
Pesticide residues in food - 2002 - Joint FAO/WHO Meeting on Pesticide Residues
First draft prepared by
D.B. McGregor
Toxicology Evaluation Consultants, Lyon, France
Esfenvalerate [(S)-alpha-cyano-3-phenoxybenzyl (S)-2-(4-chlorophenyl)-3-methylbutyrate], a synthetic pyrethroid insecticide, is one of the four isomers ([2S, alphaS], [2S, alphaR], [2R, alphaS] and [2R, alphaR]) found in fenvalerate in approximately equal proportions. Esfenvalerate ([2S, alphaS]) is the biologically active component of fenvalerate. As for all the synthetic pyrethroids, the insecticidal action of esfenvalerate is due to its interaction with sodium ion channels in the axons of the target species. Fenvalerate was evaluated toxicologically by the Joint Meeting in 1979, 1981, 1982, 1984 and 1986 (Annex 1, references 32, 36, 38, 42 and 47). An ADI of 0–0.02 mg/kg bw was established in 1986.
Esfenvalerate has not previously been evaluated by the Joint FAO/WHO Meeting on Pesticide Residues.
The positions of the radiolabel in the compounds used in the studies of metabolism are shown in Figure 1, and the metabolic pathways of esfenvalerate in mammals are shown in Figure 2.
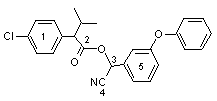
Figure 1. Positions of radiolabel in studies of the metabolism of esfenvalerate and fenvalerate
1, [14C-chlorophenyl]; 2, [14C-carbonyl]; 3, [14C-benzylic]; 4, [14C-cyano]; 5, [14C-phenoxybenzyl]
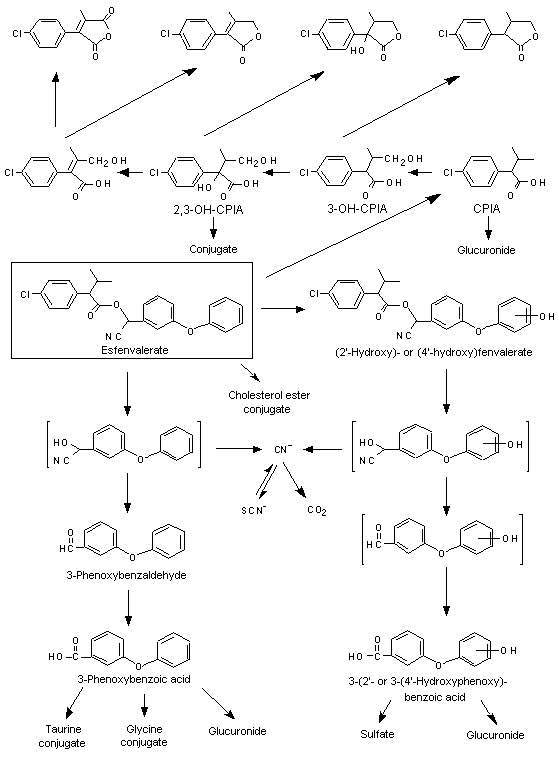
Figure 2. Metabolic pathways of esfenvalerate in mammals
CPIA, 2-(4-chlorophenyl)isovaleric acid
Rodents
Single oral doses of [14C-cyano]fenvalerate at 20 µmol/kg bw or of [14C-cyano]-, [14C-benzyl]- and [14C-carbonyl]-[2S,alphaRS]fenvalerate, [14C-carbonyl]-2-(4-chlorophenyl)isovaleric acid (CPIA), [14C]potassium thiocyanate (KS[14CN]) and [14C]potassium cyanide (K[14CN]) at five daily doses of 4 µmol/kg bw suspended in 10% Tween 80 were administered to male Sprague-Dawley rats. Urine, faeces and expired CO2 were collected daily for 6 and 14 days after administration of the single dose and for 6 days after the last of the five daily doses. Tissue samples were obtained 6 and 14 days after dosing and analysed by thin-layer chromatography (TLC), autoradiography and liquid scintillation counting.
Similar excretion patterns were seen after single and repeated administration, except with KS[14CN] and K[14CN] (Tables 1 and 2). The administered radioactivity was almost completely recovered from urine and faeces within 2 days of dosing with [14C-carbonyl]- and [14C-benzyl]-[2S,alphaRS]fenvalerate (92% and 94%, respectively). None of the radioactivity administered as fenvalerate or [2S,alphaRS]fenvalerate was excreted as 14CO2. KS[14CN] and K[14CN] were excreted more slowly, with small amounts excreted as CO2 in expired air (15% and 7.5%, respectively). The concentrations of residues in tissues after dosing with [14C-carbonyl]- or[14C-benzyl]-[2S,alphaRS]fenvalerate were generally equivalent to < 0.3 mg/kg 3 days after a single dose and had declined to 0.1 mg/kg by 14 days after dosing in adrenals, blood, brain, caecum, hair, heart, intestine, kidney, liver, lung, muscle, pancreas, sciatic nerve, skin, spinal cord, spleen, stomach and testis. The only exception was fat, which contained 2 mg/kg 6 days after dosing with [14C-carbonyl]-[2S,alphaRS]fenvalerate, the concentration declining to 0.5 mg/kg 14 days after dosing.
The highest concentrations of residues of [14C-cyano]fenvalerate, [14C-cyano][2S,alphaRS]-fenvalerate, KS[14CN] and K[14CN] after the single oral dose were found in hair and skin. Hair contained the equivalent of 36 mg/kg [2S,alphaRS]fenvalerate, 41 mg/kg KCN and 120 mg/kg KSCN 6 days after dosing. At the same time, skin contained 3 mg/kg [2S,alphaRS]fenvalerate, 4.5 mg/kg KCN and 9 mg/kg KSCN. Fat and blood contained 1.2–1.4 mg/kg [2S,alphaRS]fenvalerate or fenvalerate, and blood contained 1.4–1.7 mg/kg KSCN or KCN. A similar pattern was seen after repeated dosing.
Table 1. Per cent administered radioactivity excreted by male rats 6 days after a single oral dose of 20 µmol/kg bw
|
Medium |
[14C-carbonyl] [2S,alphaRS] fenvalerate |
[14C-benzyl] [2S,alphaRS] fenvalerate |
[14C-cyano] [2S,alphaRS] fenvalerate |
[14C-cyano] fenvalerate |
[14C-carbonyl] 2- (4-chlorophenyl)-isovaleric acid |
K[14CN] |
KS[14CN] |
|
Expired air |
0 |
0 |
7.5 |
10 |
0 |
7.5 |
15 |
|
Urine |
60 |
46 |
22 |
24 |
78 |
32 |
31 |
|
Faeces |
32 |
48 |
38 |
41 |
20 |
22 |
12 |
|
Total |
92 |
94 |
66 |
75 |
98 |
62 |
59 |
From Ohkawa et al. (1979). K[14C], [14C]potassium cyanide; KS[14C], [14C]potassium thiocyanate
Table 2. Per cent administered radioactivity excreted 6 days after five consecutive daily doses of [2S]fenvalerate at 4 µmol/kg bw per day
|
Medium |
Carbonyl |
Benzyl |
Cyano |
|
Expired air |
0 |
0 |
7 |
|
Urine |
48 |
59 |
25 |
|
Faeces |
50 |
38 |
34 |
|
Total |
98 |
96 |
64 |
From Ohkawa et al. (1979)
Autoradiography 6 and 24 h after the single oral dose of [14C-cyano][2S,alphaRS]fenvalerate showed that the radioactivity was distributed throughout the body, except to the brain and spinal cord. The highest concentrations, after the gastrointestinal tract, appeared to be in liver and lung, but this conclusion was based only on visual inspection of single autoradiographs available 6 h and 24 h after dosing. At 144 h after dosing, radioactivity was detected only in hair, skin and stomach contents (the last presumably as a result of grooming). The distribution to the skin did not show regional variation, indicating that it was the result of contamination from urine. TLC of the stomach contents 144 h after the single dose of [14C-cyano][2S,alphaRS]fenvalerate showed that essentially all the radioactivity was associated with thiocyanate, at a concentration of 41 ppm. A similar pattern was seen with KS[14CN] and K[14CN]. The concentrations of [2S,alphaRS]fenvalerate in blood and liver reached a maximum (0.5 mg/kg) 3 h after a single dose of [14C-cyano][2S,alphaRS]-fenvalerate and then rapidly declined to about 0.01 mg/kg by 48 h. There appeared to be no significant difference in the excretion patterns of [2S,alphaRS]fenvalerate and fenvalerate (Ohkawa et al., 1979).
Five male and five female Sprague-Dawley rats were given [14C-chlorophenyl]- or [14C-phenoxybenzyl]fenvalerate at 2.5 or 10 mg/kg bw, [14C-chlorophenyl]- or [14C-phenoxybenzyl] esfenvalerate at 2.5 mg/kg bw or [14C-chlorophenyl]- or [14C-phenoxybenzyl] esfenvalerate mixed with the three other unlabelled isomers. The chemicals were administered orally by gavage in corn oil. Urine and faeces were collected daily for 7 days, when the rats were killed and the major tissues were collected. Excreta and tissues were analysed for radioactivity by liquid scintillation counting. There were no significant differences in the excretion of [14C-chlorophenyl]- or [14C-phenoxybenzyl]-labelled material according to dose, sex or quantities of metabolites. About 63–86% of the radioactivity was excreted 1 day after dosing, and 95–101% had been excreted by 7 days after dosing. Between 20% and 39% of the radioactivity was excreted in urine, with the remainder in the faeces. The tissue concentrations were generally low, the highest values being found in fat (180–310 µg/kg at the lower dose and 1300 µg/kg at the higher dose). The concentration of residues after treatment with fenvalerate (10 mg/kg) was generally four times higher than that after treatment with esfenvalerate (2.5 mg/kg) (Kaneko et al., 1985).
In a study of placental transfer, groups of three pregnant Sprague-Dawley rats were dosed orally with [14C-chlorophenyl]esfenvalerate at 2.5 mg/kg bw per day or [14C-chlorophenyl]-fenvalerate at 10 mg/kg bw per day in corn oil. Dosing was begun on day 13 of gestation and consisted of either a single dose of [14C]-labelled compound or three consecutive daily doses of unlabelled compound followed by two consecutive daily doses of labelled compound. The rats were killed 3, 6, 12, 24 or 48 h after the final dose. Radioactivity in the fetuses, amniotic fluid, placentas, maternal blood and ovaries was measured by liquid scintillation counting. Maximum levels of radioactivity were found within 12 h after the last dose, after which they declined rapidly. The concentration of fenvalerate residues was generally four times higher than that of esfenvalerate residues, except in maternal blood, placenta, amniotic fluid and ovary 24 h after a single dose, where the concentration was 18–31 times higher, and the ovaries 24 and 48 h after multiple doses, where the concentrations were similar. The concentrations in tissues were highest 3–6 h after the last dose and then declined rapidly. The highest concentrations of residues from both pyrethroids were found in maternal blood and ovaries and the lowest were found in the fetuses and amniotic fluid. The highest concentration of esfenvalerate equivalents found in fetuses was 150 µg/kg, 6 h after the last of the five consecutive daily doses. During the experiment, < 0.07% of the applied radioactivity was found in fetuses, indicating that there was virtually no transfer from maternal blood. There was no evidence of accumulation of esfenvalerate in fetal tissues or amniotic fluid (Shiba et al., 1985).
Five male and five female ddY mice were given [14C-chlorophenyl]- or [14C-phenoxybenzyl]-fenvalerate at a dose of 2.5 or 10 mg/kg bw, [14C-chlorophenyl]- or [14C-phenoxybenzyl]-esfenvalerate at 2.5 mg/kg bw or [14C-chlorophenyl]- or [14C-phenoxybenzyl]esfenvalerate mixed with the three other unlabelled isomers. The chemicals were administered orally by gavage in corn oil. Urine and faeces were collected daily for 7 days, when the animals were killed and major tissues excised. Excreta and tissues were analysed for radioactivity by liquid scintillation counting. In both sexes, 86–94% of the administered radioactivity was excreted within 1 day of dosing, and 94–102% was excreted within 7 days. Roughly equal quantities of radioactivity were excreted in urine and faeces. The highest concentrations of residues were detected in fat tissues, with 0.12–0.48 mg/kg after the dose of 2.5 mg/kg bw and 1.2–1.6 mg/kg after the dose of 10 mg/kg bw (Kaneko et al., 1985).
Groups of six male and six female ddY mice were fed diets containing [14C-chlorophenyl]-esfenvalerate at a concentration of 25 ppm or [14C-chlorophenyl]fenvalerate at a concentration of 25 or 100 ppm, providing estimated daily intakes of 120 µg/mouse of esfenvalerate and 110–120 µg and 420–480 µg/mouse of fenvalerate. Mice were killed after 10, 19, 24 or 28 days on the diets and after 4, 7, 21 or 28 days of untreated diet. The concentrations of residues were highest in fat (about 7 µg of esfenvalerate equivalents per gram of tissue after 24–28 days), about sevenfold lower in the adrenals, lymph nodes and skin, about 10-fold lower in liver and about 25-fold lower in blood. The concentrations of radioactivity approached a plateau after 24–28 days of feeding and declined when the mice were returned to untreated diet. The total concentrations of radioactivity in adrenals, spleen and ovaries were lower after feeding of esfenvalerate than after a similar dose of fenvalerate. In other tissues, the concentrations of total radioactivity were similar. About four times more total radioactivity was found in the tissues of animals fed fenvalerate at 100 ppm in the diet than in those at 25 ppm. No differences were found between the sexes (Isobe et al., 1985).
Groups of five male and five female ddY mice received 10 consecutive daily oral doses of [14C-chlorophenyl]esfenvalerate at 2.5 mg/kg bw or [14C-chlorophenyl]fenvalerate at 10 mg/kg bw. Excreta were collected daily for 7 days after the last dose. Most of the radioactivity (90% of that administered) was excreted within 1 day of the last dose. Excretion was 91–98% complete within 7 days, with equal amounts found in urine and faeces. Thus, repeated dosing with either esfenvalerate or fenvalerate resulted in similar excretion patterns with respect to dose or sex (Kaneko et al., 1985).
One male and one female Sprague-Dawley rats and four male and four female ddY mice were given single oral doses of [14C-carbonyl]-, [14C-benzyl]- or [14C-cyano]fenvalerate at 7 mg/kg bw or [14C-chlorophenyl]-, [14C-phenoxybenzyl]- or [14C-cyano]esfenvalerate at 4.2 mg/kg bw. Each of the labelled compounds was also administered to two male and two female rats at a dose of 30 mg/kg bw. Urine and faeces were collected daily for 6–7 days, and CO2 was collected from those rats and mice that received [14C-cyano]-labelled compound. The rats were killed 6–7 days after dosing. Excreta and tissues were analysed for metabolites by TLC, autoradiography and liquid scintillation counting. The two rats given low doses of [14C-chlorophenyl]- or [14C-phenoxybenzyl]esfenvalerate and fenvalerate excreted 93–99% of the radioactivity in urine and faeces within 6–7 days after dosing. The half-life of the radioactivity was 0.5–0.6 days. The mice excreted 95–102% of the dose within the same period, and the half-life of radioactivity was 0.5–0.6 days. The rats given [14C-chlorophenyl]- or [14C-phenoxybenzyl]esfenvalerate at 30 mg/kg bw excreted 86–97% of the dose within 6 days, with a half-life of 0.6–0.9 days. In rats given the lower doses of [14C-cyano]fenvalerate and esfenvalerate, excretion of radioactivity was complete within 6–7 days, with a half-life of 1.7–2.0 days. In similarly treated mice, excretion was 88–89% complete within 6–7 days, with a half-life of 1–1.7 days. Exhaled [14C]carbon dioxide accounted for 6–14% of the administered radioactivity in both species. The residues in the tissues of rats and mice given the lower doses of [14C-chlorophenyl]- and [14C-phenoxybenzyl]esfenvalerate and [14C-carbonyl]- and [14C-benzyl]fenvalerate were very low 6–7 days after dosing. The highest concentrations were found in fat, with 0.71–1.5 mg/kg in rats and 0.45–0.89 mg/kg in mice, expressed as equivalents. The concentrations of residues in fat of rats given esfenvalerate at 30 mg/kg bw were 4.3–8.9 mg/kg as equivalents. The concentrations in blood, hair, liver and skin of both species were < 0.36 mg/kg after the lower dose and 0.81 mg/kg after the higher dose. [14C-cyano]-labelled compounds left higher concentrations of residues in hair, skin and stomach contents than the other compounds (see Ohkawa et al., 1979, above) (Kaneko et al., 1981).
Dogs
Metabolism in dogs was studied with [14C-chlorophenyl]- or [14C-phenoxybenzyl]fenvalerate administered as a single oral dose of 1.7 mg/kg bw, dissolved in corn oil, in gelatine capsules. Excreta and blood were collected daily for 3 days and analysed for radioactivity by liquid scintillation counting. Elimination of radioactivity was rapid and occurred primarily via the urine and faeces. Three days after dosing, 32% and 56% of the [14C-chlorophenyl]fenvalerate and 37% and 42% of the [14C-phenoxybenzyl]fenvalerate had been eliminated in urine and faeces, respectively. More total radioactivity was recovered in animals given [14C-chlorophenyl]fenvalerate than in those given [14C-phenoxybenzyl]fenvalerate, and the half-lives were 1 day and 0.7 day, respectively. The maximum concentration of radioactivity in blood (approximately 1 µg/ml) was attained 2 h after dosing. The concentration of radioactivity decreased rapidly in more than two phases. The blood concentration 80 h after dosing was < 0.05 and 0.05–0.1 µg/ml in animals given [14C-chlorophenyl]- or [14C-phenoxybenzyl]fenvalerate, respectively. The concentration of the parent compound was below the level of detection (0.01 mg/l) 48 h after dosing (Kaneko et al., 1984).
The metabolites of [2S,alphaRS]fenvalerate and fenvalerate produced by male Sprague-Dawley rats were investigated by one-dimensional TLC with two solvent systems and detected by autoradiography. Metabolites were identified either by co-chromatography with authentic standards or extracted from the gel and identified with infra-red or mass spectroscopy or nuclear magnetic resonance.
More than 20 metabolites were identified. The major radioactive products in the faeces were unmetabolized fenvalerate and two ester metabolites (2’-hydroxy- and 4’-hydroxyfenvalerate). No unmetabolized fenvalerate was found in urine. The major urinary metabolites were CPIA, 3-phenoxybenzoic acid, SCN– and products of further oxidation and conjugation. As the excretion, tissue residues and metabolite excretion patterns of fenvalerate and esfenvalerate were similar, it was concluded that the two compounds were metabolized similarly. The significant metabolic reactions were oxidation at the 2 and 4 positions of the acid and at the 2’ and 4’ positions of the alcohol moiety, cleavage of the ester linkage and conversion of the cyano group to SCN– and CO2 (Ohkawa et al., 1979).
When groups of male and female Sprague-Dawley rats and ddY mice were dosed orally with 14C-labelled acid, alcohol or cyano moieties of fenvalerate and esfenvalerate, the metabolites found were essentially the same in both species, with no differences in relation to isomer, dose or sex. Several differences between species were observed in the nature and amounts of metabolites derived from the alcohol moiety: 4’-hydroxylation was greater in rats than in mice; 3-phenoxy-benzoic acid was conjugated with taurine in mice but not in rats; sulfate conjugation of 3-(4’-hydroxyphenoxy)benzoic acid was more extensive in rats than in mice; and conversion of the cyano group to SCN– was more extensive in mice than in rats. There was little difference between the species in elimination of acid- and alcohol-labelled fenvalerate and esfenvalerate, but mice eliminated the cyano-labelled compounds more quickly and more completely than rats. Higher doses of acid and alcohol labelled compounds were excreted more slowly than low doses, and the cyano labels were more slowly excreted than the acid and alcohol labels (Kaneko et al., 1981).
In a 28-day study in mice , the main metabolites in the liver and kidney of animals fed diets containing [14C-chlorophenyl]esfenvalerate were CPIA and its hydroxylated derivative. These disappeared after untreated diets were fed. These metabolites were also found in mice fed diets containing [14C-chlorophenyl]fenvalerate. In addition, CPIA cholesterol ester was found in mice given [14C-chlorophenyl]-fenvalerate but not in those given [14C-chlorophenyl]esfenvalerate. This metabolite was also present at the end of 28 days on untreated diets and accounted for the majority of the radioactivity present at that time (Isobe et al., 1985).
In a study of placental transfer in rats, metabolites were tentatively identified by co-chromatography with standards on TLC and five solvent systems, visualized by autoradiography and ultraviolet fluorescence and quantified by liquid scintillation counting. The main metabolites, CPIA, its hydroxlated derivative in its free and lactone forms, and the parent compounds were found in maternal blood and placenta and in fetuses. A trace of the CPIA cholesterol ester, derived from [14C-chlorophenyl]fenvalerate, was found in maternal blood and placenta but not fetuses. Consecutive treatment resulted in slightly higher concentrations of 3-hydroxy-CPIA in rats given esfenvalerate than those given fenvalerate and slightly higher concentrations of CPIA after dosing with fenvalerate than after esfenvalerate (Shiba et al., 1985).
In a study in dogs given a single oral dose of [14C-chlorophenyl]- or [14C-phenoxybenzyl]-fenvalerate, the excreta were analysed for metabolites, which were tentatively identified and quantified by TLC co-chromatography with standards and six solvent systems, with or without enzymatic hydrolysis. The metabolites were visualized by ultraviolet fluorescence and autoradiography. The main metabolites 0–1 day after dosing are shown in Table 3. Although less total radioactivity was recovered in dogs than in rats and mice, the disappearance pattern and half-lives in dogs and rats were similar. The species differences in metabolism were that 2’-hydroxylation of the alcohol moiety did not occur in dogs; 3-phenoxybenzyl alcohol and 3-(4’-hydroxyphenoxy)benzyl alcohol were detected only in dogs; 3-phenoxybenzoyl glycine (glycine conjugate of 3-phenoxybenzoic acid) was the predominant conjugate of the alcohol moiety in dogs but a minor one in rats; and the glucuronides of the acid moiety and its hydroxy derivatives were more prevalent in dogs (Kaneko et al., 1984).
Table 3. Metabolites (% of administered radioactivity) in excreta of dogs after a single oral dose of [14C]fenvalerate at 1.7 mg/kg bw
|
Metabolite |
Conjugation |
[14C-Chlorophenyl] |
[14C-phenoxybenzyl] |
||
|
Urine |
Faeces |
Urine |
Faeces |
||
|
Fenvalerate |
|
0.2 |
8.8 |
|
3.7 |
|
4’-Hydroxyfenvalerate |
|
|
1.8 |
|
2.3 |
|
3-Phenoxybenzyl alcohol |
|
|
|
|
1.3 |
|
3-(4’-Hydroxyphenoxy)benzyl alcohol |
Free |
|
|
|
3.3 |
|
|
Glucuronide |
|
|
0.6 |
|
|
3-Phenoxybenzoic acid |
Free |
|
|
3.1 |
1.4 |
|
|
Glucuronide |
|
|
|
0.4 |
|
|
Glycine |
|
|
8.4 |
|
|
3-(4’-Hydroxyphenoxy)benzoic acid |
Free |
|
|
2.4 |
3.5 |
|
|
Sulfate |
|
|
5.8 |
1.1 |
|
|
Glucuronide |
|
|
3.8 |
|
|
2-(4-Chlorophenyl)isovaleric acid |
Free |
2.2 |
3.2 |
|
|
|
|
Glucuronide |
27 |
|
|
|
|
2-(3-Hydroxy-4-chlorophenyl)isovaleric acid |
Free |
2.9 |
|
|
|
|
2-(3-Hydroxy-4-chlorophenyl)isovaleric acid lactone |
|
|
1.9 |
|
|
|
2-(2,3-Hydroxy-4-chlorophenyl)isovaleric acid |
|
3.4 |
|
|
|
From Kaneko et al. (1984)
The excreta of rats in an earlier comparative study were analysed for metabolites by TLC with five solvent systems, autoradiography and liquid scintillation counting, sometimes after enzyme treatment. The metabolites were tentatively identified by co-chromatography with unlabelled standards and were visualized by ultraviolet fluorescence. Metabolite excretion was similar in the two sexes. In faeces, 44–60% of the administered radioactivity from [14C-chlorophenyl]- or [14C-phenoxybenzyl]-labelled materials was excreted as the parent compound; 2.9–6.5% of [14C-chlorophenyl]-labelled material was excreted as 2’-hydroxy and 4’-hydroxy parent compound. The amount of [14C-phenoxybenzyl]labelled material that was excreted was similar to that of 4’-hydroxyfenvalerate.
Other faecal metabolites were CPIA, its 4- and 2,3-hydroxy derivatives and 2-(4-chlorophenyl)-cis-2-butenedioic acid in their free forms from the acid moiety and 3-phenoxybenzoic acid and 3-(4’-hydroxyphenoxy)benzoic acid in their free forms from the alcohol moiety. The main urinary metabolites from the acid moiety were CPIA glucuronide, 3-hydroxy-CPIA (free and lactone), 2,3-hydroxy-CPIA glucuronide and 2-(4-chlorophenyl)-cis-2-butenedioic acid anhydride. The main urinary metabolite from the alcohol moiety was 3-(4’-hydroxyphenoxy)benzoic acid sulfate (16–24% of the administered radioactivity). Other metabolites included 3-phenoxybenzoic acid (free and glycine and glucuronide conjugates), 3-(4’-hydroxyphenoxy)benzoic acid (free and glucuronide) and 3-(2’-hydroxyphenoxy)benzoic acid (free and sulfate) (Kaneko et al., 1985).
The excreta of mice dosed with [14C-acid]- and [14C-alcohol]-labelled fenvalerate and esfenvalerate, as described above, were similarly analysed for metabolites. There were no significant differences between the sexes in the metabolites excreted. A trace amount of the cholesterol ester of CPIA was found in the faeces of mice treated with [14C-chlorophenyl]fenvalerate. In faeces, 26–48% of the radioactivity from [14C-chlorophenyl]- or [14C-phenoxybenzyl]-labelled material was excreted as the parent compound, 1.4–4.4% was excreted as 4’-hydroxyfenvalerate, and 0.0–1.1% was excreted as 2’-hydroxyfenvalerate. The other faecal metabolites were the same as those found in rats. The urinary metabolites after administration of acid-labelled material consisted mainly of CPIA (free and glucuronide) and 2,3-hydroxy-CPIA glucuronide. The main metabolites of alcohol-labelled material were the taurine conjugate of 3-phenoxybenzoic acid (7.9–17% of administered radioactivity) and the free, sulfate and glucuronide forms of 3-(4’-hydroxyphenoxy)benzoic acid (total, 6.9–9.9% of administered radioactivity). The minor metabolites were predominantly the same as those found in rats (Kaneko et al., 1985).
The [2R,alphaS] isomer of fenvalerate was established as the only source of cholesteryl [2R]-2-(4-chlorophenyl)isovalerate. [14C-chlorophenyl]Fenvalerate was administered to groups of two male Sprague-Dawley rats and four male ddY mice at a single oral dose of 2.5 mg/kg bw as the [2S,alphaS], [2S,alphaR], [2R,alphaS] or [2R,alphaR] isomer of fenvalerate. Excreta were collected for 6 days, and the radioactivity in tissues and excreta was analysed by liquid scintillation counting. The concentrations of residues of [2S,alphaS], [2S,alphaR] and [2R,alphaR] isomers were low (equivalent to 100 µg/kg or less) in all tissues except fat (310–760 µg/kg) in both species, but administration of the [2R,alphaS] isomer resulted in higher concentrations in all tissues, particularly the adrenals, liver, mesenteric lymph nodes and spleen, although the concentration in fat was similar to those seen after administration of the other isomers. Thus, this lipophilic tissue residue did not occur to any appreciable extent in rats or mice given esfenvalerate (Kaneko et al., 1986).
To demonstrate this more clearly, groups of male ddY mice were fed diets containing 500 mg/kg of [2R,alphaS], [2S,alphaS] or [2R,alphaR] isomer for 1 or 2 weeks. Similar results were obtained, and further analysis by TLC showed that a lipophilic metabolite was predominant only in tissues of mice treated with the [2R,alphaS] isomer. The livers of [2R,alphaR]-treated mice contained trace amounts. The lipophilic metabolite was identified by nuclear magnetic resonance spectroscopy or TLC co-chromatography, with authentic standards and three different solvent systems, high-performance liquid chromatography and mass spectrometry and was found to be the CPIA cholesterol ester (Kaneko et al., 1986).
In an investigation of the mechanism of formation of the CPIA cholesterol ester from the [2R,alphaS] isomer, various tissues, including brain, kidney, liver, spleen, adrenals, intestines and lymph nodes, were excised from male Sprague-Dawley rats, ddY mice, beagle dogs and rhesus monkeys. A 9000 × g supernatant and pellet, microsomal fraction (100 000 × g pellet) and 100 000 × g supernatant were obtained by centrifugation. Plasma was obtained from blood by centrifugation. After incubation of each of the [14C]-labelled isomers of fenvalerate with the 9000 × g supernatants, the [2S,alphaS], [2S,aphaR] and [2R,alphaR] isomers produced the hydrolysis product CPIA only, whereas the [2R,alphaS] isomer resulted in formation of CPIA and its cholesterol ester. The production of CPIA depended on the tissue: in mice, it was formed most rapidly from the [2R,alphaS] isomer in kidney, brain and spleen, from the [2R,alphaS] and [2R,alphaR] isomers in liver and from the [2S,alphaR] and [2R,alphaR] isomers in plasma.
CPIA cholesterol ester was formed only from the [2R,alphaS] isomer in all tissues and species examined, apart from mouse kidney, which produced only a trace amount from the [2R,alphaR] isomer. There were some species differences in the enzymatic activities of various tissues, but in general the greatest activity was found in mouse tissue. Mouse kidney, brain and spleen produced the most of this ester. Free CPIA was not a substrate for formation of the cholesterol ester. The activity was destroyed by heat, inhibited by tetraethylpyrophosphate and not affected by para-chloromercuribenzoic acid. After incubation of the [2R,alphaS] isomer with subcellular fractions of mouse brain, liver, kidney and spleen, most of the CPIA cholesterol ester-forming activity was found in microsomes (Miyamoto et al., 1986).
The roles of three known routes of cholesterol ester synthesis in the formation of CPIA cholesterol ester were investigated. Mouse liver microsomes were incubated with oleic acid or CPIA with and without ATP, coenzyme A and tetraethylpyrophosphate. Although the cholesterol ester was formed, the activity was inhibited by tetraethylpyrophosphate, whereas this compound had no effect on oleic acid cholesterol ester formation, indicating that acylcoenzyme A cholesterol acyl transferase was not involved in the formation of the CPIA cholesterol ester.
In a further experiment, mouse plasma and kidney microsomes were incubated with lecithin or [2R,alphaS]fenvalerate. Lecithin cholesterol ester was obtained from plasma but not kidney microsomes, and CPIA cholesterol ester was obtained from kidney microsomes but not plasma. This showed that lecithin cholesterol acyl transferase was not involved in formation of CPIA cholesterol ester. Incubation of oleic acid, [2R,alphaS]fenvalerate or CPIA with intestinal mucosa microsomes or a 100 000 × g supernatant of intestinal mucosa and cholesterol resulted in a cholesterol ester from oleic acid only, indicating that cholesterol esterase was not involved.
In order to identify the enzyme responsible for the formation of the CPIA cholesterol ester, mouse kidney and liver microsomes were incubated with [2R,alphaS]fenvalerate only or [2R,alphaS]fenvalerate plus methanol, ethanol, n-propanol or n-butanol. In the presence of the straight-chain alcohols, the amount of cholesterol ester decreased and the amount of CPIA alkyl ester increased with increasing chain length. In addition, incubation of mouse kidney microsomes with [2R,alphaS] isomer and cholesterol or cholesterol oleate resulted in the production of CPIA cholesterol ester only from cholesterol. Therefore, the cholesterol ester is produced from free cholesterol, and alkyl alcohols also act as ‘receptors’ for CPIA. It was inferred that formation of the cholesterol ester from [2R,alphaS]fenvalerate was a transesterification reaction mediated by microsomal carboxyesterase(s). When mouse kidney microsomes were solubilized with digitonin, they retained the hydrolysis activity but did not produce the CPIA cholesterol ester. Addition of liposomes to the solubilized enzyme restored the cholesterol ester-forming activity. These findings suggest that, in microsomes, carboxyesterase(s) and cholesterol are spatially arranged so that cholesterolysis of the intermediary CPIA–enzyme complex is possible, resulting in the formation of CPIA cholesterol ester (Miyamoto et al., 1986; Kaneko et al., 1988).
The acute LD50 and LC50 values for esfenvalerate are shown in Table 4.
Table 4. Results of studies of the acute toxicity of esfenvalerate
|
Species |
Strain |
Sex |
Route and vehicle |
LD50 (mg/kg bw) or LC50 (mg/m3) |
Reference |
|
Mouse |
ICR |
Male |
Oral, in 0.5% methyl cellulosea |
320 |
Omodaka (1986) |
|
Female |
250 |
|
|||
|
Rat |
Sprague-Dawley |
Male |
Oral, in corn oila |
90 |
Omodaka (1985a) |
|
Female |
90 |
|
|||
|
Rat |
Sprague-Dawley |
Male |
Dermal, in corn oila |
> 5000 |
Omodaka (1985b) |
|
Female |
> 5000 |
||||
|
Rabbit |
New Zealand white |
Male |
Dermal, undilutedb |
> 2000 |
Maedgen (1985a) |
|
Female |
> 2000 |
|
|||
|
Rat |
Sprague-Dawley |
Male |
Inhalationa |
480 |
Kohda (1985) |
|
Female |
570 |
a Purity of total isomer, 94.5%; purity of isomer ratios Aalpha:Abeta:Balpha:Bbeta, 87.2%:7.4%:4.4%:0.6%
b Purity not stated.
Esfenvalerate was administered in methyl cellulose as a single oral dose to groups of 10 ICR mice of each sex. All deaths occurred within 24 h of dosing. The overt signs of toxicity included muscular fibrillation, tremors, decreased spontaneous activity, ataxia, limb paralysis, irregular respiration and salivation. These signs, which are typical of type-II pyrethroids, developed gradually 10 min after treatment but had disappeared in surviving animals within 2 days. There were no treatment-related effects on body weight. The only gross pathological effect, seen in some mice that died during the study, was gastric haemorrhaging. Minor gastric ulcerations were also observed in feeding studies at high doses (see section 2.2). No treatment-related pathological changes were found in any of the surviving animals. The acute oral LD50 of esfenvalerate in male and female ICR mice was 320 and 250 mg/kg bw, respectively (Omodaka, 1986).
Esfenvalerate was administered in corn oil at a single oral dose to groups of 10 Sprague-Dawley rats of each sex. All deaths occurred within 24 h of dosing. Treatment-related clinical signs occurred within 1 h of dosing and resolved within 3 days in surviving animals. The treatment-related effects included muscular fibrillation, tremors, decreased spontaneous activity, ataxia, limb paralysis, irregular breathing, dyspnoea, salivation, hyperexcitability and choreoathetotic syndrome. These symptoms are all typical of type-II pyrethroids. The only treatment-related gross pathological effect was gastric haemorrhaging, which occurred in some rats that died during the study. No gross changes were seen in surviving animals post mortem. The acute oral LD50 of esfenvalerate in both male and female Sprague Dawley rats was 88 mg/kg bw (Omodaka, 1985a).
The difference in toxicity of esfenvalerate between rats and mice may be related in part to the vehicles used. Thus, the oral LD50 of fenvalerate in rats was 450 mg/kg bw when given in dimethyl sulfoxide and > 3200 mg/kg bw when given in polyethylene glycol:water. The corresponding values for mice were 100–300 mg/kg bw and 1200 mg/kg bw (WHO, 1979).
Esfenvalerate was applied at single doses of < 5000 mg/kg bw to clipped, unabraded dorsal skin of groups of 10 Sprague-Dawley rats of each sex and kept in contact with the skin for 24 h under a semi-occlusive dressing. Treatment-related signs of toxicity were seen in animals at doses > 1000 mg/kg bw 2 h to 8 days after dosing and included muscular fibrillation, decreased spontaneous activity, ataxia, irregular breathing and urinary incontinence. These symptoms of toxicity are all typical of type-II pyrethroids. There were no deaths in the study, and no pathological changes were found at autopsy. The acute dermal LD50 of esfenvalerate to Sprague-Dawley rats was > 5000 mg/kg bw (Omodaka, 1985b).
Esfenvalerate was applied at a single dose of 2000 mg/kg bw to the clipped, unabraded dorsal skin of groups of five New Zealand white rabbits of each sex and kept in contact with the skin by means of an occlusive dressing for 24 h. One treated rabbit died. The signs of toxicity included decreased activity, ataxia, body tremors, constricted pupils, decreased defaecation and urination, diarrhoea, emaciation, muscle tremors, poor hind-limb coordination and small faeces. The acute dermal LD50 of esfenvalerate in both male and female rabbits was > 2000 mg/kg bw (Maedgen, 1985a).
Groups of 10 Sprague-Dawley rats of each sex were exposed (whole body) to corn oil mists containing six concentrations of esfenvalerate for 4 h. The actual measured concentrations in the chambers ranged from 2.4 to 1100 mg/m3, and the median aerodynamic diameter of the mist particles ranged from 0.94 to 1.1 µm. Dose-related deaths occurred at concentrations of 400 mg/m3 and above, during exposure and within 2 h of termination of exposure. All animals exposed to the highest concentration of 1100 mg/m3 died. Treatment-related clinical signs were dose-related in both time of onset and severity. At concentrations > 14 mg/m3, symptoms occurred within 30 min of exposure and lasted for up to 4 days after dosing. The signs at > 200 mg/m3 included hyperpnoea, dyspnoea, nasal discharge, urinary incontinence, lachrymation, salivation, choreoathetotic movements, tremors, aggressive sparring, ataxia and hypersensitivity to sound. Body-weight gain was comparable across groups by the end of the study. At autopsy, brown spots were seen on the lungs in all groups, including controls. Apart from autolysis (due to delay in autopsy) of the intestinal tract of animals that died during the study, histopathological examination showed no treatment-related differences from control rats. The acute inhalation LC50 values of esfenvalerate in rats were 480 and 570 mg/m3 for males and females, respectively (Kohda, 1985).
The potential of esfenvalerate to irritate the skin was evaluated according to the 1982 guidelines of the Environmental Protection Agency (USA) in three male and three female New Zealand white rabbits whose skin had been shaved 24 h before exposure. A volume of 0.5 ml of undiluted esfenvalerate was applied to one intact and one abraded skin site on each rabbit, and then occlusive dressings were applied for 4 h. Skin was examined for irritation 4.5, 24, 48 and 72 h after application. No reactions were observed on either intact or abraded sites. The primary irritation score was therefore 0, and the material was judged to be not irritating to the skin (Chazono, 1985).
The potential dermal irritancy of technical-grade esfenvalerate was evaluated in three male and three female New Zealand white rabbits whose skin had been shaved 24 h before exposure. A volume of 0.5 ml of undiluted esfenvalerate was applied to intact skin site on each rabbit, and then occlusive dressings were applied for 4 h. The skin was examined for irritation 1 h and 1, 2 and 3 days after removal of the dressing. Technical-grade esfenvalerate was minimally irritating, with a primary irritation score of 0.1 (maximum possible being 8.0) (Maedgen, 1985b).
The potential ocular irritancy of esfenvalerate was evaluated according to the 1982 Environmental Protection Agency guidelines in three male and three female New Zealand white rabbits. A volume of 0.1 ml of undiluted esfenvalerate was applied to the right eye of each rabbit, and irritation was scored 1, 24, 48 and 72 h after exposure. At 1 h, all rabbits had slight conjunctival redness, and five had slight chemosis. A moderate discharge was also present after 24 h. There were no corneal or iridial reactions. All reactions had disappeared by 48 h. The ocular irritation scores were 3.7 at 1 h and 4.7 at 24 h after treatment. The ocular irritating potential was judged to be minimal (Chazono, 1985).
The potential ocular irritancy of technical-grade esfenvalerate was evaluated in three male and three female New Zealand white rabbits. A volume of 0.1 ml of undiluted esfenvalerate was applied to the right eye of each rabbit. The eyes of a similarly treated group of three male rabbits were flushed with tap water 30 s after exposure. Eye irritation was scored 1 h and 1, 2, 3 and 7 days after exposure. Esfenvalerate was mildly irritating to unwashed eyes, with a maximum irritation score of 12 (maximum possible score, 110) 1 day after treatment. The maximum average irritation score was 10 for washed eyes and was observed 1 h after exposure (Maedgen, 1985c).
The dermal sensitization potential of esfenvalerate was evaluated in the Magnusson–Kligman maximization test in groups of 20 male Hartley guinea-pigs. Esfenvalerate was injected intradermally into each guinea-pig as a 25% solution, as this concentration had been demonstrated previously to cause slight erythema and oedema. Six points were selected for challenge injection within a clipped intrascapular area on each guinea-pig, and pairs of intradermal injections were made of: distilled water emulsified in Freund complete adjuvant; a 25% solution of esfenvalerate in corn oil or a 0.05% solution of dinitrochlorobenzene in corn oil; and a 1:1 emulsion of 50% esfenvalerate or 0.1% dinitrochlorobenzene in Freund complete adjuvant and distilled water. Control groups were similarly treated except that esfenvalerate and dinitrochlorobenzene were omitted. One week later, lint loaded with 0.4 ml of esfenvalerate or 0.5% dinitrochlorobenzene was applied to the intrascapular area and secured with tape for 48 h. For the control groups, the lint was loaded with 0.4 ml of corn oil. Two weeks after the second sensitization, the hair was clipped once more, and the guinea-pigs were challenged topically with 0.2 ml of non-irritating concentrations of esfenvalerate and dinitrochlorobenzene on lint patches, which were taped in place. After 24 h, the occlusive patches were removed, and skin reactions were scored 24 and 48 h after removal. Although the individual skin reactions were mild, the sensitization rate was 85%, leading to the conclusion that esfenvalerate is a skin sensitizer which induces a mild but high response (Chazono, 1986a).
The dermal sensitization potential of esfenvalerate was also evaluated in a Buehler test in groups of 10 male Hartley guinea-pigs. Flank hair was clipped from each guinea-pig, and a lint patch loaded with 0.5 ml of esfenvalerate (a non-irritating load) or the positive control, dinitrochlorobenzene, was applied and taped in place for 6 h. This procedure was repeated every other day for a total of nine applications. Two weeks after the final sensitization treatment, the guinea-pigs were challenged with similar, single treatments of esfenvalerate or dinitrochloro-benzene. The skin reactions were scored 24 and 48 h after removal of the patches. No skin reactions were observed in the group challenged with esfenvalerate, whereas erythema and oedema were observed in the positive control group. It was concluded that esfenvalerate did not induce sensitization under the conditions of the Buehler test (Chazono, 1986b). The difference in the result from that of Chazono (1986a) was most probably due to the greater sensitivity of the Magnusson–Kligman test.
The dermal sensitization potential of technical-grade esfenvalerate was tested in groups of five male and five female albino Duncan-Hartley guinea-pigs according to Environmental Protection Agency Pesticide Guideline PB83-153916 (1982). Flank hair was clipped from each guinea-pig and the area was depilated with a hair remover; then, 0.5 ml of test material was introduced under a lint patch, which was taped in place for 6 h. This procedure was performed on days 1, 8 and 15. The test materials were ethanol (the vehicle), 0.1% 2,4-dintirochlorobenzene in diethyl ether (the positive control) and undiluted technical-grade esfenvalerate; a fourth group of guinea-pigs was treated with undiluted technical-grade esfenvalerate as a control for irritation. On day 29, the guinea-pigs were challenged with similar, single doses of ethanol, esfenvalerate or dinitrochlorobenzene. The skin reactions were scored 24 and 48 h after removal of the patches. No sensitization reactions were induced by ethanol or undiluted technical-grade esfenvalerate, whereas erythema and oedema were observed in the positive control group. No irritation was produced by undiluted technical-grade esfenvalerate. It was concluded that technical-grade esfenvalerate did not induce sensitization under the conditions of the test (Maedgen, 1986).
Mice
In a comparison of the toxicity of esfenvalerate and fenvalerate, groups of 12 B6C3F1 mice of each sex were fed diets containing esfenvalerate (total purity of isomers, 94.5%, of which 87.2% was the Aalpha isomer, esfenvalerate) at a concentration of 0, 50, 150 or 500 ppm or fenvalerate (total purity of isomers, 95.5%; isomeric ratio Aalpha:Abeta:Balpha:Bbeta, 24:25:26:24) at a concentration of 2000 ppm for 3 months. There were no deaths during the study. Clinical signs of toxicity were seen only in animals at the highest dietary concentrations and consisted of typical type-II pyrethroid neurological responses. They were seen throughout treatment and included salivation, hypersensitivity, fibrillation, tremor, convulsion, hunched posture and unsteady gait as well as gross lesions such as alopecia, skin scabs and sores. They were more severe in the mornings. Decreased body-weight gain was seen in animals at the highest dietary concentrations of the two substances. There was no consistent change in food consumption, and water intake was only initially reduced in animals at the highest concentrations. The urinary pH was decreased, while urinary protein, ketone, bilirubin and urobiligen and specific gravity were increased in animals at the highest concentrations. In these groups, haematological changes were found which included decreased erythrocyte count, haemoglobin concentration, packed cell volume and/or mean cell volume. Decreased total protein, glucose, phospholipid, cholesterol and triglyceride concentrations and increased blood urea nitrogen and alanine and aspartate aminotransferase and lactate dehydrogenase activities were also observed in animals at the highest concentrations. Males fed esfenvalerate at 150 ppm also showed decreased glucose and triglyceride concentrations. The main difference in blood biochemistry between animals given esfenvalerate and fenvalerate was in the activity of leucine aminopeptidase, which was decreased in animals fed esfenvalerate and increased in animals fed fenvalerate.
The frequency of skin lesions, thought to be due to scratching, was increased, with, in some cases, enlargement of regional lymph nodes in animals at the highest concentration of each compound. The salivary gland weights were increased in these animals. Compound-related histopathological changes were seen in the liver, spleen, lymph nodes, thymus, skin, kidney and stomach. A decrease in fat deposits in the liver and kidneys was probably related to the decreased blood lipid levels. The animals given fenvalerate also showed an increased incidence of microgranuloma and giant-cell infiltration in the liver, spleen and lymph nodes. Slight ulcerative changes in the glandular stomach were observed in four males receiving 500 ppm of esfenvalerate but not in females at the same concentration, in males and females at lower concentrations or in any of the fenvalerate-treated mice. Examination of nervous tissue (brain, spinal cord and sciatic nerve) revealed no treatment-related tissue changes.
In summary, except for minor gastric ulcerations seen with esfenvalerate, no significant toxicological differences were found in mice fed esfenvalerate at 500 ppm and those fed fenvalerate at 2000 ppm. Esfenvalerate decreased leucine aminopeptidase activity in blood. Fenvalerate, but not esfenvalerate, at 500 ppm produced microgranulomatous changes in the liver, spleen and lymph nodes and increased leucine aminopeptidase activity in blood. The NOAEL for esfenvalerate was 50 ppm, equivalent to 10 mg/kg bw per day, on the basis of biochemical changes in the blood at 150 ppm (Koyama, 1985).
Rats
Groups of 30 Sprague-Dawley rats of each sex were fed diets containing esfenvalerate (purity not stated) at a concentration of 0, 50, 150, 300 or 500 ppm for up to 90 days. Ten rats of each sex per group were killed and assessed after 7 weeks. Deaths occurred during the first 11 weeks of the study among females at the highest concentration. Clinical signs of neurological dysfunction were noted at this concentration as early as week 1, which continued until the end of the study. In a few rats, these symptoms became severe and included body tremors, convulsions, hypersensitivity to sound, unsteady gait and jerky leg movements. Body-weight gain and food consumption were reduced in rats at 500 ppm, and males at 300 ppm also showed a reduction in body-weight gain. No treatment-related haematological or clinical chemical changes were seen, and there was no effect on urine except in volume and specific gravity in rats that consumed less food. Organ weights were unaffected by treatment, except as they reflected reduced body weights. Examinations post mortem revealed scab-covered areas at the base of the tail in some animals at 300 and 500 ppm. Microscopic examination revealed a moderate incidence of parenchymal-cell hypertrophy in the parotid salivary gland and a low incidence of parenchymal-cell hypertrophy in the pituitary glands after 7 and 13 weeks in rats at 500 ppm. Parenchymal-cell hypertrophy in the parotid salivary gland was also noted after 13 weeks in some rats at 300 ppm. The NOAEL was 150 ppm, equivalent to 7.5 mg/kg bw per day, on the basis of reduced body-weight gain in males and parenchymal-cell hypertrophy in the parotid salivary and pituitary glands in some rats at 300 ppm (Kelly, 1985).
Groups of 25 Sprague-Dawley rats of each sex were fed diets containing technical-grade esfenvalerate (purity not stated) at a concentration of 0, 75, 100, 125 or 300 ppm for up to 13 weeks. Ten rats of each sex per group were killed and assessed after 7 weeks. Treatment-related clinical signs were limited to signs of hyperactivity and jerky leg movements during the last 4 weeks of the study in rats at 300 ppm. Body-weight gain was depressed in males at 300 ppm during the first 2 weeks of the study, whereas females showed significant reductions in body-weight gain throughout the study. No treatment-related effects were found in food consumption, urine, haematological or blood chemical parameters or macroscopic appearance of tissues (no histological findings were described). A significant increase in absolute and relative kidney weights was observed in females at 300 ppm and in relative kidney weights in males at 300 ppm. The NOAEL was 125 ppm, equivalent to 6.2 mg/kg bw per day, on the basis of signs of toxicity and body-weight reduction in rats at 300 ppm (Larson, 1987).
A 21-day study of dermal toxicity was performed in rats in compliance with Environmental Protection Agency (1998) OPPTS 870.3200 guidelines. Groups of 10 Crl:CD®(SD)IGS BR rats of each sex received an application of esfenvalerate (total purity of isomers, 98.6%; proportion of esfenvalerate not given) on shaved, intact skin for 6 h/day for 21 days at a dose of 0, 25, 125, 500 or 1000 mg/kg bw per day. The application sites were wrapped continuously to prevent scratching and the consequent risk of wounding. In addition to normal daily observations for clinical signs, a battery of functional observational tests and evaluations of motor activity were applied before and again at the end of treatment. No treatment-related effects were found on body weight, food consumption, clinical signs or viability. During the first week of the study, abnormal hind-limb gait was observed in animals at doses > 125 mg/kg bw per day. Increased vocalization occurred in animals at 500 and 1000 mg/kg bw per day, and hypo- or hyper-reactivity were seen in females at 1000 mg/kg bw per day; these observations were considered to be secondary to sensory stimulation by the test compound. No treatment-related effects were found on grip strength, foot splay or functional observational parameters. Increased motor activity was observed in females, but not males, given 500 or 1000 mg/kg bw per day. The NOAEL was 25 mg/kg bw per day, on the basis of abnormal hind-limb gait at 125 mg/kg bw per day (Delker, 2000).
Dogs
Groups of six beagle dogs of each sex were fed diets containing technical-grade esfenvalerate (purity not stated; 83.8% was the Aalpha isomer, esfenvalerate) at a concentration of 0, 25, 50, 100 or 200 ppm for 1 year. The study was conducted in compliance with Environmental Protection Agency guidelines for testing of pesticides (1982). There were no deaths, overt signs of toxicity, ophthalmic changes or treatment-related changes in body weight or food consumption. At autopsy, there were no macroscopic or microscopic changes considered to be related to treatment. The NOAEL was 200 ppm, equivalent to 5 mg/kg bw per day, the highest dose tested, on the basis of the absence of treatment-related effects (Dickie et al., 1986).
Long-term studies of fenvalerate were considered to be a reliable guide to the toxicological behaviour of esfenvalerate in view of the similar metabolism and toxicity of the two compounds in short-term studies. Therefore, studies on fenvalerate are included below. It is important to remember, however, that in the existing comparative studies, the toxicity of fenvalerate (of which esfenvalerate forms approximately 25% of the enantiomers) appeared to be accounted for by the esfenvalerate content. Therefore, any NOAEL derived from experiments with fenvalerate should be reduced fourfold for application to esfenvalerate.
Mice
Groups of 80 Crl:CD-1®(ICR)BR mice of each sex were fed diets containing esfenvalerate (purity, 98.8%; Aalpha isomer, 84.8%, esfenvalerate) at a concentration of 0, 35, 150 or 350 ppm for up to 18 months. Because of excessive morbidity, mortality and self-inflicted wounding, the mice at 350 ppm were killed on days 57–58, and the data were not used in the evaluation of tumorigenicity. The study was conducted under European Commission Directive 88/302/EEC according to the data requirements of Environmental Protection Agency pesticide assessment guidelines 83-2, OECD test guideline 451 and Ministry of Agriculture, Food and Fisheries Japan Nohsan No. 4200. The mean daily intake of esfenvalerate over the course of the study by animals at 0, 35 and 150 ppm was 0, 4.3 and 18 mg/kg bw per day for males and 0, 5.7 and 25 mg/kg bw per day for females.
The main effects observed were related to sensory hyperstimulation resulting from dermal contact with the test material in the ground rodent chow, as manifested by self-mutilation, primarily in animals at 150 ppm, probably in combination with mild systemic toxicity. The survival of animals at this dose (males, 46%; females 41%) was reduced relative to that of controls (males, 70%; females, 71%) because the mice were killed in extremis after the self-trauma. The survival rate of mice at 150 ppm was 75% for males at 302 days and for females at 175 days. Body weight and body-weight gain were also reduced at 150 ppm, the mean body-weight gain being depressed at the end of the study by 19% in males and 22% in females. Ophthalmic examination revealed no effects attributable directly to the test material, although self-inflicted trauma due to treatment probably contributed to some ocular lesions. No other toxicologically important changes were observed. The changes in haematological and blood chemical parameters did not show a dose–response relationship and were of insufficient magnitude to have biological significance. Microscopic lesions observed with increased frequency or severity were non-neoplastic in nature and involved mainly the skin and eyes. As a result of skin erosion and ulceration, there was an increased frequency of epidermal hyperplasia and inflammation. Similar lesions affected the pinnae and the corneas of the eyes. Secondary to skin damage, increased, dosed-related extramedullary haematopoiesis was found in the spleen (males: 0 ppm, 22/79; 35 ppm, 25/33; 150 ppm, 48/80 (p < 0.05); females: 0 ppm, 26/80; 35 ppm, 19/29; 150 ppm, 56/80 (p < 0.05) with hyperplasia in the lymph nodes and bone marrow. These effects were considered to be secondary to the self-inflicted damage induced by the pharmacological effects of the pyrethroid, rather than to any primary toxic effect. The neoplasms observed are common in CD-1 mice, including tumours of the liver, lungs and Harderian glands and lymphomas and histiocytic sarcomas; the incidence of none, or any others, was increased. The reductions in survival resulting from losses due to self-mutilation compromised the power of the study to detect a carcinogenic effect at 150 ppm. As the survival rate at 35 ppm was acceptable in comparison with that of controls, the results at this concentration were used for evaluation. Esfenvalerate was not carcinogenic at this dose. The NOAEL was 35 ppm, equal to 4.3 mg/kg bw per day, on the basis of reductions in body-weight gain and effects that were probably secondary to the pharmacological effects of the pyrethroid, namely shin damage, extra-medullary haematopoiesis in the spleen and hyperplasia in the lymph nodes and bone marrow at 150 ppm (Ross, 1997).
Groups of 50 B6C3F1 mice of each sex were fed diets containing technical-grade fenvalerate (purity, 98%) at a concentration of 0, 0, 10, 50, 250 or 1250 ppm for 104–105 weeks. The mortality rate ranged from 19% to 56% and was largely unaffected by treatment. The mortality rate of animals at 1250 ppm was 56% for males and 54% for females, these proportions being significantly greater than in the controls, but the mortality rate of males at 10 ppm also reached 54%. There were no compound-related signs of toxicity. The body weights of males and females fed 1250 ppm were significantly lowered throughout the study, and those of females fed 250 ppm were also slightly but significantly lower from week 60 onwards. There were no significant changes in food consumption or haematological parameters. The clinical biochemical changes in mice at 1250 ppm consisted of increased aspartate aminotransferase activity and decreased serum albumin concentrations. At autopsy, there were no treatment-related macroscopic lesions or consistent changes in organ weights. Histopathology revealed no increase in the frequency of neoplastic lesions. An increased incidence of microgranulomatous changes was found in the spleen, liver and lymphatic tissue of mice at 250 and 1250 ppm and in males at 50 ppm. The NOAEL was 10 ppm, equivalent to 1.5 mg/kg bw per day, on the basis of microgranulomatous changes in the spleen, liver and lymphatic tissue of males at 50 ppm (Johnston, 1979).
Groups of 50 ddY mice of each sex were fed diets containing fenvalerate (purity, 91.4%; isomeric ratios not given) at a concentration of 0, 10 30, 100 or 300 ppm for 91 weeks for males and 87 weeks for females. The study was conducted before good laboratory practice (GLP) was in place. Haematology, blood and urine chemistry and ophthalmology were carried out only at the end of the study, as the primary objective was to assess the tumorigenic potential of fenvalerate in mice.
The survival rate at the end of the study was 30–42% for both sexes. Fenvalerate did not affect mortality, food or water consumption, body-weight gain or ophthalmological or urine parameters. There were no treatment-related, overt signs of toxicity. Slight decreases in the erythrocyte count of males at 100 and 300 ppm and in the haemoglobin concentration of females at 300 ppm were observed. The blood glucose concentration was slightly lowered in females at 100 and 300 ppm, and the activity of alanine aminotransferase was slightly increased in females at 300 ppm. There were no treatment-related changes in organ weights or macroscopic findings at autopsy. Histopathological examination revealed a dose-related increase in the incidence of granulomatous changes in the lymph nodes and liver of mice at 100 and 300 ppm and in the spleen of those at 300 ppm. There was no evidence of carcinogenicity. The NOAEL was 30 ppm, equal to 3.5 mg/kg bw per day, on the basis of a slight decrease in erythrocyte count and increased numbers of histiocytes and granulomatous changes in the liver and lymph nodes of animals at 100 ppm (Arai et al., 1981).
Rats
Groups of Sprague-Dawley rats were fed diets containing technical-grade fenvalerate (purity, 98%) at a concentration of 0, 1, 5, 25 or 250 ppm for 2 years (93 animals of each sex per group for treated groups and 183 of each sex in the control group) or 0 or 500 ppm for 6 months (22 of each sex per group). This study was not conducted according to GLP but complied to a great extent with the requirements of European Commission Directive 87/18, the main deviations being the absence of ophthalmic examinations and of histological examinations of the aorta, oesophagus and femur. After 3 and 6 months (all groups) and 12 and 18 months (groups at 0, 1, 5, 25 and 250 ppm) of treatment, 10 rats of each sex per treated group and 20 of each sex from the control group were sampled for haematology and blood and urine chemistry, after which they were killed and examined histopathologically.
The mortality rate was unaltered by treatment, and there was no treatment-related increase in the incidence of palpable masses or overt signs of toxicity. The body-weight gain of females at 500 ppm was slightly decreased during most of the treatment period of 6 months. Food consumption was unaffected by treatment. Urine analysis revealed no remarkable changes, and there were no consistent treatment-related changes in haematological or blood chemical parameters or organ weights. Slight increases were found in the incidences of benign mammary tumours in females at 5 and 250 ppm (intercurrent deaths), total mammary tumours in females at 25 ppm at the terminal kill and total mammary tumours in females at 250 ppm. These non-dose-related increases (no trend in the Peto or Cochrane-Armitage test) in mammary tumour incidence in a strain of rats in which such tumours are common and of variable incidence is unlikely to be due to treatment. In addition, there was no indication that treatment was associated with an earlier occurrence of tumours. The incidence of mammary tumours was within the range of that of other controls in the same laboratory (46–65%). The NOAEL was 250 ppm, equivalent to 12 mg/kg bw per day, on the basis of reduced body-weight gain in females at 500 ppm (Gordon & Weir, 1978).
In a supplementary study, groups of 50 Sprague-Dawley rats of each sex were fed diets containing technical-grade fenvalerate (purity, 98%) at a concentration of 0 or 1000 ppm for 2 years. The study was conducted prior to GLP but was in accordance with current procedures of quality assurance and complied to a great extent with the requirements of European Commission Directive 87/18, the main deviations being no interim kills or ophthalmic examinations and performance of urine analysis, haematology and blood chemistry only at termination.
The mortality rates were not increased, and the only overt sign of toxicity was transient hind-limb weakness in six males at 1000 ppm. Some males developed skin lesions, but these were observed in controls as well and were unrelated to treatment. Food consumption was unaffected. A decrease in body-weight gain was seen from week 16 in treated males and from week 44 in treated females until the end of the study. No consistent treatment-related changes were found in haematological, clinical chemical or urine parameters. The treated and control groups did not differ in the incidence of mammary tumours. Malignant neoplasms of mesenchymal origin (sarcomas) were observed in the subcutis and dermis of 5/51 male rats (two at terminal sacrifice, three at intercurrent deaths) at 1000 ppm and in 0/50 controls. One such lesion occurred in each of the cervical, hind-limb, thoracic, perianal and axillary regions. The characteristics of different portions of the individual tumours varied, and the neoplasms differed. They were, however, predominantly collagenous and generally contained frequent mitoses. Because the cell of origin could not be determined, they were designated ‘spindle-cell sarcomas’, which encompasses a number of malignant mesenchymal neoplasms that are difficult and sometimes impossible to diagnose definitively and include fibrosarcoma, neurofibrosarcoma, malignant fibrous histiocytoma, liposarcoma and osteosarcoma. A spindle-cell sarcoma also occurred in 1/49 female rats at 1000 ppm, with none in controls.
A review of five studies of 2 years’ duration conducted with Sprague-Dawley rats at the same laboratory revealed sarcomas of the subcutis in control males at frequencies of 2/102, 2/50, 2/50, 0/64 and 0/83. Thus, although the relationship of these tumours to fenvalerate treatment is doubtful, the possibility cannot be excluded. (Gordon & Weir, 1979). A subsequent re-evaluation of the slides from the study by a pathologist (Ito, 1981) resulted in a very different conclusion. The tumours were not diagnosed as a group of spindle-cell sarcomas but as a malignant schwannoma or perhaps a leiomyosarcoma, a malignant mesenchyoma, a fibrosarcoma and two malignant amelanotic melanomas. Thus, the incidence of each sarcoma was low (1 or 2 in 51 rats). As there has been no attempt to reconcile these diagnoses, it is difficult to accept the re-evaluation. However, even in the original evaluation, there was no accumulation of sarcomas in female rats (and there is no reason to expect a sex difference in this respect). Furthermore, there was no evidence for an increase in the incidence of these tumours in the study of Arai et al. (1980; see below), in which Wistar/SLC rats were given fenvalerate at dietary concentrations up to 1500 ppm, equivalent to 75 mg/kg bw, which is 50% higher than that used in this experiment.
Groups of 80 Wistar/SLC rats of each sex were fed diets containing fenvalerate (purity, 93.4%) at a concentration of 0, 50, 150, 500 or 1500 ppm for 104 weeks (males) or 119 weeks (females). This study was conducted prior to the institution of GLP but was in accordance with current procedures of quality assurance and complied to a great extent with the requirements of European Commission Directive 87/18, the main deviations being the fact that urine analysis, haematology, blood chemistry and ophthalmology were performed only at termination. No compound-related effects were reported on clinical signs, mortality rate, water intake, clinical chemical, haematological, urinary or ophthalmic end-points or organ weights. The body-weight gains of males at 500 and 1500 ppm and females at 1500 ppm were reduced, although interpretation of the result for males was confounded by the higher initial body weights in the control group. Histopathological examination revealed giant-cell infiltration in the liver and spleen in animals at 1500 ppm and in lymph nodes and adrenals at 500 and 1500 ppm. Reticuloendothelial-cell proliferation in the mesenteric lymph nodes was observed at the two higher doses.
The neoplastic changes found in both males and females—leukaemia, chromophobe adenoma in the pituitary and phaeochromocytoma in the adrenal—occurred in all groups, including the controls, and there were no significant differences in incidence between groups. In females, uterine and mammary tumours were observed in all groups, and, again, there were no significant differences in incidence between groups. The incidences of Leydig-cell tumours were significantly increased in several groups, the overall values being 21/75 at 0 ppm, 36/78 at 50 ppm ( p < 0.05), 27/79 at 150 ppm, 56/76 at 500 ppm ( p < 0.01) and 53/77 at 1500 ppm (p < 0.01). Testicular atrophy and/or Leydig-cell hyperplasia were also seen in many males at all dietary concentrations. The incidence of testicular atrophy was slightly higher in treated groups than in controls, but the incidence of Leydig-cell hyperplasia was highest in the controls: 0 ppm, 19/75; 50 ppm, 12/78; 150 ppm, 7/79; 500 ppm, 10/76; 1500 ppm, 3/77. Leydig-cell hyperplasia and Leydig-cell adenoma are distinguished solely on the basis of size. According to some authors (e.g., Boorman et al., 1987), an adenoma is diagnosed if the area of hyperplasia is greater than that of a seminiferous tubule, whereas other authors (e.g., McConnell et al., 1992) consider that an adenoma should be diagnosed if it is equal to or greater than three seminiferous tubules. While high incidences of these lesions are common in Fischer 344 rats, this is not usually the case in Wistar rats. Indeed, a highly variable incidence of testicular tumours (11–100%) was observed at the time of the study in control Wistar/SLC rats from the same supplier over a 6-year period. Genetic analysis of Wistar/SLC rats and comparison with other strains in Japan indicated that two Wistar strains, one of which is Wistar/SCL, are closely similar to Fischer rats. The occurrence of Leydig-cell adenomas is strongly age-dependent. Reports on Wistar/SCL rats published during two relevant periods gave the following total incidences: experiment I (1974): 13 months, 10%; 19 months, 17%; 25 months, 100%; experiment II: 14 months, 14%; 20 months, 100%; 26 months, 99%. Although the final mortality rates were generally similar across groups in the current experiment (0 ppm, 79%; 50 ppm, 75%; 150 ppm, 72%; 500 ppm, 64%; 1500 ppm, 73%), more rats at 500 and 1500 ppm were at risk at 84 weeks (i.e., about the time that the rapid increase in incidence occurs), the mortality rates at this time being: 0 ppm, 65%; 50 ppm, 59%; 150 ppm, 55%; 500 ppm, 31%; 1500 ppm, 43%. This factor could have contributed to the variations in adenoma incidence between groups. It should also be noted that in two studies described above, in which Sprague-Dawley rats were fed diets containing fenvalerate at a concentration of 250 ppm (Gordon & Weir, 1978) or 1000 ppm (Gordon & Weir, 1979) for up to 2 years, there was no increase in the incidence of Leydig-cell tumours. It was therefore concluded that there may have been accelerated growth of Leydig-cell proliferating lesions in some treated groups but that these cannot be interpreted as an increased incidence of tumours. It was concluded that fenvalerate is not carcinogenic. The NOAEL was 150 ppm, equal to 7.5 mg/kg bw per day, on the basis of reduced body-weight gains at 500 ppm (Arai et al., 1980).
Esfenvalerate was tested for genotoxicity in a range of assays, both in vitro and in vivo (Table 5). There was no evidence of genotoxicity in these assays, which included tests for gene mutation in bacteria, DNA damage and repair in HeLa cells, gene mutation in Chinese hamster lung (V79) cells, chromosomal aberrations in Chinese hamster ovary cells and micronucleus formation in bone-marrow cells of mice treated in vivo. Fenvalerate also had no genotoxic activity (see Annex 1, reference 49).
Table 5. Results of testing for genetic effects with esfenvalerate
|
End-point |
Test object |
Dose |
Result (LED/HID) |
Reference |
|
In vitro |
|
|
|
|
|
Gene mutation |
S. typhimurium TA100, TA1535, TA1537, TA1538, TA98; E. coli WP2uvrA |
5000 µg/plate |
Negativea |
Kogiso (1985a) |
|
Unscheduled DNA synthesis |
HeLa cells |
1 mnol/l (420 µg/ml) |
Negativea |
Kogiso (1986) |
|
Gene mutation |
Chinese hamster lung V79 cells, Hprt locus |
1 mmol/l (420 µg/ml) |
Negativea |
Kogiso (1985b) |
|
Chromosomal aberration |
Chinese hamster ovary cells |
0.5 mmol/l (210 µg/ml) |
Negativea |
Kogiso (1985c) |
|
In vivo |
|
|
|
|
|
Micronucleus formation |
Male ICR mouse bone-marrow cells |
150 mg/kg bw, intraperitoneally |
Negative |
Kogiso (1985d) |
Purity of total isomer, 95.5%; that of Aalpha isomer, 91.5%. LED, lowest effective dose; HID, highest ineffective dose
a In the absence and presence of an exogenous metabolic activation system from male rat liver
Rats
Groups of 30 Crl:CD®BR rats of each sex were fed diets containing esfenvalerate (total isomer content, 98.8%; Aalpha isomer, esfenvalerate, 84.8%) at a concentration of 0, 75, 100 or 350 ppm in a two-generation study of reproductive toxicity performed according to European Commission Directive 87/302 and data requirements of the Environmental Protection Agency Pesticide Assessment Guidelines 83-4, OECD Guidelines for Testing of Chemicals No. 416 and Ministry of Agriculture, Fisheries and Food, Japan, Testing Guidelines for Toxicology Testing NohSan 59, No. 4200. After 73 days of receiving the test diets, the animals were mated. A group of 30 offspring were fed diets containing the same concentrations of esfenvalerate for at least 105 days after weaning, before mating to produce a second generation. Because of progressive clinical signs in animals at 350 ppm at weaning, this dietary concentration was reduced to 150 ppm at this stage. Approximately 2 weeks after the start of weaning, all animals were treated topically with vitamin E oil to alleviate skin stimulation. The overall calculated mean daily intake of esfenvalerate during the pre-mating phase was 0, 4.2, 5.6 and 19 mg/kg bw for F0 males; 0, 5.6, 7.2 and 25 mg/kg bw for F0 females; 0, 6.0, 7.8 and 19 mg/kg bw for F1 males; and 0, 7.3, 10 and 19 mg/kg bw for F1 females.
Clinical signs of toxicity in adults fed 350 ppm included abnormal gait, alopecia, skin ulcerations and dermal lesions. One male at 350 ppm was killed in extremis. The body weights of animals fed esfenvalerate at 100 and 350 ppm in the diet were significantly lower than those of the control group. Treatment-related skin lesions were also observed in adults at 75 and 100 ppm.
Litter sizes, survival rates and body weights of pups in the F1 generation at 350 ppm were significantly reduced. The clinical signs of toxicity in these animals included abnormal gait and skin lesions. Because of these effects, the dietary concentration was reduced to 150 ppm for the second generation, but this group was terminated early because of ill health and deaths among the animals. The litter size and pup weights of F1 animals at 100 ppm were reduced relative to those of controls, and there was a corresponding decrease in survival and mean litter weights. The body weights of F1 adults at 75 ppm were slightly reduced near term when compared with control values. Offspring parameters were not affected when rats were fed esfenvalerate at 75 ppm in the diet. There were no treatment-related effects on fertility indices, mating indices or reproductive organs. The NOAEL for reproductive toxicity in adult rats and their offspring was 75 ppm, equal to 4.2 mg/kg bw per day (Biegel, 1994).
A subsequent study was conducted to determine the NOAEL for the F1 generation when avoiding skin paraesthesia, which was considered to be responsible for the decreased body weights observed at the lowest concentration of 75 ppm in the first study. Pelleted feed containing esfenvalerate (total isomer content, 97.3%; esfenvalerate content, 86.0%) at a concentration of 0, 20, 40 or 100 ppm was administered to groups of 24 Crj:CD(SD)IGS SPFrats of each sex in order to reduce dermal exposure to the test compound. Dosed feed was provided for 70 days before mating and throughout mating, gestation and lactation during the production of two litters by the parental animals. Administration was continued for 203–205 days for the F1 and F1a offspring and for 105 days for the F1b offspring after weaning.
The attempt to avoid skin paraesthesia was successful, even at the highest concentration of 100 ppm, in all generations. Body weight and food consumption were reduced for both parental animals and offspring at 100 ppm. There were no adverse effects on reproductive parameters at any concentration, although a low fertility index was found in the F1b group; as there was no clear dose-dependency, this was attributed to the advanced age of the parental animals and was considered not to be treatment related. The overall NOAEL for systemic effects was 40 ppm, equal to 2.4 mg/kg bw per day, and the NOAEL for reproductive toxicity was 100 ppm, the highest dose tested, equal to 4.7 mg/kg bw per day (Higuchi, 1999).
Rats
In a study of developmental toxicity, groups of 25 female Sprague-Dawley rats were given esfenvalerate (purity, 97.1%), in corn oil by gavage at a dose of 2.5, 5, 10 or 20 mg/kg bw per day on days 6–15 of gestation. All rats survived to the end of the study, and no internal lesions were found at autopsy. Overt signs of toxicity were seen within 4 h of dosing and consisted of typical type-II pyrethroid effects, including erratic jerking and extension of the forelimbs or hindlimbs, rapid side-to-side head movements and/or excessive grooming, high carriage, tremors, ‘goose-stepping’, ataxia, convulsions, hypersensitivity to touch, soft stools, mucoid faeces, yellow faeces, diarrhoea, salivation, yellow anogenital or urogenital matting, sneezing, red material around the mouth, eyes and nose, mydriasis and/or red or clear ocular discharges. These effects were transient and were not present the following day. They were seen at all doses, although they were more prevalent at 20 mg/kg bw per day than at lower doses. Maternal body-weight gain and food consumption were reduced during treatment with 20 mg/kg bw per day. Food consumption was also reduced in animals at 10 mg/kg bw per day. Intrauterine survival and growth were not adversely affected by any dose. External malformations and variations were seen, consisting of a few examples of microphthalmia, anophthalmia, micrognathia and micromelia, with no dose–response relationship. A single incident of encephalomeningocoele was found in the group at 20 mg/kg bw per day. No soft tissue variations were found. There was a slight increase in the incidence of a fourteenth extra, rudimentary rib, the frequencies on the basis of fetuses and (litters), respectively, being: 0 mg/kg bw per day, 15% (54%); 2.5 mg/kg bw per day, 10% (39%); 5 mg/kg bw per day, 21% (58%); 10 mg/kg bw per day, 18% (44%); and 20 mg/kg bw per day, 30% (86%). The litter-based frequency at the highest dose was significantly greater than the control frequency (p < 0.05); however, the percentage increase was within the range of other controls in the same laboratory, 0.0–39% (0.0– 95%), and was therefore considered not to be a treatment-related effect. The frequencies of other fetal skeletal malformations and variations were low and unaffected by treatment. No NOAEL for maternal toxicity could be identified, as significant toxicity was seen at 2.5 mg/kg bw per day, the lowest dose tested. The NOAEL for developmental toxicity was 20 mg/kg bw per day, the highest dose tested, on the basis of the absence of developmental effects (Nemec, 1991a).
To establish the maximum tolerated dose for pregnant rats, a pilot study was conducted in which esfenvalerate (total isomer content, 98.8%; Aalpha isomer, esfenvalerate, content, 84.8%) was administered in cottonseed oil by gavage to groups of 15 pregnant Crl:CD®BR rats daily on days 7–16 of gestation at a dose of 0, 1, 2, 3, 4, 5 or 20 mg/kg bw per day. This pilot study is more recent than the study described above. Symptoms of maternal toxicity, including abnormal gait, hind-limb spasms, diarrhoea and tremors, were seen at doses > 4 mg/kg bw per day, and maternal weight gain was reduced at 5 and 20 mg/kg bw per day. Treatment had no effect on reproductive parameters, and no signs of developmental toxicity were detected. There was no effect on fetal weight, sex ratio or the incidence of fetal abnormalities. The NOAEL for maternal toxicity was 3 mg/kg bw per day, on the basis of significant effects at 4 mg/kg bw per day. The NOAEL for developmental toxicity was 20 mg/kg bw per day, the highest dose tested, as there were no developmental effects (Murray, 1994a).
Rabbits
In a study of developmental toxicity, groups of 20 artificially inseminated New Zealand white rabbits were given esfenvalerate (purity, 97.1%), in corn oil by gavage at a dose of 0, 3, 10 or 20 mg/kg bw per day on days 7–19 of gestation. One doe at 20 mg/kg bw per day died, and the macroscopic changes suggested gastrointestinal haemorrhage. No other compound-related effects were found on maternal survival or macroscopic tissue appearance. One female each at 0, 3 and 20 mg/kg bw per day aborted, but these abortions were considered to be spontaneous. Overt signs typical of pyrethroid type-II effects were seen at all doses and included erratic jerking and extension of the limbs followed by excessive grooming, rapid side-to-side head movements and, at doses > 3 mg/kg bw per day, hypersensitivity to touch, tremors, ataxia, excessive chewing, sneezing and coughing, decreased defaecation and urination and diarrhoea. The incidence and severity of these effects were dose-related. Decreased body-weight gain was observed primarily during the initial 3 days of dosing with 10 or 20 mg/kg bw per day. Food consumption was reduced during the first 6 days of dosing with 10 or 20 mg/kg bw per day and remained depressed for the group at the highest dose. No other signs of maternal toxicity were seen. Intrauterine growth and survival were not affected at any dose. No external malformations or variations were observed. Soft-tissue malformations involving the heart and great vessels were found in one control and two fetuses at 10 mg/kg bw per day but not in litters at 3 or 20 mg/kg bw per day. Other soft-tissue variations included unilateral retrocaval ureters, accessory spleens, absent or small gall-bladders and cases in which the left carotid originated from the brachiocephalic trunk. The low incidences and pattern of distribution across groups did not suggest any relationship with treatment. No NOAEL was identified for maternal toxicity, as significant effects were observed at 3 mg/kg bw per day, the lowest dose tested. The NOAEL for developmental toxicity was 20 mg/kg bw per day, the highest dose tested, as there were no developmental effects (Nemec, 1991b).
In what was described as a pilot experiment but was, in fact, an extensive study of developmental toxicity that is more recent than that described above, esfenvalerate (total isomer content, 98.8%; Aalpha isomer, esfenvalerate, content, 84.8%) was administered in cottonseed oil by gavage to groups of 14 pregnant New Zealand white rabbits at a dose of 0, 2, 3, 4, 4.5, 5 or 20 mg/kg bw per day on days 7–19 of gestation. Symptoms of maternal toxicity including excessive grooming accompanied by head and/or paw shaking were evident at doses > 3 mg/kg bw per day. In the group at 20 mg/kg bw per day, one doe died and one aborted. Maternal weight gain was reduced at 20 mg/kg bw per day. Treatment had no effect on reproductive parameters, and no sign of developmental toxicity was detected, with no effect on fetal weight, sex ratio or the incidence of fetal abnormalities. The NOAEL for maternal toxicity was 2 mg/kg bw per day on the basis of significant effects at 3 mg/kg bw per day. The NOAEL for developmental toxicity was 20 mg/kg bw per day, the highest dose tested, on the basis of the absence of developmental effects (Murray, 1994b).
A comparative study of the neurotoxic potential of esfenvalerate (purity, 94.5%; Aalpha isomer, esfenvalerate, 87.2%) and fenvalerate (purity, 95.5%; Aalpha isomer, esfenvalerate, 24.2%) was conducted in which groups of eight rats of each sex (16 of each sex at the highest dose) were given a single oral dose of either esfenvalerate at 0, 5, 20 or 90 mg/kg bw or fenvalerate at 0, 20, 80 or 360 mg/kg bw in corn oil, and were observed for 2 weeks. At that time, the rats were perfused with 10% buffered formalin, and the brain, spinal cord and sciatic and tibial nerves were examined histologically.
Two males and one female at the highest dose of esfenvalerate and one male and four females at the highest dose of fenvalerate died within 24 h of dosing. Depressed body-weight gain and overt signs of toxicity were seen in animals of each sex at these doses with each compound. The toxic signs, which occurred within 2 h of dosing, were typical type-II pyrethroid symptoms, i.e., muscular fibrillation, ataxia, tremors, salivation, limb paralysis, irregular respiration, hypersensitivity to sound, hunched posture and urinary incontinence. All the neurological symptoms disappeared within 2 days of dosing. Muscular fibrillation, ataxia, salivation and/or hunched posture were also seen in a few rats receiving esfenvalerate at 20 mg/kg bw and fenvalerate at 80 mg/kg bw. No treatment-related signs were seen in rats at the lower doses. There were no treatment-related gross changes at necropsy. Histopathological examination revealed no changes in any brain section. Rats of each sex at the highest dose of esfenvalerate and fenvalerate had an increased incidence of slight-to-minimal axonal degeneration and/or demyelination, with Schwann-cell proliferation in proximal and distal peripheral nerves, but the incidences of these neural changes in animals at the two lower doses were comparable to those of controls. The fact that these effects were not seen at the intermediate dose, at which overt signs of toxicity were seen, suggests that they were not due to peripheral nerve damage but were more likely to be of pharmacological origin (Okuno, 1985).
The potential neurotoxicity of esfenvalerate (total purity of isomers, 98.6%; proportion of esfenvalerate not given) was evaluated in adult rats according to Environmental Protection Agency Health Effects Test Guidelines, Neurotoxicity Screening Battery, OPPTS 870.6200, 712-C-98-238 (1998). Groups of 12 young adult male and 12 female Crl:CD®(IG)BR rats were given esfenvalerate in corn oil at single doses of 0, 1.75, 1.9, 20 or 80 mg/kg bw by gavage. Clinical signs of toxicity were assessed on days 2–15. A neurobehavioural test battery, consisting of tests for motor activity and functional observational outcomes, was conducted before administration of the compound and again 7 h and 8 and 15 days after dosing. On day 16 or 17, six rats of each sex per group underwent whole-body perfusion, and tissues from those at 0 and 80 mg/kg bw were examined histologically.
Treatment-related changes in neurobehavioural parameters were observed on day 1 in females given doses > 1.9 mg/kg bw and in males given doses > 20 mg/kg bw. The observations in males included soiled fur, salivation, slow righting reflex, tremors, stereotypical grooming, abnormal gait, diarrhoea and paw shaking. In females, the neurobehavioural changes included decreased fore-limb grip strength, decreased hind-limb foot splay, salivation, tremors, stereotypical grooming, abnormal gait, diarrhoea, paw shaking, uncoordinated increased reaction to approach and touch and increased reaction to tail pinch. In addition, decreased motor activity was observed in males and females at 80 mg/kg bw on day 1. On day 2, males and females at 80 mg/kg bw continued to show abnormal gait, diarrhoea and stained fur. By day 4, all clinical signs of toxicity had resolved, and there were no treatment-related effects on neurobehavioural parameters on day 8 or 15.
There were no unscheduled deaths during the study. Decreased body-weight gain was observed in males at 20 and 80 mg/kg bw and in females at 80 mg/kg bw during test days 1–2, and males at this dose had reduced food consumption during this interval. No treatment-related morphological changes were seen in nervous tissues in either sex. The NOAEL was 1.75 mg/kg bw on the basis of tremors at higher doses (Malley, 2000a).
The potential neurotoxicity of repeated intake of esfenvalerate (purity of total isomers, 97.3%; esfenvaerate, 86.0%) was evaluated in adult rats in compliance with Environmental Protection Agency (40 CFR Part 160) and OECD (C[97]186/Final) according to OECD Guideline 424. Groups of 12 young adult male and 12 female Crl:CD®(SD)BR rats were given diets containing esfenvalerate at a concentration of 0, 40, 120 or 360 ppm, equivalent to 0, 3, 8.9 and 29 mg/kg bw per day for males and 0, 3.7, 11 and 35 mg/kg bw per day for females, for 93 days. A neurobehavioural test battery, consisting of tests for motor activity and functional observational outcomes, was conducted before administration of the compound and again during weeks 2, 5, 9 and 13 of treatment. An ophthalmic examination was carried out before dosing and again during week 13. After the tests in week 13, five rats of each sex per group underwent whole-body perfusion, and tissues from animals at 0 and 360 ppm were examined histologically for neuropathological effects.
No treatment-related deaths occurred. The only overt clinical signs that might have been attributable to treatment were skin lesions and scabbing in a small number of males at 360 ppm. Body-weight gain was significantly reduced throughout the treatment period among males and females at 360 ppm and occasionally among males at 120 ppm. Food consumption was unaffected, apart from reductions among males and females at 360 ppm during the first 8 days of treatment.
The battery of functional observational tests applied during week 2 showed significantly decreased fore-limb grip strength in males and females at 360 ppm (p < 0.05), and motor activity counts were significantly reduced among females at both 360 ppm (p < 0.01) and 120 ppm (p < 0.05). No significant differences were found between the groups at any later times. No ocular changes were observed, and there were no treatment-related morphological changes in nervous tissues from males or females at 360 ppm.
The NOAEL was 40 ppm, equivalent to 3.0 mg/kg bw per day, on the basis of decreased body-weight gain in males at higher doses (Beyrouty, 1999).
The potential neurotoxicity of repeated intake of esfenvalerate (purity of total isomers, 98.6%; proportion of esfenvalerate not given) was evaluated in adult rats according to Environmental Protection Agency Health Effects Test Guidelines, Neurotoxicity Screening Battery, OPPTS 870.6200, 712-C-98-238 (1998). Groups of 12 young adult male and 12 female Crl:CD®(IG)BR rats were given diets containing esfenvalerate at a concentration of 0, 50, 100 or 300 ppm, equivalent to 0, 3.2, 6.4 and 20 mg/kg bw per day for males and 0, 3.7, 7.3 and 23 mg/kg bw per day for females, for 91 days. A neurobehavioural test battery, consisting of tests for motor activity and functional observational outcomes, was conducted before administration of the compound and again after 4, 8 and 13 weeks of treatment. After the tests in week 13, six rats of each sex per group underwent whole-body perfusion, and tissues from those at 0 and 300 ppm were examined microscopically.
Two males at 300 ppm were killed before the end of the study because of poor condition resulting from skin wounds. Males and females at this dietary concentration showed reduced body-weight gain and reduced food consumption, as well as abnormal gait and decreased fore-limb and hind-limb grip strength. In addition, males at this dose showed increased motor activity, decreased foot splay and an increased incidence of skin sores. Male rats at 100 ppm had decreased fore-limb grip strength, but there were no effects on hind-limb grip strength, foot splay or motor activity or the 37 neurobehavioural parameters evaluated in the functional observational battery in males or females at this dose. No effects were seen in males or females at 50 ppm. There were no primary treatment-related morphological changes in nervous tissues from males or females at 300 ppm.
The NOAEL was 50 ppm, equivalent to 3.2 mg/kg bw per day, on the basis of decreased fore-limb grip strength in males at 100 ppm (Malley, 2000b).
The toxicity of fenvalerate and other pyrethroids has been reviewed (Aldridge, 1990; Vijverberg & van den Bercken, 1990; Appel & Gericke, 1993). In all species tested, high doses of pyrethroids induced toxic signs characteristic of strong excitation of the nervous system (Vijverberg & Oortgiessen, 1988). The principal action of pyrethroids on the peripheral nervous system is to induce pronounced repetitive activity. In particular, sensory organs produce trains of nerve impulses instead of single impulses, both in vitro and in vivo. With some pyrethroids, sensory nerves, motor nerve endings and muscle fibres may also show repetitive activity. Another possible effect is depolarization of membranes, leading to increased release of neurotransmitters or even blockage of excitation.
The pyrethroids can be divided into two classes on the basis of the pattern of toxic signs in rats after administration of sublethal or lethal doses. The early synthetic pyrethroids induced signs of intoxication in rats very similar to those of the natural pyrethrins. Initial tremors, aggressive behaviour and extreme sensitivity to sensor stimuli were followed by prostration, with whole-body tremors preceding death. This sequence, later designated the T-syndrome, is characteristic of non-cyano pyrethroids and may be incomplete, depending on the molecular structure and on the route of application and dose of the pyrethroid. Most of the alpha-cyano-3-phenoxybenzyl pyrethroids evoke a distinct sequence of symptoms, called the CS-syndrome. Initial pawing and burrowing behaviour is rapidly followed by profuse salivation and whole-body tremors, progressing to sinuous writhing (choreoathetosis), which gradually becomes more intense. In the final stage, clonic seizures may occur. As in the case of non-cyano pyrethroids, the symptoms may be evoked or enhanced by sensory stimuli.
Vijverberg & van den Bercken (1990) summarized the evidence for the hypothesis that the main biological activity of pyrethroids is mediated by effects on sodium channels. The stereoselective pyrethroids fix themselves to sodium channels and cause them to stay open much longer than normal, resulting in prolongation of the transient current associated with membrane depolarization. In frog myelinated nerve fibres, this prolongation can range from 6 ms with phenothrin to 1770 ms with deltamethrin. The time constant is a characteristic of each structure, and those causing the T syndrome and those causing the CS syndrome in rats can generally be distinguished on this basis. Investigations into the site of action of pyrethroids within the nervous system have not resulted in a clear distinction between central and peripheral effects. Several published results suggest that, in rats, the lethal site of pyrethroids is located peripherally, possibly in the cardiovascular or respiratory system. The effects can be attributed to modifications of presynaptic and postsynaptic sodium channels. Postsynaptic neurotransmitter responses are unaffected by concentrations of pyrethroids that cause marked sodium channel modification. At high concentrations, both insecticidal and non-insecticidal pyrethroid isomers cause non-specific suppression of the postsynaptic neurotransmitter response.
The toxicity of many pyrethroids to laboratory mammals treated orally or dermally is very low: when the lethal doses are compared to those needed to kill insects by topical application on a weight basis, the selectivity factors range from 50 to 30 000. In a comparative study of the acute oral toxicity of pyrethroids, mice appeared to be more sensitive than rats. No significant differences were found in the toxicity of five pyrethroids to male and female animals. All the cis isomers were more toxic than the corresponding trans isomers. The available evidence indicates that the large differences in the toxicity of pyrethroid between species are due to pharmacokinetics and metabolism. Similar considerations apply to the high toxicity of pyrethroids in fish, the lethal concentration being as low as micrograms per kilogram under laboratory conditions. Owing to their lipophilicity, these insecticides readily accumulate in aquatic organisms; the bioconcentration factors obtained in experiments with various aquatic species indicate that the steady-state concentration in the organism exceeds that in the surrounding water by two to four orders of magnitude.
The potential for fenvalerate and esfenvalerate to affect the male endocrine system, primarily by testicular tumorigenesis, was investigated in groups of male Slc:Wistar rats fed diets containing fenvalerate (purity, 92.9%) at a concentration of 0, 50, 150, 500 or 1500 ppm or esfenvalerate (purity, 86.0%) at a concentration of 375 ppm for 26 weeks. This study included a component in which female rats were housed in close proximity (but without contact) to subgroups of males given the lowest and highest concentrations of fenvalerate and of those given esfenvalerate. Blood samples were taken at 4-week intervals. No rats died during the study. Lower body weights and decreased food consumption relative to controls were observed in the groups given 375 ppm of esfenvalerate or 1500 ppm of fenvalerate. The serum concentrations of luteinizing hormone and testosterone were not affected to any biologically significant extent; those statistically significant changes that were observed did not show a dose–response relationship or temporal consistency. Marginal increases in relative (but not absolute) organ weights were observed during the phase of the study in which the males were housed separately from the females, as follows: testes (+5%), pituitary (+15%) and liver (+9%) at 1500 ppm of fenvalerate; and epididymides (+4%), seminal vesicles (+9%) and liver (+4%) at 375 ppm of esfenvalerate. There were no gross or histopathological changes that were considered to be related to treatment. Housing males and females in close proximity without physical contact had no effect different from those seen when males were housed alone (Yamada, 1999).
The major photodegradation product of fenvalerate, decarboxyfenvalerate [2-(3-phenoxyphenyl)-3-(4-chlorophenyl)-4-methylpentanenitrile], is less acutely toxic to mammals than fenvalerate itself.
Decarboxyfenvalerate (purity, 92%) was administered orally in corn oil as a single dose of 0 or 5000 mg/kg bw to groups of five Fischer 344 rats of each sex. As there were no deaths, the acute oral LD50 of decarboxyfenvalerate in male and female Fischer 344 rats was > 5000 mg/kg bw (Cannelongo, 1983a).
Decarboxyfenvalerate (purity, 92%) was applied undiluted at a single dose of 0 or 2 ml/kg bw to the clipped unabraded dorsal skin of five New Zealand white rabbits of each sex. The test substance was kept in contact with the skin by means of an occlusive dressing for 24 h. As there were no deaths, the acute dermal LD50 of esfenvalerate in male and female rabbits was > 2 ml/kg bw (Cannelongo, 1983b).
The potential of decarboxyfenvalerate to irritate the skin was evaluated in three male and three female New Zealand white rabbits whose skin had been shaved 24 h before exposure. A volume of 0.5 ml of undiluted decarboxyfenvalerate (purity, 92%) was applied to two intact and two abraded skin sites on each rabbit, and then occlusive dressings were applied, according to the Draize (1977) method. After 24 h, the dressings were removed, and the skin was observed for irritation 24 and 72 h and 7 days after application. The mean primary irritation score was 2.4 (maximum score, 8), an effect judged to be mildly irritating (Cannelungo, 1983c).
The potential of decarboxyfenvalerate to irritate the eye was evaluated in six male and three female New Zealand white rabbits. A volume of 0.1 ml of undiluted decarboxyfenvalerate (purity, 92%) was applied to the right eye of each rabbit. After 30 s, the exposed eyes of three of the male rabbits were flushed with tap water. Eye irritation was scored 1, 24 and 72 h and 7 and 14 days after exposure. The mean irritation scores were 7.7 and 8.7 for the washed and unwashed eyes, respectively, the maximum score being 110 (Cannelongo, 1983d).
The potential of decarboxyfenvalerate (purity not stated) to sensitize skin was tested in groups of five male and five female albino Duncan-Hartley guinea-pigs. Flank hair was clipped from each guinea-pig, and then the area was depilated with hair remover. Ethanol (the vehicle), 0.1% 2,4-dinitrochlorobenzene in diethyl ether (positive control) or undiluted decarboxyfenvalerate was applied in a volume of 0.5 ml under a lint patch, which was taped into place for 6 h, on days 1, 8 and 15. On day 29, the guinea-pigs were challenged on virgin sites with similar, single treatments with ethanol, dinitrochlorobenzene or decarboxyfenvalerate, and a fourth group of guinea-pigs was treated with undiluted decarboxyfenvalerate as a control for irritation. Skin reactions were scored 24 and 48 h after removal of the patches. No sensitization was induced in groups challenged with ethanol or undiluted decarboxyfenvalerate, whereas erythema and oedema were observed in the positive control group. No irritation was induced by undiluted decarboxyfenvalerate, which was judged not to sensitize skin under the conditions of the test (Parker, 1983).
Groups of 30 Fischer 344 rats of each sex were fed diets containing decarboxyfenvalerate (purity, 98%) at a concentration of 0, 30, 100, 300, 3000 or 10 000 ppm for up to 13 weeks. The mortality rate was not affected by treatment, but body-weight gain was significantly reduced in males at the highest concentration throughout the study and in females during the first 7 weeks. Body-weight gain was also significantly reduced in males at 3000 ppm during the first 7 weeks. Leukocyte counts were significantly reduced at weeks 7 and 13 in animals of each sex at 10 000 ppm and in females at 3000 ppm (p < 0.05, Dunnett test); similar reductions in the leukocyte count in males at 100 and 300 ppm at 13 weeks were not dose-related. The mean corpuscular volume was significantly reduced at 7 and 13 weeks in animals of each sex at 3000 and 10 000 ppm (p < 0.05; Dunnett test), except in females at 3000 ppm at 7 weeks. Some deviations in blood chemistry were noted, but they were generally inconsistent or of insufficient magnitude to be of biological importance. The absolute and relative (to body weight) weights of the liver were increased in animals of each sex at doses > 300 ppm, except in males at 300 ppm at 7 weeks. Consistent, significant increases were also seen in the absolute or relative weights of the kidneys in animals of each sex at 3000 and 10 000 ppm. The significant microscopic changes were limited to centrilobular hepatocellular hypertrophy at 10 000 ppm, minimal focal areas of hepatocellular necrosis in some rats at 3000 and 10 000 ppm and an increased incidence, but not severity, of glomerulonephrosis in animals at 10 000 ppm at 13 weeks. The NOAEL was 100 ppm, equivalent to 5 mg/kg bw per day, on the basis of increases in the absolute and relative weights of the liver in animals at 300 ppm (Parker et al., 1986).
A study of the acute oral toxicity of carboxy-fenvalerate, a metabolite of fenvalerate, in groups of 10 ddY mice of each sex gave an LD50 value of 410 mg/kg bw in both males and females (Kohda et al., 1980a). A similar study of the acute oral toxicity of carbamoyl-fenvalerate, a metabolite of fenvalerate, gave an LD50 value of > 2800 mg/kg bw in both males and females (Kohda et al., 1980b).
In a study of the potential genotoxicity of two fenvalerate metabolites, neither two lots of carboxy-fenvalerate (purity, 96.1% and 91.3%) nor carbamoyl-fenvalerate (purity, 98.4%) induced mutations in Salmonella typhimurium strain TA100, TA 98, TA1535 or TA1537 or in Escherichia coli WP2uvrA in the presence or absence of an exogenous metabolic system (Yamada & Kitamoto, 1995a,b).
The minor metabolites of esfenvalerate thus appear to be less toxic than the parent compounds.
No studies have been conducted on human exposure to esfenvalerate; however, studies of occupational exposure and volunteer studies have been carried out with fenvalerate.
In a clinical and electrophysiological assessment of 23 workers exposed to synthetic pyrethroids during research and development, 19 had experienced one or more episodes of abnormal facial sensations, which developed 0.5–3 h after exposure, with symptoms lasting up to 8 h. There were no neurological signs, and electrophysiological examination of the arms and legs of these workers showed that they were normal (LeQuesne & Maxwell, 1980). The dermal sensations reported by people exposed in particular to the alpha-cyano moiety may have been triggered by a series of impulses in sensory nerve endings, typical of pyrethroid stimulation of the nervous system by its action on the sodium channels in nerve membranes. These neurophysiological findings correlate well with the human experiences described, and indicate that the effects are transient and reversible. Additional clinical studies on fenvalerate were reported in WHO Environmental Health Criteria No. 95 (WHO, 1990).
No documented cases of human intoxication with esfenvalerate have been reported in the literature, but a report from China described 573 cases of intoxication with deltamethrin, cypermethrin or fenvalerate. In 229 cases of reported occupational exposure, the signs included burning or itching, tingling and paraesthesia. In the 344 cases of accidental ingestion, abdominal pain, nausea and vomiting were recorded. All but seven of the patients recovered within 2–3 weeks and most within 1–6 days (He et al., 1989). The role of exposure to the vehicle in these cases is unclear, and such a large number of adverse responses to alpha-cyano pyrethroids in occupational circumstances is unusual. Fenvalerate was reported previously not to produce effects in male or female volunteers (Annex 1, reference 33).
On the basis of a study of the effectiveness of methocarbamol as an antidote for esfenvalerate intoxication in rats, it was concluded that repetitive treatment with methocarbamol could reduce the pharmacological clinical signs produced by esfenvalerate in humans (Hiromori, 1986).
Studies of metabolism have been conducted in rats, mice and dogs with 14C-labelled esfenvalerate and fenvalerate. Excretion of both compounds was very rapid in rats and mice, 78–95% of the administered label being excreted within 1 day after oral administration. The concentrations of residues in tissues were generally very low; more persistent fenvalerate residues were found in mice than in other species. The metabolism of esfenvalerate in rodents was similar to that of fenvalerate. In dogs given labelled fenvalerate orally, less total radioactivity was recovered than in mice or rats, but the half-life was similar to that in rodents. The pattern of hydroxylation was different in rats and dogs, and the glycine conjugate, 3-phenoxybenzylglycine, was the major conjugate of the alcohol moiety in dogs, whereas it was a minor one in rats. Dogs also had a higher proportion of glucuronides of the acid moiety and its hydroxy derivatives. There was no evidence of accumulation of esfenvalerate or fenvalerate in fetal tissue or amniotic fluid of rats. No major sex differences were found in the metabolism of esfenvalerate or fenvalerate.
Esfenvalerate is a type II pyrethroid, a class that induces a typical syndrome characterized by choreoathetosis (coarse tremors progressing to sinuous writhing), sedation, salivation, dyspnoea and/or clonic seizures; sometimes, body tremors and prostration are seen. Such toxic signs have been observed in various species tested with esfenvalerate and are characteristic of a strong excitatory action on the nervous system, resulting from a specific interaction between esfenvalerate and the sodium channels of the nerve membranes. Series of nerve impulses are induced as a result of a change in the permeability of the membranes to sodium (repetitive effect). While the nerve endings of sensory organs are particularly sensitive to this effect, other parts of the nervous system are also affected.
The oral LD50 value of esfenvalerate in corn oil in rats was about 90 mg/kg bw, whereas the value in mice when administered in methyl cellulose was 320 mg/kg bw, but no investigation has been conducted to determine whether this is a true species difference or was due to differences in the vehicles used. Other pyrethroids, including fenvalerate, were less toxic when administered in an aqueous vehicle, than in an oily or lipophilic vehicle. The dermal LD50 was > 5000 mg/kg bw in rats and > 2000 mg/kg bw in rabbits. After inhalation, the 4-h LC50 value for rats was 480 mg/m3 in males and 570 mg/m3 in females.
Esfenvalerate was not irritating to the skin and was minimally irritating to the unwashed eyes of rabbits. It was judged to be a skin sensitizer in guinea-pigs in the Magnusson and Kligman maximization test but not in the Buehler test. WHO has classified esfenvalerate as ‘moderately hazardous’ (WHO, 2000).
Studies with repeated oral administration to mice, rats and dogs and dermal application to rats showed that the main effects of esfenvalerate are on clinical signs. These include hypersensitivity, agitation, impaired locomotor activity and reduction in body-weight gain. In the short-term studies with esfenvalerate in the diet, the NOAEL was 10 mg/kg bw per day in mice treated for 13 weeks, 6.2 mg/kg bw per day in rats treated for 13 weeks and 5 mg/kg bw per day in dogs treated for 12 months. In a 21-day study in rats treated cutaneously with esfenvalerate, the NOAEL was 25 mg/kg bw per day.
In the only long-term study with esfenvalerate, no evidence of carcinogenicity was found in mice; however, the single treated group that was suitable for evaluation received a concentration of 35 ppm in the diet, equal to 4.3 mg/kg bw per day, which was well below the maximum tolerated dose. The next highest concentration, 150 ppm, resulted in excessive self-mutilation and reduced survival, so that the results could not be used.
The Meeting was aware of five long-term studies of toxicity with fenvalerate, two in mice and three in rats. There was no evidence of carcinogenicity in mice. In the experiments in rats, increased incidences of certain neoplasms were observed in some groups: mammary tumours in one experiment without a dose–response relationship, and no increase in the incidence of this tumour type in another experiment with the same strain of rat and a higher dose; spindle-cell sarcomas (probably various types and at a low incidence even when combined) in males but not in females; and Leydig-cell adenomas in a strain of rats in which they are particularly common and occur at variable incidence. Increased incidences of these benign Leydig-cell tumours, which are rare in man, were not observed in the other experiments in rats or in mice and were consequently considered to be of little relevance, if any, to an evaluation of effects on human health. The unusual, but low, combined incidence of various sarcomas of the subcutis in males in one of the experiments in rats could not be entirely dismissed. These tumours were not, however, observed in females in the same experiment or in a different strain of rat in another experiment in which a 50% higher dose was used. In spite of these three examples of elevated tumour incidences with fenvalerate, the weight of evidence led the Meeting to the conclusion that esfenvalerate is not carcinogenic in rodents.
The NOAEL for the toxicity of fenvalerate in long-term studies in rats was 150 ppm, equivalent to 7.5 mg/kg bw per day, on the basis of a reduction of body-weight gain in males, giant-cell infiltration of lymph nodes and adrenals and reticuloendothelial-cell proliferation in the lymph nodes at 500 ppm, equivalent to 25 mg/kg bw per day. The NOAEL for the toxicity of esfenvalerate in long-term studies in mice was 35 ppm, equal to 4.3 mg/kg bw per day, on the basis of dermal damage and extramedullary haematopoiesis in the spleen at 150 ppm, equal to 18 mg/kg bw per day, in the same study.
Esfenvalerate was tested for genotoxicity in an adequate range of assays, both in vitro and in vivo. It showed no evidence of genotoxicity.
The Meeting concluded that esfenvalerate, which has been tested in mice and rats as a component of fenvalerate, is unlikely to pose a carcinogenic risk to humans.
In a three-generation study of reproductive toxicity in rats, the NOAEL for systemic toxicity was 75 ppm, equal to 4.2 mg/kg bw per day, on the basis of a reduction in body-weight gain in males and females at 100 ppm, equal to 5.6 mg/kg bw per day, during the pre-mating period. Treatment-related dermal lesions were found in adults fed powdered diets containing 75 or 100 ppm of esfenvalerate. No NOAEL was identified for the adults in this study, but the NOAEL for toxicity in offspring was 75 ppm, equal to 4.2 mg/kg bw per day, on the basis of reductions in litter size and pup body weight at 100 ppm. The NOAEL for reproductive toxicity was 100 ppm, equal to 5.6 mg/kg bw per day, the highest dose tested. A second study was conducted to identify the NOAEL for adults when cutaneous exposure was avoided by administering esfenvalerate in pelleted diets. There were no dermal lesions. The NOAEL for adults was 40 ppm, equal to 2.4 mg/kg bw per day, on the basis of reduced body weight and food consumption at 100 ppm in both the parental rats and their offspring.
In a study of developmental toxicity in rats, the NOAEL for maternal toxicity was 3 mg/kg bw per day on the basis of significant maternal toxicity (abnormal gait, hind-limb spasms, diarrhoea and tremors) at 4 mg/kg bw per day, and the NOAEL for developmental toxicity was 20 mg/kg bw per day, the highest dose tested.
In a study of developmental toxicity in rabbits, the NOAEL for maternal toxicity was 2 mg/kg bw per day on the basis of significant maternal toxicity (erratic jerking and extension of the limbs, followed by excessive grooming and rapid side-to-side head movements) at 3 mg/kg bw per day. The NOAEL for developmental toxicity was 20 mg/kg bw per day, the highest dose tested.
The results of acute and 90-day studies of neurotoxicity in rats showed that esfenvalerate does not induce neuropathological changes. The NOAEL for neurotoxicity in a study in rats given a single dose was 1.75 mg/kg bw, as tremors were induced at 1.90 mg/kg bw. The NOAEL for systemic toxicity and neurotoxicity in a 90-day study in rats was 40 ppm, equal to 3 mg/kg bw per day, on the basis of decreased motor activity and reduced body-weight gain at 120 ppm, equal to 8.9 mg/kg bw per day.
The Meeting concluded that the existing database was adequate to characterize the potential hazard of esfenvalerate to fetuses, infants and children.
An ADI of 0–0.02 mg/kg bw was established for esfenvalerate on the basis of the NOAEL of 2 mg/kg bw per day for maternal toxicity in the study of developmental toxicity in rabbits, which was supported by the NOAEL of 2.4 mg/kg bw per day in the multigeneration study of reproductive toxicity in rats and a safety factor of 100.
The Meeting established an acute RfD of 0.02 mg/kg bw on the basis of the NOAEL of 1.75 mg/kg bw in the study of acute neurotoxicity in rats and a safety factor of 100.
|
Levels relevant to risk assessment |
||||
|
Species |
Study |
Effect |
NOAEL |
LOAEL |
|
Mouse |
18-month study of toxicity and carcinogenicitya |
Toxicity |
35 ppm, equal to 4.3 mg/kg bw per day |
150 ppm, equal to 18 mg/kg bw per day |
|
Carcinogenicity |
35 ppm, equal to 4.3 mg/kg bw per dayC |
– |
||
|
Rat |
104–119-week study of toxicity and carcinogenicity with fenvaleratea |
Toxicity |
150 ppm, equivalent to 7.5 mg/kg bw per day |
500 ppm, equivalent to 25 mg/kg bw per day |
|
Carcinogenicity |
1500 ppm, equivalent to 75 mg/kg bw per dayd |
– |
||
|
Three-generation study of reproductive toxicitya |
Parental toxicity |
40 ppm, equal to 2.4 mg/kg bw per day |
100 ppm, equal to 4.7 mg/kg bw per day |
|
|
Pup toxicity |
75 ppm, equal to 4.2 mg/kg bw per day |
100 ppm, equal to 5.6 mg/kg bw per day |
||
|
Developmental toxicityb |
Maternal toxicity |
3 mg/kg bw per day |
4 mg/kg bw per day |
|
|
Pup toxicity |
20 mg/kg bw per dayd |
– |
||
|
Acute neurotoxicityb |
Neurotoxicity |
1.75 mg/kg bw |
1.9 mg/kg bw |
|
|
13-week study of neurotoxicitya |
Neurotoxicity |
40 ppm, equal to 3 mg/kg bw per day |
120 ppm, equal to 8.9 mg/kg bw per day |
|
|
Rabbit |
Developmental toxicityb |
Maternal toxicity |
2 mg/kg bw per day |
3 mg/kg bw per day |
|
Kit toxicity |
20 mg/kg bw per dayd |
– |
||
|
Dog |
1-year study of toxicitya |
Toxicity |
200 ppm, equivalent to 5 mg/kg bw per dayd |
– |
Esfenvalerate was tested, except where indicated.
a Dietary administration
b Gavage
c Only dose suitable for evaluation
d Highest dose tested
Estimate of acceptable daily intake for humans
0–0.02 mg/kg bw
Estimate of acute reference dose
0.02 mg/kg bw
Studies that would provide information useful for continued evaluation of the compound
Further observations in humans
List of end-points relevant for setting guidance values for dietary and non-dietary exposure
|
Absorption, distribution, excretion and metabolism |
|
|
Rate and extent of absorption of an oral dose |
78–95% excretion within 24 h, indicating extensive absorption |
|
Dermal absorption |
No study of direct dermal absorption available |
|
Distribution |
Distributed throughout the body. Generally very low concentrations of residues in tissues. Most extensive distribution to fat, skin, hair and stomach |
|
Potential for accumulation |
Low, due to rapid excretion |
|
Rate and extent of excretion |
78–95% excretion within 24 h |
|
Metabolism in animals |
Extensive |
|
Toxicologically significant compounds (animals, plants and environment) |
Parent |
|
|
|
|
Acute toxicity |
|
|
Rat: LD50. oral |
90 mg/kg bw |
|
Rat: LD50, dermal |
> 5000 mg/kg bw |
|
Rat: LC50, inhalation |
480 mg/m3 (4 h) |
|
Mouse: LD50, oral |
250 mg/kg bw |
|
Rabbit: LD50, dermal |
> 2000 mg/kg bw |
|
Rabbit: Skin irritation |
Not irritating |
|
Rabbit: Eye irritation |
Mildly irritating |
|
Guinea-pig: Skin sensitization |
Sensitizing in Magnusson & Kligman test |
|
|
Not sensitizing in Buehler test |
|
|
|
|
Short-term studies of toxicity |
|
|
Target/critical effect |
Clinical signs of neurotoxicity and decreased body-weight gain |
|
Lowest relevant oral NOAEL |
125 ppm, equivalent to 6.2 mg/kg bw per day (90 days, rat) |
|
Lowest relevant dermal NOAEL |
1000 mg/kg bw per day (21 days, rabbit) |
|
Lowest relevant inhalation NOAEL |
No data available |
|
Genotoxicity |
Not genotoxic |
|
|
|
|
Long-term studies of toxicity and carcinogenicity |
|
|
Target/critical effect |
Decreased body-weight gain |
|
Lowest relevant NOAEL |
35 ppm, equal to 4.3 mg/kg bw per day (18 months, mouse) |
|
Carcinogenicity |
Not carcinogenic |
|
|
|
|
Reproductive toxicity |
|
|
Target/critical effect for reproductive toxicity |
Reduced parental and offspring body weight. |
|
Lowest relevant NOAEL for reproductive toxicity |
40 ppm, equal to 2.4 mg/kg bw per day |
|
Target/critical effect for developmental toxicity |
Maternal: clinical signs of toxicity |
|
|
Developmental: none |
|
Lowest relevant NOAEL for developmental toxicity |
Maternal: 2 mg/kg bw per day |
|
|
Developmental: 20 mg/kg bw per day, highest dose tested |
|
|
|
|
Neurotoxicity |
|
|
Target/critical effect for acute neurotoxicity |
Tremors |
|
Lowest relevant NOAEL for acute neurotoxicity |
1.8 mg/kg bw |
|
Target/critical effect for 90-day neurotoxicity |
Decreased motor activity |
|
Lowest relevant NOAEL for 90-day neurotoxicity |
3.0 mg/kg/day |
|
|
|
|
Medical data |
Transient paraesthesia |
|
Summary |
Value |
Study |
Safety factor |
|
ADI |
0–0.02 mg/kg bw |
Maternal toxicity in a study of developmental toxicity in rabbits |
100 |
|
Acute RfD |
0.02 mg/kg bw |
Rat, acute neurotoxicity |
100 |
(Studies marked with * were conducted according to good laboratory practice.)
Aldridge, W.N. (1990) An assessment of the toxicological properties of pyrethroids and their neurotoxicity. Crit. Rev. Toxicol., 21, 105–127.
Appel, K.E. & Gericke, S. (1993) [Neurotoxicity and toxicokinetics of pyrethroids.] Bundesgesundhblatt, 6, 219–228 (in German).
*Arai, M., Suzuki, T., Okuno, Y., Ito, S., Murakami, M. & Seki, T. (1980) Two-year chronic toxicity study of S5602 in rats. Unpublished report No. AT-10-0278 from Sumitomo Chemical Co. Ltd, Takarazuka Research Centre, Hyogo, Japan, and Laboratory Animal Centre, Nihon Dobutsu Co., Osaka, Japan. Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
*Arai, M., Hiromori, T., Ito, Y. & Okuno, Y. (1981) Life span chronic toxicity study of S5602 in mice. Unpublished report No. AT-10-0286 from Sumitomo Chemical Co. Ltd, Laboratory of Biochemistry and Toxicology, Hyogo, Japan. Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
*Beyrouty, P. (1999) A 13-week dietary neurotoxicity study of esfenvalerate TG in the rat. Project ID 97524. ClinTrials BioResearch Ltd, Quebec, Canada. Unpublished report No. LLT-0189. Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
*Biegel, L.B. (1994) Reproductive and fertility effects with DPX-YB656-84 multigeneration reproduction study in rats. E.I. du Pont de Nemours & Co., Newark, Delaware, USA. Unpublished report (LLT-41-0169). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Boorman, G.A., Hamlin, M.H. & Eustis, S.L. (1987) Focal interstitial cell hyperplasia, testes, rat. In: Jones, T.C., Mohr, U. & Hunt, R.D., eds, Monographs on Pathology of Laboratory Animals. Genital System, Berlin: Springer, pp. 192–195.
Cannelongo, B.F. (1983a) Rat acute oral toxicity of SD 54597. 2840-82. Stillmeadow, Inc., Biological Testing Laboratory, Houston, Texas, USA. Unpublished report (AT-31-0366). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Cannelongo, B.F. (1983b) Acute dermal toxicity of SD 54597. 2841-82. Stillmeadow, Inc., Biological Testing Laboratory, Houston, Texas, USA. Unpublished report (AT-31-0366). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Cannelongo, B.F. (1983c) Primary skin irritation of SD 54597. 2843-82. Stillmeadow, Inc., Biological Testing Laboratory, Houston, Texas, USA. Unpublished report (AT-31-0366). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Cannelongo, B.F. (1983d) Rabbit eye irritation of SD 54597. 2842-82. Stillmeadow, Inc., Biological Testing Laboratory, Houston, Texas, USA. Unpublished report (AT-31-0366). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
*Chazono, Y. (1985) Primary eye and skin irritation tests with Sumi-alpha in rabbits. Unpublished report LLT-50-0001 from Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
*Chazono, Y. (1986a) Skin sensitisation of S-1844 in guinea pigs—Maximisation test. ANT 8512. Unpublished report LLT-60-0154 from Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
*Chazono, Y. (1986b) Skin sensitisation of S-1844 in guinea pigs—Buehler test. ANT 8520. Unpublished report LLT-60-0153 from Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Delker, D.A. (2000) Esfenvalerate technical: Repeated dose dermal 21-day toxicity study in rats. DuPont-4228. DuPont Haskell Laboratory, Newark, Delaware, USA. Unpublished report (LLT-0207). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Dickie, B.C. (1986) One-year oral study in dogs with MO 70616 technical. 6160-103. Hazleton Laboratories America Inc., Madison, Wisconsin, USA. Unpublished report (LLT-61-0063). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Draize, J.H. (1977) Dermal and eye toxicity tests. In: Principles and Procedures for Evaluating the Toxicity of Household Substances, Washington DC: National Academy of Sciences, pp. 48–49.
Gordon, E.B. & Weir, R.J. (1978) Lifetime feeding study in rats. LBI 2541. Litton Bionetics Inc., Kensington, Maryland, USA. Unpublished report (AT-81-0182). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Gordon, E.B. & Weir, R.J. (1979) Lifetime feeding study in rats. 20733-01. Litton Bionetics Inc., Kensington, Maryland, USA. Unpublished report (AT-91-0231). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
He, F., Wang, S., Liu, L., Chen, S., Zhang, Z. & Sun, J. (1989) Clinical manifestations and diagnosis of acute pyrethroid poisoning. Arch. Toxicol., 63, 54–58.
Higuchi, H. (1999) Reproduction study in rats with esfenvalerate (in pelleted diet). 3356. Environmental Health Science Laboratory, Sumitomo Chemical Co. Ltd Unpublished report (LLT-0192). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Hiromori, T. (1986) Methocarbamol as a therapeutic against acute intoxication with S-1844 in rats. RS-8. Sumitomo Chemical Co. Ltd, Laboratory of Biochemistry and Toxicology Research Department, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-60-0032). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Isobe, N., Kaneko, H., Yanagita, S., Saito, K., Ohe, A., Yoshitake, A. & Miyamioto, J. (1985) Comparative metabolism of esfenvalerate and fenvalerate in rats and mice. 2. 28-days dietary administration in mice. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLM-50-0008). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Ito, N. (1981) The comment on spindle cell sarcomas which were seen in the life time feeding study in rats of SD-43775 technical by Litton Bionetics LBI No. 20733-01. Unpublished report (LLM-50-0008). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Johnston, C.D. (1979) Chronic toxicity and potential carcinogenicity study in the mouse. 20738. Litton Bionetics Inc., Kensington, Maryland, USA. Unpublished report (AT-91-0230). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Kaneko, H., Ohkawa, H. & Miyamoto, J. (1981) Comparative metabolism of fenvalerate and the [2S, alphaS]-isomer in rats. J. Pestic. Sci., 6, 317–326.
Kaneko, H., Izumi, T., Matsuo, M. & Miyamoto, J. (1984) Metabolism of fenvalerate in dogs. J. Pestic. Sci., 9, 269–274.
Kaneko, H., Isobe, N., Shiba, K., Saito, K., Yanagita, S., Kitamura, N, Ohe, A., Yoshitake, A. & Miyamoto, J. (1985) Comparative metabolism of esfenvalerate and fenvalerate in rats and mice. 1. Single or 10 consecutive oral administration. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLM-50-0007). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Kaneko, H., Matsuo, M. & Miyamoto, J. (1986) Differential metabolism of fenvalerate and granuloma formation. 1. Identification of a cholesterol ester derived from a specific chiral isomer of fenvalerate. Toxicol. Appl. Pharmacol., 83, 148–156.
Kaneko, H., Takamatsu, Y., Okuno, Y., Abiko, J., Yoshitake, A. & Miyamoto, J. (1988) Substrate specificity for formation of a cholesterol ester conjugate from fenvalerate analogues and for granuloma formation. Xenobiotica, 18, 11–19.
Kelly, W.A. (1985) Subchronic feeding study of MO 70616 in the rat. 227A-101-030-84. TPS Inc., Mount Vernon, Indiana, USA. Unpublished report (LLT-51-0013). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Kogiso, S. (1985a) Reverse mutation test of S-1844 in Salmonella typhimurium and Escherichia coli. MUT 85066. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-50-0009). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Kogiso, S. (1985b) In vitro gene mutation test of S-1844 in V79 Chinese hamster cells in culture. MUT 85069. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-50-0012). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Kogiso, S. (1985c) In vitro chromosomal aberration test of S-1844 in Chinese hamster ovary cells (CHO-K1). MUT 85070. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-50-0010). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Kogiso, S. (1985d) Micronucleus test of S-1844 in mouse bone marrow cells. MUT 85067. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-50-0011). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Kogiso, S. (1986) Unscheduled DNA synthesis assay of S-1844 in HeLa cells. MUT 85068. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-60-0022). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Kohda et al. (1980a) Acute oral toxicity study of S-5602-COOH in mice. Unpublished report (AT-00-0481). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Kohda et al. (1980b) Acute oral toxicity study of S-5602-CONH2 in mice. Unpublished report (AT-00-0482). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Kohda, H. (1985) Acute inhalation toxicity of S-1844 in rats. IHT 8505. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-50-0002). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Koyama, Y. (1985) Comparative subacute toxicity in B6C3F1 mice treated with S-1844 and S-5602 for 3 months. GTS-8505. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-50-0004). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Larson, D.M. (1987) 13-week dietary admix study of MO 70616 technical in rats. 227B-102-634-86. TPS Inc., Mount Vernon, Indiana, USA. Unpublished report (LLT-71-0086). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
LeQuesne, P.M. & Maxwell, I.C. (1980) Transient facial sensory symptoms following exposure to synthetic pyrethroids: A clinical and electrophysiological assessment. Neurotoxicology, 2, 1–11.
Maedgen, J.L. (1985a) Acute dermal toxicity of MO 70616 in the rabbit. 3881-85. Stillmeadow Inc., Houston, Texas, USA. Unpublished report (LLT-61-0041). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Maedgen, J.L. (1985b) Primary skin irritation of MO 70616 in the rabbit. 3883-85. Stillmeadow Inc., Houston, Texas, USA. Unpublished report (LLT-61-0043). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Maedgen, J.L. (1985c) Eye irritation of MO 70616 in the rabbit. 3882-85. Stillmeadow Inc., Houston, Texas, USA. Unpublished report (LLT-61-0042). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Maedgen, J.L. (1986) Guinea pig skin sensitisation of MO70616 technical. 4274-86. Stillmeadow Inc., Houston, Texas, USA. Unpublished report (LLT-61-0116). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Malley, L.A. (2000a) Esfenvalerate: Acute oral neurotoxicity study in rats. DuPont-3874. E.I. du Pont de Nemours & Co., Newark, Delaware, USA. Unpublished report (LLT-0206). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Malley, L.A. (2000b) Esfenvalerate: Subchronic oral neurotoxicity study in rats. DuPont-3081. E.I. du Pont de Nemours & Co., Newark, Delaware, USA. Unpublished report (LLT-0205). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
McConnell, R.F., Westen, H.H., Ulland, B.M., Bosland, M.C. & Ward, J.M. (1992) Proliferative lesions of the testes of rats with selected examples from mice, URG-3. In: Guides for Toxicologic Pathology, Washington DC, American Registry of Pathology.
Miyamoto, J. (1976) Degradation, metabolism and toxicity of synthetic pyrethroids. Environ. Health Perspectives, 14, 15–28.
Miyamoto, J., Kaneko, H. & Takamatsu, Y. (1986) Stereoselective formation of a cholesterol ester conjugate from fenvalerate by mouse microsomal carboxyesterase(s). J. Biochem. Toxicol., 1, 79–94.
Murray, M.A. (1994a) Pilot developmental toxicity study of DPX-YB656-84 in rats. HLR 36-94. E.I. du Pont de Nemours & Co., Newark, Delaware, USA. Unpublished report (LLT-41-0167). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Murray, M.A. (1994b) Pilot developmental toxicity study of DPX-YB656-84 in rabbits. HLR 43-94. E.I. du Pont de Nemours & Co., Newark, Delaware, USA. Unpublished report (LLT-41-0168). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Nemec, M.D. (1991a) A developmental toxicity study of S-1844 in rats. WIL-118010. WIL Research Laboratories Inc., Ashland, Ohio, USA. Unpublished report (LLT-11-0143). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Nemec, M.D. (1991b) A developmental toxicity study of S-1844 in rabbits. WIL-118013. WIL Research Laboratories Inc., Ashland, Ohio, USA. Unpublished report (LLT-01-0135). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Ohkawa, H., Kaneko, H., Tsuji, H. & Miyamoto, J. (1979) Metabolism of fenvalerate (Sumicidin®) in rats. J. Pestic. Sci., 4, 143–155.
Okuno, Y. (1985) Comparative neurotoxicity of S-1844 and S-5602: Effects of a single oral administration. GTS 8509. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-50-0003). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Omodaka, H. (1985a) Acute oral toxicity of S-1844 in rats. ACT 85064. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-50-0005). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Omodaka, H. (1985b) Acute dermal toxicity of S-1844 in rats. ACT 85065. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-50-0006). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Omodaka, H. (1986) Acute oral toxicity of S-1844 in mice. ACT 85079. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLT-60-0017). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Parker, C.M. (1983) Guinea pig sensitisation study of SD 54597. 61530. Westhollow Research Center, Toxicology Laboratory, Houston, Texas, USA. Unpublished report (AT-31-0367). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Parker, C.M., Wimberly, H.C., Lam, A.S., Gardiner, T.H. & Van Gelder, G.A. (1986) Subchronic feeding study of decarboxyfenvalerate in rats. J. Toxicol. Environ. Health, 18, 77–90.
Ross, P.E. (1997) Oncogenicity study with DPX-YB656-84: Eighteen-month feeding study in mice. Haskell Laboratory Report No. 853-95, Newark, Delaware. Unpublished report (LLT-0180). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Shiba, K., Kaneko, H., Kitamura, N., Yoshitake, A. & Miyamoto, J. (1985) Comparative metabolism of esfenvalerate and fenvalerate in rats and mice. 3. Placental transfer in pregnant rats. Sumitomo Chemical Co. Ltd, Takarazuka Research Center, Hyogo, Japan. Unpublished report (LLM-50-0009). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Vijverberg, H.P.M. & van den Bercken, J. (1990) Neurotoxicological effects and the mode of action of pyrethroid insecticides. Crit. Rev. Toxicol., 21, 105–126
Vijverberg, H.P.M. & Oortgiessen, M. (1988) Steric structure and action of pyrethroids. In: Ariens, E.J., van Rensen, J.J.S. & Welling, W., eds, Stereoselectivity of Pesticides; Biological and Chemical Problems, Amsterdam: Elsevier Science Publishers.
WHO (1990) Environmental Health Criteria 95: Fenvalerate, Geneva.
WHO (2000) The WHO recommended classification of pesticides by hazard and guidelines to classification 2000–2202 (WHO/PCS/01.5). Available from the International Programme on Chemical Safety, World Health Organization, Geneva, Switzerland.
Yamada, T. (1999) Hormonal investigation for rat testicular tumorigenicity using fenvalerate and esfenvalerate. 3357. Environmental Health Science Laboratory, Sumitomo Chemical Co. Ltd. Unpublished report (LLT-0190). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Yamada, T. & Kitamoto, ? (1995a) Reverse mutation test of COOH-fenvalerate in bacterial systems. M0983. Environmental Health Science Laboratory, Sumitomo Chemical Co. Ltd. Unpublished report (AT-50-0485). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
Yamada, T. & Kitamoto, S. (1995b) Reverse mutation test of CONH2-fenvalerate in bacterial systems. M0982. Environmental Health Science Laboratory, Sumitomo Chemical Co. Ltd. Unpublished report (AT-50-0486). Submitted to WHO by Sumitomo Chemical Co. Ltd, Tokyo, Japan.
See Also: Toxicological Abbreviations Esfenvalerate (ICSC) Esfenvalerate (UKPID)