
PESTICIDE RESIDUES IN FOOD - 1997
Sponsored jointly by FAO and WHO
with the support of the International Programme
on Chemical Safety (IPCS)
TOXICOLOGICAL AND ENVIRONMENTAL
EVALUATIONS 1994
Joint meeting of the
FAO Panel of Experts on Pesticide Residues
in Food and the Environment
and the
WHO Core Assessment Group
Lyon 22 September - 1 October 1997
The summaries and evaluations contained in this book are, in most
cases, based on unpublished proprietary data submitted for the purpose
of the JMPR assessment. A registration authority should not grant a
registration on the basis of an evaluation unless it has first
received authorization for such use from the owner who submitted the
data for JMPR review or has received the data on which the summaries
are based, either from the owner of the data or from a second party
that has obtained permission from the owner of the data for this
purpose.
MALATHION
First draft prepared by
T.C. Marrs
Medical Toxicology and Environmental Health
Department of Health, London, United Kingdom
Explanation
Evaluation for acceptable daily intake
Biochemical aspects
Absorption, distribution, and excretion
Biotransformation
Effects on enzymes and other biochemical parameters
Cholinesterases
Other enzyme systems
Interactions with other organophosphates
Toxicological studies
Acute toxicity
Short-term toxicity
Long-term toxicity and carcinogenicity
Genotoxicity
Reproductive toxicity
Multigeneration reproductive toxicity
Developmental toxicity
Special studies
Dermal and ocular irritation and dermal
sensitization
Macrophage and mast cell function
Ocular function
Neurotoxicity
Antidotes
Observations in humans
Comments
Toxicological evaluation
References
Explanation
Malathion was evaluated by the JMPR in 1963, 1965, and 1966
(Annex 1, references 2, 4, and 6). An ADI of 0-0.02 mg/kg bw was
established in 1963, which was confirmed in 1965 and 1966. It was
evaluated at the present Meeting within the CCPR periodic review
programme.
Malathion is S-1,2-bis(ethoxycarbonyl)ethyl O,O-dimethyl
phosphorodithioate. It is likely that the results of earlier
toxicological studies on malathion have been substantially affected by
impurities. Of particular interest are isomalathion
[ S-1,2-bis(ethoxycarbonyl)ethyl O,S-dimethyl phosphorodithioate]
and various trialkyl phosphorothioates [for reviews, see Aldridge et
al. (1985) and Dinsdale (1992)]. These compounds are notable for their
pulmonary toxicity. Furthermore, isomalathion has a greater than
additive effect when administered with malathion, probably due to
carboxylesterase inhibition (Ryan & Fukuto, 1984). In fact,
isomalathion appears to be the major impurity of malathion and affects
the LD50 of the commercial formulation. O,O,S-Trimethyl
phosphorothioate and O,S,S-trimethyl phosphorothioate produce
disorders of blood clotting (Keadtisuke et al., 1990), and
O,O,S-trimethyl phosphorothioate produces an unusual neurotoxic
syndrome with hypophagia, weight loss, and hypothermia (Ohtako et al.,
1995).
After an epidemic of malathion poisoning among spraymen in
Pakistan (Baker et al., 1978), WHO issued specifications for malathion
water-dispersible powders, which required that a 50% powder contain no
more than 0.9% isomalathion after storage at 54 ¡C for six days (Miles
et al., 1979; WHO, 1985). Subsequently, major manufacturers, under the
auspices of FAO, adopted a code of conduct which requires that,
inter alia, the active ingredient and co-formulant of commericial
formulation be the same as those tested toxicologically (FAO, 1986).
It is likely that esterase-inhibitory activity attributed to
technical-grade malathion is due largely to the action of malaoxon
(WHO, 1986).
Evaluation for acceptable daily intake
1. Biochemical aspects
(a) Absorption, distribution, and excretion
The disposition of 14C-malathion (purity, > 98%; specific
activity; 90 µCi/mg (3.3 MBq/mg)), labelled at the carbonyl carbons of
the ethoxycarbonyl groups, was studied by Reddy et al. (1989). After a
preliminary study, 14C-malathion in corn oil was administered by
gavage as single doses of 40 or 800 mg/kg bw to groups of five male
and five female Sprague-Dawley (Crl:CD BR) rats. Disposition was also
assessed after administration of oral doses of unlabelled malathion
(purity, 94.6%) at 40 mg/kg bw per day for 15 days, followed by a 16th
dose of 14C-malathion. In the preliminary study, very little of the
radiolabel appeared in expired air, and most was eliminated within 72
h; consequently, in the main study, the animals were killed at 72 h.
Malathion was rapidly absorbed, biotransformed, and excreted,
predominantly in the urine but also in the faeces. After the low dose,
84% appeared in the urine of males and 88% in that of females within
72 h, mostly within 12 h: faecal elimination was 11 and 5.9% in males
and females, respectively. Less than 1% of the administered dose was
recovered in the tissues. At the high dose, urinary excretion was 76%
for the males and 85% for the females; faecal elimination was 14 and
6.6%, respectively. Low concentrations were present in tissues at 72
h. After the repeated doses, 85 and 88% of the label was excreted in
the urine within 72 h, mostly within the first 12 h, and faecal
elimination was 6.8% in males and 5.8% in females. Less than 1% of the
dose was present in the tissues.
Malathion, either of 94.6% purity or a 50% emulsifiable
concentrate, was labelled with 14C in one methoxy group and given
orally to male Sprague-Dawley albino rats at a dose of 280 mg/kg
active ingrdient (approximately one-tenth of the LD50). More than 90%
of the dose was excreted in the urine within 24 h; the rest of the
label was detected in the faeces, intestines, liver, and kidney, in
descending order of concentration. The disposition of the pure
malathion and the 50% emulsifiable concentrate was not significantly
different (Abou Zeid et al., 1993).
The toxicokinetics of malathion was studied by Garcia-Repetto et
al. (1995) after oral administration of a dose of 467 mg/kg bw (stated
to be one-third of the LD50) to male albino Wistar rats. A two-
compartment model was discerned, the central compartment being blood,
adipose tissue, and muscle and the peripheral compartment, brain and
liver. The half-life in blood was 1.4 ± 0.25 days. In a fatal case of
malathion poisoning, malathion and the mono- and dicarboxylic acids
were found in cardiac blood and tissues, malaoxon being found
additionally in most tissues (Morgade & Barquet, 1982).
(b) Biotransformation
In the study of Reddy et al. (1989) cited above, the metabolites
of malathion were studied by high-perfomance liquid chromatography and
gas chromatography-mass spectrometry. 14C-Malathion was excreted in
urine and faeces as the a and b monocarboxylic acids and the
dicarboxylic acid of malathion. Minor metabolites were the oxon of
malathion (malaoxon), O,O-dimethylphosphorodithioic acid,
2-mercaptosuccinic acid, fumaric acid, monoethyl fumarate,
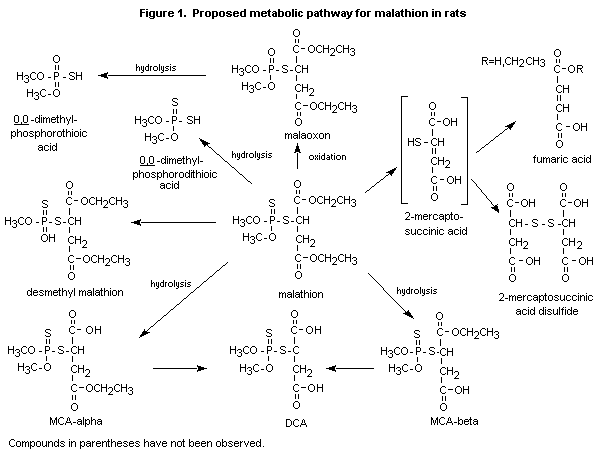
O,O-dimethylphosphorothioic acid, and desmethylmalathion. Figure 1
shows the proposed pathway for the metabolism of malathion in rats.
(c) Effects on enzymes and other biochemical parameters
(i) Cholinesterases
Groups of 27 male and 27 female Sprague-Dawley Crl:CD:BR rats
were given malathion (purity, 96.4%) by gavage in corn oil at doses of
0, 500, 1000, or 2000 mg/kg bw; 20 animals of each sex were used to
measure cholinesterase activity, and seven of each sex for
determination of neuropathological effects. Viability, clinical signs,
body weights, and the results of a functional observational battery of
tests and locomotor activity were recorded before treatment, 15 min
after treatment, and on days 7 and 14 in seven animals of each sex per
dose of those reserved for neuropathology and five of each sex at each
dose of those destined for cholinesterase measurements. Cholinesterase
activity was determined in plasma, erythrocytes, and brain regions in
five animals of each sex per group before the start of the study, 15
min after treatment, and on day 15. Similar measurements were made on
day 7, but as one male was killed in extremis, only four remained at
this time. Treatment-related clinical signs consisting of salivation
and/or anogenital staining occurred after one or two days of treatment
in all groups. Additionally reduced hindlimb extensor strength was
 seen in one male at the highest dose and decreased ambulatory and
motor activity counts in males at this dose on day 0. These males also
showed a > 20% reduction in plasma cholinesterase activity in
comparison with controls at day 7; although no reduction was seen at
1000 mg/kg bw, a marginal reduction was seen at 500 mg/kg bw. No
reductions were seen at other times. In females, reductions > 20%
were seen at both 500 and 2000 mg/kg bw at day 7 and at the latter
dose at day 15. Erythrocyte acetylcholinesterase activity was reduced
by > 20% in the males at the highest dose at day 7, while in females
it was reduced by > 20% at day 0 (500 and 2000 mg/kg bw), day 7 (1000
and 2000 mg/kg bw) and day 15 (2000 mg/kg bw only). No consistent
biologically significant depressions in brain acetylcholinesterase
activity were seen, although there were 10-20% decreases in activity
in comparison with concurrent controls, mainly in the group at the
high dose. There were no treatment-related neuropathological lesions.
There was no NOAEL, as clinical signs occurred in all groups (Lamb,
1994a).
Malathion (purity, 96.4%) was given to groups of 25 male and 25
female Sprague-Dawley Crl:CD:BR rats at dietary concentrations of 0,
50, 5000, or 20 000 ppm (equal to 0, 4, 350, or 1500 mg/kg bw per day
in males and 0, 4, 400, or 1600 mg/kg bw per day in females) for
13 weeks. Clinical signs, body weight, and food consumption were
recorded, a functional observational battery of tests was carried out,
and locomotor activity was evaluated. Plasma, erythrocyte, and
regional brain cholinesterase activities were measured in five animals
of each sex per dose before treatment, at weeks 3 and 7, and at the
end of the study. Tissues from the remaining five animals in each
group were perfused in situ, and neuropathological examinations were
carried out on the brains of the controls and animals at the high
dose. All animals survived to the end of the study.
Anogenital staining was observed in rats at the high dose, and
body-weight gain and food consumption were reduced in comparison with
controls. No treatment-related effects were seen in functional and
locomotor evaluations. Plasma cholinesterase activity was > 20% lower
than in concurrent controls in males at 20 000 ppm at all times after
the start of treatment, while the activity in rats at 5000 ppm was
reduced by 10-20%; erythrocyte acetylcholinesterase activity was
reduced by > 20% at all times in rats at doses > 5000 ppm. In the
females, reductions of > 20% were seen in plasma cholinesterase at
5000 ppm at week 7 only and at 20 000 ppm at all three times (all by
comparison with concurrent controls). Reductions in erythrocyte
acetylcholinesterase activity of > 20 % were seen at all times in
females at 5000 and 20 000 ppm. Regional brain acetylcholinesterase
activity was very variable; significant depressions in activity were
seen only in rats at the high dose. Thus, significant depressions were
seen in the olfactory lobe (by 34%) and midbrain (by 24%) and a
marginally significant depression (18%) in the brain stem of males at
week 13; in the cerebral cortex, a 26% depression in activity was seen
at week 7 only. No clinically or biologically significant depression
in activity was seen in the hippocampus or cerebellum of males. In
females, depressed brain acetylcholinesterase activity was observed
more often and frequently to a greater extent than in the males.
Depressed activity in comparison with concurrent controls was seen in
the olfactory lobe at 3 (31%), 7 (27%), and 13 weeks (50%) and in the
brainstem at 13 weeks (36%), less depression in activity being seen at
the other times. In the midbrain, depressed activity was seen at 7
(34%) and 13 weeks (40%). In the cerebral cortex, depressions were
seen at 3 (32%), 7 (40%), and 13 weeks (53%). In the hippocampus,
depressed activity occurred at all times, by 44% at 3 weeks, 38% at 7
weeks, and 47% at 13 weeks. In the cerebellum, depressions of 20% at 3
weeks and 32% at 13 weeks were seen. No effects were observed on the
absolute or relative weights of the brain or brain regions, and no
neuropathological abnormalities were observed. The NOAEL was 5000 ppm,
equal to 350 mg/kg bw per day, on the basis of the occurrence of
statistically and biologically significant inhibition of brain
acetylcholinesterase activity at the highest dose (Lamb, 1994b).
Malaoxon is a much more powerful anticholinesterase than
malathion, and very pure samples of the latter have little activity
(WHO, 1986). Thus, the IC50 values for cholinesterase inhibition in
17-day-old aggregate cultures of rat neural cells were > 12 × 10-4
mol/L for malathion and 2.8 × 10-4 mol/L for malaoxon (Segal &
Federoff, 1989). Malaoxon produces a dimethylphosphorylated
cholinesterase, however, which rapidly undergoes spontaneous
reactivation, as shown ex vivo in the blood of malaoxon-poisoned
rats, rabbits, dogs, and monkeys (Abraham & Edery, 1976).
Abou Zeid et al. (1993) showed that there was faster recovery of
serum cholinesterase activity in Sprague-Dawley rats after dermal
application of pure malathion than of the 50% emulsifiable
concentrate.
Ward et al. (1993) reported a correlation between the
anticholinesterase activity of a series of organophosphates, including
malathion and malaoxon, and their ability to compete with agonist
binding to muscarinic receptors.
(ii) Other enzyme systems
When rat microsomal suspensions were incubated with 4 mmol/L
malathion in vitro, the release of ß-glucuronidase was inhibited
(Lechener & Abdel-Rahman, 1985).
(iii) Interactions with other organophosphates
Feeding Holtzman rats with fenchlorphos potentiated the effect on
erythrocyte or brain acetylcholinesterase activity of a single
intraperitoneal challenge with malathion at 200 mg/kg bw (Murphy &
Cheever, 1968). The combined effect on dioxathion and malathion was
more or less than additive, depending on the doses used. Malathion
acts synergistically with many other organophosphates, such as
ethyl- para-nitrophenyl thionobenzenephosphonate (Frawley et al.,
1957), at substantial doses. The LD50 of malathion is markedly
reduced by co-administration of tri- ortho-tolylphosphate (Murphy et
al., 1959).
2. Toxicological studies
(a) Acute toxicity
It is likely that the results of earlier studies on malathion
were substantially affected by impurities. The LD50 values for these
impurities in rats after oral administration are: isomalathion, 89-120
mg/kg bw, O,O,S-trimethylphosphorodithioate, 450-660 mg/kg bw,
O,S,S-trimethyl-phosphorodithioate, 26-110 mg/kg bw, and
O,O,S-trimethylphophorothioate, 47-260 mg/kg bw (Aldridge et al.,
1979).
The results of studies on the acute toxicity of malathion are
given in Table 1; those on malaoxon are summarized in Table 2.
(b) Short-term toxicity
Rats
Malathion (purity, 96.4%) was administered in the diet to groups
of five male and five female albino Fischer (CDF:F-344/CrlBR) rats for
29 or 30 days at concentrations of 0, 50, 100, 500, 10 000, or 20 000
ppm, equal to 0, 5.1, 10, 52, 1000, or 2000 mg/kg bw per day for males
and 0, 5.7, 12, 58, 1100, or 2200 mg/kg bw per day for females. The
animals were observed weekly for body weight and food consumption.
Ophthalmological, haematological, and clinical chemical examinations,
including plasma and erythrocyte cholinesterase activity, were
undertaken before treatment and at termination of the study; brain
acetylcholinesterase activity was measured at termination. Animals
were autopsied at the end of treatment, and selected organs were
examined and weighed; microscopic examination of organs was carried
out only in the controls and rats at 20 000 ppm.
No deaths occurred during the study, adverse clinical signs were
not seen, and no abnormalities were present on ophthalmological or
haematological examination. A number of abnormal biochemical variables
were noted, including cholinesterase activity. That in plasma was
decreased by > 20% in animals at the two highest doses in comparison
with concurrent controls, while erythrocyte acetylcholinesterase
activity was decreased in males at 10 000 ppm (by 17%) and 20 000 ppm
(by 16%) and only slightly in females. At 10 000 ppm, brain
acetylcholinesterase activity was depressed at termination by 11% in
males and 17% in females, while at 20 000 ppm there was a 26% decrease
in males and 28% in females. Differences were seen between treated
groups in total protein and albumin concentrations, and a significant
decrease in alkaline phosphatase activity was seen in animals at the
two highest doses. Animals at 20 000 ppm had a significant decrease in
weight gain in comparison with the control group, while food
consumption was decreased only during week 1. The relative and
Table 1. Acute toxicity of malathion
Species Strain Sex Route LD50 and 95% CI, ±SEM, Purity Reference
or range (mg/kg bw, unless (%)
otherwise stated) or LC50
Mouse Swiss white F Oral 6100 > 95 Toia et al. (1980)
Mouse Swiss-Webster M,F Intraperitoneal 985 NR Menzer & Best (1968)
954-1018
Rate Wistar M Oral 2800 NR Dauterman & Main (1966)
2660-3110
Rat Osborne-Mendel NR Oral 1400 ± 100 98 Frawley (1957)
Rat Sherman M Oral 1375 NR Gaines (1969)
1206-1568
F 1000
885-1130
Rat Wistar M,F Oral 1580 92.2 Pellegrini & Santi (1972)
Rat Wistar M,F Oral 8000 98.2 Pellegrini & Santi (1972)
Rat Wistar M Oral (laboratory chow) 1090 ± 83 95 Boyd & Tanikella (1969)
Oral (26% casein) 1401 ± 99
Oral (3.5% casein) 5993 ± 138
Rat Sprague-Dawley M,F Oral 5000 ± 385 NR Terrell et al. (1978)
Rat Sprague-Dawley M Oral 3800 (3040-4750) NR Cooper & Terrell (1979a)
Rat Lac:P Oral 10 700 (9300-12 300) 99.7 Aldridge et al. (1979)
Rat Sprague-Dawley F Oral 4400 (2533-8228) NR Cooper & Terrell (1979a)
Table 1. (continued)
Species Strain Sex Route LD50 and 95% CI, ±SEM, Purity Reference
or range (mg/kg bw, unless (%)
otherwise stated) or LC50
Rat Sprague-Dawley M Oral 3200 (2651-3862) NR Cooper & Terrell (1979b)
F 3700 (2221-6164)
Rat CD Sprague- M Oral 5400 (4100-6900) 96-98 Kynoch (1985a)
Dawley-derived F 5700 (4300-7800)
Rat Albino Crl:CD M Oral 6156 (4665-8123) 96.8 Fischer (1991)
(SD)BR F 4061 (3078-5359)
Rat Wistar M Oral 734 NR Jokanovic & Maksimovic
F (1995)
Rat HSD Sprague- M Oral 8210 (6518-10 342) 99.1 Kuhn (1996)
Dawley F 8239 (6239-10 881)
Rat Sprague-Dawley M Intraperitoneal 1100 NR Murphy et al. (1959)
Rat Sherman M,F Derman (57% > 44 444 NR Gaines (1969)
emulsifiable
concentrate)
Rat CD Sprague- M,F Derman > 2000 96-98 Kynoch (1985b)
Dawley-derived
Rat Albino Wistar M,F Inhalation (4 h) > 5.2 mg/L 96-98 Jackosn et al. (1986)
Rabbit New Zealand M,F Derman 8.79 ± 0.48 NR Imlay et al. (1978)
albino
Hamster Syrian F Intraperitoneal (30% 24 00 NR Dzwonkowska &
commercial Hubner (1986)
preparation)
Table 1. (continued)
Species Strain Sex Route LD50 and 95% CI, ±SEM, Purity Reference
or range (mg/kg bw, unless (%)
otherwise stated) or LC50
Dog Mongrel NR Oral > 4000 98 Frawley (1957)
Dog Mongrel M Intraperitoneal 1.517 ml/kg bw 95 Guiti & Sadoghi (1969)
0.77-2.25
Buffalo Indian (Bubalus M Oral 100-125 NR Gupta (1984)
bubalis)
Chicken White Leghorn F Oral 775 93.6 Fletcher (1989)
610-984
NR, not reported
Table 2. Acute toxicity of malaoxon
Species Strain Sex Route LD50 and 95% CI, ±SEM, Purity Reference
or range (mg/kg bw, unless (%)
otherwise stated)
Mouse Swiss white F Oral 215 NR Toia et al. (1980)
Rat Sprague-Dawley M,F Intraperitoneal About 25 NR Brodeur & DuBois (1967)
Rat Wistar M Oral 158 NR Dauterman & Main (1966)
142-175
NR, not reported
absolute weights of the liver were increased in males at the highest
dose and in females at the two highest doses, and periportal
hepatocytic hypertrophy was seen at the two highest doses in animals
of each sex. These changes were considered to be related to treatment.
Increased relative kidney weights were observed in males at the two
highest doses and in females at the highest dose. The NOAEL was 500
ppm, equal to 52 mg/kg bw per day, on the basis of the increased
weight of the livers with histopathological changes and inhibition of
brain acetylcholinesterase activity (Daly, 1993a).
Malathion (purity, 96.4%) was administered in the diet to groups
of 10 male and 10 female albino Fischer (CDF:F-344/CrlBR) rats for 90
days at concentrations of 0, 100, 500, 5000, 10 000, or 20 000 ppm,
equal to 0, 6.6, 34, 340, 680, or 1400 mg/kg bw per day for males and
0, 7.9, 39, 380, 780, or 1600 mg/kg bw per day for females. Body
weight and food consumption were estimated before treatment and
periodically during the study. Ophthalmological, haematological, and
clinical chemical examinations, including plasma, erythrocyte, and
brain cholinesterase activities, were undertaken before treatment and
at termination of the study. The animals were killed at least 90 days
after the start of the study and autopsied. Selected organs were
examined and weighed, and all animals were examined microscopically.
One male at the high dose died during the study from unknown
cause. Anogenital staining was seen in four males and six females at
the high dose during treatment, and in animals at this dose, body
weights and weight gain were consistently lower than in the control
group; there was a decrease in food consumption only in week 1, in
contrast to greater food consumption by animals at the high dose than
by controls later in the study. Haemoglobin count and haematocrit were
decreased in males at the high dose, while the mean corpuscular volume
and mean cell haemoglobin were decreased in males at doses > 5000
ppm. In females, the erythrocyte count was marginally increased at
doses > 500 ppm, while the mean corpuscular volume was decreased at
10 000 and 20 000 ppm, and the mean cell haemoglobin was decreased at
doses > 5000 ppm. A number of abnormal biochemical variables were
noted, including cholinesterase activity. Plasma cholinesterase
activity was decreased marginally (17%) in males at 5000 ppm, while
there was clearly significant depression at the higher doses in
comparison with the values in concurrent controls. In female rats,
plasma cholinesterase activity was depressed by > 20% at doses >
5000 ppm. In the males, erythrocyte acetylcholinesterase activity was
marginally but significantly depressed at 500 ppm (by 18% in
comparison with concurrent controls) and markedly depressed at higher
doses. In the females, depression of erythrocyte acetylcholinesterase
activity by > 20% was observed at all doses. At 10 000 ppm, there was
marginally significant depression of brain acetylcholinesterase
activity at termination (by 13% in males and 17% in females), while at
20 000 ppm there was biologically significant depression, by 20%, in
males and 44% in females At 5000 ppm, there was less inhibition of
brain acetylcholinesterase activity in animals of each sex (8.8% in
males and 10% in females), which was, however, statistically
significant. Significant decreases in alkaline phosphatase activity
were seen in males at the three highest doses and in females at the
highest dose. The activity of gamma-glutamyl transpeptidase was
elevated in males at the highest dose and in females at the two
highest doses. A reduction in aspartate aminotransferase activity was
observed in females at the highest dose.
Differences in relative and absolute liver and kidney weights
were seen between groups, with associated histopathological changes.
The absolute and relative weights of the liver were increased in males
at doses > 5000 ppm and in females at the highest dose. Periportal
hepatocyte hypertrophy was seen in males at doses > 10 000 ppm and
in females at doses > 5000; these changes were considered to be
related to treatment. The absolute and relative weights of the kidney
were increased in animals of each sex at the highest dose. At 10 000
ppm, the absolute and relative kidney weights were increased in males
and the relative kidney weights in females; at 5000 ppm, the relative
kidney weights were increased in animals of each sex. Chronic
nephropathy was more severe in males at doses > 5000 ppm than in
those at lower doses or in controls, but there were no differences
between groups in the prevalence of this pathological change. The
NOAEL was 500 ppm on the basis of decreased mean corpuscular volume
and mean corpuscular haemoglobin, increased liver weights and relative
kidney weights, and chronic nephropathy in males at the next highest
dose and decreased mean corpuscular haemoglobin, hepatocytic
hypertrophy, and increased relative kidney weight in the females at
the next highest dose. There were also marginally significant
decreases in brain acetylcholinesterase activity at 5000 ppm. The
finding of a marginal increase in erythrocyte count at 500 ppm in
females is ignored. The NOAEL is equal to 34 mg/kg bw per day (Daly,
1993b).
Rabbits
Malathion (purity, 94%) was applied to the skin of groups of six
male and six female New Zealand white rabbits for 6 h per day on five
days per week for three weeks at doses of 50, 300, or 1000 mg/kg bw
per day; six males and five females were sham-treated. Effects on the
skin, organ and body weights, food consumption, clinical chemical
parameters including cholinesterase activity, and haematological
variables were evaluated; selected organs were examined
pathologically. Two males, one at 50 mg/kg bw per day and one at the
high dose, died before termination of the study,. There were no
physical alterations or changes in body or organ weights or food
consumption attributable to treatment, except for erythema and oedema
in treated animals. Decreases in plasma, erythrocyte, and brain
cholinesterase activity > 20% were seen at the highest dose in
animals of each sex; females also showed depression of erythrocyte
activity at 300 mg/kg bw per day. Brain acetylcholinesterase activity
was substantially reduced in the cerebrum and cerebellum of animals of
each sex at the highest dose. Females at 300 mg/kg bw per day had a
19% reduction in brain acetylcholinesterase activity in comparison
with concurrent controls, but this reduction was not statistically
significant. The NOAEL was 300 mg/kg bw per day on the basis of
inhibition of brain acetylcholinesterase activity at the highest dose
(Moreno, 1989).
Dogs
Malathion (purity, 92.4%) was administered to groups of three
male and three female beagles in gelatin capsules at doses of 0, 125,
250, or 500 mg/kg bw per day for 28 days. The animals were observed
twice daily, and haematological and clinical chemical measurements
were made before treatment and 15 and 29 days after the start of
treatment. Selected organs were weighed and examined post mortem.
Diarrhoea was observed at all doses, and anorexia at the highest
dose. One male at the high dose died. Weight gain was reduced at the
highest dose and marginally so at 250 mg/kg bw per day; food
consumption was reduced at the highest dose. At 15 days, serum albumin
and sodium levels were decreased in dogs at the highest dose, as were
blood urea nitrogen, aspartate aminotransferase activity, and
creatinine. Decreased plasma and erythrocyte cholinesterase activities
were seen at 15 days and at termination: plasma cholinesterase
activity was decreased by > 20% in dogs at the intermediate and high
doses at 15 days and at all doses at termination; erythrocyte
acetylcholinesterase activity was decreased by 20% at the high dose by
15 days and by 17% at all doses at termination. The LOAEL was 125
mg/kg bw per day, on the basis of reduced erythrocyte
acetylcholinesterase activity at all doses at termination of the
study, with clinical signs (diarrhoea) at all doses. No NOAEL was
identified (Fischer, 1988).
Malathion (purity, 95%) was administered in capsules to groups of
six male and six female beagles at doses of 0, 62.5, 125, or 250 mg/kg
bw per day on seven days a week for one year. The animals were
observed twice daily, with more detailed examinations weekly; they
were weighed before the start of the study, at the beginning of
treatment, weekly thereafter, and at sacrifice. Food consumption was
measured weekly, and haematological and clinical chemical variables,
including plasma and erythrocyte cholinesterase activity, were
determined before treatment, at six weeks, three months, six months,
and just before the animals were killed. Cerebellar and cerebral
acetylcholinesterase activity was determined at termination of the
study. Ophthalmological examination was carried before the start of
treatment and just before sacrifice.
No clinical signs of toxicity were observed, nor was any
abnormality seen on ophthalmological examination. No significant
difference was seen in body weight or food consumption, although
animals of each sex at the high dose showed a small decrease in mean
weight. Clinical chemistry revealed perturbations in a number of
variables. Plasma and erythrocyte cholinesterase activities were
decreased by more than 20% in animals of each sex at all doses and
times in comparison with concurrent controls. Brain
acetylcholinesterase activity was unaffected, except for some
diminution in cerebellar acetylcholinesterase activity at the highest
dose (by 16% in males and 11% in females); cerebral cholinesterase
activity was unaffected. Serum albumin, total protein, the
albumin:globulin ratio, and calcium levels were decreased and lactate
dehydrogenase increased in animals of each sex, generally only at the
high dose but occasionally in those at the intermediate dose. In
females, albumin was reduced at all doses at six weeks. Alkaline
phosphatase activity was increased in males at the high dose but not
in females. Other changes observed occasionally included low blood
urea nitrogen in animals at the high dose and decreased alanine
aminotransferase activity in those at the intermediate and high doses,
but these did not appear to be of toxicological significance.
Significantly lower calcium levels were found in animals at the high
dose at six weeks and later. Haematological examination revealed
dose-related decreases in erythrocyte and haemoglobin counts and
haematocrit. The erythrocyte and haemoglobin counts were decreased at
all times in males at the high dose, and the haematocrit was decreased
in the males at the high dose at three and six months. In the females,
erythrocyte and haemoglobin counts and haematocrit were marginally
affected at six weeks in the group at the high dose, but at three
months the haematocrits were decreased at the intermediate and high
doses and haemoglobin count at the high dose. Additionally there was
an increase in mean corpuscular volume at the high dose and in mean
corpuscular haemoglobin at the intermediate and high doses. At six
months, the erythrocyte and haemoglobin counts and haematocrit were
all decreased in females at the high dose and the erythrocyte count in
those at the intermediate dose. At termination, only a decrease in
erythrocyte count was seen in females at the highest dose. Platelet
counts were increased in males and females at all times and all doses
in comparison with concurrent controls; many of these changes were
statistically significant. Urinalysis revealed no marked changes. The
absolute liver weights of females at the low and high doses were
increased, and the relative liver weights were raised in males at the
intermediate and high doses and in females at all doses. The absolute
and relative kidney weights were raised in animals at the intermediate
and high doses. No treatment-related pathological alteration was seen
macroscopically or microscopically. The NOAEL was 125 mg/kg bw per day
on the basis of body-weight depression and haematological and clinical
chemical changes. The changes in liver weights and the reduced albumin
in females at six weeks are discounted on the grounds that there was
no morphological correlate and that there was no clear dose-response
relationship (Schellenberger & Billups, 1987).
(c) Long-term toxicity and carcinogenicity
A number of long-term studies have been carried out in mice and
rats by the US National Cancer Institute and others. Many of the
studies were reviewed by IARC (1983) and by Rueber (1985). The working
group convened by IARC concluded that the available data did not
provide evidence that malathion or malaoxon is carcinogenic in humans.
This view is in line with those of the study authors but not with
those of Rueber (1983). More modern studies are now available and are
summarized below, with a brief resumé of the earlier studies for the
sake of completeness.
Mice
Groups of 50 B6C3F1 mice of each sex were given doses of 0,
8000, or 16 000 ppm malathion admixed in the diet for 80 weeks,
equivalent to 1200 or 2400 mg/kg bw per day. The animals were killed
14-15 weeks after discountinuation of the malathion-containing diets.
Throughout the study, the mean body weights of animals of each sex
were lower than those of controls; poor food consumption,
hyperexcitability, and abdominal distention were also noted in the
second year; tremors were seen in a few female mice. Malathion was
reported not to be carcinogenic (US National Cancer Institute, 1978);
however, the slides were re-examined by Rueber (1985), who concluded
that malathion had increased the incidence of neoplasms of the liver
in male mice.
Malathion (purity, 96.4%) was administered in the diet to groups
of 65 B6C3F1 BR mice of each sex for 18 months at concentrations of 0,
100, 800, 8000, or 16 000 ppm (equal to 0, 17, 140, 1500, or 3000
mg/kg bw per day for males and 0, 21, 170, 1700, and 3500 mg/kg bw per
day for females). Animals were observed twice daily and examined in
detail weekly. Body weights were determined weekly until week 14,
fortnightly to week 26, and monthly thereafter. Plasma, erythrocyte,
and brain cholinesterase activity was determined 10 animals of each
sex from each group killed at 12 months and at termination; only
erythrocyte enzyme activity was determined in 10 mice of each sex per
group at week 36, and these mice were retained until terminal
sacrifice of the survivors at 18 months. The mice were examined
post mortem, and selected organs were processed and examined
histologically.
There was no treatment-related effect on mortality, but body
weights and food consumption were reduced in animals of each sex at
8000 and 16 000 ppm. Plasma cholinesterase activity was reduced by
> 20% in comparison with concurrent controls at 12 and 18 months in
males at 800 ppm; similar results were seen in females, except that
the reduction was 18% in those at 800 ppm by 12 months. Erythrocyte
acetylcholinesterase activity was decreased by > 20% in animals of
each sex at doses > 800 ppm at 9, 12, and 18 months, while brain
acetylcholinesterase activity was decreased in males at the highest
dose at 12 and 18 months and in those at 8000 ppm at 18 months. In
females, brain acetylcholinesterase activity was not decreased at 8000
ppm at 12 months but was decreased at 18 months; females at 16 000 ppm
showed a 20% decrease in brain acetylcholinesterase activity at
12 months and a 43% decrease at 18 months. The absolute and relative
liver weights were increased in males at the two highest doses, and
the relative liver weight was increased in females at the highest
dose. Other changes in organ weights included increased relative
kidney weights in certain groups.
Macroscopically, an increased incidence of liver nodules was seen
at the two highest doses; microscopically, effects were seen on the
liver, kidney, adrenal cortex, and bone. Liver hepatocellular
hypertrophy was observed in all animals at the two highest doses at
termination, and milder hypertrophy was seen in the animals killed at
12 months. The incidences of liver tumours in animals that survived to
termination are given in Table 3. There was a significant trend in the
incidence of adenomas in animals of each sex, and the incidence was
significantly raised by comparison with controls at the two highest
doses; the incidences in historical controls at the same laboratory
were 14-22% in males and 0-11% in females. There was no significant
trend for hepatocellular carcinoma, but the incidence was
significantly raised in the males at 100 and 8000 ppm.
Proximal tubular vacuolation seen at lower doses in the kidneys
was absent in all males at the highest dose and most of those at 8000
ppm. Female mice at 8000 and 16 000 ppm had an increased incidencse of
renal cortical mineralization. A treatment-related decrease in fibrous
osteodystrophy of the sternum observed in females at the highest dose
is of unknown significance. A treatment-related, early disappearance
of the X zone of the adrenal cortex was observed in females at 8000
and 16 000 ppm at 12 months. The overall NOAEL was 800 ppm, equal to
140 mg/kg bw per day, on the basis of inhibition of brain
acetylcholinesterase activity and an increased incidence of liver
adenomas in animals of each sex at the next highest dose (Slauter,
1994).
Rats
Malathion
Three studies were carried out by Hazleton Laboratories (Hazleton
& Holland, 1953). In the first, groups of 20 male Colworth Farm rats
were given technical-grade malathion (purity, 65%) in the diet at
doses of 0, 100, 1000, or 5000 ppm, equivalent to 5, 50, and 250 mg/kg
bw per day, for 109 weeks. Body weight and food consumption were
decreased in those at the highest dose. Depressed cholinesterase
activity was seen in rats at 5000 ppm and to a lesser extent in those
at 1000 ppm. The study was not adequate for conclusions to be drawn
about carcinogenicity. In the second study, with the same doses,
similar criticisms can be made, except that the purity of the
malathion was > 90%. A third study was carried out with male and
female Colworth Farm rats which received doses of 0, 500, 1000, 5000,
or 20 000 ppm of malathion (purity, 99%). The highest dose was lethal
to the male rats; the size of the study precluded conclusions about
carcinogenicity.
Groups of 50 male and 50 female Osborne-Mendel rats were given
technical-grade malathion (purity, 95%) in the diet at concentrations
of 4700 or 8150 ppm (equivalent to 240 and 410 mg/kg bw per day) for
80 weeks. A pooled control group consisted of 15 matched controls of
each sex and 40 untreated male and female rats from bioassays of other
chemicals. The rats were killed after 109 weeks. The body weights of
female rats receiving malathion were lower than those of controls, and
the survival times of those at the higher dose were decreased.
Malathion was reported not to be carcinogenic (US National Cancer
Institute, 1978). The slides were re-evaluated by Rueber (1985), who
Table 3. Incidences of hepatocellular tumours (%) in mice at terminal sacrifice after treatment with malathion
Tumour Dose (ppm)
Males Females
0 100 800 8000 16 000 0 100 800 8000 16 000
Adenoma 2 11.8 4.2 24.1 98.0 0 1.9 0 17.9 82.4
Carcinoma 0 11.8 4.2 11.1 2.0 1.8 0 3.8 1.9 3.9
found that the incidence of benign and malignant neoplasms at all
sites analysed together was increased in treated rats; in particular,
the incidence of carcinomas of the endocrine organs was increased, and
malignant neoplasms of the brain were observed in seven treated rats.
Rueber concluded that malathion was carcinogenic in male and female
Osborne-Mendel rats. In a re-evaluation of the slides commissioned by
the US National Toxicology Program (Huff et al., 1985), the original
interpretation that malathion is not carcinogenic was confirmed.
In a second study by the US National Cancer Institute (1979a),
malathion (purity, 95%) was fed in the diet to groups of 50 male and
50 female Fischer 344 rats at doses of 0, 2000, or 4000 ppm
(equivalent to 100 or 200 mg/kg bw per day; only 49 males at the
higher dose) for 103 weeks. The authors concluded that malathion was
not carcinogenic. When Rueber (1985) re-evaluated the slides, he found
that the incidence of benign and malignant neoplasms analysed together
was significantly increased, particularly in males. He concluded that
malathion was carcinogenic in Fischer 344 rats. A re-evaluation of the
slides commissioned by the US National Toxicology Program (Huff et
al., 1985) confirmed that malathion was not carcinogenic.
Malathion (purity, 92.1%) was given in the diet to groups of 50
male and 50 female Sprague-Dawley rats at concentrations of 0, 100,
1000, or 5000 ppm, equivalent to 5, 50, or 250 mg/kg bw per day. The
animals were observed daily throughout the study, and body weights and
food consumption were recorded at the end of weeks 1, 13, 24, 53, 79,
and 103. Blood samples for haematological examination and
determination of cholinesterase activity and urine samples for
urinalysis were collected from five rats of each sex per group in
weeks 12, 26, and 53; blood and urine were also collected at week 104,
and alanine and aspartate aminotransferase activities, urea nitrogen,
and glucose were determined additionally in blood. Brain
acetylcholinesterase activity was not determined. Animals that died,
were killed in extremis, or killed at termination were examined
post mortem.
No significant difference in food consumption or survival was
seen, and no significant intergroup differences were seen on
haematological or biochemical examination, except in cholinesterase
activity. During the first year of the study, the body weights of
animals of each sex at the highest dose were reduced, while in the
second year the body weights of those at 1000 and 5000 ppm were
depressed. Moreover, erythrocyte acetylcholinesterase activity was
reduced by > 20% in comparison with the controls in rats at the
intermediate and high doses at 3, 6, 12, and 24 months; plasma
cholinesterase activity was less affected, although there was a
depression of > 20% in the females at the high dose at 12 and 24
months. Absolute and relative liver weights were increased in male
rats at the high dose and relative kidney weights in males at the two
highest doses. Absolute brain weights were decreased in females at the
two highest doses and relative kidney weights in those at the high
dose.
Although foci of cellular alteration were recorded twice in the
livers of males at the highest doses and once in females at the
intermediate and high doses, this difference was not statistically
significant. A significant difference in sinusoidal dilatation was
found between the controls (2%) and males at the high dose (16%).
Extramedullary splenic haematopoiesis was seen more often in males at
the high dose than in controls. There was no evidence of carcinogenic
potential. The NOAEL was 100 ppm, equivalent to 5 mg/kg bw per day, on
the basis of reduced erythrocyte acetylcholinesterase activity and
body weight at the next highest dose (Rucci et al., 1980). After an
audit by the US Environmental Protection Agency (1987; Cyanamid,
1990), the Agency requested a re-evaluation of the slides. Seely
(1991) found only two treatment-related lesions: periportal
hepatocellular hypertrophy and cystic hepatocellular degeneration,
both only in male rats at the highest dose. There was no evidence of
differences in tumour incidence. The NOAEL for effects on the liver
was thus 1000 ppm (equivalent to 50 mg/kg bw per day). This
re-evaluation does not alter the overall NOAEL of 5 mg/kg bw per day
(see above).
Malathion (purity, 96.4%) was administered in the diet of groups
of 90 male and female Fischer 344 (CDF:F-344/CrlBR) rats at
concentrations of 0, 100/50, 500, 6000, or 12 000 ppm for two years;
the lowest dose was reduced from 100 to 50 ppm at week 18 because of
inhibition of erythrocyte acetylcholinesterase activity, resulting in
mean intakes over the entire study of 0, 4, 29, 360, and 740 mg/kg bw
per day for males and 0, 5, 35, 420, and 870 mg/kg bw per day for
females. Groups of 10 animals of each sex per group were killed at
three and six months, 15 of each sex per group at 12 months, and the
remainder at two years. Physical condition, ophthalmoscopic
parameters, body weight, and food consumption were determined before
treatment and at selected intervals, while electroretinography,
haematology, and clinical chemistry (including determination of
cholinesterase activity) were performed at selected intervals and on
selected animals. Selected organs from animals killed at 12 and 24
months were weighed, and the animals were examined macroscopically.
Tissues from those at the high dose and from controls, and certain
organs from animals at the low dose were examined histopathologically.
Malathion reduced the survival of males at 6000 and 12 000 ppm,
early deaths being observed from the 14th month in males at the
highest dose and from about the 20th month in those at the next lowest
dose. Survival of females at the high dose was impaired towards the
end of the study. Nephropathy and mononuclear-cell leukaemia were the
main causes of death, although the frequency of neither was
treatment-related. Anogenital staining was seen in females at the
highest dose. Decrements in body weight and weight gain were seen in
animals of each sex throughout the study at the two highest doses,
although mean food consumption was greater in these animals than in
the controls, throughout the study in the case of the males and in the
second year of the study in the case of the females. Decreases in mean
haemoglobin concentration, haematocrit, mean corpuscular volume, and
mean cell haemoglobin were seen in animals of each sex at the two
highest doses at 6, 12, and 18 months, although all parameters were
not affected at all the time intervals and there was a tendency for
improvement during the study. The mean cell haemoglobin concentration
was decreased in males at the two highest doses only at 12 months,
accompanied by an increase in platelet count.
At 3, 6, 12, and 24 months, animals of each sex showed reductions
in plasma, erythrocyte, and brain cholinesterase activity,
predominantly at the two highest doses. Thus, animals of each sex at
the highest dose had decreased plasma cholinesterase activity at all
times. In males at 6000 ppm, decreases in activity > 20% were seen at
three and six months and at termination, while there was a marginal
decrease at 12 months (83% of control value). Males at 500 ppm had a
significant decrease in activity only at termination, when the
activity was 71% of that of concurrent controls. In the females,
plasma cholinesterase activity was consistently reduced by at the two
highest doses; the activity was little affected at 500 ppm, except at
termination when there was a marginally significant decrease of 18%.
There were consistent reductions in erythrocyte acetylcholinesterase
activity at the two highest doses in males and a marginally
significant reduction at 500 ppm at termination only, when the
activity was 83% that of concurrent controls. Erythrocyte
acetylcholinesterase activity was similarly reduced in females at 6000
and 12 000 ppm. Although reductions > 20% were seen in females at 500
ppm at three months and at termination and a marginal reduction at 12
months (86% of control value), no reduction was seen at six months.
Erythrocyte acetylcholinesterase activity was also reduced in females
at the lowest dose at three months (75% of control value), so that on
day 113 the lowest dose was reduced from 100 ppm to 50 ppm. Six weeks
later, erythrocyte acetylcholinesterase activity was evaluated in 10
controls and 10 at 50 ppm and found to be comparable. Thereafter, the
activity in animals at 50 ppm was unremarkable. Brain
acetylcholinesterase activity was reduced by > 20% in males at 6000
ppm at termination. Decreases seen at the highest dose were 84% of the
control value at three months, 81% at six months, and 85% at 12
months; no determination was carried out at termination because there
were no survivors. At 6000 ppm, the activity was decreased to 88% of
the control value at three months, 88% at six months, and 89% at 12
months. In females at 12 000 ppm, the activity of brain
acetylcholinesterase was substantially reduced in comparison with that
of concurrent controls. Smaller decreases were seen at 6000 ppm at
three months (15%), six months (17%), 12 months (12%), and termination
(18%). No significant inhibition was seen at lower doses.
Alkaline phosphatase activity was reduced in comparison with
concurrent controls in animals of each sex at the two highest doses at
6 and 12 months and at the highest dose at 18 months. Aspartate
aminotransferase activity was reduced in females at doses > 500 ppm
at 12 months and at the highest dose at 18 months. Alanine
aminotransferase activity was also decreased in females at the three
highest doses at 12 months. gamma-Glutamyl transpeptidase activity was
increased consistently in males at the two highest doses from 12
months and at most intervals in females. Cholesterol content was
increased in animals of each sex at the two highest doses at 6, 12,
18, and 24 months. Increases in mean and relative liver and kidney
weights were observed in animals of each sex at the two highest doses
at the interim sacrifice, in females at 6000 and 12 000 ppm at
terminal sacrifice, and in males at 6000 ppm at terminal sacrifice.
Males also had decreased relative kidney weights at 500 ppm. Relative
spleen weight was increased in males at the two highest doses at
interim sacrifice, and absolute spleen weight was reduced in males at
6000 ppm and in females at 12 000 ppm at terminal sacrifice. Relative
and absolute thyroid and parathyroid weights were elevated in males at
the two highest doses at interim sacrifice, in males at 6000 ppm at
termination, and in females at 6000 and 12 000 ppm at termination.
Microscopic findings of significance were largely confined to
nasoturbinal tissues, kidney, and liver. Degeneration and hyperplasia
of the olfactory epithelium were seen in animals of each sex at the
two highest doses. The hyperplasia was focal, with thickening of the
epithelium and proliferation of basal cells, forming clusters in the
lamina propria. While focal degeneration was also observed in a few
controls and rats at lower doses, the hyperplasia was confined to
those at the two highest doses. In several rats, the epithelium was
replaced by ciliated and non-ciliated columnar epithelium. Subacute
and chronic inflammation and dilated and hyperplastic mucosal glands
were seen in some animals; subacute and chronic inflammation and
hyperplasia of the respiratory epithelium of the nasopharynx and
dilated mucosal glands were also seen. Like the other changes in nasal
tissues, inflammatory cells and cell debris were seen most frequently
at the two highest doses. Thus, the NOAEL for this effect was 500 ppm.
The incidence and severity of nephropathy was greater in rats at 6000
and 12 000 ppm than in controls.
A nasal turbinate adenoma was seen in one male at 6000 ppm and a
carcinoma in one male at 12 000 ppm; although the numbers observed
were small, this is a rare tumour in Fischer rats. Hepatocellular
adenomas and carcinomas were seen in some animals. In the female rats,
the prevalence of adenomas and cacinomas combined was increased at the
highest dose and that of adenomas alone at 6000 ppm (see Table 4).
Testicular interstitial-cell tumours were seen in virtually all male
rats that survived to termination. The overall NOAEL was 500 ppm,
equal to 29 mg/kg bw per day, on the basis of decreased survival and
body-weight gain, increased food consumption, changes in
haematological parameters, decreased brain acetylcholinesterase
activity, increased g-glutamyl transpeptidase activity, increased
liver, kidney, thyroid, and parathyroid weights, and degeneration and
hyperplasia of the olfactory epithelium at the next highest dose.
Although an increased incidence of liver tumours was seen in females,
malathion was not considered to be carcinogenic in view of the small
numbers of such tumours observed (Daly, 1996a).
Table 4. Prevalences of hepatocelular adenomas and carcinomas in rats at termination after treatment with malathion
Dose (ppm)
Males Females
0 100/50 500 6000 12 000 0 100/50 500 6000 12 000
No. of animals 37 41 29 14 0 38 41 41 34 20
Tumour
Adenoma 2 2 3 1 0 0 0 1 3 3
Carcinoma 1 1 0 1 0 0 1 1 0 1
Malaoxon
Malaoxon (purity, > 95%) was fed to groups of 50 Fischer 344
rats of each sex in the diet at concentrations of 0, 500, or 1000 ppm
for 103 weeks. The authors concluded that malaoxon was not
carcinogenic in rats (US National Cancer Institute, 1979b). The slides
were re-examined by Rueber (1985), who concluded that the incidence of
benign and malignant neoplasms at all sites was increased in the
treated animals. The neoplasms in question were in endocrine organs,
including the pituitary, adrenal, and thyroid glands; the incidences
of hyperplasia, adenomas, and carcinomas of C cells of the thyroid
were increased. In a re-evaluation of the slides commissioned by the
US National Toxicology Program (Huff et al., 1985), the original
conclusion that malaoxon is not carcinogenic was largely confirmed,
but there was stated to be equivocal evidence that malaoxon is
carcinogenic in that there was an increased incidence of C-cell
neoplasms of the thyroid.
Groups of 85 Fischer 344 (CDF:F-344/CrlBR) rats of each sex were
exposed to malaoxon (purity, 96.4%) admixed in the diet at
concentrations of 0, 20, 1000, or 2000 ppm (equal to 1, 57, or
110 mg/kg bw per day in males and 1, 68, or 140 mg/kg bw per day in
females); 55 animals were retained for 24 months, while 10 of each sex
per group were killed at 3, 6, and 12 months. Cholinesterase activity
was estimated in all animals. Clinical chemical and haematological
parameters were determined in all animals killed at 6 and 12 months
and in 10 animals of each sex per group of animals that were retained
at 18 months and termination. Physical observations, ophthalmoscopy,
and measurements of body weight and food consumption were carried out
before treatment and at selected intervals during the study. The
surviving rats were sacrificed at 24 months. The rats were examined
post mortem, and selected organs were weighed. Histopathological
examinations were perfomed on controls and those at the high dose at
12 and 24 months and on rats that died or were killed in extremis
during the study. Selected tissues from animals at the intermediate
and low doses were also examined.
Survival was curtailed in female rats at 1000 and 2000 ppm and in
males at 2000 ppm. The most common causes of death were pneumonitis
and mononuclear leukaemia; the occurrence of the former appeared to be
dose-related. Anogenital staining was seen in females at the highest
dose throughout the study and in males in the latter part of the
study. Treatment-related decreases in body weight and weight gain were
seen at the highest dose. Food consumption was decreased in males at
1000 and 2000 ppm. Plasma, erythrocyte, and brain cholinesterase
activity was affected by malathion. Plasma cholinesterase activity was
reduced by > 20% at all times in animals of each sex at the two
highest doses. Erythrocyte acetylcholinesterase activity was reduced
by > 20% in the same groups and in males at the lowest dose at six
months; the reductions at other times in animals of each sex at this
dose were 10-20%. Brain acetylcholinesterase activity was reduced by
18-11% in comparison with concurrent controls in males at the highest
dose and more clearly reduced in females at earlier times. There were
substantial reductions in brain acetylcholinesterase activity in
animals of each sex at the highest dose at termination of the study.
At the intermediate dose, there was a 30% decrease in brain
acetylcholinesterase activity in males at termination.
No abnormality was seen on ophthalmoscopic examination. Although
there were sporadic differences in clinical chemical measurements
between groups, none appeared to be treatment-related. The absolute
and relative liver and kidney weights were increased in males at 2000
ppm at 12 months, and the relative and absolute adrenal weights were
increased in males at this dose at two years. The absolute and
relative spleen weights of females at 2000 ppm were decreased. The
incidence of emaciation: was increased in males at 2000 ppm and in
females at 1000 and 2000 ppm. Inflammatory changes in the nasal
turbinates, lungs, and tympanic spaces, which may have been secondary
to increased disposition of food particles, were present in males at
the highest dose and females at the two highest doses. Thus, foreign
material such as food was found in the nasal lumen with inflammatory
cells and cell debris. The nasal mucosa also showed chronic
inflammatory changes and hyperplasia and hypertrophy of goblet cells.
In a small number of rats, squamous metaplasia was observed.
Degeneration of the olfactory epithelium was accompanied by focal
replacement by ciliated and non-ciliated columnar epithelium.
Mineralization of the stomach was seen in males at the two highest
doses and females at the highest dose. No treatment-related neoplasia
was observed. Interstitial tumours of the testis were present in
> 75% of the animals at all doses and were not considered to be
related to treatment. The NOAEL was 20 ppm, equal to 1 mg/kg bw per
day, on the basis of decreased food consumption and brain
acetylcholinesterase activity at termination in males and emaciation
at termination and inflammatory changes in the nasal turbinates in
females at the next highest dose (Daly, 1996b).
(d) Genotoxicity
The results of tests for the genotoxicity of malathion and
malaoxon are shown in Table 5. Four impurities in malathion,
isomalathion, O,O,S-trimethyl phosphorothioate, O,S,S-trimethyl
phosphorodithioate, and O,O,O-trimethyl phosphorothioate of > 99%
purity were tested for their potential to induce reverse mutation in
S. typhimurium TA97, TA98, and TA100 at doses of 10-1000 µg/plate.
Negative results were obtained, with and without metabolic activation
and with and without preincubation (Imamura & Talcott, 1985).
(e) Reproductive toxicity
(i) Multigeneration reproductive toxicity
In a small study in Wistar rats given malathion in the diet at
about 240 mg/kg bw, a higher incidence of ring-tail disease was seen
in treated than in control rats (Kalow & Marton, 1961).
Table 5. Results of tests for the genotoxicity of malathion and malaoxon
End-point Test system Concentration Purity Results Reference
(%)
Malathion
In vitro
Reverse mutation S. typhimurium NR NR Negativea McCann et al. (1975)
TA98, TA100,
TA1535, TA1537
Reverse mutation S. typhimurium 100-5000 µg/plate 95.2 Negativea Traul (1987)
TA98, TA100,
TA1535, TA1537,
TA1538
Reverse mutation S. typhimurium 5-300 µg/plate NR Positive Shiau et al. (1980)
TA98, TA100, (TA 1535
TA1535, TA1536, without S9)
TA1537, TA1538
Reverse mutation S. typhimurium 33-165 µg/plate NR Negativeb Pednekar et al. (1987)
TA97a, TA98, TA100
Reverse mutation S. typhimurium NR NR Negative Byeon et al. (1976)c
TA98, TA100,
TA1535, TA1538
Reverse mutation S. typhimurium 80 and 400 ppm/plate 90-95 Negativea Wong et al. (1989)
TA98, TA102,
TA1535, TA1537
Reverse mutation S. typhimurium < 5000 µg/plate NR Negativea Moriya et al. (1983)
TA98, TA100,
TA1535, TA1537,
TA1538
Table 5. (continued)
End-point Test system Concentration Purity Results Reference
(%)
Reverse mutation S. typhimurium < 10 mg/plate NR Negativea Waters et al. (1982)
TA98, TA100,
TA1535, TA1537,
TA1538
Reverse mutation S. typhimurium NR (preincubation) NR Positive Ishidate et al. (1981)
TA98, TA100, (TA 100 with
TA1537 S9)
Reverse mutation Escherischia coli NR NR Negativea Nagy et al. (1975)
Reverse mutation Escherischia coli 100-5000 µg/plate 95.2 Negativea Traul ( t 987)
Reverse mutation Escherischia coli < 5000 µg/plate NR Negativea Moriya et al. (1983)
Reverse mutation Escherischia coli < 10 mg/plate NR Negativea Waters et al. (1982)
Forward mutation Escherischia coli 2 × 20 mol/L NR Negative Mohn (1973)
Forward mutation Schizosaccharomyces NR 99 Negative Degraeve et al. (1980)
pombe
Forward mutation Schizosaccharomyces 30-182 mmol/L NR Negativea Gilot-Delhalle et al.
pombe (1983)
DNA damage Bacillus subtilis 5-300 µg/plate NR Positive Shiau et al. (1980)
rec and exc
DNA damage Bacillus subtilis rec 200 µg/plate NR Negative Shirasu et al. (1976)
DNA damage Bacillus subtilis rep NR NR Negative Waters et al. (1982)
Table 5. (continued)
End-point Test system Concentration Purity Results Reference
(%)
DNA damage Bacillus subtilis rew NR NR Negative Waters et al. (1982)
Primary DNA Saccharomyces NR NR Negative Waters et al. (1982)
damage cerevisiae
Unscheduled Primary rat 0.01-0.16 µl/ml 94 Negative Pant (1989)
DNA synthesis hepatocytes
Unscheduled Human lung NR NR Negativea Waters et al. (1982)
DNA synthesis fibroblasts
Chromosomal Chinese hamster NR NR Positivea Ishidate et al. (1981)
aberration lung fibroblasts
Chromosomal Cultured human 0.02-20 µg/ml NR Positive Balaji & Sasikali (1993)
aberrations peripheral leukocytes
Sister chromatid Cultured human 0.02-20 µg/ml NR Positive Balaji & Sasikali (1993)
exchange peripheral leukocytes
Chromosomal Cultured human 33-660 µg/ml NR Positivea Garry et al. (1990)
aberrations peripheral leukocytes
Sister chromatid Cultured human 33-660 µg/ml NR Positivea Garry et al. (1990)
exchange peripheral leukocytes
Sister chromatid Human fetal fibroblasts 2.5-40 µg/ml 99 Positive Nicholas et al. (1979)
exchange
Table 5. (continued)
End-point Test system Concentration Purity Results Reference
(%)
Sister chromatid Chinese hamster V79 10-80 µg/ml 94 Positive Chen et al. ( 1981)
exchange/cell cells (high dose)
cycle delay
Sister chromatid Chinese hamster ovary 0.03-1 mmol/L 99 Positive Nishio & Yueki (1981)
exchange cells
In vivo
Sex-linked recessive Drosophila melanogaster Feeding: adult, 50 ppm; 50% Negative Velazquez et al. (1987)
lethal mutation larva, 100 ppm ECd
Injection: adult, 10 and
25 ppm
Sex-linked recessive Drosophila melanogaster NR NR Negative Waters et at. (1982)
lethal mutation
Sex chromosome Drosophila melanogaster Feeding: 0-10 ppm 50% Negative Velazquez et al. (1987)
loss adult Injection: 0 and 5 ppm ECd
Non-disjunction Drosophila melanogaster Feeding: 0-20 ppm Ecd 50% Negative Velazquez et al. (1987)
Dominant lethal Mouse NR NR Negative Degraeve et al. (1980)
mutation
Dominant lethal Mouse 'Maximum lethal dose' NR Negative Waters et al. (1982)
mutation
Chromosomal Rat bone marrow 0.5-2.0 g/kg 94 Negative Gudi (1990)
aberration (Sprague-Dawley)
Chromosomal Syrian hamster bone 0.24-1.2 g/kg 30e,f Positive Dzwonkowska &
aberration marrow Hubner (1986)
Table 5. (continued)
End-point Test system Concentration Purity Results Reference
(%)
Chromosomal CFW mouse spermatocytes 0.45 mg/day for 30f Positive Bulsiewicz et al. (1976)
aberration 50 or 100 days
Chromosomal Mouse bone marrow and 500-2000 mg/kg bw NR Positive Salvadori et al. (1988)
aberration primary spermatocytes single dose or 5 daily
doses dermally
Malaoxon
In vitro
Reverse mutation S. typhimurium 100-10 000 µg/plate 94.4 Negativea Zeiger et al. (1988)
TA97, TA98, TA100,
TA1535, TA1537
Cell mutation Mouse lymphoma 12.5-300 nl/ml NR Positive Myhr & Caspary (1991)
tk locus L5178Y cells without S9;
equivocal with
S9
Sister chromatid Chinese hamster ovary 0.03-1 mmol/L 96 Positive Nishio & Yueki (1981)
exchange cells
Chromosomal Chinese hamster ovary > 5 mg/ml 94.4 Negative Ivett et al. (1989)
aberration cells
Sister chromatid Chinese hamster ovary > 5 mg/ml 94.4 Positivea Ivett et al. (1989)
exchange cells
In vivo
Sex-linked recessive Drosophila melanogaster Feeding: 5 ppm 94.4 Positive Foureman et al. (1994)
lethal mutation Injection: 2 ppm feeding;
negative,
injection
Table 5. (continued)
NR, not reported; S9, 9000 × g supernatant of rodent liver
a With and without metabolic activation
b With and without metabolic activation with S9 and caecal microbial extract
c In Korean; not fully evaluated
d 50% emulsifiable concentrate
e 30% commercial preparation from Organika-Azot, Poland
f Sadofos-30 (approximately 30% solution)
In a two-generation study of reproductive toxicity, groups of 25
male and 25 female CD:Sprague-Dawley-derived rats were fed diets
containing malathion (purity, 94%) at concentrations of 0, 550, 1700,
5000, or 7500 ppm, equal to 0, 43, 130, 390, or 600 mg/kg bw per day
in the F0 males and 0, 50, 150, 440, and 660 mg/kg bw per day in the
F0 females. The equivalent intakes for the F1 generation were: 43,
130, 390, or 630 mg/kg bw per day for males and 51, 150, 460, and 750
mg/kg bw per day for females. Each parent generation was mated to
produce two litters, and offspring were selected randomly from the
second (F1b) litter to be the parents of the next generation.
Offspring that were not selected, the offspring of the first litters
(F1a and F2a), and the F2b offspring were examined grossly and
discarded. One pup of each sex per F1b and F2b litter was selected
randomly, killed, and examined post mortem; abnormal tissues were
saved. The F0 and F1 adults were killed and examined post mortem,
and the reproductive organs and abnormal tissues were saved. Tissues
from the controls and animals at the high dose were examined
histologically.
Treatment had no effect on clinical signs, growth before mating,
food consumption, maternal weight gain during gestation, reproductive
performance, fertility indices, gestation length, or parturition in
the F0 and F1 parental generations. Pup sex ratio and survival were
also unaffected. Pup weight was reduced at day 21 in the F1a litters
at 5000 and 7500 ppm and in the F1b litters at 7500 ppm. Mean pup
weights in the F2a litters were comparable in all groups, except in
those at the high dose on day 21, which were decreased. Pup weights
were reduced at days 4, 7, 14, and 21 in the F2b litters at 5000 ppm
but not at 7500, except on day 21. At the highest dose, mean pup
weight at day 21 was lower than that of concurrent controls. Similar
effects were not seen at lower doses. Examination post mortem showed
no treatment-related effects. The NOAEL for reproductive toxicity was
7500 ppm, equal to 600 mg/kg bw per day, while that for developmental
toxicity was 1700 ppm, equal to 130 mg/kg bw per day (Schroeder,
1990).
In a study reported briefly, malathion (purity unspecified) was
administered to 16 male JIPMER albino rats for 12 weeks at a dose of
45 mg/kg bw per day by gavage. There were 12 appropriate controls. The
histological changes observed included interstitial oedema,
congestion, desquamation of cells lining the seminiferous tubules,
reduced numbers of spermatogonia, and absence of Leydig cells
(Balasubramanian et al., 1987).
(ii) Developmental toxicity
Female Sherman-strain rats were pair-mated with healthy adult
male rats of about the same age. On day 11 after insemination, they
were given a single intraperitoneal injection of malathion at a dose
of 700 or 900 mg/kg bw. On day 20 of gestation, the fetuses were
removed. The offspring and placentae were weighed, the numbers of
resorptions and dead animals were recorded, and half of the offspring
were examined in detail. The higher dose of malathion affected the
body weight of the dams but had no effect on the weight of the fetuses
and did not induce malformations (Kimbrough & Gaines, 1968).
Technical-grade malathion (purity unspecified) was administered
by gavage to groups of 20 female Wistar rats at doses of 0, 50, 100,
200, or 300 mg/kg bw on days 6-15 of gestation. Neither maternal nor
fetal toxicity was observed at the highest dose used (Khera et al.,
1978).
After a dose-ranging study, groups of female Crl:CD:(SD)BR rats
were given malathion (purity, 94%) by gavage on days 6-15 of gestation
at doses of 0, 200, 400, or 800 mg/kg bw per day in corn oil; the
groups consisted of 24 rats at the lowest dose and 25 at the other
doses. The animals were observed daily for clinical signs, body-weight
gain, and food consumption. After 20 days, the rats were killed and
examined for pregnancy, implantations, resorptions, live and dead
fetuses, and number of corpora lutea; the uterus was weighed, and
fetuses were examined for malformations. Cholinesterase activity was
not measured in this study. Malathion had no effect on survival, the
only early death occurring in the control group. Five rats at the
highest dose had urine staining of abdominal fur and decreased mean
weight gain and food consumption during treatment; after treatment,
the weight gain of animals at the high dose was increased in
comparison with the controls. No effect was seen on pregnancy rate or
numbers of corpora lutea, implantations, resorptions, or fetuses per
litter, fetal body weight, or sex ratio of fetuses. No fetal
abnormality attributable to treatment was observed. The NOAEL was 400
mg/kg bw per day on the basis of maternal toxicity at the highest
dose. The NOAEL for fetal toxicity was 800 mg/kg bw per day (Lochry,
1989).
Malathion (70% with 30% calcium carbonate) was administered at a
dose of 100 mg/kg bw per day on days 7-12 of gestation to seven mated
New Zealand white rabbits. Five mated rabbits received the vehicle
alone. No difference between the treated and control groups was seen
in respect of resorptions, fetal size, or external or visceral
abnormalities. The NOAEL for effects on the fetus was thus 100 mg/kg
bw per day. It is unclear from the paper whether any significant
maternal toxicity was observed (Machin & McBride, 1989).
After a range-finding study, malathion (purity, 92.4%) was
administered at doses of 25, 50, or 100 mg/kg bw per day by gavage in
corn oil to groups of 20 mated female New Zealand white rabbits on
days 6-18 of gestation; controls received the vehicle alone. The
rabbits were examined daily for mortality and for physical and
behavioural abnormalities. Body-weight gain was calculated for days
0-6, 6-12, 6-18, 18-29, and 0-29 days after the start of the study and
on days 6, 12, 18, and 29. The survivors were killed on gestation day
29 and examined post mortem. The uterus and ovaries were excised and
examined, and the number of corpora lutea recorded. The number and
position of live and dead fetuses, resorption sites, and the total
number of implantation sites was also recorded. Live fetuses were
removed, weighed, measured crown to rump, and examined for gross
external and visceral abnormalities. Fetuses were then processed and
examined for skeletal abnormalities. Cholinesterase activity was not
measured.
Although there was no statistically significant difference in
survival between the treated and controls groups, no deaths occurred
among the controls, four in the group at the low dose, three in the
group at the intermediate dose, and two in the group at the high dose.
In the last group, the deaths resulted from intrapulmonary intubation.
Maternal weight gain was reduced at the doses of 50 and 100 mg/kg bw
per day during treatment on days 6-18 of gestation. At days 12, 18,
and 29, the mean body weight of the animals at the high dose was
decreased in comparison with the controls. The mean number and percent
of resorptions was slightly increased at doses > 50 mg/kg bw per
day. There was no difference in fertility, number of corpora lutea,
implantation sites, litter size, or fetal weight or length. No other
signs of toxicity were seen in does or fetuses, nor was there any
evidence of teratogenicity. The NOEAL was 25 mg/kg bw per day for
maternal toxicity and 100 mg/kg bw per day for fetal toxicity, the
former being based on decreased weight gain at the next highest dose
and the latter on the absence of fetal toxicity at any dose (Siglin,
1985).
(f) Special studies
(i) Dermal and ocular irritation and dermal sensitization
A single semi-occluded application of malathion (purity, 96-98%)
to the skin of New Zealand white rabbits elicited slight to
well-defined, transient dermal reactions, with very slight oedema in
six animals and very slight erythema in five animals; the sixth had
well-defined erythema. The skins were all normal by day 2 (Liggett &
Parcell, 1985a).
Malathion (purity, 96-98%) produced mild conjunctival reactions
in the eyes of New Zealand white rabbits. No damage to the cornea or
iris was seen at any stage, and the eyes were normal after two days
(Liggett & Parcell, 1985b).
Malathion (purity, 96-98%) was tested in nine albino guinea-pigs;
there were 10 controls. One treated animal with respiratory distress
was killed in extremis. There was no evidence of delayed contact
hypersensitivity (Kynoch & Smith, 1985).
(ii) Macrophage and mast cell function
Repeated administration of malathion to female C57Bl/6 mice at a
dose of 1 mg/kg bw per day increased macrophage function, while 0.1
mg/kg bw per day caused mast cell degranulation (Rodgers & Xiong,
1997).
(iii) Ocular function
Malathion (purity, 98%) instilled into the eyes of Long-Evans
rats had no effect on responses evoked by a visual pattern and
produced no ophthalmological abnormality (Boyes, 1997).
(iv) Neurotoxicity
The potential of malathion (purity, 93.6%) to induce delayed
neuropathy was tested in white Leghorn hens. After determination of
the oral LD50, the ability of atropine to antagonize the effects of
malathion at doses greater than the LD50 was investigated. In the
main study, 60 birds received malathion at a dose of 1000 mg/kg bw
(1.3 times the unprotected LD50). They were given atropine sulfate
subcutaneously at 10 mg/kg bw 1 h before administration of malathion
and then at 30 mg/kg bw 15 min and 1, 3, and 5 h afterwards. A total
of 39 birds died with clinical signs consistent with cholinesterase
activity poisoning within 15 days. Three weeks after the first dose of
malathion, the survivors were dosed again, this time at 850 mg/kg bw
(1.1 times the LD50), with atropinization as above. A further seven
birds died, but the survivors recovered completely. Positive controls
were treated with tri- ortho-tolylphosphate at 500 mg/kg bw. The hens
were observed daily; body weights and food consumption were recorded
at the start of the study, and body weight was recorded thereafter at
three-day intervals. All dead birds were examined with perfusion
fixation, and the brain, spinal cord, and sciatic nerve were examined
histologically No treatment-related histopathological changes were
seen in the birds treated with malathion, whereas those treated with
tri- ortho-tolylphosphate showed changes typical of
organophosphate-induced delayed polyneuropathy in the spinal cord and
sciatic nerve. Clinical signs of delayed polyneuropathy were seen only
in the positive control birds (Fletcher, 1989).
Groups of 12 retired laying Leghorn hens were given malathion
orally at doses of 75, 150, or 300 mg/kg bw, and groups of 12
Long-Evans rats (28 rats at the high dose) received 600, 1000, or
2000 mg/kg bw. All received atropine pretreatment, and some received
subsequent treatment with atropine. Clinical assessments were carried
out. Cholinesterase and neuropathy target esterase activities were
estimated, and sections of the medulla, cervical and lumber spinal
cord, and branches of the tibial nerve were examined. Flaccid
paralysis was seen in the hens at 300 mg/kg bw for about 24 h, but
none died. There was no clinical indication of delayed polyneuropathy
in the hens treated with malathion, whereas those given
tri- ortho-tolylphosphate, mipafox, or diisopropylphosphorofluoridate
developed typical behavioural signs of neuropathy. Malathion at a dose
of 2000 mg/kg bw induced clinical signs consistent with cholinesterase
poisoning in the rats, and gait changes were observed 14-21 days after
administration at the highest dose. No treatment-related
histopathological changes were seen in the birds or rats treated with
malathion, whereas those treated with tri- ortho-tolylphosphate,
mipafox, or diisopropyl phosphorofluoridate showed changes typical of
organophosphate-induced delayed polyneuropathy. The activities of both
acetylcholinesterase and neuropathy target esterase were inhibited by
malathion. In the hens, brain acetylcholinesterase activity was
inhibited by 17 ± 3% at the lowest dose of malathion and by 76 ± 1% at
the highest; the corresponding inhibition of neuropathy target
esterase activity was 0 ± 3% and 50 ± 22%. In the rats,
acetylcholinesterase activity inhibition was 26 ± 6% at the lowest
dose and 56 ± 2% at the highest; the corresponding figures for
neuropathy target esterase inhibition were 19 ± 7% and 75 ± 5%, all
compared with concurrent controls (Ehrich et al., 1995).
Malathion and malaoxon produced nugatory inhibition of neuropathy
target esterase activity in human neuroblastoma cells (3 and 1%,
respectively) (Ehrich et al., 1994).
The ratio of the IC50 value for neuropathy target esterase to
that for acetylcholinesterase was 30 000 in murine neuroblastoma cells
and 76 000 in human cells (Ehrich et al., 1997).
(v) Antidotes
Malaoxon, the oxon analogue of malathion, inhibits cholinesterase
activity by producing a dimethylphosphoryl derivative, which is
susceptible to oxime-induced reactivation. Experimental evidence
indicates that clinically significant reactivation occurs (Hobbiger,
1973). Most authors have found significant reactivation with oximes,
but there are notable exceptions. For example, Ganendran and
Balabaskaran (1976) found little reactivation of malathion- and
malaoxon-inhibited human whole-blood cholinesterase activity. Similar
results were obtained for acetylcholinesterase activity in goat brain
(Cheema et al., 1989).
The efficacy of four pyridinium oximes, trimedoxime, obidoxime,
pralidoxime, and HI-6, in the treatment of poisoning by malathion
(purity, 96%) was tested in male Wistar rats, which were given
malathion at twice the LD50, as determined experimentally during the
study, and treated with 30 mol/kg bw of trimedoxime, obidoxime, or
HI-6 or 60 mol/kg bw of pralidoxime; atropine and diazepam therapy
were also used. Of the oximes, obidoxime was the most effective,
followed by trimoxime, and then pralidoxime and HI-6, which were
equally effective; however, better survival was achieved with HI-6 at
a dose of 150 mol/kg bw than with the other regimes. Thus, malathion
posoning can be treated with mono- and bis-pyridinium oximes
(Jokanovic & Maksimovic, 1995).
Malathion (50% emulsifiable concentrate) was administered at a
dose of 100 mg/kg bw or at a minimally lethal dose of 125 mg/kg bw to
buffalo calves (Bubalus bubalis). Pralidoxime methiodide combined
with atropine was reported to reverse the clinical evidence of
toxicity (Gupta, 1984).
Pralidoxime chloride or diacetyl monoxime at a dose of 100 mg/kg
bw intraperitoneally reversed the rise in blood glucose observed after
injection of malathion at 500 mg/kg bw to female albino rats, provided
the oximes were given immediately after the malathion. When given 15
min later, the antidotes were ineffective (Agarwal & Matin, 1981).
Obidoxime reversed malaoxon-induced inhibition of cholinesterase
activity in isolated rat diaphragm and restored the ability to sustain
tetany; moreover, obidoxime at 20 mg/kg bw together with atropine
raised the LD50 in OF mice by 5.1-fold, the comparable figure for
atropine alone being 1.7-fold (Abraham & Edery, 1976).
Obixodime has been used successfully in treating malathion
poisoning in humans (Dive et al., 1994; see below).
3. Observations in humans
Malathion and ethyl- para-nitrophenyl thionobenzenephosphonate
(purity of each unspecified) were tested in volunteer male prisoners
aged 23-36 years. In phase I of the study, four samples of blood were
taken during two weeks before the start of the study for measurements
of plasma and erythrocyte cholinesterase activity, and then malathion
was administered at a dose of 8 mg/day to five subjects for 32 days.
Phase II was begun three weeks after completion of phase I. On the two
days before its start, samples of plasma and washed erythrocytes were
taken for measurements of cholinesterase activity, and then malathion
was administered to the same five subjects at a dose of 16 mg/day for
47 days. In phase III, five new subjects were selected; plasma and
erythrocyte cholinesterase activity was determined in blood samples
drawn twice weekly for 36 days, before administration of malathion at
a dose of 24 mg/day for 56 days.
During phase I, no clinical effects were observed, and there were
no changes in blood counts or the results of urinalysis. Furthermore,
no significant depression of plasma or erythrocyte cholinesterase
activity was observed in any subject. Similarly, no clinical effects
were observed in phase II. In phase III, depression of plasma
cholinesterase activity was observed two weeks after the first
administration of malathion, the maximum depression being 25%, seen
three weeks after cessation of treatment; erythrocyte
acetylcholinesterase activity was depressed to the same extent. The
NOAEL was 6 mg/day, approximately equal to 0.27 mg/kg bw per day. No
potentiation of malathion by ethyl- para-nitrophenyl
thionobenzenephosphonate was observed (Moeller & Rider, 1962).
An epidemic of poisoning of spraymen in Pakistan was attributed
largely to contaminants, particularly iso-malathion, in the
formulation of malathion used (Baker et al., 1978).
There are numerous case reports of individual poisoning. Thus,
Dive et al. (1994), reported an instance in which an elderly woman
consumed about 100 ml of a garden preparation containing 15% malathion
in isopropyl alcohol. The preparation also contained
isopropylmalathion and O,O,S-trimethylphosphorothioate. A typical
cholinergic crisis was followed by cardiac, pulmonary, neurological,
and renal manifestations, and the patient was treated with atropine
and obidoxime. The cardiac manifestations included arrhythmia and
conduction disturbances. Mild interstitial pulmonary fibrosis was
observed in a lung biopsy sample.
Matsushita et al. (1985) reported allergic contact dermatitis in
people exposed to organophosphorus insecticides including malathion.
Thomas et al. (1990) reported a study of women exposed to
malathion during aerial spraying of large areas of the San Francisco
Bay area, USA, in 1981-82. A number of associations were found, of
which one, congenital gastrointestinal anomalies, remained
statistically significant after control for confounders.
An episode of epidemic hysteria was reported at an elementary
school in Arizona, USA, in response to the smell of malathion (Baker &
Selvey, 1992).
During a campaign to eradicate Mediterranean fruit flies with a
pesticide based on malathion, a number of cases of urticaria,
angioneurotic oedema, and non-specific rashes were reported. Of 10
subjects that received a patch test, none responded. One case of
possible immediate reaction to malathion bait was reported (Schanker
et al., 1992).
No case of organophosphate-induced delayed polyneuropathy due to
malathion has been reported in humans.
Comments
Malathion is rapidly absorbed, biotransformed, and excreted,
predominantly in the urine but also in the faeces, largely as its two
monocarboxylic acids and the dicarboxylic acid.
The oral LD50 values for malathion in laboratory rodents were
1000-10 000 mg/kg bw, the observed differences probably being due to
impurities. The most recent LD50 values tend to be higher. The
cholinesterase-inhibiting metabolite of malathion, malaoxon, has much
lower oral LD50 values of 100-220 mg/kg bw. WHO has classified
malathion as slightly hazardous (WHO, 1996).
In a study of neurotoxicity in rats receiving single doses of 0,
500, 1000, or 2000 mg/kg bw, there was no NOAEL, as clinical signs
were present at all doses. In a 13-week study of neurotoxicity, also
in rats, at dietary concentrations of 0, 50, 5000, or 20 000 ppm, the
NOAEL was 5000 ppm, equal to 350 mg/kg bw per day, on the basis of
inhibition of brain acetylcholinesterase at the highest dose.
In a 30-day study of toxicity in rats receiving malathion in the
diet at concentrations of 0, 50, 100, 500, 10 000, or 20 000 ppm, the
NOAEL was 500 ppm, equal to 52 mg/kg bw per day, on the basis of
increased liver weight and histopathological changes in the liver
(periportal hepatocyte hypertrophy) at the next highest dose.
In a 90-day study of toxicity in rats, malathion was given at
dietary concentrations of 0, 100, 500, 5000, 10 000, or 20 000 ppm.
The NOAEL was 500 ppm, equal to 34 mg/kg bw per day, on the basis of
decreased mean corpuscular volume and mean corpuscular haemoglobin,
increased liver weights and relative kidney weights, and chronic
nephropathy in males and decreased mean cell volume, hepatocyte
hypertrophy, and increased relative kidney weight in females at the
next highest dose.
A 21-day study of dermal toxicity was carried out in which
rabbits were treated with malathion at doses of 0, 50, 300, or 1000
mg/kg bw per day for 6 h per day, five days per week. The NOAEL was
300 mg/kg bw per day on the basis of inhibition of brain
acetylcholinesterase activity at the highest dose.
In a 28-day study of toxicity in dogs, malathion was fed in
gelatin capsules at doses of 0, 125, 250, or 500 mg/kg bw per day for
28 days. There was no NOAEL because of clinical signs at all doses.
In a one-year study of toxicity in dogs, malathion was
administered orally in capsules at doses of 0, 62.5, 125, or 250 mg/kg
bw per day on seven days per week. The NOAEL was 125 mg/kg bw per day
on the basis of body-weight depression and changes in haematological
and clinical chemical parameters at the highest dose.
A number of long-term studies of toxicity and carcinogenicity
have been carried out on malathion in both rats and mice. The earlier
ones were reviewed by a working group convened by the IARC, which
concluded that the available data did not provide evidence that
malathion was carcinogenic.
In an 18-month study in mice, malathion was administered at
dietary concentrations of 0, 100, 800, 8000, or 16 000 ppm. The NOAEL
was 800 ppm, equal to 140 mg/kg bw per day, on the basis of inhibition
of brain acetylcholinesterase activity at termination and an increased
incidence of liver adenomas in animals of each sex at the next highest
dose.
In a two-year study in rats, dietary concentrations of 0, 100,
1000, or 5000 ppm were used. The NOAEL was 100 ppm, equivalent to 5
mg/kg bw per day, on the basis of reduced erythrocyte
acetylcholinesterase activity and body weight. In another long-term
study in rats, malathion was given at doses of 0, 100/50, 500, 6000,
or 12 000 ppm for two years. The NOAEL was 500 ppm, equal to 29 mg/kg
bw per day, on the basis of decreased survival and body-weight gain,
changes in haematological parameters, decreased brain
acetylcholinesterase activity, increased g-glutamyl transpeptidase
activity, increased liver, kidney, and thyroid/parathyroid weights,
and changes in the olfactory epithelium at the next highest dose.
Numerous tests have been carried out for genotoxicity both in
vitro and in vivo. Most of the evidence indicates that malathion
is not genotoxic, although some studies indicate that it can produce
chromosomal aberrations and sister chromatid exchange in vitro.
There was no evidence that malathion induces chromosomal aberrations
in vivo. Malaoxon did not induce reverse mutation in bacteria, but
it caused sister chromatid exchange in two tests in mammalian cells
and induced sex-linked recessive lethal mutation in Drosophila in
vivo. The four common impurities of malathion, isomalathion,
O,O,S-trimethyl phosphorothioate, O,S,S-trimethyl
phosphorodithioate, and O,O,O-trimethyl phosphorothioate, did not
induce reverse mutation in bacteria. The Meeting concluded that
malathion is not genotoxic.
A number of studies of reproductive toxicity have been carried
out, only some of which showed NOAELs. In a study in rats, malathion
was administered by gavage to groups of pregnant animals on days 6-15
of gestation at doses of 0, 200, 400, or 800 mg/kg bw per day. The
NOAEL was 400 mg/kg bw per day on the basis of maternal toxicity at
the highest dose; no fetal toxicity was observed.
Malathion was administered orally at doses of 0, 25, 50, or 100
mg/kg bw per day to groups of pregnant rabbits on days 6-18 of
gestation. The NOAELs were 25 mg/kg bw per day for maternal toxicity
and 100 mg/kg bw per day for fetal toxicity; teratogenicity was not
seen at any dose.
A two-generation study was undertaken in rats in which malathion
was given at dietary concentrations of 0, 550, 1700, 5000, or 7500
ppm. The NOAEL was 7500 ppm, equal to 600 mg/kg bw per day, for
reproductive toxicity and 1700 ppm, equal to 130 mg/kg bw per day, for
developmental toxicity, the latter being based on reduced pup weights.
Two studies on the neurotoxicity of malathion in hens were
reviewed. In neither was there evidence that malathion can cause
delayed neuropathy, although some inhibition of neuropathy target
esterase activity was found in the brains of birds at 2000 mg/kg bw.
In a study in volunteers with doses of 8, 16, or 24 mg of
malathion per day, the NOAEL was 16 mg per day (equivalent to 0.27
mg/kg bw per day) on the basis of inhibition of plasma and erythrocyte
cholinesterase activity. Several cases of exposure to impure malathion
have been reported, none of which resulted in delayed neuropathy.
An ADI of 0-0.3 mg/kg bw was established on the basis of the
NOAEL of 29 mg/kg bw per day in the two-year study of toxicity and
carcinogenicity in rats, with a safety factor of 100. This ADI is
supported by the NOAEL of 25 mg/kg bw per day in the study of
developmental toxicity in rabbits. The alternative approach of basing
the ADI on the study in humans was not taken, as the study was old and
the material was therefore likely to contain toxic impurities.
Toxicological evaluation
Levels that cause no toxic effect
Mouse: 800 ppm, equal to 140 mg/kg bw per day (18-month study
of toxicity and carcinogenicity)
Rat: 500 ppm, equal to 29 mg/kg bw per day (two-year study
of toxicity and carcinogenicity)
1700 ppm, equal to 130 mg/kg bw per day (study of
reproductive toxicity)
400 mg/kg bw per day (maternal toxicity in a study of
developmental toxicity)
Rabbit: 25 mg/kg bw per day (maternal toxicity in a study of
developmental toxicity)
Dog: 125 mg/kg bw per day (one-year study of toxicity)
Human: 0.3 mg/kg bw per day (47-day study of toxicity)
Estimate of acceptable daily intake for humans
0-0.3 mg/kg bw
Studies that would provide information useful for continued
evaluation of the compound
Further observations in humans
References
Abou Zeid, M.M., El-Barouty, G., Abdel-Reheim, E., Blancato, J., Dary,
C., El-Sebae, A.H. & Saleh, M.A. (1993) Malathion disposition in
dermally and orally treated rats and its impact on the blood serum
acetylcholine esterase and protein profile. J. Environ. Sci.
Health, B28, 413-430.
Toxicological criteria for setting guidance values for dietary and non-dietary exposure to malathion
Human exposure Relevant route , study type, species Results, remarks
Short-term Oral, toxicity, rat LD50 = 1000-11 000 mg/kg bw
(1-7 days) Inhalation, toxicity, rat LC50 > 5.2 mg/L
Dermal irritation, rabbit Mildly irritating
Ocular irritation, rabbit Mildly irritating
Dermal sensitization, guinea-pig Not sensitizing
Medium-term Repeated oral, 90 days, rat NOAEL = 34 mg/kg bw per day: systemic toxicity
(1-26 weeks) Repeated dermal, 21 days, rabbit NOAEL = 300 mg/kg bw per day: decreased brain
acetylcholinesterase activity
Repeated oral, developmental toxicity, rabbit NOAEL = 25 mg/kg bw per day: maternal toxicity;
NOAEL = 100 mg/kg bw per day: fetal toxicity
Repeated oral, reproductive toxicity, rat NOAEL = 600 mg/kg bw per day: no parental toxicity;
NOAEL = 130 mg/kg bw per day: developmental
toxicity
Long-term Repeated oral, 2 years, rat NOAEL = 29 mg/kg bw per day: decreased survival,
(> 1 year) reduced body weight, decreased brain
acetylcholinesterase activity
Abraham, S. & Edery, H. (1976) Rapid spontaneous reactivation of
cholinesterase inhibited by malaoxon. Israel J. Med. Sci., 12, 1524.
Agarwal, R. & Matin, M.A. (1981) Effect of oximes and atropine on the
concentration of cerebral glycogen and blood glucose in
malathion-treated rats. J. Pharm. Pharmacol., 33, 795-796.
Aldridge, W.N., Miles, J.W., Mount, D.L. & Verschoyle, R.D. (1979) The
toxicological properties of impurities in malathion. Arch.
Toxicol., 42, 95-106.
Aldridge, W.N., Dinsdale, D. & Nemery, B. (1985) Some aspect of the
toxicology of trimethyl and triethyl phosphorothioates. Fundam.
Appl. Toxicol., 5, S47-S60.
Baker P & Selvey D (1992) Malathion-induced epidemic hysteria in an
elementary school. Vet. Hum. Toxicol., 34, 156-160.
Baker, E.L., Zack, M. & Miles, J.W. (1978) Epidemic malathion
poisoning in Pakistani malaria workers. Lancet, i, 31-34.
Balaji, M. & Sasikali, K. (1993) Cytogenetic effect of malathion in in
vitro culture of human peripheral blood. Mutat. Res., 301, 13-17.
Balasubramanian, K., Ratnakar, C., Ananthanarayanan, P.H. &
Balasubramanian, A. (1987) Histopathological changes in the testis of
malathion treated albino rats. Med. Sci. Res., 15, 509-510.
Boyd, E.M. & Tanikella, T.K. (1969) The acute oral toxicity of
malathion in relation to dietary protein. Arch. Toxicol., 24,
292-303.
Boyes, W.K., Hunter, E., Gary, C. & Peiffer, R.L. (1997) Topical
exposure of the eyes to the organophosphate insecticide malathion:
Lack of visual effects. Unpublished report. Submitted to WHO by the US
Environmental Protection Agency.
Brodeur, J. & DuBois, K.P. (1967) Studies on factors influencing the
acute toxicity of malathion and malaoxon in rats. Can. J. Physiol.
Pharmacol., 45, 621-631.
Bulsiewicz, H., Rozewicka, L., Januszewska, H. & Bajko, J. (1976)
Aberrations of meiotic chromosomes induced in mice with insecticides.
Folio Morphol. (Warsaw), 35, 361-363.
Byeon, W.-H., Hyun, Z.H.H. & Lee, S.Y. (1976) Salmonella/microsomal
enzyme activation system. Korean J. Microbiol., 14, 128-134.
Chen, H.H., Hsueh, J.L., Sirianni, S.R. & Huang, C.C. (1981) Induction
of sister-chromatid exchanges and cell cyle delay in cultured
mammalian cells treated with eight organophosphorus pesticides.
Mutat. Res., 88, 307-316.
Cheema, A.A., Shakoori, A.R. & Shah, F.H. (1989) Studies on
reactivation of malaoxon-inhibited acetylcholinesterase of goat-brain
by pyridine-2-aldoxime methiodide (PAM-2). Pakistan J. Zool., 21,
247-253.
Cooper, D. & Terrell, Y. (1979a) Acute oral LD50 of Fyfanon 771006 KL
(malathion) in Sprague-Dawley rats. Unpublished report (study No.
9E-4269) from Cannon Laboratories Inc., Reading, Pennsylvania, USA.
Submitted to WHO by Cheminova, Lemvig, Denmark.
Cooper, D. & Terrell, Y. (1979b) Acute oral LD50 of Fyfanon 771006 ST
L 1 AAR (malathion) in Sprague-Dawley rats. Unpublished report (study
No. 9E-4268) from Cannon Laboratories Inc., Reading, Pennsylvania,
USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Cyanamid (1990) Letter from F.G. Hess on behalf of the malathion
re-regulation task force. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Daly, I.W. (1993a) A 28-day study of malathion in the rat via dietary
administration. Unpublished report (study No. 92-3806) from
Bio/dynamics Inc, East Millstone, New Jersey, USA. Submitted to WHO by
Cheminova, Lemvig, Denmark.
Daly, I.W. (1993b) A subchronic (3-month) oral toxicity study of
malathion in the rat via dietary administration. Unpublished report
(study No. 92-3843) from Bio/dynamics Inc., East Millstone, New
Jersey, USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Daly, I.W. (1996a) A 24-month oral toxicity/oncogenicity study of
malathion in the rat via dietary administration. Unpublished report
(study No. 90-3641) from Huntingdon Life Sciences, East Millstone, New
Jersey, USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Daly, I.W. (1996b) A 24-month oral toxicity/oncogenicity study of
malaoxon in the rat via dietary administration. Unpublished report
(study No. 93-2234) from Huntingdon Life Sciences, East Millstone, New
Jersey, USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Dauterman, W.C. & Main, A.R. (1966) Relationship between acute
toxicity and in vitro inhibition and hydrolases in a series of
carbaloxy homologs. Toxicol. Appl. Pharmacol., 9, 408-418.
Degraeve, N., Gilot-Delhalle, J., Moutschen, J., Moutschen-Dahmen, M.,
Colizzi, A., Chollet, M. & Houbrechts, N. (1980) Comparison of the
mutagenic activity of organophosphorus insecticides in mouse and in
the yeast Saccharomyes pombe. Mutat. Res., 74, 201-202.
Dinsdale, D. (1992) Pulmonary toxicity of anticholinesterases. In:
Ballantyne, B. & Marrs, T.C., eds, Clinical and Experimental
Toxicology of Organophosphates and Carbamates, Oxford:
Butterworth-Heinemann, pp. 156-166.
Dive, A., Mahieu, P., van Binst, R., Hassoun, A., Lison, D., de
Bisschop, H., Nemery, B. & Lauwerys, R. (1994) Unusual manifestations
after malathion poisoning. Hum. Exp. Toxicol., 13, 271-274.
Dzwonkowska, A. & Hbner, H. (1986) Induction of chromosomal
aberrations in the Syrian hamster by insecticides tested in vivo.
Arch. Toxicol., 58, 152-156.
Ehrich, M., Correll, L. & Veronesi, B. (1994) Neuropathy target
esterase inhibition by organophosphorus esters in human neuroblastoma
cells. Neurotoxicology, 15, 309-314.
Ehrich, M., Jortner, B.S. & Padilla, S. (1995) Comparison of the
relative inhibition of acetylcholinestearse and neuropathy target
esterase in rats and hens given cholinesterase inhibitors. Fundam.
Appl. Toxicol., 24, 94-101.
Ehrich, M., Correll, L. & Veronesi, B. (1997) Acetylcholinesterase and
neuropathy target esterase inhibition in neuroblastoma cells to
distinguish organophosphorus compounds causing acute and delayed
neurotoxicity. Fundam. Appl. Toxicol., 38, 55-63
FAO (1986) International Code of Conduct on the Distribution and
Use of Pesticides, Rome: Food and Agricultural Organization of the
United Nations.
Fischer, J.E. (1988) AC 6,601: A 28-day oral toxicity study in beagle
dogs. Unpublished report (study No. AX88-3) from American Cyanamid
Co., Princeton, New Jersey, USA. Submitted to WHO by Cheminova,
Lemvig, Denmark.
Fischer, J.E. (1991) Oral LD50 study in albino rats with AC 6,601
malathion technical (Cheminova production batch). Unpublished report
No. A91-205. Submitted to WHO by Cheminova, Lemvig, Denmark.
Fletcher, D.W. (1989) 42-Day neurotoxicity study with AC 6,601
technical in mature white Leghorn hens. Unpublished report (study No.
BLAL 87 DN 109) from Bio-Life Associates Ltd, Neillsville, Wisconsin,
USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Foureman, P., Mason, J.M., Valencia, R. & Zimmering, S. (1994)
Chemical mutagenesis testing in Drosophila. X. Results of 70 coded
chemicals tested for the National Toxicology Program. Environ.
Mol. Mutag., 23, 208-227.
Frawley, J.P., Fuyat, H.N., Hagan, E.C., Blake, J.R. & Fitzhugh, O.G.
(1957) Marked potentiation of mammalian toxicity from simultaneous
administration of two anticholinesterase compounds. J. Pharmacol.
Exp. Ther., 121, 96-106.
Gaines, T.B. (1960) The acute toxicity of pesticides in rats.
Toxicol. Appl. Pharmacol., 2, 88-89.
Ganendran, A. & Balabaskaran, B. (1976) Reactivation studies on
organophosphate inhibited human cholinesterases by pralidoxime
(P-2-AM). S.E. Asian J. Trop. Med. Public Health, 7, 417-423.
Garcia-Repetto, R., Martinez, D. & Repetto, M. (1995) Malathion and
dichlorvos toxicokinetics after oral administration of malathion and
trichlorfon. Vet. Hum. Toxicol., 37, 306-309.
Garry, V.F., Nelson, R.L., Griffith, J. & Harkins, M. (1990)
Preparation for human study of pesticide applicators: Sister chromatid
exchanges and chromosome aberrations in cultured human lymphocytes
exposed to selected fumigants. Teratog. Carcinog. Mutag., 10, 21-29.
Gilot-Delhalle, J., Colizzi, A., Moutschen, J. & Moutschen-Dahmen, M.
(1983) Mutagenicity of some organophosphorus compounds at the ade6
locus of Schizosaccharomyces pombe. Mutat. Res., 117, 139-148.
Gudi, R. (1990) Acute test for chemical induction of chromosome
aberration in rat bone marrow cells in vivo with AC 6,601. Unpublished
report (study No. 0125-1531) from Sitek Research Laboratories,
Rockville, Maryland, USA. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Guiti, N. & Sadeghi, D. (1969) Acute toxicity of malathion in the
mongrel dog. Toxicol. Appl. Pharmacol., 15, 244-245.
Gupta, R.C. (1984) Acute malathion toxicosis and related enzymatic
alterations in Bubalus bubalis: Antidotal treatment with atropine,
2-PAM, and diazepam. J. Toxicol. Environ. Health, 14, 291-303.
Hazleton, L.W. & Holland, E.G. (1953) Toxicity of malathion.
Ind. Hyg. Occup. Med., 8, 399-405.
Hobbiger, F. (1973) Reactivation of phosphorylated
acetylcholinesterase. In: Eichler, O., Farah, A. & Koelle, G., eds,
Handbuch der Experimentelle Toxikologie, Berlin: Springer, pp.
921-988.
Huff, J.E., Bates, R., Eustis, S.L., Haseman, J.K. & McConnell, E.E.
(1985) Malathion and malaoxon: Histopathology reexamination of the
National Cancer Institutes carcinogenesis studies. Environ. Res.,
37, 154-173.
IARC (1983) IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Humans, Vol. 30, Miscellaneous Pesticides.
Lyon, pp. 102-129.
Imamura, T. & Talcott, R.E. (1985) Mutagenic and alkylating activities
of organophosphate impurities of commercial malathion. Mutat. Res.,
155, 1-6.
Imlay, P., Parke, G. StE. & Charles, S.J. (1978) The acute dermal LD50
of fyfanon on New Zealand albino rabbits. Unpublished report
(Laboratory No. 7E-7851) from Cannon Laboratories Inc., Reading,
Pennsylvania, USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Ishidate, M., Sofuni, T. & Yoshikawa, K. (1981) Chromsomal aberration
test in vitro as a primary screening tool for environmental mutagens
carcinogens. Gann Monogr. Cancer Res., 27, 95-108.
Ivett, J.L., Brown, B.M., Rodgers, C., Anderson, B.E., Resnick, M.A. &
Zeiger, E. (1989) Chromosomal aberrations and sister chromatid
exchange tests in Chinese hamster ovary cells in vitro. IV. Results
with 15 chemicals. Environ. Mol. Mutag., 14, 165-187.
Jackson, G.C., Hardy, C.J., Gopinath, C. & Lewis, D.J. (1986) Fyfanon
(malathion) 96/98% technical acute inhalation toxicity to rats 4-hour
exposure. Unpublished report (study No. CHV 28/8640) from Huntingdon
Research Centre Ltd, Alconbury, Huntingdon, United Kingdom. Submitted
to WHO by Cheminova, Lemvig, Denmark.
Jokanovic, M. & Maksimovic, M. (1995) A comparison of trimedoxime,
obidoxime, pralidoxime and HI-6 in the treatment of oral
organophosphorus insecticide poisoning in the rat. Arch. Toxicol.,
70, 119-123.
Kalow, W. & Martin, A. (1961) Second generation toxicity of malathion
in rats. Nature, 192, 464-465.
Keadtisuke, S., Dheranetra; W., Nakatsugawa, T. & Fukuto, T.R. (1990)
Liver damage induced in rats by malathion impurities. Toxicol.
Lett., 52, 35-46.
Khera, K.S., Whalen, C. & Trivett, G. (1978) Teratogenicity studies on
linuron, malathion, and methyoxychlor in rats. Toxicol. Appl.
Pharmacol., 45, 435-444.
Kimbrough, R.D. & Gaines, T.B. (1968) Effect of organic phosphorus
compounds and alkylating agents on the rat fetus. Arch. Environ.
Health, 16, 805-808.
Kuhn, J.O. (1996) Fyfanon purified. Final report acute oral toxicity
study in rats. Unpublished report (study No. 2586-95) from Stillmeadow
Inc., Sugar Land, Texas, USA. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Kynoch, S.R. (1985a) Acute oral toxicity to rats of malathion
(Fyfanon) technical. Unpublished report (study No. 85134D/CHV 33/AC)
from Huntingdon Research Centre Ltd, Alconbury, Huntingdon, United
Kingdom. Submitted to WHO by Cheminova, Lemvig, Denmark.
Kynoch, S.R. (1985b) Acute dermal toxicity to rats of malathion
(Fyfanon) technical. Unpublished report (study No. 851330D/CHV 33/AC)
from Huntingdon Research Centre Ltd, Alconbury, Huntingdon, United
Kingdom. Submitted to WHO by Cheminova, Lemvig, Denmark.
Kynoch, S.R. & Smith, P.A. (1985) Delayed contact hypersensitivity in
the guinea-pig with malathion (Fyfanon) technical. Unpublished report
(study No. 8666D/CHV 37/SS,) from Huntingdon Research Centre Ltd,
Alconbury, Huntingdon, United Kingdom. Submitted to WHO by Cheminova,
Lemvig, Denmark.
Lamb, I.C. (1994a) An acute neurotoxicity study of malathion in rats.
Unpublished report (study No. WIL-206005) from WIL Research
Laboratories Inc,. Ashland, USA. Submitted to WHO by Cheminova,
Lemvig, Denmark.
Lamb, I.C. (1994b) A subchronic (13-week) neurotoxicity study of
malathion in rats. Unpublished report (study No. WIL-206006) from WIL
Research Laboratories Inc., Ashland, USA. Submitted to WHO by
Cheminova, Lemvig, Denmark.
Lechener, A. & Abdel-Rahman, Y. (1985) Alterations in rat liver
microsomal enzymes following exposure to carbaryl and malathion in
combination. Arch. Environ. Contam. Toxicol., 14, 451-457.
Liggett, M.P. & Parcell, B.I. (1985a) Irritant effects on rabbit skin
of malathion (Fyfanon) technical. Unpublished report (study No.
851221D/CHV 33/SE) from Huntingdon Research Centre Ltd, Alconbury,
Huntingdon, United Kingdom. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Liggett, M.P. & Parcell, B.I. (1985b) Irritant effects on the rabbit
eye of malathion (Fyfanon) technical. Unpublished report (study No.
851214D/CHV 36/SE) from Huntingdon Research Centre Ltd, Alconbury,
Huntingdon, United Kingdom. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Lochry, E.A. (1989) A developmental toxicity study with AC 6,601 in
rats. Unpublished report (study No. 101-005) from Argus Research
Laboratories Inc., Perkasie, Pennsylvania, USA. Submitted to WHO by
Cheminova, Lemvig, Denmark.
McCann, J., Choi, E., Yamasaki, E. & Ames, B.N. (1975) Detection of
carcinogens as mutagens in the Salmonella/microsome test: Assay of 300
chemicals. Proc. Natl Acad. Sci. USA, 72, 5735- 5739.
Machin, M.G.A. & McBride, W.G. (1989) Teratology study of malathion in
the rabbit. J. Toxicol. Environ. Health, 26, 249-253.
Matsushita, A., Aoyama, K., Yoshimi, K., Fujita, Y. & Ueda, A. (1985)
Allergic contact dermatitis from organophosphorus insecticides.
Ind. Health, 23, 145-153.
Miles, J.W., Mount, D.L. & Staiger, M.A. (1979) S-Methyl isomer
content of stored malathion and fenitrothion water disbursable powders
and its relation to toxicity. J. Agric. Food Chem., 27, 421-425.
Menzer, R.E. & Best, N.H. (1968) Effect of phenobarbital on the
toxicity of several organophosphorus insecticides. Toxicol. Appl.
Pharmacol., 13, 37-42.
Moeller, H.C. & Rider, J.A. (1962) Plasma and red blood cell
cholinesterase activity as indications of the threshold of incipient
toxicity of ethyl-p-nitrophenyl thionobenzenephosphonate (EPN) and
malathion in human beings. Toxicol. Appl. Pharmacol., 4, 123-130.
Mohn, G. (1973) 5-Methyltryptophan resistance mutations in
Escherichia coli K-12. Mutat. Res., 20, 7-15.
Moreno, O.M. (1989) 21-Day dermal toxicity study with AC 6,601 in
rabbits. Unpublished report (study No. MRID 41054201) from MB Research
Laboratories, Inc., Spinnerstown, Pennsylvania, USA. Submitted to WHO
by Cheminova, Lemvig, Denmark.
Morgade, C. & Barquet, A. (1982) Body distribution of malathion and
its metabolites in a fatal poisoning by ingestion. J. Toxicol.
Environ. Health, 10, 321-325.
Moriya, M., Ohta, T., Watanabe, K., Miyazawa, T., Kato, K. & Shirasu,
Y. (1983) Further mutagenicity studies on pesticides in bacterial
reversion assay systems. Mutat. Res., 116, 185-216.
Murphy, S.D. & Cheever, K.L. (1968) Effects of feeding insecticides:
Inhibition of carboxyesterase and cholinesterase activities in rats.
Arch. Environ. Health, 17, 749-758.
Murphy, S.D., Anderson, R.L. & DuBois, K.P. (1959) Potentiation of
toxicity of malathion by triorthotolyl phosphate. Proc. Soc. Exp.
Biol. Med., 100, 483-487.
Myhr, B.C. & Caspary, W.J. (1991) Chemical mutagenesis at the
thymidine kinase locus in L5178Y mouse lymphoma cells: Results for 31
coded compounds in the National Toxicology Program. Environ. Mol.
Mutag., 18, 51-83.
Nagy, Z., Mile, I. & Antoni, F. (1975) The mutagenic effect of
pesticides on Escherischia coli WP2 try-. Acta Microbiol. Acad.
Sci. Hung., 22, 309-314.
Nicholas, A.H., Vienne, M. & van den Berghe, H. (1979) Induction of
sister-chromatid exchanges in cultured human cells by an
organophosphorus insecticide: Malathion. Mutat. Res., 67, 167-172.
Nishio, A. & Uyeki, E.M. (1981) Induction of sister chromatid
exchanges in Chinese hamster ovary cells by organophosphate
insecticides and their oxygen analogs. J. Toxicol. Environ. Health,
8, 939-946.
Ohtaka, K., Hamade, N., Yamazaki, Y., Suzuki, M. & Koizumi, A. (1995)
A direct involvement of the central nervous system in hypophagia and
inhibition of respiratory rate in rats after treatment with
O,O,S-trimethyl phosphorothioate. Arch. Toxicol., 69, 555-564.
Pant, K.J. (1989) Test for chemical induction of unscheduled DNA
synthesis in rat primary hepatocyte cultures by autoradiography with
AC 6,601. Unpublished report (study No. 0125-5100) from Sitek Research
Laboratories, Rockville, Maryland, USA. Submitted to WHO by Cheminova,
Lemvig, Denmark.
Pednekar, M.D., Ghandi, S.R. & Netrawali, M.S. (1987) Evaluation of
mutagenic activities of endosulfan, phosalone, malathion, and
permethrin, before and after metabolic activation in the Ames
Salmonella test. Bull. Environ. Contam. Toxicol., 38, 925-933.
Pellegrini, G. & Santi, R. (1972) Potentiation of toxicity of
organophosphorus compounds containing carboxylic ester functions
toward warm-blooded animals by some organophosphorus impurities.
J. Agric. Food Chem., 20, 944-950.
Reddy, V., Freeman, T. & Cannon, M. (1989) Disposition and metabolism
of 14C-labeled malathion in rats (preliminary and definitive study).
Unpublished report (study No. 9354-B) from Midwest Research Institute,
Kansas City, Missouri, USA. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Rodgers, K. & Xiong, S. (1997) Effect of administration of malathion
for 14 days on macrophage function and mast cell degranulation.
Fundam. Appl. Toxicol., 37, 95-99.
Rucci, G., Becci, P.J. & Parent, R.A. (1980) The evaluation of the
chronic toxicity effects of Cythion administered in the diet to
Sprague-Dawley rats for 24 consecutive months. Unpublished report
(Project No. 5436) from Food and Drug Research Laboratories Inc.,
Waverly, New York, USA. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Rueber, M.D. (1985) Carcinogenicity and toxicity of malathion and
malaoxon. Environ. Res., 37, 119-153.
Ryan, D.L. & Fukuto, T.R. (1984) The effect of isomalathion and
O,S,S-trimethyl phosphorothoate on the in vivo metabolism of malathion
in rats. Pestic. Biochem. Physiol., 21, 349-357.
Salvadori, D.M.F., Ribeiro, L.R., Pereira, C.A.B. & Becak, W. (1988)
Cytogenetic effects of malathion insecticide on somatic and germ cells
of mice. Mutat. Res., 204, 283-287.
Schellenberger, T.E. & Billups, L.H. (1987) One-year oral toxicity
study in purebred beagle dogs with AC 6,601. Unpublished report (study
No. 85010) from Tegeris Laboratories Inc., Laurel, Maryland, USA.
Submitted to WHO by Cheminova, Lemvig, Denmark.
Schanker, H.M., Rachelefsky, G., Siegel, S., Katz, R., Spector, S.,
Rohr, A., Rodriquiz, C., Woloshin, K. & Papanek, P.J. (1992) Immediate
and delayed type hypersensitivity to malathion. Ann. Allergy, 69,
526-528.
Schroeder, R.E. (1990) A two-generation (two litters) reproduction
study with AC 6,601 to rats. Unpublished report (study No. 87-3243)
from Bio/dynamics Inc, East Millstone, New Jersey, USA. Submitted to
WHO by Cheminova, Lemvig, Denmark.
Seely, J.C. (1991) Histopathologic evaluation: The evaluation of the
chronic effects of AC 6,601 administered in the diet to Sprague Dawley
rats for 24 consecutive months. Unpublished report (study No. 90-78)
from Pathco Inc., Research Triangle Park, North Carolina, USA.
Submitted to WHO by Cheminova, Lemvig, Denmark.
Segal, L.M. & Fedoroff, S. (1989) The acute and subchronic effects of
organophosphorus and carbamate pesticides on cholinesterase activity
in aggregate cultures of neural cells from the foetal rat brain.
Toxicol. in vitro, 3, 111-122.
Shiau, S.Y., Huff, R.A., Wells, B.C. & Felkner, I.C. (1980)
Mutagenicity and DNA-damaging activity for several pesticides tested
with Bacillus subtililis mutants. Mutat. Res., 71, 169-179.
Shirasu, Y., Moriya, M., Kato, K., Furuhashi, A. & Kada, T. (1976)
Mutagenicity screening of pesticides in the microbial system.
Mutat. Res., 40, 19-30.
Siglin, J.C. (1985) A teratology study with AC 6,601 in rabbits.
Unpublished report No. 8171 from Food and Drug Research Laboratories,
Inc., Waverly, New York, USA. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Slauter, R.W. (1994) 18-Month oral (dietary) oncogenicity study in
mice, test substance malathion. Unpublished report (study No. 668-001)
from International Research and Development Corp, Mattawan, Michigan,
USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Terrell, Y., Parke, G.StE. & Charles, S.J. (1978) Acute oral LD50 in
rats, compound: malathion technical (Fyfanon). Unpublished report
(Laboratory No. 7E-7850) from Cannon Laboratories, Inc., Reading,
Pennsylvania, USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Thomas, D., Goldharbour, M., Petitti, D., Swan, S.H., Rapperport, E. &
Hertz-Picciotto, I. (1990) Reproductive outcomes in women exposed to
malathion. Am. J. Epidemiol., 132, 794-795.
Toia, R.F., March, R.B., Umetsu, N., Mallipudi, N.M., Allahyari, R. &
Fukuto, T.R. (1980) Identification and toxicological evaluation of
impurities in technical malathion and fenthion. J. Agric. Food
Chem., 28, 605-609.
Traul, K.A. (1987) Evaluation of CL 6601 in the bacterial/microsome
mutagenicity test. Unpublished report (study No. 114) from American
Cyanamid Company, Princeton, New Jersey, USA. Submitted to WHO by
Cheminova, Lemvig, Denmark.
US Environmental Protection Agency (1987) Letter from D.S. Goldman to
R. Linkfield, American Cyanamid Co. Submitted to WHO by Cheminova,
Lemvig, Denmark.
US National Cancer Institute (1978) Bioassay of malathion for possible
carcinogenicity (NCI Technical Report 24), Department of Health,
Education, and Welfare publication (NIH) 78 824. Bethesda, Maryland:
US Department of Health, Education, and Welfare, Public Health
Service.
US National Cancer Institute (1979a) Bioassay of malathion for
possible carcinogenicity (NCI Technical Report 192), Department of
Health, Education, and Welfare publication (NIH) 79 1748. Bethesda,
Maryland: US Department of Health, Education, and Welfare, Public
Health Service.
US National Cancer Institute (1979b) Bioassay of malaoxon for possible
carcinogenicity (NCI Technical Report 135), Department of Health,
Education, and Welfare publication (NIH) 79 1390. Bethesda, Maryland:
US Department of Health, Education, and Welfare, Public Health
Service.
Velazquez, A., Creus, A., Xamena, N. & Marcos, R. (1987) Lack of
mutagenicity of the organophosphorus insecticide malathion in
Drosophila melanogaster. Environ. Mutag., 9, 343-348.
Ward, T.R., Ferris, D.J., Tilson, H.A. & Mundy, W.R. (1993)
Correlation of the anticholinesterase activity of a series of
organophosphates with their ability to compete with agonist binding to
muscarinic receptors. Toxicol. Appl. Toxicol., 122, 300-307.
Waters, M.D., Sandhu, S.S., Simmon, V.F., Mortelmans, K.E., Mitchell,
A.D., Jorgenson, T.A., Jones, D.C.L., Valencia, R. & Garrett, N.E.
(1982) Study of pesticide genotoxicity. Basic Life Sci., 21,
275-326.
WHO (1985) Specifications for Pesticides Used in Public Health, 6th
Ed., Geneva.
WHO (1986) Organophosphorus Insecticides: A General Introduction
(Environmental Health Criteria 63), Geneva.
WHO (1996) The WHO Recommended Classification of Pesticides by
Hazard and Guidelines to Classification 1996-1997 (WHO/PCS/96.3),
Geneva, International Programme on Chemical Safety.
Wong, P.K., Wai, C.C. & Liong, E. (1989) Comparative study on
mutagenicities of organophosphorus insecticides in Salmonella.
Chemosphere, 18, 2413-2422.
Zeiger, E., Anderson, B., Haworth, S., Lawlor, T. & Mortelmans, K.
(1988) Salmonella mutagenicity tests: IV. Results from the testing of
300 chemicals. Environ. Mol. Mutag., 11 (Suppl 12), 1-158.
seen in one male at the highest dose and decreased ambulatory and
motor activity counts in males at this dose on day 0. These males also
showed a > 20% reduction in plasma cholinesterase activity in
comparison with controls at day 7; although no reduction was seen at
1000 mg/kg bw, a marginal reduction was seen at 500 mg/kg bw. No
reductions were seen at other times. In females, reductions > 20%
were seen at both 500 and 2000 mg/kg bw at day 7 and at the latter
dose at day 15. Erythrocyte acetylcholinesterase activity was reduced
by > 20% in the males at the highest dose at day 7, while in females
it was reduced by > 20% at day 0 (500 and 2000 mg/kg bw), day 7 (1000
and 2000 mg/kg bw) and day 15 (2000 mg/kg bw only). No consistent
biologically significant depressions in brain acetylcholinesterase
activity were seen, although there were 10-20% decreases in activity
in comparison with concurrent controls, mainly in the group at the
high dose. There were no treatment-related neuropathological lesions.
There was no NOAEL, as clinical signs occurred in all groups (Lamb,
1994a).
Malathion (purity, 96.4%) was given to groups of 25 male and 25
female Sprague-Dawley Crl:CD:BR rats at dietary concentrations of 0,
50, 5000, or 20 000 ppm (equal to 0, 4, 350, or 1500 mg/kg bw per day
in males and 0, 4, 400, or 1600 mg/kg bw per day in females) for
13 weeks. Clinical signs, body weight, and food consumption were
recorded, a functional observational battery of tests was carried out,
and locomotor activity was evaluated. Plasma, erythrocyte, and
regional brain cholinesterase activities were measured in five animals
of each sex per dose before treatment, at weeks 3 and 7, and at the
end of the study. Tissues from the remaining five animals in each
group were perfused in situ, and neuropathological examinations were
carried out on the brains of the controls and animals at the high
dose. All animals survived to the end of the study.
Anogenital staining was observed in rats at the high dose, and
body-weight gain and food consumption were reduced in comparison with
controls. No treatment-related effects were seen in functional and
locomotor evaluations. Plasma cholinesterase activity was > 20% lower
than in concurrent controls in males at 20 000 ppm at all times after
the start of treatment, while the activity in rats at 5000 ppm was
reduced by 10-20%; erythrocyte acetylcholinesterase activity was
reduced by > 20% at all times in rats at doses > 5000 ppm. In the
females, reductions of > 20% were seen in plasma cholinesterase at
5000 ppm at week 7 only and at 20 000 ppm at all three times (all by
comparison with concurrent controls). Reductions in erythrocyte
acetylcholinesterase activity of > 20 % were seen at all times in
females at 5000 and 20 000 ppm. Regional brain acetylcholinesterase
activity was very variable; significant depressions in activity were
seen only in rats at the high dose. Thus, significant depressions were
seen in the olfactory lobe (by 34%) and midbrain (by 24%) and a
marginally significant depression (18%) in the brain stem of males at
week 13; in the cerebral cortex, a 26% depression in activity was seen
at week 7 only. No clinically or biologically significant depression
in activity was seen in the hippocampus or cerebellum of males. In
females, depressed brain acetylcholinesterase activity was observed
more often and frequently to a greater extent than in the males.
Depressed activity in comparison with concurrent controls was seen in
the olfactory lobe at 3 (31%), 7 (27%), and 13 weeks (50%) and in the
brainstem at 13 weeks (36%), less depression in activity being seen at
the other times. In the midbrain, depressed activity was seen at 7
(34%) and 13 weeks (40%). In the cerebral cortex, depressions were
seen at 3 (32%), 7 (40%), and 13 weeks (53%). In the hippocampus,
depressed activity occurred at all times, by 44% at 3 weeks, 38% at 7
weeks, and 47% at 13 weeks. In the cerebellum, depressions of 20% at 3
weeks and 32% at 13 weeks were seen. No effects were observed on the
absolute or relative weights of the brain or brain regions, and no
neuropathological abnormalities were observed. The NOAEL was 5000 ppm,
equal to 350 mg/kg bw per day, on the basis of the occurrence of
statistically and biologically significant inhibition of brain
acetylcholinesterase activity at the highest dose (Lamb, 1994b).
Malaoxon is a much more powerful anticholinesterase than
malathion, and very pure samples of the latter have little activity
(WHO, 1986). Thus, the IC50 values for cholinesterase inhibition in
17-day-old aggregate cultures of rat neural cells were > 12 × 10-4
mol/L for malathion and 2.8 × 10-4 mol/L for malaoxon (Segal &
Federoff, 1989). Malaoxon produces a dimethylphosphorylated
cholinesterase, however, which rapidly undergoes spontaneous
reactivation, as shown ex vivo in the blood of malaoxon-poisoned
rats, rabbits, dogs, and monkeys (Abraham & Edery, 1976).
Abou Zeid et al. (1993) showed that there was faster recovery of
serum cholinesterase activity in Sprague-Dawley rats after dermal
application of pure malathion than of the 50% emulsifiable
concentrate.
Ward et al. (1993) reported a correlation between the
anticholinesterase activity of a series of organophosphates, including
malathion and malaoxon, and their ability to compete with agonist
binding to muscarinic receptors.
(ii) Other enzyme systems
When rat microsomal suspensions were incubated with 4 mmol/L
malathion in vitro, the release of ß-glucuronidase was inhibited
(Lechener & Abdel-Rahman, 1985).
(iii) Interactions with other organophosphates
Feeding Holtzman rats with fenchlorphos potentiated the effect on
erythrocyte or brain acetylcholinesterase activity of a single
intraperitoneal challenge with malathion at 200 mg/kg bw (Murphy &
Cheever, 1968). The combined effect on dioxathion and malathion was
more or less than additive, depending on the doses used. Malathion
acts synergistically with many other organophosphates, such as
ethyl- para-nitrophenyl thionobenzenephosphonate (Frawley et al.,
1957), at substantial doses. The LD50 of malathion is markedly
reduced by co-administration of tri- ortho-tolylphosphate (Murphy et
al., 1959).
2. Toxicological studies
(a) Acute toxicity
It is likely that the results of earlier studies on malathion
were substantially affected by impurities. The LD50 values for these
impurities in rats after oral administration are: isomalathion, 89-120
mg/kg bw, O,O,S-trimethylphosphorodithioate, 450-660 mg/kg bw,
O,S,S-trimethyl-phosphorodithioate, 26-110 mg/kg bw, and
O,O,S-trimethylphophorothioate, 47-260 mg/kg bw (Aldridge et al.,
1979).
The results of studies on the acute toxicity of malathion are
given in Table 1; those on malaoxon are summarized in Table 2.
(b) Short-term toxicity
Rats
Malathion (purity, 96.4%) was administered in the diet to groups
of five male and five female albino Fischer (CDF:F-344/CrlBR) rats for
29 or 30 days at concentrations of 0, 50, 100, 500, 10 000, or 20 000
ppm, equal to 0, 5.1, 10, 52, 1000, or 2000 mg/kg bw per day for males
and 0, 5.7, 12, 58, 1100, or 2200 mg/kg bw per day for females. The
animals were observed weekly for body weight and food consumption.
Ophthalmological, haematological, and clinical chemical examinations,
including plasma and erythrocyte cholinesterase activity, were
undertaken before treatment and at termination of the study; brain
acetylcholinesterase activity was measured at termination. Animals
were autopsied at the end of treatment, and selected organs were
examined and weighed; microscopic examination of organs was carried
out only in the controls and rats at 20 000 ppm.
No deaths occurred during the study, adverse clinical signs were
not seen, and no abnormalities were present on ophthalmological or
haematological examination. A number of abnormal biochemical variables
were noted, including cholinesterase activity. That in plasma was
decreased by > 20% in animals at the two highest doses in comparison
with concurrent controls, while erythrocyte acetylcholinesterase
activity was decreased in males at 10 000 ppm (by 17%) and 20 000 ppm
(by 16%) and only slightly in females. At 10 000 ppm, brain
acetylcholinesterase activity was depressed at termination by 11% in
males and 17% in females, while at 20 000 ppm there was a 26% decrease
in males and 28% in females. Differences were seen between treated
groups in total protein and albumin concentrations, and a significant
decrease in alkaline phosphatase activity was seen in animals at the
two highest doses. Animals at 20 000 ppm had a significant decrease in
weight gain in comparison with the control group, while food
consumption was decreased only during week 1. The relative and
Table 1. Acute toxicity of malathion
Species Strain Sex Route LD50 and 95% CI, ±SEM, Purity Reference
or range (mg/kg bw, unless (%)
otherwise stated) or LC50
Mouse Swiss white F Oral 6100 > 95 Toia et al. (1980)
Mouse Swiss-Webster M,F Intraperitoneal 985 NR Menzer & Best (1968)
954-1018
Rate Wistar M Oral 2800 NR Dauterman & Main (1966)
2660-3110
Rat Osborne-Mendel NR Oral 1400 ± 100 98 Frawley (1957)
Rat Sherman M Oral 1375 NR Gaines (1969)
1206-1568
F 1000
885-1130
Rat Wistar M,F Oral 1580 92.2 Pellegrini & Santi (1972)
Rat Wistar M,F Oral 8000 98.2 Pellegrini & Santi (1972)
Rat Wistar M Oral (laboratory chow) 1090 ± 83 95 Boyd & Tanikella (1969)
Oral (26% casein) 1401 ± 99
Oral (3.5% casein) 5993 ± 138
Rat Sprague-Dawley M,F Oral 5000 ± 385 NR Terrell et al. (1978)
Rat Sprague-Dawley M Oral 3800 (3040-4750) NR Cooper & Terrell (1979a)
Rat Lac:P Oral 10 700 (9300-12 300) 99.7 Aldridge et al. (1979)
Rat Sprague-Dawley F Oral 4400 (2533-8228) NR Cooper & Terrell (1979a)
Table 1. (continued)
Species Strain Sex Route LD50 and 95% CI, ±SEM, Purity Reference
or range (mg/kg bw, unless (%)
otherwise stated) or LC50
Rat Sprague-Dawley M Oral 3200 (2651-3862) NR Cooper & Terrell (1979b)
F 3700 (2221-6164)
Rat CD Sprague- M Oral 5400 (4100-6900) 96-98 Kynoch (1985a)
Dawley-derived F 5700 (4300-7800)
Rat Albino Crl:CD M Oral 6156 (4665-8123) 96.8 Fischer (1991)
(SD)BR F 4061 (3078-5359)
Rat Wistar M Oral 734 NR Jokanovic & Maksimovic
F (1995)
Rat HSD Sprague- M Oral 8210 (6518-10 342) 99.1 Kuhn (1996)
Dawley F 8239 (6239-10 881)
Rat Sprague-Dawley M Intraperitoneal 1100 NR Murphy et al. (1959)
Rat Sherman M,F Derman (57% > 44 444 NR Gaines (1969)
emulsifiable
concentrate)
Rat CD Sprague- M,F Derman > 2000 96-98 Kynoch (1985b)
Dawley-derived
Rat Albino Wistar M,F Inhalation (4 h) > 5.2 mg/L 96-98 Jackosn et al. (1986)
Rabbit New Zealand M,F Derman 8.79 ± 0.48 NR Imlay et al. (1978)
albino
Hamster Syrian F Intraperitoneal (30% 24 00 NR Dzwonkowska &
commercial Hubner (1986)
preparation)
Table 1. (continued)
Species Strain Sex Route LD50 and 95% CI, ±SEM, Purity Reference
or range (mg/kg bw, unless (%)
otherwise stated) or LC50
Dog Mongrel NR Oral > 4000 98 Frawley (1957)
Dog Mongrel M Intraperitoneal 1.517 ml/kg bw 95 Guiti & Sadoghi (1969)
0.77-2.25
Buffalo Indian (Bubalus M Oral 100-125 NR Gupta (1984)
bubalis)
Chicken White Leghorn F Oral 775 93.6 Fletcher (1989)
610-984
NR, not reported
Table 2. Acute toxicity of malaoxon
Species Strain Sex Route LD50 and 95% CI, ±SEM, Purity Reference
or range (mg/kg bw, unless (%)
otherwise stated)
Mouse Swiss white F Oral 215 NR Toia et al. (1980)
Rat Sprague-Dawley M,F Intraperitoneal About 25 NR Brodeur & DuBois (1967)
Rat Wistar M Oral 158 NR Dauterman & Main (1966)
142-175
NR, not reported
absolute weights of the liver were increased in males at the highest
dose and in females at the two highest doses, and periportal
hepatocytic hypertrophy was seen at the two highest doses in animals
of each sex. These changes were considered to be related to treatment.
Increased relative kidney weights were observed in males at the two
highest doses and in females at the highest dose. The NOAEL was 500
ppm, equal to 52 mg/kg bw per day, on the basis of the increased
weight of the livers with histopathological changes and inhibition of
brain acetylcholinesterase activity (Daly, 1993a).
Malathion (purity, 96.4%) was administered in the diet to groups
of 10 male and 10 female albino Fischer (CDF:F-344/CrlBR) rats for 90
days at concentrations of 0, 100, 500, 5000, 10 000, or 20 000 ppm,
equal to 0, 6.6, 34, 340, 680, or 1400 mg/kg bw per day for males and
0, 7.9, 39, 380, 780, or 1600 mg/kg bw per day for females. Body
weight and food consumption were estimated before treatment and
periodically during the study. Ophthalmological, haematological, and
clinical chemical examinations, including plasma, erythrocyte, and
brain cholinesterase activities, were undertaken before treatment and
at termination of the study. The animals were killed at least 90 days
after the start of the study and autopsied. Selected organs were
examined and weighed, and all animals were examined microscopically.
One male at the high dose died during the study from unknown
cause. Anogenital staining was seen in four males and six females at
the high dose during treatment, and in animals at this dose, body
weights and weight gain were consistently lower than in the control
group; there was a decrease in food consumption only in week 1, in
contrast to greater food consumption by animals at the high dose than
by controls later in the study. Haemoglobin count and haematocrit were
decreased in males at the high dose, while the mean corpuscular volume
and mean cell haemoglobin were decreased in males at doses > 5000
ppm. In females, the erythrocyte count was marginally increased at
doses > 500 ppm, while the mean corpuscular volume was decreased at
10 000 and 20 000 ppm, and the mean cell haemoglobin was decreased at
doses > 5000 ppm. A number of abnormal biochemical variables were
noted, including cholinesterase activity. Plasma cholinesterase
activity was decreased marginally (17%) in males at 5000 ppm, while
there was clearly significant depression at the higher doses in
comparison with the values in concurrent controls. In female rats,
plasma cholinesterase activity was depressed by > 20% at doses >
5000 ppm. In the males, erythrocyte acetylcholinesterase activity was
marginally but significantly depressed at 500 ppm (by 18% in
comparison with concurrent controls) and markedly depressed at higher
doses. In the females, depression of erythrocyte acetylcholinesterase
activity by > 20% was observed at all doses. At 10 000 ppm, there was
marginally significant depression of brain acetylcholinesterase
activity at termination (by 13% in males and 17% in females), while at
20 000 ppm there was biologically significant depression, by 20%, in
males and 44% in females At 5000 ppm, there was less inhibition of
brain acetylcholinesterase activity in animals of each sex (8.8% in
males and 10% in females), which was, however, statistically
significant. Significant decreases in alkaline phosphatase activity
were seen in males at the three highest doses and in females at the
highest dose. The activity of gamma-glutamyl transpeptidase was
elevated in males at the highest dose and in females at the two
highest doses. A reduction in aspartate aminotransferase activity was
observed in females at the highest dose.
Differences in relative and absolute liver and kidney weights
were seen between groups, with associated histopathological changes.
The absolute and relative weights of the liver were increased in males
at doses > 5000 ppm and in females at the highest dose. Periportal
hepatocyte hypertrophy was seen in males at doses > 10 000 ppm and
in females at doses > 5000; these changes were considered to be
related to treatment. The absolute and relative weights of the kidney
were increased in animals of each sex at the highest dose. At 10 000
ppm, the absolute and relative kidney weights were increased in males
and the relative kidney weights in females; at 5000 ppm, the relative
kidney weights were increased in animals of each sex. Chronic
nephropathy was more severe in males at doses > 5000 ppm than in
those at lower doses or in controls, but there were no differences
between groups in the prevalence of this pathological change. The
NOAEL was 500 ppm on the basis of decreased mean corpuscular volume
and mean corpuscular haemoglobin, increased liver weights and relative
kidney weights, and chronic nephropathy in males at the next highest
dose and decreased mean corpuscular haemoglobin, hepatocytic
hypertrophy, and increased relative kidney weight in the females at
the next highest dose. There were also marginally significant
decreases in brain acetylcholinesterase activity at 5000 ppm. The
finding of a marginal increase in erythrocyte count at 500 ppm in
females is ignored. The NOAEL is equal to 34 mg/kg bw per day (Daly,
1993b).
Rabbits
Malathion (purity, 94%) was applied to the skin of groups of six
male and six female New Zealand white rabbits for 6 h per day on five
days per week for three weeks at doses of 50, 300, or 1000 mg/kg bw
per day; six males and five females were sham-treated. Effects on the
skin, organ and body weights, food consumption, clinical chemical
parameters including cholinesterase activity, and haematological
variables were evaluated; selected organs were examined
pathologically. Two males, one at 50 mg/kg bw per day and one at the
high dose, died before termination of the study,. There were no
physical alterations or changes in body or organ weights or food
consumption attributable to treatment, except for erythema and oedema
in treated animals. Decreases in plasma, erythrocyte, and brain
cholinesterase activity > 20% were seen at the highest dose in
animals of each sex; females also showed depression of erythrocyte
activity at 300 mg/kg bw per day. Brain acetylcholinesterase activity
was substantially reduced in the cerebrum and cerebellum of animals of
each sex at the highest dose. Females at 300 mg/kg bw per day had a
19% reduction in brain acetylcholinesterase activity in comparison
with concurrent controls, but this reduction was not statistically
significant. The NOAEL was 300 mg/kg bw per day on the basis of
inhibition of brain acetylcholinesterase activity at the highest dose
(Moreno, 1989).
Dogs
Malathion (purity, 92.4%) was administered to groups of three
male and three female beagles in gelatin capsules at doses of 0, 125,
250, or 500 mg/kg bw per day for 28 days. The animals were observed
twice daily, and haematological and clinical chemical measurements
were made before treatment and 15 and 29 days after the start of
treatment. Selected organs were weighed and examined post mortem.
Diarrhoea was observed at all doses, and anorexia at the highest
dose. One male at the high dose died. Weight gain was reduced at the
highest dose and marginally so at 250 mg/kg bw per day; food
consumption was reduced at the highest dose. At 15 days, serum albumin
and sodium levels were decreased in dogs at the highest dose, as were
blood urea nitrogen, aspartate aminotransferase activity, and
creatinine. Decreased plasma and erythrocyte cholinesterase activities
were seen at 15 days and at termination: plasma cholinesterase
activity was decreased by > 20% in dogs at the intermediate and high
doses at 15 days and at all doses at termination; erythrocyte
acetylcholinesterase activity was decreased by 20% at the high dose by
15 days and by 17% at all doses at termination. The LOAEL was 125
mg/kg bw per day, on the basis of reduced erythrocyte
acetylcholinesterase activity at all doses at termination of the
study, with clinical signs (diarrhoea) at all doses. No NOAEL was
identified (Fischer, 1988).
Malathion (purity, 95%) was administered in capsules to groups of
six male and six female beagles at doses of 0, 62.5, 125, or 250 mg/kg
bw per day on seven days a week for one year. The animals were
observed twice daily, with more detailed examinations weekly; they
were weighed before the start of the study, at the beginning of
treatment, weekly thereafter, and at sacrifice. Food consumption was
measured weekly, and haematological and clinical chemical variables,
including plasma and erythrocyte cholinesterase activity, were
determined before treatment, at six weeks, three months, six months,
and just before the animals were killed. Cerebellar and cerebral
acetylcholinesterase activity was determined at termination of the
study. Ophthalmological examination was carried before the start of
treatment and just before sacrifice.
No clinical signs of toxicity were observed, nor was any
abnormality seen on ophthalmological examination. No significant
difference was seen in body weight or food consumption, although
animals of each sex at the high dose showed a small decrease in mean
weight. Clinical chemistry revealed perturbations in a number of
variables. Plasma and erythrocyte cholinesterase activities were
decreased by more than 20% in animals of each sex at all doses and
times in comparison with concurrent controls. Brain
acetylcholinesterase activity was unaffected, except for some
diminution in cerebellar acetylcholinesterase activity at the highest
dose (by 16% in males and 11% in females); cerebral cholinesterase
activity was unaffected. Serum albumin, total protein, the
albumin:globulin ratio, and calcium levels were decreased and lactate
dehydrogenase increased in animals of each sex, generally only at the
high dose but occasionally in those at the intermediate dose. In
females, albumin was reduced at all doses at six weeks. Alkaline
phosphatase activity was increased in males at the high dose but not
in females. Other changes observed occasionally included low blood
urea nitrogen in animals at the high dose and decreased alanine
aminotransferase activity in those at the intermediate and high doses,
but these did not appear to be of toxicological significance.
Significantly lower calcium levels were found in animals at the high
dose at six weeks and later. Haematological examination revealed
dose-related decreases in erythrocyte and haemoglobin counts and
haematocrit. The erythrocyte and haemoglobin counts were decreased at
all times in males at the high dose, and the haematocrit was decreased
in the males at the high dose at three and six months. In the females,
erythrocyte and haemoglobin counts and haematocrit were marginally
affected at six weeks in the group at the high dose, but at three
months the haematocrits were decreased at the intermediate and high
doses and haemoglobin count at the high dose. Additionally there was
an increase in mean corpuscular volume at the high dose and in mean
corpuscular haemoglobin at the intermediate and high doses. At six
months, the erythrocyte and haemoglobin counts and haematocrit were
all decreased in females at the high dose and the erythrocyte count in
those at the intermediate dose. At termination, only a decrease in
erythrocyte count was seen in females at the highest dose. Platelet
counts were increased in males and females at all times and all doses
in comparison with concurrent controls; many of these changes were
statistically significant. Urinalysis revealed no marked changes. The
absolute liver weights of females at the low and high doses were
increased, and the relative liver weights were raised in males at the
intermediate and high doses and in females at all doses. The absolute
and relative kidney weights were raised in animals at the intermediate
and high doses. No treatment-related pathological alteration was seen
macroscopically or microscopically. The NOAEL was 125 mg/kg bw per day
on the basis of body-weight depression and haematological and clinical
chemical changes. The changes in liver weights and the reduced albumin
in females at six weeks are discounted on the grounds that there was
no morphological correlate and that there was no clear dose-response
relationship (Schellenberger & Billups, 1987).
(c) Long-term toxicity and carcinogenicity
A number of long-term studies have been carried out in mice and
rats by the US National Cancer Institute and others. Many of the
studies were reviewed by IARC (1983) and by Rueber (1985). The working
group convened by IARC concluded that the available data did not
provide evidence that malathion or malaoxon is carcinogenic in humans.
This view is in line with those of the study authors but not with
those of Rueber (1983). More modern studies are now available and are
summarized below, with a brief resumé of the earlier studies for the
sake of completeness.
Mice
Groups of 50 B6C3F1 mice of each sex were given doses of 0,
8000, or 16 000 ppm malathion admixed in the diet for 80 weeks,
equivalent to 1200 or 2400 mg/kg bw per day. The animals were killed
14-15 weeks after discountinuation of the malathion-containing diets.
Throughout the study, the mean body weights of animals of each sex
were lower than those of controls; poor food consumption,
hyperexcitability, and abdominal distention were also noted in the
second year; tremors were seen in a few female mice. Malathion was
reported not to be carcinogenic (US National Cancer Institute, 1978);
however, the slides were re-examined by Rueber (1985), who concluded
that malathion had increased the incidence of neoplasms of the liver
in male mice.
Malathion (purity, 96.4%) was administered in the diet to groups
of 65 B6C3F1 BR mice of each sex for 18 months at concentrations of 0,
100, 800, 8000, or 16 000 ppm (equal to 0, 17, 140, 1500, or 3000
mg/kg bw per day for males and 0, 21, 170, 1700, and 3500 mg/kg bw per
day for females). Animals were observed twice daily and examined in
detail weekly. Body weights were determined weekly until week 14,
fortnightly to week 26, and monthly thereafter. Plasma, erythrocyte,
and brain cholinesterase activity was determined 10 animals of each
sex from each group killed at 12 months and at termination; only
erythrocyte enzyme activity was determined in 10 mice of each sex per
group at week 36, and these mice were retained until terminal
sacrifice of the survivors at 18 months. The mice were examined
post mortem, and selected organs were processed and examined
histologically.
There was no treatment-related effect on mortality, but body
weights and food consumption were reduced in animals of each sex at
8000 and 16 000 ppm. Plasma cholinesterase activity was reduced by
> 20% in comparison with concurrent controls at 12 and 18 months in
males at 800 ppm; similar results were seen in females, except that
the reduction was 18% in those at 800 ppm by 12 months. Erythrocyte
acetylcholinesterase activity was decreased by > 20% in animals of
each sex at doses > 800 ppm at 9, 12, and 18 months, while brain
acetylcholinesterase activity was decreased in males at the highest
dose at 12 and 18 months and in those at 8000 ppm at 18 months. In
females, brain acetylcholinesterase activity was not decreased at 8000
ppm at 12 months but was decreased at 18 months; females at 16 000 ppm
showed a 20% decrease in brain acetylcholinesterase activity at
12 months and a 43% decrease at 18 months. The absolute and relative
liver weights were increased in males at the two highest doses, and
the relative liver weight was increased in females at the highest
dose. Other changes in organ weights included increased relative
kidney weights in certain groups.
Macroscopically, an increased incidence of liver nodules was seen
at the two highest doses; microscopically, effects were seen on the
liver, kidney, adrenal cortex, and bone. Liver hepatocellular
hypertrophy was observed in all animals at the two highest doses at
termination, and milder hypertrophy was seen in the animals killed at
12 months. The incidences of liver tumours in animals that survived to
termination are given in Table 3. There was a significant trend in the
incidence of adenomas in animals of each sex, and the incidence was
significantly raised by comparison with controls at the two highest
doses; the incidences in historical controls at the same laboratory
were 14-22% in males and 0-11% in females. There was no significant
trend for hepatocellular carcinoma, but the incidence was
significantly raised in the males at 100 and 8000 ppm.
Proximal tubular vacuolation seen at lower doses in the kidneys
was absent in all males at the highest dose and most of those at 8000
ppm. Female mice at 8000 and 16 000 ppm had an increased incidencse of
renal cortical mineralization. A treatment-related decrease in fibrous
osteodystrophy of the sternum observed in females at the highest dose
is of unknown significance. A treatment-related, early disappearance
of the X zone of the adrenal cortex was observed in females at 8000
and 16 000 ppm at 12 months. The overall NOAEL was 800 ppm, equal to
140 mg/kg bw per day, on the basis of inhibition of brain
acetylcholinesterase activity and an increased incidence of liver
adenomas in animals of each sex at the next highest dose (Slauter,
1994).
Rats
Malathion
Three studies were carried out by Hazleton Laboratories (Hazleton
& Holland, 1953). In the first, groups of 20 male Colworth Farm rats
were given technical-grade malathion (purity, 65%) in the diet at
doses of 0, 100, 1000, or 5000 ppm, equivalent to 5, 50, and 250 mg/kg
bw per day, for 109 weeks. Body weight and food consumption were
decreased in those at the highest dose. Depressed cholinesterase
activity was seen in rats at 5000 ppm and to a lesser extent in those
at 1000 ppm. The study was not adequate for conclusions to be drawn
about carcinogenicity. In the second study, with the same doses,
similar criticisms can be made, except that the purity of the
malathion was > 90%. A third study was carried out with male and
female Colworth Farm rats which received doses of 0, 500, 1000, 5000,
or 20 000 ppm of malathion (purity, 99%). The highest dose was lethal
to the male rats; the size of the study precluded conclusions about
carcinogenicity.
Groups of 50 male and 50 female Osborne-Mendel rats were given
technical-grade malathion (purity, 95%) in the diet at concentrations
of 4700 or 8150 ppm (equivalent to 240 and 410 mg/kg bw per day) for
80 weeks. A pooled control group consisted of 15 matched controls of
each sex and 40 untreated male and female rats from bioassays of other
chemicals. The rats were killed after 109 weeks. The body weights of
female rats receiving malathion were lower than those of controls, and
the survival times of those at the higher dose were decreased.
Malathion was reported not to be carcinogenic (US National Cancer
Institute, 1978). The slides were re-evaluated by Rueber (1985), who
Table 3. Incidences of hepatocellular tumours (%) in mice at terminal sacrifice after treatment with malathion
Tumour Dose (ppm)
Males Females
0 100 800 8000 16 000 0 100 800 8000 16 000
Adenoma 2 11.8 4.2 24.1 98.0 0 1.9 0 17.9 82.4
Carcinoma 0 11.8 4.2 11.1 2.0 1.8 0 3.8 1.9 3.9
found that the incidence of benign and malignant neoplasms at all
sites analysed together was increased in treated rats; in particular,
the incidence of carcinomas of the endocrine organs was increased, and
malignant neoplasms of the brain were observed in seven treated rats.
Rueber concluded that malathion was carcinogenic in male and female
Osborne-Mendel rats. In a re-evaluation of the slides commissioned by
the US National Toxicology Program (Huff et al., 1985), the original
interpretation that malathion is not carcinogenic was confirmed.
In a second study by the US National Cancer Institute (1979a),
malathion (purity, 95%) was fed in the diet to groups of 50 male and
50 female Fischer 344 rats at doses of 0, 2000, or 4000 ppm
(equivalent to 100 or 200 mg/kg bw per day; only 49 males at the
higher dose) for 103 weeks. The authors concluded that malathion was
not carcinogenic. When Rueber (1985) re-evaluated the slides, he found
that the incidence of benign and malignant neoplasms analysed together
was significantly increased, particularly in males. He concluded that
malathion was carcinogenic in Fischer 344 rats. A re-evaluation of the
slides commissioned by the US National Toxicology Program (Huff et
al., 1985) confirmed that malathion was not carcinogenic.
Malathion (purity, 92.1%) was given in the diet to groups of 50
male and 50 female Sprague-Dawley rats at concentrations of 0, 100,
1000, or 5000 ppm, equivalent to 5, 50, or 250 mg/kg bw per day. The
animals were observed daily throughout the study, and body weights and
food consumption were recorded at the end of weeks 1, 13, 24, 53, 79,
and 103. Blood samples for haematological examination and
determination of cholinesterase activity and urine samples for
urinalysis were collected from five rats of each sex per group in
weeks 12, 26, and 53; blood and urine were also collected at week 104,
and alanine and aspartate aminotransferase activities, urea nitrogen,
and glucose were determined additionally in blood. Brain
acetylcholinesterase activity was not determined. Animals that died,
were killed in extremis, or killed at termination were examined
post mortem.
No significant difference in food consumption or survival was
seen, and no significant intergroup differences were seen on
haematological or biochemical examination, except in cholinesterase
activity. During the first year of the study, the body weights of
animals of each sex at the highest dose were reduced, while in the
second year the body weights of those at 1000 and 5000 ppm were
depressed. Moreover, erythrocyte acetylcholinesterase activity was
reduced by > 20% in comparison with the controls in rats at the
intermediate and high doses at 3, 6, 12, and 24 months; plasma
cholinesterase activity was less affected, although there was a
depression of > 20% in the females at the high dose at 12 and 24
months. Absolute and relative liver weights were increased in male
rats at the high dose and relative kidney weights in males at the two
highest doses. Absolute brain weights were decreased in females at the
two highest doses and relative kidney weights in those at the high
dose.
Although foci of cellular alteration were recorded twice in the
livers of males at the highest doses and once in females at the
intermediate and high doses, this difference was not statistically
significant. A significant difference in sinusoidal dilatation was
found between the controls (2%) and males at the high dose (16%).
Extramedullary splenic haematopoiesis was seen more often in males at
the high dose than in controls. There was no evidence of carcinogenic
potential. The NOAEL was 100 ppm, equivalent to 5 mg/kg bw per day, on
the basis of reduced erythrocyte acetylcholinesterase activity and
body weight at the next highest dose (Rucci et al., 1980). After an
audit by the US Environmental Protection Agency (1987; Cyanamid,
1990), the Agency requested a re-evaluation of the slides. Seely
(1991) found only two treatment-related lesions: periportal
hepatocellular hypertrophy and cystic hepatocellular degeneration,
both only in male rats at the highest dose. There was no evidence of
differences in tumour incidence. The NOAEL for effects on the liver
was thus 1000 ppm (equivalent to 50 mg/kg bw per day). This
re-evaluation does not alter the overall NOAEL of 5 mg/kg bw per day
(see above).
Malathion (purity, 96.4%) was administered in the diet of groups
of 90 male and female Fischer 344 (CDF:F-344/CrlBR) rats at
concentrations of 0, 100/50, 500, 6000, or 12 000 ppm for two years;
the lowest dose was reduced from 100 to 50 ppm at week 18 because of
inhibition of erythrocyte acetylcholinesterase activity, resulting in
mean intakes over the entire study of 0, 4, 29, 360, and 740 mg/kg bw
per day for males and 0, 5, 35, 420, and 870 mg/kg bw per day for
females. Groups of 10 animals of each sex per group were killed at
three and six months, 15 of each sex per group at 12 months, and the
remainder at two years. Physical condition, ophthalmoscopic
parameters, body weight, and food consumption were determined before
treatment and at selected intervals, while electroretinography,
haematology, and clinical chemistry (including determination of
cholinesterase activity) were performed at selected intervals and on
selected animals. Selected organs from animals killed at 12 and 24
months were weighed, and the animals were examined macroscopically.
Tissues from those at the high dose and from controls, and certain
organs from animals at the low dose were examined histopathologically.
Malathion reduced the survival of males at 6000 and 12 000 ppm,
early deaths being observed from the 14th month in males at the
highest dose and from about the 20th month in those at the next lowest
dose. Survival of females at the high dose was impaired towards the
end of the study. Nephropathy and mononuclear-cell leukaemia were the
main causes of death, although the frequency of neither was
treatment-related. Anogenital staining was seen in females at the
highest dose. Decrements in body weight and weight gain were seen in
animals of each sex throughout the study at the two highest doses,
although mean food consumption was greater in these animals than in
the controls, throughout the study in the case of the males and in the
second year of the study in the case of the females. Decreases in mean
haemoglobin concentration, haematocrit, mean corpuscular volume, and
mean cell haemoglobin were seen in animals of each sex at the two
highest doses at 6, 12, and 18 months, although all parameters were
not affected at all the time intervals and there was a tendency for
improvement during the study. The mean cell haemoglobin concentration
was decreased in males at the two highest doses only at 12 months,
accompanied by an increase in platelet count.
At 3, 6, 12, and 24 months, animals of each sex showed reductions
in plasma, erythrocyte, and brain cholinesterase activity,
predominantly at the two highest doses. Thus, animals of each sex at
the highest dose had decreased plasma cholinesterase activity at all
times. In males at 6000 ppm, decreases in activity > 20% were seen at
three and six months and at termination, while there was a marginal
decrease at 12 months (83% of control value). Males at 500 ppm had a
significant decrease in activity only at termination, when the
activity was 71% of that of concurrent controls. In the females,
plasma cholinesterase activity was consistently reduced by at the two
highest doses; the activity was little affected at 500 ppm, except at
termination when there was a marginally significant decrease of 18%.
There were consistent reductions in erythrocyte acetylcholinesterase
activity at the two highest doses in males and a marginally
significant reduction at 500 ppm at termination only, when the
activity was 83% that of concurrent controls. Erythrocyte
acetylcholinesterase activity was similarly reduced in females at 6000
and 12 000 ppm. Although reductions > 20% were seen in females at 500
ppm at three months and at termination and a marginal reduction at 12
months (86% of control value), no reduction was seen at six months.
Erythrocyte acetylcholinesterase activity was also reduced in females
at the lowest dose at three months (75% of control value), so that on
day 113 the lowest dose was reduced from 100 ppm to 50 ppm. Six weeks
later, erythrocyte acetylcholinesterase activity was evaluated in 10
controls and 10 at 50 ppm and found to be comparable. Thereafter, the
activity in animals at 50 ppm was unremarkable. Brain
acetylcholinesterase activity was reduced by > 20% in males at 6000
ppm at termination. Decreases seen at the highest dose were 84% of the
control value at three months, 81% at six months, and 85% at 12
months; no determination was carried out at termination because there
were no survivors. At 6000 ppm, the activity was decreased to 88% of
the control value at three months, 88% at six months, and 89% at 12
months. In females at 12 000 ppm, the activity of brain
acetylcholinesterase was substantially reduced in comparison with that
of concurrent controls. Smaller decreases were seen at 6000 ppm at
three months (15%), six months (17%), 12 months (12%), and termination
(18%). No significant inhibition was seen at lower doses.
Alkaline phosphatase activity was reduced in comparison with
concurrent controls in animals of each sex at the two highest doses at
6 and 12 months and at the highest dose at 18 months. Aspartate
aminotransferase activity was reduced in females at doses > 500 ppm
at 12 months and at the highest dose at 18 months. Alanine
aminotransferase activity was also decreased in females at the three
highest doses at 12 months. gamma-Glutamyl transpeptidase activity was
increased consistently in males at the two highest doses from 12
months and at most intervals in females. Cholesterol content was
increased in animals of each sex at the two highest doses at 6, 12,
18, and 24 months. Increases in mean and relative liver and kidney
weights were observed in animals of each sex at the two highest doses
at the interim sacrifice, in females at 6000 and 12 000 ppm at
terminal sacrifice, and in males at 6000 ppm at terminal sacrifice.
Males also had decreased relative kidney weights at 500 ppm. Relative
spleen weight was increased in males at the two highest doses at
interim sacrifice, and absolute spleen weight was reduced in males at
6000 ppm and in females at 12 000 ppm at terminal sacrifice. Relative
and absolute thyroid and parathyroid weights were elevated in males at
the two highest doses at interim sacrifice, in males at 6000 ppm at
termination, and in females at 6000 and 12 000 ppm at termination.
Microscopic findings of significance were largely confined to
nasoturbinal tissues, kidney, and liver. Degeneration and hyperplasia
of the olfactory epithelium were seen in animals of each sex at the
two highest doses. The hyperplasia was focal, with thickening of the
epithelium and proliferation of basal cells, forming clusters in the
lamina propria. While focal degeneration was also observed in a few
controls and rats at lower doses, the hyperplasia was confined to
those at the two highest doses. In several rats, the epithelium was
replaced by ciliated and non-ciliated columnar epithelium. Subacute
and chronic inflammation and dilated and hyperplastic mucosal glands
were seen in some animals; subacute and chronic inflammation and
hyperplasia of the respiratory epithelium of the nasopharynx and
dilated mucosal glands were also seen. Like the other changes in nasal
tissues, inflammatory cells and cell debris were seen most frequently
at the two highest doses. Thus, the NOAEL for this effect was 500 ppm.
The incidence and severity of nephropathy was greater in rats at 6000
and 12 000 ppm than in controls.
A nasal turbinate adenoma was seen in one male at 6000 ppm and a
carcinoma in one male at 12 000 ppm; although the numbers observed
were small, this is a rare tumour in Fischer rats. Hepatocellular
adenomas and carcinomas were seen in some animals. In the female rats,
the prevalence of adenomas and cacinomas combined was increased at the
highest dose and that of adenomas alone at 6000 ppm (see Table 4).
Testicular interstitial-cell tumours were seen in virtually all male
rats that survived to termination. The overall NOAEL was 500 ppm,
equal to 29 mg/kg bw per day, on the basis of decreased survival and
body-weight gain, increased food consumption, changes in
haematological parameters, decreased brain acetylcholinesterase
activity, increased g-glutamyl transpeptidase activity, increased
liver, kidney, thyroid, and parathyroid weights, and degeneration and
hyperplasia of the olfactory epithelium at the next highest dose.
Although an increased incidence of liver tumours was seen in females,
malathion was not considered to be carcinogenic in view of the small
numbers of such tumours observed (Daly, 1996a).
Table 4. Prevalences of hepatocelular adenomas and carcinomas in rats at termination after treatment with malathion
Dose (ppm)
Males Females
0 100/50 500 6000 12 000 0 100/50 500 6000 12 000
No. of animals 37 41 29 14 0 38 41 41 34 20
Tumour
Adenoma 2 2 3 1 0 0 0 1 3 3
Carcinoma 1 1 0 1 0 0 1 1 0 1
Malaoxon
Malaoxon (purity, > 95%) was fed to groups of 50 Fischer 344
rats of each sex in the diet at concentrations of 0, 500, or 1000 ppm
for 103 weeks. The authors concluded that malaoxon was not
carcinogenic in rats (US National Cancer Institute, 1979b). The slides
were re-examined by Rueber (1985), who concluded that the incidence of
benign and malignant neoplasms at all sites was increased in the
treated animals. The neoplasms in question were in endocrine organs,
including the pituitary, adrenal, and thyroid glands; the incidences
of hyperplasia, adenomas, and carcinomas of C cells of the thyroid
were increased. In a re-evaluation of the slides commissioned by the
US National Toxicology Program (Huff et al., 1985), the original
conclusion that malaoxon is not carcinogenic was largely confirmed,
but there was stated to be equivocal evidence that malaoxon is
carcinogenic in that there was an increased incidence of C-cell
neoplasms of the thyroid.
Groups of 85 Fischer 344 (CDF:F-344/CrlBR) rats of each sex were
exposed to malaoxon (purity, 96.4%) admixed in the diet at
concentrations of 0, 20, 1000, or 2000 ppm (equal to 1, 57, or
110 mg/kg bw per day in males and 1, 68, or 140 mg/kg bw per day in
females); 55 animals were retained for 24 months, while 10 of each sex
per group were killed at 3, 6, and 12 months. Cholinesterase activity
was estimated in all animals. Clinical chemical and haematological
parameters were determined in all animals killed at 6 and 12 months
and in 10 animals of each sex per group of animals that were retained
at 18 months and termination. Physical observations, ophthalmoscopy,
and measurements of body weight and food consumption were carried out
before treatment and at selected intervals during the study. The
surviving rats were sacrificed at 24 months. The rats were examined
post mortem, and selected organs were weighed. Histopathological
examinations were perfomed on controls and those at the high dose at
12 and 24 months and on rats that died or were killed in extremis
during the study. Selected tissues from animals at the intermediate
and low doses were also examined.
Survival was curtailed in female rats at 1000 and 2000 ppm and in
males at 2000 ppm. The most common causes of death were pneumonitis
and mononuclear leukaemia; the occurrence of the former appeared to be
dose-related. Anogenital staining was seen in females at the highest
dose throughout the study and in males in the latter part of the
study. Treatment-related decreases in body weight and weight gain were
seen at the highest dose. Food consumption was decreased in males at
1000 and 2000 ppm. Plasma, erythrocyte, and brain cholinesterase
activity was affected by malathion. Plasma cholinesterase activity was
reduced by > 20% at all times in animals of each sex at the two
highest doses. Erythrocyte acetylcholinesterase activity was reduced
by > 20% in the same groups and in males at the lowest dose at six
months; the reductions at other times in animals of each sex at this
dose were 10-20%. Brain acetylcholinesterase activity was reduced by
18-11% in comparison with concurrent controls in males at the highest
dose and more clearly reduced in females at earlier times. There were
substantial reductions in brain acetylcholinesterase activity in
animals of each sex at the highest dose at termination of the study.
At the intermediate dose, there was a 30% decrease in brain
acetylcholinesterase activity in males at termination.
No abnormality was seen on ophthalmoscopic examination. Although
there were sporadic differences in clinical chemical measurements
between groups, none appeared to be treatment-related. The absolute
and relative liver and kidney weights were increased in males at 2000
ppm at 12 months, and the relative and absolute adrenal weights were
increased in males at this dose at two years. The absolute and
relative spleen weights of females at 2000 ppm were decreased. The
incidence of emaciation: was increased in males at 2000 ppm and in
females at 1000 and 2000 ppm. Inflammatory changes in the nasal
turbinates, lungs, and tympanic spaces, which may have been secondary
to increased disposition of food particles, were present in males at
the highest dose and females at the two highest doses. Thus, foreign
material such as food was found in the nasal lumen with inflammatory
cells and cell debris. The nasal mucosa also showed chronic
inflammatory changes and hyperplasia and hypertrophy of goblet cells.
In a small number of rats, squamous metaplasia was observed.
Degeneration of the olfactory epithelium was accompanied by focal
replacement by ciliated and non-ciliated columnar epithelium.
Mineralization of the stomach was seen in males at the two highest
doses and females at the highest dose. No treatment-related neoplasia
was observed. Interstitial tumours of the testis were present in
> 75% of the animals at all doses and were not considered to be
related to treatment. The NOAEL was 20 ppm, equal to 1 mg/kg bw per
day, on the basis of decreased food consumption and brain
acetylcholinesterase activity at termination in males and emaciation
at termination and inflammatory changes in the nasal turbinates in
females at the next highest dose (Daly, 1996b).
(d) Genotoxicity
The results of tests for the genotoxicity of malathion and
malaoxon are shown in Table 5. Four impurities in malathion,
isomalathion, O,O,S-trimethyl phosphorothioate, O,S,S-trimethyl
phosphorodithioate, and O,O,O-trimethyl phosphorothioate of > 99%
purity were tested for their potential to induce reverse mutation in
S. typhimurium TA97, TA98, and TA100 at doses of 10-1000 µg/plate.
Negative results were obtained, with and without metabolic activation
and with and without preincubation (Imamura & Talcott, 1985).
(e) Reproductive toxicity
(i) Multigeneration reproductive toxicity
In a small study in Wistar rats given malathion in the diet at
about 240 mg/kg bw, a higher incidence of ring-tail disease was seen
in treated than in control rats (Kalow & Marton, 1961).
Table 5. Results of tests for the genotoxicity of malathion and malaoxon
End-point Test system Concentration Purity Results Reference
(%)
Malathion
In vitro
Reverse mutation S. typhimurium NR NR Negativea McCann et al. (1975)
TA98, TA100,
TA1535, TA1537
Reverse mutation S. typhimurium 100-5000 µg/plate 95.2 Negativea Traul (1987)
TA98, TA100,
TA1535, TA1537,
TA1538
Reverse mutation S. typhimurium 5-300 µg/plate NR Positive Shiau et al. (1980)
TA98, TA100, (TA 1535
TA1535, TA1536, without S9)
TA1537, TA1538
Reverse mutation S. typhimurium 33-165 µg/plate NR Negativeb Pednekar et al. (1987)
TA97a, TA98, TA100
Reverse mutation S. typhimurium NR NR Negative Byeon et al. (1976)c
TA98, TA100,
TA1535, TA1538
Reverse mutation S. typhimurium 80 and 400 ppm/plate 90-95 Negativea Wong et al. (1989)
TA98, TA102,
TA1535, TA1537
Reverse mutation S. typhimurium < 5000 µg/plate NR Negativea Moriya et al. (1983)
TA98, TA100,
TA1535, TA1537,
TA1538
Table 5. (continued)
End-point Test system Concentration Purity Results Reference
(%)
Reverse mutation S. typhimurium < 10 mg/plate NR Negativea Waters et al. (1982)
TA98, TA100,
TA1535, TA1537,
TA1538
Reverse mutation S. typhimurium NR (preincubation) NR Positive Ishidate et al. (1981)
TA98, TA100, (TA 100 with
TA1537 S9)
Reverse mutation Escherischia coli NR NR Negativea Nagy et al. (1975)
Reverse mutation Escherischia coli 100-5000 µg/plate 95.2 Negativea Traul ( t 987)
Reverse mutation Escherischia coli < 5000 µg/plate NR Negativea Moriya et al. (1983)
Reverse mutation Escherischia coli < 10 mg/plate NR Negativea Waters et al. (1982)
Forward mutation Escherischia coli 2 × 20 mol/L NR Negative Mohn (1973)
Forward mutation Schizosaccharomyces NR 99 Negative Degraeve et al. (1980)
pombe
Forward mutation Schizosaccharomyces 30-182 mmol/L NR Negativea Gilot-Delhalle et al.
pombe (1983)
DNA damage Bacillus subtilis 5-300 µg/plate NR Positive Shiau et al. (1980)
rec and exc
DNA damage Bacillus subtilis rec 200 µg/plate NR Negative Shirasu et al. (1976)
DNA damage Bacillus subtilis rep NR NR Negative Waters et al. (1982)
Table 5. (continued)
End-point Test system Concentration Purity Results Reference
(%)
DNA damage Bacillus subtilis rew NR NR Negative Waters et al. (1982)
Primary DNA Saccharomyces NR NR Negative Waters et al. (1982)
damage cerevisiae
Unscheduled Primary rat 0.01-0.16 µl/ml 94 Negative Pant (1989)
DNA synthesis hepatocytes
Unscheduled Human lung NR NR Negativea Waters et al. (1982)
DNA synthesis fibroblasts
Chromosomal Chinese hamster NR NR Positivea Ishidate et al. (1981)
aberration lung fibroblasts
Chromosomal Cultured human 0.02-20 µg/ml NR Positive Balaji & Sasikali (1993)
aberrations peripheral leukocytes
Sister chromatid Cultured human 0.02-20 µg/ml NR Positive Balaji & Sasikali (1993)
exchange peripheral leukocytes
Chromosomal Cultured human 33-660 µg/ml NR Positivea Garry et al. (1990)
aberrations peripheral leukocytes
Sister chromatid Cultured human 33-660 µg/ml NR Positivea Garry et al. (1990)
exchange peripheral leukocytes
Sister chromatid Human fetal fibroblasts 2.5-40 µg/ml 99 Positive Nicholas et al. (1979)
exchange
Table 5. (continued)
End-point Test system Concentration Purity Results Reference
(%)
Sister chromatid Chinese hamster V79 10-80 µg/ml 94 Positive Chen et al. ( 1981)
exchange/cell cells (high dose)
cycle delay
Sister chromatid Chinese hamster ovary 0.03-1 mmol/L 99 Positive Nishio & Yueki (1981)
exchange cells
In vivo
Sex-linked recessive Drosophila melanogaster Feeding: adult, 50 ppm; 50% Negative Velazquez et al. (1987)
lethal mutation larva, 100 ppm ECd
Injection: adult, 10 and
25 ppm
Sex-linked recessive Drosophila melanogaster NR NR Negative Waters et at. (1982)
lethal mutation
Sex chromosome Drosophila melanogaster Feeding: 0-10 ppm 50% Negative Velazquez et al. (1987)
loss adult Injection: 0 and 5 ppm ECd
Non-disjunction Drosophila melanogaster Feeding: 0-20 ppm Ecd 50% Negative Velazquez et al. (1987)
Dominant lethal Mouse NR NR Negative Degraeve et al. (1980)
mutation
Dominant lethal Mouse 'Maximum lethal dose' NR Negative Waters et al. (1982)
mutation
Chromosomal Rat bone marrow 0.5-2.0 g/kg 94 Negative Gudi (1990)
aberration (Sprague-Dawley)
Chromosomal Syrian hamster bone 0.24-1.2 g/kg 30e,f Positive Dzwonkowska &
aberration marrow Hubner (1986)
Table 5. (continued)
End-point Test system Concentration Purity Results Reference
(%)
Chromosomal CFW mouse spermatocytes 0.45 mg/day for 30f Positive Bulsiewicz et al. (1976)
aberration 50 or 100 days
Chromosomal Mouse bone marrow and 500-2000 mg/kg bw NR Positive Salvadori et al. (1988)
aberration primary spermatocytes single dose or 5 daily
doses dermally
Malaoxon
In vitro
Reverse mutation S. typhimurium 100-10 000 µg/plate 94.4 Negativea Zeiger et al. (1988)
TA97, TA98, TA100,
TA1535, TA1537
Cell mutation Mouse lymphoma 12.5-300 nl/ml NR Positive Myhr & Caspary (1991)
tk locus L5178Y cells without S9;
equivocal with
S9
Sister chromatid Chinese hamster ovary 0.03-1 mmol/L 96 Positive Nishio & Yueki (1981)
exchange cells
Chromosomal Chinese hamster ovary > 5 mg/ml 94.4 Negative Ivett et al. (1989)
aberration cells
Sister chromatid Chinese hamster ovary > 5 mg/ml 94.4 Positivea Ivett et al. (1989)
exchange cells
In vivo
Sex-linked recessive Drosophila melanogaster Feeding: 5 ppm 94.4 Positive Foureman et al. (1994)
lethal mutation Injection: 2 ppm feeding;
negative,
injection
Table 5. (continued)
NR, not reported; S9, 9000 × g supernatant of rodent liver
a With and without metabolic activation
b With and without metabolic activation with S9 and caecal microbial extract
c In Korean; not fully evaluated
d 50% emulsifiable concentrate
e 30% commercial preparation from Organika-Azot, Poland
f Sadofos-30 (approximately 30% solution)
In a two-generation study of reproductive toxicity, groups of 25
male and 25 female CD:Sprague-Dawley-derived rats were fed diets
containing malathion (purity, 94%) at concentrations of 0, 550, 1700,
5000, or 7500 ppm, equal to 0, 43, 130, 390, or 600 mg/kg bw per day
in the F0 males and 0, 50, 150, 440, and 660 mg/kg bw per day in the
F0 females. The equivalent intakes for the F1 generation were: 43,
130, 390, or 630 mg/kg bw per day for males and 51, 150, 460, and 750
mg/kg bw per day for females. Each parent generation was mated to
produce two litters, and offspring were selected randomly from the
second (F1b) litter to be the parents of the next generation.
Offspring that were not selected, the offspring of the first litters
(F1a and F2a), and the F2b offspring were examined grossly and
discarded. One pup of each sex per F1b and F2b litter was selected
randomly, killed, and examined post mortem; abnormal tissues were
saved. The F0 and F1 adults were killed and examined post mortem,
and the reproductive organs and abnormal tissues were saved. Tissues
from the controls and animals at the high dose were examined
histologically.
Treatment had no effect on clinical signs, growth before mating,
food consumption, maternal weight gain during gestation, reproductive
performance, fertility indices, gestation length, or parturition in
the F0 and F1 parental generations. Pup sex ratio and survival were
also unaffected. Pup weight was reduced at day 21 in the F1a litters
at 5000 and 7500 ppm and in the F1b litters at 7500 ppm. Mean pup
weights in the F2a litters were comparable in all groups, except in
those at the high dose on day 21, which were decreased. Pup weights
were reduced at days 4, 7, 14, and 21 in the F2b litters at 5000 ppm
but not at 7500, except on day 21. At the highest dose, mean pup
weight at day 21 was lower than that of concurrent controls. Similar
effects were not seen at lower doses. Examination post mortem showed
no treatment-related effects. The NOAEL for reproductive toxicity was
7500 ppm, equal to 600 mg/kg bw per day, while that for developmental
toxicity was 1700 ppm, equal to 130 mg/kg bw per day (Schroeder,
1990).
In a study reported briefly, malathion (purity unspecified) was
administered to 16 male JIPMER albino rats for 12 weeks at a dose of
45 mg/kg bw per day by gavage. There were 12 appropriate controls. The
histological changes observed included interstitial oedema,
congestion, desquamation of cells lining the seminiferous tubules,
reduced numbers of spermatogonia, and absence of Leydig cells
(Balasubramanian et al., 1987).
(ii) Developmental toxicity
Female Sherman-strain rats were pair-mated with healthy adult
male rats of about the same age. On day 11 after insemination, they
were given a single intraperitoneal injection of malathion at a dose
of 700 or 900 mg/kg bw. On day 20 of gestation, the fetuses were
removed. The offspring and placentae were weighed, the numbers of
resorptions and dead animals were recorded, and half of the offspring
were examined in detail. The higher dose of malathion affected the
body weight of the dams but had no effect on the weight of the fetuses
and did not induce malformations (Kimbrough & Gaines, 1968).
Technical-grade malathion (purity unspecified) was administered
by gavage to groups of 20 female Wistar rats at doses of 0, 50, 100,
200, or 300 mg/kg bw on days 6-15 of gestation. Neither maternal nor
fetal toxicity was observed at the highest dose used (Khera et al.,
1978).
After a dose-ranging study, groups of female Crl:CD:(SD)BR rats
were given malathion (purity, 94%) by gavage on days 6-15 of gestation
at doses of 0, 200, 400, or 800 mg/kg bw per day in corn oil; the
groups consisted of 24 rats at the lowest dose and 25 at the other
doses. The animals were observed daily for clinical signs, body-weight
gain, and food consumption. After 20 days, the rats were killed and
examined for pregnancy, implantations, resorptions, live and dead
fetuses, and number of corpora lutea; the uterus was weighed, and
fetuses were examined for malformations. Cholinesterase activity was
not measured in this study. Malathion had no effect on survival, the
only early death occurring in the control group. Five rats at the
highest dose had urine staining of abdominal fur and decreased mean
weight gain and food consumption during treatment; after treatment,
the weight gain of animals at the high dose was increased in
comparison with the controls. No effect was seen on pregnancy rate or
numbers of corpora lutea, implantations, resorptions, or fetuses per
litter, fetal body weight, or sex ratio of fetuses. No fetal
abnormality attributable to treatment was observed. The NOAEL was 400
mg/kg bw per day on the basis of maternal toxicity at the highest
dose. The NOAEL for fetal toxicity was 800 mg/kg bw per day (Lochry,
1989).
Malathion (70% with 30% calcium carbonate) was administered at a
dose of 100 mg/kg bw per day on days 7-12 of gestation to seven mated
New Zealand white rabbits. Five mated rabbits received the vehicle
alone. No difference between the treated and control groups was seen
in respect of resorptions, fetal size, or external or visceral
abnormalities. The NOAEL for effects on the fetus was thus 100 mg/kg
bw per day. It is unclear from the paper whether any significant
maternal toxicity was observed (Machin & McBride, 1989).
After a range-finding study, malathion (purity, 92.4%) was
administered at doses of 25, 50, or 100 mg/kg bw per day by gavage in
corn oil to groups of 20 mated female New Zealand white rabbits on
days 6-18 of gestation; controls received the vehicle alone. The
rabbits were examined daily for mortality and for physical and
behavioural abnormalities. Body-weight gain was calculated for days
0-6, 6-12, 6-18, 18-29, and 0-29 days after the start of the study and
on days 6, 12, 18, and 29. The survivors were killed on gestation day
29 and examined post mortem. The uterus and ovaries were excised and
examined, and the number of corpora lutea recorded. The number and
position of live and dead fetuses, resorption sites, and the total
number of implantation sites was also recorded. Live fetuses were
removed, weighed, measured crown to rump, and examined for gross
external and visceral abnormalities. Fetuses were then processed and
examined for skeletal abnormalities. Cholinesterase activity was not
measured.
Although there was no statistically significant difference in
survival between the treated and controls groups, no deaths occurred
among the controls, four in the group at the low dose, three in the
group at the intermediate dose, and two in the group at the high dose.
In the last group, the deaths resulted from intrapulmonary intubation.
Maternal weight gain was reduced at the doses of 50 and 100 mg/kg bw
per day during treatment on days 6-18 of gestation. At days 12, 18,
and 29, the mean body weight of the animals at the high dose was
decreased in comparison with the controls. The mean number and percent
of resorptions was slightly increased at doses > 50 mg/kg bw per
day. There was no difference in fertility, number of corpora lutea,
implantation sites, litter size, or fetal weight or length. No other
signs of toxicity were seen in does or fetuses, nor was there any
evidence of teratogenicity. The NOEAL was 25 mg/kg bw per day for
maternal toxicity and 100 mg/kg bw per day for fetal toxicity, the
former being based on decreased weight gain at the next highest dose
and the latter on the absence of fetal toxicity at any dose (Siglin,
1985).
(f) Special studies
(i) Dermal and ocular irritation and dermal sensitization
A single semi-occluded application of malathion (purity, 96-98%)
to the skin of New Zealand white rabbits elicited slight to
well-defined, transient dermal reactions, with very slight oedema in
six animals and very slight erythema in five animals; the sixth had
well-defined erythema. The skins were all normal by day 2 (Liggett &
Parcell, 1985a).
Malathion (purity, 96-98%) produced mild conjunctival reactions
in the eyes of New Zealand white rabbits. No damage to the cornea or
iris was seen at any stage, and the eyes were normal after two days
(Liggett & Parcell, 1985b).
Malathion (purity, 96-98%) was tested in nine albino guinea-pigs;
there were 10 controls. One treated animal with respiratory distress
was killed in extremis. There was no evidence of delayed contact
hypersensitivity (Kynoch & Smith, 1985).
(ii) Macrophage and mast cell function
Repeated administration of malathion to female C57Bl/6 mice at a
dose of 1 mg/kg bw per day increased macrophage function, while 0.1
mg/kg bw per day caused mast cell degranulation (Rodgers & Xiong,
1997).
(iii) Ocular function
Malathion (purity, 98%) instilled into the eyes of Long-Evans
rats had no effect on responses evoked by a visual pattern and
produced no ophthalmological abnormality (Boyes, 1997).
(iv) Neurotoxicity
The potential of malathion (purity, 93.6%) to induce delayed
neuropathy was tested in white Leghorn hens. After determination of
the oral LD50, the ability of atropine to antagonize the effects of
malathion at doses greater than the LD50 was investigated. In the
main study, 60 birds received malathion at a dose of 1000 mg/kg bw
(1.3 times the unprotected LD50). They were given atropine sulfate
subcutaneously at 10 mg/kg bw 1 h before administration of malathion
and then at 30 mg/kg bw 15 min and 1, 3, and 5 h afterwards. A total
of 39 birds died with clinical signs consistent with cholinesterase
activity poisoning within 15 days. Three weeks after the first dose of
malathion, the survivors were dosed again, this time at 850 mg/kg bw
(1.1 times the LD50), with atropinization as above. A further seven
birds died, but the survivors recovered completely. Positive controls
were treated with tri- ortho-tolylphosphate at 500 mg/kg bw. The hens
were observed daily; body weights and food consumption were recorded
at the start of the study, and body weight was recorded thereafter at
three-day intervals. All dead birds were examined with perfusion
fixation, and the brain, spinal cord, and sciatic nerve were examined
histologically No treatment-related histopathological changes were
seen in the birds treated with malathion, whereas those treated with
tri- ortho-tolylphosphate showed changes typical of
organophosphate-induced delayed polyneuropathy in the spinal cord and
sciatic nerve. Clinical signs of delayed polyneuropathy were seen only
in the positive control birds (Fletcher, 1989).
Groups of 12 retired laying Leghorn hens were given malathion
orally at doses of 75, 150, or 300 mg/kg bw, and groups of 12
Long-Evans rats (28 rats at the high dose) received 600, 1000, or
2000 mg/kg bw. All received atropine pretreatment, and some received
subsequent treatment with atropine. Clinical assessments were carried
out. Cholinesterase and neuropathy target esterase activities were
estimated, and sections of the medulla, cervical and lumber spinal
cord, and branches of the tibial nerve were examined. Flaccid
paralysis was seen in the hens at 300 mg/kg bw for about 24 h, but
none died. There was no clinical indication of delayed polyneuropathy
in the hens treated with malathion, whereas those given
tri- ortho-tolylphosphate, mipafox, or diisopropylphosphorofluoridate
developed typical behavioural signs of neuropathy. Malathion at a dose
of 2000 mg/kg bw induced clinical signs consistent with cholinesterase
poisoning in the rats, and gait changes were observed 14-21 days after
administration at the highest dose. No treatment-related
histopathological changes were seen in the birds or rats treated with
malathion, whereas those treated with tri- ortho-tolylphosphate,
mipafox, or diisopropyl phosphorofluoridate showed changes typical of
organophosphate-induced delayed polyneuropathy. The activities of both
acetylcholinesterase and neuropathy target esterase were inhibited by
malathion. In the hens, brain acetylcholinesterase activity was
inhibited by 17 ± 3% at the lowest dose of malathion and by 76 ± 1% at
the highest; the corresponding inhibition of neuropathy target
esterase activity was 0 ± 3% and 50 ± 22%. In the rats,
acetylcholinesterase activity inhibition was 26 ± 6% at the lowest
dose and 56 ± 2% at the highest; the corresponding figures for
neuropathy target esterase inhibition were 19 ± 7% and 75 ± 5%, all
compared with concurrent controls (Ehrich et al., 1995).
Malathion and malaoxon produced nugatory inhibition of neuropathy
target esterase activity in human neuroblastoma cells (3 and 1%,
respectively) (Ehrich et al., 1994).
The ratio of the IC50 value for neuropathy target esterase to
that for acetylcholinesterase was 30 000 in murine neuroblastoma cells
and 76 000 in human cells (Ehrich et al., 1997).
(v) Antidotes
Malaoxon, the oxon analogue of malathion, inhibits cholinesterase
activity by producing a dimethylphosphoryl derivative, which is
susceptible to oxime-induced reactivation. Experimental evidence
indicates that clinically significant reactivation occurs (Hobbiger,
1973). Most authors have found significant reactivation with oximes,
but there are notable exceptions. For example, Ganendran and
Balabaskaran (1976) found little reactivation of malathion- and
malaoxon-inhibited human whole-blood cholinesterase activity. Similar
results were obtained for acetylcholinesterase activity in goat brain
(Cheema et al., 1989).
The efficacy of four pyridinium oximes, trimedoxime, obidoxime,
pralidoxime, and HI-6, in the treatment of poisoning by malathion
(purity, 96%) was tested in male Wistar rats, which were given
malathion at twice the LD50, as determined experimentally during the
study, and treated with 30 mol/kg bw of trimedoxime, obidoxime, or
HI-6 or 60 mol/kg bw of pralidoxime; atropine and diazepam therapy
were also used. Of the oximes, obidoxime was the most effective,
followed by trimoxime, and then pralidoxime and HI-6, which were
equally effective; however, better survival was achieved with HI-6 at
a dose of 150 mol/kg bw than with the other regimes. Thus, malathion
posoning can be treated with mono- and bis-pyridinium oximes
(Jokanovic & Maksimovic, 1995).
Malathion (50% emulsifiable concentrate) was administered at a
dose of 100 mg/kg bw or at a minimally lethal dose of 125 mg/kg bw to
buffalo calves (Bubalus bubalis). Pralidoxime methiodide combined
with atropine was reported to reverse the clinical evidence of
toxicity (Gupta, 1984).
Pralidoxime chloride or diacetyl monoxime at a dose of 100 mg/kg
bw intraperitoneally reversed the rise in blood glucose observed after
injection of malathion at 500 mg/kg bw to female albino rats, provided
the oximes were given immediately after the malathion. When given 15
min later, the antidotes were ineffective (Agarwal & Matin, 1981).
Obidoxime reversed malaoxon-induced inhibition of cholinesterase
activity in isolated rat diaphragm and restored the ability to sustain
tetany; moreover, obidoxime at 20 mg/kg bw together with atropine
raised the LD50 in OF mice by 5.1-fold, the comparable figure for
atropine alone being 1.7-fold (Abraham & Edery, 1976).
Obixodime has been used successfully in treating malathion
poisoning in humans (Dive et al., 1994; see below).
3. Observations in humans
Malathion and ethyl- para-nitrophenyl thionobenzenephosphonate
(purity of each unspecified) were tested in volunteer male prisoners
aged 23-36 years. In phase I of the study, four samples of blood were
taken during two weeks before the start of the study for measurements
of plasma and erythrocyte cholinesterase activity, and then malathion
was administered at a dose of 8 mg/day to five subjects for 32 days.
Phase II was begun three weeks after completion of phase I. On the two
days before its start, samples of plasma and washed erythrocytes were
taken for measurements of cholinesterase activity, and then malathion
was administered to the same five subjects at a dose of 16 mg/day for
47 days. In phase III, five new subjects were selected; plasma and
erythrocyte cholinesterase activity was determined in blood samples
drawn twice weekly for 36 days, before administration of malathion at
a dose of 24 mg/day for 56 days.
During phase I, no clinical effects were observed, and there were
no changes in blood counts or the results of urinalysis. Furthermore,
no significant depression of plasma or erythrocyte cholinesterase
activity was observed in any subject. Similarly, no clinical effects
were observed in phase II. In phase III, depression of plasma
cholinesterase activity was observed two weeks after the first
administration of malathion, the maximum depression being 25%, seen
three weeks after cessation of treatment; erythrocyte
acetylcholinesterase activity was depressed to the same extent. The
NOAEL was 6 mg/day, approximately equal to 0.27 mg/kg bw per day. No
potentiation of malathion by ethyl- para-nitrophenyl
thionobenzenephosphonate was observed (Moeller & Rider, 1962).
An epidemic of poisoning of spraymen in Pakistan was attributed
largely to contaminants, particularly iso-malathion, in the
formulation of malathion used (Baker et al., 1978).
There are numerous case reports of individual poisoning. Thus,
Dive et al. (1994), reported an instance in which an elderly woman
consumed about 100 ml of a garden preparation containing 15% malathion
in isopropyl alcohol. The preparation also contained
isopropylmalathion and O,O,S-trimethylphosphorothioate. A typical
cholinergic crisis was followed by cardiac, pulmonary, neurological,
and renal manifestations, and the patient was treated with atropine
and obidoxime. The cardiac manifestations included arrhythmia and
conduction disturbances. Mild interstitial pulmonary fibrosis was
observed in a lung biopsy sample.
Matsushita et al. (1985) reported allergic contact dermatitis in
people exposed to organophosphorus insecticides including malathion.
Thomas et al. (1990) reported a study of women exposed to
malathion during aerial spraying of large areas of the San Francisco
Bay area, USA, in 1981-82. A number of associations were found, of
which one, congenital gastrointestinal anomalies, remained
statistically significant after control for confounders.
An episode of epidemic hysteria was reported at an elementary
school in Arizona, USA, in response to the smell of malathion (Baker &
Selvey, 1992).
During a campaign to eradicate Mediterranean fruit flies with a
pesticide based on malathion, a number of cases of urticaria,
angioneurotic oedema, and non-specific rashes were reported. Of 10
subjects that received a patch test, none responded. One case of
possible immediate reaction to malathion bait was reported (Schanker
et al., 1992).
No case of organophosphate-induced delayed polyneuropathy due to
malathion has been reported in humans.
Comments
Malathion is rapidly absorbed, biotransformed, and excreted,
predominantly in the urine but also in the faeces, largely as its two
monocarboxylic acids and the dicarboxylic acid.
The oral LD50 values for malathion in laboratory rodents were
1000-10 000 mg/kg bw, the observed differences probably being due to
impurities. The most recent LD50 values tend to be higher. The
cholinesterase-inhibiting metabolite of malathion, malaoxon, has much
lower oral LD50 values of 100-220 mg/kg bw. WHO has classified
malathion as slightly hazardous (WHO, 1996).
In a study of neurotoxicity in rats receiving single doses of 0,
500, 1000, or 2000 mg/kg bw, there was no NOAEL, as clinical signs
were present at all doses. In a 13-week study of neurotoxicity, also
in rats, at dietary concentrations of 0, 50, 5000, or 20 000 ppm, the
NOAEL was 5000 ppm, equal to 350 mg/kg bw per day, on the basis of
inhibition of brain acetylcholinesterase at the highest dose.
In a 30-day study of toxicity in rats receiving malathion in the
diet at concentrations of 0, 50, 100, 500, 10 000, or 20 000 ppm, the
NOAEL was 500 ppm, equal to 52 mg/kg bw per day, on the basis of
increased liver weight and histopathological changes in the liver
(periportal hepatocyte hypertrophy) at the next highest dose.
In a 90-day study of toxicity in rats, malathion was given at
dietary concentrations of 0, 100, 500, 5000, 10 000, or 20 000 ppm.
The NOAEL was 500 ppm, equal to 34 mg/kg bw per day, on the basis of
decreased mean corpuscular volume and mean corpuscular haemoglobin,
increased liver weights and relative kidney weights, and chronic
nephropathy in males and decreased mean cell volume, hepatocyte
hypertrophy, and increased relative kidney weight in females at the
next highest dose.
A 21-day study of dermal toxicity was carried out in which
rabbits were treated with malathion at doses of 0, 50, 300, or 1000
mg/kg bw per day for 6 h per day, five days per week. The NOAEL was
300 mg/kg bw per day on the basis of inhibition of brain
acetylcholinesterase activity at the highest dose.
In a 28-day study of toxicity in dogs, malathion was fed in
gelatin capsules at doses of 0, 125, 250, or 500 mg/kg bw per day for
28 days. There was no NOAEL because of clinical signs at all doses.
In a one-year study of toxicity in dogs, malathion was
administered orally in capsules at doses of 0, 62.5, 125, or 250 mg/kg
bw per day on seven days per week. The NOAEL was 125 mg/kg bw per day
on the basis of body-weight depression and changes in haematological
and clinical chemical parameters at the highest dose.
A number of long-term studies of toxicity and carcinogenicity
have been carried out on malathion in both rats and mice. The earlier
ones were reviewed by a working group convened by the IARC, which
concluded that the available data did not provide evidence that
malathion was carcinogenic.
In an 18-month study in mice, malathion was administered at
dietary concentrations of 0, 100, 800, 8000, or 16 000 ppm. The NOAEL
was 800 ppm, equal to 140 mg/kg bw per day, on the basis of inhibition
of brain acetylcholinesterase activity at termination and an increased
incidence of liver adenomas in animals of each sex at the next highest
dose.
In a two-year study in rats, dietary concentrations of 0, 100,
1000, or 5000 ppm were used. The NOAEL was 100 ppm, equivalent to 5
mg/kg bw per day, on the basis of reduced erythrocyte
acetylcholinesterase activity and body weight. In another long-term
study in rats, malathion was given at doses of 0, 100/50, 500, 6000,
or 12 000 ppm for two years. The NOAEL was 500 ppm, equal to 29 mg/kg
bw per day, on the basis of decreased survival and body-weight gain,
changes in haematological parameters, decreased brain
acetylcholinesterase activity, increased g-glutamyl transpeptidase
activity, increased liver, kidney, and thyroid/parathyroid weights,
and changes in the olfactory epithelium at the next highest dose.
Numerous tests have been carried out for genotoxicity both in
vitro and in vivo. Most of the evidence indicates that malathion
is not genotoxic, although some studies indicate that it can produce
chromosomal aberrations and sister chromatid exchange in vitro.
There was no evidence that malathion induces chromosomal aberrations
in vivo. Malaoxon did not induce reverse mutation in bacteria, but
it caused sister chromatid exchange in two tests in mammalian cells
and induced sex-linked recessive lethal mutation in Drosophila in
vivo. The four common impurities of malathion, isomalathion,
O,O,S-trimethyl phosphorothioate, O,S,S-trimethyl
phosphorodithioate, and O,O,O-trimethyl phosphorothioate, did not
induce reverse mutation in bacteria. The Meeting concluded that
malathion is not genotoxic.
A number of studies of reproductive toxicity have been carried
out, only some of which showed NOAELs. In a study in rats, malathion
was administered by gavage to groups of pregnant animals on days 6-15
of gestation at doses of 0, 200, 400, or 800 mg/kg bw per day. The
NOAEL was 400 mg/kg bw per day on the basis of maternal toxicity at
the highest dose; no fetal toxicity was observed.
Malathion was administered orally at doses of 0, 25, 50, or 100
mg/kg bw per day to groups of pregnant rabbits on days 6-18 of
gestation. The NOAELs were 25 mg/kg bw per day for maternal toxicity
and 100 mg/kg bw per day for fetal toxicity; teratogenicity was not
seen at any dose.
A two-generation study was undertaken in rats in which malathion
was given at dietary concentrations of 0, 550, 1700, 5000, or 7500
ppm. The NOAEL was 7500 ppm, equal to 600 mg/kg bw per day, for
reproductive toxicity and 1700 ppm, equal to 130 mg/kg bw per day, for
developmental toxicity, the latter being based on reduced pup weights.
Two studies on the neurotoxicity of malathion in hens were
reviewed. In neither was there evidence that malathion can cause
delayed neuropathy, although some inhibition of neuropathy target
esterase activity was found in the brains of birds at 2000 mg/kg bw.
In a study in volunteers with doses of 8, 16, or 24 mg of
malathion per day, the NOAEL was 16 mg per day (equivalent to 0.27
mg/kg bw per day) on the basis of inhibition of plasma and erythrocyte
cholinesterase activity. Several cases of exposure to impure malathion
have been reported, none of which resulted in delayed neuropathy.
An ADI of 0-0.3 mg/kg bw was established on the basis of the
NOAEL of 29 mg/kg bw per day in the two-year study of toxicity and
carcinogenicity in rats, with a safety factor of 100. This ADI is
supported by the NOAEL of 25 mg/kg bw per day in the study of
developmental toxicity in rabbits. The alternative approach of basing
the ADI on the study in humans was not taken, as the study was old and
the material was therefore likely to contain toxic impurities.
Toxicological evaluation
Levels that cause no toxic effect
Mouse: 800 ppm, equal to 140 mg/kg bw per day (18-month study
of toxicity and carcinogenicity)
Rat: 500 ppm, equal to 29 mg/kg bw per day (two-year study
of toxicity and carcinogenicity)
1700 ppm, equal to 130 mg/kg bw per day (study of
reproductive toxicity)
400 mg/kg bw per day (maternal toxicity in a study of
developmental toxicity)
Rabbit: 25 mg/kg bw per day (maternal toxicity in a study of
developmental toxicity)
Dog: 125 mg/kg bw per day (one-year study of toxicity)
Human: 0.3 mg/kg bw per day (47-day study of toxicity)
Estimate of acceptable daily intake for humans
0-0.3 mg/kg bw
Studies that would provide information useful for continued
evaluation of the compound
Further observations in humans
References
Abou Zeid, M.M., El-Barouty, G., Abdel-Reheim, E., Blancato, J., Dary,
C., El-Sebae, A.H. & Saleh, M.A. (1993) Malathion disposition in
dermally and orally treated rats and its impact on the blood serum
acetylcholine esterase and protein profile. J. Environ. Sci.
Health, B28, 413-430.
Toxicological criteria for setting guidance values for dietary and non-dietary exposure to malathion
Human exposure Relevant route , study type, species Results, remarks
Short-term Oral, toxicity, rat LD50 = 1000-11 000 mg/kg bw
(1-7 days) Inhalation, toxicity, rat LC50 > 5.2 mg/L
Dermal irritation, rabbit Mildly irritating
Ocular irritation, rabbit Mildly irritating
Dermal sensitization, guinea-pig Not sensitizing
Medium-term Repeated oral, 90 days, rat NOAEL = 34 mg/kg bw per day: systemic toxicity
(1-26 weeks) Repeated dermal, 21 days, rabbit NOAEL = 300 mg/kg bw per day: decreased brain
acetylcholinesterase activity
Repeated oral, developmental toxicity, rabbit NOAEL = 25 mg/kg bw per day: maternal toxicity;
NOAEL = 100 mg/kg bw per day: fetal toxicity
Repeated oral, reproductive toxicity, rat NOAEL = 600 mg/kg bw per day: no parental toxicity;
NOAEL = 130 mg/kg bw per day: developmental
toxicity
Long-term Repeated oral, 2 years, rat NOAEL = 29 mg/kg bw per day: decreased survival,
(> 1 year) reduced body weight, decreased brain
acetylcholinesterase activity
Abraham, S. & Edery, H. (1976) Rapid spontaneous reactivation of
cholinesterase inhibited by malaoxon. Israel J. Med. Sci., 12, 1524.
Agarwal, R. & Matin, M.A. (1981) Effect of oximes and atropine on the
concentration of cerebral glycogen and blood glucose in
malathion-treated rats. J. Pharm. Pharmacol., 33, 795-796.
Aldridge, W.N., Miles, J.W., Mount, D.L. & Verschoyle, R.D. (1979) The
toxicological properties of impurities in malathion. Arch.
Toxicol., 42, 95-106.
Aldridge, W.N., Dinsdale, D. & Nemery, B. (1985) Some aspect of the
toxicology of trimethyl and triethyl phosphorothioates. Fundam.
Appl. Toxicol., 5, S47-S60.
Baker P & Selvey D (1992) Malathion-induced epidemic hysteria in an
elementary school. Vet. Hum. Toxicol., 34, 156-160.
Baker, E.L., Zack, M. & Miles, J.W. (1978) Epidemic malathion
poisoning in Pakistani malaria workers. Lancet, i, 31-34.
Balaji, M. & Sasikali, K. (1993) Cytogenetic effect of malathion in in
vitro culture of human peripheral blood. Mutat. Res., 301, 13-17.
Balasubramanian, K., Ratnakar, C., Ananthanarayanan, P.H. &
Balasubramanian, A. (1987) Histopathological changes in the testis of
malathion treated albino rats. Med. Sci. Res., 15, 509-510.
Boyd, E.M. & Tanikella, T.K. (1969) The acute oral toxicity of
malathion in relation to dietary protein. Arch. Toxicol., 24,
292-303.
Boyes, W.K., Hunter, E., Gary, C. & Peiffer, R.L. (1997) Topical
exposure of the eyes to the organophosphate insecticide malathion:
Lack of visual effects. Unpublished report. Submitted to WHO by the US
Environmental Protection Agency.
Brodeur, J. & DuBois, K.P. (1967) Studies on factors influencing the
acute toxicity of malathion and malaoxon in rats. Can. J. Physiol.
Pharmacol., 45, 621-631.
Bulsiewicz, H., Rozewicka, L., Januszewska, H. & Bajko, J. (1976)
Aberrations of meiotic chromosomes induced in mice with insecticides.
Folio Morphol. (Warsaw), 35, 361-363.
Byeon, W.-H., Hyun, Z.H.H. & Lee, S.Y. (1976) Salmonella/microsomal
enzyme activation system. Korean J. Microbiol., 14, 128-134.
Chen, H.H., Hsueh, J.L., Sirianni, S.R. & Huang, C.C. (1981) Induction
of sister-chromatid exchanges and cell cyle delay in cultured
mammalian cells treated with eight organophosphorus pesticides.
Mutat. Res., 88, 307-316.
Cheema, A.A., Shakoori, A.R. & Shah, F.H. (1989) Studies on
reactivation of malaoxon-inhibited acetylcholinesterase of goat-brain
by pyridine-2-aldoxime methiodide (PAM-2). Pakistan J. Zool., 21,
247-253.
Cooper, D. & Terrell, Y. (1979a) Acute oral LD50 of Fyfanon 771006 KL
(malathion) in Sprague-Dawley rats. Unpublished report (study No.
9E-4269) from Cannon Laboratories Inc., Reading, Pennsylvania, USA.
Submitted to WHO by Cheminova, Lemvig, Denmark.
Cooper, D. & Terrell, Y. (1979b) Acute oral LD50 of Fyfanon 771006 ST
L 1 AAR (malathion) in Sprague-Dawley rats. Unpublished report (study
No. 9E-4268) from Cannon Laboratories Inc., Reading, Pennsylvania,
USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Cyanamid (1990) Letter from F.G. Hess on behalf of the malathion
re-regulation task force. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Daly, I.W. (1993a) A 28-day study of malathion in the rat via dietary
administration. Unpublished report (study No. 92-3806) from
Bio/dynamics Inc, East Millstone, New Jersey, USA. Submitted to WHO by
Cheminova, Lemvig, Denmark.
Daly, I.W. (1993b) A subchronic (3-month) oral toxicity study of
malathion in the rat via dietary administration. Unpublished report
(study No. 92-3843) from Bio/dynamics Inc., East Millstone, New
Jersey, USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Daly, I.W. (1996a) A 24-month oral toxicity/oncogenicity study of
malathion in the rat via dietary administration. Unpublished report
(study No. 90-3641) from Huntingdon Life Sciences, East Millstone, New
Jersey, USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Daly, I.W. (1996b) A 24-month oral toxicity/oncogenicity study of
malaoxon in the rat via dietary administration. Unpublished report
(study No. 93-2234) from Huntingdon Life Sciences, East Millstone, New
Jersey, USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Dauterman, W.C. & Main, A.R. (1966) Relationship between acute
toxicity and in vitro inhibition and hydrolases in a series of
carbaloxy homologs. Toxicol. Appl. Pharmacol., 9, 408-418.
Degraeve, N., Gilot-Delhalle, J., Moutschen, J., Moutschen-Dahmen, M.,
Colizzi, A., Chollet, M. & Houbrechts, N. (1980) Comparison of the
mutagenic activity of organophosphorus insecticides in mouse and in
the yeast Saccharomyes pombe. Mutat. Res., 74, 201-202.
Dinsdale, D. (1992) Pulmonary toxicity of anticholinesterases. In:
Ballantyne, B. & Marrs, T.C., eds, Clinical and Experimental
Toxicology of Organophosphates and Carbamates, Oxford:
Butterworth-Heinemann, pp. 156-166.
Dive, A., Mahieu, P., van Binst, R., Hassoun, A., Lison, D., de
Bisschop, H., Nemery, B. & Lauwerys, R. (1994) Unusual manifestations
after malathion poisoning. Hum. Exp. Toxicol., 13, 271-274.
Dzwonkowska, A. & Hbner, H. (1986) Induction of chromosomal
aberrations in the Syrian hamster by insecticides tested in vivo.
Arch. Toxicol., 58, 152-156.
Ehrich, M., Correll, L. & Veronesi, B. (1994) Neuropathy target
esterase inhibition by organophosphorus esters in human neuroblastoma
cells. Neurotoxicology, 15, 309-314.
Ehrich, M., Jortner, B.S. & Padilla, S. (1995) Comparison of the
relative inhibition of acetylcholinestearse and neuropathy target
esterase in rats and hens given cholinesterase inhibitors. Fundam.
Appl. Toxicol., 24, 94-101.
Ehrich, M., Correll, L. & Veronesi, B. (1997) Acetylcholinesterase and
neuropathy target esterase inhibition in neuroblastoma cells to
distinguish organophosphorus compounds causing acute and delayed
neurotoxicity. Fundam. Appl. Toxicol., 38, 55-63
FAO (1986) International Code of Conduct on the Distribution and
Use of Pesticides, Rome: Food and Agricultural Organization of the
United Nations.
Fischer, J.E. (1988) AC 6,601: A 28-day oral toxicity study in beagle
dogs. Unpublished report (study No. AX88-3) from American Cyanamid
Co., Princeton, New Jersey, USA. Submitted to WHO by Cheminova,
Lemvig, Denmark.
Fischer, J.E. (1991) Oral LD50 study in albino rats with AC 6,601
malathion technical (Cheminova production batch). Unpublished report
No. A91-205. Submitted to WHO by Cheminova, Lemvig, Denmark.
Fletcher, D.W. (1989) 42-Day neurotoxicity study with AC 6,601
technical in mature white Leghorn hens. Unpublished report (study No.
BLAL 87 DN 109) from Bio-Life Associates Ltd, Neillsville, Wisconsin,
USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Foureman, P., Mason, J.M., Valencia, R. & Zimmering, S. (1994)
Chemical mutagenesis testing in Drosophila. X. Results of 70 coded
chemicals tested for the National Toxicology Program. Environ.
Mol. Mutag., 23, 208-227.
Frawley, J.P., Fuyat, H.N., Hagan, E.C., Blake, J.R. & Fitzhugh, O.G.
(1957) Marked potentiation of mammalian toxicity from simultaneous
administration of two anticholinesterase compounds. J. Pharmacol.
Exp. Ther., 121, 96-106.
Gaines, T.B. (1960) The acute toxicity of pesticides in rats.
Toxicol. Appl. Pharmacol., 2, 88-89.
Ganendran, A. & Balabaskaran, B. (1976) Reactivation studies on
organophosphate inhibited human cholinesterases by pralidoxime
(P-2-AM). S.E. Asian J. Trop. Med. Public Health, 7, 417-423.
Garcia-Repetto, R., Martinez, D. & Repetto, M. (1995) Malathion and
dichlorvos toxicokinetics after oral administration of malathion and
trichlorfon. Vet. Hum. Toxicol., 37, 306-309.
Garry, V.F., Nelson, R.L., Griffith, J. & Harkins, M. (1990)
Preparation for human study of pesticide applicators: Sister chromatid
exchanges and chromosome aberrations in cultured human lymphocytes
exposed to selected fumigants. Teratog. Carcinog. Mutag., 10, 21-29.
Gilot-Delhalle, J., Colizzi, A., Moutschen, J. & Moutschen-Dahmen, M.
(1983) Mutagenicity of some organophosphorus compounds at the ade6
locus of Schizosaccharomyces pombe. Mutat. Res., 117, 139-148.
Gudi, R. (1990) Acute test for chemical induction of chromosome
aberration in rat bone marrow cells in vivo with AC 6,601. Unpublished
report (study No. 0125-1531) from Sitek Research Laboratories,
Rockville, Maryland, USA. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Guiti, N. & Sadeghi, D. (1969) Acute toxicity of malathion in the
mongrel dog. Toxicol. Appl. Pharmacol., 15, 244-245.
Gupta, R.C. (1984) Acute malathion toxicosis and related enzymatic
alterations in Bubalus bubalis: Antidotal treatment with atropine,
2-PAM, and diazepam. J. Toxicol. Environ. Health, 14, 291-303.
Hazleton, L.W. & Holland, E.G. (1953) Toxicity of malathion.
Ind. Hyg. Occup. Med., 8, 399-405.
Hobbiger, F. (1973) Reactivation of phosphorylated
acetylcholinesterase. In: Eichler, O., Farah, A. & Koelle, G., eds,
Handbuch der Experimentelle Toxikologie, Berlin: Springer, pp.
921-988.
Huff, J.E., Bates, R., Eustis, S.L., Haseman, J.K. & McConnell, E.E.
(1985) Malathion and malaoxon: Histopathology reexamination of the
National Cancer Institutes carcinogenesis studies. Environ. Res.,
37, 154-173.
IARC (1983) IARC Monographs on the Evaluation of the Carcinogenic
Risk of Chemicals to Humans, Vol. 30, Miscellaneous Pesticides.
Lyon, pp. 102-129.
Imamura, T. & Talcott, R.E. (1985) Mutagenic and alkylating activities
of organophosphate impurities of commercial malathion. Mutat. Res.,
155, 1-6.
Imlay, P., Parke, G. StE. & Charles, S.J. (1978) The acute dermal LD50
of fyfanon on New Zealand albino rabbits. Unpublished report
(Laboratory No. 7E-7851) from Cannon Laboratories Inc., Reading,
Pennsylvania, USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Ishidate, M., Sofuni, T. & Yoshikawa, K. (1981) Chromsomal aberration
test in vitro as a primary screening tool for environmental mutagens
carcinogens. Gann Monogr. Cancer Res., 27, 95-108.
Ivett, J.L., Brown, B.M., Rodgers, C., Anderson, B.E., Resnick, M.A. &
Zeiger, E. (1989) Chromosomal aberrations and sister chromatid
exchange tests in Chinese hamster ovary cells in vitro. IV. Results
with 15 chemicals. Environ. Mol. Mutag., 14, 165-187.
Jackson, G.C., Hardy, C.J., Gopinath, C. & Lewis, D.J. (1986) Fyfanon
(malathion) 96/98% technical acute inhalation toxicity to rats 4-hour
exposure. Unpublished report (study No. CHV 28/8640) from Huntingdon
Research Centre Ltd, Alconbury, Huntingdon, United Kingdom. Submitted
to WHO by Cheminova, Lemvig, Denmark.
Jokanovic, M. & Maksimovic, M. (1995) A comparison of trimedoxime,
obidoxime, pralidoxime and HI-6 in the treatment of oral
organophosphorus insecticide poisoning in the rat. Arch. Toxicol.,
70, 119-123.
Kalow, W. & Martin, A. (1961) Second generation toxicity of malathion
in rats. Nature, 192, 464-465.
Keadtisuke, S., Dheranetra; W., Nakatsugawa, T. & Fukuto, T.R. (1990)
Liver damage induced in rats by malathion impurities. Toxicol.
Lett., 52, 35-46.
Khera, K.S., Whalen, C. & Trivett, G. (1978) Teratogenicity studies on
linuron, malathion, and methyoxychlor in rats. Toxicol. Appl.
Pharmacol., 45, 435-444.
Kimbrough, R.D. & Gaines, T.B. (1968) Effect of organic phosphorus
compounds and alkylating agents on the rat fetus. Arch. Environ.
Health, 16, 805-808.
Kuhn, J.O. (1996) Fyfanon purified. Final report acute oral toxicity
study in rats. Unpublished report (study No. 2586-95) from Stillmeadow
Inc., Sugar Land, Texas, USA. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Kynoch, S.R. (1985a) Acute oral toxicity to rats of malathion
(Fyfanon) technical. Unpublished report (study No. 85134D/CHV 33/AC)
from Huntingdon Research Centre Ltd, Alconbury, Huntingdon, United
Kingdom. Submitted to WHO by Cheminova, Lemvig, Denmark.
Kynoch, S.R. (1985b) Acute dermal toxicity to rats of malathion
(Fyfanon) technical. Unpublished report (study No. 851330D/CHV 33/AC)
from Huntingdon Research Centre Ltd, Alconbury, Huntingdon, United
Kingdom. Submitted to WHO by Cheminova, Lemvig, Denmark.
Kynoch, S.R. & Smith, P.A. (1985) Delayed contact hypersensitivity in
the guinea-pig with malathion (Fyfanon) technical. Unpublished report
(study No. 8666D/CHV 37/SS,) from Huntingdon Research Centre Ltd,
Alconbury, Huntingdon, United Kingdom. Submitted to WHO by Cheminova,
Lemvig, Denmark.
Lamb, I.C. (1994a) An acute neurotoxicity study of malathion in rats.
Unpublished report (study No. WIL-206005) from WIL Research
Laboratories Inc,. Ashland, USA. Submitted to WHO by Cheminova,
Lemvig, Denmark.
Lamb, I.C. (1994b) A subchronic (13-week) neurotoxicity study of
malathion in rats. Unpublished report (study No. WIL-206006) from WIL
Research Laboratories Inc., Ashland, USA. Submitted to WHO by
Cheminova, Lemvig, Denmark.
Lechener, A. & Abdel-Rahman, Y. (1985) Alterations in rat liver
microsomal enzymes following exposure to carbaryl and malathion in
combination. Arch. Environ. Contam. Toxicol., 14, 451-457.
Liggett, M.P. & Parcell, B.I. (1985a) Irritant effects on rabbit skin
of malathion (Fyfanon) technical. Unpublished report (study No.
851221D/CHV 33/SE) from Huntingdon Research Centre Ltd, Alconbury,
Huntingdon, United Kingdom. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Liggett, M.P. & Parcell, B.I. (1985b) Irritant effects on the rabbit
eye of malathion (Fyfanon) technical. Unpublished report (study No.
851214D/CHV 36/SE) from Huntingdon Research Centre Ltd, Alconbury,
Huntingdon, United Kingdom. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Lochry, E.A. (1989) A developmental toxicity study with AC 6,601 in
rats. Unpublished report (study No. 101-005) from Argus Research
Laboratories Inc., Perkasie, Pennsylvania, USA. Submitted to WHO by
Cheminova, Lemvig, Denmark.
McCann, J., Choi, E., Yamasaki, E. & Ames, B.N. (1975) Detection of
carcinogens as mutagens in the Salmonella/microsome test: Assay of 300
chemicals. Proc. Natl Acad. Sci. USA, 72, 5735- 5739.
Machin, M.G.A. & McBride, W.G. (1989) Teratology study of malathion in
the rabbit. J. Toxicol. Environ. Health, 26, 249-253.
Matsushita, A., Aoyama, K., Yoshimi, K., Fujita, Y. & Ueda, A. (1985)
Allergic contact dermatitis from organophosphorus insecticides.
Ind. Health, 23, 145-153.
Miles, J.W., Mount, D.L. & Staiger, M.A. (1979) S-Methyl isomer
content of stored malathion and fenitrothion water disbursable powders
and its relation to toxicity. J. Agric. Food Chem., 27, 421-425.
Menzer, R.E. & Best, N.H. (1968) Effect of phenobarbital on the
toxicity of several organophosphorus insecticides. Toxicol. Appl.
Pharmacol., 13, 37-42.
Moeller, H.C. & Rider, J.A. (1962) Plasma and red blood cell
cholinesterase activity as indications of the threshold of incipient
toxicity of ethyl-p-nitrophenyl thionobenzenephosphonate (EPN) and
malathion in human beings. Toxicol. Appl. Pharmacol., 4, 123-130.
Mohn, G. (1973) 5-Methyltryptophan resistance mutations in
Escherichia coli K-12. Mutat. Res., 20, 7-15.
Moreno, O.M. (1989) 21-Day dermal toxicity study with AC 6,601 in
rabbits. Unpublished report (study No. MRID 41054201) from MB Research
Laboratories, Inc., Spinnerstown, Pennsylvania, USA. Submitted to WHO
by Cheminova, Lemvig, Denmark.
Morgade, C. & Barquet, A. (1982) Body distribution of malathion and
its metabolites in a fatal poisoning by ingestion. J. Toxicol.
Environ. Health, 10, 321-325.
Moriya, M., Ohta, T., Watanabe, K., Miyazawa, T., Kato, K. & Shirasu,
Y. (1983) Further mutagenicity studies on pesticides in bacterial
reversion assay systems. Mutat. Res., 116, 185-216.
Murphy, S.D. & Cheever, K.L. (1968) Effects of feeding insecticides:
Inhibition of carboxyesterase and cholinesterase activities in rats.
Arch. Environ. Health, 17, 749-758.
Murphy, S.D., Anderson, R.L. & DuBois, K.P. (1959) Potentiation of
toxicity of malathion by triorthotolyl phosphate. Proc. Soc. Exp.
Biol. Med., 100, 483-487.
Myhr, B.C. & Caspary, W.J. (1991) Chemical mutagenesis at the
thymidine kinase locus in L5178Y mouse lymphoma cells: Results for 31
coded compounds in the National Toxicology Program. Environ. Mol.
Mutag., 18, 51-83.
Nagy, Z., Mile, I. & Antoni, F. (1975) The mutagenic effect of
pesticides on Escherischia coli WP2 try-. Acta Microbiol. Acad.
Sci. Hung., 22, 309-314.
Nicholas, A.H., Vienne, M. & van den Berghe, H. (1979) Induction of
sister-chromatid exchanges in cultured human cells by an
organophosphorus insecticide: Malathion. Mutat. Res., 67, 167-172.
Nishio, A. & Uyeki, E.M. (1981) Induction of sister chromatid
exchanges in Chinese hamster ovary cells by organophosphate
insecticides and their oxygen analogs. J. Toxicol. Environ. Health,
8, 939-946.
Ohtaka, K., Hamade, N., Yamazaki, Y., Suzuki, M. & Koizumi, A. (1995)
A direct involvement of the central nervous system in hypophagia and
inhibition of respiratory rate in rats after treatment with
O,O,S-trimethyl phosphorothioate. Arch. Toxicol., 69, 555-564.
Pant, K.J. (1989) Test for chemical induction of unscheduled DNA
synthesis in rat primary hepatocyte cultures by autoradiography with
AC 6,601. Unpublished report (study No. 0125-5100) from Sitek Research
Laboratories, Rockville, Maryland, USA. Submitted to WHO by Cheminova,
Lemvig, Denmark.
Pednekar, M.D., Ghandi, S.R. & Netrawali, M.S. (1987) Evaluation of
mutagenic activities of endosulfan, phosalone, malathion, and
permethrin, before and after metabolic activation in the Ames
Salmonella test. Bull. Environ. Contam. Toxicol., 38, 925-933.
Pellegrini, G. & Santi, R. (1972) Potentiation of toxicity of
organophosphorus compounds containing carboxylic ester functions
toward warm-blooded animals by some organophosphorus impurities.
J. Agric. Food Chem., 20, 944-950.
Reddy, V., Freeman, T. & Cannon, M. (1989) Disposition and metabolism
of 14C-labeled malathion in rats (preliminary and definitive study).
Unpublished report (study No. 9354-B) from Midwest Research Institute,
Kansas City, Missouri, USA. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Rodgers, K. & Xiong, S. (1997) Effect of administration of malathion
for 14 days on macrophage function and mast cell degranulation.
Fundam. Appl. Toxicol., 37, 95-99.
Rucci, G., Becci, P.J. & Parent, R.A. (1980) The evaluation of the
chronic toxicity effects of Cythion administered in the diet to
Sprague-Dawley rats for 24 consecutive months. Unpublished report
(Project No. 5436) from Food and Drug Research Laboratories Inc.,
Waverly, New York, USA. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Rueber, M.D. (1985) Carcinogenicity and toxicity of malathion and
malaoxon. Environ. Res., 37, 119-153.
Ryan, D.L. & Fukuto, T.R. (1984) The effect of isomalathion and
O,S,S-trimethyl phosphorothoate on the in vivo metabolism of malathion
in rats. Pestic. Biochem. Physiol., 21, 349-357.
Salvadori, D.M.F., Ribeiro, L.R., Pereira, C.A.B. & Becak, W. (1988)
Cytogenetic effects of malathion insecticide on somatic and germ cells
of mice. Mutat. Res., 204, 283-287.
Schellenberger, T.E. & Billups, L.H. (1987) One-year oral toxicity
study in purebred beagle dogs with AC 6,601. Unpublished report (study
No. 85010) from Tegeris Laboratories Inc., Laurel, Maryland, USA.
Submitted to WHO by Cheminova, Lemvig, Denmark.
Schanker, H.M., Rachelefsky, G., Siegel, S., Katz, R., Spector, S.,
Rohr, A., Rodriquiz, C., Woloshin, K. & Papanek, P.J. (1992) Immediate
and delayed type hypersensitivity to malathion. Ann. Allergy, 69,
526-528.
Schroeder, R.E. (1990) A two-generation (two litters) reproduction
study with AC 6,601 to rats. Unpublished report (study No. 87-3243)
from Bio/dynamics Inc, East Millstone, New Jersey, USA. Submitted to
WHO by Cheminova, Lemvig, Denmark.
Seely, J.C. (1991) Histopathologic evaluation: The evaluation of the
chronic effects of AC 6,601 administered in the diet to Sprague Dawley
rats for 24 consecutive months. Unpublished report (study No. 90-78)
from Pathco Inc., Research Triangle Park, North Carolina, USA.
Submitted to WHO by Cheminova, Lemvig, Denmark.
Segal, L.M. & Fedoroff, S. (1989) The acute and subchronic effects of
organophosphorus and carbamate pesticides on cholinesterase activity
in aggregate cultures of neural cells from the foetal rat brain.
Toxicol. in vitro, 3, 111-122.
Shiau, S.Y., Huff, R.A., Wells, B.C. & Felkner, I.C. (1980)
Mutagenicity and DNA-damaging activity for several pesticides tested
with Bacillus subtililis mutants. Mutat. Res., 71, 169-179.
Shirasu, Y., Moriya, M., Kato, K., Furuhashi, A. & Kada, T. (1976)
Mutagenicity screening of pesticides in the microbial system.
Mutat. Res., 40, 19-30.
Siglin, J.C. (1985) A teratology study with AC 6,601 in rabbits.
Unpublished report No. 8171 from Food and Drug Research Laboratories,
Inc., Waverly, New York, USA. Submitted to WHO by Cheminova, Lemvig,
Denmark.
Slauter, R.W. (1994) 18-Month oral (dietary) oncogenicity study in
mice, test substance malathion. Unpublished report (study No. 668-001)
from International Research and Development Corp, Mattawan, Michigan,
USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Terrell, Y., Parke, G.StE. & Charles, S.J. (1978) Acute oral LD50 in
rats, compound: malathion technical (Fyfanon). Unpublished report
(Laboratory No. 7E-7850) from Cannon Laboratories, Inc., Reading,
Pennsylvania, USA. Submitted to WHO by Cheminova, Lemvig, Denmark.
Thomas, D., Goldharbour, M., Petitti, D., Swan, S.H., Rapperport, E. &
Hertz-Picciotto, I. (1990) Reproductive outcomes in women exposed to
malathion. Am. J. Epidemiol., 132, 794-795.
Toia, R.F., March, R.B., Umetsu, N., Mallipudi, N.M., Allahyari, R. &
Fukuto, T.R. (1980) Identification and toxicological evaluation of
impurities in technical malathion and fenthion. J. Agric. Food
Chem., 28, 605-609.
Traul, K.A. (1987) Evaluation of CL 6601 in the bacterial/microsome
mutagenicity test. Unpublished report (study No. 114) from American
Cyanamid Company, Princeton, New Jersey, USA. Submitted to WHO by
Cheminova, Lemvig, Denmark.
US Environmental Protection Agency (1987) Letter from D.S. Goldman to
R. Linkfield, American Cyanamid Co. Submitted to WHO by Cheminova,
Lemvig, Denmark.
US National Cancer Institute (1978) Bioassay of malathion for possible
carcinogenicity (NCI Technical Report 24), Department of Health,
Education, and Welfare publication (NIH) 78 824. Bethesda, Maryland:
US Department of Health, Education, and Welfare, Public Health
Service.
US National Cancer Institute (1979a) Bioassay of malathion for
possible carcinogenicity (NCI Technical Report 192), Department of
Health, Education, and Welfare publication (NIH) 79 1748. Bethesda,
Maryland: US Department of Health, Education, and Welfare, Public
Health Service.
US National Cancer Institute (1979b) Bioassay of malaoxon for possible
carcinogenicity (NCI Technical Report 135), Department of Health,
Education, and Welfare publication (NIH) 79 1390. Bethesda, Maryland:
US Department of Health, Education, and Welfare, Public Health
Service.
Velazquez, A., Creus, A., Xamena, N. & Marcos, R. (1987) Lack of
mutagenicity of the organophosphorus insecticide malathion in
Drosophila melanogaster. Environ. Mutag., 9, 343-348.
Ward, T.R., Ferris, D.J., Tilson, H.A. & Mundy, W.R. (1993)
Correlation of the anticholinesterase activity of a series of
organophosphates with their ability to compete with agonist binding to
muscarinic receptors. Toxicol. Appl. Toxicol., 122, 300-307.
Waters, M.D., Sandhu, S.S., Simmon, V.F., Mortelmans, K.E., Mitchell,
A.D., Jorgenson, T.A., Jones, D.C.L., Valencia, R. & Garrett, N.E.
(1982) Study of pesticide genotoxicity. Basic Life Sci., 21,
275-326.
WHO (1985) Specifications for Pesticides Used in Public Health, 6th
Ed., Geneva.
WHO (1986) Organophosphorus Insecticides: A General Introduction
(Environmental Health Criteria 63), Geneva.
WHO (1996) The WHO Recommended Classification of Pesticides by
Hazard and Guidelines to Classification 1996-1997 (WHO/PCS/96.3),
Geneva, International Programme on Chemical Safety.
Wong, P.K., Wai, C.C. & Liong, E. (1989) Comparative study on
mutagenicities of organophosphorus insecticides in Salmonella.
Chemosphere, 18, 2413-2422.
Zeiger, E., Anderson, B., Haworth, S., Lawlor, T. & Mortelmans, K.
(1988) Salmonella mutagenicity tests: IV. Results from the testing of
300 chemicals. Environ. Mol. Mutag., 11 (Suppl 12), 1-158.