
ANTIMICROBIAL AGENTS
CEFTIOFUR
First draft prepared by
Dr. L. Ritter
Canadian Network of Toxicology Centres, University of Guelph
Guelph, Ontario, Canada
Dr. G. Kirby
Ontario Veterinary College, University of Guelph
Guelph, Ontario, Canada
Dr. C. Cerniglia
National Center for Toxicological Research,
Jefferson, Arkansas, USA
Explanation
Biological data
Biochemical aspects
Absorption, distribution, and excretion
Biotransformation
Toxicological studies
Acute toxicity studies
Short-term toxicity studies
Reproductive toxicity studies
Special studies on embryotoxicity and teratogenicity
Special studies on genotoxicity
Special studies on immunotoxicity
Special studies on microbiological effects
Observations in humans
Comments
Evaluation
References
1. EXPLANATION
Ceftiofur is a cephalosporin antibiotic with broad-spectrum
activity against both Gram-positive and Gram-negative bacteria
including ß-lactamase-producing bacterial strains. It inhibits
bacterial cell wall synthesis in a similar fashion to other
cephalosporins. Ceftiofur is used in the treatment of respiratory
infections in cattle and pigs. Ceftiofur had not been previously
evaluated by the Committee.
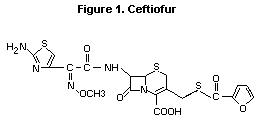
The chemical structure of ceftiofur is given in Figure 1. All
studies summarized in this monograph were performed with the sodium
salt.
Figure 1. Ceftiofur
 2. BIOLOGICAL DATA
2.1 Biochemical aspects
2.1.1 Absorption, distribution, and excretion
2.1.1.1 Rats
A group of Sprague-Dawley rats (7/sex) received single oral doses
of 14C-ceftiofur (200 mg/kg bw) in a comparative study with calves.
Approximately 55% of the total dose was recovered in the urine and the
rest was present in the faeces and GI tract. Plasma concentration at
6 h was 1 mg/kg and trace amounts of ceftiofur were present in all
tissues (i.e. liver, muscle and fat). The highest residue levels
(0.7 mg/kg) were present in kidney. The major urinary metabolite was
ceftiofursulfoxide cysteine thioester (Jaglan & Arnold, 1986a).
A study of 4 male and 4 female Sprague-Dawley rats treated
intramuscularly with 14C-ceftiofur (2 mg/kg bw) revealed that 55% of
the administered dose was excreted in the urine and about 30% in the
GI tract and faeces. The major urinary metabolite was desfuroylceftiofur
(DFC). The metabolism of ceftiofur was similar in calves administered
14C-ceftiofur (2 mg/kg bw) via the i.m. route. Unmetabolized
ceftiofur was also present in the urine (4.4-21% of total
radioactivity) (Jaglan & Arnold, 1987a).
A parallel comparative study to the one described above using
similar dosages and routes of administration in 2 rats (1 male and 1
female) and 2 calves demonstrated that acetamide conjugates of DFC
were the major urinary metabolites 1 h post-treatment (Jaglan &
Arnold, 1986b).
A study of 2 male rats treated with a single i.m. injection of
14C-ceftiofur revealed that DFC existed as complexes bound by
sulfhydryl groups to major serum proteins, albumin and alpha-1-
antitrypsin (Jaglan et al, 1987a).
A study in 8-week old Sprague-Dawley rats (7/sex) treated with
14C-ceftiofur (800 mg/kg bw/day) by oral gavage for 5 days revealed
several urinary metabolites, including DFC, ceftiofur sulfoxide, and
cysteine disulfide (Jaglan et al, 1987a). These results were similar
to those obtained following i.m. injection of ceftiofur described
above (Jaglan & Arnold, 1986a).
HPLC analysis of metabolites of 14C-ceftiofur formed by
arochlor-induced rat liver S-9 fractions in vitro revealed that DFC
was the major metabolite. Low doses (119 mg/kg bw) of ceftiofur were
completely metabolized within 15 minutes. Higher doses (857 mg/kg bw)
were converted to DFC after 60 minutes of incubation (Jaglan et al,
1987b).
2.1.1.2 Cattle
In two studies comparing the metabolism of orally administered
ceftiofur in rats, single i.m. injections of 14C-ceftiofur
(2 mg/kg bw) were given to 2 calves (sex not identified). The initial
urinary metabolite was desfuroylceftiofur formed by hydrolysis of the
thioester bond. An additional 3,3'-desfuryl ceftiofur disulfide dimer
was considered to be due to the alkaline condition in the urine of
herbivores (Jaglan & Arnold, 1987b; Jaglan et al, 1989).
A study of plasma concentrations following i.m. injections of
14ceftiofur (dose unspecified) in a heifer and a bull demonstrated
the presence of a single metabolite DFC, 1 h post-treatment. DFC
levels were undetectable after 16-24 h. DFC was due to cleavage of
thioester bond of ceftiofur (Krzeminski et al, 1985).
A study of i.m. administration of 14C-ceftiofur in a bull
revealed that 55% of the administered dose was excreted in the urine
and approximately 30% in the GI tract and faeces. The initial
metabolite in both urine and plasma was DFC. HPLC analysis of
radioactive metabolites was similar to the results found in the rat
studies (Jaglan & Arnold, 1987a). A number of metabolites were
produced, the major metabolite (87% of total urinary metabolites)
being DFC acetamide conjugates. No parent compound was observed in the
urine (Jaglan & Arnold, 1987b).
A study of lactating cows treated with 14C-ceftiofur (2.3 mg/kg
bw/day for 5 days) revealed that 32-38% of the radioactivity was
present in the milk as free metabolites. The major metabolite was
desfuroylceftiofur cysteine disulfide (DCD) representing 7-9% of the
total radioactivity. No parent compound was detected in the milk
(Jaglan et al, 1989).
A study of 4 calves (sex and breed unspecified) administered
ceftiofur intramuscularly daily for 4 days at 2 dose levels (2.2 or
4.4 mg/kg bw/day) demonstrated a plasma t1/2 of 3.5 h. Peak serum
concentration of 8.8 and 17.3 mg/ml were obtained at 2 h after doses
of 2.2 and 4.4 mg/kg bw/day, respectively. Plasma t1/2 of the
metabolite DFC was 9.7 h after i.m. administration
(Halstead et al, 1992).
Six Friesian calves (3/sex) were treated with ceftiofur according
to different protocols including one single i.m. and i.v. injection at
1 mg/kg bw, and 5 i.m. injections at 1 mg/kg bw at 24 h intervals.
Time to maximal plasma concentration following i.m. administration was
0.75 h. The t1/2 (0.07 h) was short due to rapid metabolism to DFC.
The t1/2 of DFC after i.m. and i.v. administration were similar
(9.7 and 8.6 h, respectively) (Halstead et al, 1992).
2.1.1.3 Pigs
A study (Jaglan et al, 1990) examining the profile of urinary
metabolites in pigs (number, breed and age unspecified) treated with 3
consecutive intramuscular injections of 14C-ceftiofur (5.2 mg/kg bw)
revealed a qualitatively similar profile of urinary metabolites to
that observed in rats treated with multiple oral doses of ceftiofur
(Jaglan et al, 1987a).
A study of 4- to 5-month old Yorkshire-Hampshire pigs (6/sex)
treated with 3 daily i.m. injections of 14C-ceftiofur (5.2 mg/kg bw)
produced similar results to those observed in rats and cattle. The
peak plasma levels of radioactivity (15.4 mg/kg) occurred at 2 h after
the last dose, declining to 7.0 mg/kg 12 h after the last dose. Tissue
levels in various tissues 12 h after the last dose were as follows:
lung, 2.9 mg/kg; muscle, 0.8 mg/kg; kidney, 4.5 mg/kg; GI tract 2.1,
and its contents, 5.7 mg/kg; mesentery glands, 1.9 mg/kg; turbinate,
2.7 mg/kg; tonsil, 1.7 mg/kg; brain, 0.1 mg/kg. Radioactivity in urine
and faeces accounted for 62% and 11% of the dose, respectively. Major
plasma metabolites of DFC covalently bound to proteins were identical
to those identified in rat and bovine studies. Urinary metabolites
were also similar consisting of ceftiofur and 8 metabolites including
DCD and 3,3'-desfuroylceftiofur disulfide, DFC and ceftiofur sulfoxide
cysteine thioester and an unidentified polar metabolite. The t1/2 of
DCA was 13.5 h after i.m. treatment and 12.2 h after i.v. treatment
(Yein et al, 1990).
2.1.2 Biotransformation
Metabolism of 14C-ceftiofur in cattle and rats involved rapid
cleavage of the thioester bond of ceftiofur yielding DFC and furoic
acid (Krzeminski et al, 1985; Yein et al, 1990; Banting et al, 1989).
The major urinary metabolite in cattle were desfuorylceftiofur
thiolactone, DCD, and 3,3'-desfuorylceftiofur disulfide dimer
(3,3'-DFD) formed because of the alkaline condition of urine of
herbivores. The major urinary metabolite after oral administration in
the rat was ceftiofur sulfoxide cysteine thioester due to enteric
metabolism (Jaglan & Arnold, 1987a). In rats, desfuroylceftiofur was
covalently bound to plasma proteins, principally albumin and
alpha-antitrypsin (Jaglan, et al, 1991), whereas DFC was primarily
free in calf plasma (Jaglan & Arnold, 1986b).
Studies of 14C-ceftiofur metabolism in vitro with hepatic S-9
fraction from Arochlor-1254-induced F344 rats (Jaglan et al, 1987),
and liver and kidney S-9 fractions from pigs, cattle and chickens
(Gilbertson et al, 1990), demonstrated qualitatively similar results
to the in vivo studies. In all species, DFC and its dimer were the
major metabolites of liver S-9 fractions and DCD was generated by
kidney S-9 fractions. No ceftiofur metabolite-protein complexes were
observed in vitro (Jaglan et al, 1987b).
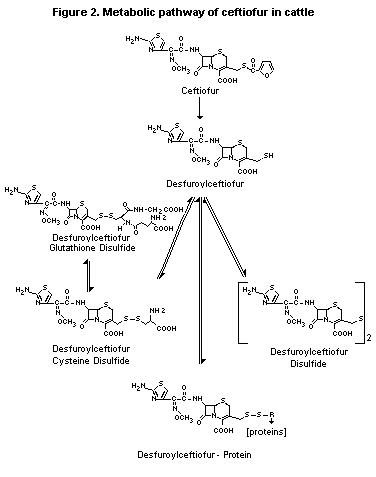
The metabolism of ceftiofur in cattle is shown in Figure 2.
2.2 Toxicological studies
2.2.1 Acute toxicity studies
2.2.1.1 Mice
The acute toxicity of ceftiofur was studied in groups of 5 female
mice per dose, which were treated via the i.v. and i.m. routes. In the
i.v. study, convulsions preceded death while in the i.m. study, mild
prostration was noted. The LD50 by the i.v. and i.m. routes were
about 2000 mg/kg bw and 3400 mg/kg bw, respectively (Berthe, 1982a).
2.2.1.2 Rats
The acute toxicity of ceftiofur was studied in female
Sprague-Dawley rats via the i.v. and i.m. routes, and in both males
and females via the oral and inhalation routes.
In an i.v. study, ceftiofur was administered at doses up to
3800 mg/kg bw. Conjunctival haemorrhage was noted during
administration of the test substance and death was preceded by
convulsion. The LD50 was 2200 mg/kg bw (Berthe, 1982a).
In an i.m. study, ceftiofur was administered at doses up to
1500 mg/kg bw. Mild prostration was noted and the LD50 was
1250 mg/kg bw (Berthe, 1982a).
In an inhalation study, ceftiofur was administered at an aerosol
concentration of 8.3 mg/litre to a group of 5 male and 5 female
Sprague-Dawley rats for a 4-h exposure period. During exposure, rats
exhibited salivation, nasal discharge and dyspnea; these signs
virtually disappeared within 1 h after exposure. Post-exposure signs
included diarrhea in 6 rats, and 1 rat exhibited a red encrusted
material around the nares. Both gross and microscopic examination did
not reveal any treatment-related changes. As none of the test animals
died either during treatment or during the post-treatment 14-day
observation period, the acute 4 h LC50 was estimated to be greater
than 8.3 mg/litre (Leong et al, 1985).
Figure 2. Metabolic pathway of ceftiofur in cattle
2. BIOLOGICAL DATA
2.1 Biochemical aspects
2.1.1 Absorption, distribution, and excretion
2.1.1.1 Rats
A group of Sprague-Dawley rats (7/sex) received single oral doses
of 14C-ceftiofur (200 mg/kg bw) in a comparative study with calves.
Approximately 55% of the total dose was recovered in the urine and the
rest was present in the faeces and GI tract. Plasma concentration at
6 h was 1 mg/kg and trace amounts of ceftiofur were present in all
tissues (i.e. liver, muscle and fat). The highest residue levels
(0.7 mg/kg) were present in kidney. The major urinary metabolite was
ceftiofursulfoxide cysteine thioester (Jaglan & Arnold, 1986a).
A study of 4 male and 4 female Sprague-Dawley rats treated
intramuscularly with 14C-ceftiofur (2 mg/kg bw) revealed that 55% of
the administered dose was excreted in the urine and about 30% in the
GI tract and faeces. The major urinary metabolite was desfuroylceftiofur
(DFC). The metabolism of ceftiofur was similar in calves administered
14C-ceftiofur (2 mg/kg bw) via the i.m. route. Unmetabolized
ceftiofur was also present in the urine (4.4-21% of total
radioactivity) (Jaglan & Arnold, 1987a).
A parallel comparative study to the one described above using
similar dosages and routes of administration in 2 rats (1 male and 1
female) and 2 calves demonstrated that acetamide conjugates of DFC
were the major urinary metabolites 1 h post-treatment (Jaglan &
Arnold, 1986b).
A study of 2 male rats treated with a single i.m. injection of
14C-ceftiofur revealed that DFC existed as complexes bound by
sulfhydryl groups to major serum proteins, albumin and alpha-1-
antitrypsin (Jaglan et al, 1987a).
A study in 8-week old Sprague-Dawley rats (7/sex) treated with
14C-ceftiofur (800 mg/kg bw/day) by oral gavage for 5 days revealed
several urinary metabolites, including DFC, ceftiofur sulfoxide, and
cysteine disulfide (Jaglan et al, 1987a). These results were similar
to those obtained following i.m. injection of ceftiofur described
above (Jaglan & Arnold, 1986a).
HPLC analysis of metabolites of 14C-ceftiofur formed by
arochlor-induced rat liver S-9 fractions in vitro revealed that DFC
was the major metabolite. Low doses (119 mg/kg bw) of ceftiofur were
completely metabolized within 15 minutes. Higher doses (857 mg/kg bw)
were converted to DFC after 60 minutes of incubation (Jaglan et al,
1987b).
2.1.1.2 Cattle
In two studies comparing the metabolism of orally administered
ceftiofur in rats, single i.m. injections of 14C-ceftiofur
(2 mg/kg bw) were given to 2 calves (sex not identified). The initial
urinary metabolite was desfuroylceftiofur formed by hydrolysis of the
thioester bond. An additional 3,3'-desfuryl ceftiofur disulfide dimer
was considered to be due to the alkaline condition in the urine of
herbivores (Jaglan & Arnold, 1987b; Jaglan et al, 1989).
A study of plasma concentrations following i.m. injections of
14ceftiofur (dose unspecified) in a heifer and a bull demonstrated
the presence of a single metabolite DFC, 1 h post-treatment. DFC
levels were undetectable after 16-24 h. DFC was due to cleavage of
thioester bond of ceftiofur (Krzeminski et al, 1985).
A study of i.m. administration of 14C-ceftiofur in a bull
revealed that 55% of the administered dose was excreted in the urine
and approximately 30% in the GI tract and faeces. The initial
metabolite in both urine and plasma was DFC. HPLC analysis of
radioactive metabolites was similar to the results found in the rat
studies (Jaglan & Arnold, 1987a). A number of metabolites were
produced, the major metabolite (87% of total urinary metabolites)
being DFC acetamide conjugates. No parent compound was observed in the
urine (Jaglan & Arnold, 1987b).
A study of lactating cows treated with 14C-ceftiofur (2.3 mg/kg
bw/day for 5 days) revealed that 32-38% of the radioactivity was
present in the milk as free metabolites. The major metabolite was
desfuroylceftiofur cysteine disulfide (DCD) representing 7-9% of the
total radioactivity. No parent compound was detected in the milk
(Jaglan et al, 1989).
A study of 4 calves (sex and breed unspecified) administered
ceftiofur intramuscularly daily for 4 days at 2 dose levels (2.2 or
4.4 mg/kg bw/day) demonstrated a plasma t1/2 of 3.5 h. Peak serum
concentration of 8.8 and 17.3 mg/ml were obtained at 2 h after doses
of 2.2 and 4.4 mg/kg bw/day, respectively. Plasma t1/2 of the
metabolite DFC was 9.7 h after i.m. administration
(Halstead et al, 1992).
Six Friesian calves (3/sex) were treated with ceftiofur according
to different protocols including one single i.m. and i.v. injection at
1 mg/kg bw, and 5 i.m. injections at 1 mg/kg bw at 24 h intervals.
Time to maximal plasma concentration following i.m. administration was
0.75 h. The t1/2 (0.07 h) was short due to rapid metabolism to DFC.
The t1/2 of DFC after i.m. and i.v. administration were similar
(9.7 and 8.6 h, respectively) (Halstead et al, 1992).
2.1.1.3 Pigs
A study (Jaglan et al, 1990) examining the profile of urinary
metabolites in pigs (number, breed and age unspecified) treated with 3
consecutive intramuscular injections of 14C-ceftiofur (5.2 mg/kg bw)
revealed a qualitatively similar profile of urinary metabolites to
that observed in rats treated with multiple oral doses of ceftiofur
(Jaglan et al, 1987a).
A study of 4- to 5-month old Yorkshire-Hampshire pigs (6/sex)
treated with 3 daily i.m. injections of 14C-ceftiofur (5.2 mg/kg bw)
produced similar results to those observed in rats and cattle. The
peak plasma levels of radioactivity (15.4 mg/kg) occurred at 2 h after
the last dose, declining to 7.0 mg/kg 12 h after the last dose. Tissue
levels in various tissues 12 h after the last dose were as follows:
lung, 2.9 mg/kg; muscle, 0.8 mg/kg; kidney, 4.5 mg/kg; GI tract 2.1,
and its contents, 5.7 mg/kg; mesentery glands, 1.9 mg/kg; turbinate,
2.7 mg/kg; tonsil, 1.7 mg/kg; brain, 0.1 mg/kg. Radioactivity in urine
and faeces accounted for 62% and 11% of the dose, respectively. Major
plasma metabolites of DFC covalently bound to proteins were identical
to those identified in rat and bovine studies. Urinary metabolites
were also similar consisting of ceftiofur and 8 metabolites including
DCD and 3,3'-desfuroylceftiofur disulfide, DFC and ceftiofur sulfoxide
cysteine thioester and an unidentified polar metabolite. The t1/2 of
DCA was 13.5 h after i.m. treatment and 12.2 h after i.v. treatment
(Yein et al, 1990).
2.1.2 Biotransformation
Metabolism of 14C-ceftiofur in cattle and rats involved rapid
cleavage of the thioester bond of ceftiofur yielding DFC and furoic
acid (Krzeminski et al, 1985; Yein et al, 1990; Banting et al, 1989).
The major urinary metabolite in cattle were desfuorylceftiofur
thiolactone, DCD, and 3,3'-desfuorylceftiofur disulfide dimer
(3,3'-DFD) formed because of the alkaline condition of urine of
herbivores. The major urinary metabolite after oral administration in
the rat was ceftiofur sulfoxide cysteine thioester due to enteric
metabolism (Jaglan & Arnold, 1987a). In rats, desfuroylceftiofur was
covalently bound to plasma proteins, principally albumin and
alpha-antitrypsin (Jaglan, et al, 1991), whereas DFC was primarily
free in calf plasma (Jaglan & Arnold, 1986b).
Studies of 14C-ceftiofur metabolism in vitro with hepatic S-9
fraction from Arochlor-1254-induced F344 rats (Jaglan et al, 1987),
and liver and kidney S-9 fractions from pigs, cattle and chickens
(Gilbertson et al, 1990), demonstrated qualitatively similar results
to the in vivo studies. In all species, DFC and its dimer were the
major metabolites of liver S-9 fractions and DCD was generated by
kidney S-9 fractions. No ceftiofur metabolite-protein complexes were
observed in vitro (Jaglan et al, 1987b).
The metabolism of ceftiofur in cattle is shown in Figure 2.
2.2 Toxicological studies
2.2.1 Acute toxicity studies
2.2.1.1 Mice
The acute toxicity of ceftiofur was studied in groups of 5 female
mice per dose, which were treated via the i.v. and i.m. routes. In the
i.v. study, convulsions preceded death while in the i.m. study, mild
prostration was noted. The LD50 by the i.v. and i.m. routes were
about 2000 mg/kg bw and 3400 mg/kg bw, respectively (Berthe, 1982a).
2.2.1.2 Rats
The acute toxicity of ceftiofur was studied in female
Sprague-Dawley rats via the i.v. and i.m. routes, and in both males
and females via the oral and inhalation routes.
In an i.v. study, ceftiofur was administered at doses up to
3800 mg/kg bw. Conjunctival haemorrhage was noted during
administration of the test substance and death was preceded by
convulsion. The LD50 was 2200 mg/kg bw (Berthe, 1982a).
In an i.m. study, ceftiofur was administered at doses up to
1500 mg/kg bw. Mild prostration was noted and the LD50 was
1250 mg/kg bw (Berthe, 1982a).
In an inhalation study, ceftiofur was administered at an aerosol
concentration of 8.3 mg/litre to a group of 5 male and 5 female
Sprague-Dawley rats for a 4-h exposure period. During exposure, rats
exhibited salivation, nasal discharge and dyspnea; these signs
virtually disappeared within 1 h after exposure. Post-exposure signs
included diarrhea in 6 rats, and 1 rat exhibited a red encrusted
material around the nares. Both gross and microscopic examination did
not reveal any treatment-related changes. As none of the test animals
died either during treatment or during the post-treatment 14-day
observation period, the acute 4 h LC50 was estimated to be greater
than 8.3 mg/litre (Leong et al, 1985).
Figure 2. Metabolic pathway of ceftiofur in cattle
 In an acute oral study, ceftiofur was administered as a single
dose of up to 7800 mg/kg bw to groups of Sprague-Dawley rats (10/sex).
Treatment-related diarrhea was noted at the 2 highest dose levels. No
other treatment-related signs were observed. As no deaths occurred at
any treatment level, the acute oral LD50 was determined to be
greater than 7800 mg/kg bw (Cole et al., 1985).
2.2.2 Short-term toxicity studies
2.2.2.1 Rats
Ceftiofur was administered i.p. to groups of Sprague-Dawley rats
(10/sex/group) at doses of 100, 200 or 400 mg/kg bw/day for 14 days.
No mortality was observed during the study. No changes were observed
in body-weight gain, food consumption or following ophthalmic
examination. Slight faecal softening was observed in animals receiving
the highest dose, and a significant increase in absolute and relative
liver weights was observed in high-dose males. The NOEL in this study
was 200 mg/kg bw/day (Berthe, 1982b).
In another study, groups of Sprague-Dawley rats (15/sex/group)
were dosed by gavage with doses of 1500, 3000 or 6000 mg/kg bw/day of
ceftiofur for 30 days. A comparable control group received water by
gavage. Clinical signs of toxicity included diarrhea at all doses
tested, and distended abdomen at the 2 highest doses. Six deaths
attributed to mechanical impactions were observed in the high-dose
group. Treatment at all dose levels caused distension of the lumen and
flattening of the mucosa of the large intestine microscopically. This
can be attributed to treatment-related alterations in the gut
bacterial flora. Body-weight gains were significantly depressed at
6000 mg/kg bw/day, but were largely unaffected at lower doses.
Significant haematologic changes were reduced erythrocyte count and
haematocrit, and reduced haemoglobin concentrations in high-dose
females only.
Treatment with the high dose also resulted in significantly
reduced serum glucose values and significant increases in urine
specific gravity. Significant dose-dependent increases in urinary
ketones were considered likely to be associated with treatment-induced
GI effects. In conclusion, ceftiofur administered orally to rats for
30 days caused GI toxicity, marked at 6000, moderate at 3000, and
minimal at 1500 mg/kg bw/day. A NOEL was not identified in this study
(Kakuk et al, 1985a).
Ceftiofur was administered by gavage to groups of Sprague-Dawley
rats (20/sex/dose) at daily doses of 30, 100, 300, 1000 or 3000 mg/kg
bw/dy for 90 days. A comparable control group received water by
gavage. The primary target organ was the GI tract. Diarrhea and
hardened stomach contents were seen clinically, and increased in
severity in a dose-dependent manner. At dose levels below 300 mg/kg
bw/day only transient diarrhea was noted. At the highest dose level,
formation of gastric concretions were observed, resulting in
mechanical obstruction and associated depression in body-weight gains.
High-dose animals were generally also associated with electrolyte
imbalance and decreased serum glucose concentration. Microscopically,
treatment-related toxicity in the high-dose group included depletion
of hepatic glycogen, and atrophy of the germinal centres of the
spleen, lymph nodes and thymus.
Urinalysis revealed a significant increase in ketones in the 1000
and 3000 mg/kg bw/day groups, as well as a lowered urine pH at doses
of 100 mg/kg bw/day or greater. Treatment also resulted in colitis in
males receiving 1000 mg/kg bw/day or greater, and in females receiving
300 mg/kg bw/day or greater.
In conclusion, oral administration of ceftiofur resulted in
diarrhea, colitis, depression in body-weight gain and in serum
glucose, and acidification of urine. The NOEL in this study was
100 mg/kg bw/day (Kakuk et al, 1985b).
2.2.2.2 Dogs
Groups of beagle dogs (4/sex/dose) were given ceftiofur at dose
levels of 300, 1000, or 3000 mg/kg bw/day in divided dose twice daily
for 51 days. Pre-treatment evaluation included physical and ophthalmic
examinations. Post-treatment evaluation included food consumption,
body weights, biochemistry, urinalysis, haematological and selected
histopathological examinations. Ophthalmic examinations were also
conducted on all test animals during week 4 of treatment and at
termination of the study.
Anaemia and thrombocytopenia were observed at all doses. Emesis,
soft stools and diarrhea were seen less frequently. Two females given
1000 mg/kg bw/day, and 2 males and 2 females given 3000 mg/kg bw/day
died. These deaths were associated with anaemia and characterized by
pale mucous membranes and increased relative spleen weights. Bone
marrow dysplasia, extramedullary haematopoiesis and thymic atrophy
were seen microscopically at all dose levels. Hepatocellular necrosis,
reported to be secondary to the anaemia, was also observed in animals
receiving 1000 mg/kg bw/day or greater. Multiple inflammatory lesions
were present in the visceral organs of test animals receiving
1000 mg/kg bw/day or more. A NOEL was not identified in this study
(Jackson et al, 1985a).
In another study, ceftiofur was administered orally by capsule to
groups of beagle dogs (5/sex/group) at doses of 10, 30, 100 or
300 mg/kg bw/day for 91 days. Physical examinations preceded
initiation of treatment. Ophthalmic examinations, urinalysis, serum
biochemistry and extensive haematological evaluations including blood
smears and differential leukocyte counts were performed on all test
animals. Coomb's tests were carried out on high-dose animals. All
animals were subjected to complete necropsy and selected
histopathological examinations.
As noted in the 51-day study in dogs, the primary site of toxic
action appeared to be the haematopoietic system. Animals at 300 mg/kg
bw/day were positive for the Coomb's test indicating the presence of
immunoglobulin on the surface of erythrocytes and some animals
developed toxic signs of severe anaemia without evidence of a
regenerative response by bone marrow until compound administration
ceased. Administration of 100 mg/kg bw/day or more was associated with
a non-progressive thrombocytopenia. Other toxic manifestations of
anaemia included depression and pale mucous membranes and tissues.
Necropsy and histopathological examinations confirmed the
treatment-related and dose-dependent anaemia at doses above 30 mg/kg
bw/day. The NOEL in this study was 30 mg/kg bw/day (Jackson et al,
1985b).
2.2.2.3 Monkeys
Ceftiofur was administered intravenously to groups of monkeys
(2/sex/group) at dose levels of 100, 200 or 400 mg/kg bw/day for 12
days. Signs of toxicity included diarrhea in all treated animals and
vomiting accompanied by tachycardia in 1 animal receiving the
200 mg/kg bw/day dose. This animal died after the 12th treatment but
had no treatment-related lesions at necropsy.
Ophthalmological examination, including intraocular pressure, was
normal in all treated animals as were results of electrocardiograms.
Although diarrhea was noted in all treated animals, concomitant weight
loss was not observed. Haematology, biochemistry and urinalysis were
all within normal limits. Histopathology revealed a nephritis,
accompanied by increased kidney weight in 1 male given the highest
dose. No other treatment-related effects were noted. A clear NOEL was
not identified in this study (Berthe, 1982c).
2.2.3 Reproductive toxicity studies
2.2.3.1 Rats
In a 2-generation fertility and general reproductive performance
study, groups of 30 male (approximately 45-day old) and 30 female
(approximately 55-day old) Sprague-Dawley rats were orally
administered ceftiofur at dosages of 0, 100, 300 or 1000 mg/kg bw/day.
Males were treated from 70 days prior to breeding, continuing for a
total of 136 days of treatment. Females were treated 14 days prior to
breeding, throughout gestation and lactation, for a total of 79 days
of treatment. The F1 generation was also retained for breeding. Body
weight, food consumption, parental survival, confirmed matings,
pregnancy rates, length of gestation, number of live offspring,
offspring survival, necropsy and histopathological findings were all
evaluated as part of this study.
All pups in the high-dose group survived and no effect on growth
was seen. No dose-dependent adverse effects on fertility, reproductive
performance or histopathological alterations in reproductive organs of
either sex in the F0 generation were observed. Alteration in
body-weight gain and enlargement of the caecum were seen in each
treated group. No treatment-related adverse effects on growth or
viability were observed in the F1 litters through weaning. There
were no abnormalities on histopathological examination of F0 and
F1 animals. The NOEL in this study was 1000 mg/kg bw/day
(Kakuk, 1985).
A 2-generation study of fertility and reproductive performance of
F1 generation rats was conducted as a continuation of the above
study. Four groups of 30 male and 30 female Sprague-Dawley F0 rats
were administered ceftiofur from the postnatal day 21 until days
145-159 for males, and days 146-160 for females. A dose-related
increase in mortality was noted in treated groups when the data from
the males and females were combined. The majority of the deaths were
attributed to accidental causes. There were no adverse effects on
fertility or reproductive performance in the F1 generation and F2
litters. Enlargement of the caecum occurred in F1 animals at
300 mg/kg bw/day or greater. In the high-dose groups, there was a
higher incidence of degenerative changes in the non-glandular stomach
(92%), and mucus hypersecretion in the glandular stomach (79%)
compared to control animals. No treatment-related histological changes
were observed in the reproductive organs of either sex at the high
dose (1000 mg/kg bw/day). The NOEL in this study was 1000 mg/kg bw/day
(Kakuk, 1986).
2.2.4 Special Studies on embryotoxicity and teratogenicity
2.2.4.1 Mice
Teratogenicity studies were conducted in mice as a second species
instead of rabbits because orally administered ceftiofur disrupts the
caecal microflora in rabbits. In a dose range-finding study, groups of
seven bred female CD-1 mice were given ceftiofur orally at doses of
1000, 2000, 4000 or 8000 mg/kg bw/day from days 6-15 of gestation. At
day 18 of gestation, uterine weight, numbers of viable fetuses,
resorptions, corpora lutea and fetal malformations were recorded.
Signs of maternal toxicity were evident at 4000 and 8000 mg/kg bw/day.
Reduced fetal body weights were recorded at 8000 mg/kg bw/day. The
NOEL for maternal toxicity was 2000 mg/kg bw/day, for fetotoxicity
4000 mg/kg bw/day, and for embryotoxicity and teratogenicity
8000 mg/kg bw/day.
A more detailed segment II oral teratogenicity study was
conducted in groups of 30 female CD-1 mice on days 6-15 of gestation
at 1000, 2000 or 4000 mg/kg bw/day. All parameters stated above were
recorded as well as extensive examination of viable fetuses for
visceral malformations, cranial and skeletal abnormalities. Increased
food consumption, distended stomach and small intestines, and enlarged
gall bladders were observed in dams in the mid- and high-dose groups.
No treatment-related effects were seen in the numbers of resorption
sites, litter size or pup weights. There were no effects on the
incidences of skeletal or visceral anomalies. The NOEL for maternal
toxicity was 1000 mg/kg bw/day, and for developmental toxicity it was
4000 mg/kg bw/day (Marks & Terry, 1993).
2.2.4.2 Rats
Groups of 24 pregnant rats (strain unspecified) were orally
administered doses of 0, 800, 1600 or 3200 mg/kg bw/day ceftiofur once
daily on days 6-15 of gestation. Observations were made daily for
signs of toxicity, and body weights were recorded on the day of
insemination, throughout the dosing period, and on day 20 when
cesarean sections were performed. At that time, the sex, weight,
number and location of viable fetuses, number and location of
resorption sites, fetal weights and gross fetal abnormalities were
determined.
Dose-related maternal toxicity (i.e. soft stools, prophyrin
staining of the eye and nares, diarrhea and blood in faeces) was
observed particularly in the high-dose group. There were no observed
adverse effects on maternal reproductive capacity and no evidence of
teratogenicity in this study. A statistically significant dose-related
decrease in mean fetal body weight, which did not exceed 7%, was
observed. The NOEL in this study was 3200 mg/kg bw/day (Shaw et al,
1985).
2.2.5 Special studies on genotoxicity
A variety of in vitro and in vivo genotoxicity assays
covering a range of endpoints were conducted with ceftiofur and the
metabolite furoic acid (Tables 1 & 2). All assays were negative except
an in vitro chromosomal aberration assay with ceftiofur, which
produced chromatid breaks, gaps and fragments in CHO cells.
Chromosomal aberrations occurred in CHO cells exposed to > 200 mg/ml
for long periods of treatment (44 h) in the absence of S9 metabolic
activation. No evidence of clastogenicity was seen following shorter
treatment times or in the presence of S9 at doses as high as
5000 mg/ml nor in chromosomal aberration assays in vivo. The
mechanism by which chromosomal aberrations were induced in vitro was
extensively investigated. Ceftiofur was profoundly cytostatic
(i.e. reducing the rate of cell division) in CHO cells under
conditions which causes chromosomal aberrations in vitro. Removal of
the drug led to reversal of cytostasis and reduction in number of
cells with aberrations. Cytotoxicity and cell lethality were not
observed in ceftiofur-treated CHO cells suggesting that cytostasis
results in chromosomal breaks and gaps due to prolongation of the cell
cycle and not by a direct effect on chromatin (Aaron, 1991).
2.2.6 Special studies on immunotoxicity
In view of the structural similarity of many ß-lactam drugs, the
possibility of immunologic cross reaction must be addressed. In order
to assess this possibility, a series of studies intended to
investigate the hypersensitivity for ß-lactam antibiotics were
developed.
The model developed was based on passive cutaneous anaphylaxis
(PCA) in the guinea-pig and was intended to determine the human safety
of residues of ceftiofur-sodium in edible tissues, including injection
site residues. In addition, because ceftiofur is structurally related
to penicillin, and because of concern that it might therefore have
antigenic determinants for penicillin, the studies also examined the
interaction between the penicillin antibody and ceftiofur.
Antibodies to benzyl penicillin G (BPG), conjugated to keyhole
limpet hemocyanin (KLH), and antibodies to ceftiofur (CEF), conjugated
to bovine gamma globulin (BGG), were prepared and assayed for PCA
activity in the guinea-pig. Reactive sera were then utilized to
passively sensitize animals prior to further challenge with conjugates
of BPG and BGG, CEF with BGG, CEF with hen egg albumin (HEA), the
deocetylcefotaxime metabolite of ceftiofur, the aminothiazolyl (atz)
side chain, common to parent drug and all metabolites, with HEA,
parent drug, free sulfhydryl metabolite (FSM) of CEF and extracts of
residue of CEF from injection site muscle and kidney from treated
animals.
The protocol involved passively sensitizing female guinea-pigs
with antibody at multiple skin sites followed by challenge 5 days
later. Dose levels utilized were selected as multiples of the
anticipated human exposure level of 0.083 mg/kg bw.
Passive cutaneous anaphylaxis occurred in guinea-pigs sensitized
with antibody to penicillin when challenged with the BGG-BPG control.
Reactions did not occur with exposure to any CEF-containing products.
Table 1. Results of genotoxicity studies on ceftiofur
Test Test object Concentration Results References
In vitro
Ames testa S. typhimurium 0.125, 0.250, 0.5, negative Mazurek & Swenson,
TA98, T100, T1535, T1537, 1.0 µg/plate 1983; Aaron, 1991
T1538
Forward Chinese hamster V-79 fibroblasts 1.0, 2.0, 4.0 negative Harbach et al., 1983
mutation assaya (HGPRT assay) µg/ml
Chromosome Chinese hamster ovary cells 211, 5000 µg/ml positive Aaron, 1991
aberration
assaya
In vivo
Micronucleus Sprague-Dawley rat bone marrow 0, 250, 500, 1000, negative Trzos et al., 1984
Test mg/kg bw
Micronucleus CD-1 mouse bone marrow 0, 250, 500, 1000 negative Aaron, 1991
Test mg/kg bw
UDS Rat hepatocytes 0, 0.03, 0.1, 0.3, negative Trzos & Swenson,
1.0 mg/ml 1984
Table 1. Results of genotoxicity studies on ceftiofur (cont'd).
Test Test object Concentration Results References
Chromosome Mouse bone marrow 450, 900, 1750 negative Aaron, 1991
aberration assay mg/kg bw
(acute)
Chromosome Mouse bone marrow 350, 700, 1400 negative Aaron, 1991
aberration assay mg/kg bw
(subchronic)
a With and without rat liver S9 fraction
Table 2. Results of genotoxicity studies on furoic acid
Test Test object Concentration Results References
In vitro
Ames test S. typhimurium 250, 500, 1000, 2000 negative Mazurek &
TA98, T100, T1535, µg/plate Zimmer, 1985
T1537, T1538
Forward Chinese hamster V-79 250, 500, 1000, 1500 negative Zimmer et al.,
mutation fibroblasts (HGPRT mg/ml 1985
assaya assay)
UDS Rat hepatocytes 1, 3, 10, 30, 100, 300, negative Harbach & Aaron,
1000 mg/ml 1991
a With or without rat liver S-9 fraction
Guinea-pigs sensitized with antibody to ceftiofur reacted to
challenge with HEA-CEF by both the i.v. and oral routes of exposure,
requiring 10 mg/kg bw by the oral route. Similarly, free sulfhydryl
metabolite caused reactions over a broad range of dose levels by both
the i.v. and oral routes. PCA reactions occurred following i.v.
challenges containing at least 0.076 µg FSM/kg bw. Reaction to the
free sulfhydryl metabolite following an oral challenge was similar to
those reported for the HEA-CEF, suggesting approximately a 1000 fold
difference in sensitivity between the i.v. and oral routes. Challenge
of guinea-pigs sensitized with antibody to ceftiofur, and administered
ceftiofur residue extracts from kidney and injection site muscle at
dose levels of 830 µg drug/kg bw failed to produce a positive
response.
These data, when taken together indicate that penicillin
antibodies do not recognize ceftiofur antigenic determinants.
Furthermore, the data also suggest that the GI tract significantly
reduces potential PCA activity. The data suggest that ceftiofur
residues at either the injection site or present in kidney are not
present in either a form or concentration which is likely to induce
PCA activity following oral exposure of animals sensitized with
ceftiofur antibodies and subsequently challenged with the residue
(Jackson et al, 1988; Brussee et al, 1989). The authors concluded
that human exposure to ceftiofur, its residues or metabolites poses
virtually no human risk because:
(a) oral challenge with extract of ceftiofur residues in sensitized
guinea-pigs did not result in positive PCA reactions;
(b) while the free sulflydryl metabolite poses the greatest risk of
eliciting a hypersensitivity reaction, this risk is indeed very
small because exposure would be restricted to the oral route
where residues are invariably bound to proteins, in very low
levels, and further inactivated in the GI tract;
(c) IgE isolated from patients with known sensitivity to pencillin
did not bind significant amounts of the ceftiofur molecule, again
implying a lack of cross reactivity.
2.2.7 Special studies on microbiological effects
Gram-positive bacterial susceptibility to ceftiofur is given in
Table 3.
Table 3. Gram-positive bacterial susceptibility to ceftiofur (µg/ml)
(Yancey et al., 1988; Klein et al., 1985)
Organism MIC50 MIC90 MICrange
Staph. intermedius 0.13 0.25 N/A
Staph. aureus N/A N/A 0.5-4.0
Staph. aureus (dog, <0.06 0.13 N/A
cat)
Staph. intermedius <0.06 <0.06 N/A
Strep. agalactiae N/A N/A <0.06-0.25
Strep. bovis N/A N/A <0.06
Strep. dysgalactiae N/A N/A <0.06-0.25
Strep. equi <0.06 <0.06 N/A
Strep. suis N/A N/A <0.06-0.5
Strep. uberis N/A N/A <0.06-0.5
Strep. <0.06 <0.06 N/A
zooepidemicus
Strep. faecalis N/A N/A >32
L. monocytogenes N/A N/A 16
R. equi 8 16 N/A
As noted in section 2.1.2, ceftiofur is rapidly degraded to
desfuroylceftiofur. This specific metabolism and the antimicrobial
activity of both the parent drug and its primary metabolite against
both Gram-positive and Gram-negative bacteria have been investigated.
The MIC values are given in Tables 4 and 5.
Table 4. Gram-positive bacterial susceptibility to ceftiofur and
desfuroylceftiofur (MIC90) (Salmon et al, 1993)
Organism Ceftiofur (µg/ml) Desfuroylceftiofur
(number tested) (µg/ml)
Strep. uberis (15) 0.03 0.5
Strep. dysgalactiae (15) <0.0039 0.03
Strep. zooepidemicus <0.0019 0.03
(48)
Strep. equi (12) <0.0019 0.03
Strep. suis (49) 0.13 0.25
Staph. aureus (10) 1.0 8.0
Staph. hyicus (14) 1.0 4.0
Staph. spp (11) 1.0 8.0
Table 5. Gram-negative bacterial susceptibility to ceftiofur and
desfuroylceftiofur (MIC90) (Salmon et al, 1994)
Organism Ceftiofur Desfuroylceftiofur
(number tested) (µg/ml) (µg/ml)
Pasteurella multocida (50)
(from Swine Resp. Dis.) <0.0039 <0.0078
Pasteurella multocida (48)
(from Bovine Resp. Dis.) <0.0039 <0.0078
Pasteurella haemolytica (42) 0.015 0.015
Haemophilus somnus (59) <0.0019 <0.0019
A. pleuropneumoniae (50) <0.0019 <0.0019
Salmonella choleraesuis (48) 1.0 1.0
E. coli (40) 0.5 0.5
Extensive investigations have also been carried out on the
in vitro activity of ceftiofur and its metabolites against cultures
of bacteria of relevance in the human GI tract. MIC values of both the
parent drug and its primary metabolites were determined against
bacterial species frequently isolated from the human intestinal tract.
The MIC values are reported in Table 6.
In vitro MIC data covering a wide range of animal and human
bacterial species were available. Fifty-eight strains commonly
isolated from the human GI tract were tested with ceftiofur and its
metabolites. The MIC values were determined by the agar dilution
technique at both high (10 6-7) and low (10 4-5) inoculum
densities. Generally, there was a 2-fold increase in the MIC values
with increasing inoculum density. Ceftiofur was always more active
than its metabolites desfuroylceftiofur, desfuroylceftiofur disulfide
and desfuroylceftiofur cysteine disulfide. Streptococcus,
Propionibacterium and Bifidobacterium were the most sensitive,
with MIC50 values of 0.016 µg/ml, 0.03 µg/ml, and 0.03 µg/ml at high
inoculum density, respectively. Bacteroides sp., Enterococcus
faecium, Eubacterium sp., and Lactobacillus sp. were least
sensitive to ceftiofur, with MIC50 values of 16 µg/ml, 128 µg/ml,
1 µg/ml, and 16 µg/ml, respectively.
Particularly noteworthy is that for most strains, metabolites of
ceftiofur were considerably less active than parent drug. The
degradation of ceftiofur residues by gut flora was also examined. The
data indicate that ceftiofur is rapidly degraded in human faecal
material incubated anaerobically, to compounds which essentially lack
microbiological activity (Hornish et al., 1994; Kotarski, 1993).
2.2.8 Observations in humans
Ceftiofur is an antimicrobial drug developed exclusively for use
in veterinary medicine and hence no direct studies in humans have been
conducted.
Ettestad et al. (1995) have recently reported on biliary
complications associated with the use of ceftriaxone, a cephalosporin
antimicrobial agent, in the treatment of unsubstantiated Lyme disease.
The authors concluded that there appeared to be a threshold for
biliary complications which required a daily dose of > 40 mg/kg
bw/day for periods of at least 1 month. It is noteworthy that
anticipated human exposure to ceftiofur through food residues is
approximately 4000 times lower than the threshold dose suggested by
the above authors.
Table 6. MIC50 values for human strains of anaerobic and facultatively anaerobic bacteria
(Thurn et al., 1994; Zurenko & Yagi, 1990; Kennedy et al., 1991; Watts et al., 1991)
Group MIC50 (µg/ml)
(no. strains tested)
ceftiofur desfuroylceftiofur desfuroylceftiofur
cysteine
disulfide
low high low high low high
Bacteroides (12 or 16) 2 16 16 64 16 128
Bifidobacterium (15) 0.25 ND 8 ND 32 ND
Clostridium (5) <.016 1 1 8 2 2
Eubacterium (13) 1 ND 128 ND 64 ND
Peptococcus and 0.25 0.5 4 16 16 32
Peptostreptococcus
(10 or 15)
Enterococcus (5 and 2) 128 ND 32 ND 8, 32 ND
Escherichia coli (7) 0.5 0.5 2 1 2 2
Proteus vulgaris (5) <.06 ND 2 ND ND ND
Lactobacillus (2 or 1) 0.5, 1 0.5, 16 2, 8 4, 128 4, ND 4, ND
ND = not determined
3. COMMENTS
Toxicological data
A range of studies on ceftiofur and its primary metabolites were
available for evaluation by the Committee, including data on
pharmacokinetics and metabolism, acute and short-term toxicity,
reproductive and developmental toxicity, genotoxicity, immunotoxicity
and microbiology.
Ceftiofur is rapidly metabolized to desfuroylceftiofur. Following
i.m. administration in the rat, approximately 55% of the dose was
excreted in the urine and about 30% in the faeces within the first
24 h. Similar results were obtained in cattle. In a separate oral
study in rats, approximately 55% of the dose was recovered in urine;
the remainder was present in the faeces and the GI tract.
Single oral doses of ceftiofur of up to 7800 mg/kg bw produced
only minimal toxicity in the rat. Toxic signs associated with repeated
oral doses in rats of up to 6000 mg/kg bw/day for 30 days were limited
to haematological changes and diarrhoea. Oral doses of up to 300 mg/kg
bw/day given to dogs for 91 days produced a reversible anaemia and
thrombocytopenia. The NOEL for treatment-related haematopoietic
effects in rats was 30 mg/kg bw/day.
In reproductive toxicity studies in rats, ceftiofur administered
at dose levels of up to 1000 mg/kg bw/day had no adverse effects on
fertility, reproductive performance or reproductive organs. Similarly,
no treatment-related effects were observed in developmental toxicity
studies in mice at doses of up to 4000 mg/kg bw/day or in rats at
doses of up to 3200 mg/kg bw/day.
A variety of in vitro and in vivo genotoxicity assays covering a
range of end-points were conducted with ceftiofur (with and without
metabolic activation with S-9 microsomal fraction) and its metabolite
furoic acid. All the assays were negative, with the exception of an
in vitro chromosomal aberration assay in the absence of metabolic
activation, but only at concentrations at which cell division was
inhibited. The Committee concluded that this finding, when taken in
conjunction with the negative in vivo chromosomal aberration
studies, was not of biological significance.
Carcinogenicity studies have not been performed on ceftiofur.
However, the Committee noted that the drug showed no evidence of
genotoxicity in a variety of assays and is not chemically related to
known carcinogens. Furthermore, it is rapidly metabolized and its
metabolites are not related to any known carcinogens. Neither
neoplastic nor preneoplastic lesions were observed in 90-day feeding
studies in rats, dogs, monkeys, or in reproductive toxicity studies
involving exposure for periods of up to 160 days in which limited
histopathological examination were carried out. Recent reports
indicate that non-genotoxic chemicals showing such a lack of toxicity
are not associated with carcinogenicity in long-term rodent toxicity
studies. Under these circumstances, the Committee concluded that
carcinogenicity studies were not necessary.
Long-term toxicity studies were not available. Even at doses
exceeding several grams/kg bw/day in rats for periods of up to 90
days, diarrhoea was the only major effect noted in rats. The Committee
concluded that allowance could be made for the absence of long-term
toxicity studies on ceftiofur by the application of an appropriate
safety factor.
The potential immunotoxicity of ceftiofur has also been
investigated. The Committee noted that penicillin antibodies do not
recognize ceftiofur antigenic determinants and that exposure to
metabolites of ceftiofur did not produce adverse reactions in
guinea-pigs sensitized to penicillin. The Committee concluded that
there is no risk of hypersensitivity reactions in humans to ceftiofur
or its residues or metabolites at the anticipated level of exposure.
Microbiological data
The potential for adverse effects on the human gut flora was
considered. In vitro MIC data covering a wide range of animal and
human bacterial species were submitted for evaluation. A total of 58
strains commonly isolated from the human GI tract were tested with
ceftiofur and its metabolites. Ceftiofur was more active than its
metabolites desfuroylceftiofur, 3,3'-desfuroylceftiofur disulfide and
desfuroylceftiofur cysteine disulfide. The Committee recognized,
however, that ceftiofur is not present as a residue because it is
extensively and rapidly metabolized, with a plasma half-life of
approximately 15 minutes in cattle and pigs. The lowest MIC50 value
reported for desfuroylceftiofur cysteine disulfide was 2 µg/ml for
Clostridium and Escherichia species.
In calculating an ADI based on antimicrobial activity, the
Committee used the formula developed at the thirty-eighth meeting of
the Committee (Annex 1, reference 97):
Concentration without
effect on human gut × Daily faecal bolus (g)
Upper limit of flora (µg/ml)
temporary ADI =
(µg/kg bw) Fraction of
oral dose × Safety factor × Weight of
bioavailable human
(60 kg)
= 2 × 150
0.1 × 1 × 60
= 50 µg/kg bw
It took the following factors into account:
* Factors to account for the range of MICs needed to allow for
sensitive bacteria, anaerobic environment, bacterial density and
pH: the most relevant sensitive species were studied under
conditions of high inoculum density. No adjustment was deemed
necessary.
* Availability: the fraction of the dose available to the gut
microflora was derived from studies of ceftiofur in humans which
showed that the drug was rapidly metabolized.
* Variability among exposed individuals: the Committee noted that a
substantial amount of data covering a variety of bacterial
strains representative of the human gut microflora was available.
In addition, it recognized that the other values selected for
this calculation was already conservative and incorporated an
adequate margin of safety. A safety factor of 1 was therefore
adopted.
4. EVALUATION
The Committee noted that the lowest NOEL based on toxicological
studies was 30 mg/kg bw/day, which was observed in the 90-day study in
dogs. It could establish an ADI of 0-60 µg/kg bw based on this NOEL
and a safety factor of 500, which would include an additional safety
factor of 5 to take account of the absence of long-term toxicity
studies. However, the Committee noted that the microbiological
end-point would give the lowest ADI and therefore established an ADI
of 0-50 µg/kg bw based on this end-point.
5. REFERENCES*
Aaron CS (1991). The Upjohn Company: TR 7228-91-036. U64279E:
Evaluation of U64279E in the In Vitro Chromosome Aberration Assay
Using Chinese Hamster Ovary (CHO) Cells.
Banting A, Mignot A, Lefebyre MA, Millerioux L, Steffan J, Gilbertson
TJ (1989). The Upjohn Company: TR 788-9760-88-018, "Plasma Profile and
Pharmacokinetic Parameters in Calves After Single (IV and IM) and
Multiple Dose Administration (IM) of Ceftiofur Sodium.
Berthe, J (1982a). Centre Des Recherches Clin-Midy, Code Nomenclature:
TO010-00, Direction Des Recherches Sanofi, Montpellier, FRANCE: Etude
de la Toxicité Aigue De CM-31916 (Etude Preliminaire).
Berthe J (1982b). Centre De Recherches Clin-Midy, Code Nomenclature:
TO020-00, Direction Des Recherches Sanofi, Montpellier, FRANCE: Etude
de la Toxicité Subaigue De CM-31916 chez le Rat Sprague-Dawley par
Voie Intraperitoneale.
Berthe J (1982c). Centre De Recherches Clin-Midy, Code Nomenclautre:
TO021-00, Direction Des Recherches Sanofi, Montpellier, FRANCE:
CM-31916 Etude de la Toxicité Subaigue chez le Macaque Par Voie
Intraveineuse.
Brussee DM, Clarke GL, Cypher JJ, Farho TG, Gilbertson TG, Hornish RE,
Jaglan PS, Miller CC (1989). Internal Memorandum, The Upjohn Company.
Cole SL, Kakuk TJ, Rop DA (1985). The Upjohn Company: TR 7263-85-002,
Acute Oral Single Dose Study in Sprague-Dawley Rats with Ceftiofur
(U-64,279E).
Ettestad PJ, Campbell GL, Welbel SF, Genese CA, Spitalny KC,
Marchetti CM, Dennis DT (1995). Biliary complications in the treatment
of unsubstantiated Lyme disease. J. of Infectious Diseases
171:356-361.
Gilbertson TJ, Roof RD, Jaglan PS (1990) The Upjohn Company:
TR 906-9760-90-001, In vitro Metabolism of 14C Ceftiofur Sodium
and Metabolites in S-9 Fractions of Livers and Kidneys of Rats, Pigs,
Cattle, and Chickens.
Halstead SL, Walker RD, Baker JC, Holland RE, Stein GE, Hauptman JG
(1992) Pharmacokinetic Evaluation of Ceftiofur in Serum, Tissue
Chamber Fluid and Bronchial Secretions from healthy Beef-Breed Calves.
Can. J. Vet. Res., 56:269-274.
* All unpublished studies were submitted to WHO by the Upjohn
Company, Kalamazoo, MI, USA
Jackson TA, Brussee DM, Cypher JJ (1988) The Upjohn Company:
TR 7220-88-026, Hypersensitivity Studies with Sodium Ceftiofur
(U-64,279E) in Hartley Albino Guinea Pigs by the Intravenous and Oral
Routes.
Jackson TA, Brussee DM, Vrbancic JP, Mulholland MP (1985a) The Upjohn
Company: TR 7263-85-077, U-64,279E; 51-Day Oral Toxicology and Drug
Safety Study in the Beagle Dog.
Jackson TA, Brussee DM, Vrbancic JP, Mulholland MP (1985b) The Upjohn
Company: TR 7263-85-079, U-64,279E; 90-Day Oral Toxicology and Drug
Safety Study in the Beagle Dog.
Jaglan PS, Adams LD, Roof RD, Reardon IM, Heinrickson RL,
Gilbertson TJ (1991) The Upjohn Company: TR 788-7926-91-001, The
Nature of Covalent Binding of Desfuroylceftiofur to Plasma Proteins of
Rats.
Jaglan, PS, Arnold, TS (1986a) The Upjohn Company: TR 788-9760-PSJ-I-
86-001, Metabolism of Ceftiofur (14C-U-64,279E) Sodium in Rats from
Oral Treatment Compared to Intramuscular Treatment of Bovine (Study
No. J-080). Part I-Disposition Study and Comparative Metabolic Profile
in the Urine of Rats and Bovine.
Jaglan PS, Arnold TS (1986b) The Upjohn Company; TR 788-9760-86-002,
Metabolism of Ceftiofur (14C U-64,279) Sodium Salt in Rats from Oral
Treatment Compared to Intramuscular Treatment of Bovine (Study
No. J-080). Part II-Comparative Metabolic Profile in Plasma of Rats
and Bovine.
Jaglan PS, Arnold TS (1987a) The Upjohn Company: TR 788-9760-86-006,
Metabolism of 14C-Ceftiofur (U-64,279E) Sodium Salt in Rats from
Intramuscular Treatment.
Jaglan PS, Arnold TS (1987b) The Upjohn Company: TR 788-9760-87-010,
Characterization of the Major Bovine Urinary Metabolites Following
Intramuscular Treatment with 14C-Ceftiofur.
Jaglan PS, Cox BL, Smart DJ, Pierce PA, Yein FS, Roof RD,
Gilbertson TJ (1989) The Upjohn Company: TR 788-9760-89-002,
Disposition and Metabolism of 14C-Ceftiofur Sodium (U64279E) in
Lactating Cows. Part II: The Nature of Milk Residues.
Jaglan PS, Kubicek MF, Cox BL, Johnson DB, Gilbertson TJ (1987a) The
Upjohn Company: TR 788-9760-87-006, Nature of Metabolites in Rats
Treated Orally with Ceftiofur from Multiple High Doses and Comparison
of the Metabolites in Liver and Kidney of Rats Versus Bovine.
Jaglan PS, Kubicek MF, Johnson DB, Stuart DJ, Mazurek JH, Wiser SK,
Aaron CS (1987b) The Upjohn Company: TR 788-9760-87-002, Metabolism of
14C-Ceftiofur (U6,4279) in vitro.
Jaglan PS, Roof RD, Yein FS, Zaya MJ, Gilbertson TJ (1990) The Upjohn
Company: TR 796-9760-89-005, Comparison of Metabolites of Ceftiofur
(U-64,279E) Sodium in The Urine and Kidneys of Pigs from intramuscular
Injection to that of Rats from Oral Doses.
Kakuk TJ, Cole SL, Rop DA (1985a) The Upjohn Company: TR 7263-85-071,
30-Day Oral Toxicity Study in Sprague-Dawley Rats with Ceftiofur
Sodium (U-64,279E).
Kakuk TJ, Cole SL, Rop DA (1985b) The Upjohn Company: TR 7263-85-075,
90-Day Oral Toxicity Study in Sprague-Dawley Rats with Ceftiofur
Sodium (U-64,279E).
Kakuk TJ (1985) The Upjohn Company: TR 7263-85-082, Two Generation
Fertility and General Reproductive Performance Study (Oral) of
Ceftiofur Sodium (U-64,279E) in Sprague-Dawley Rats. I. Fertility and
Reproductive Performance of the F0 Generation.
Kakuk TJ (1986) The Upjohn Company: TR 7263-86-031, Two Generation
Fertility and General Reproductive Performance Study (Oral) of
Ceftiofur Sodium (U-64,279E) in Sprague-Dawley Rats. II. Fertility and
Reproductive Performance of the F1 Generation.
Kennedy MJ, Yancey RJ, Kornis GI (1991) The Upjohn Company:
TR 705-7923-91-015, In vitro Activity of Ceftiofur Sodium
(U-64,279E), Desfuroylceftiofur (U-75,104) and Desfuroylceftiofur
Cystein Disulfide (U-93,112) Against Bifidobacterium spp. and
Eubacterium spp. from the Human Gastrointestinal Tract.
Klein LK, Yancey RJ, Goodenough KR, Kinney ML, Roberts BJ (1985) The
Upjohn Company: TR 705-7922-85-003, In vitro and In Vivo
Evaluation of the Monobactam Antibiotics, U70,887B and U71,689B,
Compared to Aztreonam and Ceftiofur Against Bacterial Pathogens of
Veterinary Importance.
Kotarski S (1993) Internal Memo, The Upjohn Company.
Krzeminski LF, Stuart DJ, Gosline RE, Subacz CJ, Cox BL, Reeves DR
(1985) The Upjohn Company: TR 788-9760-85-005, HPLC Assay of Bovine
Plasma and Urine Metabolites After Treatment with Carbon 14 Labeled
Ceftiofur.
Leong BKJ, Sabaitis CP, Kakuk TJ, Imlay MM (1985) The Upjohn Company:
TR 7277-85-018, Acute Four-Hour Dust Inhalation Toxicity Study on
Ceftiofur Sodium (U-64,279E) in Albino Rats.
Marks TA, Terry RD (1993) The Upjohn Company: TR 7224-93-054,
U-64279E: A Range-Finding Study (Oral) in Mice.
Salmon SA, Watts JL, Yancey RJ, Case CA (1993) The Upjohn Company:
TR 705-7923-93-007, Minimum Inhibitory Concentrations for Ceftiofur
and Desfuroylceftiofur with Isolates of Veterinary Importance.
Salmon SA, Watts JL, Case CA, Yancey RJ (1994) Minimum inhibitory
concentrations for ceftiofur and comparator antimicrobial agents
against bacterial pathogens of swine from the United States, Canada
and Denmark. TR No. 705-7923-94-020. The Upjohn Company
Shaw CI, Marks TA, Poppe SM, et al. (1985) The Upjohn Company:
TR 7259-85-011, A Segment II Teratology Study (Oral) in Rats Givn
U-64,279E
Thurn KK, Greening RC, Kotarski SF (1994) The Upjohn Company:
TR 788-7928-94-001 Minimal Inhibitory Concentrations of Ceftiofur and
its Metabolites Against Bacterial Species Frequently Isolated from the
Human Gastrointestinal Tract.
Trzos RJ, Swenson DH (1984) The Upjohn Company: TR 7268-84-018 The
primary hepatocyte unscheduled DNA synthesis (UDS) assay with U-64,279
and ultra violet light.
Trzos RJ, Swenson DH, Brown PK (1984) The Upjohn Company:
TR 7268-84-011 The micronucleus test with U-64,279 (Sanofi
cephalosporin).
Watts JL, Case CA, Yancey RJ, Kornis GI (1991) The Upjohn Company:
TR 705-7923-91-020 Evaluation of Desfuroylceftiofur-S-S-cysteine
(DCD; U-93-112) with Veterinary Pathogens.
Yancey RJ, Roberts BJ, Folz SD (1988) The Upjohn Company:
TR No. 705-7922-88-002, In vitro Activity of Ceftiofur Sodium
(U-64,279E) for Urinary and Respiratory Tract Pathogens of Companion
Animals.
Yein FS, Zaya MJ, Arnold TS, Hoffman GA, Roof RD, Dame KJ, Cox TD,
Reeves DR, Flook TF (1990) The Upjohn Company: TR 796-9760-89-002,
Absorption, Distribution, Metabolism, and Excretion of 14C-Ceftiofur
(U-64,279E) Sodium in the Swine.
Zurenko GE, Yagi BH (1990) The Upjohn Company: TR 7254-090-098
The In vitro Activity of Ceftiofur Sodium (U-64279E) and
Desfuroylceftiofur (U-75104) Against Human Bacterial Clinical
Isolates.
In an acute oral study, ceftiofur was administered as a single
dose of up to 7800 mg/kg bw to groups of Sprague-Dawley rats (10/sex).
Treatment-related diarrhea was noted at the 2 highest dose levels. No
other treatment-related signs were observed. As no deaths occurred at
any treatment level, the acute oral LD50 was determined to be
greater than 7800 mg/kg bw (Cole et al., 1985).
2.2.2 Short-term toxicity studies
2.2.2.1 Rats
Ceftiofur was administered i.p. to groups of Sprague-Dawley rats
(10/sex/group) at doses of 100, 200 or 400 mg/kg bw/day for 14 days.
No mortality was observed during the study. No changes were observed
in body-weight gain, food consumption or following ophthalmic
examination. Slight faecal softening was observed in animals receiving
the highest dose, and a significant increase in absolute and relative
liver weights was observed in high-dose males. The NOEL in this study
was 200 mg/kg bw/day (Berthe, 1982b).
In another study, groups of Sprague-Dawley rats (15/sex/group)
were dosed by gavage with doses of 1500, 3000 or 6000 mg/kg bw/day of
ceftiofur for 30 days. A comparable control group received water by
gavage. Clinical signs of toxicity included diarrhea at all doses
tested, and distended abdomen at the 2 highest doses. Six deaths
attributed to mechanical impactions were observed in the high-dose
group. Treatment at all dose levels caused distension of the lumen and
flattening of the mucosa of the large intestine microscopically. This
can be attributed to treatment-related alterations in the gut
bacterial flora. Body-weight gains were significantly depressed at
6000 mg/kg bw/day, but were largely unaffected at lower doses.
Significant haematologic changes were reduced erythrocyte count and
haematocrit, and reduced haemoglobin concentrations in high-dose
females only.
Treatment with the high dose also resulted in significantly
reduced serum glucose values and significant increases in urine
specific gravity. Significant dose-dependent increases in urinary
ketones were considered likely to be associated with treatment-induced
GI effects. In conclusion, ceftiofur administered orally to rats for
30 days caused GI toxicity, marked at 6000, moderate at 3000, and
minimal at 1500 mg/kg bw/day. A NOEL was not identified in this study
(Kakuk et al, 1985a).
Ceftiofur was administered by gavage to groups of Sprague-Dawley
rats (20/sex/dose) at daily doses of 30, 100, 300, 1000 or 3000 mg/kg
bw/dy for 90 days. A comparable control group received water by
gavage. The primary target organ was the GI tract. Diarrhea and
hardened stomach contents were seen clinically, and increased in
severity in a dose-dependent manner. At dose levels below 300 mg/kg
bw/day only transient diarrhea was noted. At the highest dose level,
formation of gastric concretions were observed, resulting in
mechanical obstruction and associated depression in body-weight gains.
High-dose animals were generally also associated with electrolyte
imbalance and decreased serum glucose concentration. Microscopically,
treatment-related toxicity in the high-dose group included depletion
of hepatic glycogen, and atrophy of the germinal centres of the
spleen, lymph nodes and thymus.
Urinalysis revealed a significant increase in ketones in the 1000
and 3000 mg/kg bw/day groups, as well as a lowered urine pH at doses
of 100 mg/kg bw/day or greater. Treatment also resulted in colitis in
males receiving 1000 mg/kg bw/day or greater, and in females receiving
300 mg/kg bw/day or greater.
In conclusion, oral administration of ceftiofur resulted in
diarrhea, colitis, depression in body-weight gain and in serum
glucose, and acidification of urine. The NOEL in this study was
100 mg/kg bw/day (Kakuk et al, 1985b).
2.2.2.2 Dogs
Groups of beagle dogs (4/sex/dose) were given ceftiofur at dose
levels of 300, 1000, or 3000 mg/kg bw/day in divided dose twice daily
for 51 days. Pre-treatment evaluation included physical and ophthalmic
examinations. Post-treatment evaluation included food consumption,
body weights, biochemistry, urinalysis, haematological and selected
histopathological examinations. Ophthalmic examinations were also
conducted on all test animals during week 4 of treatment and at
termination of the study.
Anaemia and thrombocytopenia were observed at all doses. Emesis,
soft stools and diarrhea were seen less frequently. Two females given
1000 mg/kg bw/day, and 2 males and 2 females given 3000 mg/kg bw/day
died. These deaths were associated with anaemia and characterized by
pale mucous membranes and increased relative spleen weights. Bone
marrow dysplasia, extramedullary haematopoiesis and thymic atrophy
were seen microscopically at all dose levels. Hepatocellular necrosis,
reported to be secondary to the anaemia, was also observed in animals
receiving 1000 mg/kg bw/day or greater. Multiple inflammatory lesions
were present in the visceral organs of test animals receiving
1000 mg/kg bw/day or more. A NOEL was not identified in this study
(Jackson et al, 1985a).
In another study, ceftiofur was administered orally by capsule to
groups of beagle dogs (5/sex/group) at doses of 10, 30, 100 or
300 mg/kg bw/day for 91 days. Physical examinations preceded
initiation of treatment. Ophthalmic examinations, urinalysis, serum
biochemistry and extensive haematological evaluations including blood
smears and differential leukocyte counts were performed on all test
animals. Coomb's tests were carried out on high-dose animals. All
animals were subjected to complete necropsy and selected
histopathological examinations.
As noted in the 51-day study in dogs, the primary site of toxic
action appeared to be the haematopoietic system. Animals at 300 mg/kg
bw/day were positive for the Coomb's test indicating the presence of
immunoglobulin on the surface of erythrocytes and some animals
developed toxic signs of severe anaemia without evidence of a
regenerative response by bone marrow until compound administration
ceased. Administration of 100 mg/kg bw/day or more was associated with
a non-progressive thrombocytopenia. Other toxic manifestations of
anaemia included depression and pale mucous membranes and tissues.
Necropsy and histopathological examinations confirmed the
treatment-related and dose-dependent anaemia at doses above 30 mg/kg
bw/day. The NOEL in this study was 30 mg/kg bw/day (Jackson et al,
1985b).
2.2.2.3 Monkeys
Ceftiofur was administered intravenously to groups of monkeys
(2/sex/group) at dose levels of 100, 200 or 400 mg/kg bw/day for 12
days. Signs of toxicity included diarrhea in all treated animals and
vomiting accompanied by tachycardia in 1 animal receiving the
200 mg/kg bw/day dose. This animal died after the 12th treatment but
had no treatment-related lesions at necropsy.
Ophthalmological examination, including intraocular pressure, was
normal in all treated animals as were results of electrocardiograms.
Although diarrhea was noted in all treated animals, concomitant weight
loss was not observed. Haematology, biochemistry and urinalysis were
all within normal limits. Histopathology revealed a nephritis,
accompanied by increased kidney weight in 1 male given the highest
dose. No other treatment-related effects were noted. A clear NOEL was
not identified in this study (Berthe, 1982c).
2.2.3 Reproductive toxicity studies
2.2.3.1 Rats
In a 2-generation fertility and general reproductive performance
study, groups of 30 male (approximately 45-day old) and 30 female
(approximately 55-day old) Sprague-Dawley rats were orally
administered ceftiofur at dosages of 0, 100, 300 or 1000 mg/kg bw/day.
Males were treated from 70 days prior to breeding, continuing for a
total of 136 days of treatment. Females were treated 14 days prior to
breeding, throughout gestation and lactation, for a total of 79 days
of treatment. The F1 generation was also retained for breeding. Body
weight, food consumption, parental survival, confirmed matings,
pregnancy rates, length of gestation, number of live offspring,
offspring survival, necropsy and histopathological findings were all
evaluated as part of this study.
All pups in the high-dose group survived and no effect on growth
was seen. No dose-dependent adverse effects on fertility, reproductive
performance or histopathological alterations in reproductive organs of
either sex in the F0 generation were observed. Alteration in
body-weight gain and enlargement of the caecum were seen in each
treated group. No treatment-related adverse effects on growth or
viability were observed in the F1 litters through weaning. There
were no abnormalities on histopathological examination of F0 and
F1 animals. The NOEL in this study was 1000 mg/kg bw/day
(Kakuk, 1985).
A 2-generation study of fertility and reproductive performance of
F1 generation rats was conducted as a continuation of the above
study. Four groups of 30 male and 30 female Sprague-Dawley F0 rats
were administered ceftiofur from the postnatal day 21 until days
145-159 for males, and days 146-160 for females. A dose-related
increase in mortality was noted in treated groups when the data from
the males and females were combined. The majority of the deaths were
attributed to accidental causes. There were no adverse effects on
fertility or reproductive performance in the F1 generation and F2
litters. Enlargement of the caecum occurred in F1 animals at
300 mg/kg bw/day or greater. In the high-dose groups, there was a
higher incidence of degenerative changes in the non-glandular stomach
(92%), and mucus hypersecretion in the glandular stomach (79%)
compared to control animals. No treatment-related histological changes
were observed in the reproductive organs of either sex at the high
dose (1000 mg/kg bw/day). The NOEL in this study was 1000 mg/kg bw/day
(Kakuk, 1986).
2.2.4 Special Studies on embryotoxicity and teratogenicity
2.2.4.1 Mice
Teratogenicity studies were conducted in mice as a second species
instead of rabbits because orally administered ceftiofur disrupts the
caecal microflora in rabbits. In a dose range-finding study, groups of
seven bred female CD-1 mice were given ceftiofur orally at doses of
1000, 2000, 4000 or 8000 mg/kg bw/day from days 6-15 of gestation. At
day 18 of gestation, uterine weight, numbers of viable fetuses,
resorptions, corpora lutea and fetal malformations were recorded.
Signs of maternal toxicity were evident at 4000 and 8000 mg/kg bw/day.
Reduced fetal body weights were recorded at 8000 mg/kg bw/day. The
NOEL for maternal toxicity was 2000 mg/kg bw/day, for fetotoxicity
4000 mg/kg bw/day, and for embryotoxicity and teratogenicity
8000 mg/kg bw/day.
A more detailed segment II oral teratogenicity study was
conducted in groups of 30 female CD-1 mice on days 6-15 of gestation
at 1000, 2000 or 4000 mg/kg bw/day. All parameters stated above were
recorded as well as extensive examination of viable fetuses for
visceral malformations, cranial and skeletal abnormalities. Increased
food consumption, distended stomach and small intestines, and enlarged
gall bladders were observed in dams in the mid- and high-dose groups.
No treatment-related effects were seen in the numbers of resorption
sites, litter size or pup weights. There were no effects on the
incidences of skeletal or visceral anomalies. The NOEL for maternal
toxicity was 1000 mg/kg bw/day, and for developmental toxicity it was
4000 mg/kg bw/day (Marks & Terry, 1993).
2.2.4.2 Rats
Groups of 24 pregnant rats (strain unspecified) were orally
administered doses of 0, 800, 1600 or 3200 mg/kg bw/day ceftiofur once
daily on days 6-15 of gestation. Observations were made daily for
signs of toxicity, and body weights were recorded on the day of
insemination, throughout the dosing period, and on day 20 when
cesarean sections were performed. At that time, the sex, weight,
number and location of viable fetuses, number and location of
resorption sites, fetal weights and gross fetal abnormalities were
determined.
Dose-related maternal toxicity (i.e. soft stools, prophyrin
staining of the eye and nares, diarrhea and blood in faeces) was
observed particularly in the high-dose group. There were no observed
adverse effects on maternal reproductive capacity and no evidence of
teratogenicity in this study. A statistically significant dose-related
decrease in mean fetal body weight, which did not exceed 7%, was
observed. The NOEL in this study was 3200 mg/kg bw/day (Shaw et al,
1985).
2.2.5 Special studies on genotoxicity
A variety of in vitro and in vivo genotoxicity assays
covering a range of endpoints were conducted with ceftiofur and the
metabolite furoic acid (Tables 1 & 2). All assays were negative except
an in vitro chromosomal aberration assay with ceftiofur, which
produced chromatid breaks, gaps and fragments in CHO cells.
Chromosomal aberrations occurred in CHO cells exposed to > 200 mg/ml
for long periods of treatment (44 h) in the absence of S9 metabolic
activation. No evidence of clastogenicity was seen following shorter
treatment times or in the presence of S9 at doses as high as
5000 mg/ml nor in chromosomal aberration assays in vivo. The
mechanism by which chromosomal aberrations were induced in vitro was
extensively investigated. Ceftiofur was profoundly cytostatic
(i.e. reducing the rate of cell division) in CHO cells under
conditions which causes chromosomal aberrations in vitro. Removal of
the drug led to reversal of cytostasis and reduction in number of
cells with aberrations. Cytotoxicity and cell lethality were not
observed in ceftiofur-treated CHO cells suggesting that cytostasis
results in chromosomal breaks and gaps due to prolongation of the cell
cycle and not by a direct effect on chromatin (Aaron, 1991).
2.2.6 Special studies on immunotoxicity
In view of the structural similarity of many ß-lactam drugs, the
possibility of immunologic cross reaction must be addressed. In order
to assess this possibility, a series of studies intended to
investigate the hypersensitivity for ß-lactam antibiotics were
developed.
The model developed was based on passive cutaneous anaphylaxis
(PCA) in the guinea-pig and was intended to determine the human safety
of residues of ceftiofur-sodium in edible tissues, including injection
site residues. In addition, because ceftiofur is structurally related
to penicillin, and because of concern that it might therefore have
antigenic determinants for penicillin, the studies also examined the
interaction between the penicillin antibody and ceftiofur.
Antibodies to benzyl penicillin G (BPG), conjugated to keyhole
limpet hemocyanin (KLH), and antibodies to ceftiofur (CEF), conjugated
to bovine gamma globulin (BGG), were prepared and assayed for PCA
activity in the guinea-pig. Reactive sera were then utilized to
passively sensitize animals prior to further challenge with conjugates
of BPG and BGG, CEF with BGG, CEF with hen egg albumin (HEA), the
deocetylcefotaxime metabolite of ceftiofur, the aminothiazolyl (atz)
side chain, common to parent drug and all metabolites, with HEA,
parent drug, free sulfhydryl metabolite (FSM) of CEF and extracts of
residue of CEF from injection site muscle and kidney from treated
animals.
The protocol involved passively sensitizing female guinea-pigs
with antibody at multiple skin sites followed by challenge 5 days
later. Dose levels utilized were selected as multiples of the
anticipated human exposure level of 0.083 mg/kg bw.
Passive cutaneous anaphylaxis occurred in guinea-pigs sensitized
with antibody to penicillin when challenged with the BGG-BPG control.
Reactions did not occur with exposure to any CEF-containing products.
Table 1. Results of genotoxicity studies on ceftiofur
Test Test object Concentration Results References
In vitro
Ames testa S. typhimurium 0.125, 0.250, 0.5, negative Mazurek & Swenson,
TA98, T100, T1535, T1537, 1.0 µg/plate 1983; Aaron, 1991
T1538
Forward Chinese hamster V-79 fibroblasts 1.0, 2.0, 4.0 negative Harbach et al., 1983
mutation assaya (HGPRT assay) µg/ml
Chromosome Chinese hamster ovary cells 211, 5000 µg/ml positive Aaron, 1991
aberration
assaya
In vivo
Micronucleus Sprague-Dawley rat bone marrow 0, 250, 500, 1000, negative Trzos et al., 1984
Test mg/kg bw
Micronucleus CD-1 mouse bone marrow 0, 250, 500, 1000 negative Aaron, 1991
Test mg/kg bw
UDS Rat hepatocytes 0, 0.03, 0.1, 0.3, negative Trzos & Swenson,
1.0 mg/ml 1984
Table 1. Results of genotoxicity studies on ceftiofur (cont'd).
Test Test object Concentration Results References
Chromosome Mouse bone marrow 450, 900, 1750 negative Aaron, 1991
aberration assay mg/kg bw
(acute)
Chromosome Mouse bone marrow 350, 700, 1400 negative Aaron, 1991
aberration assay mg/kg bw
(subchronic)
a With and without rat liver S9 fraction
Table 2. Results of genotoxicity studies on furoic acid
Test Test object Concentration Results References
In vitro
Ames test S. typhimurium 250, 500, 1000, 2000 negative Mazurek &
TA98, T100, T1535, µg/plate Zimmer, 1985
T1537, T1538
Forward Chinese hamster V-79 250, 500, 1000, 1500 negative Zimmer et al.,
mutation fibroblasts (HGPRT mg/ml 1985
assaya assay)
UDS Rat hepatocytes 1, 3, 10, 30, 100, 300, negative Harbach & Aaron,
1000 mg/ml 1991
a With or without rat liver S-9 fraction
Guinea-pigs sensitized with antibody to ceftiofur reacted to
challenge with HEA-CEF by both the i.v. and oral routes of exposure,
requiring 10 mg/kg bw by the oral route. Similarly, free sulfhydryl
metabolite caused reactions over a broad range of dose levels by both
the i.v. and oral routes. PCA reactions occurred following i.v.
challenges containing at least 0.076 µg FSM/kg bw. Reaction to the
free sulfhydryl metabolite following an oral challenge was similar to
those reported for the HEA-CEF, suggesting approximately a 1000 fold
difference in sensitivity between the i.v. and oral routes. Challenge
of guinea-pigs sensitized with antibody to ceftiofur, and administered
ceftiofur residue extracts from kidney and injection site muscle at
dose levels of 830 µg drug/kg bw failed to produce a positive
response.
These data, when taken together indicate that penicillin
antibodies do not recognize ceftiofur antigenic determinants.
Furthermore, the data also suggest that the GI tract significantly
reduces potential PCA activity. The data suggest that ceftiofur
residues at either the injection site or present in kidney are not
present in either a form or concentration which is likely to induce
PCA activity following oral exposure of animals sensitized with
ceftiofur antibodies and subsequently challenged with the residue
(Jackson et al, 1988; Brussee et al, 1989). The authors concluded
that human exposure to ceftiofur, its residues or metabolites poses
virtually no human risk because:
(a) oral challenge with extract of ceftiofur residues in sensitized
guinea-pigs did not result in positive PCA reactions;
(b) while the free sulflydryl metabolite poses the greatest risk of
eliciting a hypersensitivity reaction, this risk is indeed very
small because exposure would be restricted to the oral route
where residues are invariably bound to proteins, in very low
levels, and further inactivated in the GI tract;
(c) IgE isolated from patients with known sensitivity to pencillin
did not bind significant amounts of the ceftiofur molecule, again
implying a lack of cross reactivity.
2.2.7 Special studies on microbiological effects
Gram-positive bacterial susceptibility to ceftiofur is given in
Table 3.
Table 3. Gram-positive bacterial susceptibility to ceftiofur (µg/ml)
(Yancey et al., 1988; Klein et al., 1985)
Organism MIC50 MIC90 MICrange
Staph. intermedius 0.13 0.25 N/A
Staph. aureus N/A N/A 0.5-4.0
Staph. aureus (dog, <0.06 0.13 N/A
cat)
Staph. intermedius <0.06 <0.06 N/A
Strep. agalactiae N/A N/A <0.06-0.25
Strep. bovis N/A N/A <0.06
Strep. dysgalactiae N/A N/A <0.06-0.25
Strep. equi <0.06 <0.06 N/A
Strep. suis N/A N/A <0.06-0.5
Strep. uberis N/A N/A <0.06-0.5
Strep. <0.06 <0.06 N/A
zooepidemicus
Strep. faecalis N/A N/A >32
L. monocytogenes N/A N/A 16
R. equi 8 16 N/A
As noted in section 2.1.2, ceftiofur is rapidly degraded to
desfuroylceftiofur. This specific metabolism and the antimicrobial
activity of both the parent drug and its primary metabolite against
both Gram-positive and Gram-negative bacteria have been investigated.
The MIC values are given in Tables 4 and 5.
Table 4. Gram-positive bacterial susceptibility to ceftiofur and
desfuroylceftiofur (MIC90) (Salmon et al, 1993)
Organism Ceftiofur (µg/ml) Desfuroylceftiofur
(number tested) (µg/ml)
Strep. uberis (15) 0.03 0.5
Strep. dysgalactiae (15) <0.0039 0.03
Strep. zooepidemicus <0.0019 0.03
(48)
Strep. equi (12) <0.0019 0.03
Strep. suis (49) 0.13 0.25
Staph. aureus (10) 1.0 8.0
Staph. hyicus (14) 1.0 4.0
Staph. spp (11) 1.0 8.0
Table 5. Gram-negative bacterial susceptibility to ceftiofur and
desfuroylceftiofur (MIC90) (Salmon et al, 1994)
Organism Ceftiofur Desfuroylceftiofur
(number tested) (µg/ml) (µg/ml)
Pasteurella multocida (50)
(from Swine Resp. Dis.) <0.0039 <0.0078
Pasteurella multocida (48)
(from Bovine Resp. Dis.) <0.0039 <0.0078
Pasteurella haemolytica (42) 0.015 0.015
Haemophilus somnus (59) <0.0019 <0.0019
A. pleuropneumoniae (50) <0.0019 <0.0019
Salmonella choleraesuis (48) 1.0 1.0
E. coli (40) 0.5 0.5
Extensive investigations have also been carried out on the
in vitro activity of ceftiofur and its metabolites against cultures
of bacteria of relevance in the human GI tract. MIC values of both the
parent drug and its primary metabolites were determined against
bacterial species frequently isolated from the human intestinal tract.
The MIC values are reported in Table 6.
In vitro MIC data covering a wide range of animal and human
bacterial species were available. Fifty-eight strains commonly
isolated from the human GI tract were tested with ceftiofur and its
metabolites. The MIC values were determined by the agar dilution
technique at both high (10 6-7) and low (10 4-5) inoculum
densities. Generally, there was a 2-fold increase in the MIC values
with increasing inoculum density. Ceftiofur was always more active
than its metabolites desfuroylceftiofur, desfuroylceftiofur disulfide
and desfuroylceftiofur cysteine disulfide. Streptococcus,
Propionibacterium and Bifidobacterium were the most sensitive,
with MIC50 values of 0.016 µg/ml, 0.03 µg/ml, and 0.03 µg/ml at high
inoculum density, respectively. Bacteroides sp., Enterococcus
faecium, Eubacterium sp., and Lactobacillus sp. were least
sensitive to ceftiofur, with MIC50 values of 16 µg/ml, 128 µg/ml,
1 µg/ml, and 16 µg/ml, respectively.
Particularly noteworthy is that for most strains, metabolites of
ceftiofur were considerably less active than parent drug. The
degradation of ceftiofur residues by gut flora was also examined. The
data indicate that ceftiofur is rapidly degraded in human faecal
material incubated anaerobically, to compounds which essentially lack
microbiological activity (Hornish et al., 1994; Kotarski, 1993).
2.2.8 Observations in humans
Ceftiofur is an antimicrobial drug developed exclusively for use
in veterinary medicine and hence no direct studies in humans have been
conducted.
Ettestad et al. (1995) have recently reported on biliary
complications associated with the use of ceftriaxone, a cephalosporin
antimicrobial agent, in the treatment of unsubstantiated Lyme disease.
The authors concluded that there appeared to be a threshold for
biliary complications which required a daily dose of > 40 mg/kg
bw/day for periods of at least 1 month. It is noteworthy that
anticipated human exposure to ceftiofur through food residues is
approximately 4000 times lower than the threshold dose suggested by
the above authors.
Table 6. MIC50 values for human strains of anaerobic and facultatively anaerobic bacteria
(Thurn et al., 1994; Zurenko & Yagi, 1990; Kennedy et al., 1991; Watts et al., 1991)
Group MIC50 (µg/ml)
(no. strains tested)
ceftiofur desfuroylceftiofur desfuroylceftiofur
cysteine
disulfide
low high low high low high
Bacteroides (12 or 16) 2 16 16 64 16 128
Bifidobacterium (15) 0.25 ND 8 ND 32 ND
Clostridium (5) <.016 1 1 8 2 2
Eubacterium (13) 1 ND 128 ND 64 ND
Peptococcus and 0.25 0.5 4 16 16 32
Peptostreptococcus
(10 or 15)
Enterococcus (5 and 2) 128 ND 32 ND 8, 32 ND
Escherichia coli (7) 0.5 0.5 2 1 2 2
Proteus vulgaris (5) <.06 ND 2 ND ND ND
Lactobacillus (2 or 1) 0.5, 1 0.5, 16 2, 8 4, 128 4, ND 4, ND
ND = not determined
3. COMMENTS
Toxicological data
A range of studies on ceftiofur and its primary metabolites were
available for evaluation by the Committee, including data on
pharmacokinetics and metabolism, acute and short-term toxicity,
reproductive and developmental toxicity, genotoxicity, immunotoxicity
and microbiology.
Ceftiofur is rapidly metabolized to desfuroylceftiofur. Following
i.m. administration in the rat, approximately 55% of the dose was
excreted in the urine and about 30% in the faeces within the first
24 h. Similar results were obtained in cattle. In a separate oral
study in rats, approximately 55% of the dose was recovered in urine;
the remainder was present in the faeces and the GI tract.
Single oral doses of ceftiofur of up to 7800 mg/kg bw produced
only minimal toxicity in the rat. Toxic signs associated with repeated
oral doses in rats of up to 6000 mg/kg bw/day for 30 days were limited
to haematological changes and diarrhoea. Oral doses of up to 300 mg/kg
bw/day given to dogs for 91 days produced a reversible anaemia and
thrombocytopenia. The NOEL for treatment-related haematopoietic
effects in rats was 30 mg/kg bw/day.
In reproductive toxicity studies in rats, ceftiofur administered
at dose levels of up to 1000 mg/kg bw/day had no adverse effects on
fertility, reproductive performance or reproductive organs. Similarly,
no treatment-related effects were observed in developmental toxicity
studies in mice at doses of up to 4000 mg/kg bw/day or in rats at
doses of up to 3200 mg/kg bw/day.
A variety of in vitro and in vivo genotoxicity assays covering a
range of end-points were conducted with ceftiofur (with and without
metabolic activation with S-9 microsomal fraction) and its metabolite
furoic acid. All the assays were negative, with the exception of an
in vitro chromosomal aberration assay in the absence of metabolic
activation, but only at concentrations at which cell division was
inhibited. The Committee concluded that this finding, when taken in
conjunction with the negative in vivo chromosomal aberration
studies, was not of biological significance.
Carcinogenicity studies have not been performed on ceftiofur.
However, the Committee noted that the drug showed no evidence of
genotoxicity in a variety of assays and is not chemically related to
known carcinogens. Furthermore, it is rapidly metabolized and its
metabolites are not related to any known carcinogens. Neither
neoplastic nor preneoplastic lesions were observed in 90-day feeding
studies in rats, dogs, monkeys, or in reproductive toxicity studies
involving exposure for periods of up to 160 days in which limited
histopathological examination were carried out. Recent reports
indicate that non-genotoxic chemicals showing such a lack of toxicity
are not associated with carcinogenicity in long-term rodent toxicity
studies. Under these circumstances, the Committee concluded that
carcinogenicity studies were not necessary.
Long-term toxicity studies were not available. Even at doses
exceeding several grams/kg bw/day in rats for periods of up to 90
days, diarrhoea was the only major effect noted in rats. The Committee
concluded that allowance could be made for the absence of long-term
toxicity studies on ceftiofur by the application of an appropriate
safety factor.
The potential immunotoxicity of ceftiofur has also been
investigated. The Committee noted that penicillin antibodies do not
recognize ceftiofur antigenic determinants and that exposure to
metabolites of ceftiofur did not produce adverse reactions in
guinea-pigs sensitized to penicillin. The Committee concluded that
there is no risk of hypersensitivity reactions in humans to ceftiofur
or its residues or metabolites at the anticipated level of exposure.
Microbiological data
The potential for adverse effects on the human gut flora was
considered. In vitro MIC data covering a wide range of animal and
human bacterial species were submitted for evaluation. A total of 58
strains commonly isolated from the human GI tract were tested with
ceftiofur and its metabolites. Ceftiofur was more active than its
metabolites desfuroylceftiofur, 3,3'-desfuroylceftiofur disulfide and
desfuroylceftiofur cysteine disulfide. The Committee recognized,
however, that ceftiofur is not present as a residue because it is
extensively and rapidly metabolized, with a plasma half-life of
approximately 15 minutes in cattle and pigs. The lowest MIC50 value
reported for desfuroylceftiofur cysteine disulfide was 2 µg/ml for
Clostridium and Escherichia species.
In calculating an ADI based on antimicrobial activity, the
Committee used the formula developed at the thirty-eighth meeting of
the Committee (Annex 1, reference 97):
Concentration without
effect on human gut × Daily faecal bolus (g)
Upper limit of flora (µg/ml)
temporary ADI =
(µg/kg bw) Fraction of
oral dose × Safety factor × Weight of
bioavailable human
(60 kg)
= 2 × 150
0.1 × 1 × 60
= 50 µg/kg bw
It took the following factors into account:
* Factors to account for the range of MICs needed to allow for
sensitive bacteria, anaerobic environment, bacterial density and
pH: the most relevant sensitive species were studied under
conditions of high inoculum density. No adjustment was deemed
necessary.
* Availability: the fraction of the dose available to the gut
microflora was derived from studies of ceftiofur in humans which
showed that the drug was rapidly metabolized.
* Variability among exposed individuals: the Committee noted that a
substantial amount of data covering a variety of bacterial
strains representative of the human gut microflora was available.
In addition, it recognized that the other values selected for
this calculation was already conservative and incorporated an
adequate margin of safety. A safety factor of 1 was therefore
adopted.
4. EVALUATION
The Committee noted that the lowest NOEL based on toxicological
studies was 30 mg/kg bw/day, which was observed in the 90-day study in
dogs. It could establish an ADI of 0-60 µg/kg bw based on this NOEL
and a safety factor of 500, which would include an additional safety
factor of 5 to take account of the absence of long-term toxicity
studies. However, the Committee noted that the microbiological
end-point would give the lowest ADI and therefore established an ADI
of 0-50 µg/kg bw based on this end-point.
5. REFERENCES*
Aaron CS (1991). The Upjohn Company: TR 7228-91-036. U64279E:
Evaluation of U64279E in the In Vitro Chromosome Aberration Assay
Using Chinese Hamster Ovary (CHO) Cells.
Banting A, Mignot A, Lefebyre MA, Millerioux L, Steffan J, Gilbertson
TJ (1989). The Upjohn Company: TR 788-9760-88-018, "Plasma Profile and
Pharmacokinetic Parameters in Calves After Single (IV and IM) and
Multiple Dose Administration (IM) of Ceftiofur Sodium.
Berthe, J (1982a). Centre Des Recherches Clin-Midy, Code Nomenclature:
TO010-00, Direction Des Recherches Sanofi, Montpellier, FRANCE: Etude
de la Toxicité Aigue De CM-31916 (Etude Preliminaire).
Berthe J (1982b). Centre De Recherches Clin-Midy, Code Nomenclature:
TO020-00, Direction Des Recherches Sanofi, Montpellier, FRANCE: Etude
de la Toxicité Subaigue De CM-31916 chez le Rat Sprague-Dawley par
Voie Intraperitoneale.
Berthe J (1982c). Centre De Recherches Clin-Midy, Code Nomenclautre:
TO021-00, Direction Des Recherches Sanofi, Montpellier, FRANCE:
CM-31916 Etude de la Toxicité Subaigue chez le Macaque Par Voie
Intraveineuse.
Brussee DM, Clarke GL, Cypher JJ, Farho TG, Gilbertson TG, Hornish RE,
Jaglan PS, Miller CC (1989). Internal Memorandum, The Upjohn Company.
Cole SL, Kakuk TJ, Rop DA (1985). The Upjohn Company: TR 7263-85-002,
Acute Oral Single Dose Study in Sprague-Dawley Rats with Ceftiofur
(U-64,279E).
Ettestad PJ, Campbell GL, Welbel SF, Genese CA, Spitalny KC,
Marchetti CM, Dennis DT (1995). Biliary complications in the treatment
of unsubstantiated Lyme disease. J. of Infectious Diseases
171:356-361.
Gilbertson TJ, Roof RD, Jaglan PS (1990) The Upjohn Company:
TR 906-9760-90-001, In vitro Metabolism of 14C Ceftiofur Sodium
and Metabolites in S-9 Fractions of Livers and Kidneys of Rats, Pigs,
Cattle, and Chickens.
Halstead SL, Walker RD, Baker JC, Holland RE, Stein GE, Hauptman JG
(1992) Pharmacokinetic Evaluation of Ceftiofur in Serum, Tissue
Chamber Fluid and Bronchial Secretions from healthy Beef-Breed Calves.
Can. J. Vet. Res., 56:269-274.
* All unpublished studies were submitted to WHO by the Upjohn
Company, Kalamazoo, MI, USA
Jackson TA, Brussee DM, Cypher JJ (1988) The Upjohn Company:
TR 7220-88-026, Hypersensitivity Studies with Sodium Ceftiofur
(U-64,279E) in Hartley Albino Guinea Pigs by the Intravenous and Oral
Routes.
Jackson TA, Brussee DM, Vrbancic JP, Mulholland MP (1985a) The Upjohn
Company: TR 7263-85-077, U-64,279E; 51-Day Oral Toxicology and Drug
Safety Study in the Beagle Dog.
Jackson TA, Brussee DM, Vrbancic JP, Mulholland MP (1985b) The Upjohn
Company: TR 7263-85-079, U-64,279E; 90-Day Oral Toxicology and Drug
Safety Study in the Beagle Dog.
Jaglan PS, Adams LD, Roof RD, Reardon IM, Heinrickson RL,
Gilbertson TJ (1991) The Upjohn Company: TR 788-7926-91-001, The
Nature of Covalent Binding of Desfuroylceftiofur to Plasma Proteins of
Rats.
Jaglan, PS, Arnold, TS (1986a) The Upjohn Company: TR 788-9760-PSJ-I-
86-001, Metabolism of Ceftiofur (14C-U-64,279E) Sodium in Rats from
Oral Treatment Compared to Intramuscular Treatment of Bovine (Study
No. J-080). Part I-Disposition Study and Comparative Metabolic Profile
in the Urine of Rats and Bovine.
Jaglan PS, Arnold TS (1986b) The Upjohn Company; TR 788-9760-86-002,
Metabolism of Ceftiofur (14C U-64,279) Sodium Salt in Rats from Oral
Treatment Compared to Intramuscular Treatment of Bovine (Study
No. J-080). Part II-Comparative Metabolic Profile in Plasma of Rats
and Bovine.
Jaglan PS, Arnold TS (1987a) The Upjohn Company: TR 788-9760-86-006,
Metabolism of 14C-Ceftiofur (U-64,279E) Sodium Salt in Rats from
Intramuscular Treatment.
Jaglan PS, Arnold TS (1987b) The Upjohn Company: TR 788-9760-87-010,
Characterization of the Major Bovine Urinary Metabolites Following
Intramuscular Treatment with 14C-Ceftiofur.
Jaglan PS, Cox BL, Smart DJ, Pierce PA, Yein FS, Roof RD,
Gilbertson TJ (1989) The Upjohn Company: TR 788-9760-89-002,
Disposition and Metabolism of 14C-Ceftiofur Sodium (U64279E) in
Lactating Cows. Part II: The Nature of Milk Residues.
Jaglan PS, Kubicek MF, Cox BL, Johnson DB, Gilbertson TJ (1987a) The
Upjohn Company: TR 788-9760-87-006, Nature of Metabolites in Rats
Treated Orally with Ceftiofur from Multiple High Doses and Comparison
of the Metabolites in Liver and Kidney of Rats Versus Bovine.
Jaglan PS, Kubicek MF, Johnson DB, Stuart DJ, Mazurek JH, Wiser SK,
Aaron CS (1987b) The Upjohn Company: TR 788-9760-87-002, Metabolism of
14C-Ceftiofur (U6,4279) in vitro.
Jaglan PS, Roof RD, Yein FS, Zaya MJ, Gilbertson TJ (1990) The Upjohn
Company: TR 796-9760-89-005, Comparison of Metabolites of Ceftiofur
(U-64,279E) Sodium in The Urine and Kidneys of Pigs from intramuscular
Injection to that of Rats from Oral Doses.
Kakuk TJ, Cole SL, Rop DA (1985a) The Upjohn Company: TR 7263-85-071,
30-Day Oral Toxicity Study in Sprague-Dawley Rats with Ceftiofur
Sodium (U-64,279E).
Kakuk TJ, Cole SL, Rop DA (1985b) The Upjohn Company: TR 7263-85-075,
90-Day Oral Toxicity Study in Sprague-Dawley Rats with Ceftiofur
Sodium (U-64,279E).
Kakuk TJ (1985) The Upjohn Company: TR 7263-85-082, Two Generation
Fertility and General Reproductive Performance Study (Oral) of
Ceftiofur Sodium (U-64,279E) in Sprague-Dawley Rats. I. Fertility and
Reproductive Performance of the F0 Generation.
Kakuk TJ (1986) The Upjohn Company: TR 7263-86-031, Two Generation
Fertility and General Reproductive Performance Study (Oral) of
Ceftiofur Sodium (U-64,279E) in Sprague-Dawley Rats. II. Fertility and
Reproductive Performance of the F1 Generation.
Kennedy MJ, Yancey RJ, Kornis GI (1991) The Upjohn Company:
TR 705-7923-91-015, In vitro Activity of Ceftiofur Sodium
(U-64,279E), Desfuroylceftiofur (U-75,104) and Desfuroylceftiofur
Cystein Disulfide (U-93,112) Against Bifidobacterium spp. and
Eubacterium spp. from the Human Gastrointestinal Tract.
Klein LK, Yancey RJ, Goodenough KR, Kinney ML, Roberts BJ (1985) The
Upjohn Company: TR 705-7922-85-003, In vitro and In Vivo
Evaluation of the Monobactam Antibiotics, U70,887B and U71,689B,
Compared to Aztreonam and Ceftiofur Against Bacterial Pathogens of
Veterinary Importance.
Kotarski S (1993) Internal Memo, The Upjohn Company.
Krzeminski LF, Stuart DJ, Gosline RE, Subacz CJ, Cox BL, Reeves DR
(1985) The Upjohn Company: TR 788-9760-85-005, HPLC Assay of Bovine
Plasma and Urine Metabolites After Treatment with Carbon 14 Labeled
Ceftiofur.
Leong BKJ, Sabaitis CP, Kakuk TJ, Imlay MM (1985) The Upjohn Company:
TR 7277-85-018, Acute Four-Hour Dust Inhalation Toxicity Study on
Ceftiofur Sodium (U-64,279E) in Albino Rats.
Marks TA, Terry RD (1993) The Upjohn Company: TR 7224-93-054,
U-64279E: A Range-Finding Study (Oral) in Mice.
Salmon SA, Watts JL, Yancey RJ, Case CA (1993) The Upjohn Company:
TR 705-7923-93-007, Minimum Inhibitory Concentrations for Ceftiofur
and Desfuroylceftiofur with Isolates of Veterinary Importance.
Salmon SA, Watts JL, Case CA, Yancey RJ (1994) Minimum inhibitory
concentrations for ceftiofur and comparator antimicrobial agents
against bacterial pathogens of swine from the United States, Canada
and Denmark. TR No. 705-7923-94-020. The Upjohn Company
Shaw CI, Marks TA, Poppe SM, et al. (1985) The Upjohn Company:
TR 7259-85-011, A Segment II Teratology Study (Oral) in Rats Givn
U-64,279E
Thurn KK, Greening RC, Kotarski SF (1994) The Upjohn Company:
TR 788-7928-94-001 Minimal Inhibitory Concentrations of Ceftiofur and
its Metabolites Against Bacterial Species Frequently Isolated from the
Human Gastrointestinal Tract.
Trzos RJ, Swenson DH (1984) The Upjohn Company: TR 7268-84-018 The
primary hepatocyte unscheduled DNA synthesis (UDS) assay with U-64,279
and ultra violet light.
Trzos RJ, Swenson DH, Brown PK (1984) The Upjohn Company:
TR 7268-84-011 The micronucleus test with U-64,279 (Sanofi
cephalosporin).
Watts JL, Case CA, Yancey RJ, Kornis GI (1991) The Upjohn Company:
TR 705-7923-91-020 Evaluation of Desfuroylceftiofur-S-S-cysteine
(DCD; U-93-112) with Veterinary Pathogens.
Yancey RJ, Roberts BJ, Folz SD (1988) The Upjohn Company:
TR No. 705-7922-88-002, In vitro Activity of Ceftiofur Sodium
(U-64,279E) for Urinary and Respiratory Tract Pathogens of Companion
Animals.
Yein FS, Zaya MJ, Arnold TS, Hoffman GA, Roof RD, Dame KJ, Cox TD,
Reeves DR, Flook TF (1990) The Upjohn Company: TR 796-9760-89-002,
Absorption, Distribution, Metabolism, and Excretion of 14C-Ceftiofur
(U-64,279E) Sodium in the Swine.
Zurenko GE, Yagi BH (1990) The Upjohn Company: TR 7254-090-098
The In vitro Activity of Ceftiofur Sodium (U-64279E) and
Desfuroylceftiofur (U-75104) Against Human Bacterial Clinical
Isolates.