
DICOFOL
First draft prepared by A. Clevenger
Office of Pesticide Programs
US Environmental Protection Agency
Washington, DC, USA
EXPLANATION
Dicofol is an acaracide which is structurally similar to DDT.
Dicofol was previously evaluated by the Joint Meeting in 1968 (Annex
1, reference 10). An ADI of 0-0.025 mg/kg bw was allocated, based on
a NOAEL of 50 ppm in the diet, equivalent to 2.5 mg/kg bw/day in the
rat.
Since the last review a number of studies have been submitted
including studies using a purer form of dicofol corresponding to
current product purity. The purer form is generally > 95% dicofol
(80-85% p,p'-dicofol and 15-20% o,p'-dicofol) which contains less
than 0.1% DDT-related (DDTr) impurities (i.e. DDT, alpha-chloro-DDT,
DDE, and DDD). Relevant portions of the previous monograph have been
incorporated into this toxicological monograph.
EVALUATION FOR ACCEPTABLE DAILY INTAKE
BIOLOGICAL DATA
Biochemical aspects
Absorption, distribution, and excretion
Mice
The disposition of dicofol was studied using groups of 3 male
NIH mice given a single oral dose of 25 mg/kg bw of 3H-p,p'-
dicofol (3H-ring labeled; isomer composition unknown). Blood,
urine, faeces, and tissues (fat, liver, kidney, lung, brain, spleen,
heart) were collected over 4 days. Approximately 60% of the
administered dose was eliminated within 4 days primarily in the
faeces. Faecal excretion accounted for 40% of the administered dose
whereas urinary excretion accounted for 20%. Peak tissue
concentrations were reached within 24-48 h. The highest
concentrations of radiolabel were found in adipose tissue followed
by liver, kidney, lung, heart, blood plasma, brain, whole blood, and
spleen. Concentrations dropped rapidly over 4 days except in adipose
tissue (Kaneshima et al., 1980).
Rats
Upon ingestion of dicofol by mammals, storage of the compound
occurs in the adipose tissue. Rats were fed dicofol at a level of 32
ppm in their diet for 12 weeks. After eight weeks the level of the
compound in the fat had reached equilibrium at concentrations of 25
ppm for the males and 70 ppm for the females. After 12 weeks,
dicofol was withdrawn from the diet and the level of stored material
declined. The rate of decline was greater for the male animals than
for the females. By 14 weeks after withdrawal the level of dicofol
in the fat was zero for the males but still remained at about 6 ppm
for the females. Feeding with higher or lower dose levels also
showed that dicofol was stored in the fat of the female rat to a
greater degree than in the male (Smith et al., 1959).
The pharmacokinetics of p,p'-dicofol and o,p'-dicofol were
studied in female Crl:CD BR rats (groups of 4) given a single oral
dose of 50 mg/kg bw of 14C-o,p'-dicofol or 14C-p,p'-dicofol
(uniformly ring labelled). Blood, urine, faeces, and tissues (fat,
liver, adrenal gland, thyroid) were collected over 10 days. The two
isomers showed similar distribution and excretion patterns, but
p,p'-dicofol was much more persistent in the body than o,p'-dicofol.
Both isomers were excreted primarily in the faeces, but o,p-dicofol
was excreted more rapidly. Over 90% of the o,p'-dicofol administered
dose was excreted within 2 days (and essentially all eliminated by
10 days) compared to 40% of the p,p'-dicofol administered dose (80%
excreted in 10 days).
Peak tissue concentrations were reached within 6 h in most
tissues and after 1-2 days in fat. Both isomers showed a high
affinity for adipose tissue. At the time of peak concentrations,
approximately 51% of p,p'-dicofol radiolabel and 26% of o,p'-dicofol
radiolabel were in body fat (assuming fat is 7% of body-weight).
Tissue concentrations of both isomers were similar initially, but
concentrations of o,p'-dicofol radiolabel declined more rapidly than
p,p'-dicofol radiolabel. After 10 days, concentrations of p,p'-
dicofol radiolabel were: fat, 144 ppm; adrenal gland, 30 ppm;
thyroid, 16 ppm; liver, 6 ppm; whole blood, 1 ppm. In comparison,
o,p'-dicofol radiolabel concentrations were: fat, 3 ppm; adrenal
gland and thyroid, 1 ppm; blood, 0.6 ppm; liver, 0.5 ppm.
Elimination half-lives were estimated to be 1.5-4 day for o,p-
dicofol and 4-7 day for p,p'-dicofol (DiDonato et al., 1987).
In a study of similar design, the disposition of p,p'-dicofol
was studied in male and female Sprague-Dawley rats (groups of 4/sex)
given a single oral dose of 50 mg 14C-p,p'-dicofol/kg bw
(uniformly labelled ring). Blood, urine, faeces, and tissues (liver,
kidneys, fat) were collected over 7 days. Males and females excreted
78% and 51%, respectively, of the administered dose in 7 days.
Faecal excretion accounted for 32-61% of the administered dose with
the remainder (16-19%) excreted in urine. Adipose tissue contained
the highest concentration of radiolabel followed by liver, kidneys,
and blood. Tissue concentrations were much higher in females than
males. Adipose tissue concent-rations were 5-7 times higher and
liver concentrations 3-4 times higher in females than males. After 7
days, fat concentrations were 148 ppm in females versus 30 ppm in
males, and liver concentrations were 8 ppm in females versus 2 ppm
in males (Tillman & Mazza, 1986).
The disposition of dicofol and DDT were compared following a
single oral dose. Male and female Sprague-Dawley rats (groups of 1-
4/sex) were given a single dose of 50 mg 14C-p,p'-dicofol or
14C-p,p'-DDT/kg bw (uniformly ring labelled). Blood, urine,
faeces, and tissues were collected over 8 days. DDT and dicofol
radiolabel showed qualitatively similar distribution and elimination
patterns, but DDT was more persistent in the body than dicofol. Both
distributed preferentially to adipose tissue and were eliminated
mainly in the faeces. Essentially all of the dicofol dose was
excreted within 8 days compared to 80% of the DDT dose. The highest
concentrations of both compounds were found in adipose tissue and
adrenal glands. After 8 days, DDT-radiolabel in fat was 275 ppm
whereas dicofol-radiolabel was negligible. Dicofol-derived
radiolabel was eliminated from the tissues more rapidly than DDT-
derived radiolabel. Females generally had higher tissue levels than
males. Elimination half-lives were estimated to be 30 h for dicofol
in males and females and 55-95 h for DDT (Steigerwalt et al.,
1984a).
The overall excretion rate of dicofol in this study was
considerably more rapid than the overall excretion reported in two
other single dose studies in rats (DiDonato et al., 1986; Tillman
& Mazza, 1986).
The disposition of dicofol and DDT were compared following
multiple oral doses using female Sprague-Dawley rats (groups of 1-2)
given daily doses of 0.5 mg 14C-p,p'-dicofol or 14C-p,p'-DDT/kg
bw (uniformly ring labelled) for 16 consecutive days. Blood, urine,
faeces, and tissues were collected during treatment and over 16 days
after exposure. As in the single dose comparison study, DDT and
dicofol radiolabel showed qualitatively similar distribution and
elimination patterns, but DDT was more persistent. Dicofol
radiolabel was excreted approximately twice as fast as DDT
radiolabel. Approximately 75% of the dicofol dose was excreted
within 16 days compared to 40% of the DDT dose. Both were eliminated
mainly in the faeces. Concentrations in tissues, such as fat, liver,
and adrenal glands were comparable during treatment, but dicofol
radiolabel was eliminated from these tissues more rapidly. Fat
concentrations of DDT radiolabel increased and peaked post-exposure
whereas dicofol radiolabel began declining when exposure ceased.
After 16 days, DDT-radiolabel in fat was twice that of dicofol-
radiolabel (38 vs. 13 ppm). Elimination half-lives were estimated to
be 6-14 days for dicofol and 7-24 days for DDT (Steigerwalt et
al., 1984b).
Groups of 10 male Wistar rats were given daily oral doses of 63
mg/kg bw (1/20 LD50 of dicofol (84.8% purity)) for 40 days. Blood,
urine, faeces, and tissues were collected over the treatment period
and analyzed by TLC and GC. Twenty-eight percent of the administered
dose was eliminated as dicofol and 99% of dicofol was excreted in
the faeces. At day 40, adipose tissue contained the highest
concentration of dicofol (69 ppm) with lesser amounts in muscle (17
ppm), lung (16 ppm), testes (13 ppm), liver (9 ppm), kidney (9 ppm),
brain (8 ppm), and heart (7 ppm) (Brown et al., 1969; Brown &
Casida, 1987).
Groups of 5 male Wistar rats were given a single i.p. dose of
376 mg/kg bw (1/5 LD50 of dicofol (84.8% purity)). Blood, tissues,
urine, and faeces were collected over 4 days. Peak tissue
concentrations were reached within 32-40 h except for adipose tissue
which had not reached its peak concentration. After 4 days, adipose
tissue contained the highest concentration (77 ppm) of dicofol
followed by testes (8 ppm), liver (7 ppm), muscle (7 ppm), kidney (4
ppm), lung (4 ppm), heart (3 ppm), blood (1.5 ppm), and brain (0.9
ppm) (Brown et al., 1969; Brown & Casida, 1987).
Humans
The dicofol metabolite dichlorobenzilic acid (DCBA) was
measured in the urine of 4 workers involved in the mixing/loading or
application of dicofol (3.0 pounds/acre in 500 gallons of water) to
citrus crops for 10 consecutive days. Urine samples were obtained
over 4 days beginning 6 days after exposure. Because of previous use
of chlorobenzilate, pre-exposure DCBA excretion rates were not zero.
Mean daily DCBA excretion was 19-42 g/day over the exposure period.
The variation correlated with the difference in estimated dermal
dose (2.7-13 mg/day). The percent dermal dose excreted as DCBA was
estimated to be 0.25%. The half-life for DCBA excretion in the urine
was estimated to be 7 days (Nigg et al., 1991).
Biotransformation
Mice
Groups of 2-3 male Swiss-Webster mice were administered a
single intra-peritoneal dose of 30 mg/kg bw of radiolabelled
dicofol, alpha-chloro-DDT, dichlorobenzidine (DCB), or DDT (phenyl
C14, high chemical purity). One hour later, mice were sacrificed
and tissues were collected and analyzed for metabolites. The
proposed metabolic scheme in mice is consistent with the scheme
shown for rats in Figure 1. In mice, dicofol was converted to DCD
(same as FW-152), dichlorobenzophenone (DCBP), and
dichlorobenzhydrol (DCBH) based on analyses of brain, fat, and
liver. These three metabolites represented 33%, 30%, and 7%,
respectively, of the radiolabel in the liver. Administered DCD was
also metabolized to DCBP and DCBH. The reduction of DCBP to DCBH was
suggested to be the rate-limiting step in dicofol metabolism. The
metabolic pattern observed in the mouse in vivo was similar to
results obtained with rat liver microsomes under anaerobic
conditions and in the presence of NADPH. DDE (1,1-dichloro-2'2-
bis(p-chlorophenyl ethylene) was not detected as a metabolite of
dicofol or DCD.
Under the same experimental conditions, alpha-chloro-DDT, an
impurity of technical dicofol, was metabolically dechlorinated to
DDE in mouse liver (50% of radiolabel in liver) and rat liver
microsomes. The conversion of alpha-chloro-DDT to DDE also occurred
in vitro in the presence of reduced haematin. The alpha-chloro-DDT
impurity in technical dicofol may be a source of DDE detected in
tissues. The authors proposed that in vivo metabolic
dechlorination of dicofol and alpha-chloro-DDT involves a reduced
porphyrin in liver microsomes (Brown & Casida, 1987).
Rats
The metabolism of dicofol was studied in Sprague-Dawley rats
(groups of 4/sex) given a single oral dose of 50 mg/kg bw of 14C-
p,p'-dicofol (uniformly labelled ring). The proposed metabolic
scheme for dicofol in rats is shown in Figure 1. In the faeces, most
of the extracted radiolabel was present as FW-152 and DCBH in males
(50-70% combined) and FW-152 and OH-DCBP in females (50-60%
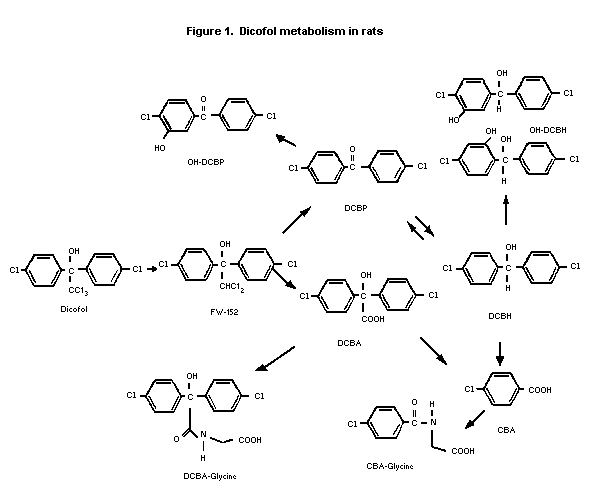 combined). Faeces contained lesser amounts of dicofol and OH-
DCBH/DCBA-glycine. In urine, the radiolabel was mostly DCBH-glycine
and OH-DCBP/DCBH (25-40% combined) in both sexes. Urine contained
smaller amounts of CBA-glycine, DCBA, OH-DCBH/CBA. About 20% of
faeces radio-label and 30-40% of urine radiolabel were unidentified.
In adipose tissue, most of the extracted radiolabel was present
as the parent compound (80-90%), with smaller amounts of DCBP and
FW-152 identified in both sexes. In the liver, most of the extracted
radiolabel was FW-152 (70-80%) in both sexes with lesser amounts of
DCBP, dicofol, and DCBH.
Small amounts of material in faeces co-chromatographed with
DDE. Additional analyses by HPLC determined that 0.27% of the
extracted radiolabel in faeces was actually DDE. Additional analyses
of fat and liver detected small amounts of DDE in one fat sample
(0.2% of extract, 0.34 ppm tissue concentration) and two liver
samples (0.25%-0.34% of extract, 0.018-0.29 ppm tissue
concentration). The 14C dosing solution was reported to contain
0.01% DDE (Tillman & Mazza, 1986).
DDE, DDT, and alpha-chloro-DDT impurities may account for the
small amounts of DDE in tissues. The latter impurity has been shown
to be converted to DDE in rat liver microsomes and mouse liver in
vivo (Brown & Casida, 1987).
Following a single i.p. dose of 376 mg/kg bw of technical
dicofol (84.8% pure) in male Wistar rats, the parent compound and a
metabolite, DCBP, were quantified in blood. 4,4-Dichlorobenzhydrol
(DCBH) was detected but not quantified. DCBP was also detected in
tissues following exposure for 40 days to 63 mg/kg bw/day. Small
amounts of DDE were detected (Brown et al., 1969, 1971).
Effects on enzymes and other biochemical parameters
Mice
The hepatic mixed-function oxidase (MFO)-inducing effect of
dicofol was studied. Groups of 4 male and 4 female CD-1 mice were
administered 3 daily oral doses of 1.4, 4.4, 14.9, 42.8, or 151
mg/kg bw of technical dicofol (87.6% purity). MFO activity in liver
microsomal cell fractions was determined by O-demethylation of p-
nitroanisole. Relative liver weight was increased at the high-dose
in both sexes and at 42.8 mg/kg bw in males. MFO activity was
increased 22%-43% in females at 14.9 mg/kg bw and above. MFO
activity was unaffected in males (Steigerwalt et al., 1984c).
The MFO-inducing effects of technical dicofol, dicofol isomers,
and technical dicofol impurities were compared in B6C3F1 male
mice. Groups of 4 mice were administered the test material in the
diet daily for two weeks. Technical dicofol (87.5% purity) was
administered at 0, 8, 25, 80, 250, or 800 ppm. The two highest doses
bracketed the doses producing liver tumours in male B6C3F1
mice. p,p'-Dicofol was administered at 0, 6, 20, 63, 195, or 625
ppm. Liver MFO activity was measured by the following enzyme assays:
p-nitroanisole O-demethylation, aminopyrine N-demethylation, and
aniline hydroxylation. Technical dicofol depressed body-weight at 80
ppm and above and increased liver weight at 250 ppm and above. p,p'-
Dicofol depressed body-weight and increased liver weight at 625 ppm.
Technical dicofol increased MFO activity at 250 and 800 ppm. p,p'-
Dicofol increased MFO activity at 63 ppm and above. A comparison of
dose-response curves indicated p,p'-dicofol was equal to or slightly
less potent than technical dicofol at comparable concentrations of
active ingredient. Administration of 37 ppm of o,p'-dicofol, ED-8
isomers, DDE isomers, and up to 195 ppm of DCBP isomers produced no
toxicity and had no effect on MFO activity indicating these
constituents of technical dicofol do not play a disproportionate
role in induction of MFO activity. The authors concluded that p,p'-
dicofol was responsible for a large majority but not all the
induction of liver MFO activity produced by technical dicofol
(Steigerwalt et al., 1984d).
Rats
MFO induction activity was studied using groups of 6 male
Sprague-Dawley rats given 4 daily intraperitoneal doses of pure
dicofol (98.8% pure; 81.4% p,p'-, 18.6% o,p'-), technical dicofol
(85% pure; 69.2% p,p', 15.8% o,p'; 15% impurities including DDTr),
pure DDT (99% pure; 81.4% p,p'-,18.6% o,p'-), phenobarbital, or -
naphthoflavone. Dose concentrations ranged from 1.5 to 59 mM (2.2 to
103 mg/kg bw). Liver MFO activity was measured by the following
enzyme assays: cytochrome C reductase, aminopyrine N-demethylase,
ethoxycoumarin O-deethylase, microsomal epoxide hydrolase, cytosolic
epoxide hydrolase, and glutathione-S-transferase. Technical and pure
dicofol and DDT induced MFO activity in a pattern consistent with
phenobarbital-type induction. At a concentration of 59 mM (87.4
mg/kg bw), pure dicofol increased microsomal protein 1.7-fold and
cytochrome P-450 activities 2- to 3-fold. Equimolar doses of
technical dicofol and pure dicofol produced comparable responses,
and dicofol was equal in potency to DDT of equivalent isomer
composition (Narloch et al., 1987).
MFO induction activity was assessed using groups of 6 male
Wistar rats administered dicofol (described as "pure") daily in the
diet for two weeks at 0, 2, 5, 10, 20, 50, or 200 ppm. MFO activity
in liver microsomal cell fractions was determined by aniline
hydroxylase, aminopyrine demethylase, and hexobarbital oxidase
activities. Dicofol at concentrations of 10 ppm and above increased
MFO activity. Aminopyrine demethylase activity showed the greatest
induction with activity increased 2- to 5.7-fold. p,p'-DDT increased
the activity of this enzyme 2.4- to 7.6-fold over the same dose
range. Dicofol ranked after heptachlor, DDT, chlorfenson, and
dieldrin in capacity for inducing MFO enzymes (Den Tonkelaar & Van
Esch, 1974).
Dicofol inhibited gap junctional intercellular communication in
two systems: Chinese hamster V79 metabolic cooperation assay and
scrape-loading/dye transfer assay in WB-F344 rat liver epithelial
cells. Dicofol (1000 ppm in the diet for 11 weeks) enhanced the
development of gamma-glutamyltranspeptidase-positive hepatic foci in
nitrosamine-initiated male Sprague-Dawley rats (Flodstrom et al.,
1990).
Dogs
The effect of dicofol on plasma 17-hydroxy-corticosteroids in
the dog was determined in two dogs which were fed 300 ppm or 900 ppm
dicofol over two separate periods of one to two months' duration.
The ability of the adrenal cortex to elaborate
17-hydroxy-corticosteroids in response to ACTH stimulation was
slightly reduced at the 300 ppm level and markedly reduced at the
900 ppm level. The results also showed that following this treatment
with dicofol, the ability of the adrenal gland to return to the
pre-treatment level of response to ACTH proceeded slowly and,
possibly, incompletely (Smith et al., 1959).
Toxicological studies
Acute toxicity studies
The acute toxicity of technical dicofol is summarized in Table
1. Common signs of toxicity include decreased spontaneous motor
activity, ataxia, passiveness, somnolence, prostration, and
occasionally tremors. In cats, dicofol given i.v. had no convulsive
activity but produced cardiovascular effects consisting of prolonged
arrhythmia and hypertension at sublethal doses and ventricular
fibrillation at a lethal dose.
Table 1. Acute toxicity of dicofol
LD50 LC50
Species Strain Sex Route (mg/kg bw) (mg/l) Reference
Mouse CRJ:CD-1 M oral 669 Onishi (1989)
(ICR) F 6751
Rat Charles River CD M oral 595 Krzywicki & Bonin
F 5871 (1985a)
? M oral 809 Smith et al. (1959);
F 6842 AnnexI: 11
Wistar M oral 14952,4 Brown et al. 1969
Charles River CD M
F dermal (24-hr >5000 Krzywicki & Bonin,
exp) >50001 (1985b)
Wistar M&F i.p. 11153-11502,4 deGroot (1974);
M Brown et al. (1969)
Crl:CDBR M inhalation >5 Fisher & Hagan,
F (4 hr exp) >51 (1987)
Rabbit ? M oral 18102 Smith et al. 1959;
AnnexI: 11
New Zeeland, F dermal (24-hr >25001 Krzywicki & Bonin
white exp) (1985b)
Cat ? M i.v. <203 Joy (1976)
Dog ? M&F oral >40002 Smith et al. (1959);
Annex I: 11
1 Purity of technical dicofol was 94-96%, <0.1% DDTr.
2 Purity of technical dicofol was 80-85%.
3 Purity of technical dicofol was unspecified.
4 The observation period was 7 days only.
Short-term toxicity studies
Mice
Groups of 10 CD-1 (ICR) mice/sex received technical dicofol
(95.6% pure; < 0.1% DDTr) in the diet daily for 13 weeks at 0, 10,
125, 250, 500, or 1000 ppm, (equal to 1.6, 18, 38, 84, and 180 mg/kg
bw/day for males and 2.1, 29, 56, 110, and 190 mg/kg bw/day for
females). At 125 ppm, final body-weight was reduced in females,
hepatic mixed function oxidase (MFO) activity was increased in both
sexes, and absolute and relative liver weight was increased in
females. Liver cell hypertrophy in both sexes, SGPT in females, and
kidney weight in females were increased at 250 ppm. Findings at 500
and 1000 ppm only included increased plasma proteins and lipids,
degenerative changes in the kidney of females, adrenal cortex
hypertrophy, and hepatocellular necrosis and vacuolation. The NOAEL
was 10 ppm, equal to 2.1 mg/kg bw/day based on reduced weight, liver
enlargement, and increased hepatic MFO activity at 125 ppm (Goldman
& Harris, 1986).
In a dose-range finding study for a carcinogenicity study,
groups of 10 male B6C3F1 mice received technical dicofol (>
95% pure) in the diet daily for 13 weeks at 0, 250, 500, or 750 ppm
(equivalent to 36, 71, or 107 mg/kg bw/day). At 500 and 750 ppm,
final body-weight, overall food consumption, and heart weight were
reduced. Liver histopathology was evident at all dose levels.
Hepatic changes were characterized by centrilobular hypertrophy,
eosinophilic and vitreous liver cells, and polynuclear cells. In the
high-dose group, entire liver lobules were vitreous in some cases. A
NOAEL was not identified in this study. Histological changes in the
liver were observed at all dose levels (Sato et al., 1987).
Rats
Dicofol was fed to groups, each containing 10 male and 10
female rats, for 90 days at dietary concentrations of 0, 20, 100,
500, 1250 or 2500 ppm. Survival was adversely affected at 1250 ppm
and above. Growth was inhibited at 100 ppm and higher in the females
but only at 1250 ppm in the males. Increased liver to body weight
ratios occurred in the survivors in both sexes. Liver lesions were
the most consistent histopathological finding, but were only of
scattered incidence at dose levels below 1250 ppm (Smith et al.,
1959).
Groups of 10 Crl-CD(SD) rats/sex received technical dicofol
(95.6% pure, < 0.1% DDTr) in the diet daily for 13 weeks at 0, 1,
10, 100, 500, or 1500 ppm. ( equal to 0.07, 0.64, 6.5, 32, or 96
mg/kg bw/day for males and 0.08, 0.78, 7.8, 36, or 110 mg/kg bw/day
for females). The highest dose of 1500 ppm produced mortality,
ataxia, and lethargy. At 500 ppm and above in both sexes, body-
weight and overall food consumption were reduced, liver weight was
increased, blood corticosterone levels were decreased, and the
incidence of adrenal cortex vacuolation was increased. At 100 ppm,
hepatic MFO activity and the incidence of liver hypertrophy were
increased. The incidence and severity of thyroid follicular cell
hypertrophy (minimal to marked) was increased in males at 10 ppm and
above and in females at 500 and 1500 ppm. The pathologist considered
the thyroid finding of uncertain significance because it is a
relatively non-specific change that has been associated with
environmental factors such as low temperature and stress. The NOAEL
was 1 ppm, equal to 0.07 mg/kg bw/day based on the increase in
thyroid follicular epithelial hypertrophy in males (Goldman et
al., 1986).
Groups of 10 Wistar rats/sex received technical dicofol (74%
pure) in the diet daily for 13 weeks at 0, 50, 200, 1000, or 3000
ppm (equivalent to 2.5, 10, 50, or 150 mg/kg bw/day). All animals
receiving the high-dose died within five weeks. All dose levels
adversely affected body-weight (final weight reduced 10-40%). Food
consumption was not measured. Absolute liver weight was increased in
high-dose males and in females receiving 200 ppm and higher.
Histopathological changes in the liver, described as SER whorls and
V101-cells, were observed in males and females at 200 and 1000 ppm.
V101-cells were described as enlarged hepatocytes with enlarged
nuclei, some hyperchromatic or with unbalanced chromatic
distribution. A basophilic granulation was usually seen in the
periphery of the enlarged cell with the remainder of the cytoplasm
containing fine granules and having eosinophilic character. An
additional observation in high-dose females was increased thyroid
weight. No microscopic changes in the thyroid were found. A NOAEL
was not identified in this study (Verschuuren et al., 1973).
Groups of 6 Crl:CD BR rats/sex received dermal applications (6
h/day, 5 days/week) of the formulation Kelthane MF-B (44.8% dicofol)
at doses of 1, 2.5, 4, or 40 mg active ingredient/kg bw/day for 4
weeks. Control groups received either dermal application of
distilled water or the formulation vehicle (vehicle dose of 53 mg/kg
bw). The vehicle and all dose levels caused skin irritation
attributable to the formulation vehicle. During the third week of
treatment, males receiving 40 mg/kg bw/day experienced a reduction
in absolute body-weight (10%) and body-weight gain (20%). Effects on
the liver were observed at the high-dose in both sexes. SGPT was
slightly elevated in high-dose males. Liver weight relative to body-
weight was increased for high-dose males and females. Minimal
hypertrophy of centrilobular hepatocytes was observed in 5/6 males
and 6/6 females receiving 40 mg/kg bw/day compared to none in
controls. The enlarged hepatocytes were characterized by
eosinophilic cytoplasm. High-dose males also showed increased
severity of multifocal inflammation of the liver. Single-cell
necrosis was observed in some foci. A systemic NOAEL of 4 mg/kg
bw/day was determined based on reduced body-weight and liver effects
at 40 mg/kg bw/day (Lampe & Baldwin, 1990).
Dogs
Groups of six beagle dogs/sex received technical dicofol (93.3%
pure; < 0.1% DDTr) in the diet daily for 13 weeks at 0, 10, 100,
300, or 1000 ppm equal to 0.29, 3.3, 9.9, or 26 mg/kg bw/day for
males and 0.31, 3.4, 9.8, and 27 mg/kg bw/day for females. Clinical
laboratory tests on blood, urinalysis, and physiological
measurements (i.e. electrocardiogram, heart rate, and body
temperature) were conducted prior to treatment, after 4 weeks, and
prior to study termination. The high-dose produced mortality in 5/6
males and 5/6 females. Both sexes receiving 300 and 1000 ppm
exhibited signs of toxicity such as laboured breathing, excessive
salivation, inactivity, incoordination, dehydration, and red-tinged
diarrhoea. Body-weight and food consumption were unaffected at 300
ppm and below. Clinical chemistry findings were consistent with
liver injury at 300 and 1000 ppm and there were effects on adrenal
gland function at 100 ppm and above. In both sexes receiving 1000
ppm, serum enzymes (SAP, SGPT) were increased and serum proteins
(albumin, total protein) were decreased. In 300 ppm females,
alkaline phosphatase was increased four-fold and albumin was
slightly decreased. Baseline cortisol blood levels were normal, but
cortisol response to ACTH challenge (20 units of ACTH; cortisol
measured 30 and 90 minutes after challenge) was markedly decreased
(50-75%) in both sexes at 100 ppm and above. Electrocardiograms
suggested treatment-related prolongation of the QT and PR intervals
in dogs receiving 300 or 1000 ppm. Liver weight was increased in
males at 300 ppm and in females at 1000 ppm. Microscopic changes
were notable only at the high-dose. Findings consisted of single
cell necrosis and mononuclear cell infiltrates in the liver of both
sexes, gastrointestinal haemorrhagic enteritis and congestion in
both sexes, and myocardial necrosis in one male. An additional
observation was oligospermatogenesis observed in three middle-dose
(300 ppm) males and five high-dose (1000 ppm) males. The NOAEL was
10 ppm, equal to 0.29 mg/kg bw/day, based on reduced cortisol
response to ACTH challenge at 100 ppm (Shellenberger, 1986).
Groups of six beagle dogs/sex received technical dicofol (93.3%
pure; < 0.1% DDTr) in the diet daily for 52 weeks at 0, 5, 30, or
180 ppm (equal to 0.12, 0.82, or 5.7 mg/kg bw/day for males and
0.13, 0.85, or 5.4 mg/kg bw/day for females). Adverse findings
occurred only at the high dose and were confined to the liver and
adrenal glands. Slightly elevated serum alkaline phosphatase and
reduced albumin were suggestive of mild liver injury in both sexes
at the high dose. Baseline cortisol blood levels were normal, but
cortisol response to ACTH challenge (20 units of ACTH; cortisol
measured 30 and 90 minutes after challenge) was markedly decreased
(about 50%) in high-dose males and females. Liver weight relative to
body-weight and brain weight was increased in males. Minimal to mild
hepatocellular hypertrophy was observed in 5/6 males and 5/6 females
receiving the high-dose compared to none in control or lower-dose
groups. No treatment-related microscopic changes in the adrenal
gland were found. The NOAEL was 30 ppm, equal to 0.82 mg/kg bw/day
based on histological and clinical chemistry indices of an effect on
the liver and reduced cortisol response to ACTH challenge at 180 ppm
(Tegeris & Shellenberger, 1988).
Groups each containing three dogs were given dicofol at 100,
300 or 900 ppm for one year. Survival was affected only at 900 ppm.
Body-weight gain was normal and haematological and histological
observations revealed no pathological effects (Smith et al.,
1959).
Rabbits
The formulation Kelthane MF (40.7% dicofol) was tested in
rabbits by the dermal route. Groups of 6 male and 6 female New
Zeeland white rabbits received dermal applications (6 h/day, 5
days/week) of the formulation at doses of 4.1, 10.2, or 61.1 mg
active ingredient/kg bw for 4 weeks. Control groups (6/sex) received
dermal applications of distilled water or the formulation vehicle
(concentration equal to the vehicle concentration of the high dose).
The vehicle and all dose levels of the test material caused dermal
irritation attributable to the formulation vehicle. Reduced body-
weight at the high- (males and females) and middle-doses (males) was
the only other sign of toxicity. Overall body-weight gain was
reduced 60-65% in high-dose males and females and reduced 56% in
middle-dose males compared to water controls. These groups also
showed consistently lower weight gain than vehicle controls. A NOAEL
of 4.1 mg/kg bw based on reduced weight gain at 10.2 mg/kg bw and
above (Bonin et al., 1986).
Long-term toxicity/carcinogenicity studies
Mice
Groups of 50 B6C3F1 mice/sex were administered technical
dicofol (90% pure, < 1% DDTr) in the diet daily for 78 weeks and
the basal diet for an additional 14 weeks. Purity of the test
material was initially reported as 40-60% but later analyses of the
test material (and a lot sample) indicated 87-93% purity (A.M.
Rothman, 1981. Personal communication). Male mice received time-
weighted average diet concentrations of 260 or 530 ppm and female
mice received time-weighted average concentrations of 120 or 240
ppm, equivalent to 40 or 80 mg/kg bw/day for males and 18 or 36
mg/kg bw/day for females. Groups of 20 male and 20 female control
mice received untreated diets for 91 weeks. At the end of the study,
survival rates were 35, 76, and 76% for males and 95, 84, and 96%
for females administered the control, low-dose, and high-dose,
respectively. Body-weights of treated males were comparable to that
of controls but weights of low- and high-dose females were lower
than controls from week 40 to the end of the study. Food consumption
data were not reported. No clinical signs or non-neoplastic lesions
were related to dicofol treatment. A dose-related increase in the
incidence of liver adenomas was observed in male mice. Based on a
re-read of the slides using updated diagnostic criteria, the
incidence of liver tumours for the control, low-dose, and high-dose
groups were 0, 27, and 49% for hepatocellular adenomas,
respectively; 11, 25, and 19% for hepatocellular carcinomas,
respectively; and 11, 52, and 68% for hepatocellular adenomas and
carcinomas combined, respectively (R.R. Maronpot, Personal
communication). The re-read resulted in reclassification of a large
number (about 50% at low-dose and 75% at high-dose) of carcinomas as
adenomas. The majority of tumours reported in the 1978 by NCI were
carcinomas (NCI, 1978).
Rats
Groups containing equal numbers of male and female rats were
fed 0, 2, 5, 10, 15 or 20 ppm dicofol in their diets for 55 weeks.
Growth, survival and liver to body weight ratios were not affected
at any dose level (Smith et al., 1959).
Dicofol was fed to 60 groups, each containing 10 male and 10
female rats, at dietary levels of 0, 20, 100, 250, 500 or 1000 ppm
dicofol for two years. Growth depression occurred in male rats at
500 and 1000 ppm, and in female rats progressively with increasing
dietary concentrations at 250, 500 and 1000 ppm. Growth depression
after three months, recorded in female rats at 20 ppm (but not at
100 ppm), was not observed at a later time. Absolute organ weights
showed no significant differences from the controls, with the
exception of an increase in the case of the livers and kidneys of
the female rats fed 1000 ppm. Organ to body weight ratios were
significantly increased for the liver at 250 ppm and for the liver,
kidney and heart at 500 ppm in females, but only for the liver at
500 ppm in males. Histopathological findings were confined to
hydropic changes in the liver which were regarded as reversible
(Larson, 1957).
Groups of 60 Crl-CD BR rats/sex received technical dicofol
(93.3% pure, < 0.1% DDTr) in the diet daily for 24 months at 0, 5,
50, or 250 ppm (equal to 0.22, 2.2 or 11 mg/kg bw/day for males and
0.27, 2.7 or 14 mg/kg bw/day for females). Additional groups
(10/sex/dose) were treated for 3, 12, and 18 months. Survival was
unaffected, and no clinical signs were related to treatment. Body-
weight was reduced 15-25% at 250 ppm in both sexes. Overall food
consumption was reduced 12% in females receiving 250 ppm. Hepatic
MFO activity, measured by aminopyrine N-demethylation after 3 and 12
months, was increased at 50 and 250 ppm. Blood levels of
corticosterone and thyroid hormones (T3, T4, TSH) were normal.
Relative liver weight was increased 19% at 50 and 250 ppm in males
and 35% at 250 ppm in females. Gross changes in the liver (i.e.
prominent lobular architecture, focal discoloration) were seen at 50
and 250 ppm. At the terminal sacrifice, the incidence and severity
of histopathological changes in the liver and adrenal gland were
increased at 50 and 250 ppm. Liver cell changes included minimal to
marked centrilobular hypertrophy, centrilobular and mid-zonal
vacuolation, and cellular alteration of the eosinophilic type. The
incidences of centrilobular hypertrophy were 0/58, 0/57, 35/60, and
52/58 in males and 0/59, 0/61, 42/60, and 56/59 in females at the 0,
5, 50, and 250 ppm dose levels, respectively. Eosinophilic cellular
alteration appeared to be increased in low-dose females at the 24-
month sacrifice; however, this was unaccompanied by hypertrophic
cells observed at the higher doses. Focal hepatocellular hyperplasia
was increased in high-dose females. Diffuse vacuolation of adrenal
cortical cells in the zona fasciculata and zona reticularis was
increased primarily at the 250 ppm dose level at the terminal
sacrifice and at 50 and 250 ppm at the 18-month sacrifice. At the
terminal sacrifice an increase in chronic cystitis of the urinary
bladder was noted in high-dose females. In the liver and adrenal
gland, microscopic changes were observed at all sacrifice times. No
treatment-related changes in the thyroid were observed at any time
point. No neoplastic lesions were associated with dicofol treatment.
The NOAEL was 5 ppm, equal to 0.22 mg/kg bw/day, based on
histopathological changes in the liver and adrenal gland at 50 and
250 ppm (Hazelton & Harris, 1989).
Groups of 50 Osborne-Mendel rats/sex were administered
technical dicofol (90% pure, < 1% DDTr) in the diet daily for 78
weeks then a basal diet during a 34-week observation period. Purity
of the test material was initially reported as 40-60% but later
analyses of the test material (and a lot sample) indicated 87-93%
purity (A.M. Rothman, Personal communication, 1981). Male rats
received time-weighted average diet concentrations of 470 or 940 ppm
and female rats received constant diet concentrations of 380 or 760
ppm equivalent to 24 or 47 mg/kg bw/day for males and 19 or 38 mg/kg
bw/day for females. Groups of 20 male and 20 female control rats
received untreated diets for 110 weeks. Survival rates at 100 weeks
were 55, 64, and 72% for males and 80, 92, and 88% for females
administered the control, low-dose, and high-dose, respectively.
Body-weights of low- and high-dose males and females were lower than
control weights throughout the treatment period. Food consumption
data were not reported. No treatment-related clinical signs were
observed. No neoplastic or nonneoplastic lesions were associated
with dicofol treatment (NCI, 1978).
Reproduction studies
Mice
Groups of varying numbers of mice were maintained throughout
five generations on dietary levels of 0, 7, 25, 100, 225 or 500 ppm
dicofol. At 500 ppm the litter sizes, average weight of the pups and
the fertility, viability and lactation indices were lower than for
the control group. However, all these parameters were normal at 225
ppm and below (Brown, 1967a).
Rats
Four groups each of 27 male and 27 female rats were fed dietary
levels of 0, 100, 500 or 1000 ppm dicofol in a two-generation
reproduction study. There were no Flb pups surviving at 21 days
when the original parents were fed 500 or 1000 ppm dicofol. Litter
size from the 1000 ppm group was similar to the control, but overall
mortality in the pups was greater. Considerable reduction in
fertility of the animals fed 500 and 1000 ppm dicofol was evident.
No congenital defects were observed in any of the F2a or F2b
animals (Brown, 1965).
Groups of rats were maintained on diets containing 25 or 75 ppm
dicofol through a three-generation study. The average number of pups
born per litter to parents receiving 75 ppm was slightly lower than
for the controls. There were no compound-related effects relative to
body weight, fertility, gestation, viability or lactation indices at
either level, nor were there any congenital abnormalities evident in
either the viable or the still-born pups (Brown, 1967b).
Dicofol technical (93.3% pure) was administered to Crl:CD BR
rats over two generations (one-two litter study) at 5, 25, 125, or
250 ppm in the diet equal to 0.5, 2.1, 10 or 21 mg/kg bw/day for
males and 0.5, 2.2, 11 or 18 mg/kg bw/day for females. The first
parental (P1) animals were treated for 10 weeks prior to mating,
during mating, during pregnancy, and through weaning of the F1
offspring. Selected F1 offspring (P2) were treated during
growth, mating, the production of two F2 litters (F2a, F2b),
and until the second F2 litter was weaned. During the pre-mating
period and gestation, P1 females receiving 125 or 250 ppm showed
reduced body-weight gain and food consumption. Treatment-related
histological changes were observed in the liver, ovaries, and
adrenal glands of P1 and P2 rats. The most prominent liver
change was minimal to moderately severe hypertrophy of centrilobular
hepatocytes accompanied by centrilobular to mid-zonal vacuolation in
P1 and P2 males and females. The response was more severe in
males than females. The incidence in P2 males was 0/25, 1/25,
14/25, 24/25, and 25/25 in 0, 5, 25, 125, and 250 ppm groups,
respectively. Focal eosinophilic cellular alteration was increased
in P2 male (6/25) and female (8/25) rats at 250 ppm and P2
females at 125 ppm (6/25) compared to controls (1/25 in males; 0/25
in females). At 250 ppm, there was an increase in bile duct
hyperplasia in P1 and P2 females. Vacuolation of the ovary was
increased at 250 ppm in P1 females and at 25 ppm and above in P2
females. The incidences in P2 females were 1/25, 1/25, 6/25, 5/25,
and 18/25 in 0, 5, 25, 125, and 250 ppm groups, respectively. The
change was characterized by an increase in the size and/or number of
vacuoles in the cytoplasm of ovarian stromal cells. The
morphological change was described as compatible with enhanced
steroidogenic activity. The incidence of hypertrophy and/or
vacuolation of the adrenal cortex was increased in P1 and P2
females receiving 125 ppm (P1, 7/25; P2, 8/25) and 250 ppm
(P1, 23/25; P2, 25/25) compared to controls (P1 and P2,
0/25). The change was characterized by diffuse enlargement and
increased amounts of finely vacuolated cytoplasm or prominent large
vacuoles in the cells of the inner cortex.
Reproductive performance of P1 and P2 rats was unaffected.
Offspring toxicity was observed in F1 and F2 pups at 125 and 250
ppm. Viability was reduced in F1 pups at 250 ppm and F2 pups at
125 and 250 ppm. Reduced survival was primarily due to deaths during
days 0-4 of lactation. At 250 ppm, growth of F1 and F2 pups was
reduced during lactation. The NOAEL based on reproductive parameters
was 25 ppm, equal to 2.1 mg/kg bw/day. The NOAEL for parental
toxicity was 5 ppm equal to 0.5 mg/kg bw/day, based on
histopathological changes in the liver and ovaries at 25 ppm and
above. The ovarian effect was considered compatible with enhanced
steroidogenic activity (Solomon & Kulwich, 1991).
Special studies on embryo/fetotoxicity
Rats
The teratogenicity of dicofol was studied in Crl:COBS CD (SD)BR
rats. Dicofol (95.6% pure) was administered on days 6-15 of
gestation by oral gavage to groups of 25 mated females rats at doses
of 0, 0.25, 2.5, or 25 mg/kg bw/day. Controls received corn oil.
Rats were sacrificed on day 20. During the treatment period, a
majority (21/25) of the high-dose group frequently exhibited
excessive salivation as did one-fifth (5/25) of the middle-dose
group (on one to three occasions). This clinical sign was not
observed in the dose range-finding study in which 8 rats/sex/dose
were given doses of 1, 5, 20, 60 or 180 mg/kg bw/day, except in one
animal (180 mg/kg bw/day group) on one day. Body-weight gain and
food consumption were reduced at the high-dose (25 mg/kg bw/day)
during the treatment period; a rebound increase was observed post-
treatment. Liver weight relative to body-weight was increased (7%)
at the high-dose. A histological change in the liver, consisting of
centrilobular hepatocyte hypertrophy (minimal to slight), was
observed in 17/25 of the high-dose group versus none in the control
or lower dose groups. Dicofol had no-observable-effect on the
offspring.
The NOAEL for maternal toxicity was 0.25 mg/kg bw/day based on
clinical signs of toxicity (salivation) at 2.5 mg/kg bw/day and
above. The NOAEL for embryo-fetal toxicity and teratogenicity was 25
mg/kg bw/day based on no-observable-effect on the offspring at the
highest dose tested (Hoberman & Christian, 1986b).
Rabbits
The teratogenicity of dicofol was studied in New Zeeland white
rabbits. Dicofol (95.6% pure) was administered on days 7-19 of
gestation by oral gavage to groups of 20 artificially inseminated
females at doses of 0, 0.4, 4, or 40 mg/kg bw/day. The control
received the aqueous methylcellulose vehicle. Rabbits were
sacrificed on day 29. Maternal toxicity was produced by the 4 and 40
mg/kg bw/day doses. The high-dose group experienced clinical signs
(abnormal faeces), weight loss, and reduced food consumption during
the treatment period. Although body-weight showed a rebound increase
after treatment, overall body-weight gain was depressed (42%).
Relative liver weight expressed to body-weight was increased (20%)
at the high dose. The incidence of eosinophilic, hyaline material in
centrilobular hepatocytes was increased at the 4 mg/kg bw/day (2/19)
and 40 mg/kg bw/day (8/20) dose levels compared to controls (0/20).
Diffuse vacuolation of hepatocytes was observed in 6/20 of the high-
dose group compared to 0/20 of controls. An increased incidence of
abortion was observed at the high dose (high dose, 4/19; control,
1/18). Dicofol treatment had no other effect on the developing
offspring.
The NOAEL for maternal toxicity was 0.4 mg/kg bw/day based on
histopathological changes in the liver at 4 mg/kg bw/day and above.
The NOAEL for teratogenicity was 40 mg/kg bw/day based on no
observable effect on the offspring at the highest dose tested
(Hoberman & Christian, 1986a). The incidence of abortion was
increased at the high-dose (4/19) compared to concurrent controls
(1/18) and historical controls (up to 1/14 to 2/15 with an outlier
of 1/4). The high incidence may be related to maternal toxicity, but
a direct developmental effect cannot be excluded. The NOAEL for
embryo-fetal toxicity was therefore 4 mg/kg bw/day based on the
increased incidence of abortion at 40 mg/kg bw/day.
Special studies on eye and skin irritation and hypersensitivity
Technical dicofol is reported to be irritating to the skin but
non-irritating to the eye (Baldwin & Hurt, 1985).
Technical dicofol produced delayed contact hypersensitivity in
guinea-pigs (Bonin & Hazelton, 1987).
Special studies on genotoxicity
Results of representative genotoxicity studies are shown in
Table 2. Dicofol has been overwhelmingly negative in assays for
point mutation, chromosomal aberration, unscheduled DNA synthesis,
and sister chromatid exchange. Occasional positive findings have not
been substantiated by other studies.
Observations in humans
In 1979, 78 incidents of Kelthane(R) exposure were reported
by the US Environmental Protection Agency Pesticide Incident
Monitoring System. Fourteen cases involved dicofol alone and 8 of
these reported symptoms. One case involved dicofol ingestion (amount
unspecified) leading to nausea, dizziness, and vomiting. Three cases
involved inhalation exposure resulting in dizziness, weakness, and
vomiting in two cases and sinus congestion in the third. Two cases
involved dermal exposure (amount unspecified) resulting in skin
irritation in one case and rash (allergic reaction) in the other
(USEPA, 1979).
In a case report, a 12-year-old male was accidentally exposed
to dicofol when he fell from a bicycle into a puddle of spilled
undiluted dicofol formulation (470 g/l; 50-gal. drum). The skin was
abraded and clothing contaminated. The patient had initial symptoms
of nausea, dizziness, disorientation, confusion, lethargy, and
headache. The patient demonstrated horizontal nystagmus and impaired
balance. These symptoms resolved within three weeks. Three weeks
after the incident, serum dicofol levels were 1.1 g/l and adipose
tissue levels were 0.153 g/kg (analytical methods unspecified). No
dicofol was detected in serum 16 weeks after the exposure. Following
persistent emotional difficulties, the patient underwent a
neuropsychological evaluation eight months after the exposure, which
showed impairment of certain cognitive functions including auditory
attention, immediate memory, and ability to selectively inhibit
inappropriate responses. A pre-exposure neuropsychological analysis
was unavailable for comparison (Lessenger & Riley, 1991).
Table 2. Results of genotoxicity assays on dicofol
Test system Test object Concentration of dicofol Purity Results Reference
Ames test (1) S. typhimurium 5-5000 µg/plate dissolved in 95.6% Negative (2) Higginbotham & Byers
TA98, TA100, TA1535, TA1537 DMSO (1985)
S. typhimurium 1-1000 µg/plate dissolved in 89.9% Negative Shirasu et al. (1980)
TA98, TA100, TA1535, TA1537, DMSO
TA1538
E. coli mutation assay (1) E. coli, WP2 hcr 1-5000 µg/plate dissolved in 89.9% Negative Shirasu et al. (1980)
DMSO
B. subtilis rec-assay B. subtilis, H17, M45 20-2000 µg/disk dissolved in 89.9% Negative Shirasu et al. (1980)
DMSO
CHO/HGPRT mutation assay Chinese hamster ovary cells 3-20 µg/ml dissolved in DMSO 95.6% Negative Foxall (1986)
(1) (CHO-K1-BH4)
Sex-linked recessive lethal D. melangaster 10 000 ppm, feeding and 34.8% Negative Woodruff et al. (1985)
mutation injection
Unscheduled DNA synthesis Male rat (F-344) primary culture 0.025-0.5 µg/ml in DMSO 95.6% Negative (3) Foxall & Byers (1986)
hepatocytes
In vitro sister chromatid Chinese hamster ovary cells 5-500 µg/ml ? Negative Galloway et al. (1987)
exchange (1) (CHO-W-Bl)
In vitro cytogenetics (1) Chinese hamster ovary cells 7.5-20 µg/ml dissolved in DMSO 95.6% Negative Ivett & Myhr (1986)
(CHO-WBL)
50-500 µg/ml ? Negative Galloway et al. (1987)
Chinese hamster ovary cells
(CHO-W-Bl)
In vivo cytogenetics Male CRL:COBS-CD(SD) rat, 47.8-478 mg/kg bw orally X 1 89.6% Negative (4) Sames & Doolittle
bone marrow (1986)
Table 2 (continued)
(1) Both with and without metabolic activation
(2) No positive control in nonactivated assay
(3) Unable to verify cytotoxicity
(4) No evidence presented (e.g., miototic index) to demonstrate test material reached the target tissue. A maximum tolerated dose may not
have been used.
COMMENTS
Dicofol was extensively absorbed from the gastrointestinal
tract. At near steady-state conditions, the highest tissue
concentrations were found in adipose tissue followed by the adrenal
glands, thyroid, and liver. The p,p'-dicofol isomer, the main
component of technical dicofol, was more persistent in the body than
the o,p'-isomer. Female rats tended to retain dicofol to a greater
extent than males. Dicofol and DDT showed a similar pattern of
distribution and elimination. Dicofol is more polar and therefore
less persistent in the body.
In rats, dicofol was excreted as polar metabolites, primarily
in the faeces, but with lesser amounts in the urine. Metabolism
involved dechlorination and oxidation of the ethanol moiety and
hydroxylation of the aromatic rings. In adipose tissue, the parent
compound was predominant. The metabolic profile was similar in mice.
Dicofol had moderate acute oral toxicity. It produces signs of
toxicity consistent with CNS depression. WHO has classified dicofol
as slightly hazardous (WHO, 1992).
In a 13-week study in mice using dietary concentrations of 0,
10, 125, 250, 500, or 1000 ppm in the diet, the NOAEL was 10 ppm,
equal to 2.1 mg/kg bw/day, based on reduced body-weight, liver
enlargement, and increased hepatic mixed function oxidase (MFO)
activity. In another 13-week study in mice using dietary
concentrations of 0, 250, 500, or 750 ppm, liver histopathology,
including centrilobular hypertrophy and eosinophilia of heptocytes,
was observed at all dose levels.
In a 13-week study in rats at dietary concentrations of 0, 1,
10, 100, 500, or 1500 ppm, the NOAEL was 1 ppm, equal to 0.07 mg/kg
bw/day. Although the incidence and severity of thyroid follicular
epithelial hypertrophy was increased in males at 10 ppm and above,
this thyroid effect was not found in a second 13-week study using
dietary concentrations of 0, 50, 200, 1000, or 3000 ppm.
In a 13-week study in dogs using dietary concentrations of 0,
10, 100, 300, or 1000 ppm in the diet, the NOAEL was 10 ppm, equal
to 0.29 mg/kg bw/day. At 100 ppm, equal to 3.3 mg/kg bw/day,
cortisol response to ACTH was reduced. A 1-year dog study used
dietary levels of 0, 5, 30, or 180 ppm was performed to better
define the NOAEL. The NOAEL was 30 ppm, equal to 0.82 mg/kg bw/day,
based on liver changes and reduced cortisol response to ACTH at 180
ppm, equal to 5.7 mg/kg bw/day.
In a 78-week carcinogenicity study in mice using time-weighted
average concentrations of 260 or 530 ppm for males and 120 or 240
ppm for females, dicofol produced an increased incidence of liver
adenomas and adenomas/carcinomas combined in male mice at 260 and
530 ppm, equivalent to 40 and 80 mg/kg bw/day. Dicofol was not
carcinogenic in female mice.
In a two-year study in rats using dietary concentrations of 0,
5, 50, or 250 ppm in the diet, the NOAEL was 5 ppm, equal to 0.22
mg/kg bw/day, based on histopathological changes in the liver and
vacuolation of adrenal cortical cells at 50 ppm, equal to 2.2 mg/kg
bw/day. No treatment-related changes in the thyroid or in the
incidence of neoplasia were observed. There was no evidence of
carcinogenicity in a 78-week carcinogenicity study in rats using
time-weighted average concentrations of 470 or 940 ppm (24 or 47
mg/kg bw/day) for males and 380 or 760 ppm (19 or 38 mg/kg bw/day)
for females. Dicofol was not carcinogenic in rats.
In a two-generation reproduction study in rats using dietary
concentrations of 5, 25, 125, or 250 ppm in the diet, the NOAEL was
5 ppm, equal to 0.5 mg/kg bw/day, based on an increased incidence of
ovarian stromal cell hypertrophy and hepatocellular changes at 25
ppm. Offspring viability was reduced at 125 and 250 ppm. The NOAEL
for reproductive parameters was 25 ppm, equal to 2.1 mg/kg bw/day.
In a teratology study in rats using gavage doses of 0, 0.25,
2.5, or 25 mg/kg bw/day, the NOAEL for maternal toxicity was 0.25
mg/kg bw/day based on clinical signs of toxicity at 2.5 mg/kg
bw/day. The NOAEL for embryofoetal toxicity was 25 mg/kg bw/day. In
a teratology study in rabbits using gavage doses of 0, 0.4, 4, or 40
mg/kg bw/day, the NOAEL for maternal toxicity was 0.4 mg/kg bw/day
based on histopathological changes in the liver at 4 mg/kg bw/day.
The NOAEL for embryofoetal toxicity was 4 mg/kg bw/day based on an
increased incidence of abortion at 40 mg/kg bw/day. Teratogenic
effects were not found in these studies.
After reviewing the available genotoxicity data, the Meeting
concluded that dicofol was not genotoxic.
The Meeting concluded, after consideration of the liver tumours
in male mice found in the long-term studies together with the
genotoxicity data, that dicofol did not present a carcinogenic
hazard for humans.
The previous ADI was revised. A new ADI was allocated, based
upon the NOAEL of 0.22 mg/kg bw/day in the long-term study in rats,
using a safety factor of 100.
TOXICOLOGICAL EVALUATION
Level causing no toxicological effect
Mouse: 10 ppm, equal to 2.1 mg/kg bw/day (13-week study)
Rat: 5 ppm, equal to 0.22 mg/kg bw/day in males (two-year
study) 0.25 mg/kg bw/day (teratogenicity study,
maternal toxicity)
Rabbit: 0.4 mg/kg bw/day (teratogenicity study, maternal
toxicity)
Dog: 30 ppm, equal to 0.82 mg/kg bw/day (one-year study).
Estimate of acceptable daily intake for humans
0-0.002 mg/kg bw
Studies which will provide information valuable in the continued
evaluation of the compound
Further observations in humans.
REFERENCES
Baldwin, R.C. & Hurt, S.S. (1989) Dicofol technical purified skin
and eye irritation data summarized according to EEC criteria.
Unpublished report No. 85R-004A from Rohm and Haas Company, Spring
House, PA, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Bonin, R. & Hazelton, G.A. (1987) Dicofol (Kelthane technical
miticide): Delayed contact hypersensitivity study in guinea pigs.
Unpublished report No. 87R-027 from Rohm and Haas Company, Spring
House, PA, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Bonin, R., Hazelton, G.A., & Kulwich, B.A. (1986) Dicofol (Kelthane
MF miticide): 4-Week dermal toxicity study in rabbits. Unpublished
report No. 86R-047 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Brown, J.R. (1965) Toxicologic studies on the effects of kelthane in
the diet of albino rats on reproduction. Department of Physiological
Hygiene, University of Toronto. Unpublished report submitted to WHO.
Brown, J.R. (1967a) Toxicologic studies on 2,2-bis-chlorophenyl-
2,2,2-trichloroethanol, kelthane. Brown Biological Laboratories Ltd.
Unpublished report submitted to WHO.
Brown, J.R. (1967b) Three-generation reproduction study on rats
receiving technical kelthane in their diet. Department of
Physiological Hygiene, University of Toronto. Unpublished report
submitted to WHO.
Brown, J.R. (1971) Effect of dietary Kelthane on mouse and rat
reproduction. In: Tahori, A.S. (Ed.), Fate of pesticides in
environment. Proceedings of the Second International IUPAC Congress
of Pesticide Chemistry. Vol. VI, pp. 531-548. Gordon and Breach
Science Publishers, London, New York, and Paris.
Brown, J.R., Hughes, H., & Viriyanondha, S. (1969) Storage,
distribution, and metabolism of 1,1-bis(4-chlorophenyl)-2,2,2-
trichloroethanol. Toxicol. Appl. Pharmacol., 15: 30-37.
Brown, M.A. & Casida, J.E. (1987) Metabolism of a dicofol impurity
alpha-chloro-DDT, but not dicofol or dechlorodicofol, to DDE in mice
and a liver microsomal system. Xenobiotica, 17: 1169-1174.
de Groot, A.P. (1974) Determination of the acute intraperitoneal
toxicity of Kelthane technical in rats. Unpublished report No. 74RC-
1096 Central Institute for Nutrition and Food Research. Submitted to
WHO by Rohm and Haas Company, Spring House, PA, USA.
Den Tonkelaar, E.M. & Van Esch, G.J. (1974) No-effect levels of
organochlorine pesticides based on induction of microsomal liver
enzymes in short-term toxicity experiments. Toxicology, 2: 371-
380.
DiDonato, L.J., Steigerwalt, R.B., & Longacre, S.L. (1987) o,p'-
Dicofol and p,p'-dicofol: Kinetic study in female rats. Unpublished
report No. 86R-173 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Fisher, J.R. & Hagan, J.V. (1987) Dicofol (Kelthane technical
miticide) acute inhalation toxicity study in rats. Unpublished
report No. 87R-001 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Flodstrom, S., Hemming, H., Warngard, L., & Ahlborg, U.G. (1990)
Promotion of altered hepatic foci development in rat liver,
cytochrome P450 enzyme induction and inhibition of cell-cell
communication by DDT and some structurally related organohalogen
pesticides. Carcinogenesis, 11: 1413-1417.
Foxall, S. (1986) Kelthane technical CHO/HGPRT gene mutation assay.
Unpublished report No. 86R-002 from Rohm and Haas Company, Spring
House, PA, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Foxall, S. & Byers, M.J. (1986) Dicofol (Kelthane technical
miticide): In vitro unscheduled DNA synthesis assay. Unpublished
report No. 85R-202 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Galloway, S.M., Armstrong, M.J., Reuben, C., Colman, S., Brown, B.,
& Cannon, C. (1987) Chromosome aberrations and sister chromatid
exchanges in Chinese hamster ovary cells: Evaluations of 108
chemicals. Environmental and Molecular Mutagenesis, 10: 1-175.
Goldman, P.R., Bernacki, H.J., & Quinn, D.L. (1986) Kelthane: Three-
month dietary toxicity study in rats. Unpublished report No. 85R-093
from Rohm and Haas Company, Spring House, PA, USA. Submitted to WHO
by Rohm and Haas Company, Spring House, PA, USA.
Goldman, P.R. & Harris, J.C. (1986) Dicofol (Kelthane technical
miticide): Three month dietary toxicity study in mice. Unpublished
report No. 85R-104 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Hazelton, G.A. & Harris, J.C. (1989) Dicofol (Kelthane technical
miticide): 24-Month dietary chronic/oncogenic study in rats.
Unpublished report No. 89R-190 from Rohm and Haas Company, Spring
House, PA, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Higginbotham, C.A. & Byers, M.J. (1985) Dicofol (Kelthane technical
miticide): Microbial mutagen assay. Unpublished report No. 85R-0042
from Rohm and Haas Company, Spring House, PA, USA. Submitted to WHO
by Rohm and Haas Company, Spring House, PA, USA.
Hoberman, A.M. & Christian, M.S. (1986a) A developmental toxicity
study of dicofol administered via stomach tube to New Zeeland white
rabbits. Unpublished report No. 018-009 (Rohm and Haas report No.
86RC-15) from Argus Research Laboratories, Inc., Horsham,
Pennsylvania, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Hoberman, A.M. & Christian, M.S. (1986b) A developmental toxicity
study of dicofol (Kelthane technical miticide) administered via
gavage to Crl:COBS CD (SD)BR presumed pregnant rats. Unpublished
report No. 018-010 (Rohm and Haas report No. 85RC-69) from Argus
Research Laboratories, Inc., Horsham, Pennsylvania, USA. Submitted
to WHO by Rohm and Haas Company, Spring House, PA, USA.
Ivett, J.L. & Myhr, B.C. (1986) Dicofol (Kelthane technical
miticide): In vitro cytogenetic assay in Chinese hamster ovary
(CHO) cells. Unpublished report No. 20990 (Rohm and Haas report No.
85RC-0068) from Litton Bionetics, Inc., Kensington, Maryland, USA.
Submitted to WHO by Rohm and Haas Company, Spring House, PA, USA.
Joy, R.M. (1976) Convulsive properties of chlorinated hydrocarbon
insecticides in the cat central nervous system. Toxicol. Appl.
Pharmacol., 35: 95-106.
Kaneshima, H., Okui, T., Hiroshi, O., & Yamaguchi, T. (1980) Studies
on the biological fate of Kelthane. I. Distribution and excretion of
3H-Kelthane in mice. Eisei Kagaku, 26: 193-195.
Krzywicki, K. & Bonin, R. (1985a) Acute definitive oral LD50 in
rats in Kelthane technical. Unpublished report No. 85R-034A/B from
Rohm and Haas Company, Spring House, PA, USA. Submitted to WHO by
Rohm and Haas Company, Spring House, PA, USA.
Krzywicki, K. & Bonin, R. (1985b) Acute definitive dermal LD50 in
rats/rabbits in Kelthane technical. Unpublished report No. 85R-
033A/B/C/D from Rohm and Haas Company, Spring House, PA, USA.
Submitted to WHO by Rohm and Haas Company, Spring House, PA, USA.
Lampe, K.R. & Baldwin, R.C. (1990) Dicofol (Kelthane MF-B miticide)
four week dermal toxicity study in rats. Unpublished report No. 89R-
085 from Rohm and Haas Company, Spring House, PA, USA. Submitted to
WHO by Rohm and Haas Company, Spring House, PA, USA.
Larson, P.S. (1957) Two-year study on the effect of adding kelthane
to the diet of rats. Medical College of Virginia. Unpublished report
submitted to WHO.
Lessenger, J.E. & Riley, N. (1991) Neurotoxicities and behavioral
changes in a 12-year-old male exposed to dicofol, an organochlorine
pesticide.
Maronpot, R.R. (1985) Personal Communication for the Head of
Experimental Pathology, National Toxicology Program, Research
Triangle Park, North Carolina, to J.A. Moore, EPA, Washington DC.
Submitted to WHO by Rohm and Haas Co., Philadelphia, Pennsylvania,
USA.
Narloch, B.A., Lawton, M.P., Moody, D.E., Hammock, B.D., & Shull,
L.R. (1987) The effects of dicofol on induction of hepatic
microsomal metabolism in rats. Pest. Biochem. Physiol., 28: 362-
370.
NCI (1978) Bioassay of dicofol for possible carcinogenicity.
National Cancer Institute carcinogenesis technical report series No.
90.
Nigg, H.N., Stamper, J.H., Deshmukh, S.N., & Queen, R.M. (1991)
4,4'-Dichlorobenzilic acid urinary excretion by dicofol pesticide
applicators. Chemosphere, 22: 365-373.
Onishi, M. (1989) Acute oral toxicity of DICOFOL (Kelthane) TECH.B
in mice. Unpublished report No. 89RC-1025 from Shin Nippon
Biomedical Laboratories, Ltd., Kagoshima 891-13, Japan. Submitted to
WHO by Rohm and Haas Company, Spring House, PA, USA.
Rothman, A.M. (1981) Personal Communication for the Head of
Experimental Pathology, National Toxicology Program, Research
Triangle Park, North Carolina to J.A. Moore, EPA, Washington DC.
Submitted to WHO by Rohm and Haas Co., Philadelphia, Pennsylvania,
USA.
Sames, J.L. & Doolittle, D.J. (1986) Dicofol (Kelthane technical
miticide): Kelthane in vivo cytogenetic study in rats. Unpublished
report No. 85R-215 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Sato, H., Toyoda, K., Furukawa, F., Hasegawa, R., Takahashi, M., &
Hayashi, Y. (1987) Subchronic oral toxicity test of dicofol (1,1-
bis(p-chlorophenyl)-2,2,2-trichloroethanol) as the basis for the
design of a long-term carcinogenicity study in B6C3F1 mice.
Bulletin of National Institute of Hygienic Sciences 105: 42-45.
Shellenberger, T.E. (1986) Dicofol (Kelthane miticide): Three-month
dietary toxicity study in dogs. Unpublished report No. 85014 (Rohm
and Haas report No. 86RC-23) from Tegeris Laboratories Inc., Laurel,
Maryland, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Shirasu, Y., Moriya, M., & Ohta, T. (1980) Microbial mutagenicity
test of Kelthane. Unpublished report (Rohm and Haas No. 80RC-1024)
from Institute of Environmental Toxicology, Japan. Submitted to WHO
by Rohm and Haas Company, Spring House, PA, USA.
Smith, R.B., Jr., Larson, P.S., Finnegan, J.K., Haag, H.B.,
Hennigar, G.R., & Cobey, F. (1959) Toxicologic studies on 2,2- bis-
(chlorophenyl)-2,2,2-trichloroethanol. Toxicol. Appl. Pharmacol.,
1: 119-134.
Solomon, H.M. & Kulwich, B.A. (1991) Dicofol: Two-generation study
in rats. Unpublished report No. 89R-028 from Rohm and Haas Company,
Spring House, PA, USA. Submitted to WHO by Rohm and Haas Company,
Spring House, PA, USA.
Steigerwalt, R.B., Deckert, F.W., & Longacre, S.L. (1984a)
Comparative disposition of a single po dose of 14C-Kelthane and
14C-DDT in rats. Unpublished report No. 79R-130 from Rohm and Haas
Company, Spring House, PA, USA. Submitted to WHO by Rohm and Haas
Company, Spring House, PA, USA.
Steigerwalt, R.B., Deckert, F.W., & Longacre, S.L. (1984b)
Comparative disposition of multiple doses of 14C-Kelthane and
14C-DDT in female rats. Unpublished report No. 80R-174 from Rohm
and Haas Company, Spring House, PA, USA. Submitted to WHO by Rohm
and Haas Company, Spring House, PA, USA.
Steigerwalt, R.B., Deckert, F.W., & Longacre, S.L. (1984c) Mouse
liver mixed function oxidase assay for Kelthane technical - three
day po induction study. Unpublished report No. 80R-018 from Rohm and
Haas Company, Spring House, PA, USA. Submitted to WHO by Rohm and
Haas Company, Spring House, PA, USA.
Steigerwalt, R.B., Udinsky, J.R., Deckert, F.W., & Longacre, S.L.
(1984d) Liver mixed function oxidase assays for Kelthane technical,
its active ingredients, and principal technical impurities in male
B6C3F1 mice after two week dietary exposure. Unpublished
report No. 79R-167 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Tegeris, A.S. & Shellenberger, T.E. (1988) Dicofol (Kelthane
technical miticide): One year dietary toxicity study in beagle dogs.
Unpublished report No. 86014 (Rohm and Haas report No. 87RC-027)
from Tegeris Laboratories Inc., Laurel, Maryland, USA. Submitted to
WHO by Rohm and Haas Company, Spring House, PA, USA.
Tillman, A.M. & Mazza, L.S. (1986) Part I: Absorption and excretion
of 14C-dicofol in male and female rats. Part II: A metabolism
study of 14C-dicofol in male and female rats. Unpublished report
No. 31L-86-02 from Rohm and Haas Company, Spring House, PA, USA.
Submitted to WHO by Rohm and Haas Company, Spring House, PA, USA.
USEPA (1979) (US Environmental Protection Agency) pesticide incident
monitoring system. Summary of reported pesticide incidents involving
Kelthane, Report No. 173. Office of Pesticide Programs.
Verschuuren, H.G., Kroes, R., & Den Tonkelaar, E.M. (1973) Toxicity
studies on tetrasul III. Short-term comparative studies in rats with
tetrasul and structurally related acaricides. Toxicology, 1: 113-
123.
WHO (1992). The WHO recommended classification of pesticides by
hazard and guidelines to classification 1992-1993 (WHO/PCS/92.14).
Available from the International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland.
Woodruff, R.C., Mason, J.M., Valencia, R., & Zimmering, S. (1985)
Chemical mutagenesis testing in Drosophila. V. Results of 53 coded
compounds tested for the National Toxicology Program. Environmental
Mutagenesis, 7: 677-702.
combined). Faeces contained lesser amounts of dicofol and OH-
DCBH/DCBA-glycine. In urine, the radiolabel was mostly DCBH-glycine
and OH-DCBP/DCBH (25-40% combined) in both sexes. Urine contained
smaller amounts of CBA-glycine, DCBA, OH-DCBH/CBA. About 20% of
faeces radio-label and 30-40% of urine radiolabel were unidentified.
In adipose tissue, most of the extracted radiolabel was present
as the parent compound (80-90%), with smaller amounts of DCBP and
FW-152 identified in both sexes. In the liver, most of the extracted
radiolabel was FW-152 (70-80%) in both sexes with lesser amounts of
DCBP, dicofol, and DCBH.
Small amounts of material in faeces co-chromatographed with
DDE. Additional analyses by HPLC determined that 0.27% of the
extracted radiolabel in faeces was actually DDE. Additional analyses
of fat and liver detected small amounts of DDE in one fat sample
(0.2% of extract, 0.34 ppm tissue concentration) and two liver
samples (0.25%-0.34% of extract, 0.018-0.29 ppm tissue
concentration). The 14C dosing solution was reported to contain
0.01% DDE (Tillman & Mazza, 1986).
DDE, DDT, and alpha-chloro-DDT impurities may account for the
small amounts of DDE in tissues. The latter impurity has been shown
to be converted to DDE in rat liver microsomes and mouse liver in
vivo (Brown & Casida, 1987).
Following a single i.p. dose of 376 mg/kg bw of technical
dicofol (84.8% pure) in male Wistar rats, the parent compound and a
metabolite, DCBP, were quantified in blood. 4,4-Dichlorobenzhydrol
(DCBH) was detected but not quantified. DCBP was also detected in
tissues following exposure for 40 days to 63 mg/kg bw/day. Small
amounts of DDE were detected (Brown et al., 1969, 1971).
Effects on enzymes and other biochemical parameters
Mice
The hepatic mixed-function oxidase (MFO)-inducing effect of
dicofol was studied. Groups of 4 male and 4 female CD-1 mice were
administered 3 daily oral doses of 1.4, 4.4, 14.9, 42.8, or 151
mg/kg bw of technical dicofol (87.6% purity). MFO activity in liver
microsomal cell fractions was determined by O-demethylation of p-
nitroanisole. Relative liver weight was increased at the high-dose
in both sexes and at 42.8 mg/kg bw in males. MFO activity was
increased 22%-43% in females at 14.9 mg/kg bw and above. MFO
activity was unaffected in males (Steigerwalt et al., 1984c).
The MFO-inducing effects of technical dicofol, dicofol isomers,
and technical dicofol impurities were compared in B6C3F1 male
mice. Groups of 4 mice were administered the test material in the
diet daily for two weeks. Technical dicofol (87.5% purity) was
administered at 0, 8, 25, 80, 250, or 800 ppm. The two highest doses
bracketed the doses producing liver tumours in male B6C3F1
mice. p,p'-Dicofol was administered at 0, 6, 20, 63, 195, or 625
ppm. Liver MFO activity was measured by the following enzyme assays:
p-nitroanisole O-demethylation, aminopyrine N-demethylation, and
aniline hydroxylation. Technical dicofol depressed body-weight at 80
ppm and above and increased liver weight at 250 ppm and above. p,p'-
Dicofol depressed body-weight and increased liver weight at 625 ppm.
Technical dicofol increased MFO activity at 250 and 800 ppm. p,p'-
Dicofol increased MFO activity at 63 ppm and above. A comparison of
dose-response curves indicated p,p'-dicofol was equal to or slightly
less potent than technical dicofol at comparable concentrations of
active ingredient. Administration of 37 ppm of o,p'-dicofol, ED-8
isomers, DDE isomers, and up to 195 ppm of DCBP isomers produced no
toxicity and had no effect on MFO activity indicating these
constituents of technical dicofol do not play a disproportionate
role in induction of MFO activity. The authors concluded that p,p'-
dicofol was responsible for a large majority but not all the
induction of liver MFO activity produced by technical dicofol
(Steigerwalt et al., 1984d).
Rats
MFO induction activity was studied using groups of 6 male
Sprague-Dawley rats given 4 daily intraperitoneal doses of pure
dicofol (98.8% pure; 81.4% p,p'-, 18.6% o,p'-), technical dicofol
(85% pure; 69.2% p,p', 15.8% o,p'; 15% impurities including DDTr),
pure DDT (99% pure; 81.4% p,p'-,18.6% o,p'-), phenobarbital, or -
naphthoflavone. Dose concentrations ranged from 1.5 to 59 mM (2.2 to
103 mg/kg bw). Liver MFO activity was measured by the following
enzyme assays: cytochrome C reductase, aminopyrine N-demethylase,
ethoxycoumarin O-deethylase, microsomal epoxide hydrolase, cytosolic
epoxide hydrolase, and glutathione-S-transferase. Technical and pure
dicofol and DDT induced MFO activity in a pattern consistent with
phenobarbital-type induction. At a concentration of 59 mM (87.4
mg/kg bw), pure dicofol increased microsomal protein 1.7-fold and
cytochrome P-450 activities 2- to 3-fold. Equimolar doses of
technical dicofol and pure dicofol produced comparable responses,
and dicofol was equal in potency to DDT of equivalent isomer
composition (Narloch et al., 1987).
MFO induction activity was assessed using groups of 6 male
Wistar rats administered dicofol (described as "pure") daily in the
diet for two weeks at 0, 2, 5, 10, 20, 50, or 200 ppm. MFO activity
in liver microsomal cell fractions was determined by aniline
hydroxylase, aminopyrine demethylase, and hexobarbital oxidase
activities. Dicofol at concentrations of 10 ppm and above increased
MFO activity. Aminopyrine demethylase activity showed the greatest
induction with activity increased 2- to 5.7-fold. p,p'-DDT increased
the activity of this enzyme 2.4- to 7.6-fold over the same dose
range. Dicofol ranked after heptachlor, DDT, chlorfenson, and
dieldrin in capacity for inducing MFO enzymes (Den Tonkelaar & Van
Esch, 1974).
Dicofol inhibited gap junctional intercellular communication in
two systems: Chinese hamster V79 metabolic cooperation assay and
scrape-loading/dye transfer assay in WB-F344 rat liver epithelial
cells. Dicofol (1000 ppm in the diet for 11 weeks) enhanced the
development of gamma-glutamyltranspeptidase-positive hepatic foci in
nitrosamine-initiated male Sprague-Dawley rats (Flodstrom et al.,
1990).
Dogs
The effect of dicofol on plasma 17-hydroxy-corticosteroids in
the dog was determined in two dogs which were fed 300 ppm or 900 ppm
dicofol over two separate periods of one to two months' duration.
The ability of the adrenal cortex to elaborate
17-hydroxy-corticosteroids in response to ACTH stimulation was
slightly reduced at the 300 ppm level and markedly reduced at the
900 ppm level. The results also showed that following this treatment
with dicofol, the ability of the adrenal gland to return to the
pre-treatment level of response to ACTH proceeded slowly and,
possibly, incompletely (Smith et al., 1959).
Toxicological studies
Acute toxicity studies
The acute toxicity of technical dicofol is summarized in Table
1. Common signs of toxicity include decreased spontaneous motor
activity, ataxia, passiveness, somnolence, prostration, and
occasionally tremors. In cats, dicofol given i.v. had no convulsive
activity but produced cardiovascular effects consisting of prolonged
arrhythmia and hypertension at sublethal doses and ventricular
fibrillation at a lethal dose.
Table 1. Acute toxicity of dicofol
LD50 LC50
Species Strain Sex Route (mg/kg bw) (mg/l) Reference
Mouse CRJ:CD-1 M oral 669 Onishi (1989)
(ICR) F 6751
Rat Charles River CD M oral 595 Krzywicki & Bonin
F 5871 (1985a)
? M oral 809 Smith et al. (1959);
F 6842 AnnexI: 11
Wistar M oral 14952,4 Brown et al. 1969
Charles River CD M
F dermal (24-hr >5000 Krzywicki & Bonin,
exp) >50001 (1985b)
Wistar M&F i.p. 11153-11502,4 deGroot (1974);
M Brown et al. (1969)
Crl:CDBR M inhalation >5 Fisher & Hagan,
F (4 hr exp) >51 (1987)
Rabbit ? M oral 18102 Smith et al. 1959;
AnnexI: 11
New Zeeland, F dermal (24-hr >25001 Krzywicki & Bonin
white exp) (1985b)
Cat ? M i.v. <203 Joy (1976)
Dog ? M&F oral >40002 Smith et al. (1959);
Annex I: 11
1 Purity of technical dicofol was 94-96%, <0.1% DDTr.
2 Purity of technical dicofol was 80-85%.
3 Purity of technical dicofol was unspecified.
4 The observation period was 7 days only.
Short-term toxicity studies
Mice
Groups of 10 CD-1 (ICR) mice/sex received technical dicofol
(95.6% pure; < 0.1% DDTr) in the diet daily for 13 weeks at 0, 10,
125, 250, 500, or 1000 ppm, (equal to 1.6, 18, 38, 84, and 180 mg/kg
bw/day for males and 2.1, 29, 56, 110, and 190 mg/kg bw/day for
females). At 125 ppm, final body-weight was reduced in females,
hepatic mixed function oxidase (MFO) activity was increased in both
sexes, and absolute and relative liver weight was increased in
females. Liver cell hypertrophy in both sexes, SGPT in females, and
kidney weight in females were increased at 250 ppm. Findings at 500
and 1000 ppm only included increased plasma proteins and lipids,
degenerative changes in the kidney of females, adrenal cortex
hypertrophy, and hepatocellular necrosis and vacuolation. The NOAEL
was 10 ppm, equal to 2.1 mg/kg bw/day based on reduced weight, liver
enlargement, and increased hepatic MFO activity at 125 ppm (Goldman
& Harris, 1986).
In a dose-range finding study for a carcinogenicity study,
groups of 10 male B6C3F1 mice received technical dicofol (>
95% pure) in the diet daily for 13 weeks at 0, 250, 500, or 750 ppm
(equivalent to 36, 71, or 107 mg/kg bw/day). At 500 and 750 ppm,
final body-weight, overall food consumption, and heart weight were
reduced. Liver histopathology was evident at all dose levels.
Hepatic changes were characterized by centrilobular hypertrophy,
eosinophilic and vitreous liver cells, and polynuclear cells. In the
high-dose group, entire liver lobules were vitreous in some cases. A
NOAEL was not identified in this study. Histological changes in the
liver were observed at all dose levels (Sato et al., 1987).
Rats
Dicofol was fed to groups, each containing 10 male and 10
female rats, for 90 days at dietary concentrations of 0, 20, 100,
500, 1250 or 2500 ppm. Survival was adversely affected at 1250 ppm
and above. Growth was inhibited at 100 ppm and higher in the females
but only at 1250 ppm in the males. Increased liver to body weight
ratios occurred in the survivors in both sexes. Liver lesions were
the most consistent histopathological finding, but were only of
scattered incidence at dose levels below 1250 ppm (Smith et al.,
1959).
Groups of 10 Crl-CD(SD) rats/sex received technical dicofol
(95.6% pure, < 0.1% DDTr) in the diet daily for 13 weeks at 0, 1,
10, 100, 500, or 1500 ppm. ( equal to 0.07, 0.64, 6.5, 32, or 96
mg/kg bw/day for males and 0.08, 0.78, 7.8, 36, or 110 mg/kg bw/day
for females). The highest dose of 1500 ppm produced mortality,
ataxia, and lethargy. At 500 ppm and above in both sexes, body-
weight and overall food consumption were reduced, liver weight was
increased, blood corticosterone levels were decreased, and the
incidence of adrenal cortex vacuolation was increased. At 100 ppm,
hepatic MFO activity and the incidence of liver hypertrophy were
increased. The incidence and severity of thyroid follicular cell
hypertrophy (minimal to marked) was increased in males at 10 ppm and
above and in females at 500 and 1500 ppm. The pathologist considered
the thyroid finding of uncertain significance because it is a
relatively non-specific change that has been associated with
environmental factors such as low temperature and stress. The NOAEL
was 1 ppm, equal to 0.07 mg/kg bw/day based on the increase in
thyroid follicular epithelial hypertrophy in males (Goldman et
al., 1986).
Groups of 10 Wistar rats/sex received technical dicofol (74%
pure) in the diet daily for 13 weeks at 0, 50, 200, 1000, or 3000
ppm (equivalent to 2.5, 10, 50, or 150 mg/kg bw/day). All animals
receiving the high-dose died within five weeks. All dose levels
adversely affected body-weight (final weight reduced 10-40%). Food
consumption was not measured. Absolute liver weight was increased in
high-dose males and in females receiving 200 ppm and higher.
Histopathological changes in the liver, described as SER whorls and
V101-cells, were observed in males and females at 200 and 1000 ppm.
V101-cells were described as enlarged hepatocytes with enlarged
nuclei, some hyperchromatic or with unbalanced chromatic
distribution. A basophilic granulation was usually seen in the
periphery of the enlarged cell with the remainder of the cytoplasm
containing fine granules and having eosinophilic character. An
additional observation in high-dose females was increased thyroid
weight. No microscopic changes in the thyroid were found. A NOAEL
was not identified in this study (Verschuuren et al., 1973).
Groups of 6 Crl:CD BR rats/sex received dermal applications (6
h/day, 5 days/week) of the formulation Kelthane MF-B (44.8% dicofol)
at doses of 1, 2.5, 4, or 40 mg active ingredient/kg bw/day for 4
weeks. Control groups received either dermal application of
distilled water or the formulation vehicle (vehicle dose of 53 mg/kg
bw). The vehicle and all dose levels caused skin irritation
attributable to the formulation vehicle. During the third week of
treatment, males receiving 40 mg/kg bw/day experienced a reduction
in absolute body-weight (10%) and body-weight gain (20%). Effects on
the liver were observed at the high-dose in both sexes. SGPT was
slightly elevated in high-dose males. Liver weight relative to body-
weight was increased for high-dose males and females. Minimal
hypertrophy of centrilobular hepatocytes was observed in 5/6 males
and 6/6 females receiving 40 mg/kg bw/day compared to none in
controls. The enlarged hepatocytes were characterized by
eosinophilic cytoplasm. High-dose males also showed increased
severity of multifocal inflammation of the liver. Single-cell
necrosis was observed in some foci. A systemic NOAEL of 4 mg/kg
bw/day was determined based on reduced body-weight and liver effects
at 40 mg/kg bw/day (Lampe & Baldwin, 1990).
Dogs
Groups of six beagle dogs/sex received technical dicofol (93.3%
pure; < 0.1% DDTr) in the diet daily for 13 weeks at 0, 10, 100,
300, or 1000 ppm equal to 0.29, 3.3, 9.9, or 26 mg/kg bw/day for
males and 0.31, 3.4, 9.8, and 27 mg/kg bw/day for females. Clinical
laboratory tests on blood, urinalysis, and physiological
measurements (i.e. electrocardiogram, heart rate, and body
temperature) were conducted prior to treatment, after 4 weeks, and
prior to study termination. The high-dose produced mortality in 5/6
males and 5/6 females. Both sexes receiving 300 and 1000 ppm
exhibited signs of toxicity such as laboured breathing, excessive
salivation, inactivity, incoordination, dehydration, and red-tinged
diarrhoea. Body-weight and food consumption were unaffected at 300
ppm and below. Clinical chemistry findings were consistent with
liver injury at 300 and 1000 ppm and there were effects on adrenal
gland function at 100 ppm and above. In both sexes receiving 1000
ppm, serum enzymes (SAP, SGPT) were increased and serum proteins
(albumin, total protein) were decreased. In 300 ppm females,
alkaline phosphatase was increased four-fold and albumin was
slightly decreased. Baseline cortisol blood levels were normal, but
cortisol response to ACTH challenge (20 units of ACTH; cortisol
measured 30 and 90 minutes after challenge) was markedly decreased
(50-75%) in both sexes at 100 ppm and above. Electrocardiograms
suggested treatment-related prolongation of the QT and PR intervals
in dogs receiving 300 or 1000 ppm. Liver weight was increased in
males at 300 ppm and in females at 1000 ppm. Microscopic changes
were notable only at the high-dose. Findings consisted of single
cell necrosis and mononuclear cell infiltrates in the liver of both
sexes, gastrointestinal haemorrhagic enteritis and congestion in
both sexes, and myocardial necrosis in one male. An additional
observation was oligospermatogenesis observed in three middle-dose
(300 ppm) males and five high-dose (1000 ppm) males. The NOAEL was
10 ppm, equal to 0.29 mg/kg bw/day, based on reduced cortisol
response to ACTH challenge at 100 ppm (Shellenberger, 1986).
Groups of six beagle dogs/sex received technical dicofol (93.3%
pure; < 0.1% DDTr) in the diet daily for 52 weeks at 0, 5, 30, or
180 ppm (equal to 0.12, 0.82, or 5.7 mg/kg bw/day for males and
0.13, 0.85, or 5.4 mg/kg bw/day for females). Adverse findings
occurred only at the high dose and were confined to the liver and
adrenal glands. Slightly elevated serum alkaline phosphatase and
reduced albumin were suggestive of mild liver injury in both sexes
at the high dose. Baseline cortisol blood levels were normal, but
cortisol response to ACTH challenge (20 units of ACTH; cortisol
measured 30 and 90 minutes after challenge) was markedly decreased
(about 50%) in high-dose males and females. Liver weight relative to
body-weight and brain weight was increased in males. Minimal to mild
hepatocellular hypertrophy was observed in 5/6 males and 5/6 females
receiving the high-dose compared to none in control or lower-dose
groups. No treatment-related microscopic changes in the adrenal
gland were found. The NOAEL was 30 ppm, equal to 0.82 mg/kg bw/day
based on histological and clinical chemistry indices of an effect on
the liver and reduced cortisol response to ACTH challenge at 180 ppm
(Tegeris & Shellenberger, 1988).
Groups each containing three dogs were given dicofol at 100,
300 or 900 ppm for one year. Survival was affected only at 900 ppm.
Body-weight gain was normal and haematological and histological
observations revealed no pathological effects (Smith et al.,
1959).
Rabbits
The formulation Kelthane MF (40.7% dicofol) was tested in
rabbits by the dermal route. Groups of 6 male and 6 female New
Zeeland white rabbits received dermal applications (6 h/day, 5
days/week) of the formulation at doses of 4.1, 10.2, or 61.1 mg
active ingredient/kg bw for 4 weeks. Control groups (6/sex) received
dermal applications of distilled water or the formulation vehicle
(concentration equal to the vehicle concentration of the high dose).
The vehicle and all dose levels of the test material caused dermal
irritation attributable to the formulation vehicle. Reduced body-
weight at the high- (males and females) and middle-doses (males) was
the only other sign of toxicity. Overall body-weight gain was
reduced 60-65% in high-dose males and females and reduced 56% in
middle-dose males compared to water controls. These groups also
showed consistently lower weight gain than vehicle controls. A NOAEL
of 4.1 mg/kg bw based on reduced weight gain at 10.2 mg/kg bw and
above (Bonin et al., 1986).
Long-term toxicity/carcinogenicity studies
Mice
Groups of 50 B6C3F1 mice/sex were administered technical
dicofol (90% pure, < 1% DDTr) in the diet daily for 78 weeks and
the basal diet for an additional 14 weeks. Purity of the test
material was initially reported as 40-60% but later analyses of the
test material (and a lot sample) indicated 87-93% purity (A.M.
Rothman, 1981. Personal communication). Male mice received time-
weighted average diet concentrations of 260 or 530 ppm and female
mice received time-weighted average concentrations of 120 or 240
ppm, equivalent to 40 or 80 mg/kg bw/day for males and 18 or 36
mg/kg bw/day for females. Groups of 20 male and 20 female control
mice received untreated diets for 91 weeks. At the end of the study,
survival rates were 35, 76, and 76% for males and 95, 84, and 96%
for females administered the control, low-dose, and high-dose,
respectively. Body-weights of treated males were comparable to that
of controls but weights of low- and high-dose females were lower
than controls from week 40 to the end of the study. Food consumption
data were not reported. No clinical signs or non-neoplastic lesions
were related to dicofol treatment. A dose-related increase in the
incidence of liver adenomas was observed in male mice. Based on a
re-read of the slides using updated diagnostic criteria, the
incidence of liver tumours for the control, low-dose, and high-dose
groups were 0, 27, and 49% for hepatocellular adenomas,
respectively; 11, 25, and 19% for hepatocellular carcinomas,
respectively; and 11, 52, and 68% for hepatocellular adenomas and
carcinomas combined, respectively (R.R. Maronpot, Personal
communication). The re-read resulted in reclassification of a large
number (about 50% at low-dose and 75% at high-dose) of carcinomas as
adenomas. The majority of tumours reported in the 1978 by NCI were
carcinomas (NCI, 1978).
Rats
Groups containing equal numbers of male and female rats were
fed 0, 2, 5, 10, 15 or 20 ppm dicofol in their diets for 55 weeks.
Growth, survival and liver to body weight ratios were not affected
at any dose level (Smith et al., 1959).
Dicofol was fed to 60 groups, each containing 10 male and 10
female rats, at dietary levels of 0, 20, 100, 250, 500 or 1000 ppm
dicofol for two years. Growth depression occurred in male rats at
500 and 1000 ppm, and in female rats progressively with increasing
dietary concentrations at 250, 500 and 1000 ppm. Growth depression
after three months, recorded in female rats at 20 ppm (but not at
100 ppm), was not observed at a later time. Absolute organ weights
showed no significant differences from the controls, with the
exception of an increase in the case of the livers and kidneys of
the female rats fed 1000 ppm. Organ to body weight ratios were
significantly increased for the liver at 250 ppm and for the liver,
kidney and heart at 500 ppm in females, but only for the liver at
500 ppm in males. Histopathological findings were confined to
hydropic changes in the liver which were regarded as reversible
(Larson, 1957).
Groups of 60 Crl-CD BR rats/sex received technical dicofol
(93.3% pure, < 0.1% DDTr) in the diet daily for 24 months at 0, 5,
50, or 250 ppm (equal to 0.22, 2.2 or 11 mg/kg bw/day for males and
0.27, 2.7 or 14 mg/kg bw/day for females). Additional groups
(10/sex/dose) were treated for 3, 12, and 18 months. Survival was
unaffected, and no clinical signs were related to treatment. Body-
weight was reduced 15-25% at 250 ppm in both sexes. Overall food
consumption was reduced 12% in females receiving 250 ppm. Hepatic
MFO activity, measured by aminopyrine N-demethylation after 3 and 12
months, was increased at 50 and 250 ppm. Blood levels of
corticosterone and thyroid hormones (T3, T4, TSH) were normal.
Relative liver weight was increased 19% at 50 and 250 ppm in males
and 35% at 250 ppm in females. Gross changes in the liver (i.e.
prominent lobular architecture, focal discoloration) were seen at 50
and 250 ppm. At the terminal sacrifice, the incidence and severity
of histopathological changes in the liver and adrenal gland were
increased at 50 and 250 ppm. Liver cell changes included minimal to
marked centrilobular hypertrophy, centrilobular and mid-zonal
vacuolation, and cellular alteration of the eosinophilic type. The
incidences of centrilobular hypertrophy were 0/58, 0/57, 35/60, and
52/58 in males and 0/59, 0/61, 42/60, and 56/59 in females at the 0,
5, 50, and 250 ppm dose levels, respectively. Eosinophilic cellular
alteration appeared to be increased in low-dose females at the 24-
month sacrifice; however, this was unaccompanied by hypertrophic
cells observed at the higher doses. Focal hepatocellular hyperplasia
was increased in high-dose females. Diffuse vacuolation of adrenal
cortical cells in the zona fasciculata and zona reticularis was
increased primarily at the 250 ppm dose level at the terminal
sacrifice and at 50 and 250 ppm at the 18-month sacrifice. At the
terminal sacrifice an increase in chronic cystitis of the urinary
bladder was noted in high-dose females. In the liver and adrenal
gland, microscopic changes were observed at all sacrifice times. No
treatment-related changes in the thyroid were observed at any time
point. No neoplastic lesions were associated with dicofol treatment.
The NOAEL was 5 ppm, equal to 0.22 mg/kg bw/day, based on
histopathological changes in the liver and adrenal gland at 50 and
250 ppm (Hazelton & Harris, 1989).
Groups of 50 Osborne-Mendel rats/sex were administered
technical dicofol (90% pure, < 1% DDTr) in the diet daily for 78
weeks then a basal diet during a 34-week observation period. Purity
of the test material was initially reported as 40-60% but later
analyses of the test material (and a lot sample) indicated 87-93%
purity (A.M. Rothman, Personal communication, 1981). Male rats
received time-weighted average diet concentrations of 470 or 940 ppm
and female rats received constant diet concentrations of 380 or 760
ppm equivalent to 24 or 47 mg/kg bw/day for males and 19 or 38 mg/kg
bw/day for females. Groups of 20 male and 20 female control rats
received untreated diets for 110 weeks. Survival rates at 100 weeks
were 55, 64, and 72% for males and 80, 92, and 88% for females
administered the control, low-dose, and high-dose, respectively.
Body-weights of low- and high-dose males and females were lower than
control weights throughout the treatment period. Food consumption
data were not reported. No treatment-related clinical signs were
observed. No neoplastic or nonneoplastic lesions were associated
with dicofol treatment (NCI, 1978).
Reproduction studies
Mice
Groups of varying numbers of mice were maintained throughout
five generations on dietary levels of 0, 7, 25, 100, 225 or 500 ppm
dicofol. At 500 ppm the litter sizes, average weight of the pups and
the fertility, viability and lactation indices were lower than for
the control group. However, all these parameters were normal at 225
ppm and below (Brown, 1967a).
Rats
Four groups each of 27 male and 27 female rats were fed dietary
levels of 0, 100, 500 or 1000 ppm dicofol in a two-generation
reproduction study. There were no Flb pups surviving at 21 days
when the original parents were fed 500 or 1000 ppm dicofol. Litter
size from the 1000 ppm group was similar to the control, but overall
mortality in the pups was greater. Considerable reduction in
fertility of the animals fed 500 and 1000 ppm dicofol was evident.
No congenital defects were observed in any of the F2a or F2b
animals (Brown, 1965).
Groups of rats were maintained on diets containing 25 or 75 ppm
dicofol through a three-generation study. The average number of pups
born per litter to parents receiving 75 ppm was slightly lower than
for the controls. There were no compound-related effects relative to
body weight, fertility, gestation, viability or lactation indices at
either level, nor were there any congenital abnormalities evident in
either the viable or the still-born pups (Brown, 1967b).
Dicofol technical (93.3% pure) was administered to Crl:CD BR
rats over two generations (one-two litter study) at 5, 25, 125, or
250 ppm in the diet equal to 0.5, 2.1, 10 or 21 mg/kg bw/day for
males and 0.5, 2.2, 11 or 18 mg/kg bw/day for females. The first
parental (P1) animals were treated for 10 weeks prior to mating,
during mating, during pregnancy, and through weaning of the F1
offspring. Selected F1 offspring (P2) were treated during
growth, mating, the production of two F2 litters (F2a, F2b),
and until the second F2 litter was weaned. During the pre-mating
period and gestation, P1 females receiving 125 or 250 ppm showed
reduced body-weight gain and food consumption. Treatment-related
histological changes were observed in the liver, ovaries, and
adrenal glands of P1 and P2 rats. The most prominent liver
change was minimal to moderately severe hypertrophy of centrilobular
hepatocytes accompanied by centrilobular to mid-zonal vacuolation in
P1 and P2 males and females. The response was more severe in
males than females. The incidence in P2 males was 0/25, 1/25,
14/25, 24/25, and 25/25 in 0, 5, 25, 125, and 250 ppm groups,
respectively. Focal eosinophilic cellular alteration was increased
in P2 male (6/25) and female (8/25) rats at 250 ppm and P2
females at 125 ppm (6/25) compared to controls (1/25 in males; 0/25
in females). At 250 ppm, there was an increase in bile duct
hyperplasia in P1 and P2 females. Vacuolation of the ovary was
increased at 250 ppm in P1 females and at 25 ppm and above in P2
females. The incidences in P2 females were 1/25, 1/25, 6/25, 5/25,
and 18/25 in 0, 5, 25, 125, and 250 ppm groups, respectively. The
change was characterized by an increase in the size and/or number of
vacuoles in the cytoplasm of ovarian stromal cells. The
morphological change was described as compatible with enhanced
steroidogenic activity. The incidence of hypertrophy and/or
vacuolation of the adrenal cortex was increased in P1 and P2
females receiving 125 ppm (P1, 7/25; P2, 8/25) and 250 ppm
(P1, 23/25; P2, 25/25) compared to controls (P1 and P2,
0/25). The change was characterized by diffuse enlargement and
increased amounts of finely vacuolated cytoplasm or prominent large
vacuoles in the cells of the inner cortex.
Reproductive performance of P1 and P2 rats was unaffected.
Offspring toxicity was observed in F1 and F2 pups at 125 and 250
ppm. Viability was reduced in F1 pups at 250 ppm and F2 pups at
125 and 250 ppm. Reduced survival was primarily due to deaths during
days 0-4 of lactation. At 250 ppm, growth of F1 and F2 pups was
reduced during lactation. The NOAEL based on reproductive parameters
was 25 ppm, equal to 2.1 mg/kg bw/day. The NOAEL for parental
toxicity was 5 ppm equal to 0.5 mg/kg bw/day, based on
histopathological changes in the liver and ovaries at 25 ppm and
above. The ovarian effect was considered compatible with enhanced
steroidogenic activity (Solomon & Kulwich, 1991).
Special studies on embryo/fetotoxicity
Rats
The teratogenicity of dicofol was studied in Crl:COBS CD (SD)BR
rats. Dicofol (95.6% pure) was administered on days 6-15 of
gestation by oral gavage to groups of 25 mated females rats at doses
of 0, 0.25, 2.5, or 25 mg/kg bw/day. Controls received corn oil.
Rats were sacrificed on day 20. During the treatment period, a
majority (21/25) of the high-dose group frequently exhibited
excessive salivation as did one-fifth (5/25) of the middle-dose
group (on one to three occasions). This clinical sign was not
observed in the dose range-finding study in which 8 rats/sex/dose
were given doses of 1, 5, 20, 60 or 180 mg/kg bw/day, except in one
animal (180 mg/kg bw/day group) on one day. Body-weight gain and
food consumption were reduced at the high-dose (25 mg/kg bw/day)
during the treatment period; a rebound increase was observed post-
treatment. Liver weight relative to body-weight was increased (7%)
at the high-dose. A histological change in the liver, consisting of
centrilobular hepatocyte hypertrophy (minimal to slight), was
observed in 17/25 of the high-dose group versus none in the control
or lower dose groups. Dicofol had no-observable-effect on the
offspring.
The NOAEL for maternal toxicity was 0.25 mg/kg bw/day based on
clinical signs of toxicity (salivation) at 2.5 mg/kg bw/day and
above. The NOAEL for embryo-fetal toxicity and teratogenicity was 25
mg/kg bw/day based on no-observable-effect on the offspring at the
highest dose tested (Hoberman & Christian, 1986b).
Rabbits
The teratogenicity of dicofol was studied in New Zeeland white
rabbits. Dicofol (95.6% pure) was administered on days 7-19 of
gestation by oral gavage to groups of 20 artificially inseminated
females at doses of 0, 0.4, 4, or 40 mg/kg bw/day. The control
received the aqueous methylcellulose vehicle. Rabbits were
sacrificed on day 29. Maternal toxicity was produced by the 4 and 40
mg/kg bw/day doses. The high-dose group experienced clinical signs
(abnormal faeces), weight loss, and reduced food consumption during
the treatment period. Although body-weight showed a rebound increase
after treatment, overall body-weight gain was depressed (42%).
Relative liver weight expressed to body-weight was increased (20%)
at the high dose. The incidence of eosinophilic, hyaline material in
centrilobular hepatocytes was increased at the 4 mg/kg bw/day (2/19)
and 40 mg/kg bw/day (8/20) dose levels compared to controls (0/20).
Diffuse vacuolation of hepatocytes was observed in 6/20 of the high-
dose group compared to 0/20 of controls. An increased incidence of
abortion was observed at the high dose (high dose, 4/19; control,
1/18). Dicofol treatment had no other effect on the developing
offspring.
The NOAEL for maternal toxicity was 0.4 mg/kg bw/day based on
histopathological changes in the liver at 4 mg/kg bw/day and above.
The NOAEL for teratogenicity was 40 mg/kg bw/day based on no
observable effect on the offspring at the highest dose tested
(Hoberman & Christian, 1986a). The incidence of abortion was
increased at the high-dose (4/19) compared to concurrent controls
(1/18) and historical controls (up to 1/14 to 2/15 with an outlier
of 1/4). The high incidence may be related to maternal toxicity, but
a direct developmental effect cannot be excluded. The NOAEL for
embryo-fetal toxicity was therefore 4 mg/kg bw/day based on the
increased incidence of abortion at 40 mg/kg bw/day.
Special studies on eye and skin irritation and hypersensitivity
Technical dicofol is reported to be irritating to the skin but
non-irritating to the eye (Baldwin & Hurt, 1985).
Technical dicofol produced delayed contact hypersensitivity in
guinea-pigs (Bonin & Hazelton, 1987).
Special studies on genotoxicity
Results of representative genotoxicity studies are shown in
Table 2. Dicofol has been overwhelmingly negative in assays for
point mutation, chromosomal aberration, unscheduled DNA synthesis,
and sister chromatid exchange. Occasional positive findings have not
been substantiated by other studies.
Observations in humans
In 1979, 78 incidents of Kelthane(R) exposure were reported
by the US Environmental Protection Agency Pesticide Incident
Monitoring System. Fourteen cases involved dicofol alone and 8 of
these reported symptoms. One case involved dicofol ingestion (amount
unspecified) leading to nausea, dizziness, and vomiting. Three cases
involved inhalation exposure resulting in dizziness, weakness, and
vomiting in two cases and sinus congestion in the third. Two cases
involved dermal exposure (amount unspecified) resulting in skin
irritation in one case and rash (allergic reaction) in the other
(USEPA, 1979).
In a case report, a 12-year-old male was accidentally exposed
to dicofol when he fell from a bicycle into a puddle of spilled
undiluted dicofol formulation (470 g/l; 50-gal. drum). The skin was
abraded and clothing contaminated. The patient had initial symptoms
of nausea, dizziness, disorientation, confusion, lethargy, and
headache. The patient demonstrated horizontal nystagmus and impaired
balance. These symptoms resolved within three weeks. Three weeks
after the incident, serum dicofol levels were 1.1 g/l and adipose
tissue levels were 0.153 g/kg (analytical methods unspecified). No
dicofol was detected in serum 16 weeks after the exposure. Following
persistent emotional difficulties, the patient underwent a
neuropsychological evaluation eight months after the exposure, which
showed impairment of certain cognitive functions including auditory
attention, immediate memory, and ability to selectively inhibit
inappropriate responses. A pre-exposure neuropsychological analysis
was unavailable for comparison (Lessenger & Riley, 1991).
Table 2. Results of genotoxicity assays on dicofol
Test system Test object Concentration of dicofol Purity Results Reference
Ames test (1) S. typhimurium 5-5000 µg/plate dissolved in 95.6% Negative (2) Higginbotham & Byers
TA98, TA100, TA1535, TA1537 DMSO (1985)
S. typhimurium 1-1000 µg/plate dissolved in 89.9% Negative Shirasu et al. (1980)
TA98, TA100, TA1535, TA1537, DMSO
TA1538
E. coli mutation assay (1) E. coli, WP2 hcr 1-5000 µg/plate dissolved in 89.9% Negative Shirasu et al. (1980)
DMSO
B. subtilis rec-assay B. subtilis, H17, M45 20-2000 µg/disk dissolved in 89.9% Negative Shirasu et al. (1980)
DMSO
CHO/HGPRT mutation assay Chinese hamster ovary cells 3-20 µg/ml dissolved in DMSO 95.6% Negative Foxall (1986)
(1) (CHO-K1-BH4)
Sex-linked recessive lethal D. melangaster 10 000 ppm, feeding and 34.8% Negative Woodruff et al. (1985)
mutation injection
Unscheduled DNA synthesis Male rat (F-344) primary culture 0.025-0.5 µg/ml in DMSO 95.6% Negative (3) Foxall & Byers (1986)
hepatocytes
In vitro sister chromatid Chinese hamster ovary cells 5-500 µg/ml ? Negative Galloway et al. (1987)
exchange (1) (CHO-W-Bl)
In vitro cytogenetics (1) Chinese hamster ovary cells 7.5-20 µg/ml dissolved in DMSO 95.6% Negative Ivett & Myhr (1986)
(CHO-WBL)
50-500 µg/ml ? Negative Galloway et al. (1987)
Chinese hamster ovary cells
(CHO-W-Bl)
In vivo cytogenetics Male CRL:COBS-CD(SD) rat, 47.8-478 mg/kg bw orally X 1 89.6% Negative (4) Sames & Doolittle
bone marrow (1986)
Table 2 (continued)
(1) Both with and without metabolic activation
(2) No positive control in nonactivated assay
(3) Unable to verify cytotoxicity
(4) No evidence presented (e.g., miototic index) to demonstrate test material reached the target tissue. A maximum tolerated dose may not
have been used.
COMMENTS
Dicofol was extensively absorbed from the gastrointestinal
tract. At near steady-state conditions, the highest tissue
concentrations were found in adipose tissue followed by the adrenal
glands, thyroid, and liver. The p,p'-dicofol isomer, the main
component of technical dicofol, was more persistent in the body than
the o,p'-isomer. Female rats tended to retain dicofol to a greater
extent than males. Dicofol and DDT showed a similar pattern of
distribution and elimination. Dicofol is more polar and therefore
less persistent in the body.
In rats, dicofol was excreted as polar metabolites, primarily
in the faeces, but with lesser amounts in the urine. Metabolism
involved dechlorination and oxidation of the ethanol moiety and
hydroxylation of the aromatic rings. In adipose tissue, the parent
compound was predominant. The metabolic profile was similar in mice.
Dicofol had moderate acute oral toxicity. It produces signs of
toxicity consistent with CNS depression. WHO has classified dicofol
as slightly hazardous (WHO, 1992).
In a 13-week study in mice using dietary concentrations of 0,
10, 125, 250, 500, or 1000 ppm in the diet, the NOAEL was 10 ppm,
equal to 2.1 mg/kg bw/day, based on reduced body-weight, liver
enlargement, and increased hepatic mixed function oxidase (MFO)
activity. In another 13-week study in mice using dietary
concentrations of 0, 250, 500, or 750 ppm, liver histopathology,
including centrilobular hypertrophy and eosinophilia of heptocytes,
was observed at all dose levels.
In a 13-week study in rats at dietary concentrations of 0, 1,
10, 100, 500, or 1500 ppm, the NOAEL was 1 ppm, equal to 0.07 mg/kg
bw/day. Although the incidence and severity of thyroid follicular
epithelial hypertrophy was increased in males at 10 ppm and above,
this thyroid effect was not found in a second 13-week study using
dietary concentrations of 0, 50, 200, 1000, or 3000 ppm.
In a 13-week study in dogs using dietary concentrations of 0,
10, 100, 300, or 1000 ppm in the diet, the NOAEL was 10 ppm, equal
to 0.29 mg/kg bw/day. At 100 ppm, equal to 3.3 mg/kg bw/day,
cortisol response to ACTH was reduced. A 1-year dog study used
dietary levels of 0, 5, 30, or 180 ppm was performed to better
define the NOAEL. The NOAEL was 30 ppm, equal to 0.82 mg/kg bw/day,
based on liver changes and reduced cortisol response to ACTH at 180
ppm, equal to 5.7 mg/kg bw/day.
In a 78-week carcinogenicity study in mice using time-weighted
average concentrations of 260 or 530 ppm for males and 120 or 240
ppm for females, dicofol produced an increased incidence of liver
adenomas and adenomas/carcinomas combined in male mice at 260 and
530 ppm, equivalent to 40 and 80 mg/kg bw/day. Dicofol was not
carcinogenic in female mice.
In a two-year study in rats using dietary concentrations of 0,
5, 50, or 250 ppm in the diet, the NOAEL was 5 ppm, equal to 0.22
mg/kg bw/day, based on histopathological changes in the liver and
vacuolation of adrenal cortical cells at 50 ppm, equal to 2.2 mg/kg
bw/day. No treatment-related changes in the thyroid or in the
incidence of neoplasia were observed. There was no evidence of
carcinogenicity in a 78-week carcinogenicity study in rats using
time-weighted average concentrations of 470 or 940 ppm (24 or 47
mg/kg bw/day) for males and 380 or 760 ppm (19 or 38 mg/kg bw/day)
for females. Dicofol was not carcinogenic in rats.
In a two-generation reproduction study in rats using dietary
concentrations of 5, 25, 125, or 250 ppm in the diet, the NOAEL was
5 ppm, equal to 0.5 mg/kg bw/day, based on an increased incidence of
ovarian stromal cell hypertrophy and hepatocellular changes at 25
ppm. Offspring viability was reduced at 125 and 250 ppm. The NOAEL
for reproductive parameters was 25 ppm, equal to 2.1 mg/kg bw/day.
In a teratology study in rats using gavage doses of 0, 0.25,
2.5, or 25 mg/kg bw/day, the NOAEL for maternal toxicity was 0.25
mg/kg bw/day based on clinical signs of toxicity at 2.5 mg/kg
bw/day. The NOAEL for embryofoetal toxicity was 25 mg/kg bw/day. In
a teratology study in rabbits using gavage doses of 0, 0.4, 4, or 40
mg/kg bw/day, the NOAEL for maternal toxicity was 0.4 mg/kg bw/day
based on histopathological changes in the liver at 4 mg/kg bw/day.
The NOAEL for embryofoetal toxicity was 4 mg/kg bw/day based on an
increased incidence of abortion at 40 mg/kg bw/day. Teratogenic
effects were not found in these studies.
After reviewing the available genotoxicity data, the Meeting
concluded that dicofol was not genotoxic.
The Meeting concluded, after consideration of the liver tumours
in male mice found in the long-term studies together with the
genotoxicity data, that dicofol did not present a carcinogenic
hazard for humans.
The previous ADI was revised. A new ADI was allocated, based
upon the NOAEL of 0.22 mg/kg bw/day in the long-term study in rats,
using a safety factor of 100.
TOXICOLOGICAL EVALUATION
Level causing no toxicological effect
Mouse: 10 ppm, equal to 2.1 mg/kg bw/day (13-week study)
Rat: 5 ppm, equal to 0.22 mg/kg bw/day in males (two-year
study) 0.25 mg/kg bw/day (teratogenicity study,
maternal toxicity)
Rabbit: 0.4 mg/kg bw/day (teratogenicity study, maternal
toxicity)
Dog: 30 ppm, equal to 0.82 mg/kg bw/day (one-year study).
Estimate of acceptable daily intake for humans
0-0.002 mg/kg bw
Studies which will provide information valuable in the continued
evaluation of the compound
Further observations in humans.
REFERENCES
Baldwin, R.C. & Hurt, S.S. (1989) Dicofol technical purified skin
and eye irritation data summarized according to EEC criteria.
Unpublished report No. 85R-004A from Rohm and Haas Company, Spring
House, PA, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Bonin, R. & Hazelton, G.A. (1987) Dicofol (Kelthane technical
miticide): Delayed contact hypersensitivity study in guinea pigs.
Unpublished report No. 87R-027 from Rohm and Haas Company, Spring
House, PA, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Bonin, R., Hazelton, G.A., & Kulwich, B.A. (1986) Dicofol (Kelthane
MF miticide): 4-Week dermal toxicity study in rabbits. Unpublished
report No. 86R-047 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Brown, J.R. (1965) Toxicologic studies on the effects of kelthane in
the diet of albino rats on reproduction. Department of Physiological
Hygiene, University of Toronto. Unpublished report submitted to WHO.
Brown, J.R. (1967a) Toxicologic studies on 2,2-bis-chlorophenyl-
2,2,2-trichloroethanol, kelthane. Brown Biological Laboratories Ltd.
Unpublished report submitted to WHO.
Brown, J.R. (1967b) Three-generation reproduction study on rats
receiving technical kelthane in their diet. Department of
Physiological Hygiene, University of Toronto. Unpublished report
submitted to WHO.
Brown, J.R. (1971) Effect of dietary Kelthane on mouse and rat
reproduction. In: Tahori, A.S. (Ed.), Fate of pesticides in
environment. Proceedings of the Second International IUPAC Congress
of Pesticide Chemistry. Vol. VI, pp. 531-548. Gordon and Breach
Science Publishers, London, New York, and Paris.
Brown, J.R., Hughes, H., & Viriyanondha, S. (1969) Storage,
distribution, and metabolism of 1,1-bis(4-chlorophenyl)-2,2,2-
trichloroethanol. Toxicol. Appl. Pharmacol., 15: 30-37.
Brown, M.A. & Casida, J.E. (1987) Metabolism of a dicofol impurity
alpha-chloro-DDT, but not dicofol or dechlorodicofol, to DDE in mice
and a liver microsomal system. Xenobiotica, 17: 1169-1174.
de Groot, A.P. (1974) Determination of the acute intraperitoneal
toxicity of Kelthane technical in rats. Unpublished report No. 74RC-
1096 Central Institute for Nutrition and Food Research. Submitted to
WHO by Rohm and Haas Company, Spring House, PA, USA.
Den Tonkelaar, E.M. & Van Esch, G.J. (1974) No-effect levels of
organochlorine pesticides based on induction of microsomal liver
enzymes in short-term toxicity experiments. Toxicology, 2: 371-
380.
DiDonato, L.J., Steigerwalt, R.B., & Longacre, S.L. (1987) o,p'-
Dicofol and p,p'-dicofol: Kinetic study in female rats. Unpublished
report No. 86R-173 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Fisher, J.R. & Hagan, J.V. (1987) Dicofol (Kelthane technical
miticide) acute inhalation toxicity study in rats. Unpublished
report No. 87R-001 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Flodstrom, S., Hemming, H., Warngard, L., & Ahlborg, U.G. (1990)
Promotion of altered hepatic foci development in rat liver,
cytochrome P450 enzyme induction and inhibition of cell-cell
communication by DDT and some structurally related organohalogen
pesticides. Carcinogenesis, 11: 1413-1417.
Foxall, S. (1986) Kelthane technical CHO/HGPRT gene mutation assay.
Unpublished report No. 86R-002 from Rohm and Haas Company, Spring
House, PA, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Foxall, S. & Byers, M.J. (1986) Dicofol (Kelthane technical
miticide): In vitro unscheduled DNA synthesis assay. Unpublished
report No. 85R-202 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Galloway, S.M., Armstrong, M.J., Reuben, C., Colman, S., Brown, B.,
& Cannon, C. (1987) Chromosome aberrations and sister chromatid
exchanges in Chinese hamster ovary cells: Evaluations of 108
chemicals. Environmental and Molecular Mutagenesis, 10: 1-175.
Goldman, P.R., Bernacki, H.J., & Quinn, D.L. (1986) Kelthane: Three-
month dietary toxicity study in rats. Unpublished report No. 85R-093
from Rohm and Haas Company, Spring House, PA, USA. Submitted to WHO
by Rohm and Haas Company, Spring House, PA, USA.
Goldman, P.R. & Harris, J.C. (1986) Dicofol (Kelthane technical
miticide): Three month dietary toxicity study in mice. Unpublished
report No. 85R-104 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Hazelton, G.A. & Harris, J.C. (1989) Dicofol (Kelthane technical
miticide): 24-Month dietary chronic/oncogenic study in rats.
Unpublished report No. 89R-190 from Rohm and Haas Company, Spring
House, PA, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Higginbotham, C.A. & Byers, M.J. (1985) Dicofol (Kelthane technical
miticide): Microbial mutagen assay. Unpublished report No. 85R-0042
from Rohm and Haas Company, Spring House, PA, USA. Submitted to WHO
by Rohm and Haas Company, Spring House, PA, USA.
Hoberman, A.M. & Christian, M.S. (1986a) A developmental toxicity
study of dicofol administered via stomach tube to New Zeeland white
rabbits. Unpublished report No. 018-009 (Rohm and Haas report No.
86RC-15) from Argus Research Laboratories, Inc., Horsham,
Pennsylvania, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Hoberman, A.M. & Christian, M.S. (1986b) A developmental toxicity
study of dicofol (Kelthane technical miticide) administered via
gavage to Crl:COBS CD (SD)BR presumed pregnant rats. Unpublished
report No. 018-010 (Rohm and Haas report No. 85RC-69) from Argus
Research Laboratories, Inc., Horsham, Pennsylvania, USA. Submitted
to WHO by Rohm and Haas Company, Spring House, PA, USA.
Ivett, J.L. & Myhr, B.C. (1986) Dicofol (Kelthane technical
miticide): In vitro cytogenetic assay in Chinese hamster ovary
(CHO) cells. Unpublished report No. 20990 (Rohm and Haas report No.
85RC-0068) from Litton Bionetics, Inc., Kensington, Maryland, USA.
Submitted to WHO by Rohm and Haas Company, Spring House, PA, USA.
Joy, R.M. (1976) Convulsive properties of chlorinated hydrocarbon
insecticides in the cat central nervous system. Toxicol. Appl.
Pharmacol., 35: 95-106.
Kaneshima, H., Okui, T., Hiroshi, O., & Yamaguchi, T. (1980) Studies
on the biological fate of Kelthane. I. Distribution and excretion of
3H-Kelthane in mice. Eisei Kagaku, 26: 193-195.
Krzywicki, K. & Bonin, R. (1985a) Acute definitive oral LD50 in
rats in Kelthane technical. Unpublished report No. 85R-034A/B from
Rohm and Haas Company, Spring House, PA, USA. Submitted to WHO by
Rohm and Haas Company, Spring House, PA, USA.
Krzywicki, K. & Bonin, R. (1985b) Acute definitive dermal LD50 in
rats/rabbits in Kelthane technical. Unpublished report No. 85R-
033A/B/C/D from Rohm and Haas Company, Spring House, PA, USA.
Submitted to WHO by Rohm and Haas Company, Spring House, PA, USA.
Lampe, K.R. & Baldwin, R.C. (1990) Dicofol (Kelthane MF-B miticide)
four week dermal toxicity study in rats. Unpublished report No. 89R-
085 from Rohm and Haas Company, Spring House, PA, USA. Submitted to
WHO by Rohm and Haas Company, Spring House, PA, USA.
Larson, P.S. (1957) Two-year study on the effect of adding kelthane
to the diet of rats. Medical College of Virginia. Unpublished report
submitted to WHO.
Lessenger, J.E. & Riley, N. (1991) Neurotoxicities and behavioral
changes in a 12-year-old male exposed to dicofol, an organochlorine
pesticide.
Maronpot, R.R. (1985) Personal Communication for the Head of
Experimental Pathology, National Toxicology Program, Research
Triangle Park, North Carolina, to J.A. Moore, EPA, Washington DC.
Submitted to WHO by Rohm and Haas Co., Philadelphia, Pennsylvania,
USA.
Narloch, B.A., Lawton, M.P., Moody, D.E., Hammock, B.D., & Shull,
L.R. (1987) The effects of dicofol on induction of hepatic
microsomal metabolism in rats. Pest. Biochem. Physiol., 28: 362-
370.
NCI (1978) Bioassay of dicofol for possible carcinogenicity.
National Cancer Institute carcinogenesis technical report series No.
90.
Nigg, H.N., Stamper, J.H., Deshmukh, S.N., & Queen, R.M. (1991)
4,4'-Dichlorobenzilic acid urinary excretion by dicofol pesticide
applicators. Chemosphere, 22: 365-373.
Onishi, M. (1989) Acute oral toxicity of DICOFOL (Kelthane) TECH.B
in mice. Unpublished report No. 89RC-1025 from Shin Nippon
Biomedical Laboratories, Ltd., Kagoshima 891-13, Japan. Submitted to
WHO by Rohm and Haas Company, Spring House, PA, USA.
Rothman, A.M. (1981) Personal Communication for the Head of
Experimental Pathology, National Toxicology Program, Research
Triangle Park, North Carolina to J.A. Moore, EPA, Washington DC.
Submitted to WHO by Rohm and Haas Co., Philadelphia, Pennsylvania,
USA.
Sames, J.L. & Doolittle, D.J. (1986) Dicofol (Kelthane technical
miticide): Kelthane in vivo cytogenetic study in rats. Unpublished
report No. 85R-215 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Sato, H., Toyoda, K., Furukawa, F., Hasegawa, R., Takahashi, M., &
Hayashi, Y. (1987) Subchronic oral toxicity test of dicofol (1,1-
bis(p-chlorophenyl)-2,2,2-trichloroethanol) as the basis for the
design of a long-term carcinogenicity study in B6C3F1 mice.
Bulletin of National Institute of Hygienic Sciences 105: 42-45.
Shellenberger, T.E. (1986) Dicofol (Kelthane miticide): Three-month
dietary toxicity study in dogs. Unpublished report No. 85014 (Rohm
and Haas report No. 86RC-23) from Tegeris Laboratories Inc., Laurel,
Maryland, USA. Submitted to WHO by Rohm and Haas Company, Spring
House, PA, USA.
Shirasu, Y., Moriya, M., & Ohta, T. (1980) Microbial mutagenicity
test of Kelthane. Unpublished report (Rohm and Haas No. 80RC-1024)
from Institute of Environmental Toxicology, Japan. Submitted to WHO
by Rohm and Haas Company, Spring House, PA, USA.
Smith, R.B., Jr., Larson, P.S., Finnegan, J.K., Haag, H.B.,
Hennigar, G.R., & Cobey, F. (1959) Toxicologic studies on 2,2- bis-
(chlorophenyl)-2,2,2-trichloroethanol. Toxicol. Appl. Pharmacol.,
1: 119-134.
Solomon, H.M. & Kulwich, B.A. (1991) Dicofol: Two-generation study
in rats. Unpublished report No. 89R-028 from Rohm and Haas Company,
Spring House, PA, USA. Submitted to WHO by Rohm and Haas Company,
Spring House, PA, USA.
Steigerwalt, R.B., Deckert, F.W., & Longacre, S.L. (1984a)
Comparative disposition of a single po dose of 14C-Kelthane and
14C-DDT in rats. Unpublished report No. 79R-130 from Rohm and Haas
Company, Spring House, PA, USA. Submitted to WHO by Rohm and Haas
Company, Spring House, PA, USA.
Steigerwalt, R.B., Deckert, F.W., & Longacre, S.L. (1984b)
Comparative disposition of multiple doses of 14C-Kelthane and
14C-DDT in female rats. Unpublished report No. 80R-174 from Rohm
and Haas Company, Spring House, PA, USA. Submitted to WHO by Rohm
and Haas Company, Spring House, PA, USA.
Steigerwalt, R.B., Deckert, F.W., & Longacre, S.L. (1984c) Mouse
liver mixed function oxidase assay for Kelthane technical - three
day po induction study. Unpublished report No. 80R-018 from Rohm and
Haas Company, Spring House, PA, USA. Submitted to WHO by Rohm and
Haas Company, Spring House, PA, USA.
Steigerwalt, R.B., Udinsky, J.R., Deckert, F.W., & Longacre, S.L.
(1984d) Liver mixed function oxidase assays for Kelthane technical,
its active ingredients, and principal technical impurities in male
B6C3F1 mice after two week dietary exposure. Unpublished
report No. 79R-167 from Rohm and Haas Company, Spring House, PA,
USA. Submitted to WHO by Rohm and Haas Company, Spring House, PA,
USA.
Tegeris, A.S. & Shellenberger, T.E. (1988) Dicofol (Kelthane
technical miticide): One year dietary toxicity study in beagle dogs.
Unpublished report No. 86014 (Rohm and Haas report No. 87RC-027)
from Tegeris Laboratories Inc., Laurel, Maryland, USA. Submitted to
WHO by Rohm and Haas Company, Spring House, PA, USA.
Tillman, A.M. & Mazza, L.S. (1986) Part I: Absorption and excretion
of 14C-dicofol in male and female rats. Part II: A metabolism
study of 14C-dicofol in male and female rats. Unpublished report
No. 31L-86-02 from Rohm and Haas Company, Spring House, PA, USA.
Submitted to WHO by Rohm and Haas Company, Spring House, PA, USA.
USEPA (1979) (US Environmental Protection Agency) pesticide incident
monitoring system. Summary of reported pesticide incidents involving
Kelthane, Report No. 173. Office of Pesticide Programs.
Verschuuren, H.G., Kroes, R., & Den Tonkelaar, E.M. (1973) Toxicity
studies on tetrasul III. Short-term comparative studies in rats with
tetrasul and structurally related acaricides. Toxicology, 1: 113-
123.
WHO (1992). The WHO recommended classification of pesticides by
hazard and guidelines to classification 1992-1993 (WHO/PCS/92.14).
Available from the International Programme on Chemical Safety, World
Health Organization, Geneva, Switzerland.
Woodruff, R.C., Mason, J.M., Valencia, R., & Zimmering, S. (1985)
Chemical mutagenesis testing in Drosophila. V. Results of 53 coded
compounds tested for the National Toxicology Program. Environmental
Mutagenesis, 7: 677-702.