.M. & Giembycz, M.A. (2000) Albuterol-induced down-regulation of Gsalpha accounts for pulmonary beta2adrenoceptor desensitization in vivo. J. Clin. Invest., 106, 125-135
Garriott, M.L. & Rexroat, M.A. (1996) The effect of ractopamine hydrochloride on the induction of reverse mutations in Salmonella typhimurium and Escherichia coli using the Ames test. Unpublished report on studies 951114AMS2000 and 951205AMS2000 from Toxicology Research Laboratories, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Garriott, M.L. & Yount, D.J. (1996) The effect of ractopamine hydrochloride on the induction of forward mutation at the thymidine kinase focus of L5178Y mouse lymphoma cells. Unpublished report on studies 951115MLA2000 and 951219MLA2000 from Toxicology Research Laboratories, Lilly Research Laboratories, Division of Eli Lilly and Company,
Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Garriott, M.L., Schwier, L.S., Parton, J.W. & Williams, G.D. (1993) The effect of ractopamine hydrochloride given orally for 2 consecutive days on the induction of micronuclei in bone marrow of ICR mice. Unpublished report on study No. 930811MNT2000 from Toxicology Research Laboratories, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Garriott, M.L., Schwier, L.S. & Parton, J.W. (1996) The effect of ractopamine hydrochloride given orally by gavage for 14 consecutive days on the induction of micronuclei in bone marrow of Fischer 344 rats. Unpublished report on studies 960610MNT2000 and 960815MNT2000 from Toxicology Research Laboratories, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Gibson, J.P., Sells, D.M., Cheng, H.C. & Yuh, L. (1987) Induction of uterine leiomyomas in mice by medroxalol and prevention by propranolol. Toxicol. Pathol., 15, 468-473.
Gopinath, C. & Gibson, W.A. (1987) Mesovarian leiomyomas in the rat. Environ. Health Perspect., 73, 107-113.
Hoffman, B.B. (2001) Catecholamines, sympathomimetic drugs, and adrenergic receptor antagonists. In: Hardman, J.G., Limbird, L.E. & Goodman Gilman, A., eds, Goodman & Gilman's The Pharmacological Basis of Therapeutics, 10th Ed., New York: McGraw-Hill Medical Publishing Division, pp. 215-268.
Hunt, T.L. (1994) Cardiovascular activity and safety of ractopamine hydrochloride: determination of a no-effect dose. Unpublished report on study No. T4V-LC-ERAA from Pharmaco LSR, Austin, Texas 78704, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Jack, D., Poynter, D. & Spurling, N.W. (1983) Beta-adrenoceptor stimulants and mesovarian leiomyomas in the rat. Toxicology, 27, 315-320.
Jackson, M.S. & Garriott, M.L. (1996) The effect of ractopamine hydrochloride on the in vitro induction of chromosome aberrations in Chinese hamster ovary cells. Unpublished report on studies 951108CAB2000 and 951206CAB2000 from Toxicology Research Laboratories, Lilly Research Laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Lewis, J.J. (1985) Antimicrobial activity of ractopamine hydrochloride. Unpublished report on study No. JJL8505 from Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Lohse, M.J., Engelhardt, S., Danner, S. & Böhm, M. (1996) Mechanisms of beta-adrenergic receptor desensitization: from molecular biology to heart failure. Basic Res. Cardiol., 91, 29-34.
Mercier, O. (1992) Test to evaluate sensitizing potential in the guinea-pig. Unpublished report on study No. 207309 from Hazleton France, Les Oncins, L'Arbresle, France. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Mills, S.E. (2002) Biological basis of ractopamine response. J. Anim. Sci., 80 (E. Suppl.), 28-32.
Mills, S.E., Kissel, J., Bidwell, C.A. & Smith, D.J. (2003a) Stereoselectivity of porcine beta-adrenergic receptors for ractopamine stereoisomers. J. Anim. Sci, 81, 122-129.
Mills, S.E., Spurlock, M.E. & Smith, D.J. (2003b) Beta-adrenergic subtypes that mediate ractopamine stimulation of lipolysis. J. Anim. Sci., 81, 662-668.
Morgan, D.J. (1990) Clinical pharmacokinetics of beta-agonists. Clin. Pharmacokinet., 18, 270-294.
Murli, H. (1996a) Mutagenicity test on ractopamine hydrochloride measuring chromosomal aberrations in human whole blood lymphocytes with and without exogenous metabolic activation. Unpublished report on study No. 17282-0-449EC from Corning Hazleton Inc. (CHV), Leesburg Pike, Vienna, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Murli, H. (1996b) Mutagenicity test on ractopamine hydrochloride (compound 031537) measuring chromosomal aberrations in vivo in mouse bone marrow cells. Unpublished report on study No. 17282-0-451 from Corning Hazleton Inc. (CHV), Leesburg Pike, Vienna, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Murli, H. (1996c) Mutagenicity test on ractopamine hydrochloride (compound 031537) measuring chromosomal aberrations in vivo in mouse bone marrow cells. Unpublished report on study No. 17282-2-451 from Corning Hazleton Inc. (CHV), Leesburg Pike, Vienna, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Murphy, A.T. & Gillespie, T.A. (1996) Analysis of mouse lymphoma cell culture media for ractopamine and ractopamine catechol using LC/MS/MS. Unpublished summary document from Toxicology Research Laboratories, Lilly Research Laboratories, A Division of Eli Lilly and Company, Indianapolis, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Newton, G.E., Azevedo, E.R. & Parker, J.D. (1999) Inotropic and sympathetic responses to the intracoronary infusion of a beta2-receptor agonist. Circulation, 99, 2402-2407.
Oberly, T.J. & Yount, D.J. (1996) Effect of ractopamine hydrochloride on the induction of forward mutation at the thymidine kinase locus of L5178Y mouse lymphoma cells: Unpublished report on special studies 960313MLA2000, 960409MLA2000, 960416MLA2000, 960423MLA2000, 960723MLA2000. 960730MLA2000, 961015MLA2000, & 961030MLA2000 from Toxicology Research Laboratories, Lilly Research Laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Sarazan, R.D., Main, B.W. & Williams, G.D. (1993) An acute cardiovascular toxicity study with ractopamine HCl (compound 031537) administered orally in the conscious instrumented beagle. Unpublished report on study No. DV0193 from Toxicology Research Laboratories, Lilly Research Laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Scola, A.M., Chong, L.K., Suvarna, S.K., Chess-Williams, R. & Peachell, P.T. (2004) Desensitisation of mast cell beta2-adrenoceptor-mediated responses by salmeterol and formoterol. Br. J. Pharmacol., 141, 163-171.
Sears, M.R. (2002) Adverse effects of beta-agonists. J. Allergy Clin. Immunol., 110, S322-S328.
Smith, D.J. (1998) The pharmacokinetics, metabolism, and tissue residues of beta-adrenergic agonists in livestock. J. Anim. Sci., 76, 173-194.
Smith, D.J. & Paulson, G.D. (1994) Growth characteristics of rats receiving ractopamine hydrochloride and the metabolic disposition of ractopamine hydrochloride after oral or intraperitoneal administration. J. Anim. Sci., 72, 404-414.
Smith, D.J. & Rodewald, J.M. (1994) Urinary excretion of ractopamine and its conjugated metabolites by humans. Unpublished report on study No. T4V759404 from Lilly Research Laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Smith, D.J., Giddings, J.M., Heil, V.J. & Paulson, G.D. (1995) Identification of ractopamine hydrochloride metabolites excreted in rat bile. Xenobiotica, 25, 511-520.
Staehlin, M., Simons, P., Jaeggi, K. & Wigger, N. (1983) CGP 12177: a hydrophilic beta-adrenergic receptor radioligand reveals high affinity binding of agonists to intact cells. J. Biol. Chem., 258, 3496-3502.
Williams, G.D. (1987a) Comparative bioavailability of 14C-ractopamine hydrochloride (031537, EL-737) following a single oral dose of 0.125 mg/kg in the dog and monkey. Unpublished reports Nos D04686 and P03086 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. (1987b) A chronic toxicity study of ractopamine hydrochloride administered orally to beagle dogs for one year. Unpublished report on study No. D05885 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. (1987c) A target animal safety and drug tolerance study of ractopamine hydrochloride administered in the diet of swine during the finishing period. Unpublished report on study No. T4VVX8510 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. (1989) A reproductive safety study in pigs (gilts) administered ractopamine hydrochloride in the diet during the finishing period. Unpublished report on study No. VX8713 from Toxicology Division, Lilly Research Laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. (1993) A chronic study to evaluate the toxicity of ractopamine HCl in a non-rodent species whose cardiovascular responsiveness approximates that of humans given this class of compounds. Unpublished report on study No. P05191 from Toxicology Research Laboratories, Lilly Research Laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. (1998a) An oncogenic study and companion blood level study in CD-1 mice given ractopamine hydrochloride in the diet for 21 months. Unpublished report on studies M04196, M04296 & M04396 from Toxicology Research Laboratories, Lilly Research Laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. (1998b) An oncogenic study in Fischer 344 rats given ractopamine hydrochloride in the diet for 2 years. Unpublished report on studies R16696 & R16796 from Toxicology Research Laboratories, Lilly Research Laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. & Hoyt, J.A. (1987) An eleven-month two-generation reproduction study, including a teratology segment, in CD rats maintained on diets containing ractopamine hydrochloride (EL-737, Compound 31537). Unpublished report on studies R11385 and R18985 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. & Stone, D.N. (1987a) Special study: The effect of subchronic administration of ractopamine hydrochloride on heart rate and electrocardiographic waveforms in conscious rhesus monkeys. Unpublished report on study No. P02186 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. & Stone, D.N. (1987b) Special study: the hemodynamic effects of intravenous administration of ractopamine hydrochloride in anesthetized dogs. Unpublished report on study No. DC1485 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. & Stone, D.N. (1987c) Special study: the hemodynamic effects of intravenous administration of ractopamine hydrochloride in anesthetized monkeys. Unpublished report on study No. P07585 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. & Stone, D.N. (1987d) Special study: the effects of intravenous administration of ractopamine hydrochloride compared in conscious and anesthetized monkeys. Unpublished report on study No. P07685 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D. & Shoufler, R. (1993) A subchronic toxicity study of ractopamine hydrochloride given by nasogastric gavage to rhesus monkeys for 6 weeks. Unpublished report on study No. P00691 from Toxicology Research Laboratories, Lilly Research Laboratories, A Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D., Oberly, T.J., Bewsey, B.J. & Probst, G.S. (1984a). The effect of 31537 (EL-737) on the induction of forward mutation at the thymidine kinase locus of L517Y mouse lymphoma cells. Unpublished report No. 840627MLA2000 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D., Negilski, D.S. & Markey, T.F. (1984b) The acute dermal, ocular, and inhalation toxicity of compound 31537 (EL-737). Unpublished reports Nos B-D-82-84, B-E-108-84 and R-H-47-84 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D., Cochrane, R.L. & Meyerhoff, R.D. (1986a) The toxicity of ractopamine hydrochloride (EL-737, compound 31537) to juvenile mallards in a five-day dietary study. Unpublished report on study No. A00986 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D., Cochrane, R.L. & Meyerhoff, R.D. (1986b) The toxicity of ractopamine hydrochloride (EL-737, compound 31537) to bobwhite in a 14-day Acute oral study. Unpublished report on study No. A00586 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D., Cochrane, R.L. & Meyerhoff, R.D. (1986c) The toxicity of ractopamine hydrochloride (EL-737, compound 31537) to juvenile bobwhite in a five-day dietary study. Unpublished report on study No. A01186 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D., Grothe, D.W. & Francis, P.C. (1986d) The acute toxicity of ractopamine hydrochloride (EL-737, compound 31537) to rainbow trout (Salmo gairdneri) in a static test system. Unpublished report on study No. F03286 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D., Grothe, D.W. & Francis, P.C. (1986e) The acute toxicity of ractopamine hydrochloride (EL-737, compound 31537) to bluegill (Lepomis macrochirus) in a static test system. Unpublished report on study No. F03186 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D., Grothe, D.W. & Francis, P.C. (1986f) The acute toxicity of ractopamine hydrochloride (EL-737, compound 31537) to daphnia magna in a static test system. Unpublished report on study No. C00786 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D., Pohland, R.C. & Byrd, T.K. (1987) Bioavailability of radiocarbon following the administration of single oral doses of 14C-EL-737 (31537) to F344/N Hsd BR rats. Unpublished reports Nos R03985, R04085, & R04185 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Williams, G.D., Williams, P.D. & Colbert, W.E. (1989) In vitro studies of ractopamine hydrochloride in the smooth muscle of Sprague Dawley rats and Hartley albino guinea pigs and cardiac muscle of Hartley albino guinea pigs. Unpublished reports Nos PM8945 and PM 8946 from Toxicology Division, Lilly Research Laboratories, Division of Eli Lilly and Company, Greenfield, IN, USA. Submitted to WHO by Elanco Animal Health, Division of Eli Lilly and Company, Indianapolis, IN, USA.
Xiao, R.P., Cheng, H., Zou, Y., Kuschel, M. & Lattaka, E.G. (1999) Recent advances in cardiac beta2-adrenergic signal transduction. Circ. Res., 85, 1092-1100.
See Also:
Toxicological Abbreviations
Ractopamine (WHO Food Additives Series 31)
RACTOPAMINE (JECFA Evaluation)